Atrial Fibrillation: Epidemiology, Pathophysiology, and Clinical Outcomes (original) (raw)
. Author manuscript; available in PMC: 2018 Apr 28.
Abstract
The last three decades have been characterized by an exponential growth in knowledge and advances in the clinical treatment of atrial fibrillation (AF). It is now known that AF genesis requires a vulnerable atrial substrate and that the formation and composition of this substrate may vary depending on comorbid conditions, genetics, sex, and other factors. Population-based studies have identified numerous factors that modify the atrial substrate and increase AF susceptibility. To date, genetic studies have reported seventeen independent signals for AF at fourteen genomic regions. Studies have established that advanced age, male sex, and European ancestry are prominent AF risk factors. Other modifiable risk factors, including sedentary lifestyle, smoking, obesity, diabetes mellitus, obstructive sleep apnea, and elevated blood pressure predispose to AF and each factor has been shown to induce structural and electrical remodeling of the atria. Both heart failure and myocardial infarction increase risk of AF and vice versa creating a feed forward loop that increases mortality. Other cardiovascular outcomes attributed to AF, including stroke and thromboembolism, are well established and epidemiology studies have championed therapeutics that mitigate these adverse outcomes. However, the role of anticoagulation for preventing dementia attributed to AF is less established. Our review is a comprehensive examination of the epidemiological data associating unmodifiable and modifiable risk factors for AF and of the pathophysiological evidence supporting the mechanistic link between each risk factor and AF genesis. Our review also critically examines the epidemiological data on clinical outcomes attributed to AF and summarizes current evidence linking each outcome with AF.
Keywords: atrial fibrillation, epidemiology, risk factors, prognosis, pathophysiology
Introduction
The association of an irregular pulse and mitral stenosis was first described by Robert Adams in 1827, but it was not until the turn of the 20th century when William Einthoven invented electrocardiography that atrial fibrillation (AF) was first recorded on the electrocardiogram.1 Its pathogenesis and clinical importance gained enhanced appreciation in the 1990’s when early community-based studies including the Framingham Heart Study (FHS)2–4 provided critical epidemiological data on associated risk factors (RF) and clinical outcomes. These associations empowered scientists and clinicians by focusing their attention on specific disease models. Over the last 3 decades an explosion of research has yielded progress in the clinical treatment of AF at a time when AF is reaching epidemic proportions.
Our review provides an overview of the pathogenesis of non-valvular AF and a comprehensive examination of the epidemiological data associating various RFs with AF. For each RF we highlight key population studies supporting its association and critically review data on how the RF may lead to the development of the AF substrate and AF genesis. Lastly, we review clinical outcomes associated with AF and discuss possible mechanisms linking these associations. Our review focuses on the epidemiology and pathophysiology of AF rather than its clinical treatment.
1. Pathophysiology and Natural History AF
AF is characterized by high frequency excitation of the atrium that results in both dyssynchronous atrial contraction and irregularity of ventricular excitation. Whereas AF may occur in the absence of known structural or electrophysiological abnormalities, epidemiological association studies are increasingly identifying comorbid conditions, many of which have been shown to cause structural and histopathological changes that form a unique AF substrate or atrial cardiomyopathy.5
1.1 AF Initiation: Ectopic Firing
The prevailing hypothesis of AF genesis is that rapid triggering initiates propagating reentrant waves in a vulnerable atrial substrate. The relative importance of the initiating trigger may decrease as the AF substrate progresses and AF becomes more stabilized. Haissaguerre and colleagues first identified focal ectopic firing arising from myocyte sleeves within the pulmonary veins (PV) in patients with paroxysmal AF; ablation of these ectopic foci reduced AF burden, demonstrating their role in AF genesis.6 (Figure 1A) It is now known that the PVs have unique electrical properties and a complex fiber architecture that promote reentry and ectopic activity to initiate AF.7 Autopsy studies have identified pacemaker cells, transitional cells, and Purkinje cells within the PVs.8 The molecular basis for PV triggers has been primarily attributed to abnormal calcium Ca2+ handling. A diastolic leak of Ca2+ from the sarcoplasmic reticulum (SR) activates an inward Na+ current via Na+Ca2+ exchanger resulting in spontaneous myocyte depolarization (early or delayed afterdepolarization). Hyperphosphorylation of key regulatory proteins and enzymes including protein kinase A, calmodulin kinase II, phospholamban, and the ryanodine receptor type 2 are important contributors to SR Ca2+ overload and diastolic membrane instability.9, 10 A reentrant mechanism for PV triggers also has been described. Decremental conduction and repolarization heterogeneity within the PV enable localized reentry and may foster a focal driver for AF.11
Figure 1.
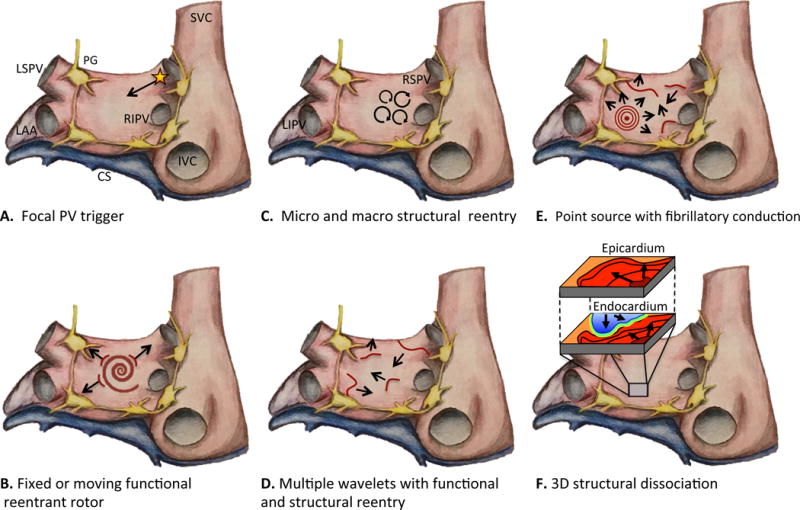
Rendering of left and right atrium showing various mechanisms of AF. A. Focal trigger arising from muscle sleeve of PV propagating into left atrium and initiating AF in the vulnerable substrate. B. Fixed or moving spiral rotor, a result of functional reentry, acts as a driver for AF. C. Circus movement around anatomic structures or scar generating micro and macro reentrant circuits. D. Perpetual propagation of multiple simultaneous wavelets mediated by both functional and structural reentry. E. Point source with fibrillatory conduction acting as driver for persistence of AF. F. Electrical dissociation between myocardial layers enabling reentry in three-dimensional construct. Abbreviations: SVC, superior vena cava; IVC, inferior vena cava; LAA, left atrial appendage; LSPV, left superior; RIPV, right inferior pulmonary vein; RSPV, right superior pulmonary vein; LIPV, left inferior pulmonary vein; CS, coronary sinus; PG, parasympathetic ganglia (yellow).
1.2 AF Perpetuation: Reentry
Whereas triggers are required for AF initiation, a vulnerable atrial substrate is equally important. Structural, architectural, and electrophysiological atrial abnormalities promote the perpetuation of AF by stabilizing reentry. The mechanism of reentry in AF remains controversial with two dominant hypotheses including reentrant rotors12, 13 or multiple independent wavelets.14 (Figure 1B–E) Advances in electroanatomic mapping and ablation technologies have yielded increasing evidence supporting the former mechanism.15, 16 Recent data supporting a third hypothesis, the double layer hypothesis, suggests that electrical dissociation of epicardial and endocardial layers also may facilitate reentry.17, 18 (Figure 1D)
For perpetuation of functional reentry, the propagating wavefront must complete one circus movement in a time period long enough for atrial tissue within that circuit to recover excitability (refractory period, ERP). Thus, slow conduction velocity and a short ERP promote reentry. Both reduce wavelength size increasing the likelihood of multiple simultaneous reentrant circuits and AF perpetuation.
Atrial substrates that promote reentry are characterized by abnormalities of the atrial cardiomyocyte, fibrotic changes, and alterations in the interstitial matrix with primarily non-collagen deposits.5 These molecular and histologic changes impair normal anisotropic conduction (fibrosis and/or reduced cell coupling) and may shorten atrial ERP. For example, in familial AF, congenital abnormalities that lead to a gain in K+ channel function shorten ERP of atrial cardiomyocytes; whereas in heart failure (HF) a combination of atrial fibrosis and alterations in cardiomyocyte function result in both a slowing of conduction velocity and shortening of ERP. Thus, the development of and characterization of the vulnerable atrial substrate is specific to the predisposing AF RF.
1.3 Natural History
For decades the prevailing notion was that AF began with paroxysmal episodes that increased in frequency and duration causing progression to more persistent AF subtypes. This so-called “AF begets AF” postulate was based on early experimental data in goats showing that tachycardia induces electrophysiologic atrial remodeling resulting in persistence of AF.19 Regional heterogeneity and shortening of the atrial ERP20, 21 occurs within 30 minutes of tachycardia onset and is a result of adaptation to intracellular Ca2+ overload.22 In the FHS, only 10% of participants remained free of AF 2-years after incident AF and recurrent (26%) or sustained AF (34%) was common.23 But other studies suggest that this abiding notion is not ubiquitous. In the Canadian Registry of Atrial Fibrillation (CARAF) progression of paroxysmal AF to more persistent (chronic) AF subtype was 8.6% at 1-year and 24.7% by 5 years.24 The Euro Heart Survey followed 5,333 patients with AF for one year and found that 80% of patients with paroxysmal AF remained paroxysmal while 30% of patients with persistent AF progressed to permanent.25 Studies in patients with pacemakers allow for more robust assessment of AF burden and have shown that the majority of patients (54–76%) with paroxysmal AF remain paroxysmal.26, 27 One study showed that only 24% of patients with paroxysmal AF progressed to persistent AF in one year and that there was a progressive pattern of increasing arrhythmia burden in these patients except in the days prior to the development of persistent AF supporting the mechanism of tachy-mediated atrial remodeling.27
The most remarkable observation is that persistent AF may spontaneously switch to paroxysmal subtype;26 highlighting the complex natural history of AF, the limitations of experimental data, and the existing uncertainty in the mechanisms and factors that govern the clinical course of AF. In addition, the natural history of AF may change over time as the RFs contributing to AF onset shift in prevalence and severity (e.g. less smoking and lower blood pressures, higher prevalence of obesity), and primary (e.g., better hypertension control) and secondary (anticoagulation) prevention treatments evolve.28 Finally, the means for quantifying and assessing AF burden over time in various population studies is inconsistent leading to ascertainment bias and challenges in predicting the natural history of AF subtypes.
2 Risk Factors for Developing AF
2.1 Unmodifiable Risk Factors
2.1.1 Genetics
Epidemiology
Near the start of the millennium rare familial forms of AF were identified and loci were mapped to 10q22-24,29 6q14-16,30 and 11p15-5.31 Subsequent population-based studies showed that family history of AF is associated with a 40% increased risk of first-degree relatives developing AF.32, 33 The recognition of the heritability of AF in the general population has propelled the search for associated genetic loci.34
Classic Mendelian genetics and candidate gene approaches have been used to define the familial basis of AF. To date, at least fifteen AF-causing mutations in K+ channel genes or accessory subunit have been identified,34 including mutations in ABCC9 (IKATP), HCN4 (If), KCNA5 (IKur), KCND3 (IKs), KCNE1 (IKs), KCNE2 (IKs), KCNE3 (IKs), KCNE4 (IKs), KCNE5 (IKs), KCNH2 (IKr), KCNJ2 (IK1), KCNJ5 (IKAch), KCNJ8 (IKATP), KCNN3 (IAHP), and KCNQ1 (IKs). Gain-of-function mutations increase repolarizing K+ current shortening the action potential duration (APD) and atrial refractoriness. Loss-of function mutations delay repolarization and promote Ca2+ mediated after-depolarization that trigger AF.35, 36 Six variations in Na+ channel genes have been identified and these include SCN1B, SCN2B, SCN3B, SCN4B, SCN5A, and SCN10A.34 Gain-of-function mutations may increase AF vulnerability by increasing cellular hyperexcitability,37 whereas loss-of function mutations shorten ERP and slow conduction.38 Other important AF-causing genetic variants include mutations in the gap junctional protein-coding gene GJA5 that diminishes cell-cell coupling and promotes reentry by slowing conduction velocity and shortening the wavelength.39
Instead of isolating a specific AF-causing gene, genome-wide association studies (GWAS) seek to scan the entire genome for disease-related genetic variants in single-nucleotide polymorphisms (SNPs). The first GWAS for AF identified SNP rs2200733 located on chromosome 4q25 upstream of PITX2 in an Icelandic population, which was strongly replicated in samples from Sweden, United States, and Hong Kong.40 A meta-analysis of three AF susceptibility loci (4q25, 1q21, 16q22) showed that the chromosome 4q25 SNP rs2200733 was associated with 30% increased risk of recurrent atrial tachycardia following AF ablation.41 In a separate meta-analysis this locus was associated with 38% increased risk of cardioembolic stroke.42
In experimental mouse models, the Pitx2 gene, encodes a transcription factor important for the embryonic organogenesis of the asymmetric organs placed in the left side including the heart.43 Moreover, Pitx2c is involved in the formation of the PV muscle sleeves,44 the most common site of triggered activity in AF. Loss of function of Pitx2c gene plays a role in the development of AF, and the differentiation, proliferation, and expansion of pulmonary myocardial cells.45, 46
In separate GWAS, the SNP rs2106261 near gene ZFHX3 identified on locus 16q22 was associated with increased AF risk. A knockdown of ZFHX3 dysregulates Ca2+, shortens the APD, and promotes arrhythmia susceptibility.47
AFGen consortium was formed in 2008 to increase statistical power for identifying loci associated with AF.48 To date 17 independent susceptibility signals for AF at 14 genomic regions have been identified. These include KCNN3, PRRX1, CAV1, SYNE2, C9orf3, HCN4, and MYOZ1.49, 50 The identification of genes related to AF is still in an early stage but in the future may allow for the assessment of an individuals’ AF risk and the discovery of novel therapeutic targets.
2.1.2 Age
Epidemiology
Advancing age is the most prominent RF for AF.28 Understanding the influence of age on AF risk is important for assessing how changes in life expectancy will affect the prevalence of AF.
Though the prevalence of AF varies among different ethnic populations, epidemiological studies have consistently found a step-wise increase in the prevalence of AF with advancing age.51–53 For example, a population-based multicenter cohort study reported age-specific rates in individuals aged 65–74 and 75–84 years of 3.4 (95% confidence interval (CI), 1.4–7.0) and 8.6 (95% CI, 4.6–14.9) for Chinese, 4.9 (95% CI, 3.1–7.3) and 10.6 (95% CI, 7.2–15.1) for non-Hispanic blacks, 7.3 (95% CI, 4.7–10.7) and 9.4 (95% CI, 5.9–14.4) for Hispanics, and 13.4 (95% CI, 10.6–16.7) and 19.6 (95% CI, 15.6–24.3) for non-Hispanic whites, respectively.51
The incidence of AF also increases with advancing age. In a Scottish study the incidence rates per 1000 persons was 0.5 for age 45–54 years, 1.1 for age 55–64, 3.2 for age 65–74, 6.2 for age 75–84, and 7.7 for age ≥85 years.53
The FHS examined temporal trends in AF RFs. During the last 5 decades, age was observed to be the greatest RF for AF as compared with other RFs including male sex, body mass index (BMI), diabetes, smoking, alcohol consumption, systolic blood pressure, hypertension treatment, left ventricular hypertrophy, heart murmur, HF, and myocardial infarction.28 In the most recent time-period studied (1998–2007), participant age of 60–69, 70–79, and 80–89 years was associated with 4.98, 7.35, and 9.33 fold risk of AF, respectively, compared with individuals aged 50–59 years.28 Accordingly, a step-wise increment in age has been incorporated in AF risk prediction scores.54, 55 Among AF patients, those with age <65 years also have been found to be healthier, have a different AF RF-profile, and less in-hospital deaths compared with those age ≥65 years.56
2.1.2 Sex Differences
Epidemiology
There is now greater recognition that epidemiology of AF differs between men and women.57 The age-adjusted incidence of AF is higher in men compared to women in North American and European populations. In the FHS, the AF incidence (per 1,000 person-years) was 3.8 in men and 1.6 in women.28 The Olmsted County Minnesota Study58 and the Rotterdam Study59 reported the AF incidence (per 1,000 person-years) in men to be 4.7 and 11.5, respectively, compared to 2.7 and 8.9 in women. A higher incidence of AF in men also is observed in Asian populations; although less data are available.60, 61 Similarly, the age-adjusted prevalence of AF is higher in men than in women in North American and European populations. Among the Medicare beneficiaries of adults aged ≥65 years, the prevalence was 10.3% in men and 7.4% in women.62 The higher prevalence of AF in men is also observed globally in both high-income and low and middle-income countries.60 However, there are less consistent data in Asian countries with some studies showing higher prevalence in men;61, 63, 64 whereas others showed no sex difference.52, 65, 66
In North American and European populations, the lifetime risk for AF is similar between men and women despite higher AF incidence in men owing to their shorter life expectancy.57 The FHS reported the lifetime risks to develop AF at age 40 years were 26% for men and 23% for women.67 In the Rotterdam study, the lifetime risks at age 55 years were 23.8% for men and 22.2% for women.59 On the contrary, among Chinese adults, the lifetime risk was consistently lower in men compared to women across all age groups.52
Underlying RFs of AF and taller stature in men largely explain higher AF incidence in men.57 The CHARGE-AF Consortium reported that after adjusting for AF-related RFs, male sex was no longer an independent RF for AF.54 The population-attributable risks of the RFs for AF differ by sex.57 The population-attributable risk for AF of coronary disease is higher in men68 whereas the population-attributable risks of elevated systolic blood pressure57 and valvular disease68 are higher in women.
