Structural basis of TPR mediated oligomerization and activation of oncogenic fusion kinases (original) (raw)
. Author manuscript; available in PMC: 2018 Jun 6.
Published in final edited form as: Structure. 2017 May 18;25(6):867–877.e3. doi: 10.1016/j.str.2017.04.015
Summary
The nuclear pore complex subunit TPR is found in at least five different oncogenic fusion kinases, including TPR-MET, yet how TPR fusions promote activation of kinases and their oncogenic activities remains poorly understood. Here we report the crystal structure of TPR(2-142), the MET fusion partner of oncogenic TPR-MET. TPR(2-142) contains a continuous 124-residue α-helix that forms an anti-parallel tetramer from two leucine zipper-containing parallel coiled coils. Remarkably, single mutations cause strikingly different conformations of the coiled coil, indicating its highly dynamic nature. We further show that fusion of TPR(2-142) to the MET intracellular domain strongly and selectively stabilizes the αG-helix of the MET kinase domain and mutations of only the TPR leucine zipper residues at the junction to MET, but not other leucine zipper residues, abolish kinase activation. Together, these results provide critical insight into the TPR structure and its ability to induce dimerization and activation of fusion kinases.
Keywords: receptor tyrosine kinase, oncogenic fusion kinase, MET, receptor dimerization
eTOC
Pal et al. present a high resolution structure of the oligomeric protein TPR. TPR fusion with cell-signaling protein kinases by chromosomal translocations can generate potent oncogenes, in which TPR induces kinase hyperactivation. The structure provides insights into TPR conformational constraints required for oncogenic kinase activation.
Introduction
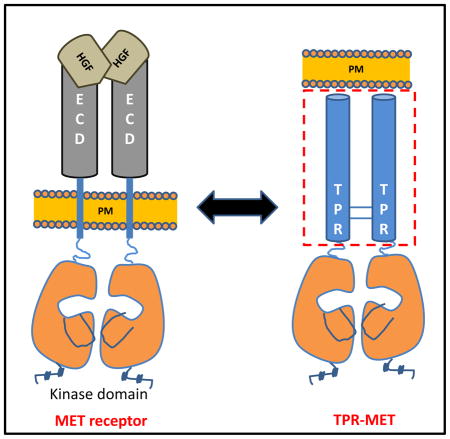
Oncogenic mutations that result in gene fusion-mediated activation of receptor tyrosine kinases (RTKs) have been found in many malignancies and tumors (Lamorte and Park, 2001; Rodrigues and Park, 1994). From a recent pan-cancer analysis of tumor sample transcriptomes, almost 3% of the samples contained oncogenic, constitutively active kinase fusions (Stransky et al., 2014). For instance, MET receptor tyrosine kinase, the receptor for Hepatocyte Growth Factor (HGF), was first identified in the context of the oncogenic fusion protein, TPR-MET (Cooper et al., 1984; Park et al., 1986), a fusion of the Met kinase domain with the oligomerization domain of the nuclear pore complex subunit TPR. A genomic rearrangement mediated by the carcinogen N-methyl-N′-nitrosoguanidine directed the fusion between the upstream region of TPR locus 1q25 (Translocated Promoter Region) to the downstream sequence of MET kinase domain 7q31 (Fig. 1A) in a human osteogenic sarcoma cell line (Dean et al., 1987; Park et al., 1987). This translocation resulted in a complete replacement of the extracellular and transmembrane domain (Fig. 1B) of MET by the N-terminal oligomerization domain of TPR. This domain mediates the stable dimerization of the MET kinase domain in a ligand independent manner, making it a constitutively active, cytosolic oncogene that lacks all self-regulatory mechanisms (Rodrigues and Park, 1993). Importantly, while ligand-mediated receptor dimerization or reorganization of constitutively dimerized receptors is required for RTK activation, RTK dimerization itself may be insufficient for activation, but rather may require topological constraints to induce formation of an active kinase conformation in the intracellular kinase domain (Arkhipov et al., 2013; Endres et al., 2014). In the absence of any full length RTK crystal structure, the exact mechanisms of coupling remain incompletely understood.
Figure 1. Schematic representation of the TPR-MET chromosomal translocation.

**(A)**Structural and functional domains of MET receptor, Translocated promoter region (TPR), and the TPR(1-142)-MET(1010-1390) fusion protein. Chromosomal translocation mediated fusion between the Translocated Promoter Region (TPR) and the MET receptor genes are indicated by blue lines. Translocation results in expression of a cytosolic, constitutively active TPR-MET oncogene. (B) MET consists of a large extracellular region that contains a Sema domain, a cysteine-rich (CR) domain, and four immunoglobulin (IG) domains followed by a single transmembrane helix and the intracellular juxtamembrane and kinase domains. Hepatocyte growth factor (HGF)-regulated MET dimerization in the extracellular region transmits a signal for kinase domain (KD) autophosphorylation and downstream signaling. TPR-MET lacks the MET regulatory extracellular domain and forms a dimeric, constitutively active fusion kinase oncogene.
The nuclear pore complex (NPC) is a 120 MDa complex consisting of multiple copies of ~30 different nucleoporins that has critical functions in nucleocytoplasmic transport, chromatin organization, and regulation of gene expression and DNA repair (Beck and Hurt, 2017). TPR is a 270 kDa component of the basket subcomplex on the nuclear side of the NPC. It consists of a ~1600 residue coiled coil-rich N-terminal domain and an 800 amino acid highly acidic and posttranslationally modified C-terminus (Nakano et al., 2010; Rajanala et al., 2014). The N-terminal domain tethers TPR via Nup153 to the NCP and transiently interacts with the spindle checkpoint Mad1 and the motor proteins dynein and dynactin, consistent with a function of TPR as a spindle checkpoint regulator (Nakano et al., 2010).
