Endothelial nitric oxide synthase in red blood cells: Key to a new erythrocrine function? (original) (raw)
Abstract
Red blood cells (RBC) have been considered almost exclusively as a transporter of metabolic gases and nutrients for the tissues. It is an accepted dogma that RBCs take up and inactivate endothelium-derived NO via rapid reaction with oxyhemoglobin to form methemoglobin and nitrate, thereby limiting NO available for vasodilatation. Yet it has also been shown that RBCs not only act as “NO sinks”, but exert an erythrocrine function – i.e an endocrine function of RBC – by synthesizing, transporting and releasing NO metabolic products and ATP, thereby potentially controlling systemic NO bioavailability and vascular tone. Recent work from our and others laboratory demonstrated that human RBCs carry an active type 3, endothelial NO synthase (eNOS), constitutively producing NO under normoxic conditions, the activity of which is compromised in patients with coronary artery disease. In this review we aim to discuss the potential role of red cell eNOS in RBC signaling and function, and to critically revise evidence to this date showing a role of non-endothelial circulating eNOS in cardiovascular pathophysiology.
Keywords: Red blood cells, Cardiovascular disease, Nitric oxide, eNOS
Graphical abstract
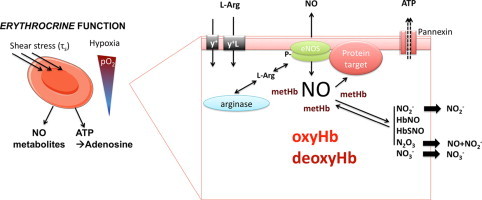
Highlights
- •
We define erythrocrine function the ability of RBC to secret signaling entities or transmitter momlecules. - •
Erythocrine function include scavenging, transporting and producing NO metabolites and ATP. - •
There is in vitro and in vivo evidence of a role of red cell eNOS in signaling and erythrocrine function. - •
Red cell eNOS and erythocrine function might be involved in organ protection. - •
Further studies should address the role of red cell eNOS/RBC signaling in cardiovascular health.
Nitric oxide (NO) is a short-lived signaling/regulatory product of the endothelium that is critically important for vascular health [1]. The first biological activity attributed to NO was the ability to induce relaxation of preconstricted aortic vascular preparations, and for as long as its chemical nature remained unknown it was defined as “endothelium-derived relaxing factor” (EDRF)[2], [3], [4]. NO is now recognized to play a fundamental role in the control of vascular tone and blood flow [5] and to represent a central signaling entity in the cardiovascular system [6]; moreover, it is also a critical player in neurotransmission [7], host defense and inflammatory processes [8].
NO is enzymatically produced by NO synthases (NOS, EC 1.14.13.39), heme-containing proteins catalyzing the five-electron oxidation of the guanidino nitrogen of l-arginine to NO and citrulline. This process requires oxygen and a number of cofactors including calcium, calmodulin, NADPH, flavin mononucleotide (FMN), flavin adenine dinucleotide (FAD), and tetrahydrobiopterin [6], [9]. The proposed reaction mechanism involves electron transfer from the flavin-bindings site via calmodulin to the heme group, where the oxidation of one of the guanidino nitrogens of l-arginine to the intermediate product, N-hydroxy-l-arginine takes place [10]. Tetrahydrobiopterin appears to be important in maintaining the enzyme in its active dimeric form [9], [10].
In mammals there are three distinct isoforms of NOS, encoded by three different genes. The human loci are defined as NOS1 (Gene ID 4842) coding for the neuronal nitric oxide synthase (nNOS), NOS2 (Gene ID 4843) for the inducible nitric oxide synthase (iNOS) and NOS3 (Gene ID: 4846) for endothelial nitric oxide synthase (eNOS). Whereas iNOS typically produces high-output NO levels and participates in host defense, inflammatory stress and airway epithelial NO formation, the constitutively expressed isoforms, nNOS and eNOS, are lower NO output systems that are important for physiological processes such as neuronal signaling, inhibition of the hemostatic system, vasodilation and blood pressure control [9], [10].
The constitutively expressed enzymes eNOS and nNOS are activated after stimulation of specific receptors by various agonists (e.g., acetycholine, bradykinin, serotonin, adenosine, ADP:ATP, histamine, and thrombin), with consecutive increase of intracellular free Ca2+. The binding of Ca2+ to the Ca2+/calmodulin subunit of these proteins activates the enzyme to produce NO. In vascular tissue, the binding of NO to the Fe2+-heme of soluble guanylyl cyclase (sGC) activates this enzyme to produce cyclic guanosine-3-5-monophosphate (cGMP) from guanosine-5-triphosphate (GTP). The signal is then transduced to downstream elements of the signaling cascade, including cGMP-dependent protein kinases, cGMP-gated cation channels, and cGMP-regulated phosphodiesterase [11].
In healthy blood vessels, eNOS-derived NO contributes to the regulation of blood flow and blood pressure, is an inhibitor of platelet activation and aggregation as well as leukocyte adhesion and migration [12]. Furthermore, endothelial eNOS appears to contribute to the formation of bioactive circulating NO metabolites [13], [14], [15], [16], [17] (Fig. 1) that participate in important endocrine activities such as hypoxic vasodilation, blood pressure regulation, gene expression and cytoprotection following myocardial infarction [18], [19], [20], [21], [22], [23], [24], [25], [26], [27]. Mice genetically deficient in endothelial nitric oxide synthase (eNOS-/-) are hypertensive and have lower circulating nitrite levels, demonstrating the importance of constitutively produced NO to blood pressure regulation and vascular homeostasis [14], [28], [29], [30].
Fig. 1.
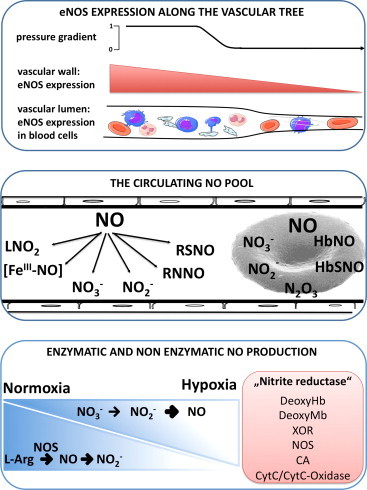
Upper panel: The expression of eNOS in the endothelium of the vessels decreases along the vascular tree, while blood cell eNOS carried by all main blood populations is equally distributed. Middle panel: In the blood compartment a circulating pool of NO metabolites is formed and transported in plasma and RBC. Bottom panel: NO can be synthesized enzymatically from L-arginine under normoxic conditions in a reaction catalyzed by a NOS, and under hypoxic conditions from nitrite by the nitrate reductase activity of proteins including deoxyhemoglobin (deoxyHb), deoxymyoglobin (deoxyMb), xantine oxidoreductase (XOR), carbonic anydrase (CA), and cytochrome C (cytC)/cytC-oxidase. eNOS, endothelial nitric oxide synthase; LNO2, nitrated lipids; RSNO, nitrosothiols; RNNO, nitrosamines.
Conventional wisdom holds that the pleiotropic effects of eNOS are primarily determined by the enzyme located in the endothelium. In addition to endothelial cells, most circulating blood cells – including all main leukocyte subpopulations [31], [32], [33], [34], [35], platelet-rich plasma [36], and circulating blood microparticles [37], but not purified platelets [38], [39] also carries eNOS transcript and/or protein. Recent work from our laboratory demonstrated that RBCs also carry a functional eNOS [32], [40]. Production of NO by hemoglobin-containing RBCs under normoxic conditions by red cell eNOS, the expression of which is decreased in cardiovascular disease [32], [41], appears paradoxical in view of earlier reports suggesting a major role of these blood cells in NO inactivation, inviting a major rethink.
In this review we will discuss the potential role of red cell eNOS in RBC signaling and function and summarize the evidence to date that suggests a role for non-endothelial circulating eNOS in cardiovascular pathophysiology.
Red blood cells in control of the vascular NO pool
It is an accepted dogma that RBCs take up and inactivate endothelium-derived NO via rapid reaction with oxyhemoglobin (oxyHb, FeIIO2Hb) to form methemoglobin (metHb, FeIIIHb) and nitrate, thereby limiting NO available for vasodilatation.
| FeIIO2Hb+NO→FeIIIHb+NO3− | (1) |
|---|
Yet it has also been shown that RBCs not only act as “NO sinks” but synthesize, store, and transport NO metabolic products (Fig. 1), and that hemoglobin plays a central role in these biochemical processes [42]. Under hypoxic conditions in particular, it has been demonstrated that RBCs induce NO-dependent vasorelaxation {Cosby, 2003 #411;Webb, 2008 #448;Crawford, 2006 #518}. Mechanisms of release and potential sources of NO in RBCs are still a matter of debate, but candidates include iron–nitrosyl–hemoglobin [44], S-nitrosohemoglobin [15], [45], [46], and nitrite [47], [48]. The latter may form NO either via deoxyhemoglobin (deoxyHb, FeIIHb) [22], [49] or xanthine oxidoreductase (XOR)-mediated reduction [43], or via spontaneous [50] and carbonic anhydrase-facilitated disproportionation [51]. Most of these processes show a clear oxygen-dependence, and several are favored by low oxygen tensions. The relative contribution of either mechanism to NO formation varies with oxygen partial pressure along the vascular tree [52], [53].