2.1.3 Racial Differences
Many of the early population-based studies in AF were limited by racial diversity. However, in the last decade there has been a determined initiative to better understand the racial and ethnic differences in AF prevalence, pathophysiology, and outcomes.
Numerous studies demonstrated that AF is less prevalent in individuals of African as compared with European ancestry.51, 69–73 These data appear counterintuitive given the higher prevalence of traditional AF RFs in African Americans (AAs) than whites.69,74 The Candidate-Gene Association Resource Study69 found that among AAs the risk of AF was independently associated with increasing percentage of European ancestry (hazard ratio (HR), 1.13; 95% CI, 1.03–1.23). Adjusted associations showed that with every 10% increase in European ancestry there was a 16%–20% increased risk of AF. The data are consistent with the hypothesis that either African ancestry has protective effect against AF or European ancestry enhances AF susceptibility.
Candidate SNP analysis performed in two cohorts, showed that the rs10824026 SNP located on the 10q22 genetic locus accounted for 11.4%–31.7% increased AF risk in white individuals as compared with AAs.75 Furthermore, the minor allele G of the SNP confers low AF risk49 and is more common in AAs (37.7–37.8%) as compared with white individuals (15.5–16.4%).75 In a separate study that combined 3 cohorts with European and AA descent, an intronic SNP rs4845625 of the IL6R gene was associated with AF in white individuals (relative risk, 0.9; 95% CI, 0.85–0.95) and in AAs (relative risk, 0.86; 95% CI, 0.72–1.03 with borderline significance p = 0.09).76 Finally, the SNP rs4611994 on chromosome 4 near PITX2 was associated with AF risk in AAs (HR, 1.4; 95% CI, 1.16–1.69) and this chromosomal locus is associated with AF in white individuals.40 Although traditional AF RFs are well recognized, there is increasing data that race, ethnicity, and attributed genetic ancestral variants may play a significant role in modulating AF susceptibility.
More recently the Multi-Ethnic Study of Atherosclerosis (MESA) reported the prevalence of AF in Hispanic and Asians residing in the United States. The age- and sex-adjusted incidence rates per 1000 person-years of AF were 6.1 in Hispanics and 3.9 in Asians as compared with 11.2 in white and 5.8 in AA individuals.51 These data are comparable to The Healthcare Cost and Utilization Project that reported that Hispanics and Asians had multivariable-adjusted lower risk of AF (HR, 0.78; 95% CI, 0.77–0.79) as compared with whites.74 Both studies showed that a larger proportion of AF in non-whites was attributed to the greater presence of traditional RFs particularly hypertension as compared with whites.
2.2 Modifiable Risk Factors
2.2.1 Physical Activity and Sedentary Lifestyle
Epidemiology
The relationship between level of physical activity and risk of AF has been described as nonlinear.77–79 Sedentary lifestyle is associated with higher risk of AF,80 but paradoxically extreme levels of physical activity also are associated with increased AF risk.81–83
A large retrospective cohort study of 64,561 patients showed a graded and inverse relationship between cardiorespiratory fitness (CRF) as objectively assessed with treadmill testing. The incidence rate of AF in patients with lowest CRF was 18.8% as compared to 3.7% in those with highest CRF. Every 1-MET increase in CRF was associated with a dose-dependent 7% reduction in AF risk.84 A meta-analysis of pooled data from seven studies showed that sedentary lifestyle was associated with increased risk of AF (odds ratio (OR), 2.47; 95%CI, 1.25–3.7) compared to moderate or intense physical activity.83
Interesting and seemingly contradictory, male endurance athletes are at increased AF risk. In a prospective case-control study, individuals who had performed <2000 hours of lifetime-accumulated high-intensity exercise had an attenuated risk of lone AF (OR, 0.38; 95%CI, 0.12–0.98) as compared with sedentary individuals. But the AF risk in those with ≥2000 hours of high-intensity exercise was increased (OR, 3.88; 95%CI, 1.55–9.73).77 The type of exercise modulates AF risk with endurance sports, such as marathon running77 or long distance cross-country skiing,81 conferring the highest AF risk. A meta-analysis showed that there was over a 5-fold increased risk of AF in athletes than non-athlete referents.85
Sex-differences in the association of physical activity with AF have been identified. In men a U-shaped association of AF risk and physical activity was observed where moderate physical activity was found to confer lowest risk (OR, 0.81; 95% CI, 0.26–1.00) and intense activity conferred the highest risk (OR, 3.30; 95% CI, 1.97–4.63). In women, however, the association was inverse and linear. Increasing physical activity was associated with progressive and decreasing AF risk with ORs of 0.91 (95% CI, 0.77–0.97) and 0.72 (95% CI, 0.57–0.88) for moderate and intense activity, respectively.83
Pathophysiology
Sedentary lifestyle is known to increase risk of AF RFs including hypertension,86 obesity,86 and diabetes87. Obstructive sleep apnea (OSA) is common in obese individuals and has been associated with sedentary lifestyle.88 These conditions have been shown to independently induce structural and electrical remodeling of the atrium. Physical inactivity also increases systemic inflammation,89 which may induce atrial remodeling and has been associated with AF.90–94 Finally, sedentary lifestyle is associated with autonomic dysfunction and elevated sympathetic tone, which enhances afterdepolarization triggering and AF susceptibility.
In endurance athletes, the pathogenesis of AF has been attributed to two primary mechanisms. First, increased vagal tone in these individuals95, 96 may shorten and increase the dispersion of atrial ERP promoting PV firing and localized reentry. Second, long-term endurance training causes progressive cardiac remodeling including left atrial enlargement, which may promote AF.97–99 Atrial fibrosis and increased AF susceptibility has been observed in a rat model of prolonged, intensive exercise.100
2.2.2 Smoking
Epidemiology
Smoking is associated with incident AF.54, 101 The FHS showed that within the last 50 years, the frequency of smoking among participants with new-onset AF has decreased. Between 1998–2007, only 12.7% of AF-affected participants were smokers as compared to 15.6% in the prior decade.28
The Rotterdam Study found that both former and current smoking were equally associated with increased AF risk.102 In the Atherosclerosis Risk in Communities Study (ARIC) study, the multivariable-adjusted incidence of AF was 1.58 times higher in ever smokers (former and current) and two-fold higher (HR 2.05) in current smokers as compared with non-smokers.101 A dose-response association was observed with increasing cigarette-years.101 In the CHARGE-AF consortium, incident AF was 1.44 times higher in current smokers as compared with nonsmokers.54 Smoking is a RF for AF across various races and ethnicities.54, 101, 103
Finally, secondhand tobacco exposure also has been associated with risk of AF.104 Exposure during gestational development or early childhood is associated with approximately 40% increased risk of AF.105
Pathophysiology
Smoking is thought to increase AF susceptibility through indirect and direct mechanisms. Smoking may increase myocardial ischemia by increasing systemic catecholamine and myocardial work, reducing oxygen carrying capacity, and promoting coronary vasoconstriction.106 In addition, smoking accelerates atherosclerosis through effects on lipids, endothelial function, oxidative stress, inflammation, and thrombosis.107 These effects may indirectly increase AF susceptibility by predisposing to atrial ischemia, myocardial infarction, and HF. Reduced lung function and chronic obstructive pulmonary disease also increase vulnerability to AF.108
Smoking and nicotine also have been shown to directly contribute to the AF substrate. In a case-control study of patients undergoing coronary artery bypass, the volume of atrial fibrosis in smokers was shown to be dose-dependent, and in non-smokers nicotine was shown to induce a pattern of collagen type III expression in atrial tissue culture that was similar to that observed in smokers.109 In a dog model, nicotine induced interstitial fibrosis and increased AF susceptibility.110 The profibrotic effect of nicotine was attributed to down regulation of atrial micro RNAs, miR-133, and miR-590, which in turn increased transforming growth factors (TGF-b1 and TGFb2) and connective tissue growth factor. Finally, nicotine prolongs the APD by blocking the inward rectifier potassium channel (Ik1 and Kirs)111 and reduces the transient outward potassium current (Ito).112 Prolongation in the APD may increase arrhythmia susceptibility, but the pro-arrhythmic effect of nicotine has not been confirmed.
2.2.3 Obesity
Epidemiology
Both obesity and elevated BMI predispose to established AF RFs including hypertension,113 diabetes mellitus,114 myocardial infarction,115 left ventricular hypertrophy,116 left atrial enlargement,117 left ventricular diastolic dysfunction,118 HF,119 and OSA.2 However, when accounting for these concomitant conditions, population studies show that obesity and elevated BMI independently increase risk for AF.
Numerous population-based studies have shown an association between elevated BMI and increased risk of AF.120–122 A meta-analyses of 5 studies found that obesity confers a 49% increased risk of developing AF.123 A dose-response relationship was observed with each 1 unit increase in BMI associated with a 3%–4.7% increase in AF risk.120, 124, 125 Other measures of obesity including abdominal circumference and total fat mass have been associated with 13%–16% increase in AF risk per 1 SD over 10-year follow-up.126 Importantly, obesity is a powerful predictor of incident AF even when regression analyses have been adjusted for OSA, which commonly co-exists in such patients.125
Pathophysiology
Excess AF risk associated with obesity has been attributed to left atrial enlargement,120 increased left ventricular mass,116 and diastolic dysfunction.118, 127, 128 In a sheep model of obesity, increasing weight correlated with increased left atrial volume and pressure, ventricular mass, and pericardial fat. Histologic analysis revealed that myocardial lipidosis, fibrosis, and inflammatory infiltrates increased progressively with increasing weight. These pathologic changes were associated with decreased conduction velocity and increased AF.129 Whereas no change in atrial ERP was observed in the sheep model, a clinical study of 63 patients undergoing PV isolation reported that elevated BMI was associated with short atrial ERP and slower atrial conduction velocity,130 properties that promote reentry.
Pericardial fat also has been implicated in the pathogenesis of the obesity-AF relation. Cross-sectional studies have shown that pericardial fat is associated with prevalence, severity, and recurrence of AF.131, 132 A recent meta-analysis of 23 studies correlated epicardial adipose tissue (EAT) with AF after adjusting for traditional RFs (OR, 1.47 per standard deviation EAT increase; 95% CI, 1.17–1.84).133 A local paracrine effect of EAT mediated by inflammatory cytokines,134 growth and remodeling factors,135 angiogenic factors, and adipocytokines may lead to the development of the AF substrate.135, 136 EAT location on CT imaging correlates with high dominant excitation frequency during electoanatomic mapping in patients undergoing AF ablation.137
2.2.4 Diabetes Mellitus
Epidemiology
The FHS showed that men and women with diabetes had a 40% and 60% increased risk of AF, respectively.2 Level of blood glucose may be more predictive than actual diagnosis of diabetes in older adults.138 A meta-analysis of cohort and case-control studies found that patients with diabetes or impaired glucose homeostasis had a 34% greater risk of AF than individuals without diabetes.139 A causal association is supported by evidence that worse glycemic control and longer duration of diabetes are associated with increased AF risk.140 The estimated risk of AF increases by 3% per additional year of diabetes. The risk of AF in patients with diabetes for >10 years was 64% but only 7% in those with diabetes ≤5 years.
Pathophysiology
Glucose intolerance and insulin resistance appear to mediate the development of the AF substrate.141 The molecular mechanism by which insulin resistance alters cardiac structure is complex and involves impaired mitochondrial function and oxidative stress, which alter the transcription and translation processes essential for cardiac adaptation.142,143 In a rat model of diabetes, prolonged intra-atrial conduction time and diffuse interstitial fibrosis was observed, predisposing to increased arrhythmogenicity.144 In patients undergoing AF ablation, abnormal glucose metabolism was associated with prolonged atrial activation time and reduced bipolar voltages with electroanatomic mapping, a finding consistent with atrial fibrosis or scar.145 Finally, autonomic dysfunction also has been implicated.146
2.2.5 Obstructive Sleep Apnea
Epidemiology
OSA is highly prevalent147 and has been associated with other AF RFs including hypertension, diabetes, coronary heart disease, myocardial infarction, and HF.148 The Sleep Heart Health Study found a 4-fold increase in the prevalence of AF with OSA and one third of participants had arrhythmia during sleep.149 The Olmsted County Study similarly found that OSA and its severity strongly predicted 5-year incidence of AF (HR, 2.18; 95% CI, 1.34–3.54). In older individuals only the magnitude of nocturnal oxygen desaturation was predictive of AF.150 Similarly, a meta-analysis of five of prospective studies reported that OSA was associated with about a two-fold increased odds of post-operative AF.151 Patients with OSA have a higher recurrence of AF after cardioversion152 and catheter ablation (relative risk, 1.25; 95% CI, 1.08–1.45).153
The impact of OSA on AF outcomes was studied in the ORBIT-AF registry.154 Patients with OSA had more severe symptoms and were at higher risk of hospitalization (HR, 1.12; 95% CI, 1.03–1.22) than those without OSA, but had similar mortality, risk of stroke, or myocardial infarction. Patients with OSA who were treated with CPAP were less likely to progress to permanent AF subtype than those who were untreated (HR, 0.66; 95% CI, 0.46–0.94).
Pathophysiology
Electroanatomic mapping in patients undergoing AF ablation has been used to characterize the AF substrate associated with OSA.155 Observed structural changes included increased atrial size and expansive areas of low voltage or electrical silence, which indicate fibrosis, loss of atrial myocardium, or electrical uncoupling. Prolonged and regional disparities in atrial conduction also were seen. AF associated with OSA tends to be refractory to cardioversion and catheter ablation particularly in patients with untreated OSA, highlighting the expansive atrial remodeling associated with OSA.152 In a rat model of AF, OSA was associated with atrial conduction slowing attributed to connexin-43 down-regulation and increased atrial fibrosis. Such atrial remodeling promoted the persistence of AF.156
Several mechanisms may account for the development of AF and the AF substrate in patients with OSA. First, surges of sympathetic activity induced by hypoxia and the chemoreflex near the end of an apneic episode result in transient blood pressure rises.157 Second, vigorous inspiratory efforts during apnea accentuate the fluctuation of intrathoracic pressure increasing left atrial volume (stretch) and pressure.158 Third, an increase in oxidative stress signaling159 and systemic inflammatory mediators160 may promote atrial remodeling. Fourth, hypercapnia acutely prolongs ERP and slows conduction velocity, but with return of eucapnia delayed recovery of conduction has been associated with increased AF vulnerability.161 Fifth, negative tracheal pressure shortens atrial ERP and atrial monophasic action potential via vagal stimulation, which enhances AF inducibility.162
2.2.6 High Blood Pressure
Epidemiology
In the FHS the RF-adjusted OR for AF was 1.5 and 1.4 in men and women with hypertension, respectively.163 Later studies including the FHS found a limited association with mean arterial pressure, but found that pulse pressure was highly predictive of AF risk.164 The CHARGE-AF consortium observed that both systolic and diastolic blood pressure were predictive of AF risk.54 In addition, systolic blood pressure that approaches the upper limit of normal is associated with increased AF risk in healthy, middle aged men165 and women.166 The Women’s Health Study also showed that when an individual’s blood pressure remained elevated at follow-up visits, the risk of AF was higher as compared to those whose subsequent blood pressure recordings were lower suggesting a role for secondary prevention.166 The recent 50-year analysis of the FHS showed that while the rate of treated hypertension increased and severe hypertension became less prevalent, the population-attributed risk of AF was unaffected suggesting that anti-hypertensive therapy does not completely eliminate the elevated AF risk associated with hypertension.28
Pathophysiology
Increased left atrial size is a well-established, independent predictor of AF,163, 167 but other pathologic features of chronic hypertension including left ventricular hypertrophy163, 167, 168 and impaired diastolic dysfunction169 are also associated with AF. Common to all is an elevated left ventricular end-diastolic pressure, which increases left atrial pressure and volume. Atrial remodeling is associated with slower and more heterogeneous atrial conduction and increased PV firing. In addition, increased left atrial mass supports multiple reentry circuits.
Studies of animal models of hypertension have reported that early left atrial remodeling is characterized by atrial dilation with hypertrophy, reduced atrial ejection fraction, increased refractoriness, and prominent inflammatory infiltrates.170 Chronically, interstitial fibrosis and conduction slowing and heterogeneity are observed.170, 171 Moreover, increased atrial apoptosis has been observed.171 Electrophysiology studies in patients with chronically treated hypertension but without AF, have shown global conduction slowing, regionally delayed conduction in the crista teminalis and increased AF inducibility.172
Animal studies have suggested that the renin-angiotensin-aldosterone system (RAAS), which stimulates myocyte hypertrophy and intracellular fibrosis, also may contribute to atrial remodeling.173,174, 175 While upstream RAAS blockage was effective in animal models for reducing AF remodeling, two separate meta-analyses have reported that the benefit of RAAS blockade was limited to patients with HF or left ventricular hypertrophy.176, 177
3 Clinical Outcomes
3.1 Stroke
Epidemiology
AF is associated with increased risk of stroke and transient ischemic attack;178, 179 furthermore, AF-related strokes increase the risk of long-term disability or death.3 In the FHS the attributable risk of AF for stroke was 1.5% among 50–59 years olds whereas among 80–89 years olds it was 23.5%.179 Often AF is asymptomatic and consequently subclinical; however, in patients with implanted cardiac devices including pacemaker and defibrillators the burden of AF including subclinical AF may be accurately assessed. Atrial tachyarrhythmia (atrial rate >190 beats per minute) for longer than 6 minutes has been associated with an increased risk of clinical AF (HR, 5.56; 95% CI, 3.78–8.17) and ischemic stroke (HR, 2.50; 95% CI, 1.28–4.89).180
The risk of stroke with AF is variable and is modulated by other RFs including age ≥65, hypertension, diabetes, prior stroke/transient ischemic attack/thromboembolism history, vascular disease, HF, and female sex.181–183 Stroke risk models incorporating these RFs have been validated.184
Strokes in AF patients are associated with increased morbidity and mortality. In the Copenhagen Stroke Study, compared with individuals without AF, patients with AF had higher rates of in-hospital death (OR, 1.7; 95% CI, 1.2–2.5), longer hospital stay (50 days verse 40 days, p <0.001), and lower rates of discharge home (versus care facility, OR, 0.60; 95% CI, 0.44–0.85).185 Moreover, the infarct was larger and more commonly involved the cerebral cortex in patients with AF. The same study also showed that the odds for silent infarcts were similar for patients with AF and non-AF (OR, 0.99; 95% CI, 0.65–1.5).185 Correspondingly, the FHS showed increased 30-day mortality in AF-associated strokes than non-AF strokes (OR, 1.84; 95%CI, 1.04–3.27). Individuals with AF had worse 1-year survival following stroke and increased risk of stroke recurrences compared to those with non-AF strokes.3
Pathophysiology
Thrombogenesis in AF is not fully elucidated. A confluence of factors including blood stasis, endothelial dysfunction, and prothrombotic state has been implicated.
