PAKs inhibitors ameliorate schizophrenia-associated dendritic spine deterioration in vitro and in vivo during late adolescence (original) (raw)
Significance
Drug discovery in psychiatry has been limited to chemical modifications of compounds originally discovered serendipitously. Therefore, more mechanism-oriented strategies of drug discovery for mental disorders are awaited. Schizophrenia is a devastating mental disorder with synaptic disconnectivity involved in its pathophysiology. In this study, we studied a biological pathway underlying synaptic disturbance and examined whether p21-activated kinase inhibitors ameliorate the pathology in vitro and in vivo. The beneficial effects of these inhibitors reported here may provide us with an opportunity for drug discovery in major mental illnesses with synaptic disturbance.
Keywords: mechanism-oriented drug discovery, synapse protection
Abstract
Drug discovery in psychiatry has been limited to chemical modifications of compounds originally discovered serendipitously. Therefore, more mechanism-oriented strategies of drug discovery for mental disorders are awaited. Schizophrenia is a devastating mental disorder with synaptic disconnectivity involved in its pathophysiology. Reduction in the dendritic spine density is a major alteration that has been reproducibly reported in the cerebral cortex of patients with schizophrenia. Disrupted-in-Schizophrenia-1 (DISC1), a factor that influences endophenotypes underlying schizophrenia and several other neuropsychiatric disorders, has a regulatory role in the postsynaptic density in association with the NMDA-type glutamate receptor, Kalirin-7, and Rac1. Prolonged knockdown of DISC1 leads to synaptic deterioration, reminiscent of the synaptic pathology of schizophrenia. Thus, we tested the effects of novel inhibitors to p21-activated kinases (PAKs), major targets of Rac1, on synaptic deterioration elicited by knockdown expression of DISC1. These compounds not only significantly ameliorated the synaptic deterioration triggered by DISC1 knockdown but also partially reversed the size of deteriorated synapses in culture. One of these PAK inhibitors prevented progressive synaptic deterioration in adolescence as shown by in vivo two-photon imaging and ameliorated a behavioral deficit in prepulse inhibition in adulthood in a DISC1 knockdown mouse model. The efficacy of PAK inhibitors may have implications in drug discovery for schizophrenia and related neuropsychiatric disorders in general.
A major limitation in current treatment strategies in psychiatry is that drugs used clinically are derivatives of chemicals originally discovered serendipitously (1). Thus, in the past several years, enormous efforts have been made to discover and develop more mechanism-oriented drugs.
Genetic susceptibility factors for schizophrenia that have been identified in the past 10 y have aided understanding of the pathology at the molecular level. Although some conflicting outcomes among different methodologies lead to continuous debate, several factors, including TCF4 (the transcription factor 4), erythroblastic leukemia viral oncogene 4 (ErbB4), zinc finger protein 804A (ZNF804a), and DISC1, are likely to enhance susceptibility to schizophrenia (2–6). Studies that explore molecular and functional interactomes have suggested that a cluster of such factors (e.g., molecular pathways) contributes significantly to functions of the glutamatergic synapse, in accordance with the classic concept that schizophrenia is a disorder of connectivity (7–9).
Several groups have reproducibly reported that DISC1 is expressed in the glutamatergic postsynapses (10–12). In primary neuron culture, prolonged DISC1 knockdown results in the synaptic deterioration (decrease in spine density and size) after transient outgrowth in the pathological course (10). Mouse models that carry two independent Disc1 point mutations show decreased spine density in the glutamatergic synapse in the frontal cortex (13). Synaptic pathology has been frequently observed in brains from patients with schizophrenia (14–16). Furthermore, deficits in the glutamatergic neurotransmission in the pathology of schizophrenia are also demonstrated by studies of brain imaging, neurochemistry, and neuropharmacology (17). Thus, synaptic deterioration elicited by DISC1 knockdown might serve as a cellular model that represents, at least in part, a common pathophysiology of schizophrenia (10, 18).
Thus far, more than one mechanism has been proposed in regard to the regulation of synaptic plasticity and maintenance by DISC1 (10, 19, 20). For example, DISC1 negatively regulates access of Kalirin-7 (Kal-7) to a small GTPase protein Rac1 and contributes to proper control of Rac1 activation and synaptic maintenance: this mechanism participates in the spine change triggered by NMDA-R activation (10). Thus, we hypothesized that modulating the activity of p21-activated kinase (PAK), a key downstream molecule of Rac1 (21, 22), with chemical inhibitors may rescue the synaptic pathology elicited by DISC1 knockdown in primary neuron culture in vitro as well as in the prefrontal cortex in vivo.
Results
DISC1 Knockdown Affects NMDA Receptor-Dependent Synaptic Plasticity.
We previously observed that activation of NMDA receptor (NMDA-R) affects protein interactions involving DISC1 and Kal-7 at the biochemical level by using the withdrawal of amino-5-phosphonovaleric acid (APV), a potent inhibitor of the receptor (10, 23). Thus, the present study further reports its characterization at the cell biological and physiological levels. Time lapse imaging indicated that the majority of the spines immediately underwent enlargement by almost twofold upon NMDA-R activation, which was followed by gradual and partial decrease in size, leading to sustained spine enlargement in neurons with pretreatment of control shRNA (Fig. 1_A_ and Movie S1). In contrast, the spines in neurons pretreated with DISC1 shRNA displayed gradual shrinkage upon NMDA-R activation (Fig. 1_A_ and Movie S2). These structural changes of the spine correlated with the amplitude and frequency of miniature excitatory postsynaptic currents (mEPSC) (Fig. 1_B_). To further study a response upon APV withdrawal at the single spine level, uncaging-evoked excitatory postsynaptic currents (uEPSC) were measured by uncaging 4-methoxy-7-nitroindolinyl-caged-l-glutamate on each spine head. The spines in neurons pretreated with control shRNA displayed an increase in the amplitude of uEPSC upon APV withdrawal, whereas the spines in neurons with DISC1 shRNA showed a clear reduction in amplitude (Fig. 1_C_). These results suggest that knockdown of DISC1 in neuron cultures disturbs plasticity and maintenance of the glutamatergic synapse.