In addition, RBCs are thought to contribute to the regulation of systemic NO bioavailability by releasing ATP when subjected to hypoxia or shear stress, which seems to be dependent on the activation of erythrocytic pannexin-1 channels [54], [55], [56] inducing eNOS-dependent vasorelaxation and an increase in blood flow [57], [58], [59], [60], [61].
The history of red cell eNOS
The presence of a NOS-like activity in RBCs had been a matter of controversy for some time, and doubts about its functional significance, isoform identity and disease relevance had been put forward by different authors.
Immunostaining of human, mouse, and rat RBCs or of RBC membranes (“ghosts”) indicated that RBCs carry a protein containing epitopes of a NOS3 [62], [63], [64], [65]. In some reports, a positive staining with anti-iNOS antibodies [64], [65] was detected, while others suggested that RBCs might express a novel NOS isoform [62], [64]. Enrichment of the protein from human and murine lysates by cross-linking of specific antibody anti-eNOS to magnetic beads allowed us to confirm by mass spectrometry the chemical identity of the protein as NOS3 isoform 1, i.e. the same protein that is expressed in endothelial cells [32]. Further confirmation of identity was the absence of the protein in RBCs obtained from eNOS-/- mice [66] and the Ca2+/calmodulin dependent conversion of l-arginine to citrulline by the isolated protein [32] and RBC membrane preparations [66]. Thus, from these studies it is evident that both human and mouse RBCs carry a catalytically active eNOS.
Doubts about the functional significance of NOS activity in RBCs are mainly linked to the fact that RBCs carry high concentrations of oxyHb, corresponding to a concentration of 10 mM heme [42]. Considering that oxygenated hemoglobin is an highly efficient NO scavenger one might predict that eNOS-derived NO produced in the red cell must be scavenged as soon as it is produced; according to this logic, red cell eNOS should only have a limited, if any functional significance in adult RBCs. Yet, this is not what one finds “in real life”. NOS activity in RBCs has been studied under normoxic conditions by measuring the formation of NO and NO metabolites (nitrite, nitrate and RXNO) in the supernatant of whole RBCs [40], [63], [67], [68] and by measuring the conversion of l-3H-arginine to l-3H-citrulline in the presence of cofactors, i.e. NADPH, THB, Ca2+/Calmodulin [69], [70]. Addition of NOS inhibitors decreased accumulation of NO metabolites [40], [63], [67] and citrulline production [69], [70], while increasing arginine availability by inhibition of arginase increased release of nitrate [68]. However, two reports failed to measure citrulline formation or NO metabolite accumulation from l-arginine in RBC lysates [65] or changes in 15N/14N nitrite or nitrate ratio [71]. Nevertheless, another report confirmed that human RBC converts l-15N-arginine in l-15N-citrulline using liquid-chromatography tandem mass spectrometry [41].
One wonders why there are so many discrepant data sets regarding the presence and activity of an (e)NOS in RBCs. Some discrepancy may be due to methodological issues encountered in performing experiments with RBCs, but in some cases the discrepancy rests with differences in interpretation rather than experimental results. The main methodological issues encountered in working with RBCs comprise (but are not limited to) their fragility as compared to other cells, accompanied by their dependence on the presence of glucose as a sole source for intracellular ATP, and the presence of overwhelming concentrations of hemoglobin. The latter can pose a real challenge for biochemical or enzymatic assays. Preparation of cell lysates, or membrane preparations, defined as “ghosts”, to get around this problem might contribute to loss of cellular structures, critical protein–protein interaction and cofactors important for catalytic activity [72]. Measuring changes in 15N/14N-nitrite or 15N/14N-nitrate ratios after incubation with 15N l-arginine [71] in the presence of high concentrations of hemoglobin may not necessarily reflect true NOS activity because the chemistry of formation of nitrite and nitrate from NO may be dependent on other competing reactions involving NO/NO metabolites and oxy/deoxy/methemoglobin (metHb, FeIIIHb) occurring, which could conceivably affect not only NO but also nitrite and nitrate levels. These considerations include, but are not limited to the oxyHb-dependent conversion of NO and nitrite to nitrate [42], the presence of ceruloplasmin [73] and/or other plasma proteins converting NO into nitrite, and the enzymatic conversion of nitrite back to NO (for example catalyzed by XOR [43]). Another factor to consider is the presence of arginase in RBCs [65], [68], competing for the NOS substrate l-arginine. Kang et al. claimed that whole RBCs do not convert 3H l-arginine into citrulline but rather into ornitine, and, therefore, suggested that Metha et al. did not measure NOS activity, but arginase activity instead [65]. Metha et al. responded by saying that Kang et al. used RBC lysates instead of whole cells, thus loosing structural compartmentalized integrity of the cell [72]. More recent studies showed that increase in l-arginine availability by inhibition of arginase increase nitrate levels in the supernatant of whole blood or RBC preparations [40], [68], and that NOS inhibition on top of arginase inhibition decrease it [68], indicating a cross talk between the two enzymes.
Using flow cytometry in combination with the NO-imaging probe DAF-FM DA [74] we find that all blood cells form intracellular NO, with a rank order of monocytes>neutrophils>lymphocytes>RBCs>platelets [32]. Constitutive normoxic NO formation was abolished by NOS inhibition and intracellular NO scavenging, confirmed by laser-scanning microscopy and unequivocally validated by detection of the DAF-FM reaction product with NO using HPLC and LC-MS/MS [32], [74]. We found that isolated immunoprecipitated red cell eNOS is capable of producing 9.82 fmol/min l-citrulline under optimal substrate/cofactor supply conditions and, interpolating the areas of the DAF-FM-T related peaks observed in DAF-FM-DA loaded RBCs, we estimated that the average concentration of DAF-FM-T in these samples was 64±12 nM (_n_=19). Assuming the reaction stoichiometry to be 1:1, we therefore estimate that the amounts of total NO and/or nitrosating equivalents produced by a single RBC within 30 min at RT correspond to at least 3.2±106 fmoles under nomoxic conditions.
A central challenge of any hypothesis proposing a role of RBC-derived NO in human (patho)physiology is to understand how NO formed by these cells can escape irreversible dioxygenation reaction with oxyHb (reaction 1), which is a very rapid reaction. A “metabolon complex” of deoxyHb, AE/band 3, carbonic anhydrase, aquaporin, and Rh-protein channels was proposed to explain nitrite protonation and may serve to facilitate the export of NO or its metabolites [75]. The localization of eNOS-immunoreactivity on the cytoplasmic side of the RBC membrane, as detected by immunogold-labeling and electron microscopy imaging [40], supports the notion that the RBC membrane may play a central role in this process, possibly by effectively separating NO production, signaling and scavenging machinery from one another by compartmentalization. Local formation of metHb by reaction of eNOS-derived NO may protect further produced NO from degradation by oxyHb, allowing NO to interact with target proteins in the immediate vicinity of eNOS to exert intracellular signaling function, leading to activation of NO signaling pathways (Fig. 2). An example of such a target might be the Pannexin-1 channel, which has been shown to be regulated by nitrosation reactions [76]. The observation that NO formation can be detected in the vicinity of abundant oxyHb would seem to warrant a careful reassessment of the universally accepted paradigm that RBC in normoxic conditions should be considered just a “sink” of NO.
Fig. 2.
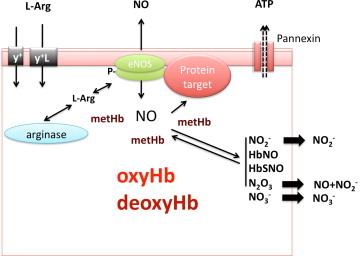
Potential role of red cell eNOS in signalling. L-arginine is imported into the RBC via the cationic amino acid transporters y+/CAT and/or y+L. Both localization of eNOS on the cytoplasmic side of the RBC membrane and protein-protein interactions may play a central role in effectively separating eNOS from arginase, as well as NO production from NO scavenging by oxyhemoglobin (oxyHb). Local formation of methemoglobin (metHb) by reaction of eNOS-derived NO may protect further produced NO from degradation by oxyHb, allowing NO to interact with target proteins in the immediate vicinity of eNOS to exert intracellular signaling function, leading to activation of NO signaling pathways. An example of such a target might be the Pannexin-1 channel.