Attention has focused on the left atrial appendage (LAA). Animal models of AF demonstrate atrial contractile dysfunction from reduced myofibrillar sensitivity to Ca2+ 186 and intracellular Ca2+ transients.187 Clinically, reduced LAA emptying velocity on transesophageal echocardiography is associated with presence of spontaneous echo contrast, increased LAA thrombus, and stroke.188 In vitro studies have attributed spontaneous echo contrast to erythrocytes and fibrinogen interaction under low flow and shear stress.189 The role of LAA in AF-related stroke is further supported by efficacy of LAA closure devices for reducing strokes in patients with AF.190
Observational studies have revealed abnormalities in coagulation in patients with AF related strokes. Increases in prothrombin fragment and thrombin-antithrombin complexes have been observed in patients with AF-related strokes191 and in those with transesophageal spontaneous echo contrast.192 Other hemostatic factors have been implicated in contributing to the hypercoagulable state including fibrinogen, D-dimer, factor VIII, and von Willebrand factor.191, 193–196 However, in an adjusted model, the FHS showed that such abnormalities are ascribed to AF RFs and presence of cardiovascular disease rather than AF alone.197 Finally, inflammation may mediate endothelial dysfunction and hypercoagulability.
3.2 Extracranial Systemic Thromboembolism
Epidemiology
Compared to AF-related stroke, relatively less is known about epidemiology of extracranial systemic embolism events (SEEs). Data from the Danish Atrial Fibrillation Cohort showed an association between hospital diagnosis of AF and increased relative risk of SEEs in men (relative risk, 4.0; 95% CI, 3.5–4.6) and women (relative risk, 5.7; 95% CI, 5.1–6.3). Nearly half of SEEs occurred in patients between the ages of 70 and 79 years. The highest risk period for SEE was during the first year of incident AF, which is consistent with stroke data.198
More contemporary data are derived from a pooled analysis of four AF antiplatelet and anticoagulation trials with 37,973 patients from >40 countries.199 During the mean follow-up of 2.4 years, there were 221 SEEs accounting for 11.5% of clinically apparent embolic events. The incidence rates per 100 person-years for SEE and stroke were 0.24 and 1.92, respectively. Anatomically, SEEs were more likely to involve the lower extremities (58%) and mesenteric circulation (22%) while involvement of splenic and renal circulation was less common. Finally, increased morbidity and mortality was associated with SEE as 64% of patients required an interventional procedure or amputation and 24% died within 30 days.
Pathophysiology
The mechanisms underlying thromboembolism with SEE are similar to that of AF-related strokes.
3.3 Dementia
Epidemiology
Whereas dementia and AF share similar RFs including advancing age, obesity, diabetes, and hypertension, AF is associated with an adjusted increased risk of cognitive impairment,200 201 dementia,200, 202–205 Alzheimer’s dementia,206 and vascular dementia205 in patients with and without a history of stroke. In patients with normal baseline cognitive function and no history of stroke, meta-analysis of eight studies found a significantly increased risk of incident dementia in those with AF (HR, 1.42; 95% CI, 1.17–1.72).203 In patients with history of stroke, two meta-analyses have shown that AF is associated with ~ 2.5-fold adjusted risk of cognitive impairment and dementia.200, 202 Twenty-year follow-up of the Rotterdam Study reported AF patients < 67 years of age had greatest risk of dementia. The dementia risk increased with AF duration (exposure); whereas there was no increased risk associated with AF duration in those ≥ 67 years of age.204
Pathophysiology
Multiple mechanisms may explain the association of AF and dementia. Nearly one third of patients with AF have been observed to have silent brain infarcts on brain magnetic resonance imaging207 and micro thromboembolisms with covert infarction have been implicated as one possible mechanism. The FHS showed that over the last three decades, the incidence of dementia including dementia associated with AF has decreased.205 During this time period there has been improved use of anticoagulation in individuals with AF supporting the hypothesis that anticoagulation may reduce AF-associated dementia. This supposition is supported by a retrospective study of patients receiving long-term warfarin showing that incident dementia was 2.4 times higher in individuals with AF versus those without AF and that the dementia risk in those with AF and non-AF was significantly mitigated by increasing time in therapeutic range.208
A second possible mechanism may involve cerebral hypoperfusion associated with AF.209, 210 Interestingly one study indicated that the effect of AF on cerebral perfusion was most pronounced in younger patients (<50 years) and that no difference was observed in those >65 year of age209 consistent with epidemiological data showing age-dependent dementia risk in AF patients.204
3.4 Heart Failure
Epidemiology
HF is both a RF and an adverse clinical cardiovascular outcome associated with AF. The association was first recognized in the 1940s211 and it is now established that HF and AF often co-exist,2 predispose to the other,212 and share common RFs including hypertension, diabetes, coronary disease, and valvular disease.
HF as a Risk Factor for AF
In major HF trials, the prevalence of AF in patients with HF ranges from 13%–27%213–215 and the prevalence increases with increasing New York Heart Association Functional Class.216 In the FHS, HF was associated with 4.5-fold risk of AF in men and 5.9-fold risk in woman.2 Other epidemiological studies showed 2.67–3.37 fold risk of AF associated with HF.217, 218 Incremental reductions in systolic function are associated with increasing AF risk.167 HF with preserved ejection fraction also confers an increased risk of developing AF (HR adjusted for age and sex, 3.75; 95% CI, 2.19–6.40) with grade IV diastolic dysfunction as assessed with echocardiography conferring the highest risk.169
HF as an Outcome Associated with AF
The FHS uniquely reported the joint incidence of AF and HF and their temporal relationship. Among 931 participants with HF, 24% had prior or concurrent AF and 17% subsequently developed AF. One fifth of participants had AF and HF detected on the same day,212 demonstrating the closely interlinked pathophysiology. The incidence of first HF in FHS participants with AF is 33 per 1000-person years,212 which is comparable to that observed in the Danish nationwide cohort study.219 In a contemporary FHS cohort the incidence of HF was markedly higher in participants with AF than the incidence of AF in those with antecedent HF.220 In other words, AF begets HF more than HF begets AF.
The association of AF on HF subtype has also been reported. AF precedes HF with preserved ejection fraction (HFpEF) more commonly than HF with reduced ejection fraction (HFrEF). Among patients with prevalent AF, the adjusted HRs of incident HFpEF and HFrEF were 2.34 (95% CI, 1.48–3.70) and 1.32 (95% CI, 0.83–2.10), respectively.220 The finding that AF is more predictive of HFpEF is consistent with increased incident AF in patients with HFpEF versus HFrEF and suggests shared common mechanisms.169
In the FHS, the combination of AF and HF was associated with reduced survival.212 Among patients with prevalent AF, incident HF was associated with increased all-cause mortality compared to those without HF (HR, 1.25; 95% CI, 1.04–1.51).220 In one of the largest, worldwide studies, HF was found to be the leading cause of death one year after new-onset of AF accounting for nearly a third of all deaths.221 A meta-analysis showed a significantly higher all-cause mortality in AF patient with HFrEF than those with HFpEF (relative risk, 1.23; 95% CI, 1.12–1.36) despite similar risk of stroke and HF hospitalizations.222
Pathophysiology
The strong association of HF and AF has been attributed to shared mechanisms that lead to neurohormonal and proinflammatory activation, which induces myocardial inflammation and fibrosis. The atrial substrate with HF is characterized by atrial fibrosis and abnormalities in Ca2+ handling. It is distinct from the electrophysiologic changes associated with AF-induced atrial remodeling.223 Studies in dogs224 and humans225 indicate that HF induces an AF-susceptible atrial substrate without significantly altering atrial ERP (except at rapid rates) or ERP heterogeneity. These findings differ from the “AF begets AF” model with reduced atrial ERP. Histologic studies in HF dogs revealed structural changes including interstitial fibrosis with cellular hypertrophy, degeneration, and loss. These changes were associated with regions of delayed conduction and AF susceptibility. In human, extensive atrial fibrosis has been observed at autopsy in patients with dilated cardiomyopathy226 and areas of low voltage or electrical silence (scar) and fractionated or delayed potentials (slow conduction) have been identified during electrophysiology studies in patients with HF (but no AF).225 Neurohormonal activation is central to the generation of the AF substrate with HF and the milieu of profibrotic mediators are induced though angiotensin-dependent and independent pathways.227 Meta-analyses demonstrate benefit of upstream RAAS inhibition in AF patients with HF, but not in AF patients with other comorbidities; highlighting the unique pathophysiology of AF with HF.176, 177 Oxidized calmodulin-dependent protein kinase II has been shown to be a molecular signal that is increased by RAAS activation and is a common promoter of sinus node dysfunction, AF, and HF; thus reducing this kinase with RAAS block may explain the benefit of upstream therapy in patients with both AF and HF.228, 229
Increased trigger activity also is associated with HF and may increase risk of AF in the vulnerable substrate by generating a rapid burst of ectopic firing or maintaining a focal driver. In dogs, HF prolongs the APD and increases the phosphorylation of key regulatory kinases and phosphatases including calmodulin-dependent protein kinase II. The net effect of these HF-mediated changes is to enhance Ca2+ uptake into the SR promoting after-depolarization initiation.186 Similarly, HF has been shown to increase calcium sparks in cardiomyocytes isolated from the PVs of rabbits.230
Finally, tachycardia and shortening of diastolic filling time associated with irregular ventricular activation with AF further impair diastolic relaxation and promote clinical HF, which further induces atrial remodeling leading to the perpetuation of AF.
3.5 Myocardial Infarction
Epidemiology
As with HF there is a bidirectional relationship between AF and myocardial infarction (MI). Coronary heart disease is associated with an increased risk of AF,231 but AF also is associated with increased risk of MI.232 In the REGARDS cohort AF was associated with a 2-fold increased risk of MI,233 that was greater in women (HR, 2.16; 95% CI, 1.41–3.31) versus men (HR, 1.39; 95% CI, 0.91–2.10) and AAs (HR, 2.53; 95% CI, 1.67–3.86) versus whites (HR, 1.26; 95% CI, 0.83–1.93). The Olmsted study reported similar unadjusted risk of coronary ischemic event, but after adjusting for age the incidence was higher in men than woman.234 Among those with newly diagnosed AF, the risk of death after coronary ischemic events was higher in woman than men (HR, 2.99; 95% CI, 2.53–3.53 versus HR, 2.33; 95% CI, 1.94–2.81). Interestingly, as observed with strokes and SEEs, the event rate of coronary ischemic events was highest within the first year of incident AF (4.7%, 95% CI, 3.9–5.6) and subsequently declined to 2.5% per year.
Pathophysiology
The mechanisms linking AF to MI are not completely understood. First, both AF and MI have overlapping RFs that may lead to the development of AF and MI in parallel. For instance, both AF and coronary heart disease are associated with proinflammatory and prothombotic states. Second, MI in AF may be attributed to coronary artery thromboembolism. Third, myocardial ischemia may arise from supply-demand mismatch in the setting of tachycardia associated with AF. Fourth, MI may lead to left ventricular remodeling that may predispose to AF.
3.6 Venous Thromboembolism
Epidemiology
Increased BMI, obesity, and smoking are associated with venous thromboembolism (VTE)235–238 as well as AF. Potential direct causal relationship between AF and VTE has been proposed, but needs to be studied further.239, 240
A few studies have reported increased risk of VTE in AF and vice versa. In a retrospective cohort study based on the national administrative database in Taiwan, the risks of VTE (adjusted HR, 1.74; 95% CI, 1.36–2.24) and pulmonary embolism (adjusted HR, 2.18; 95% CI, 1.51–3.15) were both higher in the AF group compared to non-AF referents.241 A Norwegian administrative database study reported that AF was associated with an increased risk of pulmonary embolism (adjusted HR, 1.83; 95% CI, 1.16–2.90) but not VTE (adjusted HR, 1.04; 95% CI, 0.64–1.68).239 In addition, the same study found that individuals with incident VTE were subsequently at a higher risk of developing incident AF (adjusted HR, 1.63; 95% CI, 1.22–2.17) compared to those without VTE.240 It should be noted that in both the Taiwanese and Norwegian cohorts, a significant proportion of individuals had pre-exiting RFs for VTE including lower extremity fracture, recent surgery, cancer, and immobility.239, 241 In comparison to individuals with AF or VTE alone, those with AF and VTE were older and had higher mean BMI. In the Norwegian study, the mean age and BMI were 64 years and 26.9 kg/m2 for AF alone, 57 years and 26.7 kg/m2 for VTE alone, and 68 years and 29.2 kg/m2 for AF and VTE combined.240
Pathophysiology
The mechanisms underlying the AF-VTE association are inexplicable. Several studies have shown that AF is associated with a hypercoagulable state attributed to elevated hemostatic factors including fibrinogen, D-dimer, prothombin fragment, factor VIII, and von Willebrand factor;191, 193–196 however, many of these studies were not adjusted for co-existing cardiovascular RF. In an adjusted model, the FHS showed no significant difference in levels of fibrinogen, von Willebrand factor, or tissue plasminogen activator suggesting that co-existing RFs rather than AF may explain elevated thrombotic risk.
3.7 Mortality
Epidemiology
The FHS was one of the first studies to report that AF had a multivariable-adjusted association with increased risk of death.4 In addition, the study observed a significant interaction, such that AF diminished the survival advantage generally enjoyed by women; the multivariable-adjusted OR for death in men and women was 1.5 and 1.9, respectively. At 10-year follow up, 61.5% of men with AF between 55 to 74 years of age had died compared to 30.0% of men in the same age group without AF. A similar trend was found in women with 57.6% of those with AF dying by 10-year follow up as compared to 20.9% in those without AF. The increased risk was consistent across all decades of age from 55–95 years. Even in individuals without clinical evidence of cardiovascular or valvular disease at baseline, AF was associated with a 2-fold increased risk of death.4 A retrospective study of Medicare beneficiaries 65 years or older showed that death was the most frequent AF outcome with an incidence of 19.5% at 1 year and 48.8% at 5 years after initial diagnosis.242 A recent systematic review and meta-analysis of 64 studies that included 1,009,501 patients with 149,746 (14.8%) having AF, found a pooled relative risk of death that was 1.6 (95% CI, 1.39–1.53); although there was marked heterogeneity.243 In fourteen studies cardiovascular mortality was assessed and the pooled relative risk associated with AF was 2.03 (95% CI, 1.79–2.3).
It is noteworthy that there is growing evidence that AF is associated with an increased risk of sudden cardiac death (SCD). Pooled-analysis of the ARIC and Cardiovascular Health Study cohorts showed that AF was associated with more than a doubling of the risk of SCD SCD compared to participants without AF (HR, 2.47; 95% CI, 1.95–3.13).244 A meta-analysis of 7 studies found the relative risk of SCD was 1.88 (95% CI, 1.36–2.6); although significant study heterogeneity was present.243 In the RE-LY trial, 37.4% of all deaths and 60.4% of cardiac deaths were attributed to either SCD or death due to progressive HF.245 For comparison only 9.8% of all deaths were attributed to stroke or hemorrhage.
A meta-analysis of anti-thrombotic studies has shown that oral anticoagulation reduced risk of all-cause mortality with an absolute risk reduction of 1.6% as compared with control or placebo.246 In anticoagulated patients with AF increased mortality is largely driven by cardiovascular causes rather than non-hemorrhagic stroke or systemic embolism.245, 247–249 Strokes comprised a small portion of deaths from AF. In the ROCKET-AF trial cardiovascular deaths occurred over two times more often than strokes.247 Predictors of higher all-cause mortality included HF (HR, 1.51; 95% CI, 1.33–1.70) and age ≥75 (HR, 1.69; 95% CI, 1.51–1.90). Thus further advances in anticoagulation strategies may have little effect on improving overall mortality in AF.
Conclusion
Over the last 50 years, the FHS and other epidemiological studies have yielded a breadth of data associating various RFs with risk of AF and providing insight into their mechanistic link to AF genesis. However, many questions remain. Will genetic studies improve AF risk assessment, identify novel therapeutic targets, and help guide treatment strategies for both primary and secondary prevention of AF? By what degree does RF modification alter the atrial substrate, AF burden, and clinical outcomes? What are the target goals for RF modification and how will genetics alter these targets? Ongoing and future epidemiological, translational, and clinical studies may provide insight into these unanswered questions and improve clinical outcomes in patients with AF.
Figure 2.