Fig. 1.

DISC1 knockdown affects NMDA receptor (NMDA-R)-dependent synaptic plasticity. (A) Time lapse imaging showing structural plasticity of spine upon APV withdrawal (APV WD). Whereas spines on neurons with control shRNA (control RNAi, arrowheads) underwent spine enlargement, spines with DISC1 shRNA (DISC1 RNAi, arrows) were reduced in size upon NMDA-R activation. The absolute values of spine size and relative size upon APV WD are shown, which suggests that synaptic response between control and DISC1 RNAi-treated neuron are significantly different. (B) Changes of mEPSC upon APV WD in neurons with or without DISC1 RNAi. **P < 0.01, ***P < 0.001 compared with t = 0 min. (C) Changes of uEPSC showing functional plasticity upon APV WD in neurons with or without DISC1 RNAi. APV withdrawal induced spine enlargement and enhancement in synaptic transmission in control neuron (spines 1 and 2), whereas in contrast, there were spine shrinkage and reduction in synaptic transmission in DISC1 RNAi-treated neuron (spines 3 and 4). Changes in uEPSC amplitude (Δamplitude) in each individual spine are shown (control RNAi, n = 16; DISC1 RNAi, n = 16). ***P < 0.001.
Chemical Inhibitors of PAK Block DISC1 Knockdown-Elicited Synaptic Changes Associated with NMDA-R Activation.
We previously reported a biochemical mechanism in which Rac1/PAK1 cascade comes downstream of DISC1 in the spine (10). However, this notion has not been functionally validated yet. To prove this concept experimentally, we use three newly generated PAK inhibitors in this study (Fig. 2). Twenty minutes after APV withdrawal (e.g., acute NMDA-R activation), consistent with the observations in the time lapse examination shown in Fig. 1, we observed a significant decrease in the spine size with DISC1 shRNA, whereas the spine size in controls was increased (Fig. 3 A and B). Treatment with these newly synthesized PAK inhibitors (FRAX355, FRAX486, and FRAX120: 250, 500, and 500 nM, respectively; for 1 h, starting from 40 min before APV withdrawal) prevented the reduction in spine size in the presence of DISC1 shRNA (Fig. 3 B_–_D). Similar protective effects were observed in the presence of W-7 (calmodulin inhibitor) and KN-62 (CaMKs inhibitor) (Fig. 3 B_–_D). In addition, treatment with FRAX355 and FRAX486 as well as W-7 and KN-62 rescued the DISC1 shRNA-treated neurons from a marked decrease in spine density (Fig. 3 B_–_D). FRAX 120 mildly protected neurons from the reduced spine density, but the effects were less complete. Different from W-7 and KN-62, FRAX355 and FRAX486 did not completely block the NMDA-R activation-mediated spine enlargement in the presence of control RNAi, suggesting that these FRAX compounds act less aversely to the synaptic plasticity (Fig. 3 B, C, and E).
Fig. 2.

Characteristics of PAK inhibitors. Chemical structure of three major compounds (A) and pharmacological properties (B).
Fig. 3.

Chemical inhibitors of PAK block DISC1 knockdown-elicited synaptic changes associated with NMDA-R activation. (A) Experimental design. Inhibitors with concentrations of 10 μM for W-7, 10 μM for KN-62, 250 nM for FRAX355, 500 nM for FRAX120, or 500 nM for FRAX486 were added 1 h before APV WD, followed by incubation of APV WD solution containing the inhibitor at same concentration for 20 min. (B_–_E) Morphometry spine analysis showing the effect of each compound on APV WD-induced spine structural change. The absolute values of spine size and density and the relative size and density after APV WD in the presence of each compound [∆size (%) and ∆density (%)] are shown. Beneficial effects of PAK inhibitors to the shrinkage of spine size were shown. In particular, FRAX355 and FRAX486 displayed beneficial influences on spine density without robustly affecting synaptic plasticity. **P < 0.01. ***P < 0.001. (Scale bar, 5 μm.)
PAK Inhibitors Block Development of Dendritic Spine Defects Induced by Prolonged Knockdown of DISC1.
We then hypothesized that these PAK inhibitors could also prevent spine deterioration due to prolonged DISC1 knockdown, in which the DISC1-Kal-7-Rac1 cascade was involved (10). Thus, we added PAK inhibitors to the culture concurrently with DISC1 or control shRNA application (Fig. 4_A_). To determine the protective effect on dendritic spines, we measured spine size and density in the presence of various concentrations of these compounds. All three PAK inhibitors (Fig. 2) completely rescued spine shrinkage resulting from prolonged DISC1 knockdown in a dose-dependent manner (Fig. 4 B and C and Figs. S1 A and B and S2). The PAK inhibitors had little effect on healthy spines because no deteriorating effects were observed in neurons with control shRNA up to the doses more than one hundred times higher than the effective doses for synaptic protection against DISC1 shRNA (Fig. S3). This implies that these compounds have extremely wide therapeutic index windows (dose ratio of beneficial/toxic effects).
Fig. 4.
