Structural mechanism of integrin inactivation by filamin (original) (raw)
. Author manuscript; available in PMC: 2015 Nov 1.
Published in final edited form as: Nat Struct Mol Biol. 2015 Apr 6;22(5):383–389. doi: 10.1038/nsmb.2999
Abstract
The activation of heterodimeric (α/β) integrin is crucial for regulating cell adhesion. Binding of talin to the cytoplasmic face of integrin activates the receptor, but how integrin is properly maintained in resting state to counterbalance its activation for regulating adhesion dynamics remains obscure. We report the structure of cytoplasmic domain of human integrin αIIbβ3 bound to its inhibitor, the immunoglobin repeat 21 of filamin A (FLNa-Ig21). The structure reveals an unexpected ternary complex where FLNa-Ig21 not only binds to previously predicted C-terminus of integrin β3 cytoplasmic tail (CT) but also engages N-terminal helices of αIIb and β3 CTs to stabilize an inter-CT clasp that helps restrain the integrin in a resting state. Combined with functional data, the structure reveals a novel mechanism of filamin-mediated retention of inactive integrin, suggesting a new framework for understanding regulation of integrin activation and adhesion.
Keywords: Integrin, filamin, cell adhesion, NMR
Integrins are a family of major cell surface receptors that mediate essentially every life process by orchestrating adhesion of cells to extracellular matrix (ECM) and promoting many dynamic cell adhesion processes such as migration, spreading, and survival1. Integrins are obligate (α/β) heterodimers with each subunit composed of a large ligand binding domain (ectodomain), a single transmembrane domain, and a short cytoplasmic tail (CT). Integrins can exist in either resting or active state with the latter being capable of binding to ECM and triggering cell-ECM adhesion. The resting state integrin is often depicted as a “naked” form, which may bind integrin activators, e.g., talin, and then convert into the active state via conformational change1–2. However, recent studies have suggested that a more complex mechanism can exist in which critical intracellular integrin inactivators may mediate integrin deactivation or retention of the inactive receptor during cyclical cell adhesion processes such as migration (For most recent review, see ref 3). The underlying molecular basis as to how the inactivators engage integrin to control the dynamic equilibrium between the resting and active state of the receptor remains elusive. One widely proposed mechanism is the competition between inactivator and activator for binding to an overlapping binding site on integrin CT2–3. For example, filamin was shown to compete with talin for binding to an overlapping site at the integrin β CT C-terminus4. It was also proposed that an inactivator may engage inactive state of integrin3 but no information is available as to how such interaction occurs at atomic level and inhibits integrin activation.
The focus of this study is on the integrin inactivator filamin - a large actin cross-linking protein (280kDa) that is known to regulate the cytoskeleton and many dynamic cell adhesion responses including cell migration, spreading, and proliferation5. Filamin contains two N-terminal actin binding domains followed by 24 contiguous immunoglobulin-like (Ig) repeats that engage many protein binding partners. Filamin Ig repeat 21 was previously shown to bind integrin and inhibit the receptor activation4, 6–7. Consistently, ablation or decreased expression of filamin was found to enhance integrin-mediated cell-substrate adhesion in multiple different cell lines4,6,8–10 whereas strengthened filamin-integrin interaction inhibits integrin-ligand interaction and cell migration11. Here, using NMR spectroscopy, we set out to determine the solution structure of platelet integrin αIIbβ3 cytoplasmic domain bound to filamin A Ig repeat 21 (FLNa-Ig21). Surprisingly, the structure reveals a ternary complex where FLNa-Ig21 not only binds to previously predicted C-terminal site of integrin β3 cytoplasmic tail (CT), which was thought to block the talin binding, but also engages two N-terminal helices of αIIb and β3 CTs, which stabilizes an inter-CT clasp that helps restrain the integrin at resting state. The results reveal a novel mechanism of filamin-mediated retention of integrin at a resting state. They also provide a new framework for understanding the dynamic regulation of integrin activation crucial for mediating diverse cell adhesion-dependent physiological and pathological processes.
Results
FLNa-Ig21 binds to both integrin αIIb and β3 CTs
Previous studies showed that filamin recognizes the C-terminus of integrin β CTs4,8,11–12. However, a detailed structural characterization of how filamin may engage the complete integrin cytoplasmic face has not been reported. To address this issue, we decided to use NMR to analyze the filamin A binding to αIIbβ3 – the prototypic integrin whose CT complex has been characterized before13. We first performed heteronuclear single quantum correlation (HSQC) experiment to examine the binding of filamin A Ig repeat 21 (FLNa-Ig21) to 15N-labeled β3 CT K716-T762 (Fig. 1a). As expected, FLNa-Ig21 induced chemical shift changes of the C-terminal integrin β3 CT (β3-C). However, surprisingly, FLNa-Ig21 also induced spectral perturbation (line-broadening) of the N-terminal membrane-proximal region of β3 CT (β3-MP), suggesting that FLNa-Ig21 not only binds to β3-C but also to β3-MP. To further investigate this unexpected binding mode, we designed a construct containing β3-MP (K716-W739, β3-N) but lacking β3-C. Supplementary Fig. 1a shows that purified 15N-labeled β3-N indeed bound to FLNa-Ig21. Surface plasmon resonance (SPR) experiments revealed KD~223µM (Supplementary Fig. 1b). Consistently, SPR experiments also produced sensorgrams of full length β3-CT binding to FLNa-Ig21 that could fit into a two-site binding model with KD1~4.9µM and KD2~150µM respectively (Fig. 1b). The latter may correspond to the β3-N-FLNa-Ig21 interaction (Supplementary Fig 1b) whereas the former may reflect the β3-C-FLNa-Ig21 interaction. This two site binding mode is remarkably reminiscent of the integrin activator talin-F3 binding to integrin β CT13–15, yet FLNa-Ig21 and talin-F3 have completely opposite effects on integrin activation2–3. Supplementary Fig. 1c shows that β3-N, which is bound to FLNa-Ig21, exhibits helical conformation, suggesting that while β3-C occupies the known groove of C and D strands (CD groove) of FLNa-Ig21 to form β-sheet4,8, β3-N helix may dock onto FLNa-Ig21 in a different and nonexclusive mode. To gain more definitive evidence for this binding mode, we designed a FLNa-Ig21-β3-C chimera based on the crystal structure of FLNa-Ig21-β7-C complex (PDB code 2BRQ, see details in the method section). This construct allows β3-C to tightly occupy the CD groove of FLNa-Ig21 as the strand (confirmed in supplementary Fig 1d), which would prevent potential displacement by β3-N if the two β3 CT fragments bound to FLNa-Ig21 in a mutually exclusive manner. Supplementary Fig 1e shows that β3-N, which binds to FLNa-Ig21 weakly, still induced chemical shift changes of FLNa-Ig21 in the 15N-labeled FLNa-Ig21-β3-C chimera, thus providing strong evidence that β3-C and β3-N bind to FLNa-Ig21 in nonexclusive manner.