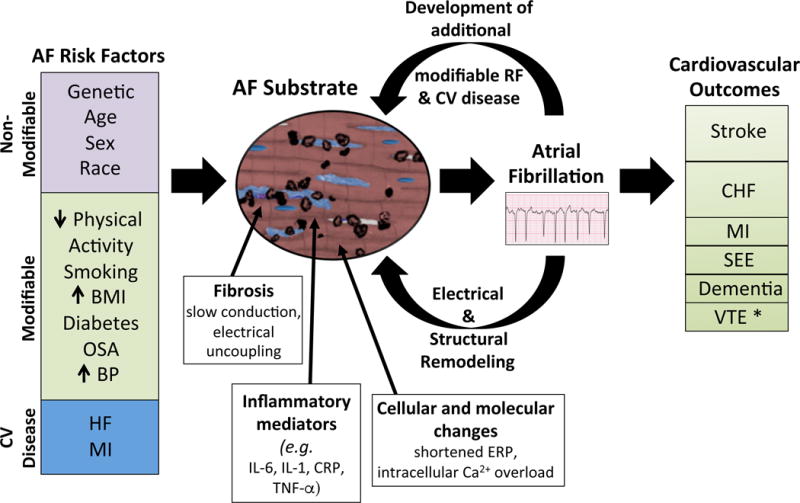
AF risk factors induce structural and histopathological changes to the atrium that are characterized by fibrosis, inflammation, and cellular and molecular changes. Such changes increase susceptibility to AF. Persistent AF further induces electrical and structural remodeling that promotes perpetuation of AF. AF also may lead to the development of additional AF risk factors that further alters the atrial substrate. Finally, AF is associated with several clinical outcomes. *There are limited data supporting the association.
Acknowledgments
Source of Funding:
This work was supported by Velux Foundation and Boston University School of Medicine and the National Heart, Lung, and Blood Institute’s Framingham Heart Study (contract: NIH/NHLBI N01-HC25195l; HHSN268201500001I; 2R01HL092577; 1R01HL128914; 1P50HL120163).
Abbreviations and acronyms
AA
African Americans
AF
Atrial fibrillation
APD
Action potential duration
ARIC
Atherosclerosis Risk in Communities Study
BMI
Body mass index
CI
Confidence interval
CRF
Cardiorespiratory fitness
EAT
Epicardial adipose tissue
ERP
Effective refractory period
FHS
Framingham Heart Study
GWAS
Genome-wide association study
HF
Heart failure
HFpEF
Heart failure with preserved ejection fraction
HFrEF
Heart failure with reduced ejection fraction
HR
Hazard ratio
LAA
Left atrial appendage
MESA
Multi-Ethnic Study of Atherosclerosis
MI
Myocardial infarction
OR
Odds ratio
OSA
Obstructive sleep apnea
PV
Pulmonary vein
RAAS
Renin-angiotensin-aldosterone system
RF
Risk factor
SCD
Sudden cardiac death
SEE
Systemic embolism events
SNP
Single-nucleotide polymorphism
SR
Sarcoplasmic reticulum
VTE
Venous thromboembolism
Footnotes
Disclosures:
Laila Staerk has received research funding from Boehringer Ingelheim.
Jason A. Sherer: none
Darae Ko: none
Emelia J. Benjamin: none
Robert H. Helm: none
References
- 1.Lip GY, Beevers DG. ABC of atrial fibrillation. History, epidemiology, and importance of atrial fibrillation. BMJ. 1995;311:1361–3. doi: 10.1136/bmj.311.7016.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benjamin EJ, Levy D, Vaziri SM, D’Agostino RB, Belanger AJ, Wolf PA. Independent risk factors for atrial fibrillation in a population-based cohort. The Framingham Heart Study. Jama. 1994;271:840–4. [PubMed] [Google Scholar]
- 3.Lin HJ, Wolf PA, Kelly-Hayes M, Beiser AS, Kase CS, Benjamin EJ, D’Agostino RB. Stroke severity in atrial fibrillation. The Framingham Study. Stroke. 1996;27:1760–4. doi: 10.1161/01.str.27.10.1760. [DOI] [PubMed] [Google Scholar]
- 4.Benjamin EJ, Wolf PA, D’Agostino RB, Silbershatz H, Kannel WB, Levy D. Impact of atrial fibrillation on the risk of death: the Framingham Heart Study. Circulation. 1998;98:946–52. doi: 10.1161/01.cir.98.10.946. [DOI] [PubMed] [Google Scholar]
- 5.Goette A, Kalman JM, Aguinaga L, et al. EHRA/HRS/APHRS/SOLAECE expert consensus on Atrial cardiomyopathies: definition, characterization, and clinical implication. Europace. 2016 doi: 10.1093/europace/euw161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haissaguerre M, Jais P, Shah DC, Takahashi A, Hocini M, Quiniou G, Garrigue S, Le Mouroux A, Le Metayer P, Clementy J. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med. 1998;339:659–66. doi: 10.1056/NEJM199809033391003. [DOI] [PubMed] [Google Scholar]
- 7.Hocini M, Ho SY, Kawara T, Linnenbank AC, Potse M, Shah D, Jais P, Janse MJ, Haissaguerre M, De Bakker JM. Electrical conduction in canine pulmonary veins: electrophysiological and anatomic correlation. Circulation. 2002;105:2442–8. doi: 10.1161/01.cir.0000016062.80020.11. [DOI] [PubMed] [Google Scholar]
- 8.Perez-Lugones A, McMahon JT, Ratliff NB, Saliba WI, Schweikert RA, Marrouche NF, Saad EB, Navia JL, McCarthy PM, Tchou P, Gillinov AM, Natale A. Evidence of specialized conduction cells in human pulmonary veins of patients with atrial fibrillation. J Cardiovasc Electrophysiol. 2003;14:803–9. doi: 10.1046/j.1540-8167.2003.03075.x. [DOI] [PubMed] [Google Scholar]
- 9.El-Armouche A, Boknik P, Eschenhagen T, Carrier L, Knaut M, Ravens U, Dobrev D. Molecular determinants of altered Ca2+ handling in human chronic atrial fibrillation. Circulation. 2006;114:670–80. doi: 10.1161/CIRCULATIONAHA.106.636845. [DOI] [PubMed] [Google Scholar]
- 10.Vest JA, Wehrens XH, Reiken SR, Lehnart SE, Dobrev D, Chandra P, Danilo P, Ravens U, Rosen MR, Marks AR. Defective cardiac ryanodine receptor regulation during atrial fibrillation. Circulation. 2005;111:2025–32. doi: 10.1161/01.CIR.0000162461.67140.4C. [DOI] [PubMed] [Google Scholar]
- 11.Arora R, Verheule S, Scott L, Navarrete A, Katari V, Wilson E, Vaz D, Olgin JE. Arrhythmogenic substrate of the pulmonary veins assessed by high-resolution optical mapping. Circulation. 2003;107:1816–21. doi: 10.1161/01.CIR.0000058461.86339.7E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schuessler RB, Grayson TM, Bromberg BI, Cox JL, Boineau JP. Cholinergically mediated tachyarrhythmias induced by a single extrastimulus in the isolated canine right atrium. Circ Res. 1992;71:1254–67. doi: 10.1161/01.res.71.5.1254. [DOI] [PubMed] [Google Scholar]
- 13.Mandapati R, Skanes A, Chen J, Berenfeld O, Jalife J. Stable microreentrant sources as a mechanism of atrial fibrillation in the isolated sheep heart. Circulation. 2000;101:194–9. doi: 10.1161/01.cir.101.2.194. [DOI] [PubMed] [Google Scholar]
- 14.Moe GK, Abildskov JA. Atrial fibrillation as a self-sustaining arrhythmia independent of focal discharge. Am Heart J. 1959;58:59–70. doi: 10.1016/0002-8703(59)90274-1. [DOI] [PubMed] [Google Scholar]
- 15.Pappone C, Rosanio S, Oreto G, Tocchi M, Gugliotta F, Vicedomini G, Salvati A, Dicandia C, Mazzone P, Santinelli V, Gulletta S, Chierchia S. Circumferential radiofrequency ablation of pulmonary vein ostia: A new anatomic approach for curing atrial fibrillation. Circulation. 2000;102:2619–28. doi: 10.1161/01.cir.102.21.2619. [DOI] [PubMed] [Google Scholar]
- 16.Miller JM, Kowal RC, Swarup V, et al. Initial independent outcomes from focal impulse and rotor modulation ablation for atrial fibrillation: multicenter FIRM registry. J Cardiovasc Electrophysiol. 2014;25:921–9. doi: 10.1111/jce.12474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Allessie MA, de Groot NM, Houben RP, Schotten U, Boersma E, Smeets JL, Crijns HJ. Electropathological substrate of long-standing persistent atrial fibrillation in patients with structural heart disease: longitudinal dissociation. Circ Arrhythm Electrophysiol. 2010;3:606–15. doi: 10.1161/CIRCEP.109.910125. [DOI] [PubMed] [Google Scholar]
- 18.Eckstein J, Maesen B, Linz D, Zeemering S, van Hunnik A, Verheule S, Allessie M, Schotten U. Time course and mechanisms of endo-epicardial electrical dissociation during atrial fibrillation in the goat. Cardiovasc Res. 2011;89:816–24. doi: 10.1093/cvr/cvq336. [DOI] [PubMed] [Google Scholar]
- 19.Wijffels MC, Kirchhof CJ, Dorland R, Allessie MA. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation. 1995;92:1954–68. doi: 10.1161/01.cir.92.7.1954. [DOI] [PubMed] [Google Scholar]
- 20.Morillo CA, Klein GJ, Jones DL, Guiraudon CM. Chronic rapid atrial pacing. Structural, functional, and electrophysiological characteristics of a new model of sustained atrial fibrillation. Circulation. 1995;91:1588–95. doi: 10.1161/01.cir.91.5.1588. [DOI] [PubMed] [Google Scholar]
- 21.Fareh S, Villemaire C, Nattel S. Importance of refractoriness heterogeneity in the enhanced vulnerability to atrial fibrillation induction caused by tachycardia-induced atrial electrical remodeling. Circulation. 1998;98:2202–9. doi: 10.1161/01.cir.98.20.2202. [DOI] [PubMed] [Google Scholar]
- 22.Goette A, Honeycutt C, Langberg JJ. Electrical remodeling in atrial fibrillation. Time course and mechanisms. Circulation. 1996;94:2968–74. doi: 10.1161/01.cir.94.11.2968. [DOI] [PubMed] [Google Scholar]
- 23.Lubitz SA, Moser C, Sullivan L, et al. Atrial fibrillation patterns and risks of subsequent stroke, heart failure, or death in the community. J Am Heart Assoc. 2013;2:e000126. doi: 10.1161/JAHA.113.000126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kerr CR, Humphries KH, Talajic M, Klein GJ, Connolly SJ, Green M, Boone J, Sheldon R, Dorian P, Newman D. Progression to chronic atrial fibrillation after the initial diagnosis of paroxysmal atrial fibrillation: results from the Canadian Registry of Atrial Fibrillation. Am Heart J. 2005;149:489–96. doi: 10.1016/j.ahj.2004.09.053. [DOI] [PubMed] [Google Scholar]
- 25.Nieuwlaat R, Prins MH, Le Heuzey JY, Vardas PE, Aliot E, Santini M, Cobbe SM, Widdershoven JW, Baur LH, Levy S, Crijns HJ. Prognosis, disease progression, and treatment of atrial fibrillation patients during 1 year: follow-up of the Euro Heart Survey on atrial fibrillation. Eur Heart J. 2008;29:1181–9. doi: 10.1093/eurheartj/ehn139. [DOI] [PubMed] [Google Scholar]
- 26.Veasey RA, Sugihara C, Sandhu K, Dhillon G, Freemantle N, Furniss SS, Sulke AN. The natural history of atrial fibrillation in patients with permanent pacemakers: is atrial fibrillation a progressive disease? J Interv Card Electrophysiol. 2015;44:23–30. doi: 10.1007/s10840-015-0029-x. [DOI] [PubMed] [Google Scholar]
- 27.Saksena S, Hettrick DA, Koehler JL, Grammatico A, Padeletti L. Progression of paroxysmal atrial fibrillation to persistent atrial fibrillation in patients with bradyarrhythmias. Am Heart J. 2007;154:884–92. doi: 10.1016/j.ahj.2007.06.045. [DOI] [PubMed] [Google Scholar]
- 28.Schnabel RB, Yin X, Gona P, et al. 50 year trends in atrial fibrillation prevalence, incidence, risk factors, and mortality in the Framingham Heart Study: a cohort study. Lancet. 2015;386:154–62. doi: 10.1016/S0140-6736(14)61774-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brugada R, Tapscott T, Czernuszewicz GZ, Marian AJ, Iglesias A, Mont L, Brugada J, Girona J, Domingo A, Bachinski LL, Roberts R. Identification of a genetic locus for familial atrial fibrillation. N Engl J Med. 1997;336:905–11. doi: 10.1056/NEJM199703273361302. [DOI] [PubMed] [Google Scholar]
- 30.Ellinor PT, Shin JT, Moore RK, Yoerger DM, MacRae CA. Locus for atrial fibrillation maps to chromosome 6q14-16. Circulation. 2003;107:2880–3. doi: 10.1161/01.CIR.0000077910.80718.49. [DOI] [PubMed] [Google Scholar]
- 31.Chen YH, Xu SJ, Bendahhou S, et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science. 2003;299:251–4. doi: 10.1126/science.1077771. [DOI] [PubMed] [Google Scholar]
- 32.Fox CS, Parise H, D’Agostino RB, Sr, Lloyd-Jones DM, Vasan RS, Wang TJ, Levy D, Wolf PA, Benjamin EJ. Parental atrial fibrillation as a risk factor for atrial fibrillation in offspring. Jama. 2004;291:2851–5. doi: 10.1001/jama.291.23.2851. [DOI] [PubMed] [Google Scholar]
- 33.Arnar DO, Thorvaldsson S, Manolio TA, Thorgeirsson G, Kristjansson K, Hakonarson H, Stefansson K. Familial aggregation of atrial fibrillation in Iceland. Eur Heart J. 2006;27:708–12. doi: 10.1093/eurheartj/ehi727. [DOI] [PubMed] [Google Scholar]
- 34.Christophersen IE, Ellinor PT. Genetics of atrial fibrillation: from families to genomes. J Hum Genet. 2016;61:61–70. doi: 10.1038/jhg.2015.44. [DOI] [PubMed] [Google Scholar]
- 35.Zellerhoff S, Pistulli R, Monnig G, et al. Atrial Arrhythmias in long-QT syndrome under daily life conditions: a nested case control study. J Cardiovasc Electrophysiol. 2009;20:401–7. doi: 10.1111/j.1540-8167.2008.01339.x. [DOI] [PubMed] [Google Scholar]
- 36.Olson TM, Alekseev AE, Liu XK, Park S, Zingman LV, Bienengraeber M, Sattiraju S, Ballew JD, Jahangir A, Terzic A. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum Mol Genet. 2006;15:2185–91. doi: 10.1093/hmg/ddl143. [DOI] [PubMed] [Google Scholar]
- 37.Li Q, Huang H, Liu G, Lam K, Rutberg J, Green MS, Birnie DH, Lemery R, Chahine M, Gollob MH. Gain-of-function mutation of Nav1.5 in atrial fibrillation enhances cellular excitability and lowers the threshold for action potential firing. Biochem Biophys Res Commun. 2009;380:132–7. doi: 10.1016/j.bbrc.2009.01.052. [DOI] [PubMed] [Google Scholar]
- 38.Savio-Galimberti E, Weeke P, Muhammad R, et al. SCN10A/Nav1.8 modulation of peak and late sodium currents in patients with early onset atrial fibrillation. Cardiovasc Res. 2014;104:355–63. doi: 10.1093/cvr/cvu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gollob MH, Jones DL, Krahn AD, et al. Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. The New England journal of medicine. 2006;354:2677–88. doi: 10.1056/NEJMoa052800. [DOI] [PubMed] [Google Scholar]
- 40.Gudbjartsson DF, Arnar DO, Helgadottir A, et al. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature. 2007;448:353–7. doi: 10.1038/nature06007. [DOI] [PubMed] [Google Scholar]
- 41.Shoemaker MB, Bollmann A, Lubitz SA, et al. Common genetic variants and response to atrial fibrillation ablation. Circ Arrhythm Electrophysiol. 2015;8:296–302. doi: 10.1161/CIRCEP.114.001909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cao YY, Ma F, Wang Y, Wang DW, Ding H. Rs2200733 and rs10033464 on chromosome 4q25 confer risk of cardioembolic stroke: an updated meta-analysis. Molecular biology reports. 2013;40:5977–85. doi: 10.1007/s11033-013-2707-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Logan M, Pagan-Westphal SM, Smith DM, Paganessi L, Tabin CJ. The transcription factor Pitx2 mediates situs-specific morphogenesis in response to left-right asymmetric signals. Cell. 1998;94:307–17. doi: 10.1016/s0092-8674(00)81474-9. [DOI] [PubMed] [Google Scholar]
- 44.Mommersteeg MT, Brown NA, Prall OW, de Gier-de Vries C, Harvey RP, Moorman AF, Christoffels VM. Pitx2c and Nkx2-5 are required for the formation and identity of the pulmonary myocardium. Circulation research. 2007;101:902–9. doi: 10.1161/CIRCRESAHA.107.161182. [DOI] [PubMed] [Google Scholar]
- 45.Aguirre LA, Alonso ME, Badia-Careaga C, Rollan I, Arias C, Fernandez-Minan A, Lopez-Jimenez E, Aranega A, Gomez-Skarmeta JL, Franco D, Manzanares M. Long-range regulatory interactions at the 4q25 atrial fibrillation risk locus involve PITX2c and ENPEP. BMC biology. 2015;13:26. doi: 10.1186/s12915-015-0138-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chinchilla A, Daimi H, Lozano-Velasco E, Dominguez JN, Caballero R, Delpon E, Tamargo J, Cinca J, Hove-Madsen L, Aranega AE, Franco D. PITX2 insufficiency leads to atrial electrical and structural remodeling linked to arrhythmogenesis. Circulation Cardiovascular genetics. 2011;4:269–79. doi: 10.1161/CIRCGENETICS.110.958116. [DOI] [PubMed] [Google Scholar]
- 47.Kao YH, Hsu JC, Chen YC, Lin YK, Lkhagva B, Chen SA, Chen YJ. ZFHX3 knockdown increases arrhythmogenesis and dysregulates calcium homeostasis in HL-1 atrial myocytes. International journal of cardiology. 2016;210:85–92. doi: 10.1016/j.ijcard.2016.02.091. [DOI] [PubMed] [Google Scholar]