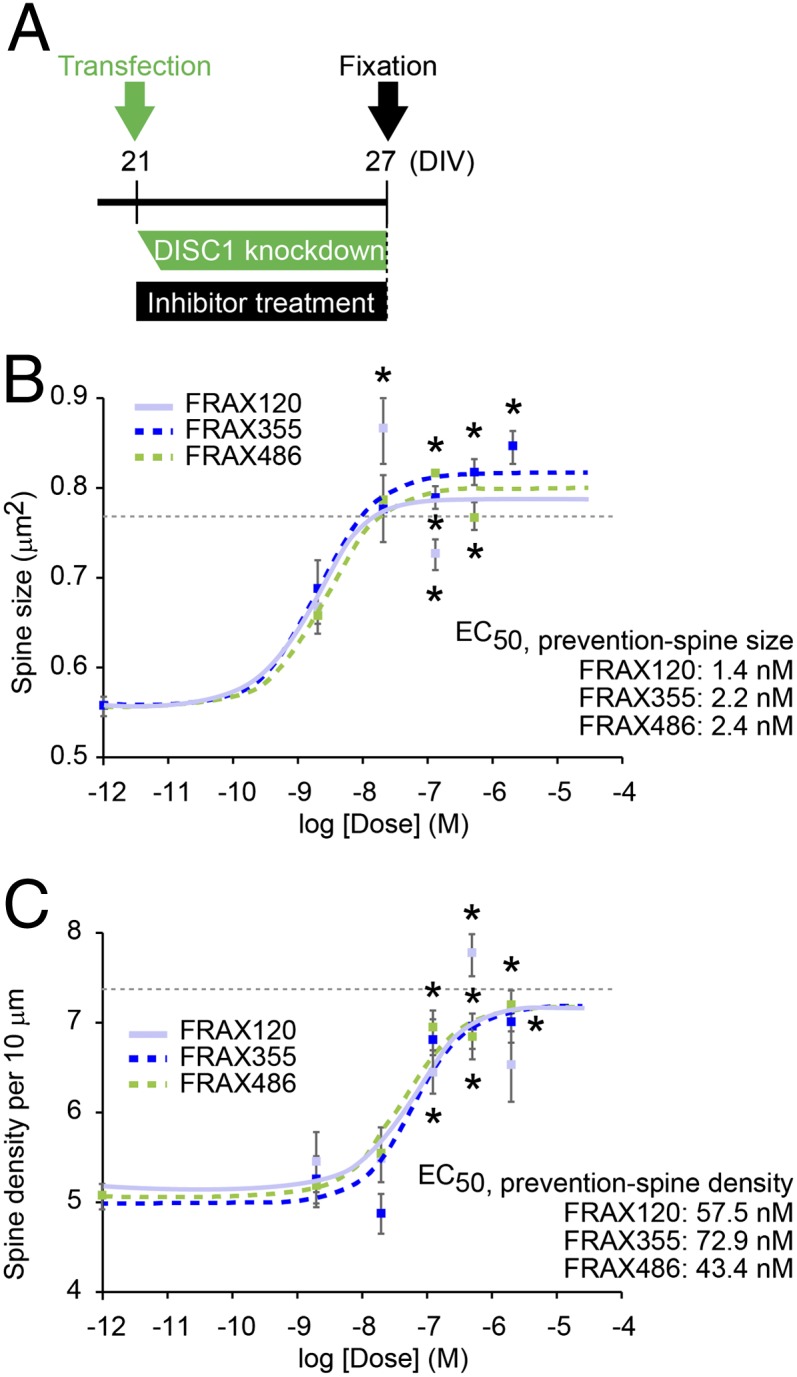
PAK inhibitors prevent DISC1 RNAi-induced spine deterioration (prophylactic effect). (A) Experimental design. PAK inhibitors were added at the time of transfection for DISC1 RNAi and incubated for 6 d before fixation. (B and C) Dose–response curves of FRAX120, FRAX355, and FRAX486 for protective effects on the spine size (B) and spine density (C) in the neurons with DISC1 shRNA (DISC1 RNAi) are shown. Asterisks indicate significant differences between the spines with vehicle control and those with PAK inhibitor, which imply therapeutic effective dose of each compound *P < 0.05. The dotted lines indicate the average of the spine size (B) and the spine density (C) in cells with control shRNA (baselines). “Veh” indicates “Vehicle only” in dose–response curve. _EC_50, half-maximum effective concentration in this prophylactic paradigm, is calculated.
PAK Inhibitors Reverse Preexisting Dendritic Spine Shrinkage Elicited by Prolonged DISC1 Knockdown.
In most therapeutic settings, drugs aimed at treating a disease should be able to reverse an already existing defect rather than simply block the development of deficits. To reconstruct this situation in vitro, we examined whether the PAK inhibitors could reverse preexisting dendritic spine defects induced by DISC1 knockdown (Fig. 5_A_ and Figs. S1 A and C and S4). Cortical neuronal cultures were pretreated with DISC1 or control shRNA for 5 d, a time frame that we had previously shown to be sufficient for full expression of the dendritic spine defects, and then tested the effects of the PAK inhibitors (Fig. 5_A_). All three compounds had dose-dependent beneficial effects on the spine size: FRAX355 and FRAX486 reversed the reduced size of dendritic spines, and although FRAX120 did not completely reverse the size to wild-type levels, it did show some beneficial effects (Fig. 5_B_). In contrast, no compound was able to restore the spine density that had been already reduced by preexisting DISC1 knockdown (Fig. 5_C_). All three compounds even at high concentrations had a negligible effect on the spine structure of neurons with control shRNA under these conditions (Fig. S5).
Fig. 5.