Pathophysiological significance of red cell eNOS–control of erythrocrine function?
The ability of RBCs to secrete bioactive molecules including NO metabolites and ATP appears to be an important aspect of their function. This “erythrocrine function1”, characterizing the exocrine activity of RBC, might play an important role in pathophysiology.
Some evidence of a role of red cell eNOS in controlling the erythrocrine function exists from in vitro studies. NOS inhibitors abolish the anti-platelet effects of RBCs in vitro [40], [69] and decrease the level of NO products released by isolated RBCs [13], [40], [63]. Perfusion of isolated lungs with washed RBCs in the presence of l-NAME increased pulmonary vascular resistance, as compared to control cells without inhibitor [78]. Perfusion with whole blood or washed RBCs in normoxia protect isolated perfused hearts from ischemia/reperfusion injury in a NOS and arginase-dependent fashion [68], [70]. Moreover, both the presence of eNOS in blood and the inhibition of arginase to increase l-arginine availability reduces infarct size and left ventricular dysfunction in an ex vivo model ischemia/reperfusion [68].
Other studies suggest that activity of a red cell eNOS may be involved in the regulation of RBC deformability [40], [79], [80]. Interestingly, a link between RBC deformation and ATP release was also demonstrated [56], [57] and nitrite was shown to induce ATP release under hypoxic conditions from RBC [81], probably by releasing NO. Considering this data, it is tempting to speculate that red cell eNOS might be involved in regulating ATP release under normoxic conditions. However, a link between red cell eNOS-derived NO production and ATP is still awaiting experimental verification.
In vivo evidence indicating a red cell eNOS-dependent control of this erythrocrine function and its pathophysiological consequences in vivo was derived by analyzing the phenotype of chimera obtained by bone marrow transplantation from eNOS-/- mice into lethally irradiated wild type (WT) mice (Fig. 3) [66], [82]. These mice carry blood cells that lack eNOS, and are characterized by a significant decrease in circulating nitrite and nitrate levels in blood, as well as higher blood pressure, as compared to the controls [66]. After depleting these mice from leukocytes or platelets, the differences between the chimeras lacking blood cell eNOS and the wild type counterpart persisted. These results demonstrated for the first time that beside the enzyme that is expressed in the endothelium, also circulating blood cell eNOS plays a role in the control of vascular tone and blood pressure; furthermore, they suggest that RBCs, the most abundant cell population in blood, might be involved in mediating those effects.
Fig. 3.
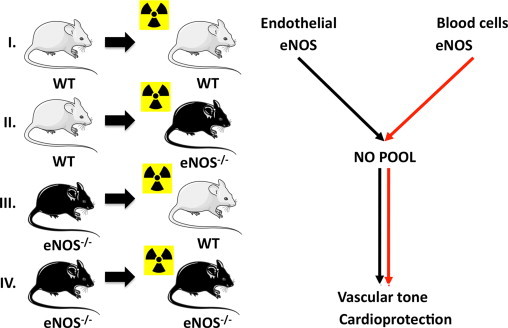
Creation in vivo chimera models for studying the role of blood cell eNOS. To analyze the role of blood cell eNOS we created chimeras carrying eNOS in the blood only (group II) or lacking eNOS in the blood only (group III) and respective controls (groups I, IV) by cross-transplanting the bone marrow of wild type (WT) or eNOS knockout (eNOS-/-) mice into lethal irradiated mice. We found that also blood cell eNOS play a role in control of vascular tone and cardioprotection.
If eNOS in blood indeed contributed to blood pressure regulation and nitrite production, then it is likely to also have effects under regenerative conditions, such as tissue repair following myocardial ischemia or stroke. In an acute model of myocardial ischemia/reperfusion we found that chimeras lacking blood cell eNOS had increased infarct size, resulting in decreased ejection fraction and increased end systolic volume after 60 min ischemia and 24 h of reperfusion [82]. Application of the NOS inhibitor ethylenethiourea (ETU) during ischemia and the first 5 min of reperfusion was associated with larger infarct size in mice carrying eNOS in blood, whereas infarct size in mice lacking blood cell eNOS was unaffected [82]. RBCs from chimera lacking blood cell eNOS have decreased deformability [82], which may lead to decreased perfusion capacity of blood.
Taken together, these results show that circulating blood cell eNOS plays a role in the control of the circulating pool of NO metabolites, and further suggests a modulating role in ischemia/reperfusion injury (Fig. 4). An earlier study by Ii and colleagues [83] may also be of relevance in this context. That study evaluated similar cross-transplantation experiments with bone marrow recipient mice subjected to experimental myocardial infarction. Cardioprotection was observed in the eNOS-/- mice receiving WT bone marrow, which at the time was ascribed to NO released by EPCs that had been incorporated into the ischemic myocardium. However, in that study only a small percentage of EPCs are incorporated in the ischemic myocardium, suggesting that the cardioprotection may have also originated from circulating blood cell-derived eNOS activity or nitrite. The blood cell(s) populations responsible for cardioprotection have still to be identified.
Fig. 4.
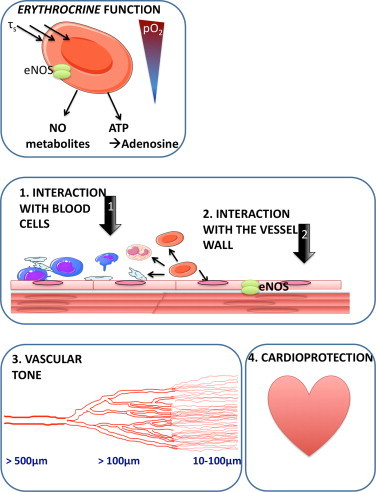
Pathophysiological significance of red cell eNOS. An accumulating body of evidence identifies RBCs as active contributors to vascular homeostasis and cardioprotection. These effects might be due to the ability of RBCs to secrete vasoactive and cardioprotective molecules, a property which we here define as erythrocrine function, and might be dependent on the activity of red cell eNOS. Further studies are required to unravel its nature, regulation and pathophysiological significance.
The use of bone marrow chimeric mice to analyze the pathophysiological role of red cell eNOS has profound methodological limitations, including irradiation-dependent activation of inflammatory pathways (which may increase the expression of an inducible NOS), the possibility of protein transfer from the blood to the endothelium (due to housing of circulating endothelial progenitor cells), the presence of low levels of circulating blood cells from the recipient, and – most importantly – the lack of erythroid lineage targeting specificity. Therefore, in these studies we could not discriminate the blood cell population(s) carrying eNOS that might affect nitrite levels, the tone of resistance arteries or exerting cardioprotective effects. These limitations can be only overcome by applying more specific genetic approaches, i.e. by using conditional mutant mice lacking or expressing eNOS in the erythroid lineage only.
Nevertheless, these are the first studies to definitively establish a role for circulating blood cell eNOS in the regulation of nitrite homeostasis, blood pressure and cardioprotection in an ischemia/reperfusion injury model under physiological conditions, and point to a possible role of red cell eNOS in this context. These findings challenge our conventional perspectives on NO and NO metabolite/nitrite signaling in blood, suggesting a more holistic regulation of cardiovascular homeostasis, with contributions from both the endothelium and circulating blood cells. In addition to the involvement of NO itself, other groups have demonstrated that circulating nitrite is a signaling molecule [18], and can be reduced to form NO, and thereby contribute to the regulation of hypoxic vasodilation, cytoprotection following ischemia/reperfusion events, adaptation to hypoxia and vascular angiogenesis [21], [24], [26], [53], [84], [85], [86], [87], [88], [89], [90], [91].
Thus, it is likely that both, direct NO formation and signaling as well as nitrite-mediated signaling, contribute to the beneficial effects of blood cell eNOS in blood pressure control and cardioprotection (Fig. 4). The existence of a functional blood-borne eNOS opens the door to studies addressing the function and dysfunction of blood eNOS in health and disease, such as a role in red blood cell enzymopathies, hemoglobinopathies and membranopathies, as well as in infectious diseases such as malaria.