Figure 1.

Interaction of FLNa-Ig21 with αIIbβ3 CTs. (a) Selected region of HSQC of 0.1mM 15N-labeled β3-CT in the absence (black) and presence (red) of 0.1mM FLNa-Ig21. Strongly perturbed residues are labeled in the spectrum of the free β3-CT. (b) Representative real time SPR sensorgrams of the binding between β3-CT and FLNa-Ig21 (n=2). The sensorgrams were fitted into a two-site binding model with KD1~4.9µM and KD2~150µM respectively. (c) Selected regions of HSQC of 0.1mM 15N-labeled αIIb-CT in the absence (black) and presence (red) of 0.1mM FLNa-Ig21. (d) Representative real-time SPR sensorgrams of the binding between αIIb-CT and FLNa-Ig21 (n=2). The real-time binding curves were fitted using a global fitting algorithm to a 1:1 binding model, resulting in the KD~229µM.
Given that FLNa-Ig21 engages inactive integrin4,6 where αIIb CT and β3 CT associate to form a clasp13,16, the forgoing findings led us wonder if FLNa-Ig21, while binding to β3-CT, might also interact with αIIb-CT. Interestingly, both NMR (Fig. 1c) and SPR (Fig. 1d) experiments showed that FLNa-Ig21 binds to αIIb CT with KD~229µM. Like β3-N, the FLNa-Ig21-bound αIIb-CT also adopts helical conformation in its N-terminal region (Supplementary Fig. 1f–1g).
FLNa-Ig21 stabilizes the αIIb-β3 CT complex
The binding of FLNa-Ig21 to both αIIb and β3 CTs suggests formation of a ternary complex. To examine this possibility, we performed a series of independent binding experiments. (i) Pull-down experiments (Fig. 2a and Supplementary Data Set 1), which showed that while β3 CT fused to maltose binding protein failed to pull-down αIIb CT due to low affinity (KD~368µM for the αIIb-CT-β3-CT complex, see supplementary Fig 2a), it was able to pull-down αIIb CT in the presence of FLNa-Ig21. (ii) SPR experiments (Fig. 2b), which showed that αIIb-CT could readily bind to FLNa-Ig21 pre-saturated β3-CT. (iii) NMR experiments (Fig. 2c), which showed that while FLNa-Ig21 induced chemical shift changes of 15N-labeled αIIb-CT, addition of β3-N induced more spectral changes, indicative of additional binding event. To more definitively confirm the formation of the ternary complex, we designed an FLNa-Ig21-β3-CT chimera construct as we did for FLNa-Ig21-β3-C (supplementary Fig 1d) so that β3 CT bound tightly to FLNa-Ig21. As expected, this FLNa-Ig21-β3-CT chimera still retained the capacity to bind to αIIb-CT (Fig. 2d), indicating that β3-CT and αIIb-CT bind to two distinct regions in FLNa-Ig21. Pull-down experiments further demonstrated that αIIb-CT exhibited stronger binding to FLNa-Ig21-β3-CT chimera than to FLNa-Ig21 (Fig. 2e). Correspondingly, SPR experiments revealed a KD of ~19µM for the former (Fig. 2f) as compared to KD~229µM for the latter (Fig. 1d). The increased affinity is obviously due to the ternary binding of αIIb-CT to FLNa-Ig21 and β3-CT, respectively. Overall, these data indicated that FLNa-Ig21 promotes the αIIb CT-β3 CT complex formation.
Figure 2.

FLNa-Ig21 promotes the ternary complex formation with αIIb-CT and β3-CT. (a) Pull down experiment showing that while MBP-β3 failed to pull down His-tagged αIIb-CT, it did so in the presence of FLNa-Ig21; (b) Co-injection on β3 CT surface. 300µM FLNa-Ig21 followed by 300µM FLNa-Ig21 (green sensorgram). 300µM FLNa-Ig21 followed by 300µM FLNa-Ig21 plus 300µM αIIb CT (red sensorgram). n=2; (c) Selected regions of HSQC spectra of 0.1mM 15N-labeled αIIb-CT in the absence (black) and presence of 0.1mM FLNa-Ig21 (red), and presence of 0.1mM FLNa-Ig21 and 0.2mM β3-N; (d) Selected regions of HSQC spectra of 0.1mM 15N-labeled αIIb-CT in the absence (black) and presence of 0.1mM FLNa-Ig21-β3-CT chimera (red). (e) Pull down experiment showing that while GST-FLNa-Ig21 failed to pull down αIIb CT due to low affinity, GST-FLNa-Ig21-β3 CT chimera effectively pulled down αIIb CT. (f) SPR experiment showing that FLNa-Ig21-β3 CT binds to αIIb CT at KD~19µM. n=2.
Structure of FLNa-Ig21-αIIb CT-β3 CT ternary complex
To obtain a definitive 3D atomic view of how FLNa-Ig21 engages both αIIb-CT and β3 CT, we set out to solve the total structure of the ternary complex. β3-CT has low solubility13 and substantial line-broadening problems upon binding to FLNa-Ig21 (Fig. 1a). We therefore screened β3-CT mutants and found that a L717K-L718K mutant exhibited substantially less line-broadening yet maintained high solubility without affecting the binding of FLNa-Ig21 (supplementary Fig.2b and 2c) and still promoted the ternary complex formation (supplementary Fig. 2d). An array of NMR-based heteronuclear and homonuclear experiments were carried out for resonance and NOE assignments of the ternary complex (see the method section). Complex interfaces were first defined by definitive intermolecular NOEs (Supplementary Fig. 2e) and chemical shift mapping data, and further validated by mutagenesis data (Supplementary Table 1). A superposition of 20 lowest energy structures is shown in Fig. 3a (see statistics in Table 1). FLNa-Ig21 adopts the canonical Ig fold as previously reported (Fig. 3b)4,7–8. However, interestingly, while the CD groove of FLNa-Ig21 binds canonically to β3-C Y747-F754 as a strand, an extended surface involving N-terminal short helix, BC loop, C strand, and FG loop grabs both αIIb-N and β3-N helix, support the formation of an αIIb-N-β3-N helical clasp (Fig. 3b). Fig. 3c highlights critical interface residues involved in the FLNa-Ig21-αIIb-N-β3-N network (see also supplementary Fig. 3). From αIIb CT to FLNa-Ig21, the interactions are predominantly hydrophilic including several potential hydrogen bond and salt-bridge pairs: αIIb W988 backbone-FLNa-Ig21 N2312 side chain CONH2, αIIb K994 NH3+-E2313 backbone carbonyl group, αIIb R995 backbone carbonyl-FLNa-Ig21 E2313 carboxyl group, αIIb R997 guanidyl group-FLNa-Ig21 E2276 carboxyl group. From β3 CT to FLNa-Ig21, β3-N helix utilizes I719 and T720 CβH-CγH3 to form a hydrophobic core with FLNa-Ig21 A2268 whereas β3 T720 hydroxyl CβOH group forms potential hydrogen-bond with backbone amide of FLNa-Ig21 G2269. These contacts properly orient the αIIb-N and β3-N helices to form an inter-helical clasp involving hydrophobic contact between αIIb W988-β3 I719, potential hydrogen bond between αIIb R995 quanidyl group-T720 CβOH, and salt-bridge between αIIb R995-β3-D723. The latter has been widely presumed to be crucial for restraining the resting integrins13,16.