The physiological MET agonist HGF controls several important biological processes in development and morphogenesis, including cell proliferation and migration, by activating the MET signaling pathway through HGF-induced Met dimerization (Birchmeier et al., 2003; Peschard and Park, 2007; Tolbert et al., 2010; Tolbert et al., 2007). In addition to constitutive dimerization, the absence of the extracellular domain of MET in TPR-MET bypasses negative regulation of the kinase domain, and the absence of the docking site for the ubiquitin ligase Cbl bypasses receptor internalization and signal desensitization (Mak et al., 2007). Lack of these additional levels of regulation is a likely reason for the high transforming activity and invasiveness of TPR-MET (Merlin et al., 2009; Peschard and Park, 2003). With the discovery of TPR-MET as fusion kinase almost 30 years ago, it has become an important prototype to investigate the mechanism of RTK-derived oncogene generation by translocation. Subsequently, several other TPR based fusion kinases were reported, including the RTK fusion kinases TPR-TRK1 (Greco et al., 1992), TPR-ALK (Choi et al., 2014), and TPR-FGFR1(Malli et al., 2016) and the serine/threonine fusion kinase TPR-RAF (King et al., 1988)
Kinase activation in the context of TPR-RTK fusion proteins provides a potent tool to study the oligomeric kinase domain conformation(s) required for kinase activity in the context of a non-membrane protein. MET dimerization allows trans-autophosphorylation of Tyr365 and Tyr366 (corresponds to Tyr1234 and Tyr1235 in the full length receptor) in the activation loop of the kinase domain to catalytically activate MET, followed by further phosphorylation of Tyr482 and Tyr489 (corresponds to Tyr1349 and Tyr1356 in the full length receptor) in the domain C-terminal to the kinase domain (C-tail). These phosphotyrosines in the C-tail provide docking sites for downstream regulators of HGF signaling, including Grb-2, phospholipase Cγ, SHP-2, PI3-K, and Grab-1 (Birchmeier et al., 2003; Furge et al., 2000). In addition to positioning the kinase domains for trans-phosphorylation, oligomerization itself may also stabilize a catalysis-favorable kinase conformation (Hubbard et al., 1998; Zhang et al., 2006) and protect against dephosphorylation (Shimizu et al., 2001). The mechanism of how TPR promotes oligomerization and activation of oncogenic fusion kinase remains elusive. Here we describe the crystal structure of the TPR(2-142) fusion fragment, which is found to form an antiparallel coiled-coil tetramer and provides the molecular basis for the capability of TPR to promote dimerization and constitutive activation of MET and other oncogenic kinases.
Results
TPR(2-142) forms a parallel coiled coil dimer that further folds into antiparallel tetramers
We generated selenomethionine (SeMe)-labeled variants of TPR(2-142) to solve its crystal structure by multi-wavelength anomalous dispersion (MAD). Due to the absence of natural methionine residues in the coiled coil region, we generated a set of mutant proteins in which we replaced phenylalanine, isoleucine, or leucine residues with methionine (F28M, F46M, F55M, L127M, F55M/F113M, and F113M/I101M/F113M). Out of these, the triple mutant F55M/I101M/F113M provided sufficient anomalous signal to allow us to solve the phase problem and to determine its structure at a resolution of 2.75 Å (Table S1). To our surprise, the full structure model did not allow us to determine the structure of wildtype TPR(2-142) by the molecular replacement method, indicating that the structures of the wildtype and triple mutant proteins differed substantially. We therefore used the initial model of the triple mutant protein to determine in a stepwise manner the structures of the F55M/F113M double mutant, F113M single mutant, and finally the wildtype protein (Table S1, see Methods).
Wildtype TPR(2-142) is arranged as a tetramer in the asymmetric unit, in which two parallel dimers associate in an antiparallel orientation (Fig. 2A). Each monomeric chain contains a 124 amino acid single continuous α-helix (α3, Fig. 2A). This helix is capped by an 18 amino acid N-terminal head structure that is formed by two short helices (α1: residues 4-8, and α2: residues 11-16) and connecting loops that curve back onto α1 in a fold resembling the head of a walking stick (Fig. 2B). This head structure is stabilized by a hydrophobic core formed by V8, V21, F28 and six leucine residues (L5, L9, L14, L17, L25, and L29) and by charge interactions on the outside of the fold (intra-chain: A2-Q6, E10-E13, R11-N15, K18-Q22; Fig. 2B). Earlier studies have shown that hydrophobic interaction networks at the end of coiled coils are an important determinant of protein stability (Aurora and Rose, 1998; Ermolenko et al., 2002). Inter-chain hydrogen bond and ionic-interactions of E13 in the cap with S20 and K24 in α3, respectively, provide further stability (Fig. 2B). The residues from 19-142 form the 175.6 Å long left-handed α3 coiled coil helix (Fig. 2C–F and Fig. S1). In addition to the standard i→i+4 hydrogen bonds, each individual helix of the coiled coil is further stabilized by several intra-chain ionic interactions (e.g., K26-D30, E43-K46, R114-E117). These i→i+3/i→i+4 intra-helical salt bridges can markedly improve the stability of coiled coils (Burkhard et al., 2002; Burkhard et al., 2001).
Figure 2. Structural organization of dimeric TPR (See also Figures S1 and S2).

(A) Crystal structure of TPR(2-142). TPR forms a parallel coiled coil dimer with chain A colored in green and chain B in cyan that further oligomerizes to form antiparallel tetramers (red and yellow chains). (B) Ribbon diagram of the helical caps of Chain A (green) and Chain B (cyan) with hydrophobic and ionic interactions indicated by dashed lines. (C–F) Stick representations of dimerization interface residues in the N-terminal coiled coil, the first leucine zipper (residues 77-100, D), the second leucine zipper (residues 120-141, E), and the middle region with a C75-C75 disulfide bond found in one of the tetramers in the asymmetric unit. The side chains of ‘a’ and ‘d’ residues in heptad (a-b-c-d-e-f-g) repeats of leucine zippers are displayed. (G) Disulfide crosslinking validation of the parallel arrangement of the TPR dimer. Cells transfected with TPR mutant constructs were treated with H2O2 and lysed, lysates separated by non-reducing SDS PAGE, and TPR and TPR crosslinking adducts detected by immunoblotting.
The C-terminus of the coiled coil is stabilized by two leucine zipper motifs
3–5% of all amino acids in proteins form coiled coils, which play crucial roles in homo- and hetero-oligomerization (Wolf et al., 1997). The basic arrangements of amino acids in these domains are heptad repeats (a-b-c-d-e-f-g). The nonpolar interface residues at the a and d positions are the main affinity contributors, while solvent exposed polar residues at the e and g positions are responsible for specificity and oligomerization of helices (Mason and Arndt, 2004) (Figures 2C–E and S1, S2). The N-terminal half of the TPR coiled coil forms Van-der-Waals monomer-monomer interchain interactions through residues at the a and d positions, with the exception of Q32, H43, and S64, which form hydrogen bonds (Fig. 2C). The distance between the two strands becomes too large in the middle of the N-terminal coiled coil for Van-der-Waals interactions. In this region the interaction periodicity shifts by one residue (a “skip” (Lupas and Gruber, 2005), a′-b′-c′-d′-e′-f′-g′;): Q53 (7 amino acids distance to F46 and 8 amino acids distance to L61) on one strand of the dimer forms Van-der-Waals bonds with Y54 (8 amino acids distance to F46 and 7 amino acids distance to L61) on the other strand (Fig. 2C and Fig. S1).
The second half of the coiled coil forms two leucine zipper motifs, which span residues 78-99 and 120-134, respectively (Fig. 2D, E, and Fig. S2). Earlier studies have shown that a triple leucine to valine mutation (L78V/L85V/L92V) in the first leucine zipper, but not in the second leucine zipper (L120V/L127V/L134V), disrupts the dimer and abolishes foci formation (Rodrigues and Park, 1993), suggesting that the first leucine zipper critically contributes to dimerization and tumorigenicity, and that the leucine residues cannot be functionally replaced by valine residues. Consistent with its importance for dimerization, leucine zipper 1 is conserved across species (Fig. S2). We validated the parallel orientation of TPR monomers in solution by disulfide crosslinking, which has strict requirements on the distance and orientation between the cysteine sulfur atoms (Dombkowski and Crippen, 2000). We introduced cysteine mutations into seven residues spanning the entire coiled-coil region, whose Cα-Cα distances in the parallel dimer structure are within 5.1 to 7.3 Å distance. As shown in Fig. 2G, all seven mutant proteins formed disulfide crosslinking adducts, whereas a mutant protein with 9.3 Å Cα-Cα distance failed to form detectable crosslinks, thus supporting the parallel dimer conformation observed in the structure.