Impaired red cell eNOS function in cardiovascular disease
Impaired endothelial function, decreased eNOS activity, and/or NO bioavailability are conditions strongly related to cardiovascular disease [13], [16]. Perhaps one of the most surprising results from our more recent studies [32], which have recently been confirmed by an independent group [41], is that expression and activity of circulating eNOS in RBCs [32], or plasma microparticles [37] is decreased in patients with endothelial dysfunction as compared to healthy controls. Thus, red cell eNOS expression and activity is decreased in patients with coronary artery disease as compared to age matched healthy controls and significantly correlates with flow-mediated dilation, a diagnostic marker of endothelial function and eNOS activity [32]. Similarly, eNOS levels and activity are decreased in microparticles from patients with peripheral arterial occlusive disease as compared to young healthy controls [37]. Moreover, eNOS could not be detected in circulating angiogenic cells from coronary artery disease (CAD) patients, and both chemokinesis and chemotaxis to VEGF were decreased compared with healthy circulating angiogenic cells [92]. Thus, a systemic circulating eNOS deficiency and/or dysfunction appears to prevail in patients with cardiovascular disease the consequences of which are not limited to impaired vascular function, but may also affect function of blood cells and vascular homeostasis.
Clinical perspective
RBCs are typically considered as shuttles of respiratory gases and nutrients for tissues, less so as compartments important to vascular integrity. However, RBC size distribution [93], [94], [95], number [96], [97], [98], [99] and integrity [100] appear to profoundly affect cardiovascular morbidity and mortality as demonstrated in recent clinical studies with large patients cohorts. Patients with CAD and concomitant anemia have a poorer prognosis after myocardial infarction, percutaneous coronary intervention, and coronary artery bypass grafting, and are more prone to developing heart failure with fatal outcomes [96], [97], [98]. Surprisingly, erythropoietin treatment fails to improve diagnosis [99], indicating that a compromised gas exchange/nutrient transport capacity of blood is insufficient to explain this outcome. In a cohort of 40,000 patients baseline levels of hemoglobin have been found to correlate with mortality after myocardial infarction according to a J-shaped relationship in that a mild decrease in baseline hemoglobin concentration (12–14 g/l) corresponds to a strong increase in mortality after acute cardiovascular events [101]; these associations are difficult to explain by a decrease in the capacity of oxygen delivery to tissues.
The issues surrounding appropriate storage of blood (blood banking) and the negative effects associated with the transfusion of “old” blood or RBCs preparations have increasingly attracted attention over the years and become a matter of scientific discussion and somewhat of a “hot topic” in translational research lately [102], [103]. In a retrospective study including 6000 patients Koch et al. showed that subjects receiving RBC units older than 2 weeks were more likely to die than subjects receiving newer blood [100]. Although this study is not without controversy [102], the damaging effects of transfusion with older blood units had already been observed in earlier retrospective studies [103], [104]; there, however, they have been attributed to changes in RBC oxygen transport capacity, RBC fragility and hemolysis, and pro-inflammatory effects. Basic science studies have shown that increased membrane fragility of “old” RBCs may also lead to formation of hemoglobin-rich microparticles [105], [106], [107], as well as a decrease in intracellular ATP, NO metabolites, and l-arginine, and an increase in reactive oxygen species and oxidative damage [108], [109], [110], thus potentially affecting erythrocrine function. This leads to an overall decrease in RBC deformability, a decrease in the RBC oxygen-carrying capacity and tissue perfusion, and importantly to systemic effects of free/microparticle hemoglobin-derived decrease in NO bioavailability, including endothelial dysfunction, increases in blood pressure, vasospasm and a prothrombogenic milieu. Whether and how red cell eNOS activity and/or signaling is involved in changes of RBC function in disease has not been investigated so far. While red cell eNOS does not seem to play a role in RBC membrane fragility in either fresh or stored blood [111], a decrease of red cell eNOS activity may still be involved in the regulation of RBC lifespan [112] and RBC deformability [40], [79], [80]. Future studies are required to address these important questions and close those open loops.
Summary and outlook
An accumulating body of evidence demonstrates that not only endothelial cells, but also blood cells contribute to the NO-dependent regulation of vascular homeostasis, and identifies RBCs as active contributors to vascular homeostasis and integrity. These effects might be due to the ability of RBCs to secrete vasoactive and cardioprotective molecules, a property, which we here define as erythrocrine function. Further studies are required to unravel its nature, regulation and pathophysiological function.
Taken together, a picture emerges according to which RBCs contribute to vascular homeostasis, independent of their “classical role” as transporters of oxygen, energy substrates and nutrients. Given the contribution RBCs make to the circulating NO pool, their role in hypoxic vasodilation and their overall significance for organ protection further studies would seem to be warranted to address their functional significance for the regulation of cardiovascular function in health and disease.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (DFG 405/5-1 and FOR809 TP7 Me1821/3-1), the Anton Betz Stiftung, (26/2010), the Susanne Bunnenberg Stiftung at Düsseldorf Heart Center to M.K., and the Forschungskommission of the Medical Faculty of the Heinrich Heine University of Düsseldorf to MCK.
The authors wish to thank all their collaborators in this project, and all scientists we discussed with about red cell eNOS, and in particular Dario Vitturi and Jesus Tejero Bravo for discussions about the potential role of metHb in compartmentalization of NO synthesis. A special thank goes to Mark Gladwin, and Martin Feelisch for inspiring exchange of views about NO production in RBC.
Footnotes
☆
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
1
Note: The ability RBCs to release analgesic peptides was defined as “erythrocrine”, or exocrine activity of erythrocytes [77]. In this review we adopt this term in a more general sense to define the ability of RBCs to secrete signaling entities or transmitter molecules including amino acids, nucleotides gases and other mediators able to participate in cell-cell communications as well as to control biochemical processes.
References
- 1.Moncada S., Palmer R.M.J., Higgs E.A. Nitric oxide, biology pathophysiology and pharmacology. Pharmacol. Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 2.Furchgott R.F., Zawadzki J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- 3.Ignarro L.J. Biological actions and properties of endothelium-derived nitric oxide formed and released from artery and vein. Circ. Res. 1989;65:1–21. doi: 10.1161/01.res.65.1.1. [DOI] [PubMed] [Google Scholar]
- 4.Palmer R.M., Ferrige A.G., Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327:524–526. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- 5.Vallance P., Collier J., Moncada S. Effects of endothelium-derived nitric oxide on peripheral arteriolar tone in man. Lancet. 1989;2:997–1000. doi: 10.1016/s0140-6736(89)91013-1. [DOI] [PubMed] [Google Scholar]