Figure 3.
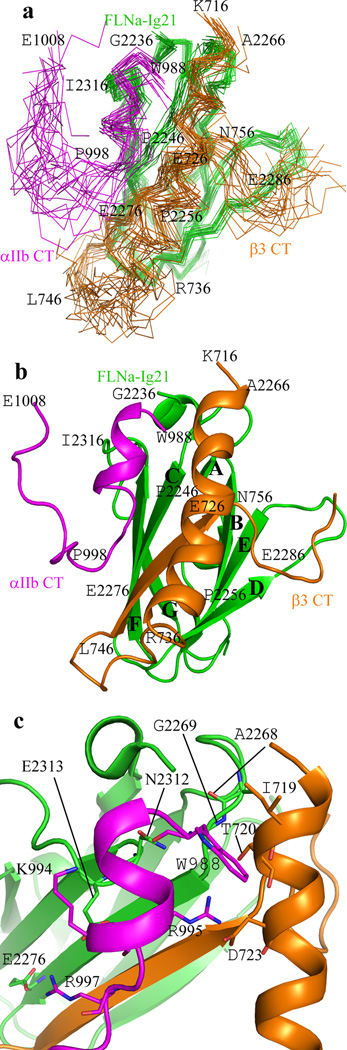
Structure of FLNa-Ig21-αIIb-CT-β3-CT ternary complex. (a) Superposition of 20 calculated FLNa-Ig21(green)-αIIb-CT(pink)-β3-CT(brown) complex structures with lowest energies, showing a well-defined structure. (b) Corresponding cartoon representation of the structure in (A) with the lowest energy. (c) Expanded regions of the complex interface based on (b).
Table 1.
NMR and refinement statistics for FLNa-Ig21-αIIb-CT-β3-CT complex structure
| FLNa-Ig21-αIIb-CT-β3-CT | |
|---|---|
| NMR distance and dihedral constraints | |
| Distance constraints | |
| Total NOE | |
| Intra-residue | 425 |
| Inter-residue | |
| Sequential (|i – j | = 1) |
| Medium-range (|i – j | < 4) |
| Long-range (|i – j | > 5) |
| Intermolecular | 31 |
| Hydrogen bonds | 157 |
| Total dihedral angle restraints | |
| ϕ | 31 |
| ψ | 31 |
| Structure statistics | |
| Violations (mean ± s.d.) | |
| Distance constraints (Å) | 0.08265 ± 0.00728 |
| Dihedral angle constraints (°) | 0.67119 ± 0.07380 |
| Max. dihedral angle violation (°) | 0.839 |
| Max. distance constraint violation (Å) | 0.211 |
| Deviations from idealized geometry | |
| Bond lengths (Å) | 0.00383 ± 0.00007 |
| Bond angles (°) | 0.48340 ± 0.00741 |
| Impropers (°) | 0.37462 ± 0.00897 |
| Average pairwise r.m.s. deviation** (Å) | |
| Heavy | 1.478 ± 0.198 |
| Backbone | 1.013 ± 0.212 |
The specific FLNa-Ig21 interfaces with αIIb CT and β3 CT are consistent with extensive mutagenesis data (supplementary Table 1). Notably, supplementary Fig.4a and 4b show that FLNa-Ig21 E2276A mutation, while retaining the binary FLNa-Ig21-β3-CT interaction, disrupted the FLNa-Ig21-αIIb-CT interaction. Also, the αIIb CT K994E-R997E mutation abolished the αIIb CT binding to WT FLNa-Ig21 (Supplementary Fig. 4c) without disrupting the αIIb-β3 CT interface (Fig. 3c and Supplementary Fig. 4d). Supplementary Fig. 4e shows that FLNa-Ig21 A2268K specifically disrupted the FLNa-Ig21 interaction with β3-N while retaining the binary FLNa-Ig21 binding to αIIb CT (Supplementary Fig. 4f).
FLNa-Ig21 promotes αIIbβ3 transmembrane heterodimerization
Examination of the structure of the FLNa-Ig21-αIIbβ3 CT complex reveals that FLNa-Ig21 would be situated near the juxta-transmembrane region when bound to the integrin CT clasp (Fig. 4a). In support of this prediction, Fig.4b and 4c show that FLNa-Ig21 binds weakly to large unilamellar vesicle (LUV) as judged by small chemical shift changes of H2239, K2240, and R2242. These residues form a positively charged surface with the integrin transmembrane border residues αIIb-CT K989 and β3-CT K716, providing a topology for how FLNa-Ig21 is engaged with the membrane-proximal regions of αIIbβ3 on the membrane surface (Fig. 4a). To further examine how filamin may engage integrin in a membrane environment, we performed the HSQC experiment of 15N-labled αIIb transmembrane-cytoplasmic domain (TMCD) bound to unlabeled β3 TMCD in the absence and presence of FLNa-Ig21. As previously reported17, we found that a dynamic equilibrium exists between free αIIb (or β3) TMCD and αIIb TMCD complexed with β3 TMCD (Fig.4d and 4e). Remarkably, FLNa-Ig21 increased the population of the αIIb-β3 TMCD complex in bicelles as judged by the signal intensity enhancement of the complex signal and the signal reduction of the free form (Figs. 4d–4f), demonstrating the role of filamin in promoting the integrin transmembrane heterodimerization by stabilizing the CT clasp.
Figure 4.