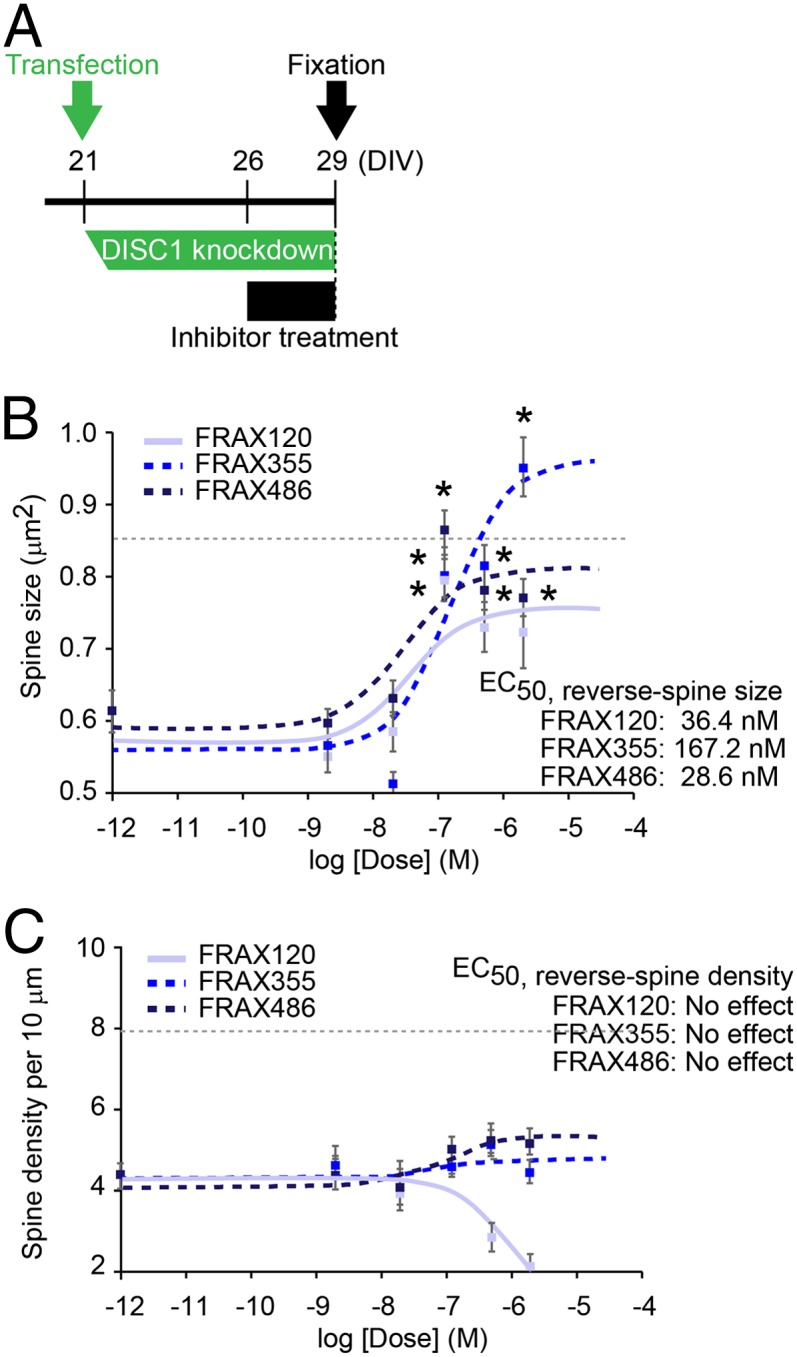
Chemical inhibitors of PAK reverse already existing spine deterioration triggered by DISC1 RNAi (treatment effect). (A) Experimental design. PAK inhibitors were added 5 d after the transfection of either control or DISC1 RNAi, followed by 3 d incubation before fixation. (B and C) Dose–response curves of FRAX120, FRAX355, and FRAX486 for reversal effect on spine size (B) and spine density (C) in the neuron with DISC1 shRNA are shown. Asterisks indicate the significant differences between the spines with vehicle control and those with PAK inhibitor *P < 0.05. The dotted lines indicate the average of the spine size (B) and the spine density (C) in cells with control shRNA (baselines). “Veh” indicates “Vehicle only” in dose–response curve. _EC_50, half-maximum effective concentration in this treatment paradigm, is calculated.
The PAK Inhibitor Ameliorates Adolescent Synapse Loss in the Prefrontal Cortex and Adult Behavior Change in a DISC1 Knockdown Mouse Model.
To validate the beneficial effects of PAK inhibition on the synapse in vivo, we administered FRAX486 into DISC1-knockdown C57BL/6 mice during adolescence (Fig. 6_A_). In this model, by in utero electroporation, DISC1 shRNA is selectively targeted to the cells in a lineage of the pyramidal neurons in the layer II/III of the prefrontal cortex (24, 25). Among the PAK inhibitors tested above, FRAX486 displayed the highest penetrance of blood–brain barrier (Fig. 2_B_). Beneficial influences of this compound were examined at both synaptic and behavioral levels. Compared with control shRNA, DISC1 shRNA led to a marked reduction in the spine density at both postnatal days 35 and 60 (P35 and P60), which was further augmented at P60 (Fig. 6_B_). These observations are in accordance with representative mouse models with genetic mutations in Disc1 that display synaptic deterioration in the adult forebrain (13). Daily administration of FRAX486, but not that of vehicle, between P35 and P60 blocked the exacerbated spine loss during adolescence (Fig. 6_B_). In addition to the significant blockade of spine elimination, we observed a trend of enhanced spine generation by treatment with FRAX486 (Fig. 6_B_). Finally, we examined whether adolescent treatment of FRAX486 might have beneficial influences on aberrant behaviors in the DISC1 knockdown model: FRAX486 treatment ameliorated a deficit in prepulse inhibition in adulthood (Fig. 6_C_).
Fig. 6.

The PAK inhibitor (FRAX486) ameliorates adolescent synapse loss in the prefrontal cortex and adult behavior change in a DISC1 knockdown mouse model. (A) Experimental design. DISC1 or control shRNA was transferred at embryonic day 14.5 (E14.5). Mice with shRNA were subjected to cranial window surgery at postnatal day 34 (P34). Two-photon imaging (2P) was performed both at P35 and P60. Location of cranial window and shRNA distribution (in green) at P60 were shown. Prepulse inhibition (PPI) was tested just before the two-photon imaging at P60. (B) Spine analyses. DISC1 shRNA led to a marked reduction in the spine density at both postnatal days 35 and 60 (P35 and P60), which was further augmented at P60 (**P < 0.01; ***P < 0.001). Daily administration of FRAX486, but not that of vehicle, between P35 and P60 blocked the exacerbated spine loss during adolescence. The effects of FRAX486 were significant in blocking spine elimination (***P < 0.001). In addition, a trend of enhanced spine generation (P = 0.07) was observed by treatment with FRAX486. Spines surrounded by yellow and green dots indicate eliminated (indicated as E) and generated (indicated as G) spines between P35 and P60, respectively. (Scale bar, 2 μm.) (C) Performance in PPI at P60. Deficit in PPI in DISC1-knockdown mice was significantly, although partially, ameliorated by FRAX486 (n = 12 per each group). *P < 0.05, **P < 0.01.