- 6.Moncada S. Nitric oxide in the vasculature: physiology and pathophysiology. Ann. New York Acad. Sci. 1997;811:60–67. doi: 10.1111/j.1749-6632.1997.tb51989.x. (discussion 67–69) [DOI] [PubMed] [Google Scholar]
- 7.Bredt D.S., Snyder S.H. Nitric oxide: a physiologic messenger molecule. Annu. Rev. Biochem. 1994;63:175–195. doi: 10.1146/annurev.bi.63.070194.001135. [DOI] [PubMed] [Google Scholar]
- 8.Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- 9.Griffith O.W., Stuehr D.J. Nitric oxide synthases: properties and catalytic mechanism. Annu. Rev. Physiol. 1995;57:707–734. doi: 10.1146/annurev.ph.57.030195.003423. [DOI] [PubMed] [Google Scholar]
- 10.Stuehr D.J. Enzymes of the l-arginine to nitric oxide pathway. J. Nutr. 2004;134:2748S–2751S. doi: 10.1093/jn/134.10.2748S. (discussion 2765S–2767S) [DOI] [PubMed] [Google Scholar]
- 11.Denninger J.W., Marletta M.A. Guanylate cyclase and the NO/cGMP signaling pathway. Biochim. Biophys. Acta. 1999;1411:334–350. doi: 10.1016/s0005-2728(99)00024-9. [DOI] [PubMed] [Google Scholar]
- 12.Sessa W.C. eNOS at a glance. J. Cell Sci. 2004;117:2427–2429. doi: 10.1242/jcs.01165. [DOI] [PubMed] [Google Scholar]
- 13.Heiss C., Lauer T., Dejam A., Kleinbongard P., Hamada S., Rassaf T., Matern S., Feelisch M., Kelm M. Plasma nitroso compounds are decreased in patients with endothelial dysfunction. J. Am. Coll. Cardiol. 2006;47:573–579. doi: 10.1016/j.jacc.2005.06.089. [DOI] [PubMed] [Google Scholar]
- 14.Kleinbongard P., Dejam A., Lauer T., Rassaf T., Schindler A., Picker O., Scheeren T., Godecke A., Schrader J., Schulz R., Heusch G., Schaub G.A., Bryan N.S., Feelisch M., Kelm M. Plasma nitrite reflects constitutive nitric oxide synthase activity in mammals. Free Radic. Biol. Med. 2003;35:790–796. doi: 10.1016/s0891-5849(03)00406-4. [DOI] [PubMed] [Google Scholar]
- 15.Rassaf T., Bryan N.S., Maloney R.E., Specian V., Kelm M., Kalyanaraman B., Rodriguez J., Feelisch M. NO adducts in mammalian red blood cells: too much or too little? Nat Med. 2003;9:481–482. doi: 10.1038/nm0503-481. [DOI] [PubMed] [Google Scholar]
- 16.Rassaf T., Heiss C., Hendgen-Cotta U., Balzer J., Matern S., Kleinbongard P., Lee A., Lauer T., Kelm M. Plasma nitrite reserve and endothelial function in the human forearm circulation. Free Radic. Biol. Med. 2006;41:295–301. doi: 10.1016/j.freeradbiomed.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 17.Milsom A.B., Fernandez B.O., Garcia-Saura M.F., Rodriguez J., Feelisch M. Contributions of nitric oxide synthases, dietary nitrite/nitrate, and other sources to the formation of NO signaling products. Antioxid. Redox Signal. 2012;17:422–432. doi: 10.1089/ars.2011.4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bryan N.S., Fernandez B.O., Bauer S.M., Garcia-Saura M.F., Milsom A.B., Rassaf T., Maloney R.E., Bharti A., Rodriguez J., Feelisch M. Nitrite is a signaling molecule and regulator of gene expression in mammalian tissues. Nature Chem. Biol. 2005;1:290–297. doi: 10.1038/nchembio734. [DOI] [PubMed] [Google Scholar]
- 19.Crawford J.H., Isbell T.S., Huang Z., Shiva S., Chacko B.K., Schechter A.N., rley-Usmar V.M., Kerby J.D., Lang J.D., Jr, Kraus D., Ho C., Gladwin M.T., Patel R.P. Hypoxia, red blood cells, and nitrite regulate NO-dependent hypoxic vasodilation. Blood. 2006;107:566–574. doi: 10.1182/blood-2005-07-2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Faassen E.E., Bahrami S., Feelisch M., Hogg N., Kelm M., Kim-Shapiro D.B., Kozlov A.V., Li H., Lundberg J.O., Mason R., Nohl H., Rassaf T., Samouilov A., Slama-Schwok A., Shiva S., Vanin A.F., Weitzberg E., Zweier J., Gladwin M.T. Nitrite as regulator of hypoxic signaling in mammalian physiology. Med. Res. Rev. 2009;29:683–741. doi: 10.1002/med.20151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Totzeck M., Hendgen-Cotta U.B., Luedike P., Berenbrink M., Klare J.P., Steinhoff H.J., Semmler D., Shiva S., Williams D., Kipar A., Gladwin M.T., Schrader J., Kelm M., Cossins A.R., Rassaf T. Nitrite regulates hypoxic vasodilation via myoglobin-dependent nitric oxide generation. Circulation. 2012;126:325–334. doi: 10.1161/CIRCULATIONAHA.111.087155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cosby K., Partovi K.S., Crawford J.H., Patel R.P., Reiter C.D., Martyr S., Yang B.K., Waclawiw M.A., Zalos G., Xu X., Huang K.T., Shields H., Kim-Shapiro D.B., Schechter A.N., Cannon Iii R.O., Gladwin M.T. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat. Med. 2003;9:1498–1505. doi: 10.1038/nm954. [DOI] [PubMed] [Google Scholar]
- 23.Gladwin M.T., Shelhamer J.H., Schechter A.N., Pease-Fye M.E., Waclawiw M.A., Panza J.A., Ognibene F.P., Cannon Iii R.O. Role of circulating nitrite and S-nitrosohemoglobin in the regulation of regional blood flow in humans. Proc. Natl. Acad. Sci. USA. 2000;97:11482–11487. doi: 10.1073/pnas.97.21.11482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lundberg J.O., Gladwin M.T., Ahluwalia A., Benjamin N., Bryan N.S., Butler A., Cabrales P., Fago A., Feelisch M., Ford P.C., Freeman B.A., Frenneaux M., Friedman J., Kelm M., Kevil C.G., Kim-Shapiro D.B., Kozlov A.V., Lancaster J.R., Jr, Lefer D.J., McColl K., McCurry K., Patel R.P., Petersson J., Rassaf T., Reutov V.P., Richter-Addo G.B., Schechter A., Shiva S., Tsuchiya K., van Faassen E.E., Webb A.J., Zuckerbraun B.S., Zweier J.L., Weitzberg E. Nitrate and nitrite in biology, nutrition and therapeutics. Nat. Chem. Biol. 2009;5:865–869. doi: 10.1038/nchembio.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shiva S., Gladwin M.T. Nitrite mediates cytoprotection after ischemia/reperfusion by modulating mitochondrial function. Basic Res. Cardiol. 2009;104:113–119. doi: 10.1007/s00395-009-0009-3. [DOI] [PubMed] [Google Scholar]
- 26.Hendgen-Cotta U.B., Merx M.W., Shiva S., Schmitz J., Becher S., Klare J.P., Steinhoff H.J., Goedecke A., Schrader J., Gladwin M.T., Kelm M., Rassaf T. Nitrite reductase activity of myoglobin regulates respiration and cellular viability in myocardial ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA. 2008;105:10256–10261. doi: 10.1073/pnas.0801336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murillo D., Kamga C., Mo L., Shiva S. Nitrite as a mediator of ischemic preconditioning and cytoprotection. Nitric Oxide. 2011;25:70–80. doi: 10.1016/j.niox.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang P.L., Huang Z., Mashimo H., Bloch K.D., Moskowitz M.A., Bevan J.A., Fishman M.C. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- 29.Shesely E.G., Maeda N., Kim H.S., Desai K.M., Krege J.H., Laubach V.E., Sherman P.A., Sessa W.C., Smithies O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA. 1996;93:13176–13181. doi: 10.1073/pnas.93.23.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Godecke A., Decking U.K., Ding Z., Hirchenhain J., Bidmon H.J., Godecke S., Schrader J. Coronary hemodynamics in endothelial NO synthase knockout mice. Circ. Res. 1998;82:186–194. doi: 10.1161/01.res.82.2.186. [DOI] [PubMed] [Google Scholar]
- 31.Sase K., Michel T. Expression of constitutive endothelial nitric oxide synthase in human blood platelets. Life Sci. 1995;57:2049–2055. doi: 10.1016/0024-3205(95)02191-k. [DOI] [PubMed] [Google Scholar]
- 32.Cortese-Krott M.M., Rodriguez-Mateos A., Sansone R., Kuhnle G.G., Thasian-Sivarajah S., Krenz T., Horn P., Krisp C., Wolters D., Heiss C., Kroncke K.D., Hogg N., Feelisch M., Kelm M. Human red blood cells at work: identification and visualization of erythrocytic eNOS activity in health and disease. Blood. 2012;120:4229–4237. doi: 10.1182/blood-2012-07-442277. [DOI] [PubMed] [Google Scholar]