- 48.http://www.afgen.org/
- 49.Ellinor PT, Lunetta KL, Albert CM, et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nature genetics. 2012;44:670–5. doi: 10.1038/ng.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tucker NR, Ellinor PT. Emerging directions in the genetics of atrial fibrillation. Circulation research. 2014;114:1469–82. doi: 10.1161/CIRCRESAHA.114.302225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rodriguez CJ, Soliman EZ, Alonso A, Swett K, Okin PM, Goff DC, Jr, Heckbert SR. Atrial fibrillation incidence and risk factors in relation to race-ethnicity and the population attributable fraction of atrial fibrillation risk factors: the Multi-Ethnic Study of Atherosclerosis. Ann Epidemiol. 2015;25:71–6. 76.e1. doi: 10.1016/j.annepidem.2014.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guo Y, Tian Y, Wang H, Si Q, Wang Y, Lip GY. Prevalence, incidence, and lifetime risk of atrial fibrillation in China: new insights into the global burden of atrial fibrillation. Chest. 2015;147:109–19. doi: 10.1378/chest.14-0321. [DOI] [PubMed] [Google Scholar]
- 53.Murphy NF, Simpson CR, Jhund PS, Stewart S, Kirkpatrick M, Chalmers J, MacIntyre K, McMurray JJ. A national survey of the prevalence, incidence, primary care burden and treatment of atrial fibrillation in Scotland. Heart. 2007;93:606–12. doi: 10.1136/hrt.2006.107573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alonso A, Krijthe BP, Aspelund T, et al. Simple risk model predicts incidence of atrial fibrillation in a racially and geographically diverse population: the CHARGE-AF consortium. J Am Heart Assoc. 2013;2:e000102. doi: 10.1161/JAHA.112.000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schnabel RB, Sullivan LM, Levy D, Pencina MJ, Massaro JM, D’Agostino RB, Sr, Newton-Cheh C, Yamamoto JF, Magnani JW, Tadros TM, Kannel WB, Wang TJ, Ellinor PT, Wolf PA, Vasan RS, Benjamin EJ. Development of a risk score for atrial fibrillation (Framingham Heart Study): a community-based cohort study. Lancet. 2009;373:739–45. doi: 10.1016/S0140-6736(09)60443-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Naderi S, Wang Y, Miller AL, Rodriguez F, Chung MK, Radford MJ, Foody JM. The impact of age on the epidemiology of atrial fibrillation hospitalizations. Am J Med. 2014;127:158.e1–7. doi: 10.1016/j.amjmed.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ko D, Rahman F, Schnabel RB, Yin X, Benjamin EJ, Christophersen IE. Atrial fibrillation in women: epidemiology, pathophysiology, presentation, and prognosis. Nat Rev Cardiol. 2016 doi: 10.1038/nrcardio.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miyasaka Y, Barnes ME, Gersh BJ, Cha SS, Bailey KR, Abhayaratna WP, Seward JB, Tsang TS. Secular trends in incidence of atrial fibrillation in Olmsted County, Minnesota, 1980 to 2000, and implications on the projections for future prevalence. Circulation. 2006;114:119–25. doi: 10.1161/CIRCULATIONAHA.105.595140. [DOI] [PubMed] [Google Scholar]
- 59.Heeringa J, van der Kuip DA, Hofman A, Kors JA, van Herpen G, Stricker BH, Stijnen T, Lip GY, Witteman JC. Prevalence, incidence and lifetime risk of atrial fibrillation: the Rotterdam study. European heart journal. 2006;27:949–53. doi: 10.1093/eurheartj/ehi825. [DOI] [PubMed] [Google Scholar]
- 60.Chugh SS, Havmoeller R, Narayanan K, et al. Worldwide epidemiology of atrial fibrillation: a Global Burden of Disease 2010 Study. Circulation. 2014;129:837–47. doi: 10.1161/CIRCULATIONAHA.113.005119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chien KL, Su TC, Hsu HC, Chang WT, Chen PC, Chen MF, Lee YT. Atrial fibrillation prevalence, incidence and risk of stroke and all-cause death among Chinese. International journal of cardiology. 2010;139:173–80. doi: 10.1016/j.ijcard.2008.10.045. [DOI] [PubMed] [Google Scholar]
- 62.Piccini JP, Hammill BG, Sinner MF, Jensen PN, Hernandez AF, Heckbert SR, Benjamin EJ, Curtis LH. Incidence and prevalence of atrial fibrillation and associated mortality among Medicare beneficiaries, 1993–2007. Circulation Cardiovascular quality and outcomes. 2012;5:85–93. doi: 10.1161/CIRCOUTCOMES.111.962688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Iguchi Y, Kimura K, Aoki J, Kobayashi K, Terasawa Y, Sakai K, Shibazaki K. Prevalence of atrial fibrillation in community-dwelling Japanese aged 40 years or older in Japan: analysis of 41,436 non-employee residents in Kurashiki-city. Circulation journal: official journal of the Japanese Circulation Society. 2008;72:909–13. doi: 10.1253/circj.72.909. [DOI] [PubMed] [Google Scholar]
- 64.Yap KB, Ng TP, Ong HY. Low prevalence of atrial fibrillation in community-dwelling Chinese aged 55 years or older in Singapore: a population-based study. Journal of electrocardiology. 2008;41:94–8. doi: 10.1016/j.jelectrocard.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 65.Li Y, Wu YF, Chen KP, Li X, Zhang X, Xie GQ, Wang FZ, Zhang S. Prevalence of atrial fibrillation in China and its risk factors. Biomedical and environmental sciences: BES. 2013;26:709–16. doi: 10.3967/0895-3988.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 66.Zhou Z, Hu D. An Epidemiological Study on the Prevalence of Atrial Fibrillation in the Chinese Population of Mainland China. Journal of epidemiology/Japan Epidemiological Association. 2008;18:209–216. doi: 10.2188/jea.JE2008021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lloyd-Jones DM, Wang TJ, Leip EP, Larson MG, Levy D, Vasan RS, D’Agostino RB, Massaro JM, Beiser A, Wolf PA, Benjamin EJ. Lifetime Risk for Development of Atrial Fibrillation: The Framingham Heart Study. Circulation. 2004;110:1042–1046. doi: 10.1161/01.CIR.0000140263.20897.42. [DOI] [PubMed] [Google Scholar]
- 68.Benjamin EJ, Levy D, Vaziri SM, D’Agostino RB, Belanger AJ, Wolf PA. Independent risk factors for atrial fibrillation in a population-based cohort. The Framingham Heart Study. Jama. 1994;271:840–4. [PubMed] [Google Scholar]
- 69.Marcus GM, Alonso A, Peralta CA, et al. European ancestry as a risk factor for atrial fibrillation in African Americans. Circulation. 2010;122:2009–15. doi: 10.1161/CIRCULATIONAHA.110.958306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sacco RL, Boden-Albala B, Abel G, Lin IF, Elkind M, Hauser WA, Paik MC, Shea S. Race-ethnic disparities in the impact of stroke risk factors: the northern Manhattan stroke study. Stroke. 2001;32:1725–31. doi: 10.1161/01.str.32.8.1725. [DOI] [PubMed] [Google Scholar]
- 71.Magnani JW, Norby FL, Agarwal SK, Soliman EZ, Chen LY, Loehr LR, Alonso A. Racial Differences in Atrial Fibrillation-Related Cardiovascular Disease and Mortality: The Atherosclerosis Risk in Communities (ARIC) Study. JAMA Cardiol. 2016;1:433–41. doi: 10.1001/jamacardio.2016.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hernandez MB, Asher CR, Hernandez AV, Novaro GM. African american race and prevalence of atrial fibrillation:a meta-analysis. Cardiol Res Pract. 2012;2012:275624. doi: 10.1155/2012/275624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lahiri MK, Fang K, Lamerato L, Khan AM, Schuger CD. Effect of race on the frequency of postoperative atrial fibrillation following coronary artery bypass grafting. Am J Cardiol. 2011;107:383–6. doi: 10.1016/j.amjcard.2010.09.032. [DOI] [PubMed] [Google Scholar]
- 74.Dewland TA, Olgin JE, Vittinghoff E, Marcus GM. Incident atrial fibrillation among Asians, Hispanics, blacks, and whites. Circulation. 2013;128:2470–7. doi: 10.1161/CIRCULATIONAHA.113.002449. [DOI] [PubMed] [Google Scholar]
- 75.Roberts JD, Hu D, Heckbert SR, et al. Genetic Investigation Into the Differential Risk of Atrial Fibrillation Among Black and White Individuals. JAMA Cardiol. 2016;1:442–50. doi: 10.1001/jamacardio.2016.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schnabel RB, Kerr KF, Lubitz SA, et al. Large-scale candidate gene analysis in whites and African Americans identifies IL6R polymorphism in relation to atrial fibrillation: the National Heart, Lung, and Blood Institute’s Candidate Gene Association Resource (CARe) project. Circ Cardiovasc Genet. 2011;4:557–64. doi: 10.1161/CIRCGENETICS.110.959197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Calvo N, Ramos P, Montserrat S, et al. Emerging risk factors and the dose-response relationship between physical activity and lone atrial fibrillation: a prospective case-control study. Europace: European pacing, arrhythmias, and cardiac electrophysiology: journal of the working groups on cardiac pacing, arrhythmias, and cardiac cellular electrophysiology of the European Society of Cardiology. 2016;18:57–63. doi: 10.1093/europace/euv216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Drca N, Wolk A, Jensen-Urstad M, Larsson SC. Atrial fibrillation is associated with different levels of physical activity levels at different ages in men. Heart (British Cardiac Society) 2014;100:1037–42. doi: 10.1136/heartjnl-2013-305304. [DOI] [PubMed] [Google Scholar]
- 79.Mozaffarian D, Furberg CD, Psaty BM, Siscovick D. Physical activity and incidence of atrial fibrillation in older adults: the cardiovascular health study. Circulation. 2008;118:800–7. doi: 10.1161/CIRCULATIONAHA.108.785626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Diouf I, Magliano DJ, Carrington MJ, Stewart S, Shaw JE. Prevalence, incidence, risk factors and treatment of atrial fibrillation in Australia: The Australian Diabetes, Obesity and Lifestyle (AusDiab) longitudinal, population cohort study. International journal of cardiology. 2016;205:127–32. doi: 10.1016/j.ijcard.2015.12.013. [DOI] [PubMed] [Google Scholar]
- 81.Andersen K, Farahmand B, Ahlbom A, Held C, Ljunghall S, Michaelsson K, Sundstrom J. Risk of arrhythmias in 52 755 long-distance cross-country skiers: a cohort study. European heart journal. 2013;34:3624–31. doi: 10.1093/eurheartj/eht188. [DOI] [PubMed] [Google Scholar]
- 82.Elosua R, Arquer A, Mont L, Sambola A, Molina L, Garcia-Moran E, Brugada J, Marrugat J. Sport practice and the risk of lone atrial fibrillation: a case-control study. Int J Cardiol. 2006;108:332–7. doi: 10.1016/j.ijcard.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 83.Mohanty S, Mohanty P, Tamaki M, Natale V, Gianni C, Trivedi C, Gokoglan Y, L DIB, Natale A. Differential Association of Exercise Intensity With Risk of Atrial Fibrillation in Men and Women: Evidence from a Meta-Analysis. Journal of cardiovascular electrophysiology. 2016;27:1021–9. doi: 10.1111/jce.13023. [DOI] [PubMed] [Google Scholar]
- 84.Qureshi WT, Alirhayim Z, Blaha MJ, Juraschek SP, Keteyian SJ, Brawner CA, Al-Mallah MH. Cardiorespiratory Fitness and Risk of Incident Atrial Fibrillation: Results From the Henry Ford Exercise Testing (FIT) Project. Circulation. 2015;131:1827–34. doi: 10.1161/CIRCULATIONAHA.114.014833. [DOI] [PubMed] [Google Scholar]
- 85.Abdulla J, Nielsen JR. Is the risk of atrial fibrillation higher in athletes than in the general population? A systematic review and meta-analysis. Europace. 2009;11:1156–9. doi: 10.1093/europace/eup197. [DOI] [PubMed] [Google Scholar]
- 86.Thorp AA, Owen N, Neuhaus M, Dunstan DW. Sedentary behaviors and subsequent health outcomes in adults a systematic review of longitudinal studies, 1996–2011. Am J Prev Med. 2011;41:207–15. doi: 10.1016/j.amepre.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 87.Hu FB, Leitzmann MF, Stampfer MJ, Colditz GA, Willett WC, Rimm EB. Physical activity and television watching in relation to risk for type 2 diabetes mellitus in men. Arch Intern Med. 2001;161:1542–8. doi: 10.1001/archinte.161.12.1542. [DOI] [PubMed] [Google Scholar]
- 88.Moreno CR, Carvalho FA, Lorenzi C, Matuzaki LS, Prezotti S, Bighetti P, Louzada FM, Lorenzi-Filho G. High risk for obstructive sleep apnea in truck drivers estimated by the Berlin questionnaire: prevalence and associated factors. Chronobiol Int. 2004;21:871–9. doi: 10.1081/cbi-200036880. [DOI] [PubMed] [Google Scholar]
- 89.Allison MA, Jensky NE, Marshall SJ, Bertoni AG, Cushman M. Sedentary behavior and adiposity-associated inflammation: the Multi-Ethnic Study of Atherosclerosis. Am J Prev Med. 2012;42:8–13. doi: 10.1016/j.amepre.2011.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rienstra M, Sun JX, Magnani JW, Sinner MF, Lubitz SA, Sullivan LM, Ellinor PT, Benjamin EJ. White blood cell count and risk of incident atrial fibrillation (from the Framingham Heart Study) Am J Cardiol. 2012;109:533–7. doi: 10.1016/j.amjcard.2011.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Aviles RJ, Martin DO, Apperson-Hansen C, Houghtaling PL, Rautaharju P, Kronmal RA, Tracy RP, Van Wagoner DR, Psaty BM, Lauer MS, Chung MK. Inflammation as a risk factor for atrial fibrillation. Circulation. 2003;108:3006–10. doi: 10.1161/01.CIR.0000103131.70301.4F. [DOI] [PubMed] [Google Scholar]
- 92.Li J, Solus J, Chen Q, Rho YH, Milne G, Stein CM, Darbar D. Role of inflammation and oxidative stress in atrial fibrillation. Heart Rhythm. 2010;7:438–44. doi: 10.1016/j.hrthm.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chung MK, Martin DO, Sprecher D, Wazni O, Kanderian A, Carnes CA, Bauer JA, Tchou PJ, Niebauer MJ, Natale A, Van Wagoner DR. C-reactive protein elevation in patients with atrial arrhythmias: inflammatory mechanisms and persistence of atrial fibrillation. Circulation. 2001;104:2886–91. doi: 10.1161/hc4901.101760. [DOI] [PubMed] [Google Scholar]
- 94.Conen D, Ridker PM, Everett BM, Tedrow UB, Rose L, Cook NR, Buring JE, Albert CM. A multimarker approach to assess the influence of inflammation on the incidence of atrial fibrillation in women. Eur Heart J. 2010;31:1730–6. doi: 10.1093/eurheartj/ehq146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mont L, Sambola A, Brugada J, Vacca M, Marrugat J, Elosua R, Pare C, Azqueta M, Sanz G. Long-lasting sport practice and lone atrial fibrillation. Eur Heart J. 2002;23:477–82. doi: 10.1053/euhj.2001.2802. [DOI] [PubMed] [Google Scholar]
- 96.Elosua R, Arquer A, Mont L, Sambola A, Molina L, Garcia-Moran E, Brugada J, Marrugat J. Sport practice and the risk of lone atrial fibrillation: a case-control study. International journal of cardiology. 2006;108:332–7. doi: 10.1016/j.ijcard.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 97.D’Andrea A, Riegler L, Cocchia R, et al. Left atrial volume index in highly trained athletes. Am Heart J. 2010;159:1155–61. doi: 10.1016/j.ahj.2010.03.036. [DOI] [PubMed] [Google Scholar]
- 98.Molina L, Mont L, Marrugat J, Berruezo A, Brugada J, Bruguera J, Rebato C, Elosua R. Long-term endurance sport practice increases the incidence of lone atrial fibrillation in men: a follow-up study. Europace. 2008;10:618–23. doi: 10.1093/europace/eun071. [DOI] [PubMed] [Google Scholar]