Non-mutated TPR-MET can also form a disulfide bond upon cell lysis (Rodrigues and Park, 1993), which could be mediated by the disulfide bond formed by C75- C75 at the middle of the coiled coil of one of the dimers in the crystal (Fig. 2F). However, it remains unclear, whether the propensity for C75-C75 disulfide bond formation is biologically relevant.
Charge interactions and distribution mediate formation of antiparallel tetramers
The two dimer chains further associate into a tetrameric conformation in an anti-parallel orientation (Fig. 3A). Tetramers are also the predominant forms in solution as determined by size exclusion chromatography for both TPR(2-142) and TPR-MET(2-493) (Fig. 3B). To validate the TPR tetramer orientation and overall structure in solution, we introduced cysteine mutations into two dimer–dimer interface residue pairs with suitable Cα-Cα distances (Q52–A108: 6.9 Å and E56–R105: 7.7 Å), as well as into one interface residue pair with a longer Cα-Cα distance (K59C/D104C: 9.8 Å). In agreement with the crystal structure, the E56C/R105C and especially the Q52C/A108C mutants formed highly efficiently disulfide bonds, whereas K59C/D104C formed disulfide bonds with only low efficiency (Fig. 3C). In a parallel dimer–dimer orientation, the same residue pairs would be too far apart to form disulfide bonds, therefore unambiguously confirming the antiparallel orientation of two dimers in the tetramer complex.
Figure 3. Tetrameric organization of TPR (See also Figures S3).

(A) TPR(2-142) tetramer in ribbon presentation, in which one dimer is overlaid with a transparent surface electrostatic potential map. Blue colored regions indicate positive charge and red colored regions negative charge (see charge potential heat map at bar below). The four monomers are colored green, cyan, magenta and yellow. (B) Tetramer distribution of TPR(2-142) wildtype and TPR-F55M/F113M(2-142) determined by size exclusion chromatography. (C) Validation of the antiparallel tetramer arrangement by in cell disulfide crosslinking between indicated residues of chain A and chain C. Lysates of H2O2-treated FLAG-TPR-MET expressing cells were analyzed by non-reducing SDS PAGE and immunoblotting with anti-FLAG antibody. (D) Parallel tetramer orientation of TPR(2-142) F113M. (E) Dimeric arrangement of TPR(2-142) F55M/F113M.
Tetrameric conformation of the oligomerization domains has also been observed for the BCR-ABL (Zhao et al., 2002) and SAM-EphB2 (Thanos et al., 1999) fusion kinases. Tetramerization is mediated by interactions between polar residues on the solvent exposed dimer surfaces (Fig. 4A). In addition to complementary charges at individual interface residue pairs, the whole N-terminal and C-terminal halves of the TPR coiled coil dimer have distinct negative and positive charge potentials, respectively, which could determine the antiparallel orientation of the tetramer complex (Fig. 3A). Charge reversal mutations in clusters of TPR dimer–dimer interface residues destabilized purified TPR, but did not affect MET-activation-dependent autophosphorylation (Fig. 4B, C). To gain insight into tetramer stability, we isolated TPR tetramer by size exclusion chromatography, confirmed its size by multi-angle static light scattering (MALS), and then reloaded dilutions of the tetramer fraction onto the size column. The broad elution peak corresponding to 193 kDa suggests that TPR-MET at 520 nM in the non-aggregaged fraction is predominantly (>75%) tetrameric. At 52 and 26 nM TPR-MET, this peak slightly shifted towards a larger elution volume, indicating that the tetramer population mildly decreased (<20%), demonstrating that the tetramer is highly stable with a tetramerization constant that is below the lowest concentration that we could test by size exclusion chromatography (<26 nM; Fig. 5).
Figure 4. The TPR dimer-dimer interface is mediated largely by ionic interactions.

(A.) Cartoon structure of the dimer-dimer interface between chain A (purple) and chain C (magenta). Key interface residues are shown in stick presentation. (B). Size column profiles of TPR-MET WT and TPR mutant proteins with charge reversal mutations of tetramer interface residues. Elution of size standards is indicated by arrows. The molecular weight of the monomeric TPR-MET fusion protein is 55.6 kDa, indicating that the peaks correspond to aggregated (~122 ml elution volume) and predominantly tetrameric (~169 ml) TPR-MET. (C). Immunoblot of lysates from AD293 cells expressing TPR-MET wildtype and tetramer interface mutants. TPR activity of mutant proteins was probed with antibodies that recognize the TPR kinase domain phosphorylated at Y1234 and Y1235 (pMET).
Figure 5. TPR-MET forms a stable tetramer.

Size exclusion chromatogram of TPR-MET at different concentrations. Blue line: chromatogram of TPR-MET Ni eluate, which resolved two major peaks and allowed us to isolate the predominantly tetramer peak at ~169 ml elution volume and a concentration of 520 nM protein. We confirmed tetramer formation in the 169 ml eluate by MALS (calculated molecular weight: 193 kDa), and reloaded at lower concentrations onto the size exclusion column, resulting in only slightly right-shifted peaks at concentrations of 52 nM (brown line) and 26 nM (red line), indicating stable tetramer formation.
Replacements of F55 and F113 with methionine induce striking changes in TPR oligomer conformations
To our surprise, while the structures of TPR F55M/I101M/F113M and TPR F55M/F113M are essentially superimposable (Fig. S3), the structures of the TPR F55M/F113M double and F113M single mutant dramatically differ from each other and from wildtype TPR. While they all eluted as tetramers from gel filtration columns (Fig. 3B), wildtype TPR formed antiparallel tetramers, F113M TPR formed parallel tetramers, and F113M/F55M TPR formed dimers (Figs. 3A, D, E). Surprisingly the side chains of F113, whose mutation switches the tetrameric conformation from antiparallel to parallel, faces the inside of the dimer and not the dimer-dimer interface (Fig. 3D). F113 significantly contributes to monomer-monomer interaction by stabilizing the F113-I109-Q110 Van-der-Waals interaction network, suggesting that rearrangements of the monomer-monomer interaction may cause altered dimer conformations that in turn affect the dimer-dimer interaction. Indeed, mutation of F113 to M causes a displacement of the residue by 3.2 Å and a striking change in the dimer conformation with an RMSD of 5.51 Å. In contrast, F55 faces the dimer-dimer interaction surface (Fig. 3D) consistent with a possible role in dimer–dimer interaction, and its mutation to M affects the oligomeric conformation in the crystal, which shows a dimeric arrangement (Fig. 3E), but not in solution (Fig. 3B). The conformational changes are most pronounced at the N-cap as well as the C terminus with overall RMSD values of 5.98 Å for F55M/F113M TPR vs. F113M TPR, and 5.93 Å for F55M/F113M TPR vs. wildtype TPR. This is a reminiscent of a single mutation-induced change in the GCN4 coiled coil oligomerization domain, which changes from a parallel tetramer to the anti-parallel configuration (Yadav et al., 2006).