A topology of FLNa-Ig21 bound to the αIIbβ3 CT heterodimer at the inner membrane surface. (a) Superposition of FLNa-Ig21-αIIbβ3-CT complex with αIIbβ3 transmembrane-cytoplasmic heterodimer (PDB code 2KNC) showing a cluster of positively charged residues H2239, K2240, and R2242 from FLNa-Ig21, αIIb-CT K989, and β3-CT K716 at the transmembrane-cytoplasmic border; (b). Selected regions of HSQC of 0.1mM 15N-labeled FLNa-Ig21 in the absence (black) and presence (red) of 1mM LUV vesicle showing the selective perturbation of the membrane binding site of FLNa-Ig21 involving the highlighted residues in Fig. 4A; (c) HSQC data indicating that FLNa-Ig21 K2240A mutation disrupts the FLNa-Ig21-membrane interaction. (d) Selective M987 signal from HSQC spectra of 0.1mM 15N-labeled αIIb TMCD bound to unlabeled β3 TMCD (1:1) in the absence (black) and presence (red) of equimolar FLNa-Ig21. The spectra were collected in 50mM POPC/POPS/DHPC (q=0.3) bicelle in 25mM HEPES, 5%(v/v) D2O, 0.02% NaN3, pH 7.4. (e). Selective G991 peak from the same HSQC spectra in (d) changed in the same trend as M987 in (d). (f). The intensity ratio (complex/free form) of M987 and G991 from the HSQC spectra of (d) and (e) in the absence (red) and presence of FLNa-Ig21.
FLNa-Ig21 mediates retention of inactive integrin
The forgoing combined biochemical and structural data strongly indicate that FLNa-Ig21 stabilizes inactive integrin by favoring integrin CT clasp formation. This is consistent with previous functional data where transfection of integrin αIIbβ3 into CHO cells led to basal level integrin activation whereas co-transfection of FLNa-Ig21 with αIIbβ3 inhibited such integrin activation6. To gain functional evidence for this possibility, we performed a series of mutagenesis experiments. Fig. 5a shows that FLNa-Ig21 E2276A and A2268K mutations, which expressed similarly as WT FLNa-Ig21 (supplementary Fig. 4g) but disrupted the FLNa-Ig21 interaction with the integrin αIIb and β3 membrane-proximal helices respectively (supplementary Fig.3a and 3e), had reduced capacity in inactivating integrin as compared to wild type FLNa-Ig21. Interestingly, the αIIb (K994E-R997E) mutation that disrupts the αIIb binding to FLNa-Ig21 (supplementary Fig. 3c) caused substantial constitutive enhancement of integrin αIIb(K994E-R997E)β3 activity as compared to the WT integrin (Fig. 5b). Since endogenous filamin is likely bound to and stabilizes inactive integrin, disrupting the filamin-αIIb CT interaction by this mutation would clearly destabilize the integrin CT clasp and shift the equilibrium to the active state of integrin, thus leading to the enhancement of integrin activation. Consistent with this interpretation, the PAC1 binding of αIIb(K994E-R997E)β3 mutant was less inhibited (~20%) than WT integrin (~40%) by FLNa-Ig21 (Fig. 5b). These functional data provide strong supporting evidence that FLNa-Ig21 mediates retention of integrin in an inactive state.
Figure 5.

Functional evidence of FLNa-Ig21-mediated inhibition on integrin activation. Data are normalized for differences in surface expression of αIIbβ3 integrin as measured by 2G12 antibody. In all experiments, EGFP or EGFP-tagged proteins are transiently expressed with αIIbβ3. RFI values are Mean±S.E.M of fold change in PAC-1 binding over transiently expressed αIIbβ3 and EGFP (scaled to 1), from multiple independent experiments. Error bars in all experiments represent standard error of mean. (a) Integrin activation assay showing the different effects of talin head (talin-H), FLNa-Ig21, and the FLNa-Ig21 mutants (E2276A, F21EA; A2268K, F21AK) on the activation state of the transiently expressed αIIbβ3 (n=4). For each experiment, 3 transfections were set up that were processed independently as 3 biological replicants. The difference between vector (EGFP) and FLNa-Ig21 is significant (p<0.001). The difference between vector (EGFP) and E2276A (F21EA) or A2268K (F21AK) is not significant (p>0.05). (b) Integrin activation assay showing that transiently expressed αIIb-K994E-R997E-β3 mutant, αIIb(KREE)β3, exhibits substantially enhanced PAC-1 binding as compared to the WT integrin (p<0.05). N=2; 2 transfections for each condition were set up that were processed independently as 3 biological replicates. (c). Integrin activation assay showing that FLNa-Ig9 (p<0.05), Ig12 (p<0.001), Ig17 (p<0.001), Ig19 (p<0.001) exert inhibitory effect on the PAC-1 binding of the transiently expressed αIIbβ3. FLNa-Ig4 did not have significant inhibitory effect (p>0.05). n=2; 2 transfections for each condition were set up that were processed independently as 3 biological replicates.
Multiple filamin repeats inhibit integrin activation
In addition to FLNa-Ig21, multiple repeats in filamin were previously found to bind integrin β CT12. This led us to wonder if they function similarly as FLNa-Ig21. Supplementary Figs. 5a–h show that just like FLNa-Ig21 (Fig. 1), FLNa Ig 9, 12, 17, and 19 all bind to both αIIb CT and β3-N. FLNa-Ig4 is unstable12, preventing its precise measurement of binding to integrin CTs. Remarkably, Fig. 5c shows that FLNa-Ig9, 12, 17, and 19 all exerted the inhibitory effect on integrin activation. FLNa-Ig4 did not have significant effect likely due to its structural instability or weaker binding to integrin. Interestingly, the small hydrophilic and/or positively charged region that may bind to membrane as shown in FLNa-Ig21 containing H2239, K2240, and R2242, is highly conserved among these repeats12. Based on biochemical and NMR data, we previously proposed that full length filamin may locally enrich inactive integrins via multiple similar repeats12. The data in Fig. 5c provides functional evidence to support this mechanism, which may spatiotemporally favor integrins to bind multivalent ligands such as fibrinogen and fibronectin upon integrin activation.