- 99.Hoogsteen J, Hoogeveen A, Schaffers H, Wijn PF, van der Wall EE. Left atrial and ventricular dimensions in highly trained cyclists. Int J Cardiovasc Imaging. 2003;19:211–7. doi: 10.1023/a:1023684430671. [DOI] [PubMed] [Google Scholar]
- 100.Benito B, Gay-Jordi G, Serrano-Mollar A, Guasch E, Shi Y, Tardif JC, Brugada J, Nattel S, Mont L. Cardiac arrhythmogenic remodeling in a rat model of long-term intensive exercise training. Circulation. 2011;123:13–22. doi: 10.1161/CIRCULATIONAHA.110.938282. [DOI] [PubMed] [Google Scholar]
- 101.Chamberlain AM, Agarwal SK, Folsom AR, Duval S, Soliman EZ, Ambrose M, Eberly LE, Alonso A. Smoking and incidence of atrial fibrillation: results from the Atherosclerosis Risk in Communities (ARIC) study. Heart rhythm: the official journal of the Heart Rhythm Society. 2011;8:1160–6. doi: 10.1016/j.hrthm.2011.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Heeringa J, Kors JA, Hofman A, van Rooij FJ, Witteman JC. Cigarette smoking and risk of atrial fibrillation: the Rotterdam Study. Am Heart J. 2008;156:1163–9. doi: 10.1016/j.ahj.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 103.Suzuki S, Otsuka T, Sagara K, et al. Association between smoking habits and the first-time appearance of atrial fibrillation in Japanese patients: Evidence from the Shinken Database. Journal of cardiology. 2015;66:73–9. doi: 10.1016/j.jjcc.2014.09.010. [DOI] [PubMed] [Google Scholar]
- 104.O’Neal WT, Qureshi WT, Judd SE, McClure LA, Cushman M, Howard VJ, Howard G, Soliman EZ. Environmental Tobacco Smoke and Atrial Fibrillation: The REasons for Geographic And Racial Differences in Stroke (REGARDS) Study. Journal of occupational and environmental medicine. 2015;57:1154–8. doi: 10.1097/JOM.0000000000000565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dixit S, Pletcher MJ, Vittinghoff E, Imburgia K, Maguire C, Whitman IR, Glantz SA, Olgin JE, Marcus GM. Secondhand smoke and atrial fibrillation: Data from the Health eHeart Study. Heart rhythm: the official journal of the Heart Rhythm Society. 2016;13:3–9. doi: 10.1016/j.hrthm.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Moliterno DJ, Willard JE, Lange RA, Negus BH, Boehrer JD, Glamann DB, Landau C, Rossen JD, Winniford MD, Hillis LD. Coronary-artery vasoconstriction induced by cocaine, cigarette smoking, or both. N Engl J Med. 1994;330:454–9. doi: 10.1056/NEJM199402173300702. [DOI] [PubMed] [Google Scholar]
- 107.Levitzky YS, Guo CY, Rong J, Larson MG, Walter RE, Keaney JF, Jr, Sutherland PA, Vasan A, Lipinska I, Evans JC, Benjamin EJ. Relation of smoking status to a panel of inflammatory markers: the framingham offspring. Atherosclerosis. 2008;201:217–24. doi: 10.1016/j.atherosclerosis.2007.12.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Buch P, Friberg J, Scharling H, Lange P, Prescott E. Reduced lung function and risk of atrial fibrillation in the Copenhagen City Heart Study. Eur Respir J. 2003;21:1012–6. doi: 10.1183/09031936.03.00051502. [DOI] [PubMed] [Google Scholar]
- 109.Goette A, Lendeckel U, Kuchenbecker A, Bukowska A, Peters B, Klein HU, Huth C, Rocken C. Cigarette smoking induces atrial fibrosis in humans via nicotine. Heart. 2007;93:1056–63. doi: 10.1136/hrt.2005.087171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shan H, Zhang Y, Lu Y, Zhang Y, Pan Z, Cai B, Wang N, Li X, Feng T, Hong Y, Yang B. Downregulation of miR-133 and miR-590 contributes to nicotine-induced atrial remodelling in canines. Cardiovasc Res. 2009;83:465–72. doi: 10.1093/cvr/cvp130. [DOI] [PubMed] [Google Scholar]
- 111.Wang H, Yang B, Zhang L, Xu D, Wang Z. Direct block of inward rectifier potassium channels by nicotine. Toxicol Appl Pharmacol. 2000;164:97–101. doi: 10.1006/taap.2000.8896. [DOI] [PubMed] [Google Scholar]
- 112.Wang H, Shi H, Zhang L, Pourrier M, Yang B, Nattel S, Wang Z. Nicotine is a potent blocker of the cardiac A-type K(+) channels. Effects on cloned Kv4.3 channels and native transient outward current. Circulation. 2000;102:1165–71. doi: 10.1161/01.cir.102.10.1165. [DOI] [PubMed] [Google Scholar]
- 113.Stamler R, Stamler J, Riedlinger WF, Algera G, Roberts RH. Weight and blood pressure. Findings in hypertension screening of 1 million Americans. Jama. 1978;240:1607–10. doi: 10.1001/jama.240.15.1607. [DOI] [PubMed] [Google Scholar]
- 114.Mokdad AH, Ford ES, Bowman BA, Dietz WH, Vinicor F, Bales VS, Marks JS. Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. Jama. 2003;289:76–9. doi: 10.1001/jama.289.1.76. [DOI] [PubMed] [Google Scholar]
- 115.Yusuf S, Hawken S, Ounpuu S, Bautista L, Franzosi MG, Commerford P, Lang CC, Rumboldt Z, Onen CL, Lisheng L, Tanomsup S, Wangai P, Jr, Razak F, Sharma AM, Anand SS, Investigators IS Obesity and the risk of myocardial infarction in 27,000 participants from 52 countries: a case-control study. Lancet. 2005;366:1640–9. doi: 10.1016/S0140-6736(05)67663-5. [DOI] [PubMed] [Google Scholar]
- 116.Lauer MS, Anderson KM, Kannel WB, Levy D. The impact of obesity on left ventricular mass and geometry. The Framingham Heart Study. Jama. 1991;266:231–6. [PubMed] [Google Scholar]
- 117.Gottdiener JS, Reda DJ, Williams DW, Materson BJ. Left atrial size in hypertensive men: influence of obesity, race and age. Department of Veterans Affairs Cooperative Study Group on Antihypertensive Agents. J Am Coll Cardiol. 1997;29:651–8. doi: 10.1016/s0735-1097(96)00554-2. [DOI] [PubMed] [Google Scholar]
- 118.Powell BD, Redfield MM, Bybee KA, Freeman WK, Rihal CS. Association of obesity with left ventricular remodeling and diastolic dysfunction in patients without coronary artery disease. Am J Cardiol. 2006;98:116–20. doi: 10.1016/j.amjcard.2006.01.063. [DOI] [PubMed] [Google Scholar]
- 119.Kenchaiah S, Evans JC, Levy D, Wilson PW, Benjamin EJ, Larson MG, Kannel WB, Vasan RS. Obesity and the risk of heart failure. N Engl J Med. 2002;347:305–13. doi: 10.1056/NEJMoa020245. [DOI] [PubMed] [Google Scholar]
- 120.Wang TJ, Parise H, Levy D, D’Agostino RB, Sr, Wolf PA, Vasan RS, Benjamin EJ. Obesity and the risk of new-onset atrial fibrillation. Jama. 2004;292:2471–7. doi: 10.1001/jama.292.20.2471. [DOI] [PubMed] [Google Scholar]
- 121.Frost L, Hune LJ, Vestergaard P. Overweight and obesity as risk factors for atrial fibrillation or flutter: the Danish Diet, Cancer, and Health Study. Am J Med. 2005;118:489–95. doi: 10.1016/j.amjmed.2005.01.031. [DOI] [PubMed] [Google Scholar]
- 122.Murphy NF, MacIntyre K, Stewart S, Hart CL, Hole D, McMurray JJ. Long-term cardiovascular consequences of obesity: 20-year follow-up of more than 15 000 middle-aged men and women (the Renfrew-Paisley study) Eur Heart J. 2006;27:96–106. doi: 10.1093/eurheartj/ehi506. [DOI] [PubMed] [Google Scholar]
- 123.Wanahita N, Messerli FH, Bangalore S, Gami AS, Somers VK, Steinberg JS. Atrial fibrillation and obesity–results of a meta-analysis. American heart journal. 2008;155:310–5. doi: 10.1016/j.ahj.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 124.Tedrow UB, Conen D, Ridker PM, Cook NR, Koplan BA, Manson JE, Buring JE, Albert CM. The long- and short-term impact of elevated body mass index on the risk of new atrial fibrillation the WHS (women’s health study) J Am Coll Cardiol. 2010;55:2319–27. doi: 10.1016/j.jacc.2010.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gami AS, Hodge DO, Herges RM, Olson EJ, Nykodym J, Kara T, Somers VK. Obstructive sleep apnea, obesity, and the risk of incident atrial fibrillation. J Am Coll Cardiol. 2007;49:565–71. doi: 10.1016/j.jacc.2006.08.060. [DOI] [PubMed] [Google Scholar]
- 126.Aronis KN, Wang N, Phillips CL, Benjamin EJ, Marcus GM, Newman AB, Rodondi N, Satterfield S, Harris TB, Magnani JW, Health ABCs Associations of obesity and body fat distribution with incident atrial fibrillation in the biracial health aging and body composition cohort of older adults. Am Heart J. 2015;170:498–505 e2. doi: 10.1016/j.ahj.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tsang TS, Barnes ME, Gersh BJ, Bailey KR, Seward JB. Left atrial volume as a morphophysiologic expression of left ventricular diastolic dysfunction and relation to cardiovascular risk burden. Am J Cardiol. 2002;90:1284–9. doi: 10.1016/s0002-9149(02)02864-3. [DOI] [PubMed] [Google Scholar]
- 128.Pritchett AM, Mahoney DW, Jacobsen SJ, Rodeheffer RJ, Karon BL, Redfield MM. Diastolic dysfunction and left atrial volume: a population-based study. J Am Coll Cardiol. 2005;45:87–92. doi: 10.1016/j.jacc.2004.09.054. [DOI] [PubMed] [Google Scholar]
- 129.Abed HS, Samuel CS, Lau DH, et al. Obesity results in progressive atrial structural and electrical remodeling: implications for atrial fibrillation. Heart Rhythm. 2013;10:90–100. doi: 10.1016/j.hrthm.2012.08.043. [DOI] [PubMed] [Google Scholar]
- 130.Munger TM, Dong YX, Masaki M, et al. Electrophysiological and hemodynamic characteristics associated with obesity in patients with atrial fibrillation. J Am Coll Cardiol. 2012;60:851–60. doi: 10.1016/j.jacc.2012.03.042. [DOI] [PubMed] [Google Scholar]
- 131.Al Chekakie MO, Welles CC, Metoyer R, Ibrahim A, Shapira AR, Cytron J, Santucci P, Wilber DJ, Akar JG. Pericardial fat is independently associated with human atrial fibrillation. J Am Coll Cardiol. 2010;56:784–8. doi: 10.1016/j.jacc.2010.03.071. [DOI] [PubMed] [Google Scholar]
- 132.Thanassoulis G, Massaro JM, O’Donnell CJ, Hoffmann U, Levy D, Ellinor PT, Wang TJ, Schnabel RB, Vasan RS, Fox CS, Benjamin EJ. Pericardial fat is associated with prevalent atrial fibrillation: the Framingham Heart Study. Circ Arrhythm Electrophysiol. 2010;3:345–50. doi: 10.1161/CIRCEP.109.912055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Agbaedeng T, Mahajan R, Munawar D, Elliott A, Twomey D, Kurmar S, Lau D, Sanders P. Meta-Analysis of Effects of Epicardial Fat on Atrial Fibrillation and Ablation Outcome. Heart, Lung and Circulation. 2016;25:S150. [Google Scholar]
- 134.Karastergiou K, Evans I, Ogston N, Miheisi N, Nair D, Kaski JC, Jahangiri M, Mohamed-Ali V. Epicardial adipokines in obesity and coronary artery disease induce atherogenic changes in monocytes and endothelial cells. Arterioscler Thromb Vasc Biol. 2010;30:1340–6. doi: 10.1161/ATVBAHA.110.204719. [DOI] [PubMed] [Google Scholar]
- 135.Greulich S, Maxhera B, Vandenplas G, de Wiza DH, Smiris K, Mueller H, Heinrichs J, Blumensatt M, Cuvelier C, Akhyari P, Ruige JB, Ouwens DM, Eckel J. Secretory products from epicardial adipose tissue of patients with type 2 diabetes mellitus induce cardiomyocyte dysfunction. Circulation. 2012;126:2324–34. doi: 10.1161/CIRCULATIONAHA.111.039586. [DOI] [PubMed] [Google Scholar]
- 136.Venteclef N, Guglielmi V, Balse E, Gaborit B, Cotillard A, Atassi F, Amour J, Leprince P, Dutour A, Clement K, Hatem SN. Human epicardial adipose tissue induces fibrosis of the atrial myocardium through the secretion of adipo-fibrokines. Eur Heart J. 2015;36:795–805a. doi: 10.1093/eurheartj/eht099. [DOI] [PubMed] [Google Scholar]
- 137.Nagashima K, Okumura Y, Watanabe I, Nakai T, Ohkubo K, Kofune M, Mano H, Sonoda K, Hiro T, Nikaido M, Hirayama A. Does location of epicardial adipose tissue correspond to endocardial high dominant frequency or complex fractionated atrial electrogram sites during atrial fibrillation? Circ Arrhythm Electrophysiol. 2012;5:676–83. doi: 10.1161/CIRCEP.112.971200. [DOI] [PubMed] [Google Scholar]
- 138.Psaty BM, Manolio TA, Kuller LH, Kronmal RA, Cushman M, Fried LP, White R, Furberg CD, Rautaharju PM. Incidence of and risk factors for atrial fibrillation in older adults. Circulation. 1997;96:2455–61. doi: 10.1161/01.cir.96.7.2455. [DOI] [PubMed] [Google Scholar]
- 139.Huxley RR, Filion KB, Konety S, Alonso A. Meta-analysis of cohort and case-control studies of type 2 diabetes mellitus and risk of atrial fibrillation. Am J Cardiol. 2011;108:56–62. doi: 10.1016/j.amjcard.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Dublin S, Glazer NL, Smith NL, Psaty BM, Lumley T, Wiggins KL, Page RL, Heckbert SR. Diabetes mellitus, glycemic control, and risk of atrial fibrillation. J Gen Intern Med. 2010;25:853–8. doi: 10.1007/s11606-010-1340-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rutter MK, Parise H, Benjamin EJ, Levy D, Larson MG, Meigs JB, Nesto RW, Wilson PW, Vasan RS. Impact of glucose intolerance and insulin resistance on cardiac structure and function: sex-related differences in the Framingham Heart Study. Circulation. 2003;107:448–54. doi: 10.1161/01.cir.0000045671.62860.98. [DOI] [PubMed] [Google Scholar]
- 142.Anderson EJ, Kypson AP, Rodriguez E, Anderson CA, Lehr EJ, Neufer PD. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J Am Coll Cardiol. 2009;54:1891–8. doi: 10.1016/j.jacc.2009.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kato T, Yamashita T, Sekiguchi A, Tsuneda T, Sagara K, Takamura M, Kaneko S, Aizawa T, Fu LT. AGEs-RAGE system mediates atrial structural remodeling in the diabetic rat. J Cardiovasc Electrophysiol. 2008;19:415–20. doi: 10.1111/j.1540-8167.2007.01037.x. [DOI] [PubMed] [Google Scholar]
- 144.Kato T, Yamashita T, Sekiguchi A, Sagara K, Takamura M, Takata S, Kaneko S, Aizawa T, Fu LT. What are arrhythmogenic substrates in diabetic rat atria? J Cardiovasc Electrophysiol. 2006;17:890–4. doi: 10.1111/j.1540-8167.2006.00528.x. [DOI] [PubMed] [Google Scholar]
- 145.Chao TF, Suenari K, Chang SL, Lin YJ, Lo LW, Hu YF, Tuan TC, Tai CT, Tsao HM, Li CH, Ueng KC, Wu TJ, Chen SA. Atrial substrate properties and outcome of catheter ablation in patients with paroxysmal atrial fibrillation associated with diabetes mellitus or impaired fasting glucose. Am J Cardiol. 2010;106:1615–20. doi: 10.1016/j.amjcard.2010.07.038. [DOI] [PubMed] [Google Scholar]
- 146.Otake H, Suzuki H, Honda T, Maruyama Y. Influences of autonomic nervous system on atrial arrhythmogenic substrates and the incidence of atrial fibrillation in diabetic heart. Int Heart J. 2009;50:627–41. doi: 10.1536/ihj.50.627. [DOI] [PubMed] [Google Scholar]
- 147.Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med. 1993;328:1230–5. doi: 10.1056/NEJM199304293281704. [DOI] [PubMed] [Google Scholar]
- 148.Shamsuzzaman AS, Gersh BJ, Somers VK. Obstructive sleep apnea: implications for cardiac and vascular disease. Jama. 2003;290:1906–14. doi: 10.1001/jama.290.14.1906. [DOI] [PubMed] [Google Scholar]
- 149.Mehra R, Benjamin EJ, Shahar E, Gottlieb DJ, Nawabit R, Kirchner HL, Sahadevan J, Redline S. Association of nocturnal arrhythmias with sleep-disordered breathing: The Sleep Heart Health Study. American journal of respiratory and critical care medicine. 2006;173:910–6. doi: 10.1164/rccm.200509-1442OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gami AS, Hodge DO, Herges RM, Olson EJ, Nykodym J, Kara T, Somers VK. Obstructive sleep apnea, obesity, and the risk of incident atrial fibrillation. Journal of the American College of Cardiology. 2007;49:565–71. doi: 10.1016/j.jacc.2006.08.060. [DOI] [PubMed] [Google Scholar]