- 33.Aubry J.P., Dugas N., Lecoanet-Henchoz S., Ouaaz F., Zhao H., Delfraissy J.F., Graber P., Kolb J.P., Dugas B., Bonnefoy J.Y. The 25-kDa soluble CD23 activates type III constitutive nitric oxide-synthase activity via CD11b and CD11c expressed by human monocytes. J. Immunol. 1997;159:614–622. [PubMed] [Google Scholar]
- 34.Mühl H., Pfeilschifter J. Endothelial nitric oxide synthase: a determinant of TNFalpha production by human monocytes/macrophages. Biochem. Biophys. Res. Commun. 2003;310:677–680. doi: 10.1016/j.bbrc.2003.09.039. [DOI] [PubMed] [Google Scholar]
- 35.Saluja R., Jyoti A., Chatterjee M., Habib S., Verma A., Mitra K., Barthwal M.K., Bajpai V.K., Dikshit M. Molecular and biochemical characterization of nitric oxide synthase isoforms and their intracellular distribution in human peripheral blood mononuclear cells. Biochim. Biophys. Acta. 2011;1813:1700–1707. doi: 10.1016/j.bbamcr.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 36.Radomski M.W., Palmer R.M., Moncada S. An l-arginine/nitric oxide pathway present in human platelets regulates aggregation. Proc. Natl. Acad. Sci. USA. 1990;87:5193–5197. doi: 10.1073/pnas.87.13.5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Horn P., Cortese-Krott M.M., Amabile N., Hundsdorfer C., Kroncke K.D., Kelm M., Heiss C. Circulating microparticles carry a functional endothelial nitric oxide synthase that is decreased in patients with endothelial dysfunction. J. Am. Heart Assoc. 2013;2:e003764. doi: 10.1161/JAHA.112.003764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gambaryan S., Kobsar A., Hartmann S., Birschmann I., Kuhlencordt P.J., MÜLler Esterl W., Lohmann S.M., Walter U. NO‐synthase‐/NO‐independent regulation of human and murine platelet soluble guanylyl cyclase activity. J. Thromb. Haemost. 2008;6:1376–1384. doi: 10.1111/j.1538-7836.2008.03014.x. [DOI] [PubMed] [Google Scholar]
- 39.Ozüyaman B., Gödecke A., Küsters S., Kirchhoff E., Scharf R.E., Schrader J. Endothelial nitric oxide synthase plays a minor role in inhibition of arterial thrombus formation. Thromb. Haemost. 2005 doi: 10.1160/TH03-09-0588. [DOI] [PubMed] [Google Scholar]
- 40.Kleinbongard P., Schulz R., Rassaf T., Lauer T., Dejam A., Jax T., Kumara I., Gharini P., Kabanova S., Ozuyaman B., Schnurch H.G., Godecke A., Weber A.A., Robenek M., Robenek H., Bloch W., Rosen P., Kelm M. Red blood cells express a functional endothelial nitric oxide synthase. Blood. 2006;107:2943–2951. doi: 10.1182/blood-2005-10-3992. [DOI] [PubMed] [Google Scholar]
- 41.Eligini S., Porro B., Lualdi A., Squellerio I., Veglia F., Chiorino E., Crisci M., Garlasche A., Giovannardi M., Werba J.P., Tremoli E., Cavalca V. Nitric oxide synthetic pathway in red blood cells is impaired in coronary artery disease. PloS one. 2013;8:e66945. doi: 10.1371/journal.pone.0066945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Helms C., Kim-Shapiro D.B. Hemoglobin-mediated nitric oxide signaling. Free Radic. Biol. Med. 2013;61c:464–472. doi: 10.1016/j.freeradbiomed.2013.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Webb A., Milsom A., Rathod K., Chu W., Qureshi S., Lovell M., Lecomte F., Perrett D., Raimondo C., Khoshbin E., Ahmed Z., Uppal R., Benjamin N., Hobbs A., Ahluwalia A. Mechanisms underlying erythrocyte and endothelial nitrite reduction to nitric oxide in hypoxia: role for xanthine oxidoreductase and endothelial nitric oxide synthase. Circ. Res. 2008;103:957–964. doi: 10.1161/CIRCRESAHA.108.175810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Herold S. The outer-sphere oxidation of nitrosyliron(II)hemoglobin by peroxynitrite leads to the release of nitrogen monoxide. Inorg. Chem. 2004;43:3783–3785. doi: 10.1021/ic035340a. [DOI] [PubMed] [Google Scholar]
- 45.Jia L., Bonaventura C., Bonaventura J., Stamler J.S. S-nitrosohaemoglobin: a dynamic activity of blood involved in vascular control. Nature. 1996;380:221–226. doi: 10.1038/380221a0. [DOI] [PubMed] [Google Scholar]
- 46.Gladwin M.T., Lancaster J.R., Jr., Freeman B.A., Schechter A.N. Nitric oxide’s reactions with hemoglobin: a view through the SNO-storm. Nat. Med. 2003;9:496–500. doi: 10.1038/nm0503-496. [DOI] [PubMed] [Google Scholar]
- 47.Dejam A., Hunter C.J., Pelletier M.M., Hsu L.L., Machado R.F., Shiva S., Power G.G., Kelm M., Gladwin M.T., Schechter A.N. Erythrocytes are the major intravascular storage sites of nitrite in human blood. Blood. 2005;106:734–739. doi: 10.1182/blood-2005-02-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vitturi D.A., Teng X., Toledo J.C., Matalon S., Lancaster J.R., Jr., Patel R.P. Regulation of nitrite transport in red blood cells by hemoglobin oxygen fractional saturation. Am. J. Physiol. Heart Circ. Physiol. 2009;296:H1398–H1407. doi: 10.1152/ajpheart.01303.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nagababu E., Ramasamy S., Albernethy R., Rifkind M. Active nitric oxide produced in the red cell under hypoxic conditions by deoxyhemoglobin-mediated nitrite reduction. J. Biol. Chem. 2003;278:46349–46356. doi: 10.1074/jbc.M307572200. [DOI] [PubMed] [Google Scholar]
- 50.Zweier J.L., Wang P., Samouilov A., Kuppusamy P. Enzyme-independent formation of nitric oxide in biological tissues. Nat. Med. 1995;1:804–809. doi: 10.1038/nm0895-804. [DOI] [PubMed] [Google Scholar]
- 51.Aamand R., Dalsgaard T., Jensen F.B., Simonsen U., Roepstorff A., Fago A. Generation of nitric oxide from nitrite by carbonic anhydrase: a possible link between metabolic activity and vasodilation. Am. J. Physiol. Heart Circ. Physiol. 2009;297:H2068–H2074. doi: 10.1152/ajpheart.00525.2009. [DOI] [PubMed] [Google Scholar]
- 52.Patel R.P., Hogg N., Kim-Shapiro D.B. The potential role of the red blood cell in nitrite-dependent regulation of blood flow. Cardiovasc. Res. 2011;89:507–515. doi: 10.1093/cvr/cvq323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Owusu B.Y., Stapley R., Patel R.P. Nitric oxide formation versus scavenging: the red blood cell balancing act. J. Physiol. 2012;590:4993–5000. doi: 10.1113/jphysiol.2012.234906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Locovei S., Bao L., Dahl G. Pannexin 1 in erythrocytes: function without a gap. Proc. Natl. Acad. Sci. USA. 2006;103:7655–7659. doi: 10.1073/pnas.0601037103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sridharan M., Adderley S.P., Bowles E.A., Egan T.M., Stephenson A.H., Ellsworth M.L., Sprague R.S. Pannexin 1 is the conduit for low oxygen tension-induced ATP release from human erythrocytes. AJP: Heart Circ. Physiol. 2010;299:H1146–H1152. doi: 10.1152/ajpheart.00301.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Forsyth A.M., Wan J., Owrutsky P.D., Abkarian M., Stone H.A. Multiscale approach to link red blood cell dynamics, shear viscosity, and ATP release. Proc. Natl. Acad. Sci. 2011;108:10986–10991. doi: 10.1073/pnas.1101315108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wan J., Forsyth A.M., Stone H.A. Red blood cell dynamics: from cell deformation to ATP release. Integr. Biol. (Camb) 2011;3:972–981. doi: 10.1039/c1ib00044f. [DOI] [PubMed] [Google Scholar]
- 58.Forsyth A.M., Wan J., Owrutsky P.D., Abkarian M., Stone H.A. Multiscale approach to link red blood cell dynamics, shear viscosity, and ATP release. Proc. Natl. Acad. Sci. USA. 2011;108:10986–10991. doi: 10.1073/pnas.1101315108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ellsworth M.L., Forrester T., Ellis C.G., Dietrich H.H. The erythrocyte as a regulator of vascular tone. Am. J. Physiol. Heart Circ. Physiol. 1995;269:H2155–H2161. doi: 10.1152/ajpheart.1995.269.6.H2155. [DOI] [PubMed] [Google Scholar]