Discussion
While emerging evidence has indicated the existence of integrin inactivators in dynamic regulation of integrin function3, the underlying molecular principle of these inactivators remained elusive. In this study, we have succeeded in determining the NMR structure of FLNa-Ig21 bound to integrin αIIbβ3 CT heterodimer. Our structure reveals for the first time that an inactivator may bind and stabilize an inactive integrin by stabilizing the integrin CT clasp, which is known to be crucial for restraining the resting state integrin13–19. It was widely thought that inactive integrin could be purified in fully “naked” state, but recent structural studies have revealed that purified integrin αIIbβ3 from resting state platelets contains a subpopulation of active state probably reflecting the intrinsic conformational equilibrium between inactive (bent) and active state (extended) of the integrin (see Fig S1E vs 1F in ref 20). This was reflected in our activation assay in Fig 5a where transiently expressed integrin αIIbβ3 exhibited basal level PAC1 binding. The basal level of integrin activation is unlikely totally caused by talin since endogenous talin is supposed to be autoinhibited21–23 and randomly distributed in the unstimulated cells24. Rather, we believe that the activation is at least partially caused by a subpopulation of transiently expressed “naked” active integrin that is in equilibrium with inactive integrin. Our interpretation is supported by several lines of evidence: (i) inhibition of this activation by filamin that probably shifts the equilibrium to inactive state of integrin (Fig. 5a). As shown before, the same assay does not reveal an inhibitory effect of filamin on the talin-mediated integrin activation (see Fig 2a in ref 25) probably due to much higher affinity of talin binding to integrin and membrane than filamin; (ii) specific point mutation in filamin, which disrupted its binding to αIIb CT (Supplementary Fig. 4a–b) without affecting talin binding to β3 CT, impaired the ability of filamin to inhibit integrin activation (Fig. 5a); (iii) point mutation in αIIb CT (K994E-R997E), which disrupted the filamin-αIIb CT interaction but not the talin-β3 CT interaction, enhanced constitutive integrin activation and filamin had reduced capacity to inhibit the mutant integrin (Fig. 5b).
Upon cellular stimulation, talin is known to rapidly localize to the plasma membrane24. By contrast, filamin is mostly situated near plasma membrane in unstimulated cells and then rapidly redistributes into the cytosol upon integrin activation26. Such reciprocal redistribution is consistent with our findings, supporting a model that in unstimulated cells, filamin stabilizes the integrin CT clasp to prevent spontaneous activation of the receptor whereas cellular stimulation induces membrane targeting and unmasking of talin to bind and activate integrin. The membrane-localized talin in its active state may then activate integrin by directly competing with filamin. Such competition was previously suggested to occur at an overlapping binding site in the C-terminus of integrin β CT4. However, our structure suggests an important revision of this competition mechanism where talin would compete with filamin for binding to both the C-terminal and membrane-proximal regions of β CT (Fig. 6a vs Fig. 6b). In addition to this talin-filamin competition pathway, other pathways may exist at certain cellular conditions. For example, migfilin, a cytoskeletal adaptor, was previously shown to bind filamin and displace it from integrin, thereby facilitating integrin activation6,12. Based on our data, such displacement would destabilize the integrin CT clasp, thus favoring the equilibrium shift from resting to active state of the receptor. The displacement may further promote the talin binding to integrin β CT and induce the integrin CT unclasping13,16, leading to enhanced integrin activation. Interestingly, migfilin was shown to be recruited by kindlin27– a well-established integrin co-activator that also binds to integrin β CT2,3. Mice with the ablation of migfilin are viable, although with defects in migration28 and bone remodeling29. It remains to be determined if there exist additional filamin binding proteins that may act similarly as migfilin to regulate integrin activation.
Figure 6.

Comparison of FLNa-Ig21 bound integrin αIIbβ3 CT complex (a) with talin-F3 bound β1 CT (b) (derived from PDB code 3G9W) showing how FLNa-Ig21 may compete with both β3-MP and β3-C for binding to talin-F3. Note that the C-terminus of β CT is β-strand (brown) when bound to FLNa-Ig21 (a) versus an extended loop when bound to talin-F3 (b), and thus the two binding events are mutually exclusive. Also binding of talin-F3 with β3-MP (yellow helix) in (b) would also cause steric clash with FLNa-Ig21 bound to the β3-MP (Cyan + Brown) in (a). The helical region in brown is involved in the interaction with FLNa-Ig21.
Lau et al previously suggested that the membrane-proximal regions of αIIbβ3 CTs are embedded in the membrane in the absence of any regulators17. Using membrane-mimetic bicelles, the authors showed that αIIb membrane-proximal CT adopts a reverse turn that packs against the β3 membrane-proximal CT helix17 (supplementary Fig 6a). Such structure is different from our filamin bound αIIbβ3 CT complex where both αIIb and β3 membrane-proximal regions adopt helical conformation and pack against each other (supplementary Fig 6b). The structural difference may be due to that the αIIbβ3 TMCD structure by Lau et al17 was determined in the presence of bicelles lacking filamin whereas ours was determined in cytosolic condition containing filamin. Interestingly, our structure is similar to other structures previously determined in cytosolic state13 (supplementary Fig 6c) and in organic solvent19 in the absence of filamin (supplementary Fig. 6d) and our data demonstrated that filamin stabilizes these structures, notably the membrane-proximal CT clasp. It is clear that the membrane-proximal regions, if inserted into the membrane as suggested before17 (supplementary Fig. 6a), would be inaccessible to filamin and possibly other cytosolic integrin inactivators such as Sharpin30 and CIB31. Indeed, our data showed that FLNa-Ig21 only weakly interacts with the membrane surface via polar interactions (Fig.4b and 4c), allowing the filamin interaction with the αIIbβ3 CT complex in the cytosolic state (Fig. 2). The cytosolic engagement of filamin not only stabilizes the αIIbβ3 CT complex (Fig. 2) but further promotes the integrin transmembrane heterodimerization in membrane bicelles (Fig 4d–4f). Given that FLNa-Ig21 only has weak polar interaction with the membrane surface instead of inserting into the membrane (Fig. 4b), we conclude that in the filamin-stabilized inactive integrin heterodimer, the membrane-proximal regions of αIIbβ3 CTs are within the cytosol and not buried in the membrane. It is possible that upon release of filamin by migfilin or by talin or other potential regulators, these regions may find the hydrophobic environment of the membrane more favorable and insert into the membrane, thereby triggering conformational change and ultimate activation of integrin. Further investigation is clearly necessary to elucidate this possibility.
In summary, we have determined, to our knowledge, the first 3D structure of an integrin inactivator bound to the resting state integrin α/β CT heterodimer. The structure suggests a novel mechanism where filamin favors retention of integrin in an inactive state. Such filamin-stabilized inactive integrin would prevent spontaneous integrin activation in resting cells, and the pathological consequences of spontaneous integrin activation such as thrombosis and tumor metastasis9. It may also allow tight control of interconversion between the resting and active state of integrin, which is necessary for dynamic regulation of cyclic processes such as cell migration. Given that multiple filamin repeats were found to bind and inhibit integrin (Fig. 5c), it is also possible that filamin clusters inactive integrins12 and that the removal of filamin from clustered integrins may spatiotemporally promote their binding to multivalent ligands such as fibrinogen and fibronectin. Finally, while the structural mechanisms of other integrin inactivators such as Sharpin, CIB, and MDG12–3 remain to be determined, the fact that they all bind to the membrane-proximal region of integrin CTs suggests that they may share a common binding mode favoring the conformation of the resting state of integrins and/or preventing the receptor binding to talin. Recent studies on Sharpin have provided excellent functional evidence for such possibility30. Hopefully more data will be reported in near future, allowing a thorough understanding of how these integrin inactivators regulate dynamic adhesion and migration of a variety of cells.
Online Methods
Plasmid constructs, protein/peptide preparation, and NMR sample preparation
Human FLNa-Ig9, 12, 17, 19, and 21 were cloned into pGST parallel vector with glutathione S-transferase as fusion tag and expressed in E.coli BL21 (DE3) as previously described7. For FLNa-Ig21-β3-C and FLNa-Ig21-β3-CT chimera proteins, cDNAs of either β3-C or β3 CT were inserted into GST-FLNa-Ig21 plasmid construct at FLNa-Ig21’s N-terminus using QuikChange Site-Directed Mutagenesis Kit. Ten extra amino acids (GASGSGASGS) were included between Ig21 and β3 as a linker to provide flexibility for binding between FLNa-Ig21 and β3. Other FLNa-Ig21 mutants and αIIb K994E/R997E mutant were also generated using QuikChange Site-Directed Mutagenesis kit. αIIb (E960-E1008)/β3 (K689-T762) encompassing complete transmembrane and cytoplasmic domain (TMCD) were expressed and purified as described before19. Cells were induced at room temperature for 16 hrs with 0.6mM IPTG, after A600 reached 0.6. The protein was purified on glutathione sepharose 4B resin to homogeneity and cleaved with TEV to release the fusion partners. The protein mixtures were further loaded onto the glutathione sepharose 4B resin to separate GST from the desired protein, and the eluted fractions were then subjected to gel filtration to remove trace GST to yield homogeneous FLNa-Ig21. The cDNA of integrin αIIb cytoplasmic tail (CT) (W988-E1008) and β3 membrane proximal region (β3-N) (K716-T762) were inserted into pET31b vector (Novagen, Inc.) that fuses small peptides to an insoluble protein ketosteroid isomerase (KSI). The fused peptides were expressed as an inclusion body with KSI. Purification of the peptides including the cleavage of KSI by CNBr was performed according to the manufacturer’s instructions. Unlabeled αIIb-CT and β3-N peptides were synthesized by Biotechnology core of Lerner Research Institute. All integrin peptides were further purified by reverse phase HPLC. Protein mutagenesis was conducted using a QuikChange Site-Directed Mutagenesis Kit from Stratagene. Isotope-labeled proteins or peptides were prepared as described before12.
All HSQC binding experiments used 0.1mM 15N-labeled proteins. In the case of FLNa-Ig21/membrane binding, LUV membrane was prepared as described previously19. For structure determination, six NMR samples were prepared for the multi-dimensional NMR experiments in 25mM sodium phosphate pH 6.4, 5mM NaCl, 1mM TCEP: (1) 1mM 15N/13C-labeled FLNa-Ig21 in the presence of 1mM unlabeled L717K/L718K β3 CT and 1mM unlabeled αIIb CT; (2) 1mM 15N-labeled L717K/L718K β3-CT in the presence of 1mM unlabeled FLNa Ig21 and 1mM unlabeled αIIb CT; (3) 2mM unlabeled αIIb-CT in the presence of 0.1mM GST-FLNa-Ig21-β3-CT chimera; (4) 1mM 15N/2H-labeled FLNa-Ig21 in the presence of 1mM unlabeled L717K/L718K β3-CT; (5) 1mM 15N/2H-labeled FLNa-Ig21 in the presence of 1mM unlabeled αIIb-CT; (6) 2mM unlabeled β3-N in the presence of 0.1mM GST-FLNa-Ig21.
NMR experiments
All NMR experiments were performed on Bruker 600MHz, 800MHz, and 900MHz spectrometers each equipped with a triple resonance probe. All experiments were performed at 25°C in 25mM Sodium phosphate pH 6.4, 5mM NaCl. Standard triple resonance experiments were used for assigning FLNa-Ig21 and β3-CT whereas 2D NOESY/TOCSY were used to assign β3-N and αIIb-CT. For structural analyses, the following NOESY experiments were performed: (A) 3D 13C-15N edited NOESY experiment (120 ms mixing time) using sample (1); (B) 3D 15N edited NOESY experiment (120 ms mixing time) using sample (2); (C) 2D transferred NOE experiment (400 ms mixing time) using sample (3); (D) 2D transferred NOE experiment (400 ms mixing time) using sample and sample (6); (E) 3D 15N edited NOESY experiments (150 ms mixing time) using sample (4); (F) 3D 15N edited NOESY experiments (150 ms mixing time) using sample (5). For 2D transferred NOE experiments, additional mixing times 50ms and 150ms were also run to identify potential spin diffusion effects. Spectra were processed by NMRPipe32, and visualized and analyzed by PIPP33 and SPARKY (T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco). PASA software was used in the FLNa-Ig21’s sequential assignment34. 100% of backbone resonances were assigned for all subunits including FLNa-Ig21, αIIb CT, and β3 CT. The side chain assignments are FLNa-Ig21 (93%), αIIb CT (89%), and β3 CT (88%). For NOEs, 398 were assigned between side chains, 856 were assigned between backbones and side chains, and 82 were assigned between backbones. 5 unambiguous intermolecular backbone-backbone NOEs and 20 unambiguous intermolecular NOEs between backbones (one subunit) to side chains (another subunit) were assigned.
For chemical shift mapping, weighted shifts of 1H-15N HSQC spectra were calculated using the equation: Δδ(HN,N) = ((ΔδHNWHN)2 + (ΔδNWN)2))1/2, where WHN and WN are weighting factors for the HN and N shifts, respectively (WHN =1; WN = 0.154), and Δδ = δbound – δfree.
Structure determination
X-plor-NIH35 was used for calculating the structure. NOE constraints were obtained using various NOESY experiments. Dihedral angle restraints for FLNa-Ig21 derived from the TALOS program36. After we calculated a preliminary structural model using intermolecular NOE distance restraints and ambiguous chemical shift mapping-based constraints, we designed structure-based mutations in the interfaces of the ternary complex to cross-validate the structure using HSQC experiments. Mutation sites that either disrupt or weaken the interactions were used as ambiguous restraints to further refine the structure based on the protocol by Clore and Schieters, 200337. Hydrogen-Deuterium exchange-based experimental hydrogen bonding constraints were also used to further refine the secondary structure at the final stage of the structure calculations. Structure quality was evaluated using the program PROCHECK38.
Pull down experiments
β3-CT fused to maltose binding protein (MBP-β3) was incubated with maltose beads (%) in a falcon tube for 20 minutes at 4°C. Then, beads were transferred into a mini column and subjected to extensive washing (at least 20 times bead volume) to remove unbound MBP-β3. Beads were transferred into clean falcon tubes and incubated with binding partners (either His-tagged αIIb-CT alone or FLNa-Ig21 mixed with His-tagged αIIb-CT) for 20 minutes at 4°C. Then, beads were transferred into mini columns and subjected to extensive washing (at least 20 times bead volume) to remove unbound ligands. The beads were mixed with SDS and boiled for 5 minutes at 100 °C, and subjected to electrophoresis. His-tagged αIIb-CT was visualized by immunoblotting with a 6×His antibody (Cell Signaling). αIIb pull down by GST-FLNa-Ig21 or GST-FLNa-Ig21-β3 CT chimera used the same method as above but visualized by Coomassie blue staining.
Surface Plasmon Resonance (SPR) experiment
All SPR experiments were conducted on the BIAcore 3000 system at the Molecular Biotechnology core of the Lerner Research Institute. The experiments were carried out at 25°C, using the buffer, 25 mM phosphate buffer, 5 mM NaCl, 1 mM TCEP (pH 6.4), supplemented with 0.1% BSA for blocking non-specific binding. β3-CT or β3-N or αIIb-CT) was immobilized on CM5 sensor chips (GE Healthcare) for about 50–100 resonance units (RU). Samples of binding partners or analytes were diluted in the running buffer and injected at a flow rate of 20 ml/min over the chip surface. Binding surfaces were regenerated to remove bound analyte by the following buffer flow for 2.5 minutes at 50 ml/min. This regeneration condition removed analyte completely but retained the surface binding capacity of the chip. Kinetic constants were calculated by global fitting of the data to a 1:1 Langmuir binding model, and Heterogeneous Ligand-Parallel Reactions model after subtracting the control surface, using the BIAevaluation software, version 4.0.1. Spike artifacts between association and dissociation phases from this subtraction were excluded from the fitting.
Integrin activation assay
The detailed procedure for measuring integrin activation and its inhibition by FLNa-Ig21 was described previously6. Briefly, EGFP vector, EGFP-fused proteins including talin-head (Talin-H), wild type FLNa-Ig4, Ig12, Ig17, Ig19, and Ig21, FLNa-Ig21 E2276A, and FLNa-Ig21 A2268K mutants were transfected into CHO cells transiently expressing αIIbβ3 (in pcDNA3.1+ vector) using Lipofectamine 2000 according to the manufacturer’s instructions. Their effects on integrin αIIbβ3 activation were assessed by binding of αIIbβ3 activation-specific PAC-1 monoclonal antibody, which was measured by flow cytometry as previously described6. The integrin activation experiments using αIIb(K994E/R997E)β3 mutant were performed in the same way as WT integrin. Variations in the level of the αIIbβ3 expression were monitored with the mouse 2G12 monoclonal antibody39. Differences in the activation of αIIbβ3 integrin measured with PAC-1 were normalized to differences in surface expression of αIIbβ3 integrin measured with 2G12 antibody. Flow cytometric data are represented as median fluorescence intensity (Relative fluorescence Intensity, RFI).
Supplementary Material
1
2
3
4
5
6
Acknowledgements
We wish to thank Xiaolun Zhang, Yan-Qing Ma, Jun-He Ma, Saurav Misra for useful discussions and technical assistance. This work was supported by US National Institutes of Health grants to JQ (GM062823), VY (DK102020), and EFP (HL073311).
Footnotes
Accession codes: The complete chemical shift assignment tables have been deposited in BMRB (25176). Detailed NOE table, along with the PDB coordinates has also been deposited in PDB bank (PDB code 2mtp).
Author Contributions: J.L and J.Q. conceived the study. J.L. performed all NMR and biochemical studies with the assistance of J.Y. and S.I. M.D. performed all functional experiments. All authors were involved in data interpretation and discussion. J.L. and J.Q. wrote the manuscript with contributions from all other authors.
References
- 1.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110(6):673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 2.Ye F, Lagarrigue F, Ginsberg MH. Snapshot: Talin and the modular nature of the integrin adhesome. Cell. 2014;156(6):1340–1340. doi: 10.1016/j.cell.2014.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bouvard D, Pouwels J, De Franceschi N, Ivaska J. Integrin inactivators: balancing cellular functions in vitro and in vivo. Nat Rev Mol Cell Biol. 2013;14(7):430–442. doi: 10.1038/nrm3599. [DOI] [PubMed] [Google Scholar]
- 4.Kiema T, et al. The molecular basis of filamin binding to integrins and competition with talin. Mol Cell. 2006;21(3):337–347. doi: 10.1016/j.molcel.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 5.Stossel TP, et al. Filamins as integrators of cell mechanics and signalling. Nat Rev Mol Cell Biol. 2001;2(2):138–145. doi: 10.1038/35052082. [DOI] [PubMed] [Google Scholar]
- 6.Das M, Ithychanda SS, Qin J, Plow EF. Migfilin and filamin as regulators of integrin activation in endothelial cells and neutrophils. PLoS One. 2011;6(10):e26355. doi: 10.1371/journal.pone.0026355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ithychanda SS, et al. Migfilin, a molecular switch in regulation of integrin activation. J Biol Chem. 2009;284(7):4713–4722. doi: 10.1074/jbc.M807719200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takala H, et al. beta 2 integrin phosphorylation on Thr758 acts as a molecular switch to regulate 14-3-3 and filamin binding. Blood. 2008;112(5):1853–1862. doi: 10.1182/blood-2007-12-127795. [DOI] [PubMed] [Google Scholar]
- 9.Xu Y, et al. Filamin A regulates focal adhesion disassembly and suppresses breast cancer cell migration and invasion. J Exp Med. 2010;207(11):2421–2437. doi: 10.1084/jem.20100433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun C, Forster C, Nakamura F, Glogauer M. Filamin-A regulates neutrophil uropod retraction through RhoA during chemotaxis. PLoS One. 2013;8(10):e79009. doi: 10.1371/journal.pone.0079009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calderwood DA, et al. Increased filamin binding to beta-integrin cytoplasmic domains inhibits cell migration. Nat Cell Biol. 2001;3(12):1060–1068. doi: 10.1038/ncb1201-1060. [DOI] [PubMed] [Google Scholar]
- 12.Ithychanda SS, et al. Identification and characterization of multiple similar filamin repeats: implication on filamin-mediated receptor clustering and cross-talking. J. Biol. Chem. 2009;284:35113–35121. doi: 10.1074/jbc.M109.060954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vinogradova O, et al. A structural mechanism of integrin alpha(IIb)beta(3) "inside-out" activation as regulated by its cytoplasmic face. Cell. 2002;110(5):587–597. doi: 10.1016/s0092-8674(02)00906-6. [DOI] [PubMed] [Google Scholar]
- 14.Wegener KL, et al. Structural basis of integrin activation by talin. Cell. 2007;128(1):171–182. doi: 10.1016/j.cell.2006.10.048. [DOI] [PubMed] [Google Scholar]
- 15.Anthis NJ, et al. The structure of an integrin/talin complex reveals the basis of inside-out signal transduction. EMBO J. 2009;28(22):3623–3632. doi: 10.1038/emboj.2009.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim M, Carman CV, Springer TA. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science. 2003;301(5640):1720–1725. doi: 10.1126/science.1084174. [DOI] [PubMed] [Google Scholar]
- 17.Lau TL, Kim C, Ginsberg MH, Ulmer TS. The structure of the integrin alphaIIbbeta3 transmembrane complex explains integrin transmembrane signalling. EMBO J. 2009;28(9):1351–1361. doi: 10.1038/emboj.2009.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu J, Luo BH, Barth P, Schonbrun J, Baker D, Springer TA. The structure of a receptor with two associating transmembrane domains on the cell surface:integrin alphaIIbbeta3. Mol Cell. 2009;34(2):234–249. doi: 10.1016/j.molcel.2009.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang, et al. Structure of an integrin αIIbβ3 transmembrane-cytoplasmic heterocomplex provides insight into integrin activation. Proc. Natl. Acad. Sci. 2009;106(42):17729–17734. doi: 10.1073/pnas.0909589106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi, et al. Three-dimensional reconstruction of intact human integrin αIIbβ3: new implications for activation-dependent ligand binding. Blood. 2013;122:4165–4171. doi: 10.1182/blood-2013-04-499194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goksoy E, et al. Structural basis for the autoinhibition of talin in regulating integrin activation. Mol Cell. 2008;31(1):124–133. doi: 10.1016/j.molcel.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song X, et al. A novel membrane-dependent on/off switch mechanism of talin FERM domain at sites of cell adhesion. Cell Res. 2012;22(11):1533–1545. doi: 10.1038/cr.2012.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goult BT, et al. Structural studies on full-length talin1 reveal a compact auto-inhibited dimer: implications for talin activation. J Struct Biol. 2013;184(1):21–32. doi: 10.1016/j.jsb.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beckerle MC, Miller DE, Bertagnolli ME, Locke SJ. Activation-dependent redistribution of the adhesion plaque protein, talin, in intact human platelets. J Cell Biol. 1989;109(6 Pt 2):3333–3346. doi: 10.1083/jcb.109.6.3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma YQ, Qin J, Wu C, Plow EF. Kindlin-2 (Mig-2): a co-activator of beta3 integrins. J. Cell. Biol. 2008;181:439–446. doi: 10.1083/jcb.200710196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pena E, Padro T, Molins B, Vilahur G, Badimon L. Proteomic signature of thrombin-activated platelets after in vivo nitric oxide-donor treatment: coordinated inhibition of signaling (phosphatidylinositol 3-kinase-gamma, 14-3-3zeta, and growth factor receptor-bound protein 2) and cytoskeleton protein translocation. Arterioscler Thromb Vasc Biol. 2011;31(11):2560–2569. doi: 10.1161/ATVBAHA.111.231852. [DOI] [PubMed] [Google Scholar]
- 27.Tu Y, Wu S, Shi X, Chen K, Wu C. Migfilin and Mig-2 link focal adhesions to filamin and the actin cytoskeleton and function in cell shape modulation. Cell. 2003;113(1):37–47. doi: 10.1016/s0092-8674(03)00163-6. [DOI] [PubMed] [Google Scholar]
- 28.Moik DV, Janbandhu VC, Fässler R. Loss of migfilin expression has no overt consequences on murine development and homeostasis. J Cell Sci. 2011;124(Pt 3):414–421. doi: 10.1242/jcs.075960. [DOI] [PubMed] [Google Scholar]
- 29.Xiao, et al. Critical role of filamin-binding LIM protein 1 (FBLP-1)/migfilin in regulation of bone remodeling. J Biol Chem. 2012;287(25):21450–21460. doi: 10.1074/jbc.M111.331249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rantala JK, et al. SHARPIN is an endogenous inhibitor of beta1-integrin activation. Nat Cell Biol. 2011;13(11):1315–1324. doi: 10.1038/ncb2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Freeman TC, Jr, et al. Identification of novel integrin binding partners for calcium and integrin binding protein 1 (CIB1): structural and thermodynamic basis of CIB1 promiscuity. Biochemistry. 2013;52(40):7082–7090. doi: 10.1021/bi400678y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Delaglio F, et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6(3):277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 33.Garrett DS, Powers R, Gronenborn AM, Clore GM. A common sense approach to peak picking in two-, three-, and four-dimensional spectra using automatic computer analysis of contour diagrams. J Magn Reson. 1991;213(2):357–363. doi: 10.1016/j.jmr.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 34.Xu Y, Wang X, Yang J, Vaynberg J, Qin J. PASA-a program for automated protein NMR backbone signal assignment by pattern-filtering approach. J Biomol NMR. 2006;34(1):41–56. doi: 10.1007/s10858-005-5358-0. [DOI] [PubMed] [Google Scholar]
- 35.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160(1):65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 36.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13(3):289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 37.Clore GM, Schwieters CD. Docking of protein-protein complexes on the basis of highly ambiguous intermolecular distance restraints derived from 1H/15N chemical shift mapping and backbone 15N-1H residual dipolar couplings using conjoined rigid body/torsion angle dynamics. J Am Chem Soc. 2003;125(10):2902–2912. doi: 10.1021/ja028893d. [DOI] [PubMed] [Google Scholar]
- 38.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993;26:283–291. [Google Scholar]
- 39.Woods VL, Jr, Oh EH, Mason D, McMillan R. Autoantibodies against the platelet glycoprotein IIb/IIIa complex in patients with chronic ITP. Blood. 1984;63:368–375. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1
2
3
4
5
6