TPR fusion induces protection of the αG-helix of the MET kinase domain
To gain insight into TPR fusion-mediated changes of the surface accessibility of the MET kinase domain, we purified both the isolated monomeric intracellular domain of human MET (amino acids 959-1390) as well as the oncogenic, dimeric TPR(2-142)-MET(1010-1390) fusion protein for hydrogen deuterium exchange mass spectrometry (HDX) analysis. HDX measures the rate at which amide hydrogen atoms of the protein backbone are exchanged with deuterium in deuterated buffers. Alterations in deuterium exchange are highly indicative of local changes in solvent accessibility and hence protein interactions and conformational rearrangements (Chalmers et al., 2006; Chalmers et al., 2007; Chalmers et al., 2011). The HDX profiles of both proteins are highly similar, with the exception of a mild protection against hydrogen exchange of the top and bottom of the kinase domain and a dramatic protection of the surface-accessible αG-helix upon fusion of the MET intracellular domain to TPR(2-124) (Fig. 6A and Fig. S4). Protection of αG is not due to the proposed concomitant change from the inactive to active MET kinase domain conformation. Rather, structures of the isolated kinase domain in active (PDB: 3Q6U) and inactive (PDB: 2WD1 and 2G15) monomeric states show that conformation of the αG-helix only subtly changes upon kinase activation in the absence of stable kinase dimerization (Rickert et al., 2011; Wang et al., 2006) (Fig. 6B). The HDX protection thus suggests that fusion of the TPR dimer induces protein interaction of the MET kinase domain αG-helix, possibly by kinase domain dimerization.
Figure 6. Hydrogen deuterium exchange mass spectrometry (HDX) analysis of monomeric and tetrameric MET kinase domains (See also Figures S4).

(A) MET kinase HDX perturbation map. Heat map of the difference in hydrogen deuterium exchange between the monomeric MET kinase domain and the tetrameric domain in the context of TPR(2-142)-MET overlaid on the MET kinase domain structure (PDB: 3Q6U). (B) Structure overlay of MET kinase domain in active (PDB: 3Q6U, green), inhibitor-bound inactive (PDB: 2WD1, orange), and non-phosphorylated inactive (PDB: 2G15, grey) conformations.
Mutations at the TPR/MET junction compromise MET activity
In TPR-MET, TPR(2-142) is fused to the MET intracellular domain lacking the first 51 amino acids of the juxtamembrane domain. It is currently unknown whether dimerization of the TPR fusion partner is sufficient for MET activation or whether activation requires a specific junction in order to correctly orient the kinase domains for trans-autophosphorylation and autoactivation. To address this question, we introduced alanine mutations into leucine residues of either of the two TPR(2-142) leucine zippers. While tetramerization/dimerization of _E. coli_-expressed, recombinant TPR-MET was severely affected by most leucine zipper mutations, including mutations of each of the four leucine residues of the second zipper (Fig. S5), cell-based coimmunoprecipitation experiments indicated that the first leucine zipper is more important for dimerization in mammalian cell (Rodrigues and Park, 1993). Yet, only mutation of the two most C-terminal leucine residues of the second leucine zipper (L127 and L134A), alone or in combination with other leucine mutations, strongly reduced or abolished MET autophosphorylation (Fig. 7A). Therefore, loss of TPR-MET phosphorylation depends on the position of local dimer destabilization because only mutations in second leucine zipper residues at the TPR/MET junction had severe effects on MET autophosphorylation.
Figure 7. Mutational analysis of the TPR leucine zipper and the MET juxtamembrane domain (See also Figures S5).

(A) Leucine residues of the two TPR leucine zippers were mutated individually or in combination. Wildtype and mutant total (MET) and active, phosphorylated (pMET) TPR-MET were detected by immunoblotting. (B) Autophosphorylation of TPR-MET variants with truncations in the MET juxtamembrane domain. Left: Schematic of constructs analyzed. Right: Immunoblot probed with antibodies for total (MET) and active, phosphorylated (pMET) TPR-MET.
We also tested the effects of three different truncations of the TPR juxtamembrane region at the TPR-MET border. None of the three truncations reduced MET autophosphorylation. On the contrary, relative MET autophosphorylation was increased with longer deletion of the juxtamembrane domain (Figure 6C). Collectively, these data indicate that the kinase activity of the TPR-MET fusion protein critically depends on the second dimerization motif of TPR at the MET junction site and the linker length between the juxtamembrane domain and the MET kinase domain.
Discussion
Chromosomal translocations of TPR have resulted in cytoplasmic oncogenic fusion proteins, in which TPR fragments are covalently fused to the intracellular domains of the MET, TRK1, RAF, and ALK protein kinases. Oncogenicity is due to constitutive kinase activity from the fusion proteins, which form constitutive dimers or tetramers through the stable oligomeric state of TPR. TPR dimers are thought to orient the intracellular RTK juxtamembrane segment and kinase domain in a manner that mimics that of RTKs activated by agonist-induced dimerization of their extracellular domains. Knowledge of the molecular architecture of the TPR oligomer and understanding of the conformational constraints required for kinase activation may therefore provide important insight into the activation mechanisms of both oncogenic fusion kinases as well as of wildtype full length RTKs.
Our crystal structures demonstrate that TPR(2-142) forms parallel homodimers that are further arranged into antiparallel tetramers, whose conformations we have validated by size exclusion chromatography, charge reversal mutations, and disulfide crosslinking. The oligomeric structures of the F55M and F55M/F113M TPR mutant proteins demonstrate the remarkable conformational flexibility of TPR, which may be an important contributor to its ability to activate multiple protein kinases. Moreover, kinase activity in the case of the TPR-MET fusion protein critically depends on the junction between TPR(2-142) and the MET intracellular domain, because only mutations of the two leucine zipper residues (L127 and L134) closest to the junction markedly decreased kinase activation, even though these mutant proteins still appear to dimerize/oligomerize in cells. These results confirm that dimerization of the MET intracellular domain is insufficient for kinase activation and that rather the dimer has to be topologically constrained to allow autoactivation. Similarly, deletions in the juxtamembrane domain increased MET kinase activity in the context of TPR-MET, which is reminiscent of the autoinhibitory interaction between juxtamembrane and kinase domains for several RTKs, including those of the Eph, PDGF, Musk, and insulin receptor families (Griffith et al., 2004; Hubbard et al., 1998; Mol et al., 2004; Wybenga-Groot et al., 2001). Finally, our HDX profiles have identified a striking, highly selective stabilization of the αG helix of the MET kinase domain upon dimerization through TPR fusion. Importantly, activation of the monomeric MET kinase domain through activation loop phosphorylation in vitro does not change the conformation of the αG-helix, suggesting that the αG-helix may be part of the currently unknown dimerization interface whose induced formation is critical for RTK autoactivation. Computational docking of the isolated MET kinase domain dimer without any imposed constraints indicates that TPR-MET can adopt antiparallel tetramers in which the αG helix of one kinase domain forms part of the dimerization interface, and the tyrosine phospho-acceptor residues Y1234/Y1235 in the activation loop of one monomer can insert into the catalytic cleft of the other monomer. The antiparallel TPR tetramer structure of parallel dimers is therefore sterically compatible with a TPR-MET tetramer structure that allows transautophosphorylation (Fig. S6). While the exact mechanism of RTK autoactivation will require the structure of a full length RTK in agonist-bound state, our data presented here provide insight into constraints for RTK activation.