- 60.Ellsworth M.L., Sprague R.S. Regulation of blood flow distribution in skeletal muscle: role of erythrocyte-released ATP. J. Physiol. 2012;590:4985–4991. doi: 10.1113/jphysiol.2012.233106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gonzalez-Alonso J. ATP as a mediator of erythrocyte-dependent regulation of skeletal muscle blood flow and oxygen delivery in humans. J. Physiol. 2012;590:5001–5013. doi: 10.1113/jphysiol.2012.235002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bhattacharya S., Chakraborty P.S., Basu R.S., Kahn N.N., Sinha A.K. Purification and properties of insulin-activated nitric oxide synthase from human erythrocyte membranes. Arch. Physiol. Biochem. 2001;109:441–449. doi: 10.1076/apab.109.5.441.8042. [DOI] [PubMed] [Google Scholar]
- 63.Mihov D., Vogel J., Gassmann M., Bogdanova A. Erythropoietin activates nitric oxide synthase in murine erythrocytes. Am. J. Physiol. Cell Physiol. 2009;297:C378–C388. doi: 10.1152/ajpcell.00543.2008. [DOI] [PubMed] [Google Scholar]
- 64.Jubelin B.C., Gierman J.L. Erythrocytes may synthesize their own nitric oxide. AJH. 1996;9:1214–1219. doi: 10.1016/S0895-7061(96)00257-9. [DOI] [PubMed] [Google Scholar]
- 65.Kang E.S., Ford K., Grokulsky G., Wang Y.B., Chiang T.M., Acchiardo S.R. Normal circulating adult human red blood cells contain inactive NOS proteins. J. Lab Clin. Med. 2000;135:444–451. doi: 10.1067/mlc.2000.106805. [DOI] [PubMed] [Google Scholar]
- 66.Wood K.C., Cortese-Krott M.M., Kovacic J.C., Noguchi A., Liu V.B., Wang X., Raghavachari N., Boehm M., Kato G.J., Kelm M., Gladwin M.T. Circulating blood endothelial nitric oxide synthase contributes to the regulation of systemic blood pressure and nitrite homeostasis. Arterioscler Thromb. Vasc. Biol. 2013;33:1861–1871. doi: 10.1161/ATVBAHA.112.301068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Deliconstantinos G., Villiotou V., Stavrides J.C., Salemes N., Gogas J. Nitric oxide and peroxynitrite production by human erythrocytes: a causative factor of toxic anemia in breast cancer patients. Anticancer Res. 1995;15:1435–1446. [PubMed] [Google Scholar]
- 68.Yang J., Gonon A.T., Sjoquist P.O., Lundberg J.O., Pernow J. Arginase regulates red blood cell nitric oxide synthase and export of cardioprotective nitric oxide bioactivity. Proc. Natl. Acad. Sci. USA. 2013;110:15049–15054. doi: 10.1073/pnas.1307058110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen L.Y., Mehta J.L. Evidence for the presence of l-arginine-nitric oxide pathway in human red blood cells: relevance in the effects of red blood cells on platelet function. J. Cardiovasc. Pharmacol. 1998;32:57–61. doi: 10.1097/00005344-199807000-00009. [DOI] [PubMed] [Google Scholar]
- 70.Yang B.C., Nichols W.W., Mehta J.L. Cardioprotective Effects of Red Blood Cells on Ischemia and Reperfusion Injury in Isolated Rat Heart: Release of Nitric Oxide as a Potential Mechanism. J. Cardiovasc. Pharmacol. Therapeut. 1996;1:297–306. doi: 10.1177/107424849600100405. [DOI] [PubMed] [Google Scholar]
- 71.Böhmer A., Beckmann B., Sandmann J., Tsikas D. Doubts concerning functional endothelial nitric oxide synthase in human erythrocytes. Blood. 2012;119:1322–1323. doi: 10.1182/blood-2011-11-393124. [DOI] [PubMed] [Google Scholar]
- 72.Metha J.L., Metha P., Li D. Nitric oxide synthase in adult red blood cells: vestige of an earlier age or a biologically active enzyme? J. Lab Clin. Med. 2000;135:430–431. doi: 10.1067/mlc.2000.106802. [DOI] [PubMed] [Google Scholar]
- 73.Shiva S., Wang X., Ringwood L.A., Xu X., Yuditskaya S., Annavajjhala V., Miyajima H., Hogg N., Harris Z.L., Gladwin M.T. Ceruloplasmin is a NO oxidase and nitrite synthase that determines endocrine NO homeostasis. Nat. Chem. Biol. 2006;2:486–493. doi: 10.1038/nchembio813. [DOI] [PubMed] [Google Scholar]
- 74.Cortese-Krott M.M., Rodriguez-Mateos A., Kuhnle G.G., Brown G., Feelisch M., Kelm M. A multilevel analytical approach for detection and visualization of intracellular NO production and nitrosation events using diaminofluoresceins. Free Radic. Biol. Med. 2012;53:2146–2158. doi: 10.1016/j.freeradbiomed.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 75.Gladwin M.T., Schechter A., Kim-Shapiro D.B., Patel R., Hogg N., Shiva S., Cannon R., Kelm M., Wink D., Espey M., Oldfield E., Pluta R., Freeman B., Lancaster J., Feelisch M., Lundberg J.O. The emerging biology of the nitrite anion in signaling, blood flow and hypoxic nitric oxide homeostasis. Nat. Chem. Biol. 2005;1:308–314. doi: 10.1038/nchembio1105-308. [DOI] [PubMed] [Google Scholar]
- 76.Lohman A.W., Weaver J.L., Billaud M., Sandilos J.K., Griffiths R., Straub A.C., Penuela S., Leitinger N., Laird D.W., Bayliss D.A., Isakson B.E. S-nitrosylation inhibits pannexin 1 channel function. J. Biol. Chem. 2012;287:39602–39612. doi: 10.1074/jbc.M112.397976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Song C.Z., Wang Q.W., Song C.C. Erythrocyte-based analgesic peptides. Regul. Pept. 2013;180:58–61. doi: 10.1016/j.regpep.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 78.Sprague R.S., Stephenson A.H., Dimmitt R.A., Weintraub N.L., Branch C.A., McMurdo L., Lonigro A.J. Effect of l-NAME on pressure-flow relationships in isolated rabbit lungs: role of red blood cells. Am. J. Physiol. 1995;269:H1941–H1948. doi: 10.1152/ajpheart.1995.269.6.H1941. [DOI] [PubMed] [Google Scholar]
- 79.Bor-Kucukatay M., Wenby R.B., Meiselman H.J., Baskurt O.K. Effects of nitric oxide on red blood cell deformability. Am. J. Physiol. Heart Circul. Physiol. 2003;284:H1577–H1584. doi: 10.1152/ajpheart.00665.2002. [DOI] [PubMed] [Google Scholar]
- 80.Horn P., Cortese-Krott M.M., Keymel S., Kumara I., Burghoff S., Schrader J., Kelm M., Kleinbongard P. Nitric oxide influences red blood cell velocity independently of changes in the vascular tone. Free Radic. Res. 2011;45:653–661. doi: 10.3109/10715762.2011.574288. [DOI] [PubMed] [Google Scholar]
- 81.Cao Z., Bell J.B., Mohanty J.G., Nagababu E., Rifkind J.M. Nitrite enhances RBC hypoxic ATP synthesis and the release of ATP into the vasculature: a new mechanism for nitrite-induced vasodilation. Am. J. Physiol. Heart Circ. Physiol. 2009;297:H1494–H1503. doi: 10.1152/ajpheart.01233.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Merx M.W., van de Sandt G.S., Cortese-Krott A.M., Ohlig M.M., Stern J., Rassaf M., Gödecke T., Gladwin A., Kelm M.T., M. Depletion of circulating blood NOS3 increases severity of myocardial infarction and left ventricular dysfunction. Basic Res. Cardiol. 2013;109(1):398–408. doi: 10.1007/s00395-013-0398-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ii M., Nishimura H., Iwakura A., Wecker A., Eaton E., Asahara T., Losordo D.W. Endothelial progenitor cells are rapidly recruited to myocardium and mediate protective effect of ischemic preconditioning via “imported” nitric oxide synthase activity. Circulation. 2005;111:1114–1120. doi: 10.1161/01.CIR.0000157144.24888.7E. [DOI] [PubMed] [Google Scholar]
- 84.Hendgen-Cotta U.B., Luedike P., Totzeck M., Kropp M., Schicho A., Stock P., Rammos C., Niessen M., Heiss C., Lundberg J.O., Weitzberg E., Kelm M., Rassaf T. Dietary nitrate supplementation improves revascularization in chronic ischemia. Circulation. 2012;126:1983–1992. doi: 10.1161/CIRCULATIONAHA.112.112912. [DOI] [PubMed] [Google Scholar]
- 85.Levett D.Z., Fernandez B.O., Riley H.L., Martin D.S., Mitchell K., Leckstrom C.A., Ince C., Whipp B.J., Mythen M.G., Montgomery H.E., Grocott M.P., Feelisch M. The role of nitrogen oxides in human adaptation to hypoxia. Scient. Rep. 2011;1:109. doi: 10.1038/srep00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Umbrello M., Dyson A., Feelisch M., Singer M. The key role of nitric oxide in hypoxia: hypoxic vasodilation and energy supply-demand matching. Antioxid Redox Signal. 2013;19:1690–1710. doi: 10.1089/ars.2012.4979. [DOI] [PubMed] [Google Scholar]