- 151.Qaddoura A, Kabali C, Drew D, van Oosten EM, Michael KA, Redfearn DP, Simpson CS, Baranchuk A. Obstructive sleep apnea as a predictor of atrial fibrillation after coronary artery bypass grafting: a systematic review and meta-analysis. Can J Cardiol. 2014;30:1516–22. doi: 10.1016/j.cjca.2014.10.014. [DOI] [PubMed] [Google Scholar]
- 152.Kanagala R, Murali NS, Friedman PA, Ammash NM, Gersh BJ, Ballman KV, Shamsuzzaman AS, Somers VK. Obstructive sleep apnea and the recurrence of atrial fibrillation. Circulation. 2003;107:2589–94. doi: 10.1161/01.CIR.0000068337.25994.21. [DOI] [PubMed] [Google Scholar]
- 153.Ng CY, Liu T, Shehata M, Stevens S, Chugh SS, Wang X. Meta-analysis of obstructive sleep apnea as predictor of atrial fibrillation recurrence after catheter ablation. Am J Cardiol. 2011;108:47–51. doi: 10.1016/j.amjcard.2011.02.343. [DOI] [PubMed] [Google Scholar]
- 154.Holmqvist F, Guan N, Zhu Z, et al. Impact of obstructive sleep apnea and continuous positive airway pressure therapy on outcomes in patients with atrial fibrillation-Results from the Outcomes Registry for Better Informed Treatment of Atrial Fibrillation (ORBIT-AF) Am Heart J. 2015;169:647–654 e2. doi: 10.1016/j.ahj.2014.12.024. [DOI] [PubMed] [Google Scholar]
- 155.Dimitri H, Ng M, Brooks AG, Kuklik P, Stiles MK, Lau DH, Antic N, Thornton A, Saint DA, McEvoy D, Antic R, Kalman JM, Sanders P. Atrial remodeling in obstructive sleep apnea: implications for atrial fibrillation. Heart Rhythm. 2012;9:321–7. doi: 10.1016/j.hrthm.2011.10.017. [DOI] [PubMed] [Google Scholar]
- 156.Iwasaki YK, Kato T, Xiong F, Shi YF, Naud P, Maguy A, Mizuno K, Tardif JC, Comtois P, Nattel S. Atrial fibrillation promotion with long-term repetitive obstructive sleep apnea in a rat model. J Am Coll Cardiol. 2014;64:2013–23. doi: 10.1016/j.jacc.2014.05.077. [DOI] [PubMed] [Google Scholar]
- 157.Leuenberger U, Jacob E, Sweer L, Waravdekar N, Zwillich C, Sinoway L. Surges of muscle sympathetic nerve activity during obstructive apnea are linked to hypoxemia. J Appl Physiol (1985) 1995;79:581–8. doi: 10.1152/jappl.1995.79.2.581. [DOI] [PubMed] [Google Scholar]
- 158.Orban M, Bruce CJ, Pressman GS, Leinveber P, Romero-Corral A, Korinek J, Konecny T, Villarraga HR, Kara T, Caples SM, Somers VK. Dynamic changes of left ventricular performance and left atrial volume induced by the mueller maneuver in healthy young adults and implications for obstructive sleep apnea, atrial fibrillation, and heart failure. Am J Cardiol. 2008;102:1557–61. doi: 10.1016/j.amjcard.2008.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ntalapascha M, Makris D, Kyparos A, Tsilioni I, Kostikas K, Gourgoulianis K, Kouretas D, Zakynthinos E. Oxidative stress in patients with obstructive sleep apnea syndrome. Sleep Breath. 2013;17:549–55. doi: 10.1007/s11325-012-0718-y. [DOI] [PubMed] [Google Scholar]
- 160.Shamsuzzaman AS, Winnicki M, Lanfranchi P, Wolk R, Kara T, Accurso V, Somers VK. Elevated C-reactive protein in patients with obstructive sleep apnea. Circulation. 2002;105:2462–4. doi: 10.1161/01.cir.0000018948.95175.03. [DOI] [PubMed] [Google Scholar]
- 161.Stevenson IH, Roberts-Thomson KC, Kistler PM, Edwards GA, Spence S, Sanders P, Kalman JM. Atrial electrophysiology is altered by acute hypercapnia but not hypoxemia: implications for promotion of atrial fibrillation in pulmonary disease and sleep apnea. Heart Rhythm. 2010;7:1263–70. doi: 10.1016/j.hrthm.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 162.Linz D, Schotten U, Neuberger HR, Bohm M, Wirth K. Negative tracheal pressure during obstructive respiratory events promotes atrial fibrillation by vagal activation. Heart Rhythm. 2011;8:1436–43. doi: 10.1016/j.hrthm.2011.03.053. [DOI] [PubMed] [Google Scholar]
- 163.Kannel WB, Wolf PA, Benjamin EJ, Levy D. Prevalence, incidence, prognosis, and predisposing conditions for atrial fibrillation: population-based estimates. Am J Cardiol. 1998;82:2N–9N. doi: 10.1016/s0002-9149(98)00583-9. [DOI] [PubMed] [Google Scholar]
- 164.Mitchell GF, Vasan RS, Keyes MJ, Parise H, Wang TJ, Larson MG, D’Agostino RB, Sr, Kannel WB, Levy D, Benjamin EJ. Pulse pressure and risk of new-onset atrial fibrillation. Jama. 2007;297:709–15. doi: 10.1001/jama.297.7.709. [DOI] [PubMed] [Google Scholar]
- 165.Grundvold I, Skretteberg PT, Liestol K, Erikssen G, Kjeldsen SE, Arnesen H, Erikssen J, Bodegard J. Upper normal blood pressures predict incident atrial fibrillation in healthy middle-aged men: a 35-year follow-up study. Hypertension. 2012;59:198–204. doi: 10.1161/HYPERTENSIONAHA.111.179713. [DOI] [PubMed] [Google Scholar]
- 166.Conen D, Tedrow UB, Koplan BA, Glynn RJ, Buring JE, Albert CM. Influence of systolic and diastolic blood pressure on the risk of incident atrial fibrillation in women. Circulation. 2009;119:2146–52. doi: 10.1161/CIRCULATIONAHA.108.830042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Vaziri SM, Larson MG, Benjamin EJ, Levy D. Echocardiographic predictors of nonrheumatic atrial fibrillation. The Framingham Heart Study. Circulation. 1994;89:724–30. doi: 10.1161/01.cir.89.2.724. [DOI] [PubMed] [Google Scholar]
- 168.Verdecchia P, Reboldi G, Gattobigio R, Bentivoglio M, Borgioni C, Angeli F, Carluccio E, Sardone MG, Porcellati C. Atrial fibrillation in hypertension: predictors and outcome. Hypertension. 2003;41:218–23. doi: 10.1161/01.hyp.0000052830.02773.e4. [DOI] [PubMed] [Google Scholar]
- 169.Tsang TS, Gersh BJ, Appleton CP, Tajik AJ, Barnes ME, Bailey KR, Oh JK, Leibson C, Montgomery SC, Seward JB. Left ventricular diastolic dysfunction as a predictor of the first diagnosed nonvalvular atrial fibrillation in 840 elderly men and women. J Am Coll Cardiol. 2002;40:1636–44. doi: 10.1016/s0735-1097(02)02373-2. [DOI] [PubMed] [Google Scholar]
- 170.Lau DH, Mackenzie L, Kelly DJ, et al. Short-term hypertension is associated with the development of atrial fibrillation substrate: a study in an ovine hypertensive model. Heart Rhythm. 2010;7:396–404. doi: 10.1016/j.hrthm.2009.11.031. [DOI] [PubMed] [Google Scholar]
- 171.Kistler PM, Sanders P, Dodic M, Spence SJ, Samuel CS, Zhao C, Charles JA, Edwards GA, Kalman JM. Atrial electrical and structural abnormalities in an ovine model of chronic blood pressure elevation after prenatal corticosteroid exposure: implications for development of atrial fibrillation. Eur Heart J. 2006;27:3045–56. doi: 10.1093/eurheartj/ehl360. [DOI] [PubMed] [Google Scholar]
- 172.Medi C, Kalman JM, Spence SJ, Teh AW, Lee G, Bader I, Kaye DM, Kistler PM. Atrial electrical and structural changes associated with longstanding hypertension in humans: implications for the substrate for atrial fibrillation. J Cardiovasc Electrophysiol. 2011;22:1317–24. doi: 10.1111/j.1540-8167.2011.02125.x. [DOI] [PubMed] [Google Scholar]
- 173.Nakashima H, Kumagai K, Urata H, Gondo N, Ideishi M, Arakawa K. Angiotensin II antagonist prevents electrical remodeling in atrial fibrillation. Circulation. 2000;101:2612–7. doi: 10.1161/01.cir.101.22.2612. [DOI] [PubMed] [Google Scholar]
- 174.Li D, Shinagawa K, Pang L, Leung TK, Cardin S, Wang Z, Nattel S. Effects of angiotensin-converting enzyme inhibition on the development of the atrial fibrillation substrate in dogs with ventricular tachypacing-induced congestive heart failure. Circulation. 2001;104:2608–14. doi: 10.1161/hc4601.099402. [DOI] [PubMed] [Google Scholar]
- 175.Shi Y, Li D, Tardif JC, Nattel S. Enalapril effects on atrial remodeling and atrial fibrillation in experimental congestive heart failure. Cardiovasc Res. 2002;54:456–61. doi: 10.1016/s0008-6363(02)00243-2. [DOI] [PubMed] [Google Scholar]
- 176.Healey JS, Baranchuk A, Crystal E, Morillo CA, Garfinkle M, Yusuf S, Connolly SJ. Prevention of atrial fibrillation with angiotensin-converting enzyme inhibitors and angiotensin receptor blockers: a meta-analysis. J Am Coll Cardiol. 2005;45:1832–9. doi: 10.1016/j.jacc.2004.11.070. [DOI] [PubMed] [Google Scholar]
- 177.Schneider MP, Hua TA, Bohm M, Wachtell K, Kjeldsen SE, Schmieder RE. Prevention of atrial fibrillation by Renin-Angiotensin system inhibition a meta-analysis. J Am Coll Cardiol. 2010;55:2299–307. doi: 10.1016/j.jacc.2010.01.043. [DOI] [PubMed] [Google Scholar]
- 178.McAllen PM, Marshall J. Cardiac dysrhythmia and transient cerebral ischaemic attacks. Lancet (London, England) 1973;1:1212–4. doi: 10.1016/s0140-6736(73)90527-8. [DOI] [PubMed] [Google Scholar]
- 179.Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke. 1991;22:983–8. doi: 10.1161/01.str.22.8.983. [DOI] [PubMed] [Google Scholar]
- 180.Healey JS, Connolly SJ, Gold MR, Israel CW, Van Gelder IC, Capucci A, Lau CP, Fain E, Yang S, Bailleul C, Morillo CA, Carlson M, Themeles E, Kaufman ES, Hohnloser SH. Subclinical atrial fibrillation and the risk of stroke. N Engl J Med. 2012;366:120–9. doi: 10.1056/NEJMoa1105575. [DOI] [PubMed] [Google Scholar]
- 181.Risk factors for stroke and efficacy of antithrombotic therapy in atrial fibrillation. Analysis of pooled data from five randomized controlled trials. Arch Intern Med. 1994;154:1449–57. [PubMed] [Google Scholar]
- 182.Patients with nonvalvular atrial fibrillation at low risk of stroke during treatment with aspirin: Stroke Prevention in Atrial Fibrillation III Study. The SPAF III Writing Committee for the Stroke Prevention in Atrial Fibrillation Investigators. Jama. 1998;279:1273–7. [PubMed] [Google Scholar]
- 183.Hart RG, Pearce LA, McBride R, Rothbart RM, Asinger RW. Factors associated with ischemic stroke during aspirin therapy in atrial fibrillation: analysis of 2012 participants in the SPAF I–III clinical trials. The Stroke Prevention in Atrial Fibrillation (SPAF) Investigators. Stroke. 1999;30:1223–9. doi: 10.1161/01.str.30.6.1223. [DOI] [PubMed] [Google Scholar]
- 184.Olesen JB, Lip GY, Hansen ML, Hansen PR, Tolstrup JS, Lindhardsen J, Selmer C, Ahlehoff O, Olsen AM, Gislason GH, Torp-Pedersen C. Validation of risk stratification schemes for predicting stroke and thromboembolism in patients with atrial fibrillation: nationwide cohort study. Bmj. 2011;342:d124. doi: 10.1136/bmj.d124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Jorgensen HS, Nakayama H, Reith J, Raaschou HO, Olsen TS. Acute stroke with atrial fibrillation. The Copenhagen Stroke Study. Stroke. 1996;27:1765–9. doi: 10.1161/01.str.27.10.1765. [DOI] [PubMed] [Google Scholar]
- 186.Yeh YH, Wakili R, Qi XY, Chartier D, Boknik P, Kaab S, Ravens U, Coutu P, Dobrev D, Nattel S. Calcium-handling abnormalities underlying atrial arrhythmogenesis and contractile dysfunction in dogs with congestive heart failure. Circ Arrhythm Electrophysiol. 2008;1:93–102. doi: 10.1161/CIRCEP.107.754788. [DOI] [PubMed] [Google Scholar]
- 187.Kneller J, Sun H, Leblanc N, Nattel S. Remodeling of Ca(2+)-handling by atrial tachycardia: evidence for a role in loss of rate-adaptation. Cardiovasc Res. 2002;54:416–26. doi: 10.1016/s0008-6363(02)00274-2. [DOI] [PubMed] [Google Scholar]
- 188.Goldman ME, Pearce LA, Hart RG, Zabalgoitia M, Asinger RW, Safford R, Halperin JL. Pathophysiologic correlates of thromboembolism in nonvalvular atrial fibrillation: I. Reduced flow velocity in the left atrial appendage (The Stroke Prevention in Atrial Fibrillation [SPAF-III] study) J Am Soc Echocardiogr. 1999;12:1080–7. doi: 10.1016/s0894-7317(99)70105-7. [DOI] [PubMed] [Google Scholar]
- 189.Merino A, Hauptman P, Badimon L, Badimon JJ, Cohen M, Fuster V, Goldman M. Echocardiographic “smoke” is produced by an interaction of erythrocytes and plasma proteins modulated by shear forces. J Am Coll Cardiol. 1992;20:1661–8. doi: 10.1016/0735-1097(92)90463-w. [DOI] [PubMed] [Google Scholar]
- 190.Holmes DR, Reddy VY, Turi ZG, Doshi SK, Sievert H, Buchbinder M, Mullin CM, Sick P, Investigators PA Percutaneous closure of the left atrial appendage versus warfarin therapy for prevention of stroke in patients with atrial fibrillation: a randomised non-inferiority trial. Lancet. 2009;374:534–42. doi: 10.1016/S0140-6736(09)61343-X. [DOI] [PubMed] [Google Scholar]
- 191.Turgut N, Akdemir O, Turgut B, Demir M, Ekuklu G, Vural O, Ozbay G, Utku U. Hypercoagulopathy in stroke patients with nonvalvular atrial fibrillation: hematologic and cardiologic investigations. Clin Appl Thromb Hemost. 2006;12:15–20. doi: 10.1177/107602960601200104. [DOI] [PubMed] [Google Scholar]
- 192.Tsai LM, Chen JH, Tsao CJ. Relation of left atrial spontaneous echo contrast with prethrombotic state in atrial fibrillation associated with systemic hypertension, idiopathic dilated cardiomyopathy, or no identifiable cause (lone) Am J Cardiol. 1998;81:1249–52. doi: 10.1016/s0002-9149(98)00131-3. [DOI] [PubMed] [Google Scholar]
- 193.Mondillo S, Sabatini L, Agricola E, Ammaturo T, Guerrini F, Barbati R, Pastore M, Fineschi D, Nami R. Correlation between left atrial size, prothrombotic state and markers of endothelial dysfunction in patients with lone chronic nonrheumatic atrial fibrillation. Int J Cardiol. 2000;75:227–32. doi: 10.1016/s0167-5273(00)00336-3. [DOI] [PubMed] [Google Scholar]
- 194.Conway DS, Heeringa J, Van Der Kuip DA, Chin BS, Hofman A, Witteman JC, Lip GY. Atrial fibrillation and the prothrombotic state in the elderly: the Rotterdam Study. Stroke. 2003;34:413–7. doi: 10.1161/01.str.0000051728.85133.32. [DOI] [PubMed] [Google Scholar]
- 195.Lip GY, Lowe GD, Rumley A, Dunn FG. Fibrinogen and fibrin D-dimer levels in paroxysmal atrial fibrillation: evidence for intermediate elevated levels of intravascular thrombogenesis. Am Heart J. 1996;131:724–30. doi: 10.1016/s0002-8703(96)90278-1. [DOI] [PubMed] [Google Scholar]
- 196.Kahn SR, Solymoss S, Flegel KM. Nonvalvular atrial fibrillation: evidence for a prothrombotic state. CMAJ. 1997;157:673–81. [PMC free article] [PubMed] [Google Scholar]
- 197.Feng D, D’Agostino RB, Silbershatz H, Lipinska I, Massaro J, Levy D, Benjamin EJ, Wolf PA, Tofler GH. Hemostatic state and atrial fibrillation (the Framingham Offspring Study) Am J Cardiol. 2001;87:168–71. doi: 10.1016/s0002-9149(00)01310-2. [DOI] [PubMed] [Google Scholar]
- 198.Wolf PA, Kannel WB, McGee DL, Meeks SL, Bharucha NE, McNamara PM. Duration of atrial fibrillation and imminence of stroke: the Framingham study. Stroke. 1983;14:664–7. doi: 10.1161/01.str.14.5.664. [DOI] [PubMed] [Google Scholar]
- 199.Bekwelem W, Connolly SJ, Halperin JL, Adabag S, Duval S, Chrolavicius S, Pogue J, Ezekowitz MD, Eikelboom JW, Wallentin LG, Yusuf S, Hirsch AT. Extracranial Systemic Embolic Events in Patients With Nonvalvular Atrial Fibrillation: Incidence, Risk Factors, and Outcomes. Circulation. 2015;132:796–803. doi: 10.1161/CIRCULATIONAHA.114.013243. [DOI] [PubMed] [Google Scholar]