STAR METHODS
Contact for Reagent and Resource sharing
Further information and requests for reagents may be directed to, and will be fullfiilled by the Lead Contact Karsten Melcher (Karsten.Melcher@vai.org) and H. Eric Xu (Eric.Xu@vai.org).
Experimental models and Subject Details
NIH 3T3 and AD 293 cells culture
NIH 3T3 and AD 293 cells were cultured in DMEM media containing 10% fetal bovine serum (FBS)
Methods Details
Molecular cloning
The gene sequence encoding residues 2-142 of the dimerization domain of TPR-MET fusion kinase was PCR amplified and cloned into a modified pETDuet (Novagen) vector that provides in frame coding sequence for an N-terminal His6-MBP (Maltose Binding Protein)-Sumo tag fusion. For Se-Met expression of TPR, seven different methionine mutant constructs (F28M, F46M, F55M, F113M, L127M, F113M/F55M, and F113M/F55M/I100M) were generated by site directed mutagenesis of wildtype constructs using the QuickChange method (Agilent Technologies). The TPR-MET-His8 construct was generated by fusing the gene encoding TPR(1-142) to cytoplasmic MET(1010-1360) using overlap PCR. The TPR-MET orf was then amplified using a reverse primer that includes the His8 coding sequence and cloned into the pETDuet vector for expression. All expression and mutation constructs were confirmed by DNA sequencing.
For cross-linking studies and immunoblot studies of TPR-MET proteins, TPR-MET(1-523) was PCR-amplified and cloned into pcDNA6.1 and pcDNA3.1. For crosslinking studies, individual cysteine mutations were introduced into the pcDNA6.1-TPR-MET wildtype construct using the QuickChange method (Agilent Technologies).
Protein expression and purification
Plasmids of pETDuet-His6-MBP-SUMO-TPR(2-142) wildtype and methionine mutants were expressed in E. coli BL21 (DE3) and E. coli BL21 (B834) cells (Novagen), respectively. Protein expression was induced with 0.1 mM isopropyl-β-D-thiogalactopyranoside at 16 °C overnight. Cells were harvested and lysed in buffer A [50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 10% (v/v) glycerol, 25 mM imidazole]. After homogenization at 10,000 psi using an APV2000 cell homogenizer (SPX Corporation), the lysate was centrifuged for 30 min at 20,000g. The supernatant was loaded on a 50 ml Ni-chelating HP-Sepharose column (GE Healthcare), and the fusion protein was eluted in buffer A with 250 mM imidazole. The peak fractions were pooled and dialyzed against 20 mM Tris/HCl (pH 7.5), 150 mM NaCl, and 5% glycerol in the presence of Ulp1 Sumo-protease at a protease/protein ratio of 1:1000 at 4 °C overnight. The cleaved His6-MBP-Sumo tag and uncleaved fusion protein were removed by passing through a 50 Ni-chelating HP-Sepharose column (GE Healthcare), and the TPR(2-142) in the flow-through was further purified by gel filtration chromatography through a HiLoad 26/60 Superdex 200 column. Ion exchange chromatography was performed to further enhance the purity of TPR(2-142) using a 5 ml Q column (GE Healthcare).
The pETDuet-TPR-MET(1-523)-His8 construct was expressed in E. coli BL21 (DE3) cells (Novagen) under the same conditions as for the His6-MBP-SUMO-TPR(2-142) construct. The cells were lysed in lysis buffer (50 mM Tris pH8.0, 250 mM NaCl, 25 mM imidazole, 1 % CHAPS and 10 % glycerol) at 10,000 psi using an APV2000 cell homogenizer (SPX Corporation). The lysate was centrifuged for 30 min at 20,000g and the supernatant was loaded onto a 50 ml Ni-chelating HP-Sepharose column (GE Healthcare). The target protein was eluted in buffer A with 250 mM imidazole. The peak fractions were concentrated and purified by gel filtration through a 300 ml Hi-load 26/60 Superdex 200 column (GE Healthcare).
Crystallization, data collection and structure determination
The purified native protein was concentrated by ultrafiltration to 8 mg/ml and crystals were grown at 22 °C by the sitting drop vapor diffusion method. For SeMet constructs, TPR (F113M), TPR (F55M, F113M), TPR (F55M, I100M, F113M), the proteins were concentrated to 9 mg/ml, 12 mg/ml and 12 mg/ml, respectively. Initial crystallization trials were conducted using commercially available Hampton Research screens. Diffracting native protein crystals were manually optimized using the sitting-drop method in mother liquor of 0.1 M Tris/HCl (pH 8.0), 150 mM sodium malonate, 29% w/v polyethylene glycol 1000. Similarly, best crystals for TPR (F113M) were grown in well solution containing 0.1 M HEPES (pH 7.25), 28% w/v polyethylene glycol 1000. TPR (F55M, F113M) crystals were further optimized in well solution containing 0.1 M Bis-Tris (pH 6.25) and 16% w/v polyethylene glycol 1500. The TPR (F55M, I100M, and F113M) crystals were grown in well solution of 0.1 M HEPES (pH 7.5) and 30% w/v polyethylene glycol 1000. Diffraction data for native and SeMet crystals were collected at LS-CAT beamlines 21-ID-F/G/D (Advanced Photon Source) and data were processed using HKL2000. The phase information was obtained by multi-wavelength anomalous dispersion (MAD) from the TPR (F55M/I100M/F113M) crystal in which Autosol identified 13 selenium positions with occupancies of more than 0.5. Subsequently, TPR (WT), TPR (F113M) and TPR (F55M, F113M) structures were determined by the molecular replacement method using an initial model generated from TPR (F55M, I100M, F113M) MAD data. All models were built using COOT (Emsley and Cowtan, 2004) and refined using several cycles of Refmac (Murshudov et al., 1997) and Phenix (Terwilliger et al., 2008) refine programs. Data collection and refinement statistics are summarized in Table S1. All structure figures were prepared using PyMol (www.pymol.org). Electrostatic surface potential calculations were performed with APBS (Baker et al., 2001).