- 87.Cosby K., Partovi K.S., Crawford J.H., Patel R.P., Reiter C.D., Martyr S., Yang B.K., Waclawiw M.A., Zalos G., Xu X., Huang K.T., Shields H., Kim-Shapiro D.B., Schechter A.N., Cannon R.O., 3rd, Gladwin M.T. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat. Med. 2003;9:1498–1505. doi: 10.1038/nm954. [DOI] [PubMed] [Google Scholar]
- 88.Maher A.R., Milsom A.B., Gunaruwan P., Abozgula K., Ahmed I., Weaver R.A., Thomas P., Ashrafian H., Born G.V., James P.E., Frenneaux M.P. Hypoxic modulation of exogenous nitrite-induced vasodilation in humans. Circulation. 2008;117:670–677. doi: 10.1161/CIRCULATIONAHA.107.719591. [DOI] [PubMed] [Google Scholar]
- 89.Duranski M.R., Greer J.J., Dejam A., Jaganmohan S., Hogg N., Langsotn W., Patel R.P., Yet S.F., Wang X., Kevil C.G., Gladwin M.T., Lefer D.J. Cytoprotective effects of nitirite during in vivo ischemia-reperfusion of the heart and liver. J. Clin. Invest. 2005;115:1232–1240. doi: 10.1172/JCI22493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dezfulian C., Raat N., Shiva S., Gladwin M.T. Role of the anion nitrite in ischemia-reperfusion cytorprotection and therapeutics. Cardiovasc. Res. 2007;75:327–338. doi: 10.1016/j.cardiores.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Frerart F., Lobysheva I., Gallez B., Dessy C., Feron O. Vascular caveolin deficiency supports the angiogenic effects of nitrite, a major end product of nitric oxide metabolism in tumors. Mol. Cancer Res. MCR. 2009;7:1056–1063. doi: 10.1158/1541-7786.MCR-08-0388. [DOI] [PubMed] [Google Scholar]
- 92.Heiss C., Schanz A., Amabile N., Jahn S., Chen Q., Wong M.L., Rassaf T., Heinen Y., Cortese-Krott M., Grossman W., Yeghiazarians Y., Springer M.L. Nitric oxide synthase expression and functional response to nitric oxide are both important modulators of circulating angiogenic cell response to angiogenic stimuli. Arterioscler. Thromb. Vasc. Biol. 2010;30:2212–2218. doi: 10.1161/ATVBAHA.110.211581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Felker G.M., Allen L.A., Pocock S.J., Shaw L.K., McMurray J.J., Pfeffer M.A., Swedberg K., Wang D., Yusuf S., Michelson E.L., Granger C.B. Red cell distribution width as a novel prognostic marker in heart failure: data from the CHARM Program and the Duke Databank. J. Am. Coll. Cardiol. 2007;50:40–47. doi: 10.1016/j.jacc.2007.02.067. [DOI] [PubMed] [Google Scholar]
- 94.Patel K.V., Semba R.D., Ferrucci L., Newman A.B., Fried L.P., Wallace R.B., Bandinelli S., Phillips C.S., Yu B., Connelly S., Shlipak M.G., Chaves P.H., Launer L.J., Ershler W.B., Harris T.B., Longo D.L., Guralnik J.M. Red cell distribution width and mortality in older adults: a meta-analysis. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2010;65:258–265. doi: 10.1093/gerona/glp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tonelli M., Sacks F., Arnold M., Moye L., Davis B., Pfeffer M. Cholesterol, f. t. Investigators, R. E. T. Relation between red Blood cell distribution width and cardiovascular event rate in people with coronary disease. Circulation. 2008;117:163–168. doi: 10.1161/CIRCULATIONAHA.107.727545. [DOI] [PubMed] [Google Scholar]
- 96.Sabatine M.S., Morrow D.A., Giugliano R.P., Burton P.B., Murphy S.A., McCabe C.H., Gibson C.M., Braunwald E. Association of hemoglobin levels with clinical outcomes in acute coronary syndromes. Circulation. 2005;111:2042–2049. doi: 10.1161/01.CIR.0000162477.70955.5F. [DOI] [PubMed] [Google Scholar]
- 97.Kulier A., Levin J., Moser R., Rumpold-Seitlinger G., Tudor I.C., Snyder-Ramos S.A., Moehnle P., Mangano D.T. Impact of preoperative anemia on outcome in patients undergoing coronary artery bypass graft surgery. Circulation. 2007;116:471–479. doi: 10.1161/CIRCULATIONAHA.106.653501. [DOI] [PubMed] [Google Scholar]
- 98.Anand I.S. Anemia and chronic heart failure implications and treatment options. J. Am. Coll. Cardiol. 2008;52:501–511. doi: 10.1016/j.jacc.2008.04.044. [DOI] [PubMed] [Google Scholar]
- 99.Najjar S.S., Rao S.V., Melloni C., Raman S.V., Povsic T.J., Melton L., Barsness G.W., Prather K., Heitner J.F., Kilaru R., Gruberg L., Hasselblad V., Greenbaum A.B., Patel M., Kim R.J., Talan M., Ferrucci L., Longo D.L., Lakatta E.G., Harrington R.A. Intravenous erythropoietin in patients with ST-segment elevation myocardial infarction: REVEAL: a randomized controlled trial. JAMA: J. Am. Med. Assoc. 2011;305:1863–1872. doi: 10.1001/jama.2011.592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Koch C.G., Li L., Sessler D.I., Figueroa P., Hoeltge G.A., Mihaljevic T., Blackstone E.H. Duration of red-cell storage and complications after cardiac surgery. New Engl. J. Med. 2008;358:1229–1239. doi: 10.1056/NEJMoa070403. [DOI] [PubMed] [Google Scholar]
- 101.Bassand J.P., Afzal R., Eikelboom J., Wallentin L., Peters R., Budaj A., Fox K.A., Joyner C.D., Chrolavicius S., Granger C.B., Mehta S., Yusuf S. Relationship between baseline haemoglobin and major bleeding complications in acute coronary syndromes. Eur. Heart J. 2010;31:50–58. doi: 10.1093/eurheartj/ehp401. [DOI] [PubMed] [Google Scholar]
- 102.Lee J.S., Gladwin M.T. Bad blood: The risks of red cell storage. Nat. Med. 2010;16:381–382. doi: 10.1038/nm0410-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tinmouth A., Fergusson D., Yee I.C., Hebert P.C. Clinical consequences of red cell storage in the critically ill. Transfusion. 2006;46:2014–2027. doi: 10.1111/j.1537-2995.2006.01026.x. [DOI] [PubMed] [Google Scholar]
- 104.Marik P.E., Corwin H.L. Efficacy of red blood cell transfusion in the critically ill: a systematic review of the literature. Critical Care Med. 2008;36:2667–2674. doi: 10.1097/CCM.0b013e3181844677. [DOI] [PubMed] [Google Scholar]
- 105.Donadee C., Raat N.J., Kanias T., Tejero J., Lee J.S., Kelley E.E., Zhao X., Liu C., Reynolds H., Azarov I., Frizzell S., Meyer E.M., Donnenberg A.D., Qu L., Triulzi D., Kim-Shapiro D.B., Gladwin M.T. Nitric oxide scavenging by red blood cell microparticles and cell-free hemoglobin as a mechanism for the red cell storage lesion. Circulation. 2011;124:465–476. doi: 10.1161/CIRCULATIONAHA.110.008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gladwin M.T., Kim-Shapiro D.B. Storage lesion in banked blood due to hemolysis-dependent disruption of nitric oxide homeostasis. Curr. Opin. Hematol. 2009;16:515–523. doi: 10.1097/MOH.0b013e32833157f4. [DOI] [PubMed] [Google Scholar]
- 107.Liu C., Zhao W., Christ G.J., Gladwin M.T., Kim-Shapiro D.B. Nitric oxide scavenging by red cell microparticles. Free Radic. Biol. Med. 2013;65:1164–1173. doi: 10.1016/j.freeradbiomed.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bennett-Guerrero E., Veldman T.H., Doctor A., Telen M.J., Ortel T.L., Reid T.S., Mulherin M.A., Zhu H., Buck R.D., Califf R.M., McMahon T.J. Evolution of adverse changes in stored RBCs. Proc. Natl. Acad. Sci. USA. 2007;104:17063–17068. doi: 10.1073/pnas.0708160104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Doctor A., Spinella P. Effect of processing and storage on red blood cell function in vivo. Semin. Perinatol. 2012;36:248–259. doi: 10.1053/j.semperi.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hare G.M., Tsui A.K., Crawford J.H., Patel R.P. Is methemoglobin an inert bystander, biomarker or a mediator of oxidative stress-the example of anemia? Redox Biol. 2013;1:65–69. doi: 10.1016/j.redox.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kanias T., Wang L., Lippert A., Kim-Shapiro D.B., Gladwin M.T. Red blood cell endothelial nitric oxide synthase does not modulate red blood cell storage hemolysis. Transfusion. 2013;53:981–989. doi: 10.1111/j.1537-2995.2012.03850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lang K.S., Lang P.A., Bauer C., Duranton C., Wieder T., Huber S.M., Lang F. Mechanisms of suicidal erythrocyte death. Cell Physiol Biochem. 2005;15:195–202. doi: 10.1159/000086406. [DOI] [PubMed] [Google Scholar]