- 200.Kalantarian S, Ruskin JN. Cognitive impairment associated with atrial fibrillation–in response. Ann Intern Med. 2013;158:849. doi: 10.7326/0003-4819-158-11-201306040-00016. [DOI] [PubMed] [Google Scholar]
- 201.Marzona I, O’Donnell M, Teo K, Gao P, Anderson C, Bosch J, Yusuf S. Increased risk of cognitive and functional decline in patients with atrial fibrillation: results of the ONTARGET and TRANSCEND studies. CMAJ. 2012;184:E329–36. doi: 10.1503/cmaj.111173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Kwok CS, Loke YK, Hale R, Potter JF, Myint PK. Atrial fibrillation and incidence of dementia: a systematic review and meta-analysis. Neurology. 2011;76:914–22. doi: 10.1212/WNL.0b013e31820f2e38. [DOI] [PubMed] [Google Scholar]
- 203.Santangeli P, Di Biase L, Bai R, Mohanty S, Pump A, Cereceda Brantes M, Horton R, Burkhardt JD, Lakkireddy D, Reddy YM, Casella M, Dello Russo A, Tondo C, Natale A. Atrial fibrillation and the risk of incident dementia: a meta-analysis. Heart Rhythm. 2012;9:1761–8. doi: 10.1016/j.hrthm.2012.07.026. [DOI] [PubMed] [Google Scholar]
- 204.de Bruijn RF, Heeringa J, Wolters FJ, Franco OH, Stricker BH, Hofman A, Koudstaal PJ, Ikram MA. Association Between Atrial Fibrillation and Dementia in the General Population. JAMA Neurol. 2015;72:1288–94. doi: 10.1001/jamaneurol.2015.2161. [DOI] [PubMed] [Google Scholar]
- 205.Satizabal C, Beiser AS, Seshadri S. Incidence of Dementia over Three Decades in the Framingham Heart Study. N Engl J Med. 2016;375:93–4. doi: 10.1056/NEJMc1604823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Dublin S, Anderson ML, Haneuse SJ, Heckbert SR, Crane PK, Breitner JC, McCormick W, Bowen JD, Teri L, McCurry SM, Larson EB. Atrial fibrillation and risk of dementia: a prospective cohort study. J Am Geriatr Soc. 2011;59:1369–75. doi: 10.1111/j.1532-5415.2011.03508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Hara M, Ooie T, Yufu K, Tsunematsu Y, Kusakabe T, Ooga M, Saikawa T, Sakata T. Silent cortical strokes associated with atrial fibrillation. Clin Cardiol. 1995;18:573–4. doi: 10.1002/clc.4960181008. [DOI] [PubMed] [Google Scholar]
- 208.Bunch TJ, May HT, Bair TL, Crandall BG, Cutler MJ, Day JD, Jacobs V, Mallender C, Osborn JS, Stevens SM, Weiss JP, Woller SC. Atrial Fibrillation Patients Treated With Long-Term Warfarin Anticoagulation Have Higher Rates of All Dementia Types Compared With Patients Receiving Long-Term Warfarin for Other Indications. J Am Heart Assoc. 2016;5 doi: 10.1161/JAHA.116.003932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Lavy S, Stern S, Melamed E, Cooper G, Keren A, Levy P. Effect of chronic atrial fibrillation on regional cerebral blood flow. Stroke. 1980;11:35–8. doi: 10.1161/01.str.11.1.35. [DOI] [PubMed] [Google Scholar]
- 210.Petersen P, Kastrup J, Videbaek R, Boysen G. Cerebral blood flow before and after cardioversion of atrial fibrillation. J Cereb Blood Flow Metab. 1989;9:422–5. doi: 10.1038/jcbfm.1989.62. [DOI] [PubMed] [Google Scholar]
- 211.Phillips E, Levine SA. Auricular fibrillation without other evidence of heart disease; a cause of reversible heart failure. Am J Med. 1949;7:478–89. doi: 10.1016/0002-9343(49)90397-6. [DOI] [PubMed] [Google Scholar]
- 212.Wang TJ, Larson MG, Levy D, Vasan RS, Leip EP, Wolf PA, D’Agostino RB, Murabito JM, Kannel WB, Benjamin EJ. Temporal relations of atrial fibrillation and congestive heart failure and their joint influence on mortality: the Framingham Heart Study. Circulation. 2003;107:2920–5. doi: 10.1161/01.CIR.0000072767.89944.6E. [DOI] [PubMed] [Google Scholar]
- 213.Middlekauff HR, Stevenson WG, Stevenson LW. Prognostic significance of atrial fibrillation in advanced heart failure. A study of 390 patients. Circulation. 1991;84:40–8. doi: 10.1161/01.cir.84.1.40. [DOI] [PubMed] [Google Scholar]
- 214.Torp-Pedersen C, Moller M, Bloch-Thomsen PE, Kober L, Sandoe E, Egstrup K, Agner E, Carlsen J, Videbaek J, Marchant B, Camm AJ. Dofetilide in patients with congestive heart failure and left ventricular dysfunction. Danish Investigations of Arrhythmia and Mortality on Dofetilide Study Group. N Engl J Med. 1999;341:857–65. doi: 10.1056/NEJM199909163411201. [DOI] [PubMed] [Google Scholar]
- 215.Carson PE, Johnson GR, Dunkman WB, Fletcher RD, Farrell L, Cohn JN. The influence of atrial fibrillation on prognosis in mild to moderate heart failure. The V-HeFT Studies. The V-HeFT VA Cooperative Studies Group. Circulation. 1993;87:VI102–10. [PubMed] [Google Scholar]
- 216.Camm AJ, Savelieva I. Atrial fibrillation: advances and perspectives. Lead Article. 1912;13:183. [Google Scholar]
- 217.Furberg CD, Psaty BM, Manolio TA, Gardin JM, Smith VE, Rautaharju PM. Prevalence of atrial fibrillation in elderly subjects (the Cardiovascular Health Study) Am J Cardiol. 1994;74:236–41. doi: 10.1016/0002-9149(94)90363-8. [DOI] [PubMed] [Google Scholar]
- 218.Krahn AD, Manfreda J, Tate RB, Mathewson FA, Cuddy TE. The natural history of atrial fibrillation: incidence, risk factors, and prognosis in the Manitoba Follow-Up Study. Am J Med. 1995;98:476–84. doi: 10.1016/S0002-9343(99)80348-9. [DOI] [PubMed] [Google Scholar]
- 219.Schmidt M, Ulrichsen SP, Pedersen L, Botker HE, Nielsen JC, Sorensen HT. 30-year nationwide trends in incidence of atrial fibrillation in Denmark and associated 5-year risk of heart failure, stroke, and death. Int J Cardiol. 2016;225:30–36. doi: 10.1016/j.ijcard.2016.09.071. [DOI] [PubMed] [Google Scholar]
- 220.Santhanakrishnan R, Wang N, Larson MG, Magnani JW, McManus DD, Lubitz SA, Ellinor PT, Cheng S, Vasan RS, Lee DS, Wang TJ, Levy D, Benjamin EJ, Ho JE. Atrial Fibrillation Begets Heart Failure and Vice Versa: Temporal Associations and Differences in Preserved Versus Reduced Ejection Fraction. Circulation. 2016;133:484–92. doi: 10.1161/CIRCULATIONAHA.115.018614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Healey JS, Oldgren J, Ezekowitz M, et al. Occurrence of death and stroke in patients in 47 countries 1 year after presenting with atrial fibrillation: a cohort study. Lancet. 2016;388:1161–9. doi: 10.1016/S0140-6736(16)30968-0. [DOI] [PubMed] [Google Scholar]
- 222.Kotecha D, Chudasama R, Lane DA, Kirchhof P, Lip GY. Atrial fibrillation and heart failure due to reduced versus preserved ejection fraction: A systematic review and meta-analysis of death and adverse outcomes. Int J Cardiol. 2016;203:660–6. doi: 10.1016/j.ijcard.2015.10.220. [DOI] [PubMed] [Google Scholar]
- 223.Li D, Melnyk P, Feng J, Wang Z, Petrecca K, Shrier A, Nattel S. Effects of experimental heart failure on atrial cellular and ionic electrophysiology. Circulation. 2000;101:2631–8. doi: 10.1161/01.cir.101.22.2631. [DOI] [PubMed] [Google Scholar]
- 224.Li D, Fareh S, Leung TK, Nattel S. Promotion of atrial fibrillation by heart failure in dogs: atrial remodeling of a different sort. Circulation. 1999;100:87–95. doi: 10.1161/01.cir.100.1.87. [DOI] [PubMed] [Google Scholar]
- 225.Sanders P, Morton JB, Davidson NC, Spence SJ, Vohra JK, Sparks PB, Kalman JM. Electrical remodeling of the atria in congestive heart failure: electrophysiological and electroanatomic mapping in humans. Circulation. 2003;108:1461–8. doi: 10.1161/01.CIR.0000090688.49283.67. [DOI] [PubMed] [Google Scholar]
- 226.Ohtani K, Yutani C, Nagata S, Koretsune Y, Hori M, Kamada T. High prevalence of atrial fibrosis in patients with dilated cardiomyopathy. J Am Coll Cardiol. 1995;25:1162–9. doi: 10.1016/0735-1097(94)00529-y. [DOI] [PubMed] [Google Scholar]
- 227.Cardin S, Li D, Thorin-Trescases N, Leung TK, Thorin E, Nattel S. Evolution of the atrial fibrillation substrate in experimental congestive heart failure: angiotensin-dependent and -independent pathways. Cardiovasc Res. 2003;60:315–25. doi: 10.1016/j.cardiores.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 228.Swaminathan PD, Purohit A, Soni S, et al. Oxidized CaMKII causes cardiac sinus node dysfunction in mice. J Clin Invest. 2011;121:3277–88. doi: 10.1172/JCI57833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Purohit A, Rokita AG, Guan X, et al. Oxidized Ca(2+)/calmodulin-dependent protein kinase II triggers atrial fibrillation. Circulation. 2013;128:1748–57. doi: 10.1161/CIRCULATIONAHA.113.003313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Chang SL, Chen YC, Yeh YH, Lin YK, Wu TJ, Lin CI, Chen SA, Chen YJ. Heart failure enhanced pulmonary vein arrhythmogenesis and dysregulated sodium and calcium homeostasis with increased calcium sparks. J Cardiovasc Electrophysiol. 2011;22:1378–86. doi: 10.1111/j.1540-8167.2011.02126.x. [DOI] [PubMed] [Google Scholar]
- 231.Kannel WB, Abbott RD, Savage DD, McNamara PM. Coronary heart disease and atrial fibrillation: the Framingham Study. Am Heart J. 1983;106:389–96. doi: 10.1016/0002-8703(83)90208-9. [DOI] [PubMed] [Google Scholar]
- 232.Violi F, Soliman EZ, Pignatelli P, Pastori D. Atrial Fibrillation and Myocardial Infarction: A Systematic Review and Appraisal of Pathophysiologic Mechanisms. J Am Heart Assoc. 2016;5 doi: 10.1161/JAHA.116.003347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Soliman EZ, Safford MM, Muntner P, Khodneva Y, Dawood FZ, Zakai NA, Thacker EL, Judd S, Howard VJ, Howard G, Herrington DM, Cushman M. Atrial fibrillation and the risk of myocardial infarction. JAMA Intern Med. 2014;174:107–14. doi: 10.1001/jamainternmed.2013.11912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Miyasaka Y, Barnes ME, Gersh BJ, Cha SS, Bailey KR, Seward JB, Iwasaka T, Tsang TS. Coronary ischemic events after first atrial fibrillation: risk and survival. Am J Med. 2007;120:357–63. doi: 10.1016/j.amjmed.2006.06.042. [DOI] [PubMed] [Google Scholar]
- 235.Holst AG, Jensen G, Prescott E. Risk factors for venous thromboembolism: results from the Copenhagen City Heart Study. Circulation. 2010;121:1896–903. doi: 10.1161/CIRCULATIONAHA.109.921460. [DOI] [PubMed] [Google Scholar]
- 236.Braekkan SK, Hald EM, Mathiesen EB, Njolstad I, Wilsgaard T, Rosendaal FR, Hansen JB. Competing risk of atherosclerotic risk factors for arterial and venous thrombosis in a general population: the Tromso study. Arterioscler Thromb Vasc Biol. 2012;32:487–91. doi: 10.1161/ATVBAHA.111.237545. [DOI] [PubMed] [Google Scholar]
- 237.Wattanakit K, Lutsey PL, Bell EJ, Gornik H, Cushman M, Heckbert SR, Rosamond WD, Folsom AR. Association between cardiovascular disease risk factors and occurrence of venous thromboembolism. A time-dependent analysis. Thromb Haemost. 2012;108:508–15. doi: 10.1160/TH11-10-0726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Mahmoodi BK, Cushman M, Anne Naess I, et al. Association of Traditional Cardiovascular Risk Factors With Venous Thromboembolism: An Individual Participant Data Meta-Analysis of Prospective Studies. Circulation. 2017;135:7–16. doi: 10.1161/CIRCULATIONAHA.116.024507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Enga KF, Rye-Holmboe I, Hald EM, Lochen ML, Mathiesen EB, Njolstad I, Wilsgaard T, Braekkan SK, Hansen JB. Atrial fibrillation and future risk of venous thromboembolism:the Tromso study. Journal of thrombosis and haemostasis: JTH. 2015;13:10–6. doi: 10.1111/jth.12762. [DOI] [PubMed] [Google Scholar]
- 240.Hald EM, Enga KF, Lochen ML, Mathiesen EB, Njolstad I, Wilsgaard T, Braekkan SK, Hansen JB. Venous thromboembolism increases the risk of atrial fibrillation: the Tromso study. Journal of the American Heart Association. 2014;3:e000483. doi: 10.1161/JAHA.113.000483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Wang CC, Lin CL, Wang GJ, Chang CT, Sung FC, Kao CH. Atrial fibrillation associated with increased risk of venous thromboembolism. A population-based cohort study. Thrombosis and haemostasis. 2015;113:185–92. doi: 10.1160/TH14-05-0405. [DOI] [PubMed] [Google Scholar]
- 242.Piccini JP, Hammill BG, Sinner MF, Hernandez AF, Walkey AJ, Benjamin EJ, Curtis LH, Heckbert SR. Clinical course of atrial fibrillation in older adults: the importance of cardiovascular events beyond stroke. Eur Heart J. 2014;35:250–6. doi: 10.1093/eurheartj/eht483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Odutayo A, Wong CX, Hsiao AJ, Hopewell S, Altman DG, Emdin CA. Atrial fibrillation and risks of cardiovascular disease, renal disease, and death: systematic review and meta-analysis. Bmj. 2016;354:i4482. doi: 10.1136/bmj.i4482. [DOI] [PubMed] [Google Scholar]
- 244.Chen LY, Sotoodehnia N, Buzkova P, Lopez FL, Yee LM, Heckbert SR, Prineas R, Soliman EZ, Adabag S, Konety S, Folsom AR, Siscovick D, Alonso A. Atrial fibrillation and the risk of sudden cardiac death: the atherosclerosis risk in communities study and cardiovascular health study. JAMA Intern Med. 2013;173:29–35. doi: 10.1001/2013.jamainternmed.744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Marijon E, Le Heuzey JY, Connolly S, Yang S, Pogue J, Brueckmann M, Eikelboom J, Themeles E, Ezekowitz M, Wallentin L, Yusuf S, Investigators R-L Causes of death and influencing factors in patients with atrial fibrillation: a competing-risk analysis from the randomized evaluation of long-term anticoagulant therapy study. Circulation. 2013;128:2192–201. doi: 10.1161/CIRCULATIONAHA.112.000491. [DOI] [PubMed] [Google Scholar]
- 246.Hart RG, Pearce LA, Aguilar MI. Meta-analysis: antithrombotic therapy to prevent stroke in patients who have nonvalvular atrial fibrillation. Ann Intern Med. 2007;146:857–67. doi: 10.7326/0003-4819-146-12-200706190-00007. [DOI] [PubMed] [Google Scholar]
- 247.Pokorney SD, Piccini JP, Stevens SR, Patel MR, Pieper KS, Halperin JL, Breithardt G, Singer DE, Hankey GJ, Hacke W, Becker RC, Berkowitz SD, Nessel CC, Mahaffey KW, Fox KA, Califf RM. Cause of Death and Predictors of All-Cause Mortality in Anticoagulated Patients With Nonvalvular Atrial Fibrillation: Data From ROCKET AF. J Am Heart Assoc. 2016;5:e002197. doi: 10.1161/JAHA.115.002197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Larsen TB, Rasmussen LH, Skjoth F, Due KM, Callreus T, Rosenzweig M, Lip GY. Efficacy and safety of dabigatran etexilate and warfarin in “real-world” patients with atrial fibrillation: a prospective nationwide cohort study. J Am Coll Cardiol. 2013;61:2264–73. doi: 10.1016/j.jacc.2013.03.020. [DOI] [PubMed] [Google Scholar]
- 249.Proietti M, Laroche C, Opolski G, Maggioni AP, Boriani G, Lip GY, Investigators AFGP ’Real-world’ atrial fibrillation management in Europe: observations from the 2-year follow-up of the EURObservational Research Programme-Atrial Fibrillation General Registry Pilot Phase. Europace. 2016 doi: 10.1093/europace/euw112. [DOI] [PubMed] [Google Scholar]