In-cell disulfide bond crosslinking
The TPR(2-142)-MET(1-522) orf was amplified and cloned with and without C-terminal FLAG tag into pcDNA3.1. Then cysteine mutations were systematically introduced into the TPR portion of the TPR-MET expression construct and constructs transfected into AD293 cells. The cells were split and plated one day before transfection in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) cell-culture medium. The cells were co-transfected with 100 ng TPR-MET-FLAG and 100 ng TPR-MET expression constructs using Lipofectamine 2000 at a 1:2 ratio of DNA:Lipofectamine. The transfected cells were allowed to grow for two days. H2O2 to a concentration of 1 mM was freshly added to the cell-culture medium and cells incubated for 5 mins at room temperature. Following the H2O2 treatment, the media were aspirated and cells incubated with 100 μl of CellLytic M (Sigma C2978) for 10 min at room temperature. The cell lysates were harvested in 1.5 ml microcentrifuge tubes and centrifuged at 16,000g for 5 min. Lysate supernatants were mixed with SDS loading buffer lacking reducing agent for 5 min at room temperature and separated by SDS PAGE for immunoblot blot analysis, using horseradish peroxidase–conjugated anti-FLAG (Sigma M2) antibody.
Hydrogen deuterium exchange mass spectrometry
TPR-MET(2-523) and MET(956-1390) were incubated in 20 mM Tris pH 8.0, 150 mM NaCl at 4 °C for 10s, 30s, 60s, 900s and 3,600s followed by quenching of the deuterium exchange reaction with 3 M urea, 1% TFA at 4 °C. The protein solutions were passed through an in-line immobilized pepsin column at 200 μl min−1 (0.1% v/v TFA, 15 °C) and the resulting peptides were trapped on a C8 trap column (Hypersil Gold, Thermo scientific, CA). The bound peptides were gradient-eluted (5–50% CH3CN w/v and 0.3% w/v formic acid) across a 2 mm × 50 mm C18-HPLC column (Hypersil Gold, Thermo Scientific, CA) for 5 min at 4 °C. Eluted peptides were subjected to electrospray ionization directly coupled to a high resolution Orbitrap QExactive mass spectrometer (Thermo Scientific, CA). Each HDX experiment was carried out in triplicate and the intensity weighted average m/z value (centroid) of each peptide isotopic envelope was calculated with HDX Workbench software (Pascal et al., 2012). The values for percentage of deuterium exchange representing each peptide were narrowed to a single value by averaging.
Immunoblotting
pcDNA6.1 TPR-MET wildtype and mutant constructs were transfected in NIH-3T3 cells previously cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) cell-culture medium. After 24–36 hours of expression, the cells were lysed in radioimmunoprecipitation assay (RIPA) buffer and lysate supernatants were separated on SDS-PAGE gel for immunoblot analysis. Total MET was probed with anti-MET-19S mouse monoclonal antibody (Faletto et al., 1992) and phosphorylated MET was probed with anti-rabbit phospho-Met (Tyr1234/1235; Cell Signaling) antibody.
Molecular Docking of Tyrosine kinase domain
Automatic protein docking server ZDOCK version 3.0.2 (Pierce BG et al., 2011) was used to generate the dimer model of the MET tyrosine kinase domain. The docking was performed using two different structures of the kinase domain (PDB: 3Q6U & 2WD1) independent of probable interface data from HDX-MS analysis. ZDOCK provided the 10 lowest energy models. Among those we identified a parallel kinase dimer conformation that satisfied the binding interface data from HDX-MS analysis. The final model of MET dimer and TPR was generated in PyMol with a 10 Å distance between the C termini of TPR and N-termini of the MET kinase domains.
DATA AND SOFTWARE AVAILABILITY
All structures solved in this work have been deposited into the public database and are accessible via the following Protein Data Bank (PDB) accession codes: 5T06.pdb for wildtype TPR; 5T07.pdb for F113M TPR; 5T05.pdb for F55M, F113M TPR; and 5TV5.pdb for F55M, I101M, F113M TPR.
Supplementary Material
supplement
Table 1.
X-ray crystallographic data collection and model refinement
| Data collection | Native | TPR (F113M) | TPR (F55M, F113M) | TPR (F55M, I101, F113M) |
|---|---|---|---|---|
| Detector type/source | Rayonix CCD/APS | Rayonix CCD/APS | Rayonix CCD/APS | Rayonix CCD/APS |
| Space group | p1211 | C121 | C121 | C2 |
| a, b, c (Å) | 76.79, 34.86, 113.47 | 164.49, 51.19, 113.80 | 74.19, 51.59, 95.00 | 74.35, 50.67, 94.65 |
| α, β, γ (°) | 90.00, 91.84. 90.00 | 90.00, 129.32, 90.00 | 90.00, 107.84, 90.00 | 90.00, 108.01, 90.00 |
| Resolution range (Å) | 46.3-2.5 | 47.49-2.4 | 21.85-2.18 | 50-2.75 |
| Wavelength (Å) | 0.97872 | 0.97872 | 1.07812 | 0.97622 |
| Measured reflections | 141866 | 210682 | 97778 | 751118 |
| Unique reflections | 20228 | 28341 | 17251 | 9404 |
| Completeness | 94.5 % | 97.5 % | 96.9 % | 99.2 % |
| Mean[I/σ(I)] | 10.3 (34.3) | 11.2 (45.7) | 15.8 (30.9) | 39.3 (6.59) |
| Rmerge (%) | 14.9 % | 13 % | 7.9 % | 8 % |
| Refinement | ||||
| Resolution range (Å) | 46.3 – 2.7 | 44 – 2.6 | 21.4-2.5 | 41.18-2.75 |
| No. of reflections working/test | 16902/842 | 22233/1174 | 11481/544 | 8040/807 |
| R (working set; %) | 23.92 % | 23.62% | 23.18 % | 24.15 % |
| R free (%) | 28.82 % | 28.09 % | 28.20 % | 30.27 % |
| Protein atoms | 4779 | 4824 | 2463 | 2383 |
| r.m.s.d. in bond length (Å) | 0.002 | 0.005 | 0.004 | 0.003 |
| r.m.s.d. in bond angle (°) | 0.490 | 0.733 | 0.722 | 0.572 |
| Ramachandran plot | ||||
| Most favored regions (%) | 100 % | 99.82 % | 100 % | 100 % |
| Allowed | 0.00 % | 0.18 % | 0.00 % | 0.00 % |
| Outliers | 0.00 % | 0.00 % | 0.00 % | 0.00 % |
Highlights.