Discussion
This study was designed to determine whether newly developed inhibitors of PAKs would protect against deterioration of dendritic spines elicited by knockdown of DISC1 in vitro and in vivo. PAK inhibitors were tested to block spine deterioration due to prolonged overactivation of Rac1, which is elicited by DISC1 knockdown (10, 26, 27). We found that these inhibitors blocked development of defects in the spines and also partially reversed preexisting spine shrinkage elicited by prolonged DISC1 knockdown in the cortical culture. Different from CaMK/calmodulin inhibitors, FRAX355 and FRAX486 did not completely block the NMDA-R activation-mediated spine enlargement, suggesting that these PAK inhibitors act less aversely to the synaptic plasticity. Moreover, the beneficial effects were observed with very low concentrations of the inhibitors with virtually no toxicity. We also demonstrated that the beneficial effects of these PAK inhibitors were from distinct chemical series, but their biological responses were commonly relevant to PAKs. Thus, although we cannot entirely preclude activity contributions from other kinases, we believe that PAKs are the primary drivers of the biological response.
The genetic role of DISC1 in sporadic cases of schizophrenia is now under debate (28, 29). In contrast, biological studies of DISC1 have been rigorously taken place based on solid discovery of the DISC1 gene in the original Scottish pedigree, indicating that the DISC1 pathway can underlie the endophenotypes relevant to a wide range of psychiatric disorders, in particular, schizophrenia and depression (2). DISC1 interacts with many proteins, and its regulatory roles in synaptic plasticity may be multiple (10, 12, 19, 20). It remains elusive how these multiple mechanisms interact with each other for the overall synaptic phenotypes. Nonetheless, the present study by using good pharmacological agents proposes an idea that the cascade of DISC1 involving Rac1-PAKs may play a pivotal role in these mechanisms in vitro and in vivo.
There is precedence in neurological disorders for which cell models with modulation of genetic factors provide important templates for drug discovery at the initial step. For example, by using cells inducibly expressing mutant Huntingtin and other disease factors, systematic compound screenings have provided some promising leads that are also tested and proven as beneficial in animal models (30, 31). The biological validity is considered more in the second step, whereas the pragmatic efficiency rather than the validity is emphasized in the first step. In the present study, we initially used rat primary neuron culture as the platform in testing the compounds and then further validated the notion in vivo.
Aberrant synaptic pruning during adolescence has been suggested in the pathology of schizophrenia (7, 32–34). In accordance with this hypothesis, dynamic changes in brain morphology and glutamate signaling have also been reported in recent onset schizophrenia cases and subjects in the prodromal stages of schizophrenia (35, 36). In this study, by using two-photon spine imaging, we demonstrated that administration of a PAK inhibitor FRAX486 in late adolescence is sufficient to block deteriorating spine loss and effective in preventing an adult behavioral deficit associated with schizophrenia. Although the concept of preventive medication is of interest, substantial side effects of neuroleptics currently available, such as their effects on metabolism, hamper their use to the prodromal subjects (37). Given that the PAK inhibitor displayed beneficial effects at a low dose without eliciting major adverse effects, we may consider PAK inhibitor as a good target of this early intervention strategy.
There are many other neuropsychiatric disorders with synaptic changes that might benefit from these compounds. The Tonegawa laboratory previously published that PAK inhibition and knockout are protective against synaptic deterioration in an animal model for Fragile X syndrome (38, 39). In addition, several lines of evidence have suggested the involvement of PAKs in Alzheimer’s disease and mental retardation (40–43). Studies that aim to identify rare variants associated with neuropsychiatric disorders may further reveal PAK family genes as genetic factors. Thus, consideration of these compounds in many other neuropsychiatric disorders may also be an important subject in future studies.
Because PAKs are widely expressed in both neuronal and nonneuronal tissues, unexpected effects on nonneuronal tissues might be a potential concern, especially on carcinogenesis. As far as we are aware, PAKs are regarded as therapeutic targets in cancer and immune/allergy-related conditions (44, 45). Although this question requires careful consideration, we expect minimal adverse effects of PAK inhibitors when we target neuropsychiatric disorders.
Materials and Methods
Ethical Observance.
All research, including use and care of animals as well as DNA and genetic engineering work, were approved by The Johns Hopkins University and the University of Tokyo.
Plasmid Construction and Transfection.
H1-RNA polymerase III promoter-driven shRNA against DISC1, which also carries EGFP downstream of the CMV promoter, was previously described (10). Cultured cells were transfected with plasmids by use of LipofectAMINE2000 (Invitrogen). In primary rat cortical neuron culture, 2 μg of pSuper-Venus RNAi were transfected into 1.5 × 105 cells.
Neuron Culture and Treatment.
Dissociated rat cortical neuron cultures were prepared as described previously (10). The procedures for uncaging and electrophysiological experiments are described in SI Materials and Methods. Briefly, for measuring uEPSC, cells were perfused with amino-5-phosphonovaleric acid extracellular solution (APV-ECS) supplemented with 5 mM MNI-glutamate (Tocris Bioscience). Two-photon uncaging laser (720 nm, mode-locked Ti-Sapphire laser) was delivered to the spine head to photolyse the caged-glutamate for 2 ms. Intensity of uncaging laser was 20 mW at the back aperture of the objective lens. Withdrawal of DL-APV (Ascent Scientific) was used for acute chemical activation of NMDA receptors (23, 46). Briefly, 200 μM APV was added to the culture dish 2 d before APV withdrawal (APV WD) and was maintained in the medium. For treatment of APV WD, cells were preincubated in artificial cerebrospinal fluid (ACSF) (in mM: 125 NaCl, 2.5 KCl, 26.2 NaHCO3, 1 NaH2PO4, 11 glucose, 5 Hepes, 2.5 CaCl2, and 1.25 MgCl2) with 200 μM APV for 1 h at 37 °C. Coverslips were then washed in withdrawal medium (ACSF plus 30 μM d-serine, 100 μM picrotoxin, and 1 μM strychnine) and transferred into new withdrawal medium for 20 min. Cells were then immediately fixed for immunofluorescence, and spine morphology was determined as previously described. At least two independent analyses, usually three, were carried out while blinded to transfection conditions until statistical analysis had been done (Fig. S1). Antibodies to GFP (Invitrogen) and Alexa 488-conjugated secondary antibodies (Invitrogen) were used.
PAK Inhibitors.
The small-molecule pyridopyrimidinone inhibitors FRAX355 and FRAX486 were designed through a traditional structure activity relationship approach starting from a distinct chemical series identified in a high throughput screening (HTS) campaign. To find inhibitors of group 1 PAKs, we screened a kinase-focused library of 12,000 compounds. Hits were identified that met the criteria threshold of >50% inhibition at 10 μM. All hits were followed up with a 7 pt dose–response. The results of the HTS campaign identified a class of unsubstituted pyridopyrimidinones that demonstrated moderate group 1 inhibition. Further optimization identified FRAX355 and FRAX486 as group 1 PAK inhibitors. FRAX120 was identified from published literature and is a reported inhibitor of PAKs.
PAK biochemical assays were performed using the Z’Lyte Kinase assay platform (Invitrogen) according to the manufacturer’s protocol. Briefly, the assay system consisted of 50 mM Hepes (pH 7.5); 0.01% BRIJ-35; 10 mM MgCl2; 1 mM EGTA; 2 μM substrate Ser/Thr19 peptide for PAK1 and Ser/Thr20 peptide for PAK2, PAK3, and PAK4 (Proprietary Life Technologies Sequence); PAK enzymes (2.42–30.8 ng for PAK1, 0.29–6 ng for PAK2, 1.5–20 ng for PAK3 and 0.1–0.86 ng for PAK4; actual enzyme amounts depended on lot activity of the enzyme preparation); test compound in DMSO, with the final DMSO concentration in the assay reaction 1%; and ATP at _K_m apparent (50 μM ATP for PAK1 assay, 75 μM ATP for PAK2 assay, 100 μM ATP for PAK3 assay, and 5 μM ATP for PAK4 assay). The total assay volume was 10 μL. Reaction mixtures were incubated at room temperature for 1 h. Following the kinase reaction, 5 μL of development solution A (Life technologies) was added, and the reaction mixture was incubated for an additional 1 h at room temperature. Plates were analyzed in a standard fluorescence plate reader (Tecan) using an excitation wavelength of 400 nm and emission wavelengths of 445 nm and 520 nm. Inhibition of kinase reaction was determined by emission ratio: emission at 445 nm/emission at 520 nm. Pharmacokinetic data for the compounds FRAX120, FRAX355, and FRAX486 were obtained using fasted male C57BL/6 mice with the following conditions: For FRAX486, i.v. dose was 3 mg/kg using a 1 mg/mL solution in 20% (wt/vol) 2-hydroxypropyl-β-cyclodextrin in water, and per oral administration (o.s.) (PO) dose was 30 mg/kg in a 3 mg/mL solution in water. For FRAX355, i.v. dose was 3 mg/kg using a 0.6 mg/mL solution in 10% (vol/vol) dimetylacetamide (DMA) in water, and PO dose was 10 mg/kg in a 2 mg/mL solution in 10% (vol/vol) DMA in water. For FRAX120, i.v. dose was 3 mg/kg using a 0.1 mg/mL solution in 10% (vol/vol) DMA in saline, and PO dose was 10 mg/kg in a 2 mg/mL solution of 20% (vol/vol) PEG400, 1% (vol/vol) Tween80 and 79% (vol/vol) [0.5% (wt/vol) carboxymethyl cellulose in water]. For the in vivo experiment, FRAX486 was intraperitoneally administered [10 μg/BW (g)] once daily from P35 to P60, which provides brain levels at >175 nM.
In Utero Electroporation.
This procedure was performed according the published protocol with minor modifications (24, 25). Briefly, pregnant C57BL/6 mice were anesthetized at embryonic day 14.5 (E14.5) by isoflurane, and shRNA plasmids (control or DISC1 shRNA) together with CAG::Venus vector were injected into the bilateral ventricles. Electrode pulses (35V, 50 ms) were charged bilaterally. Pups that expressed strong Venus fluorescence in the prefrontal cortex of both hemispheres were used for further experiments.
Cranial Window Surgery and in Vivo Two-Photon Spine Imaging.
Mice at postnatal day 35 (P35) were anesthetized by isoflurane with i.p. injection of mannitol (4 μg/g of body weight) and dexamethasone (7 μg/g of body weight) for prevention of brain swelling, and a s.c. injection of ketoprofen (40 μg/g of body weight). The skull was exposed over the area corresponding to the prefrontal cortex, according to the stereotactic coordinates. A drill (φ 1.8 mm) was used to generate open skull window, which corresponded to the size of a circular glass coverslip (φ 1.8 mm, <0.1 mm thickness) (Matsunami Glass). The rim of the coverslip was mounted by dental cement. In vivo two-photon imaging was performed using an upright microscope (BX61WI; Olympus) equipped with a FV1000 laser scanning microscope system (FV1000; Olympus) with a water-immersion objective lens (60×, N.A. 1.0). Femtosecond-pulse Ti-sapphire lasers (MaiTai) were used at 950 nm for imaging. For 3D reconstruction of dendrite images, XY images with 0.8-μm step size were stacked by summation of fluorescence values at each pixel.
Statistics.
All data are means ± SEM of values and representative of two to three separate experiments (Fig. S1_A_). One-way ANOVAs were used, followed by Scheffé post hoc for multiple comparisons. Statistical analyses were performed using SPSS 11.0 software (*P < 0.05; **P < 0.01; ***P < 0.001).
Supplementary Material
Supporting Information
Acknowledgments
We thank Dr. Pamela Talalay for critical reading of this manuscript and Ms. Yukiko Lema for manuscript and figure preparation. This work was supported by Grants MH-084018 (to A.S.), MH-094268 from the Silvio O. Conte Center (to A.S.), MH-069853 (to A.S.), MH-085226 (to A.S.), MH-088753 (to A.S.), and MH-092443 (to A.S.) and grants from Stanley (to A.S.), the S & R Foundation (to A.S.), the RUSK Foundation (to A.S.), National Alliance for Research on Schizophrenia and Depression (to A.S., and A.H.-T.), The Johns Hopkins University Brain Science Institute (to A.S.), the Maryland Stem Cell Research Fund (to A.S.), Japan Society for the Promotion of Science (to Y.A. and A.H.-T.), Howard Hughes Medical Institute (to R.L.H.), and Presto (to A.H.-T.).
Footnotes
Conflict of interest statement: B.V., S.G.D., and D.A.C. are former employees of Afraxis, Inc.
This article is a PNAS Direct Submission.
References
- 1.Ibrahim HM, Tamminga CA. Schizophrenia: Treatment targets beyond monoamine systems. Annu Rev Pharmacol Toxicol. 2011;51:189–209. doi: 10.1146/annurev.pharmtox.010909.105851. [DOI] [PubMed] [Google Scholar]
- 2.Brandon NJ, Sawa A. Linking neurodevelopmental and synaptic theories of mental illness through DISC1. Nat Rev Neurosci. 2011;12(12):707–722. doi: 10.1038/nrn3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bray NJ, Leweke FM, Kapur S, Meyer-Lindenberg A. The neurobiology of schizophrenia: New leads and avenues for treatment. Curr Opin Neurobiol. 2010;20(6):810–815. doi: 10.1016/j.conb.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 4.Stefansson H, et al. Genetic Risk and Outcome in Psychosis (GROUP) Common variants conferring risk of schizophrenia. Nature. 2009;460(7256):744–747. doi: 10.1038/nature08186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kirov G, et al. The penetrance of copy number variations for schizophrenia and developmental delay. Biol Psychiatry. 2014;75(5):378–385. doi: 10.1016/j.biopsych.2013.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stefanis NC, et al. Schizophrenia candidate gene ERBB4: Covert routes of vulnerability to psychosis detected at the population level. Schizophr Bull. 2013;39(2):349–357. doi: 10.1093/schbul/sbr169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hayashi-Takagi A, Sawa A. Disturbed synaptic connectivity in schizophrenia: Convergence of genetic risk factors during neurodevelopment. Brain Res Bull. 2010;83(3-4):140–146. doi: 10.1016/j.brainresbull.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 8.Javitt DC, Spencer KM, Thaker GK, Winterer G, Hajós M. Neurophysiological biomarkers for drug development in schizophrenia. Nat Rev Drug Discov. 2008;7(1):68–83. doi: 10.1038/nrd2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lips ES, et al. International Schizophrenia Consortium Functional gene group analysis identifies synaptic gene groups as risk factor for schizophrenia. Mol Psychiatry. 2012;17(10):996–1006. doi: 10.1038/mp.2011.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hayashi-Takagi A, et al. Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat Neurosci. 2010;13(3):327–332. doi: 10.1038/nn.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kirkpatrick B, et al. DISC1 immunoreactivity at the light and ultrastructural level in the human neocortex. J Comp Neurol. 2006;497(3):436–450. doi: 10.1002/cne.21007. [DOI] [PubMed] [Google Scholar]
- 12.Paspalas CD, Wang M, Arnsten AF. Constellation of HCN channels and cAMP regulating proteins in dendritic spines of the primate prefrontal cortex: Potential substrate for working memory deficits in schizophrenia. Cereb Cortex. 2013;23(7):1643–1654. doi: 10.1093/cercor/bhs152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee FH, et al. Disc1 point mutations in mice affect development of the cerebral cortex. J Neurosci. 2011;31(9):3197–3206. doi: 10.1523/JNEUROSCI.4219-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry. 2000;57(1):65–73. doi: 10.1001/archpsyc.57.1.65. [DOI] [PubMed] [Google Scholar]
- 15.Selemon LD, Goldman-Rakic PS. The reduced neuropil hypothesis: A circuit based model of schizophrenia. Biol Psychiatry. 1999;45(1):17–25. doi: 10.1016/s0006-3223(98)00281-9. [DOI] [PubMed] [Google Scholar]
- 16.Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci. 2011;14(3):285–293. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McGuire P, Howes OD, Stone J, Fusar-Poli P. Functional neuroimaging in schizophrenia: diagnosis and drug discovery. Trends Pharmacol Sci. 2008;29(2):91–98. doi: 10.1016/j.tips.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 18.Ramsey AJ, et al. Impaired NMDA receptor transmission alters striatal synapses and DISC1 protein in an age-dependent manner. Proc Natl Acad Sci USA. 2011;108(14):5795–5800. doi: 10.1073/pnas.1012621108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei J, et al. Regulation of N-methyl-D-aspartate receptors by disrupted-in-schizophrenia-1. Biol Psychiatry. 2014;75(5):414–424. doi: 10.1016/j.biopsych.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Q, et al. The psychiatric disease risk factors DISC1 and TNIK interact to regulate synapse composition and function. Mol Psychiatry. 2011;16(10):1006–1023. doi: 10.1038/mp.2010.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Penzes P, Cahill ME, Jones KA, Srivastava DP. Convergent CaMK and RacGEF signals control dendritic structure and function. Trends Cell Biol. 2008;18(9):405–413. doi: 10.1016/j.tcb.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 22.Rex CS, et al. Different Rho GTPase-dependent signaling pathways initiate sequential steps in the consolidation of long-term potentiation. J Cell Biol. 2009;186(1):85–97. doi: 10.1083/jcb.200901084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie Z, et al. Kalirin-7 controls activity-dependent structural and functional plasticity of dendritic spines. Neuron. 2007;56(4):640–656. doi: 10.1016/j.neuron.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Niwa M, et al. Knockdown of DISC1 by in utero gene transfer disturbs postnatal dopaminergic maturation in the frontal cortex and leads to adult behavioral deficits. Neuron. 2010;65(4):480–489. doi: 10.1016/j.neuron.2010.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kubo K, et al. Migration defects by DISC1 knockdown in C57BL/6, 129X1/SvJ, and ICR strains via in utero gene transfer and virus-mediated RNAi. Biochem Biophys Res Commun. 2010;400(4):631–637. doi: 10.1016/j.bbrc.2010.08.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luo L, et al. Differential effects of the Rac GTPase on Purkinje cell axons and dendritic trunks and spines. Nature. 1996;379(6568):837–840. doi: 10.1038/379837a0. [DOI] [PubMed] [Google Scholar]
- 27.Tashiro A, Minden A, Yuste R. Regulation of dendritic spine morphology by the rho family of small GTPases: Antagonistic roles of Rac and Rho. Cereb Cortex. 2000;10(10):927–938. doi: 10.1093/cercor/10.10.927. [DOI] [PubMed] [Google Scholar]
- 28.Sullivan PF. Questions about DISC1 as a genetic risk factor for schizophrenia. Mol Psychiatry. 2013;18(10):1050–1052. doi: 10.1038/mp.2012.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Porteous DJ, et al. DISC1 as a genetic risk factor for schizophrenia and related major mental illness: Response to Sullivan. Mol Psychiatry. 2014;19(2):141–143. doi: 10.1038/mp.2013.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang W, et al. Compounds blocking mutant huntingtin toxicity identified using a Huntington’s disease neuronal cell model. Neurobiol Dis. 2005;20(2):500–508. doi: 10.1016/j.nbd.2005.03.026. [DOI] [PubMed] [Google Scholar]
- 31.Masuda N, et al. Tiagabine is neuroprotective in the N171-82Q and R6/2 mouse models of Huntington’s disease. Neurobiol Dis. 2008;30(3):293–302. doi: 10.1016/j.nbd.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feinberg I. Schizophrenia: Caused by a fault in programmed synaptic elimination during adolescence? J Psychiatr Res. 1982-1983;17(4):319–334. doi: 10.1016/0022-3956(82)90038-3. [DOI] [PubMed] [Google Scholar]
- 33.McGlashan TH, Hoffman RE. Schizophrenia as a disorder of developmentally reduced synaptic connectivity. Arch Gen Psychiatry. 2000;57(7):637–648. doi: 10.1001/archpsyc.57.7.637. [DOI] [PubMed] [Google Scholar]
- 34.Seshadri S, Zeledon M, Sawa A. Synapse-specific contributions in the cortical pathology of schizophrenia. Neurobiol Dis. 2013;53:26–35. doi: 10.1016/j.nbd.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hayashi-Takagi A, Barker PB, Sawa A. Readdressing synaptic pruning theory for schizophrenia: Combination of brain imaging and cell biology. Commun Integr Biol. 2011;4(2):211–212. doi: 10.4161/cib.4.2.14492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Andreasen NC, et al. Progressive brain change in schizophrenia: A prospective longitudinal study of first-episode schizophrenia. Biol Psychiatry. 2011;70(7):672–679. doi: 10.1016/j.biopsych.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Francey SM, et al. Who needs antipsychotic medication in the earliest stages of psychosis? A reconsideration of benefits, risks, neurobiology and ethics in the era of early intervention. Schizophr Res. 2010;119(1-3):1–10. doi: 10.1016/j.schres.2010.02.1071. [DOI] [PubMed] [Google Scholar]
- 38.Hayashi ML, et al. Inhibition of p21-activated kinase rescues symptoms of fragile X syndrome in mice. Proc Natl Acad Sci USA. 2007;104(27):11489–11494. doi: 10.1073/pnas.0705003104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dolan BM, et al. Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by the small-molecule PAK inhibitor FRAX486. Proc Natl Acad Sci USA. 2013;110(14):5671–5676. doi: 10.1073/pnas.1219383110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen LY, et al. Physiological activation of synaptic Rac>PAK (p-21 activated kinase) signaling is defective in a mouse model of fragile X syndrome. J Neurosci. 2010;30(33):10977–10984. doi: 10.1523/JNEUROSCI.1077-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Allen KM, et al. PAK3 mutation in nonsyndromic X-linked mental retardation. Nat Genet. 1998;20(1):25–30. doi: 10.1038/1675. [DOI] [PubMed] [Google Scholar]
- 42.Zhao L, et al. Role of p21-activated kinase pathway defects in the cognitive deficits of Alzheimer disease. Nat Neurosci. 2006;9(2):234–242. doi: 10.1038/nn1630. [DOI] [PubMed] [Google Scholar]
- 43.Ramakers GJ. Rho proteins, mental retardation and the cellular basis of cognition. Trends Neurosci. 2002;25(4):191–199. doi: 10.1016/s0166-2236(00)02118-4. [DOI] [PubMed] [Google Scholar]
- 44.Van den Broeke C, Radu M, Chernoff J, Favoreel HW. An emerging role for p21-activated kinases (Paks) in viral infections. Trends Cell Biol. 2010;20(3):160–169. doi: 10.1016/j.tcb.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumar R, Gururaj AE, Barnes CJ. p21-activated kinases in cancer. Nat Rev Cancer. 2006;6(6):459–471. doi: 10.1038/nrc1892. [DOI] [PubMed] [Google Scholar]
- 46.Xie Z, Huganir RL, Penzes P. Activity-dependent dendritic spine structural plasticity is regulated by small GTPase Rap1 and its target AF-6. Neuron. 2005;48(4):605–618. doi: 10.1016/j.neuron.2005.09.027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information