- Genomic translocation can generate a highly oncogenic TPR(2-142)-MET fusion kinase
- TPR(2-142) forms an extremely long continuous helix that folds into a tetramer
- Mutations of TPR leucine zipper residues at the junction to MET inactivate TPR-MET
- TPR conformationally stabilizes the αG helix of the MET kinase domain in TPR-MET
Acknowledgments
This research was supported by the Van Andel Research Institute (H.E.X. and K.M.), the National Institutes of Health (R01 GM102545 and GM102545 to K.M. and DK071662 H.E.X.), the National Natural Science Foundation of China (31300245 to H.E.X.), Ministry of Science and Technology (China) grants 2012ZX09301001 and 2012CB910403, 2013CB910600, XDB08020303, 2013ZX09507001 (H.E.X), and Amway (China) (H.E.X.). We thank Michelle Martin for administrative support and staff members of the Life Science Collaborative Access Team of the Advanced Photon Source (APS) for assistance in data collection at the beam lines of sector 21, which is in part funded by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). Use of APS was supported by the Office of Science of the US Department of Energy, under Contract No. DE-AC02-06CH11357. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Lead contact for manuscript handling: Karsten.Melcher@vai.org
Contributions
K.M. and H.E.X. conceived the project and designed experiments, K.P., A.B., X.E.Z., Q.P., D.P.M., S.Y., R.P.G., and G.VdW. performed and/or interpreted experiments. K.P., K.M., and H.E.X. wrote the manuscript with support from all authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arkhipov A, Shan Y, Das R, Endres NF, Eastwood MP, Wemmer DE, Kuriyan J, Shaw DE. Architecture and membrane interactions of the EGF receptor. Cell. 2013;152:557–569. doi: 10.1016/j.cell.2012.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurora R, Rose GD. Helix capping. Protein science: a publication of the Protein Society. 1998;7:21–38. doi: 10.1002/pro.5560070103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems:application to microtubules and the ribosome. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck M, Hurt E. The nuclear pore complex: understanding its function through structural insight. Nature reviews Molecular cell biology. 2017;18:73–89. doi: 10.1038/nrm.2016.147. [DOI] [PubMed] [Google Scholar]
- Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nature reviews Molecular cell biology. 2003;4:915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- Burkhard P, Ivaninskii S, Lustig A. Improving coiled-coil stability by optimizing ionic interactions. Journal of molecular biology. 2002;318:901–910. doi: 10.1016/S0022-2836(02)00114-6. [DOI] [PubMed] [Google Scholar]
- Burkhard P, Stetefeld J, Strelkov SV. Coiled coils: a highly versatile protein folding motif. Trends in cell biology. 2001;11:82–88. doi: 10.1016/s0962-8924(00)01898-5. [DOI] [PubMed] [Google Scholar]
- Chalmers MJ, Busby SA, Pascal BD, He Y, Hendrickson CL, Marshall AG, Griffin PR. Probing protein ligand interactions by automated hydrogen/deuterium exchange mass spectrometry. Anal Chem. 2006;78:1005–1014. doi: 10.1021/ac051294f. [DOI] [PubMed] [Google Scholar]
- Chalmers MJ, Busby SA, Pascal BD, Southern MR, Griffin PR. A two-stage differential hydrogen deuterium exchange method for the rapid characterization of protein/ligand interactions. J Biomol Tech. 2007;18:194–204. [PMC free article] [PubMed] [Google Scholar]
- Chalmers MJ, Busby SA, Pascal BD, West GM, Griffin PR. Differential hydrogen/deuterium exchange mass spectrometry analysis of protein-ligand interactions. Expert Rev Proteomics. 2011;8:43–59. doi: 10.1586/epr.10.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YL, Lira ME, Hong M, Kim RN, Choi SJ, Song JY, Pandy K, Mann DL, Stahl JA, Peckham HE, et al. A novel fusion of TPR and ALK in lung adenocarcinoma. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer. 2014;9:563–566. doi: 10.1097/JTO.0000000000000093. [DOI] [PubMed] [Google Scholar]
- Cooper CS, Park M, Blair DG, Tainsky MA, Huebner K, Croce CM, Vande Woude GF. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature. 1984;311:29–33. doi: 10.1038/311029a0. [DOI] [PubMed] [Google Scholar]
- Dean M, Park M, Vande Woude GF. Characterization of the rearranged tpr-met oncogene breakpoint. Molecular and cellular biology. 1987;7:921–924. doi: 10.1128/mcb.7.2.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombkowski AA, Crippen GM. Disulfide recognition in an optimized threading potential. Protein Eng. 2000;13:679–689. doi: 10.1093/protein/13.10.679. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Endres NF, Barros T, Cantor AJ, Kuriyan J. Emerging concepts in the regulation of the EGF receptor and other receptor tyrosine kinases. Trends Biochem Sci. 2014;39:437–446. doi: 10.1016/j.tibs.2014.08.001. [DOI] [PubMed] [Google Scholar]
- Ermolenko DN, Thomas ST, Aurora R, Gronenborn AM, Makhatadze GI. Hydrophobic interactions at the Ccap position of the C-capping motif of alpha-helices. Journal of molecular biology. 2002;322:123–135. doi: 10.1016/s0022-2836(02)00734-9. [DOI] [PubMed] [Google Scholar]
- Faletto DL, Tsarfaty I, Kmiecik TE, Gonzatti M, Suzuki T, Vande Woude GF. Evidence for non-covalent clusters of the c-met proto-oncogene product. Oncogene. 1992;7:1149–1157. [PubMed] [Google Scholar]
- Furge KA, Zhang YW, Vande Woude GF. Met receptor tyrosine kinase: enhanced signaling through adapter proteins. Oncogene. 2000;19:5582–5589. doi: 10.1038/sj.onc.1203859. [DOI] [PubMed] [Google Scholar]
- Greco A, Pierotti MA, Bongarzone I, Pagliardini S, Lanzi C, Della Porta G. TRK-T1 is a novel oncogene formed by the fusion of TPR and TRK genes in human papillary thyroid carcinomas. Oncogene. 1992;7:237–242. [PubMed] [Google Scholar]
- Griffith J, Black J, Faerman C, Swenson L, Wynn M, Lu F, Lippke J, Saxena K. The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol Cell. 2004;13:169–178. doi: 10.1016/s1097-2765(03)00505-7. [DOI] [PubMed] [Google Scholar]
- Hubbard SR, Mohammadi M, Schlessinger J. Autoregulatory mechanisms in protein-tyrosine kinases. The Journal of biological chemistry. 1998;273:11987–11990. doi: 10.1074/jbc.273.20.11987. [DOI] [PubMed] [Google Scholar]
- King HW, Tempest PR, Merrifield KR, Rance AJ. tpr homologues activate met and raf. Oncogene. 1988;2:617–619. [PubMed] [Google Scholar]
- Lamorte L, Park M. The receptor tyrosine kinases: role in cancer progression. Surgical oncology clinics of North America. 2001;10:271–288. viii. [PubMed] [Google Scholar]
- Lupas AN, Gruber M. The structure of alpha-helical coiled coils. Adv Protein Chem. 2005;70:37–78. doi: 10.1016/S0065-3233(05)70003-6. [DOI] [PubMed] [Google Scholar]
- Mak HH, Peschard P, Lin T, Naujokas MA, Zuo D, Park M. Oncogenic activation of the Met receptor tyrosine kinase fusion protein, Tpr-Met, involves exclusion from the endocytic degradative pathway. Oncogene. 2007;26:7213–7221. doi: 10.1038/sj.onc.1210522. [DOI] [PubMed] [Google Scholar]
- Malli T, Buxhofer-Ausch V, Rammer M, Erdel M, Kranewitter W, Rumpold H, Marschon R, Deutschbauer S, Simonitsch-Klupp I, Valent P, et al. Functional characterization, localization, and inhibitor sensitivity of the TPR-FGFR1 fusion in 8p11 myeloproliferative syndrome. Genes Chromosomes Cancer. 2016;55:60–68. doi: 10.1002/gcc.22311. [DOI] [PubMed] [Google Scholar]
- Mason JM, Arndt KM. Coiled coil domains: stability, specificity, and biological implications. Chembiochem: a European journal of chemical biology. 2004;5:170–176. doi: 10.1002/cbic.200300781. [DOI] [PubMed] [Google Scholar]
- Merlin S, Pietronave S, Locarno D, Valente G, Follenzi A, Prat M. Deletion of the ectodomain unleashes the transforming, invasive, and tumorigenic potential of the MET oncogene. Cancer science. 2009;100:633–638. doi: 10.1111/j.1349-7006.2008.01079.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mol CD, Dougan DR, Schneider TR, Skene RJ, Kraus ML, Scheibe DN, Snell GP, Zou H, Sang BC, Wilson KP. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. The Journal of biological chemistry. 2004;279:31655–31663. doi: 10.1074/jbc.M403319200. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nakano H, Funasaka T, Hashizume C, Wong RW. Nucleoporin translocated promoter region (Tpr) associates with dynein complex, preventing chromosome lagging formation during mitosis. The Journal of biological chemistry. 2010;285:10841–10849. doi: 10.1074/jbc.M110.105890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M, Dean M, Cooper CS, Schmidt M, O’Brien SJ, Blair DG, Vande Woude GF. Mechanism of met oncogene activation. Cell. 1986;45:895–904. doi: 10.1016/0092-8674(86)90564-7. [DOI] [PubMed] [Google Scholar]
- Park M, Dean M, Kaul K, Braun MJ, Gonda MA, Vande Woude G. Sequence of MET protooncogene cDNA has features characteristic of the tyrosine kinase family of growth-factor receptors. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:6379–6383. doi: 10.1073/pnas.84.18.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascal BD, Willis S, Lauer JL, Landgraf RR, West GM, Marciano D, Novick S, Goswami D, Chalmers MJ, Griffin PR. HDX workbench: software for the analysis of H/D exchange MS data. Journal of the American Society for Mass Spectrometry. 2012;23:1512–1521. doi: 10.1007/s13361-012-0419-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschard P, Park M. Escape from Cbl-mediated downregulation: a recurrent theme for oncogenic deregulation of receptor tyrosine kinases. Cancer cell. 2003;3:519–523. doi: 10.1016/s1535-6108(03)00136-3. [DOI] [PubMed] [Google Scholar]
- Peschard P, Park M. From Tpr-Met to Met, tumorigenesis and tubes. Oncogene. 2007;26:1276–1285. doi: 10.1038/sj.onc.1210201. [DOI] [PubMed] [Google Scholar]
- Rajanala K, Sarkar A, Jhingan GD, Priyadarshini R, Jalan M, Sengupta S, Nandicoori VK. Phosphorylation of nucleoporin Tpr governs its differential localization and is required for its mitotic function. J Cell Sci. 2014;127:3505–3520. doi: 10.1242/jcs.149112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickert KW, Patel SB, Allison TJ, Byrne NJ, Darke PL, Ford RE, Guerin DJ, Hall DL, Kornienko M, Lu J, et al. Structural basis for selective small molecule kinase inhibition of activated c-Met. The Journal of biological chemistry. 2011;286:11218–11225. doi: 10.1074/jbc.M110.204404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues GA, Park M. Dimerization mediated through a leucine zipper activates the oncogenic potential of the met receptor tyrosine kinase. Molecular and cellular biology. 1993;13:6711–6722. doi: 10.1128/mcb.13.11.6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues GA, Park M. Oncogenic activation of tyrosine kinases. Current opinion in genetics & development. 1994;4:15–24. doi: 10.1016/0959-437x(94)90086-8. [DOI] [PubMed] [Google Scholar]
- Shimizu A, Persson C, Heldin CH, Ostman A. Ligand stimulation reduces platelet-derived growth factor beta-receptor susceptibility to tyrosine dephosphorylation. The Journal of biological chemistry. 2001;276:27749–27752. doi: 10.1074/jbc.C100286200. [DOI] [PubMed] [Google Scholar]
- Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nature communications. 2014;5:4846. doi: 10.1038/ncomms5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Zwart PH, Hung LW, Read RJ, Adams PD. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr D Biol Crystallogr. 2008;64:61–69. doi: 10.1107/S090744490705024X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanos CD, Goodwill KE, Bowie JU. Oligomeric structure of the human EphB2 receptor SAM domain. Science. 1999;283:833–836. doi: 10.1126/science.283.5403.833. [DOI] [PubMed] [Google Scholar]
- Tolbert WD, Daugherty-Holtrop J, Gherardi E, Vande Woude G, Xu HE. Structural basis for agonism and antagonism of hepatocyte growth factor. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:13264–13269. doi: 10.1073/pnas.1005183107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolbert WD, Daugherty J, Gao C, Xie Q, Miranti C, Gherardi E, Vande Woude G, Xu HE. A mechanistic basis for converting a receptor tyrosine kinase agonist to an antagonist. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:14592–14597. doi: 10.1073/pnas.0704290104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Marimuthu A, Tsai J, Kumar A, Krupka HI, Zhang C, Powell B, Suzuki Y, Nguyen H, Tabrizizad M, et al. Structural characterization of autoinhibited c-Met kinase produced by coexpression in bacteria with phosphatase. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:3563–3568. doi: 10.1073/pnas.0600048103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf E, Kim PS, Berger B. MultiCoil: a program for predicting two- and three-stranded coiled coils. Protein science: a publication of the Protein Society. 1997;6:1179–1189. doi: 10.1002/pro.5560060606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wybenga-Groot LE, Baskin B, Ong SH, Tong J, Pawson T, Sicheri F. Structural basis for autoinhibition of the Ephb2 receptor tyrosine kinase by the unphosphorylated juxtamembrane region. Cell. 2001;106:745–757. doi: 10.1016/s0092-8674(01)00496-2. [DOI] [PubMed] [Google Scholar]
- Yadav MK, Leman LJ, Price DJ, Brooks CL, 3rd, Stout CD, Ghadiri MR. Coiled coils at the edge of configurational heterogeneity. Structural analyses of parallel and antiparallel homotetrameric coiled coils reveal configurational sensitivity to a single solvent-exposed amino acid substitution. Biochemistry. 2006;45:4463–4473. doi: 10.1021/bi060092q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- Zhao X, Ghaffari S, Lodish H, Malashkevich VN, Kim PS. Structure of the Bcr-Abl oncoprotein oligomerization domain. Nature structural biology. 2002;9:117–120. doi: 10.1038/nsb747. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplement