NLRP12-PANoptosome activates PANoptosis and pathology in response to heme and PAMPs (original) (raw)
. Author manuscript; available in PMC: 2024 Jun 22.
Published in final edited form as: Cell. 2023 Jun 1;186(13):2783–2801.e20. doi: 10.1016/j.cell.2023.05.005
SUMMARY
Cytosolic innate immune sensors are critical for host defense and form complexes, such as inflammasomes and PANoptosomes, that induce inflammatory cell death. The sensor NLRP12 is associated with infectious and inflammatory diseases, but its activating triggers and roles in cell death and inflammation remain unclear. Here, we discovered that NLRP12 drives inflammasome and PANoptosome activation, cell death and inflammation in response to heme plus PAMPs or TNF. TLR2/4-mediated signaling induced Nlrp12 expression. NLRP12 formed an inflammasome to induce maturation of IL-1β and IL-18, and the inflammasome served as an integral component of a larger NLRP12-PANoptosome that drove inflammatory cell death through caspase-8/RIPK3. Deletion of Nlrp12 protected mice from acute kidney injury and lethality in a hemolytic model. Overall, we identified NLRP12 as an essential cytosolic sensor for heme plus PAMPs-mediated PANoptosis, inflammation and pathology, suggesting NLRP12 and molecules in this pathway are potential drug targets for hemolytic and inflammatory diseases.
Keywords: NLRP12, IRF1, heme, hemolysis, PAMP, DAMP, Pam3CSK4, LPS, TNF, phenylhydrazine, inflammasome, inflammatory cell death, pyroptosis, apoptosis, necroptosis, gasdermin E, gasdermin D, caspase, caspase-1, caspase-8, caspase-3, caspase-7, MLKL, RIPK1, RIPK3, TLRs, TLR2, TLR4
Graphical Abstract
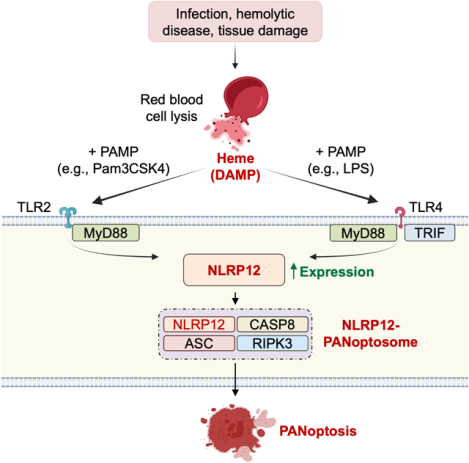
In Brief
NLRP12 is an immune sensor that activates inflammatory host responses when it senses heme and pathogen-associated molecular danger signals indicative of hemolytic diseases.
INTRODUCTION
Innate immune sensors called pattern recognition receptors (PRRs)1–3 play a key role in protecting the host from invading pathogens and sterile insults. Sensors can either act as direct receptors and bind specific pathogen-associated molecular patterns (PAMPs) or endogenous damage-associated molecular patterns (DAMPs), or they can respond to homeostatic perturbations caused by PAMPs and DAMPs. Both sensing strategies result in the activation of innate immune signaling pathways, which include nuclear factor-κB (NF-κB), mitogen activated protein kinase (MAPK) and type I interferon (IFN) pathways4.
One sub-family of cytosolic PRRs called nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) play diverse roles in activating inflammasomes and cell death as well as inflammation4–6. Additionally, these inflammasomes can act as integral components of larger cell death-inducing complexes called PANoptosomes, which integrate components from multiple cell death pathways and drive a form of lytic, innate immune inflammatory cell death called PANoptosis7–10. While a few NLRs are relatively well-characterized, little is known about the functions of many other sensors in this family.
While engagement of PRRs, including NLRs, is often beneficial and can reduce pathogen burden in the infected host11, excess activation can also lead to pathogenic inflammation, cytokine storms, tissue damage and DAMP release12–14. Together, PAMPs and DAMPs released during infections and inflammatory conditions can further induce multi-organ failure and mortality. However, the innate immune sensors involved in detecting the collective release of PAMPs and DAMPs and their role in activating inflammasomes, PANoptosomes and inflammatory responses to contribute to disease pathogenesis remain poorly defined.
One central DAMP that can act as a danger signal across diseases, including in many infections as well as inflammatory and hemolytic diseases, is heme. When red blood cells are damaged or lysed, heme is released into the bloodstream and can activate innate immunity. This activation leads to the production of proinflammatory cytokines and the recruitment of immune cells to the site of infection, contributing to the host’s defense against the pathogen. However, excess heme can also have harmful effects, leading to tissue damage and organ dysfunction. In hemolytic diseases, such as hereditary spherocytosis and sickle cell anemia, the release of heme from lysed red blood cells contributes to chronic inflammation and tissue damage, leading to further hemolysis and a feed-forward cycle of inflammation and tissue damage that exacerbates disease15–17. Given the role of heme in driving pathology, there is a critical need to identify therapeutic targets within the heme innate immune sensing pathway to mitigate these harmful effects. However, the molecular mechanisms of heme sensing and the subsequent innate immune signaling that drives pathology remain unclear.
In this study, we found that the combination of heme plus PAMPs or the inflammatory cytokine TNF mimicked infection to induce inflammation. We identified NLRP12 as the innate immune cytosolic sensor responsible for the inflammasome and PANoptosome activation, inflammatory cell death and inflammation in response to heme plus PAMPs. Upstream, the Toll-like receptors (TLRs) TLR2 and TLR4 induced signaling to upregulate Nlrp12 expression. Moreover, _Nlrp12_−/− mice were significantly protected from acute kidney injury and lethality in a hemolytic model, further implicating NLRP12 in disease pathogenesis. Overall, our results identified NLRP12 as an essential cytosolic sensor for heme plus PAMPs that forms a PANoptosome, with the inflammasome as an integral component, and drives inflammatory cell death and pathology in hemolytic conditions, highlighting the potential for NLRP12 and molecules in its activation pathway as future drug targets in disease.
RESULTS
Identification of unique PAMP and DAMP combinations that induce innate immune cell death
Innate immunity is activated by pathogens, PAMPs or DAMPs. Enhanced immune activation can lead to cellular damage, tissue destruction and organ injury, as well as the release of DAMPs. PRRs have classically been studied for their ability to sense a specific PAMP or DAMP18. However, combined activation of PRRs by multiple PAMPs and DAMPs together has not been well characterized. We hypothesized that the presence of multiple stimuli would mimic infection and drive innate immunity and inflammatory cell death. To study the involvement of multiple PAMPs in cell death and search for combinations that could mimic infection, we tested lipopolysaccharide (LPS) and Pam3CSK4 (Pam3), which mimic bacterial infection, and polyinosinic:polycytidylic acid (poly(I:C)) and Resiquimod (R848), which mimic viral infection. Each of these PAMPs activate different signaling pathways and are expected to have unique effects on the cells; therefore, we screened several to identify conserved and distinct phenotypes. To test whether individual PAMPs alone could induce cell death, we treated bone marrow-derived macrophages (BMDMs) with Pam3, poly(I:C), LPS or R848. Minimal cell death occurred with these triggers (Figures S1A and S1C). However, combining PAMPs, as would be expected to occur in natural infection, induced robust cell death in many combinations (Figures 1A, 1B and S1A). Because tissue damage and DAMP release often also occur during infections and inflammatory conditions, we sought to determine the involvement of DAMPs in cell death. To assess this, we used DAMPs associated with infections and inflammatory diseases, high-mobility group box 1 protein (HMGB1), monosodium urate (MSU), S100A8/A9 and heme. Similar to the response to individual PAMPs, individual DAMPs associated with infections and inflammatory diseases, high-mobility group box 1 protein (HMGB1), monosodium urate (MSU), S100A8/A9 and heme, did not induce cell death in BMDMs (Figures S1B and S1C). Moreover, combinations of DAMPs also did not induce cell death (Figures 1B and S1D).
Figure 1. Identification of unique PAMP and DAMP combinations that induce inflammatory cell death.
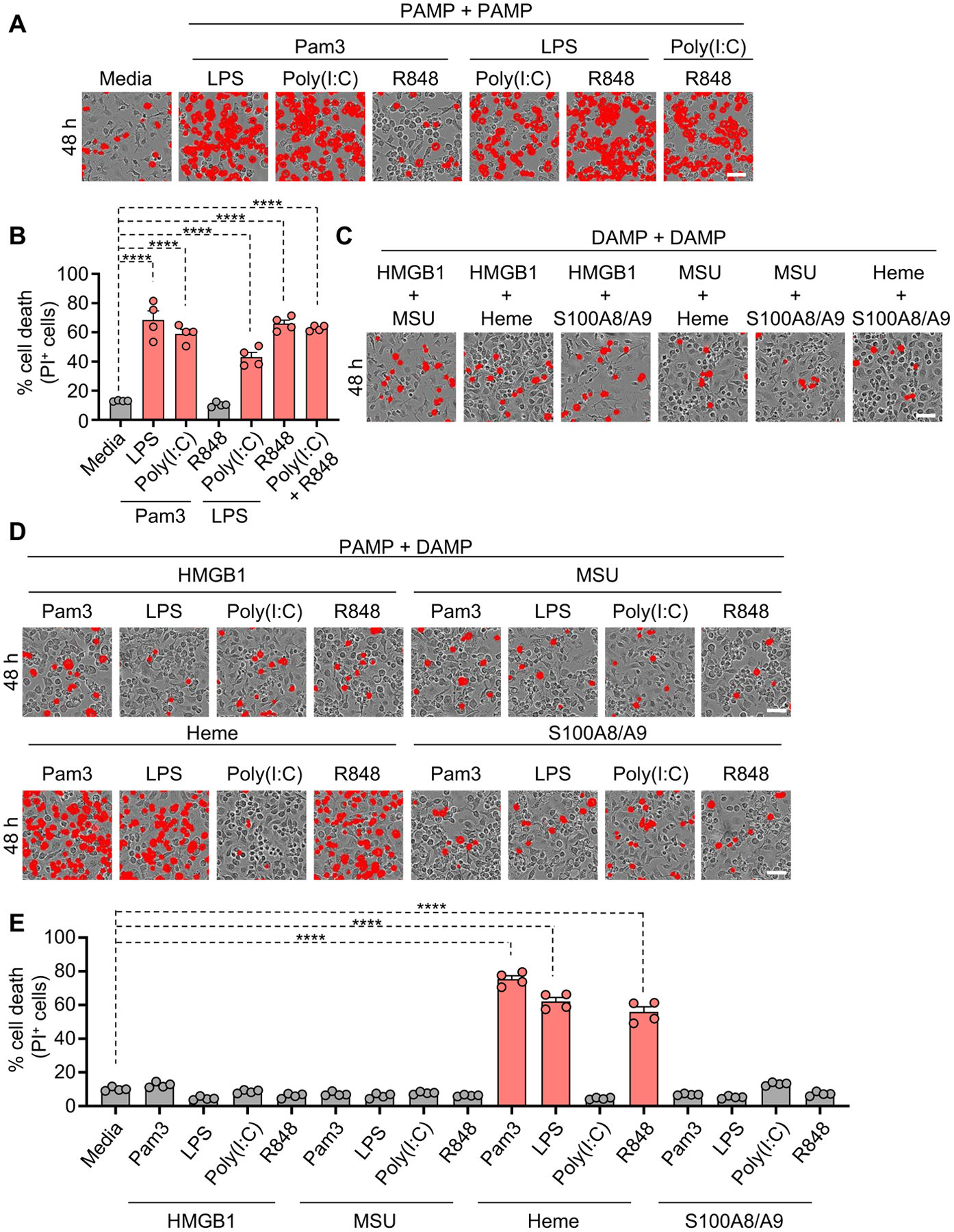
(A and B) Representative images of cell death (A) and quantification showing percentage of cell death (B) in wildtype (WT) bone marrow-derived macrophages (BMDMs) in response to media or treatment with combinations of Pam3CSK4 (Pam3) plus lipopolysaccharide (LPS), polyinosinic:polycytidylic acid (poly(I:C)) or Resiquimod (R848); or LPS plus poly(I:C) or R848; or poly(I:C) plus R848 for 48 h. (C) Representative images of cell death in WT BMDMs treated with combinations of high-mobility group box 1 protein (HMGB1) plus monosodium urate (MSU), heme or S100A8/A9; or MSU plus heme or S100A8/A9; or heme plus S100A8/A9 for 48 h. (D and E) Representative images of cell death (D) and quantification showing percentage of cell death (E) in WT BMDMs treated with combinations of HMGB1 plus Pam3, LPS, poly(I:C) or R848; MSU plus Pam3, LPS, poly(I:C) or R848; heme plus Pam3, LPS, poly(I:C) or R848; or S100A8/A9 plus Pam3, LPS, poly(I:C) or R848 for 48 h. Scale bar = 50 μm (A, C, D). Data are representative of at least three independent experiments. Data are shown as mean ± SEM (B, E). Analysis was performed using the one-way ANOVA (B, E). ****P < 0.0001.
See also Figures S1 and S2.
We next sought to determine whether combinations of PAMPs with DAMPs could induce cell death. Robust cell death occurred specifically in response to the combinations of heme with Pam3, LPS or R848, but not in other combinations tested, including heme with poly(I:C) (Figures 1D and 1E). Given the robust cell death observed with heme and PAMP combinations, we tested whether this cell death was dependent on iron, a metabolite of heme. However, neither the iron scavenger deferoxamine (DFO) nor the ferroptosis inhibitor ferrostatin-1 (Fer-1) inhibited the cell death induced by heme plus Pam3 (Figures S1E and S1F), suggesting iron is not responsible for the heme-mediated cell death. Together, these results indicate that signaling driven by specific PAMP combinations and the combination of heme with PAMPs could induce robust cell death.
Heme plus PAMPs induces inflammatory cell death
While heme is known to induce cell death19 and activate the NLRP3 inflammasome in low serum conditions20,21, the cytosolic sensors and molecular mechanisms involved under more physiologic conditions remain unclear. Therefore, we next elucidated the biochemical features of the cell death induced in response to heme alone, Pam3 alone and heme plus Pam3 in comparison to the canonical NLRP3 inflammasome trigger LPS plus ATP. Caspase-1 activation was increased in response to heme plus Pam3 treatment in comparison to heme alone or Pam3 alone, suggesting inflammasome activation was occurring, but this activation was minimal compared with that observed in response to LPS plus ATP treatment (Figure S2A). Downstream of inflammasome activation, gasdermin D (GSDMD) can be processed to release its pore-forming N-terminus as a P30 fragment to execute cell death22–24. GSDMD was cleaved to this P30 fragment in response to LPS plus ATP, but not in response to heme plus Pam3 treatment (Figure S2A). Another member of the gasdermin family, gasdermin E (GSDME), has been shown to induce cell death under specific conditions25; GSDME cleavage was observed in response to heme plus Pam3 treatment, but not in response to LPS plus ATP (Figure S2A). In addition, apoptotic caspases, caspase-8, -3 and -7, were cleaved in BMDMs treated with heme plus Pam3 more so than in response to LPS plus ATP treatment (Figure S2A). Previous studies have shown that activation of caspase-3 and -7 can inactivate GSDMD by processing it to produce a P20 fragment26–28, which was observed in response to heme plus Pam3, but not in response to LPS plus ATP (Figure S2A). Furthermore, MLKL was phosphorylated (pMLKL) in response to heme plus Pam3 (Figure S2A). Next, we assessed the biochemical activation of cell death in response to PAMP plus PAMP-mediated cell death to determine whether this followed the same pathway as heme plus Pam3-mediated cell death. Robust activation of GSDME, caspase-8, -3, and -7 and pMLKL were observed in response to Pam3 plus LPS, Pam3 plus poly(I:C), LPS plus poly(I:C), LPS plus R848 and R848 plus poly(I:C) treatments (Figure S2B). Collectively, these data suggest that synergistic signaling from heme plus PAMPs and PAMP plus PAMP co-treatment induces multiple cell death markers indicative of PANoptosis, rather than activating pyroptosis, apoptosis or necroptosis.
NLRP12 regulates inflammatory cell death induced by heme plus PAMPs
Cytosolic sensors, and particularly some NLRP proteins, are implicated in the induction of cell death and disease4–6. However, the roles of many NLRPs in innate immunity and cell death remain unclear. Since we observed robust cell death in response to heme plus PAMP combinations, we sought to understand which NLRPs were differentially expressed in hemolytic diseases, where heme is released, by re-analyzing publicly available datasets for patients with malaria or sickle cell disease (SCD)29–32. Earlier studies have reported that heme can induce NLRP3 inflammasome activation and IL-1β secretion20,21, and the expression of NLRP3 was significantly upregulated in CD71+ cells from patients with malaria and in SCD monocytes (Figure 2A). In addition to NLRP3, NLRP12 was also highly upregulated in these samples, whereas NLRP6 was only significantly upregulated in CD71+ bone marrow cells from patients with malaria, and NLRP2 and NLRP1 were not significantly upregulated (Figure 2A).
Figure 2. NLRP12 regulates inflammatory cell death, PANoptosis, induced by heme plus PAMPs.
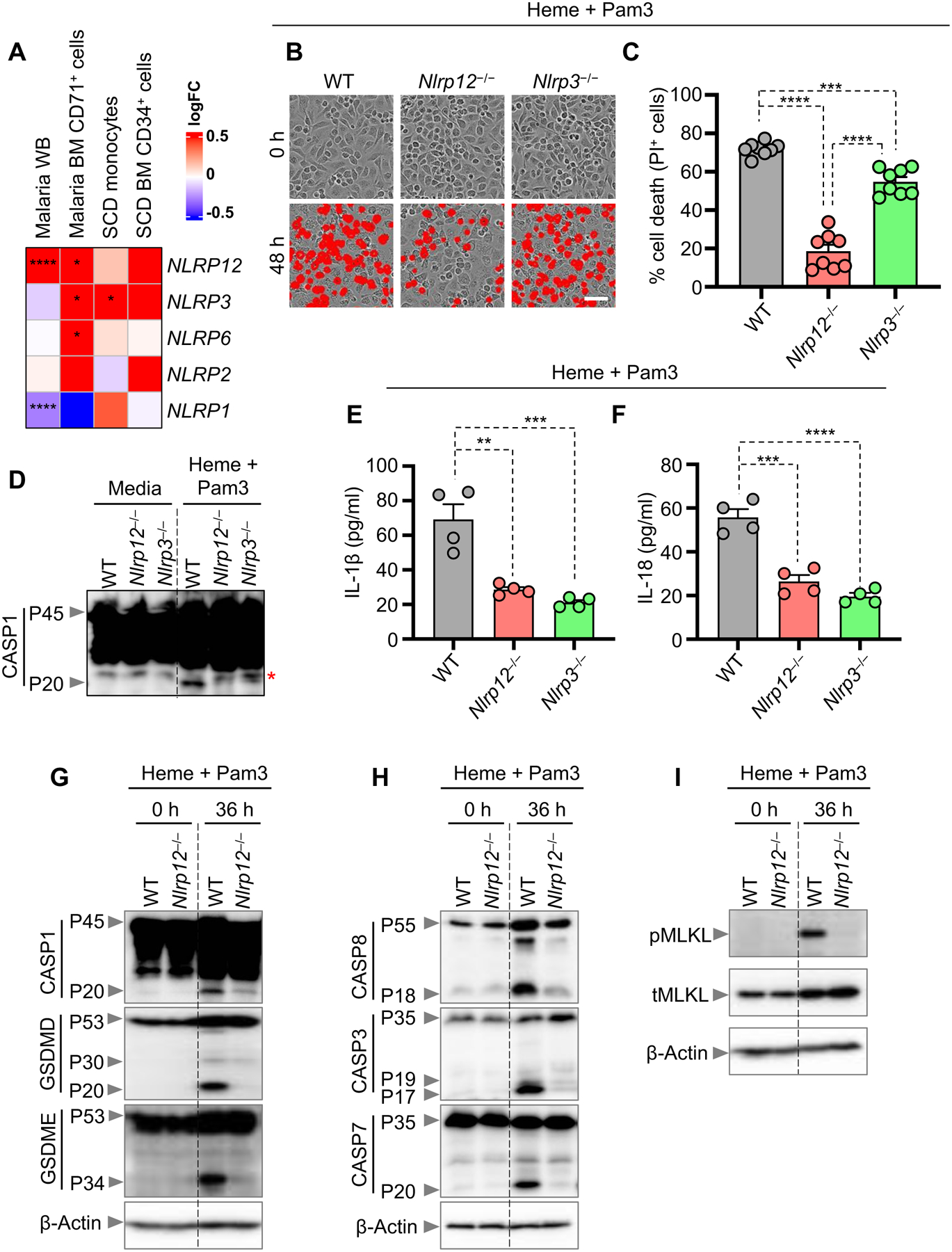
(A) Heatmap depicting the expression profile of NLRPs in malaria whole blood (WB), malaria bone marrow (BM) CD71+ cells, sickle cell disease (SCD) monocytes and SCD BM CD34+ cells compared to healthy controls. (B and C) Representative images of cell death (B) and quantification showing percentage of cell death (C) in wildtype (WT), _Nlrp12_−/− and _Nlrp3_−/− bone marrow-derived macrophages (BMDMs) treated with heme plus Pam3CSK4 (Pam3) for 48 h. (D) Immunoblot showing pro- (P45) and activated (P20) caspase-1 (CASP1) in WT, _Nlrp12_−/−and _Nlrp3_−/− BMDMs at 0 h (media control) or treated with heme plus Pam3 for 42 h. (E–F) Measurement of IL-1β (E) and IL-18 (F) release from the supernatant of WT, _Nlrp12_−/− and _Nlrp3_−/− BMDMs treated with heme plus Pam3 for 36 h. (G–I) Immunoblot analysis of (G) pro- (P45) and activated (P20) CASP1, pro- (P53), activated (P30) and inactivated (P20) gasdermin D (GSDMD) and pro- (P53) and activated (P34) gasdermin E (GSDME); (H) pro- (P55) and cleaved caspase-8 (CASP8; P18), pro- (P35) and cleaved caspase-3 (CASP3; P19 and P17); pro- (P35) and cleaved caspase-7 (CASP7; P20), and (I) phosphorylated MLKL (pMLKL) and total MLKL (tMLKL) in WT and _Nlrp12_−/− BMDMs at 0 h or treated with heme plus Pam3 for 36 h. β-actin was used as a loading control. Scale bar = 50 μm (B). Data are representative of at least three independent experiments. Data are shown as mean ± SEM (C, E, F). Analysis was performed using the non-parametric Wilcox test (A) or the one-way ANOVA (C, E, F). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
See also Figure S3.
To determine whether these upregulated NLRPs played a role in inflammatory cell death in response to heme, we evaluated cell death in NLRP12-, NLRP3- and NLRP6- deficient BMDMs stimulated with heme plus Pam3. Similar levels of cell death occurred in wildtype (WT) and _Nlrp6_−/− BMDMs in response to heme plus Pam3 (Figures S3A and S3B). Loss of NLRP3 provided a partial reduction in cell death, whereas loss of NLRP12 provided significantly more protection from cell death in response to heme plus Pam3 (Figures 2B and 2C). To further confirm the role of NLRP12 in driving cell death in response to heme plus PAMPs, we treated WT and _Nlrp12_−/− BMDMs with heme plus LPS and heme plus R848. Consistent with observations in response to heme plus Pam3 stimulation, _Nlrp12_−/− BMDMs were significantly protected from cell death during heme plus LPS and heme plus R848 treatment (Figures S3C–S3F). Previous studies have also shown that cell death, characterized by activation of necroptotic and apoptotic molecules, can be induced by heme plus TNF19,33. We therefore sought to determine whether NLRP12 was also important for the cell death induced by heme plus TNF. _Nlrp12_−/− BMDMs were significantly protected from cell death in response to heme plus TNF (Figures S3G and S3H). Excess circulating TNF is a feature of both infectious and inflammatory diseases34, suggesting that NLRP12 could have a role in heme-mediated cell death in both infections and inflammatory disease conditions.
To assess whether the NLRP12-mediated cell death we observed in murine macrophages in response to heme plus PAMP stimulation also occurred in human cells, we used primary human monocytes differentiated to macrophages and siRNA knockdown of NLRP12 expression. Heme plus PAMP stimulation induced cell death in human macrophages treated with a control siRNA, while there was a significant decrease in cell death in macrophages treated with the _NLRP12-_targeting siRNA (Figures S3I–S3K). To determine whether NLRP12 was also involved in PAMP plus PAMP-induced cell death, we stimulated WT and _Nlrp12_−/− BMDMs with poly(I:C) plus Pam3 or poly(I:C) plus R848. However, there was no significant difference in cell death in _Nlrp12_−/− and WT BMDMs (Figures S3L–S3O). Together, these data suggest that NLRP12 is specifically required to drive inflammatory cell death in response to heme plus PAMPs in both murine and human cells.
Previous studies have reported that NLRP3 regulates inflammasome activation in response to heme20,21. Furthermore, inflammasomes can act as integral components of PANoptosomes to drive PANoptosis7–10. We therefore investigated inflammasome activation in response to heme plus PAMP treatment in WT, Nlrp12 −/− and Nlrp3 −/− BMDMs. Loss of either NLRP12 or NLRP3 was sufficient to prevent caspase-1 cleavage in response to heme plus Pam3 (Figure 2D). Moreover, inflammasome-dependent cytokine release was observed in WT BMDMs in response to heme plus Pam3 treatment, with significant reductions in both Nlrp12 −/− and Nlrp3 −/− BMDMs (Figures 2E and 2F). Together, these results indicate that both NLRP12 and NLRP3 play crucial roles in inflammasome activation and cytokine release in response to heme plus PAMP stimulation.
Next, we determined how the loss of NLRP12 molecularly impacted cell death. In response to treatment with heme plus Pam3, cleavage of caspase-1, GSDMD (especially the P20 form), GSDME, caspases-8, -3, and -7 was decreased, and phosphorylation of MLKL was reduced in _Nlrp12_−/− BMDMs compared to WT BMDMs (Figures 2G–2I), suggesting that NLRP12 regulates the activation of the inflammasome and PANoptotic inflammatory cell death molecules to control cell death in response to heme plus PAMPs.
NLRP12 drives caspase-8/RIPK3-dependent cell death induced by heme plus PAMPs
Because we observed NLRP12-dependent activation of multiple cell death proteins in response to heme plus PAMPs, including caspases-1, -3, -7 and MLKL (Figures 2G–2I), we sought to determine the contribution of each of these cell death proteins to the overall cell death phenotype. There was no significant reduction in cell death between WT and _Casp1_−/−, _Casp3_−/−, _Casp7_−/− or _Ripk3_−/− BMDMs upon heme plus Pam3 stimulation (Figures S4A and S4B), indicating that there could be redundancy among these cell death molecules in the NLRP12-dependent cell death.Furthermore, we evaluated the contribution of gasdermins in response to heme plus PAMPs and found a consistent partial reduction in cell death in Gsdme −/− and Gsdmd −/− Gsdme −/− BMDMs compared with WT BMDMs in response to heme plus Pam3 stimulation (Figures S4C and S4D), suggesting that GSDME is involved in the execution of heme-mediated inflammatory cell death, but that other executioners are also likely playing a role.
To further assess the role of other cell death proteins in this phenotype, we treated WT BMDMs with the RIPK1 inhibitor, Necrostatin 2 racemate (Nec-1), or the combination of Nec-1 with the pan-caspase inhibitor, z-VAD-FMK (zVAD). Nec-1 treatment partially reduced cell death in response to heme plus Pam3 compared to PBS control, and the addition of zVAD further reduced the cell death (Figures S4E and S4F), indicating that RIPK1 and caspases are both important for cell death in response to heme plus Pam3.
Since we observed significant cell death protection in zVAD and Nec-1–treated BMDMs upon heme plus Pam3 stimulation (Figures S4E and S4F), we sought to use a genetic model to confirm these findings using _Casp1_−/−_Casp8_−/−_Ripk3_−/− (referred to as triple knock-out [TKO]) BMDMs, where key cell death components were absent. There was significant protection from cell death in TKO BMDMs compared to WT BMDMs in response to heme plus Pam3 or heme plus LPS (Figures 3A–3D). Consistent with cell death protection, cleavage of GSDMD (P20 form), GSDME, caspases-3 and -7 and phosphorylation of MLKL were reduced in TKO BMDMs compared to WT BMDMs upon heme plus Pam3 or heme plus LPS stimulation (Figures 3E–3J). Moreover, _Casp8_−/−_Ripk3_−/− BMDMs (referred to as double knock-out [DKO]) provided significant protection from cell death (Figures S4G and S4H), similar to that observed in _Casp1_−/−_Casp8_−/−_Ripk3_−/− BMDMs in response to heme plus Pam3 treatment (Figures 3A and 3B). These results suggest that NLRP12-mediated PANoptosis is occurring.
Figure 3. NLRP12 drives caspase-1/caspase-8/RIPK3-dependent PANoptosis induced by heme plus PAMPs.
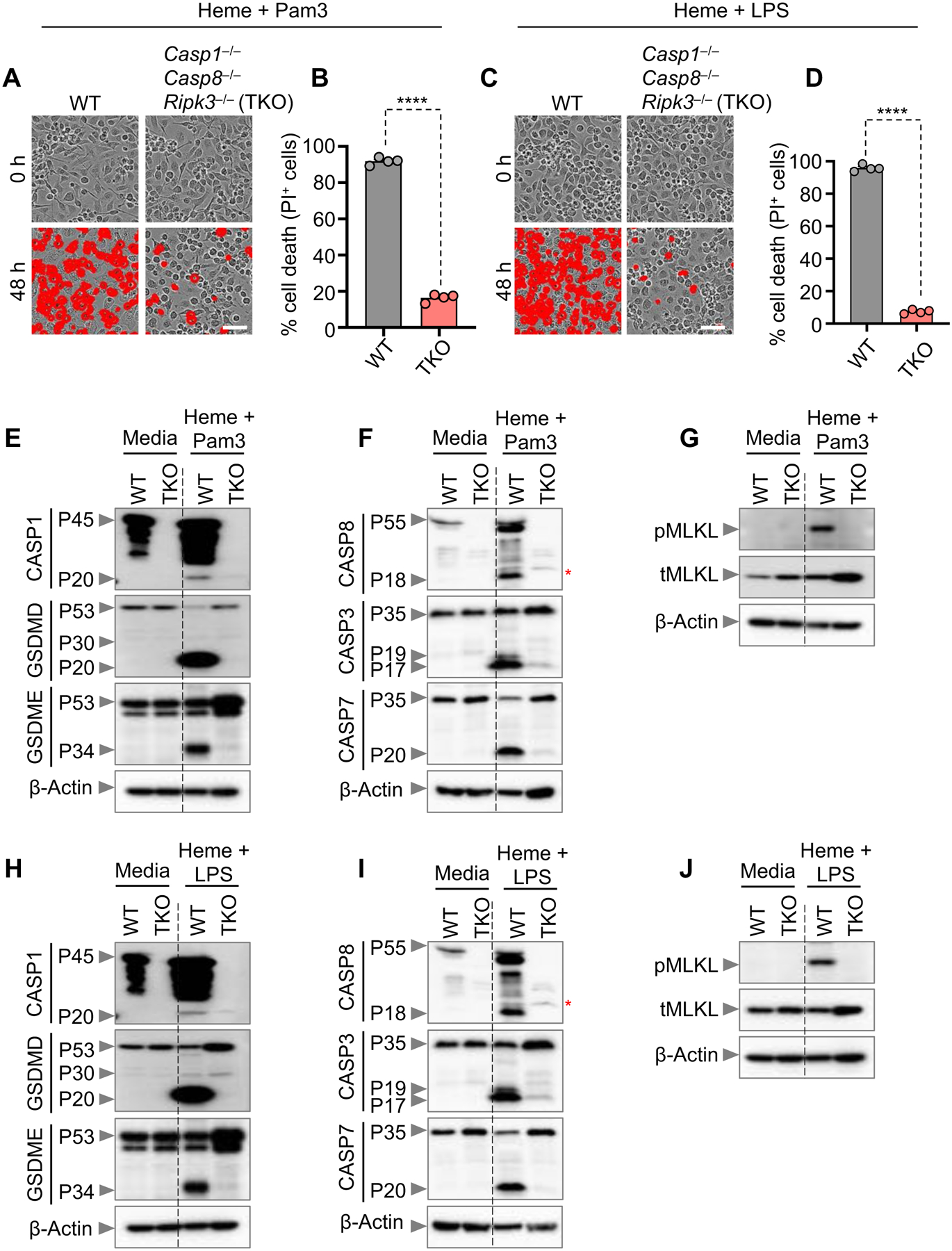
(A–D) Representative images of cell death (A and C) and quantification showing percentage of cell death (B and D) in wildtype (WT) and _Casp1_−/−_Casp8_−/−_Ripk3_−/− (TKO) bone marrow-derived macrophages (BMDMs) stimulated with heme plus Pam3CSK4 (Pam3) (A and B) or heme plus lipopolysaccharide (LPS) (C and D) for 0 h and 48 h. Quantification is shown for the 48 h timepoint. (E–J) Immunoblot analysis of (E and H) pro- (P45) and activated (P20) caspase-1 (CASP1), pro- (P53), activated (P30) and inactivated (P20) gasdermin D (GSDMD) and pro- (P53) and activated (P34) gasdermin E (GSDME); (F and I) pro- (P55) and cleaved caspase-8 (CASP8; P18), pro- (P35) and cleaved caspase-3 (CASP3; P19 and P17) and pro- (P35) and cleaved caspase-7 (CASP7; P20); and (G and J) phosphorylated MLKL (pMLKL) and total MLKL (tMLKL) in WT and TKO BMDMs treated with heme plus Pam3 (E–G) or heme plus LPS (H–J) for 36 h. β-actin was used as a loading control. Scale bar = 50 μm (A and C). Data are representative of at least three independent experiments. Data are shown as mean ± SEM (B and D). Analysis was performed using the unpaired t test (B and D). ****P < 0.0001. Red asterisk denotes nonspecific band (F and I).
See also Figure S4.
Moreover, to determine which caspases are involved in cleaving gasdermins in response to heme plus PAMP stimulation, we assessed gasdermin cleavage in different caspase-deficient BMDMs. Cleavage of GSDMD to its inactivated P20 form was notably reduced in Casp8 −/− Ripk3 −/− BMDMs, while there was little to no difference between WT, caspase-1–, caspase-3– and caspase-7–deficient BMDMs (Figures S4I). In the case of GSMDE cleavage, cleavage was partially reduced in caspase-3–deficient BMDMs compared to WT BMDMs (Figures S4I); however, this cleavage was further reduced in Casp8 −/− Ripk3 −/− BMDMs, similar to that observed for GSDMD P20 cleavage (Figures S4I).
We next evaluated the impact of loss of the critical cell death molecules on inflammasome-dependent cytokine production. We selected the Casp8 −/− Ripk3 −/− BMDMs for comparison due to the important role observed for caspase-8 in gasdermin cleavage and the induction of cell death, which was not observed for the other caspases tested. The release of IL-1β and IL-18 was significantly reduced in Casp8 −/− Ripk3 −/− BMDMs (Figures S4J and S4K). Taken together, these results suggest that caspase-8 is a key component for NLRP12-mediated PANoptosis, processing of gasdermins and release of inflammatory cytokines in response to heme plus PAMP stimulation.
NLRP12 interacts with cell death molecules to form a multiprotein PANoptosome complex that drives inflammatory cell death
Caspase-1 activation is a hallmark of inflammasome activation. Additionally, NLRP12 has been suggested to act as an inflammasome sensor during Yersinia pestis or Plasmodium chabaudi infections35,36. However, NLRP12 also has inflammasome-independent functions to dampen NF-κB and ERK activation during inflammation and Salmonella infection37,38, and loss of NLRP12 in mice results in increased susceptibility to colon inflammation, colorectal tumor development and atypical neuroinflammation37,39,40. Therefore, we sought to evaluate the role of NLRP12 in inflammasome formation and activity in response to heme plus PAMPs. The formation of ASC specks, a key microscopy feature of inflammasome activation, was increased in WT BMDMs upon heme plus Pam3 treatment when compared to untreated BMDMs (Figures 4A and 4B). Moreover, there was a significant reduction in ASC speck formation in _Nlrp12_−/− BMDMs compared to WT BMDMs (Figures 4A and 4B), indicating that NLRP12 is required for inflammasome assembly under these conditions. Together, these data suggest that NLRP12 regulates inflammasome formation in response to heme plus PAMPs.
Figure 4. NLRP12 expression and formation of the PANoptosome through interactions among cell death molecules drive inflammatory cell death.
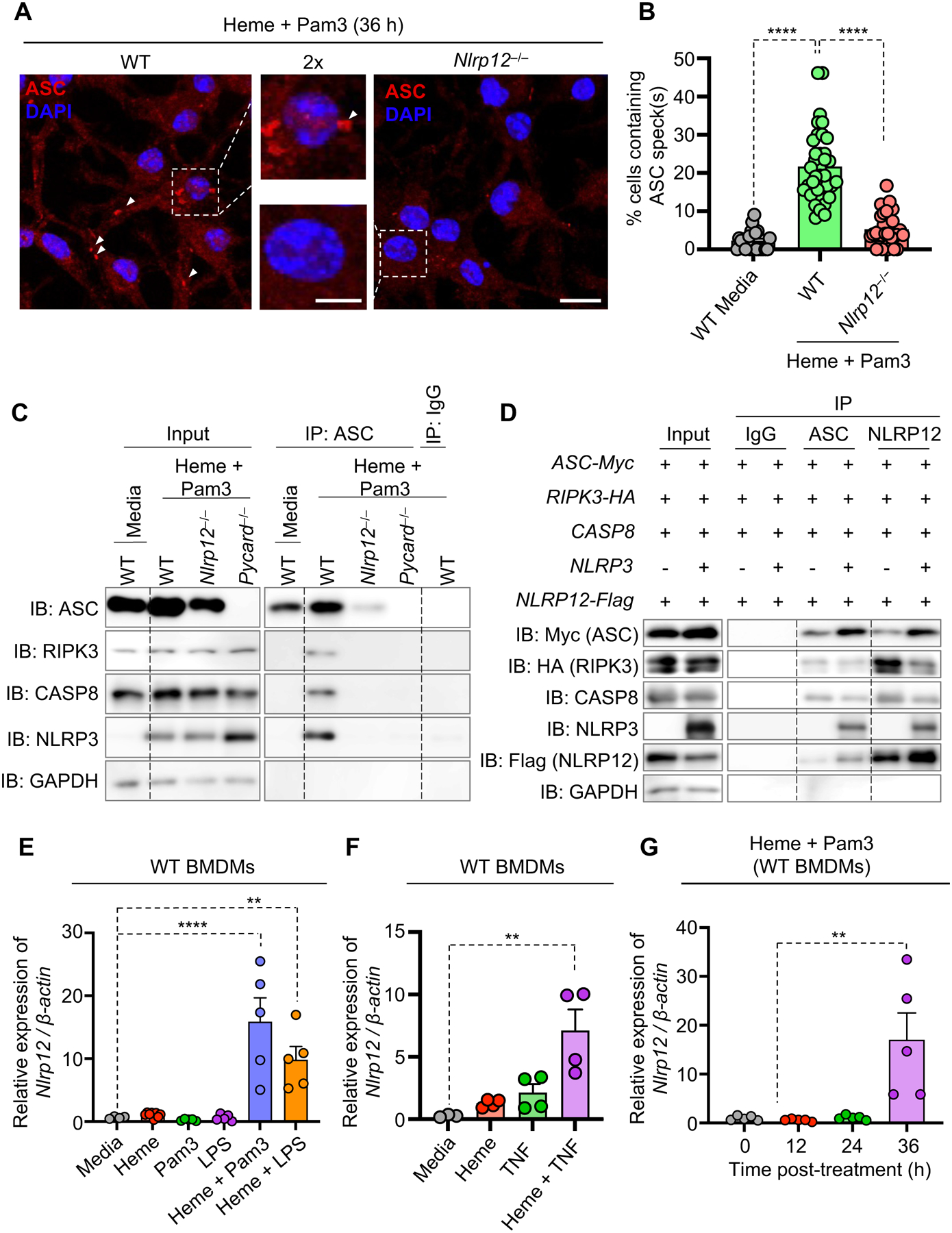
(A and B) Representative images of ASC specks (A) and quantification showing percentage of cell containing ASC specks (B) in wildtype (WT) and _Nlrp12_−/− bone marrow-derived macrophages (BMDMs) treated with heme plus Pam3CSK4 (Pam3) for 36 h. White arrows indicate ASC speck formation (A). Unstimulated WT BMDMs were used as a media control. (C) Immunoblot analysis of ASC, RIPK3, caspase-8 (CASP8) and NLRP3 following immunoprecipitation (IP) with anti-ASC or IgG control antibodies in WT, _Nlrp12_−/− and _Pycard_−/− BMDMs after 28 h of heme plus Pam3 treatment. (D) Immunoblot analysis of Myc-tagged ASC, HA-tagged RIPK3, CASP8, NLRP3, and Flag-tagged NLRP12 following IP with anti-ASC or anti-Flag (NLRP12) antibodies from lysates of 239T cells in an overexpression system. GAPDH was used as a loading control (C and D). (E) Measurement of the relative expression of Nlrp12 mRNA normalized to β-Actin expression in WT BMDMs treated with media, heme, Pam3, lipopolysaccharide (LPS), heme plus Pam3 or heme plus LPS for 36 h. (F) Measurement of the relative expression of Nlrp12 mRNA normalized to β-Actin expression in WT BMDMs treated with media, heme, TNF or heme plus TNF for 42 h. (G) Measurement of the relative expression of Nlrp12 mRNA normalized to β-Actin expression in WT BMDMs treated with heme plus Pam3 for 0, 12, 24 and 36 h. Scale bar = 10 μm (A), and 2× scale bar = 5 μm (A). Data are representative of at least three independent experiments. Data are shown as mean ± SEM (B and E–G). Analysis was performed using the one-way ANOVA (B and E–G). **P < 0.01, ****P < 0.0001.
See also Figure S5.
In addition to the inflammasome, we observed that caspase-8 played an important role in driving the NLRP12-mediated inflammatory cell death in response to heme plus PAMP treatment (Figures 3A–3D, S4G and S4H). Therefore, we next sought to determine whether there was an interaction between NLRP12 and caspase-8. Immunoprecipitation in WT, Nlrp12 −/− and Pycard −/− BMDMs following heme plus Pam3 treatment showed the formation of a multiprotein PANoptosome complex containing ASC, RIPK3, caspase-8 and NLRP3 in WT BMDMs; the formation of this complex was abolished in the absence of NLRP12 (Figure 4C), indicating that the NLRP12 inflammasome is an integral component of this multiprotein PANoptosome complex to drive inflammatory cell death in response to heme plus PAMP stimulation. To further confirm that NLRP12 is part of the PANoptosome, we overexpressed human NLRP12, NLRP3, ASC, caspase-8 and RIPK3 in 293T cells and performed immunoprecipitation using both the ASC and NLRP12 proteins as bait. Again, a multiprotein complex containing ASC, NLRP12, NLRP3, caspase-8 and RIPK3 was identified, and the complex was still formed in the absence of NLRP3 (Figure 4D), indicating that NLRP12 forms a PANoptosome that contains the inflammasome along with caspase-8 and other cell death molecules to execute cell death in response to heme plus PAMPs.
Regulation of the expression of NLRP12 controls inflammatory cell death and disease pathology
We previously observed that NLRP12 expression was upregulated in patients with multiple hemolytic diseases (Figure 2A). To further understand the regulation of NLRP12 expression and how this relates to disease pathology, we first measured the expression of Nlrp12 in BMDMs. Nlrp12 was significantly upregulated in response to heme plus PAMPs and heme plus TNF treatment (Figures 4E and 4F). Moreover, Nlrp12 expression was increased at the 36 h timepoint but not at 12 h or 24 h timepoints post-treatment in response to heme plus Pam3 (Figure 4G). Consistent with the timing of Nlrp12 expression, the cell death began at 30–32 h post-treatment (Figure S5A), suggesting that the expression of Nlrp12 is correlated with cell death. Moreover, Nlrp12 expression increased only in BMDMs treated with the combination of heme plus PAMPs or heme plus TNF, but not in BMDMs treated with heme, Pam3, LPS or TNF alone (Figures 4E and 4F), indicating that heme plus PAMPs or heme plus TNF together mediate the signaling required to induce Nlrp12 expression.
To further understand the role of NLRP12 in human cells during conditions where heme and PAMPs are present, we re-analyzed a publicly available single cell transcriptomics dataset from patients with hemolytic diseases41. NLRP12 expression was increased in erythroid, myeloid and hematopoietic stem cells in patients with beta thalassemia or SCD when compared with cells derived from healthy control bone marrow (Figures S5B and S5C). Together, these results suggest that NLRP12 expression in multiple cell types is associated with hemolytic disease. In addition to hemolytic diseases, heme is known to be released during infections and inflammatory diseases due to hemorrhagic conditions and tissue damage. Since we observed increased expression of NLRP12 in hemolytic diseases (Figures 2A, S5B and S5C), we next sought to determine the expression profile of NLRPs in infections and pandemic diseases associated with hemorrhagic conditions. NLRP12 was highly upregulated in patients infected with Crimean-Congo hemorrhagic fever virus (CCHFV) and macaques infected with Marburg virus (Figures S5D and S5E). Hemolysis has been observed in several other infections, including SARS-CoV-2 infection, which is associated with hemolytic anemia42,43; influenza virus infection, where the virus or hemagglutinin glycoproteins can cause hemolysis44,45; and pneumonia and systemic inflammatory response syndrome (SIRS), where hemolysis and release of free heme have been found to drive severe pathology and morbidity46–49. The expression of NLRP12 was also increased in patients with COVID-19, influenza, pneumonia and SIRS (Figures S5F–S5I), further suggesting an association between NLRP12 expression and hemorrhagic and pandemic diseases.
TLR2 and TLR4 regulate NLRP12 expression and inflammatory cell death
We next sought to understand the upstream signaling pathways involved in the regulation of NLRP12 expression and the induction of NLRP12-mediated cell death. Membrane-bound TLRs are cell surface receptors that are likely to be the first point of contact for extracellular stressors, and their activation can induce diverse downstream signaling cascades. TLR2 has been previously shown to be activated in response to Pam350,51, while TLR4 is activated by LPS52,53 and TLR2/4 are activated by heme54,55. Both TLR2 and TLR4 were required to drive inflammatory cell death in response to heme plus Pam3 stimulation (Figures 5A and 5B). MyD88, an important downstream signaling adaptor for both TLR2 and TLR456, was also required to drive inflammatory cell death in response to heme plus PAMP stimulation (Figures 5C and 5D). Together, these data suggest that TLR2/4 and MyD88 signaling are required to drive inflammatory cell death in response to heme plus PAMP stimulation.
Figure 5. TLR2 and TLR4 regulate IRF1 expression to drive NLRP12-dependent PANoptosis.
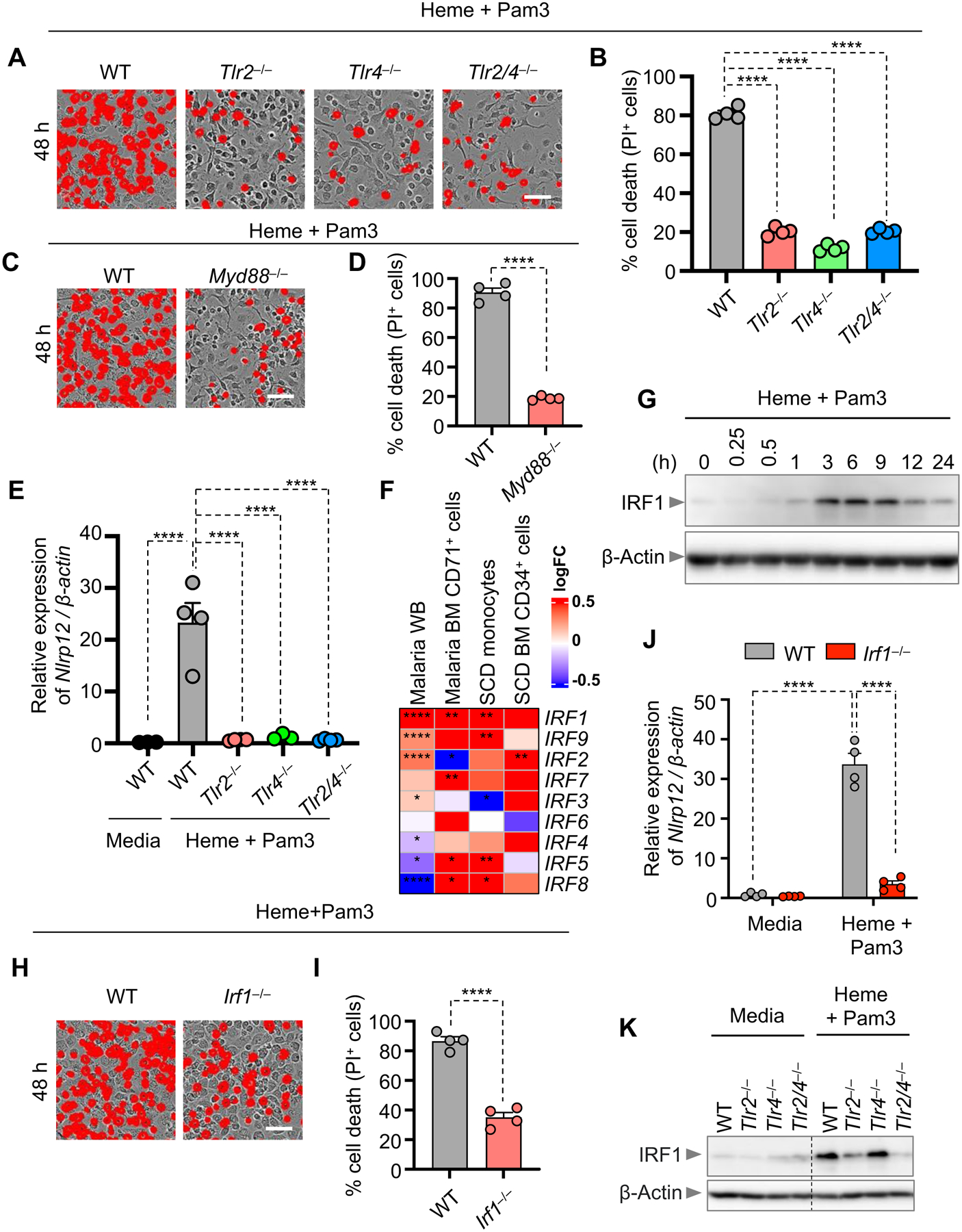
(A and B) Representative images of cell death (A) and quantification showing percentage of cell death (B) in wildtype (WT) and _Tlr2_−/−, _Tlr4_−/− and _Tlr2_−/−/_Tlr4_−/− (Tlr2/_4_−/−) bone marrow-derived macrophages (BMDMs) stimulated with heme plus Pam3CSK4 (Pam3) for 48 h. (C and D) Representative images of cell death (C) and quantification showing percentage of cell death (D) in WT and _Myd88_−/− BMDMs stimulated with heme plus Pam3 for 48 h. (E) Measurement of the relative expression of Nlrp12 mRNA normalized to β-Actin expression in WT and _Tlr2_−/−, _Tlr4_−/− and Tlr2/_4_−/− BMDMs stimulated with heme plus Pam3 for 36 h. (F) Heatmap depicting the expression profile of interferon regulatory factors (IRFs) in malaria whole blood (WB), malaria bone marrow (BM) CD71+ cells, sickle cell disease (SCD) monocytes and SCD BM CD34+ cells compared to healthy controls. (G) Immunoblot analysis of IRF1 in WT BMDMs treated with heme plus Pam3 for 0 h, 0.25 h, 0.5 h, 1 h, 3 h, 6 h, 9 h, 12 h and 24 h. β-actin was used as a loading control. (H and I) Representative images of cell death (H) and quantification showing percentage of cell death (I) in WT and _Irf1_−/− BMDMs stimulated with heme plus Pam3 for 48 h. (J) Measurement of the relative expression of Nlrp12 mRNA normalized to β-Actin expression in WT and _Irf1_−/− BMDMs treated with media or heme plus Pam3 for 36 h. (K) Immunoblot analysis of IRF1 in WT and _Tlr2_−/−, _Tlr4_−/− and Tlr2/_4_−/− BMDMs stimulated with heme plus Pam3 for 3 h or in media control. β-actin was used as a loading control. Scale bar = 50 μm (A, C and H). Data are representative of at least three independent experiments. Data are shown as mean ± SEM (B, D, E, I and J). Analysis was performed using the one-way ANOVA (B, E and J), the t test (D and I) or the non-parametric Wilcox test (F). *P < 0.05, **P < 0.01, ****P < 0.0001.
See also Figure S6.
We then assessed whether TLRs were required to upregulate NLRP12. Nlrp12 expression was robustly upregulated in WT cells, but not in _Tlr2_−/−, _Tlr4_−/− or _Tlr2_−/−_Tlr4_−/− (_Tlr2/4_−/−) BMDMs in response to heme plus PAMP stimulation (Figure 5E), suggesting that TLR2- and TLR4-mediated signalling are required for the upregulation of NLRP12.
In addition to a role for TLRs, previous reports have demonstrated connections between both NLRP12 and hemolytic diseases with IFN signaling. NLRP12 reduces the IFN response to RNA viruses, and loss of Nlrp12 increases resistance to vesicular stomatitis virus infection in mice57. Moreover, the IFN-inducible transcription factor IRF1 can drive severe pathology and mortality in mice during Plasmodium berghei infection58,59. Therefore, we investigated the potential involvement of IRFs in hemolytic diseases by re-analyzing publicly available datasets for patients with malaria or SCD29–32. IRF1 was the most highly upregulated IRF in patients with malaria or SCD hemolytic diseases (Figure 5F). Furthermore, IRF1 expression increased in response to heme plus Pam3 treatment, starting early at 3 h post-treatment, and gradually decreasing after 9 h (Figure 5G). _Irf1_−/− BMDMs had significantly reduced cell death (Figures 5H and 5I) and Nlrp12 expression (Figure 5J) compared with WT BMDMs in response to heme plus Pam3 treatment, further suggesting a critical role for IRF1 in regulating NLRP12 expression to control cell death. To further elucidate the molecular mechanisms by which TLR2/4 and IRF1 regulated NLRP12 expression in response to heme plus PAMP stimulation, we assessed the activation of downstream signalling pathways in WT, _Tlr2_−/−, _Tlr4_−/− and _Tlr2/4_−/− BMDMs. NF-κB and ERK signalling were markedly reduced, as was expression of IRF1, in TLR2- and TLR4-deficient cells in response to heme plus Pam3 (Figures 5K and S6A). Together, these data suggest that heme plus PAMP signalling via their specific TLRs activates NF-κB and ERK signalling to induce IRF1 expression and ultimately upregulate NLRP12 expression to drive inflammatory cell death.
To further understand the regulation of NLRP12-mediated cell death, we next assessed whether the effect of NLRP12 on cell death was linked to heme oxygenase-1 (HO-1), which is upregulated in response to heme and is critical in the heme detoxifying process (Figure S6B)33,60. HO-1 expression increased in response to heme plus PAMPs (Figures S6C and S6D); however, there was no difference in HO-1 expression between WT and _Nlrp12_−/− BMDMs (Figures S6C and S6D). Furthermore, treatment with the HO-1 inhibitor zinc protoporphyrin IX had no significant effect on the magnitude of the cell death in response to heme plus PAMPs (Figures S6E and S6F), suggesting that HO-1 is not involved in the NLRP12-mediated cell death.
In addition to TLR2/4 and IRF1 regulation of heme-mediated cell death, an earlier report suggested that heme exposure can trigger cell death in macrophages through the generation of reactive oxygen species (ROS)19. The production of mitochondrial ROS (mitoROS) was increased in WT BMDMs in response to heme plus PAMP stimulation (Figures 6A and 6B). Treatment with the well-known ROS scavenger N-acetyl L-cysteine (NAC)61 provided significant protection against cell death (Figures 6C and 6D), suggesting a mechanistic connection between ROS production and the induction of cell death. Furthermore, Nlrp12 expression was significantly reduced in NAC-treated BMDMs compared to PBS-treated control BMDMs in response to heme plus PAMP stimulation (Figures 6E). Together, these data suggest that heme plus PAMP stimulation induces the production of mitoROS, which contributes to the induction of Nlrp12 expression to drive cell death.
Figure 6. ROS generation downstream of TLR signaling drives heme plus PAMP-mediated inflammatory cell death.
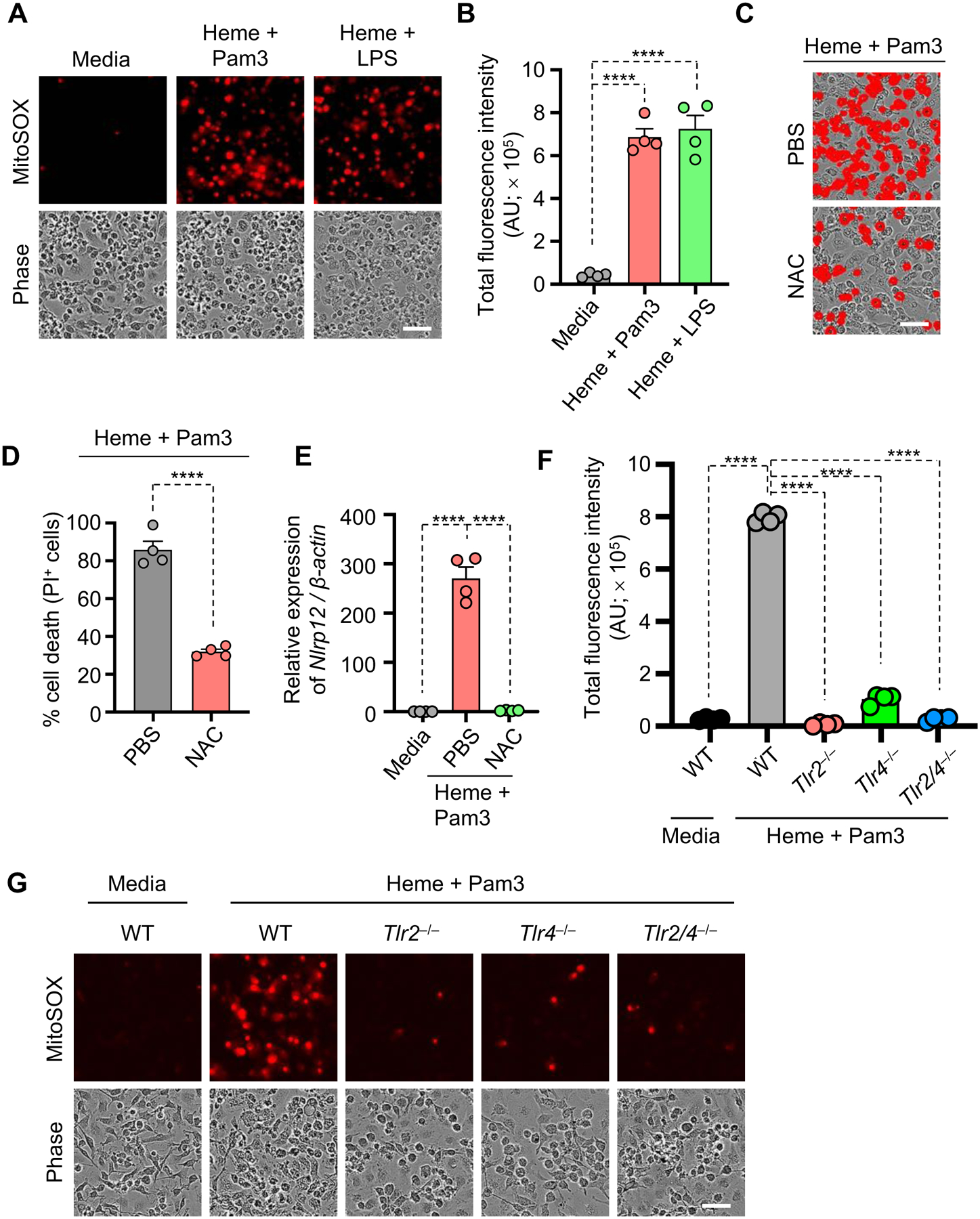
(A and B) Representative images of MitoSOX red (Top) and phase contrast imaging (Bottom) (A) and quantification showing total fluorescence intensity (B) of panel (A) in wildtype (WT) bone marrow-derived macrophages (BMDMs) stimulated with heme plus Pam3CSK4 (Pam3) or heme plus lipopolysaccharide (LPS) for 36 h. (C and D) Representative images of cell death (C) and quantification showing percentage of cell death (D) in WT BMDMs stimulated with heme plus Pam3 with and without N-acetyl cysteine (NAC) treatment for 48 h. (E) Measurement of the relative expression of Nlrp12 mRNA normalized to β-Actin expression in WT BMDMs treated with media or heme plus Pam3 with and without NAC treatment for 48 h. (F and G) Representative images of MitoSOX red (Top) and phase contrast imaging (Bottom) (G) and quantification showing total fluorescence intensity (F) of panel (G) in WT and _Tlr2_−/−, _Tlr4_−/− and Tlr2/_4_−/− BMDMs stimulated with heme plus Pam3 for 36 h. Scale bar = 50 μm (A, C and G). Data are representative of at least three independent experiments. Data are shown as mean ± SEM (B, D–F). Analysis was performed using the t test (D) or the one-way ANOVA (B, E and F). ****P < 0.0001.
See also Figure S6.
To determine whether mitoROS regulation of Nlrp12 expression occurred upstream or downstream of TLR-mediated regulation, we assessed whether mitoROS generation was dependent on TLRs. The production of mitoROS was significantly decreased in _Tlr2_−/−, _Tlr4_−/− and _Tlr2/4_−/− BMDMs when compared to WT BMDMs (Figures 6F and 6G), indicating that TLR2 and TLR4 regulate ROS production in response to heme plus PAMP stimulation. Together, these data suggest that heme plus PAMP stimulation activates TLR2/4 to induce intracellular signalling pathways through MyD88 that trigger IRF1 expression and ROS generation, inducing Nlrp12 expression to initiate the formation of a multiprotein PANoptosome complex to execute inflammatory cell death, PANoptosis.
NLRP12 drives cell death in acute kidney injury and causes lethality in a hemolytic disease model
To directly test the role of NLRP12 in disease pathogenesis in vivo, we injected mice with phenylhydrazine (PHZ), which is known to induce hemolysis62, together with a non-lethal dose of LPS. WT mice had increased amounts of serum heme and iron upon treatment with PHZ alone and the combination of PHZ plus LPS (Figures 7A–7B), indicating that hemolysis is occurring in both groups. Previous studies have demonstrated that hemolysis and heme release can cause acute kidney injury and acute tubular necrosis63,64. Consistent with these reports, levels of kidney damage markers such as blood urea nitrogen (BUN) and creatinine were significantly increased in the PHZ plus LPS cotreatment group compared to PHZ or LPS treatments alone (Figures 7C–7D). To determine whether the observed pathogenesis was driven by NLRP12, we injected PHZ plus LPS in WT and _Nlrp12_−/− mice. _Nlrp12_−/− mice were significantly protected from lethality and had significantly reduced BUN and creatinine levels when compared to WT mice in response to the PHZ plus LPS treatment (Figures 7E–7G). However, there were no significant differences in serum hemoglobin (Hb) and iron levels between WT and _Nlrp12_−/− mice (Figures S7A and S7B), indicating that the extent of hemolysis was similar in both groups. Furthermore, histology scores were significantly higher, indicating worsened pathology, in WT animals in response to PHZ plus LPS compared to the PBS-treated control animals (Figures 7H and 7J), and the extent of damage and histology score were decreased in _Nlrp12_−/− kidney tissues compared to those from WT animals (Figures 7H and 7J). Similarly, there was a significant decrease in TUNEL-positive staining in _Nlrp12_−/− kidney tissues compared to WT kidney tissues in response to PHZ plus LPS (Figures 7I and 7K). Consistent with the TUNEL staining results, activation of caspase-3 and caspase-7 were decreased in _Nlrp12_−/− kidney tissues compared to WT kidney tissues in response to PHZ plus LPS (Figures S7C and S7D), indicating that NLRP12 regulated inflammatory cell death and tissue damage in response to PHZ plus LPS. Overall, these results show that NLRP12-mediated PANoptosis induced pathology and lethality in a hemolytic disease model, implicating NLRP12 in disease pathogenesis.
Figure 7. NLRP12 drives acute kidney injury and cell death to induce lethality in hemolytic disease model.
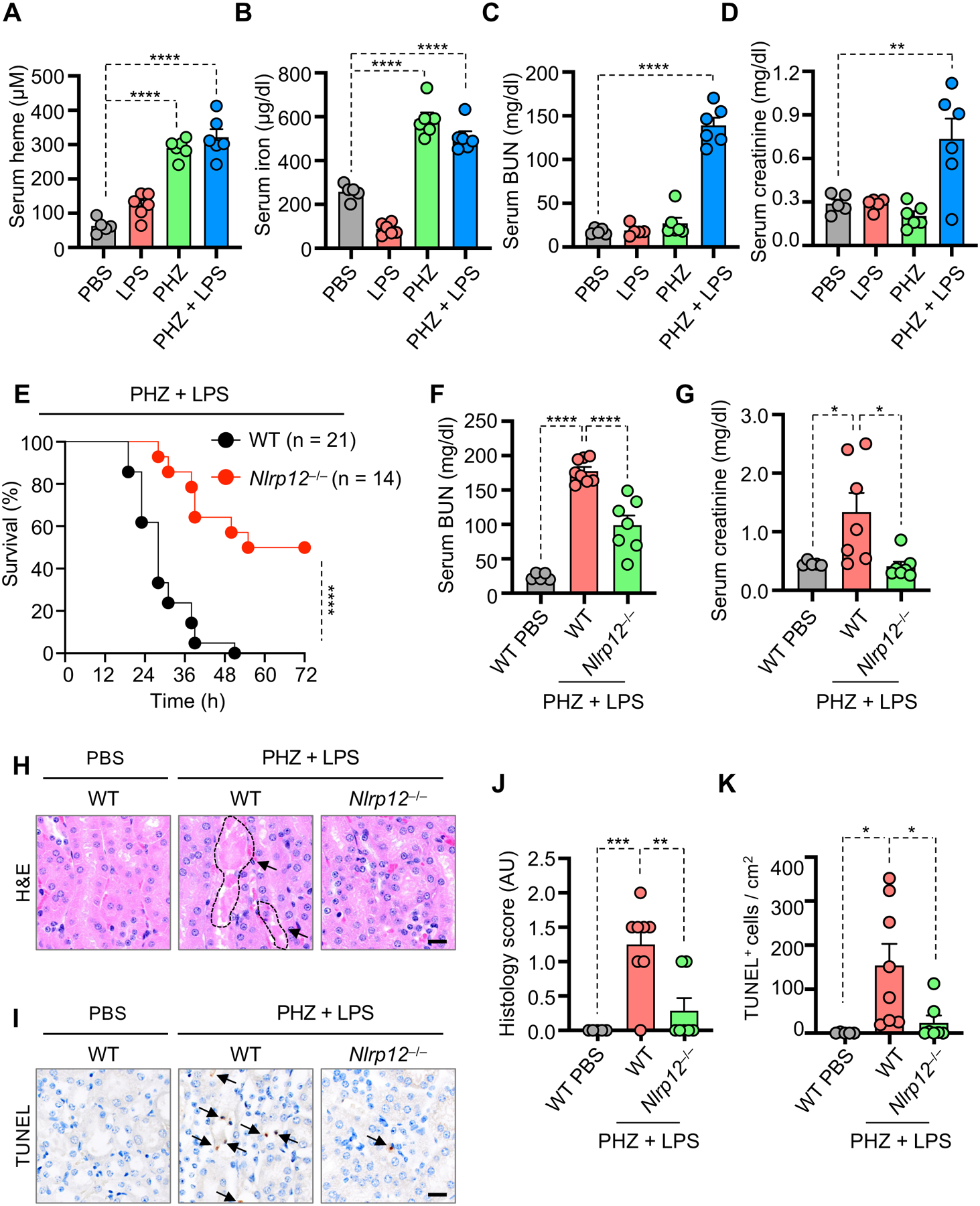
(A–D) Measurement of serum heme (A), iron (B), blood urea nitrogen (BUN) (C) and creatinine (D) in wildtype (WT) mice injected with PBS (n = 5), lipopolysaccharide (LPS) (n = 5), phenylhydrazine (PHZ) (n = 6) or PHZ and LPS (n = 6) at 26 h post-treatment. (E) Survival of 6- to 8-week-old male WT (n = 21) and _Nlrp12_−/− (n = 14) mice after intraperitoneal injection of PHZ and LPS. (F and G) Measurement of serum BUN (F) and creatinine (G) in WT and _Nlrp12_−/− mice injected with PBS or PHZ and LPS (WT PBS, n = 5; WT PHZ + LPS, n = 7–8; _Nlrp12_−/− PHZ + LPS, n = 7) at 30 h post-treatment. (H and J) Hematoxylin and eosin (H&E) staining (H) and quantification of histology score from panel H (J) in kidney tissues from WT and _Nlrp12_−/− mice injected with PBS or PHZ and LPS at 30 h post-treatment. Outlined area indicates luminal debris/hemoglobin casts (H). (I and K) TUNEL staining (I) and quantification of TUNEL positive cells from panel I (K) in kidney tissues from WT and _Nlrp12_−/− mice injected with PBS or PHZ and LPS at 30 h post-treatment. Arrows indicate TUNEL-positive cells (I). Scale bar = 20 μm (H and I). Data are representative of at least three independent experiments. Data are shown as mean ± SEM (A–D, F, G, J and K). Analysis was performed using the log-rank (Mantel-Cox) test (E) or the one-way ANOVA (A–D, F, G, J and K). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
See also Figure S7.
DISCUSSION
Given the critical role of innate immunity in detecting and counteracting infections and sterile insults, it is fundamental to the understanding of health and disease to identify the functions of innate immune sensors. NLRs are a key family of intracellular sensors involved in the regulation of inflammatory signaling in response to infection and cellular stress. Recent studies have shown pivotal roles for NLR-mediated inflammation across diverse diseases, including infections, cancers and inflammatory disorders4–6. NLRP12 is a member of this NLR family, and missense mutations in this gene are associated with inflammatory diseases in humans65,66. Although it was discovered in the 2000s, the biochemical mechanisms of NLRP12 activation and the pathways underlying its role in inflammatory diseases remain unclear, as NLRP12 can be an inflammasome sensor during Y. pestis or P. chabaudi infections35,36, but it also has inflammasome-independent functions to dampen NF-κB and ERK activation37,38 and prevent colon inflammation, colorectal tumor development and atypical neuroinflammation37,39,40.
Here, we found that NLRP12 is responsible for heme plus PAMPs-mediated inflammasome and PANoptosome activation to drive inflammatory cell death, PANoptosis. Heme is a central component of hemoglobin that can be released when red blood cells lyse to act as a key DAMP during hemolytic diseases, such as in malaria and SCD. While previous reports have shown that macrophages treated with LPS plus heme in low or no serum conditions induce rapid NLRP3-dependent IL-1β secretion20,21, here we used higher serum levels to avoid low serum stress responses and instead observed robust NLRP12- and NLRP3-dependent inflammasome activation after prolonged stimulation; however, we found that the cell death was only dependent on NLRP12. This longer stimulation allowed time for the TLR-mediated upregulation of IRF1 to drive NLRP12 expression. Understanding the regulation and activation mechanisms of inflammasomes has been historically difficult; for instance, while the NLRP1 and NLRP3 inflammasomes have been known for decades, several activation mechanisms have been proposed for each67–79, and no mechanism has yet been identified that encompasses all the activation processes observed. Additionally, increasing evidence shows that inflammasomes do not need to act alone and can integrate into larger cell death complexes called PANoptosomes7–10. Our results demonstrate that TLR2/4-mediated signaling controls the expression the expression of Nlrp12 in response to heme plus PAMPs treatment. While the direct role of NLRP12 in binding to inflammasome and PANoptosome components requires further study and is methodologically complicated by the current lack of specific murine NLRP12 antibodies, our data show that the formation of the PANoptosome is inhibited in _Nlrp12_−/− BMDMs, and that expression of NLRP12 is upregulated in hemolytic diseases such as malaria and SCD and loss of NLRP12 can reduce acute kidney damage and lethality in a murine model of hemolytic disease. Overall, our findings suggest that targeting NLRP12 or other PANoptosome components and inflammatory cell death molecules may have therapeutic potential in the treatment of diverse infections and inflammatory diseases.
Limitations of the study
Our findings show a critical role for NLRP12 in the innate immune response to heme plus PAMPs to induce inflammatory cell death, PANoptosis, and pathology. However, there are currently no specific murine NLRP12 antibodies or NLRP12 inhibitors, limiting additional biochemical characterization of NLRP12. Furthermore, our study focuses on the assessment of the cell death pathway in innate immune cells, macrophages, and it remains unknown whether this also occurs in non-immune cell types. Other innate immune NLRs, such as NLRP3, have been implicated in a wide variety of diseases. Here, we focused on the role of NLRP12 in diseases associated with hemolysis, but these analyses could be extended to other diseases to further understand how therapeutic targeting of NLRP12 or its regulatory pathway could be clinically applicable across the disease spectrum.
STAR METHODS
Resource Availability
Lead Contact
Further information and reasonable requests for reagents may be directed to, and will be fulfilled by, the lead contact Thirumala-Devi Kanneganti (thirumala-devi.kanneganti@stjude.org).
Materials Availability
All unique reagents generated in this study are available upon reasonable request from the Lead Contact.
Data and Code Availability
- The datasets generated and analyzed during the current study are contained within the manuscript and accompanying extended data figures and tables, and publicly available datasets analyzed can be found in the Gene Expression Omnibus database (GSE133181, GSE34404, GSE136046, GSE102881, GSE168532, GSE58287, GSE40012, GSE171110). Data for patients with CCHFV was obtained from Sequence Read Archive (Accession: PRJNA680886).
- This paper does not report original code.
- Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.
Experimental Model and Subject Details
Mice
_Casp1_−/−80, _Casp3_−/−81, _Casp7_−/−82, _Ripk3_−/−83, _Pycard_−/−84, _Nlrp3_−/−85, _Nlrp12_−/−40, _Nlrp6_−/−86, _Irf1_−/−87, _Ripk3_−/−_Casp8_−/−88, _Tlr2_−/−89, _Tlr4_−/−90, _Myd88_−/−91, _Gsdmd_−/−92, _Gsdme_−/−93 and _Gsdmd_−/−_Gsdme_−/− 94 mice have been previously described. _Casp1_−/−_Casp8_−/−_Ripk3_−/− mice were bred by crossing _Ripk3_−/−_Casp8_−/− 88 with _Casp1_−/− 80 mice. _Tlr2_−/−_Tlr4_−/− mice were bred by crossing _Tlr2_−/− 89 with _Tlr4_−/− 90 mice. All mice were generated on or extensively backcrossed to the C57/BL6 background. Mice were bred at the Animal Resources Center at St. Jude Children’s Research Hospital and maintained under specific pathogen-free conditions. Male age- and sex-matched 6- to 10-week-old mice were used in this study. Mice were maintained with a 12 h light/dark cycle and were fed standard chow. Animal studies were conducted under protocols approved by the St. Jude Children’s Research Hospital committee on the Use and Care of Animals. WT and _Nlrp12_−/− animals were co-housed for 2 weeks before performing in vivo experiments.
Methods Details
Bone marrow-derived macrophage (BMDM) generation
Primary mouse BMDMs from wildtype and indicated mutant mice were grown for 6 days in IMDM (12440053, Thermo Fisher Scientific) supplemented with 10% heat-inactivated fetal bovine serum (HI-FBS; S1620, Biowest), 30% L929 conditioned media, 1% non-essential amino acids (11140-050, Thermo Fisher Scientific) and 1% penicillin and streptomycin (15070-063, Thermo Fisher Scientific). BMDMs at a density of 1 × 106 cells/well in 12 well plates or 5 × 105 cells/well in 24 well plates were seeded into growth media overnight before use.
Differentiation of human monocyte-derived macrophages and siRNA transfection
Fresh human blood was collected from the apheresis rings of anonymous healthy blood donors from the blood bank of St. Jude Children’s Research Hospital, following a protocol reviewed and approved by the St. Jude IBC. Peripheral blood mononuclear cells (PBMCs) were isolated from the freshly collected blood using lymphoprep solution (cat. #07801/07811, Stemcell Technologies). Naive monocytes were purified from the PBMCs using monocyte isolation kit (EasySep direct human monocyte isolation kit, cat. #19669, Stemcell Technologies) strictly in accordance with the manufacturer’s protocol. These purified monocytes were further differentiated into monocyte-derived macrophages by culturing in RPMI 1640 media (10-040-CV, Corning) supplemented with 10% HI-FBS, 1% penicillin-streptomycin and 25 ng/ml human M-CSF (300–25, Peprotech) for 6 days in a CO2 incubator supplied with 5% CO2 in a humidified atmosphere. On day 2 and day 4, an additional 8–10 ml of media containing 25 ng/ml human M-CSF was added to the cells. On day 6, all the loosely attached and suspension cells were harvested and washed three times with PBS. The cells were resuspended in PBS at a cell density of 1 × 107 cells/ml. For siRNA-mediated knockdown of gene expression in monocyte-derived macrophages, 5 pmoles non-targeting (control) siRNA (D-001206-14-20, Horizon Discovery) or human-specific NLRP12 siRNA (m015092-00-0005, Horizon Discovery) per million cells was used for transfection by electroporation (Neon Transfection System kit, cat. #MPK5000, Thermo Fisher Scientific). Post-transfection, the cells were resuspended in RPMI medium supplemented with 10% HI-FBS, 1% penicillin-streptomycin and 25 ng/ml human M-CSF along with 0.1 ng/ml ultrapure LPS from E. coli 0111:B4 (tlrl-3pelps, InvivoGen) and 20 ng/ml human IFN-γ (300–02, Peprotech) to polarize the cells to canonical macrophages. Two days after transfection, the cells were used for stimulation.
Cell stimulation
BMDMs were stimulated in DMEM (11995-065, Gibco) containing 10% HI-FBS and 1% penicillin and streptomycin, and human monocyte-derived macrophages were stimulated in RPMI media supplemented with 10% HI-FBS, 1% penicillin-streptomycin with the following PAMPs, DAMPs and inhibitors alone or in combinations where indicated: 50 μM Hemin (heme; H9039, Sigma or or H651–9, Frontier Scientific), 0.5 μg/mL Pam3CSK4 (Pam3; tlrl-pms, InvivoGen), 15 ng/mL LPS (tlrl-3pelps, InvivoGen), 500 ng/mL R848 (tlrl-r848, InvivoGen), 100 ng/mL TNF (Peprotech, 315-01A), 1 μg/mL poly(I:C) (tlrl-picw, InvivoGen), 200 ng/mL MSU crystals (tlrl-msu, InvivoGen), 200 ng/mL recombinant mouse S100A8/S100A9 heterodimer (8916-S8–050, bio-techne), 200 ng/mL recombinant mouse HMGB1 (ab255799, Abcam), 25 μM Z-VAD(OMe)-FMK (zVAD; 14463, Cayman Chemical), 45 μM Necrostatin 2 racemate (Nec-1; S8641, Selleckchem), 50 μM Deferoxamine (DFO; 14595, Cayman Chemical), 10 μM Ferrostatin-1 (Fer-1; 17729, Cayman Chemical) and HO-1 inhibitor zinc protoporphyrin IX (HO-1i; 691550, Sigma).
Real-time imaging for cell death
The kinetics of cell death were monitored using the IncuCyte S3 or SX5 (Sartorius) live-cell analysis system. BMDMs and human primary macrophages (5 × 105 cells/well) were seeded in 24-well tissue culture plates and treated with the indicated stimuli. Cell death was measured by propidium iodide (PI; P3566, Life Technologies) incorporation following the manufacturer’s protocol. The plate was scanned for the indicated time durations where fluorescent and phase-contrast images were acquired in real-time every 1 h. PI-positive dead cells are marked with a red mask and were quantified using the software package supplied with the IncuCyte imager.
Detection of mitochondrial ROS (mitoROS)
Mitochondrial ROS was detected by staining the cells with MitoSOX red dye (M36008, Thermo Fisher Scientific), and the generation of ROS was monitored using the IncuCyte live-cell analysis system. Briefly, BMDMs were washed once with 1× PBS, and cells were treated with heme plus Pam3 in the presence of 2.5 μM MitoSOX red dye, and the images were acquired using the IncuCyte system. To scavenge mitoROS, 5 mM N-acetyl L-cysteine (NAC) (A9165, Sigma) was used in response to heme plus PAMPs treatment.
Immunoblot analysis
After appropriate treatments, cells were lysed along with culture supernatants in caspase lysis buffer (containing 10% NP-40, 25 mM DTT and 1× protease and phosphatase inhibitors (11697498001 and 04906837001, respectively, Roche)) and SDS sample loading buffer (with 2-mercaptoethanol) for probing caspase activation. For immunoblot assessment of signaling activation, culture supernatants were removed, cells were washed once with 1× Dulbecco’s PBS (DPBS; 14190-250, Thermo Fisher Scientific) and lysed in RIPA buffer and SDS sample loading buffer. Proteins were resolved on 8–12% polyacrylamide gels and transferred onto PVDF membranes (IPVH00010, Millipore) using the Trans-Blot® Turbo™ system. After blocking non-specific binding with 5% skim milk, membranes were incubated overnight with the following primary antibodies against: caspase-1 (AG-20B-0044, AdipoGen), caspase-3 (#9662, Cell Signaling Technology [CST]), cleaved caspase-3 (#9661, CST), caspase-7 (#9492, CST), cleaved caspase-7 (#9491, CST), caspase-8 (AG-20T-0138, AdipoGen), cleaved caspase-8 (#8592, CST), pMLKL (#37333, CST), tMLKL (AP14272B, Abgent), GSDMD (ab209845, Abcam), GSDME (ab215191, Abcam), IRF1 (#8478, CST), p-ERK1/2 (#9101, CST), ERK1/2 (#9102, CST), p-IκBα (#2859, CST), IκBα (#9242, CST), p-p65 (#3033, CST), p65 (#8242, CST), HO-1 (#70081, CST), GAPDH (sc-166574 HRP, Santa Cruz) and β-actin (sc-47778 HRP, Santa Cruz). Membranes were then washed and probed with the appropriate horseradish peroxidase (HRP)–conjugated secondary antibodies (anti-mouse [315-035-047] and anti-rabbit [111-035-047], Jackson ImmunoResearch Laboratories). Immunoblot images were acquired on an Amersham Imager using Immobilon® Forte Western HRP Substrate (WBLUF0500, Millipore) or SuperSignal™ West Femto Maximum Sensitivity Substrate (34096, Thermo Fisher Scientific).
Cytokine measurement
In vitro cytokines were detected in the supernatant by using multiplex ELISA (MCYTOMAG-70K, Millipore) and IL-18 ELISA (BMS618-3, Invitrogen), according to the manufacturer’s instructions.
Microarray and RNA-seq analysis
Seven datasets deposited in GEO (accession IDs: GSE3440429, GSE13604630, GSE10288131, GSE16853232, GSE5828795, GSE4001296 and GSE17111097) were used to estimate the role of NLRPs in datasets relevant for hemolytic and pandemic diseases. GSE3440429 compares whole blood RNA-seq profiles of 155 West-African children, including 94 cases undergoing symptomatic phases of Plasmodium falciparum infection and 61 age-matched controls. GSE13604630 consists of expression profiles of affinity purified CD71+ cells from three patients with Plasmodium vivax at day 1 (diagnosis visit) and day 42 after curative drug treatment (convalescence visit). From GSE10288131, RNA-seq profiles were obtained from CD34+ hematopoietic stem/progenitor cells (HSPCs) collected from the bone marrow of two healthy donors and two patients with sickle cell disease (SCD). GSE16853232 consists of transcriptomic profiles of classical monocytes from the peripheral blood of six healthy controls and 13 patients with SCD. GSE5828795 comprises temporal transcriptomic profiles from 30 peripheral blood mononuclear cells from 15 cynomolgus macaques infected with Marburg virus. The dataset includes transcriptomic profiles of 15 macaques at day 0 and three macaques post-infection at days 1, 3, 5, 7 and 9.
GSE4001296 consists of whole blood samples of patients in critical care for up to 5 days and assayed on Illumina HT-12 gene expression bead arrays. The dataset is comprised of patients with influenza A infection (n = 11), bacterial pneumonia (n = 16) or systemic inflammatory response (n = 13) along with healthy control samples (n = 36). To compare a severe disease phenotype with healthy controls while maintaining maximum patient samples per phenotype, patient samples at day 4 for influenza (n = 11), pneumonia (n = 10) and systemic inflammatory response (n = 6) were considered. GSE17111097 is comprised of whole blood transcriptomics for 44 patients with severe COVID-19 and 10 healthy controls for comparison. Additionally, a cohort of 18 patients (Sequence Read Archive: PRJNA680886)98 infected with Crimean-Congo Hemorrhagic Fever Viruses (CCHFV) was obtained, and blood transcriptomics were available for 12 patients with severe CCHFV. This allowed for a differential expression analysis for patients infected with CCHFV between the time of infection and approximately one-year post-recovery (convalescent group).
For each of these datasets, quality control steps were performed, including normalized quantiles using the ‘normalize.quantiles’ function from preprocessCore v1.58.0 package, when the counts are not normalized, followed by log2 transformation for downstream differential expression analysis. Differential expression analysis was performed using the limma v3.52.199 package in R v4.1.1. The Benjamini-Hochberg100 adjusted P value < 0.05 for GSE34404, GSE168532, GSE40012 and GSE171110 was used to determine the set of differentially expressed genes. However, owing to the small sample sizes for GSE136046 (three cases and three controls), GSE102881 (two cases and two controls) and GSE58287 (three cases for each of day 1, 3, 5, 7 and 9), P value adjustments were not made for these three datasets. A P value < 0.05 was used to estimate the set of differentially expressed genes for GSE136046 and GSE102881. The cytosolic sensors, in particular the NLRPs, were assessed to determine which were overexpressed across the disease-relevant datasets. The NLRPs were sorted based on their average fold-change across these datasets and were visualized using Heatmap from the Complex Heatmap v2.8.0101 package.
Single cell analysis
Single cell transcriptomics data were obtained from GSE13318141. This dataset consisted of single cells obtained from the CD34+ bone marrow cells of 4 normal patients (BM), 3 patients with thalassemia major (BT) and 5 patients with SCD analyzed through the 10× chromium platform. The original dataset comprised 32,389 cells of BM, 9,862 cells of BT and 16,266 cells of SCD, along with expression profiles for 33,694 genes. This dataset was analyzed using Seurat v4.1.1 package in R v4.1.1. Quality control steps were performed as suggested previously41. Cells were excluded if the number of genes detected was below 500 or the percentage of mitochondrial genes was above 5%. Log-normalization was performed using the ‘NormalizeData’ function on a scale factor as 10000. All variable genes passing the ‘vst’ filter were included for further analysis. Biological variation within the same group was removed by the ‘ScaleData’ function. Principal component analysis was performed using the ‘RunPCA’ function. The top 20 principal components (PCs) were used in downstream analysis. Louvain graph-based clustering and t-SNE based dimensionality reduction were also performed to obtain low-dimensional representations and visualize the dataset.
To determine the cell type annotation, a set of known markers and transcription factors for bone marrow cell types were used as described previously41. Each cell was annotated using the ‘AUCell_buildRankings’, ‘AUCell_calcAUC’ and ‘AUCell_exploreThresholds’ functions (in order) from the AUCell v1.18.0 package.
The final dataset consisted of 24,864 cells of BM, 6,159 cells of BT and 8,018 cells of SCD and were distributed among four cell types including: Lymphoid (Lym_P), Myeloid (G/M_P), Erythroid (M/E_P) and Multipotential Hematopoietic Stem (HSPC1) progenitor cells. A nonparametric Mann-Whitney-Wilcoxon test102 was used to compare average NLRP12 expression between BT vs BM and SCD vs BM cells bifurcated by the various cell types.
Confocal microscopy
BMDMs were seeded onto poly-D-lysine coated coverslips. After appropriate treatments, cells were washed with DPBS, fixed with 4% paraformaldehyde (15710, Electron Microscopy Sciences) and permeabilized with 0.5% Triton X-100 (T8787, Sigma). After blocking non-specific bindings with 10% normal goat serum (S-1000, Vector Laboratories), the cells were incubated overnight at 4°C with primary antibody against ASC (1:100, #04-147, Millipore). After three washes with PBS-T (0.05% Tween 20 in PBS), the coverslips were incubated with Alexa Fluor 488-conjugated secondary antibody against mouse IgG (1:1000, A11029, Invitrogen) for 2 h at room temperature. Cells were counterstained with DAPI (4′,6-Diamidino-2-Phenylindole, #40043, Biotium) to visualize nuclei, and images were acquired using Marianas spinning disc confocal system (Intelligent Imaging Innovations) comprised of an inverted AxioObserver Z.1 microscope (Carl Zeiss), CSU-W1 with SoRa (Yokogawa), Prime95B sCMOS camera (Photometrics), 405 nm, 473 nm, 561 nm, 647 nm solid state laser lines (Coherent) and a 1.4 NA 60× oil objective. Images were acquired using Slidebook software.
Immunoprecipitation
For immunoprecipitation in the overexpression system, HEK293T cells (CRL-3216, ATCC) were seeded into six-well plates and transfected with a combined total of 5 ug of the following plasmids (1:1:1:1:1, molar ratio): pcDNA3-Myc-hASC (#73952, Addgene), pEGFP-C2-hNLRP3 (#73955, Addgene), pcDNA3-hCASP8 (#11817, Addgene), pcDNA3-HA-hRIPK3 (#78804, Addgene) and pcDNA3-_hNLRP12_-FLAG (Human NLRP12 PCR product amplified using cDNA and cloned in pcDNA™3.1 (+) Mammalian Expression Vector, #V790-20, Thermo Fisher Scientific), and incubated for 24–36 h. Immunoprecipitation was performed as previously described8 with minor modifications. Briefly, cells were collected and lysed in an ice-cold lysis buffer containing 20 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Triton X-100, 10% glycerol, 1 mM Na3VO4, 2 mM PMSF, EDTA-free protease inhibitor cocktail (A32965, Thermo Fisher Scientific), phosphatase inhibitor cocktail (4906845001, Sigma) and 25 μM Z-VAD-FMK (S7023, Selleckchem). After centrifugation at 16,000 × g for 10 min, the lysates were incubated with either IgG control antibody (#3900S, CST), anti-ASC antibody (AG-25B-0006, Adipogen) or anti-NLRP12 antibody (#AP14014a, Abcepta) overnight at 4°C. The immunoprecipitated proteins were then added to washed Protein A/G magnetic beads (#88802, Thermo Fisher Scientific) and incubated for 2 h at 4°C. Subsequently, the beads were washed 4 times with lysis buffer and boiled in 2× SDS loading buffer at 100°C for 5 min. Immunoprecipitates in sample buffer were subjected to immunoblotting analysis with 0.5% of input sample and 12% of pull-down sample per lane. After blocking nonspecific binding with 5% skim milk, membranes were incubated overnight with the following primary antibodies against: Myc (#2276, CST, 1:1000), NLRP3 (#AG-20B-0014-C100, AdipoGen, 1:1000), FLAG (#14793, CST, 1:1000), HA (#2367, CST, 1:1000), human caspase-8 (#ALX-804-242, Enzo Life Science, 1:1000) and GAPDH-HRP (#166574, Santa Cruz, 1:5000).
For endogenous immunoprecipitation, 3 × 107 BMDMs were seeded and stimulated with heme plus Pam3 for 28 h. The BMDMs were then washed with ice cold PBS and scraped from the plate to collect in PBS. The cell pellet was then gently resuspended in 300 μl ice-cold lysis buffer (as described above) and incubated on a rocker at 4°C for 1 h. After centrifugation at 14,000 × g for 10 min, the lysates were incubated with either IgG control antibody (#3900S, CST) or anti-ASC antibody (AG-25B-006-C100, Adipogen) overnight at 4°C. Washed Protein A/G Magnetic beads were added and incubated for 2 h at 4°C. Subsequently, the beads were washed 5 times with lysis buffer and boiled in 2× SDS loading buffer at 100°C for 5 min. Immunoprecipitates in sample buffer were subjected to immunoblotting analysis with 0.5% of input sample and 12% of pull-down sample per lane. After blocking non-specific binding with 5% skim milk, membranes were incubated overnight with the following primary antibodies against: ASC (#2276, CST, 1:1000), RIPK3 (2283, Santa Cruz, 1:1000), caspase-8 (AG-20T-0138, Adipogen, 1:1000), NLRP3 (#AG-20B-0014-C100, AdipoGen, 1:1000), and GAPDH-HRP (#166574, Santa Cruz, 1:5000).
For both overexpression and endogenous IPs, membranes were then washed and probed with the appropriate horseradish peroxidase (HRP)–conjugated secondary antibodies (anti-mouse, anti-rabbit or anti-rat [112-035-143], Jackson ImmunoResearch Laboratories). Immunoblot images were acquired on an Amersham Imager using Immobilon® Forte Western HRP Substrate or SuperSignal™ West Femto Maximum Sensitivity Substrate.
RT-PCR analysis
Total RNA was extracted at indicated time points using TRIzol (15596026, Thermo Fisher Scientific). cDNA was synthesized with 500 ng of extracted RNA using the High-Capacity cDNA Reverse Transcription Kit (4368814, Applied Biosystems). Real-time quantitative PCR was performed with SYBR Green (4368706, Applied Biosystems) as the fluorescent reporter on an Applied Biosystems 7500 real-time PCR instrument. The mouse primer sequences used are as follows: m_Nlrp12_ - Forward primer: AAGACCGCAATGCACGATTAG; Reverse primer: TGGAGCGTTCCCACTCTACA; m_Actin_ - Forward primer: GGCTGTATTCCCCTCCATCG; Reverse primer: CCAGTTGGTAACAATGCCATGT. The human primer sequences used are as follows: h_NLRP12_ - Forward primer: ACCAGACCTTGACCGACCTT; Reverse primer: GAGGACTCGGAGTTTGCAGC; h_ACTIN_ - Forward primer: CACCATTGGCAATGAGCGGTTC; Reverse primer: AGGTCTTTGCGGATGTCCACGT.
Hemin preparation
Hemin (heme [ferriprotoporphyrin IX chloride]; H9039, Sigma or H651–9, Frontier Scientific) 100 mM stock was prepared by dissolving with filter sterilized 0.1 M NaOH and neutralizing (to pH 7.2) with 1 M HCl, as previously described103. Stocks were freshly prepared before the experiment.
In vivo injection
LPS, 1 μg/g body weight (L2630, Sigma), and phenylhydrazine, 0.125 mg/g body weight (114715, Sigma), were used for in vivo experiments. Briefly, LPS was dissolved in sterile DPBS, aliquoted and stored in −80°C until used. Phenylhydrazine was weighed and dissolved in sterile DPBS; then the pH was adjusted to 7.4 using 2 M NaOH, and phenylhydrazine was filtered through a 0.22 μm syringe filter (SLGPR33RS, Millipore). Age- and sex-matched, 6- to 8-week-old cohoused mice of the indicated genotypes were injected intraperitoneally with 1 μg/g body weight LPS alone, 0.125 mg/g body weight phenylhydrazine alone or LPS with phenylhydrazine. For LPS-phenylhydrazine combination injections, LPS was added to sterile, filtered phenylhydrazine and mixed before injection.
Histopathology
For murine samples, kidneys were fixed in 10% formalin, then processed and embedded in paraffin by standard procedures. Sections (5 μM) were stained with hematoxylin and eosin (H&E) and examined by a pathologist blinded to the experimental groups. TUNEL (terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick-end labeling) staining was performed using the Dead-End kit (Promega, PRG7130) according to the manufacturer’s instructions. TUNEL stained sections of kidney were examined, and the number of TUNEL-positive cells in the cortex of the kidneys was manually counted. These sections were then scanned on a 3DHISTECH Panoramic 250 Flash III (Epredia) to create a digital whole slide image. The total area of kidney cortex on the slides was measured using the image analysis software Halo v3.5 (Indica Labs). The manually counted number of cells was divided by the area of the cortex to obtain the final TUNEL measurement as the number of positive cells per square cm.
Clinical chemistry analysis
Serum heme was detected using a heme assay kit (MAK316, Sigma). BUN (A11A01641), creatinine (A11A01933) and iron (A11A01637) were detected using ABX Pentra 400 Reagents (HORIBA) according to the manufacturer’s instructions. Serum hemoglobin was quantified using VetHemaChemRX (Oxford Science) according to the manufacturer’s instructions.
Quantification and Statistical Analysis
GraphPad Prism 9.0 software was used for in vitro and murine in vivo data analysis. Data are shown as mean ± SEM. Statistical significance was determined by t tests (two-tailed) for two groups, one- or two-way ANOVA (with Dunnett’s or Tukey’s multiple comparisons tests) for three or more groups or log-rank (Mantel-Cox) test for survival analysis. P value < 0.05 was considered statistically significant.
Supplementary Material
1
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-caspase-3 | Cell Signaling Technology | Cat# 9662; RRID:AB 331439 |
| anti-cleaved caspase-3 | Cell Signaling Technology | Cat# 9661; RRID:AB 2341188 |
| anti-caspase-7 | Cell Signaling Technology | Cat# 9492; RRID:AB 2228313 |
| anti-cleaved caspase-7 | Cell Signaling Technology | Cat# 9491; RRID:AB 2068144 |
| anti-caspase-8 | AdipoGen | Cat# AG-20T-0138; RRID:AB 2490519 |
| anti-cleaved caspase-8 | Cell Signaling Technology | Cat# 8592; RRID:AB 10891784 |
| anti-pMLKL | Cell Signaling Technology | Cat# 37333; RRID:AB 2799112 |
| anti-tMLKL | Abgent | Cat# AP14272b; RRID:AB 11134649 |
| anti-GSDMD | Abcam | Cat# ab209845; RRID:AB 2783550 |
| anti-GSDME | Abcam | Cat# ab215191; RRID:AB 2737000 |
| anti-caspase-1 | AdipoGen | Cat# AG-20B-0044; RRID:AB 2490253 |
| anti-IRF1 | Cell Signaling Technology | Cat# 8478; RRID:AB 10949108 |
| anti-p-ERK1/2 | Cell Signaling Technology | Cat# 9101; RRID:AB 331646 |
| anti-ERK1/2 | Cell Signaling Technology | Cat# 9102; RRID:AB 330744 |
| anti-p-IκBα | Cell Signaling Technology | Cat# 2859; RRID:AB 561111 |
| anti-IκBα | Cell Signaling Technology | Cat# 9242; RRID:AB 331623 |
| anti-p-p65 | Cell Signaling Technology | Cat# 3033; RRID:AB 331284 |
| anti-p65 | Cell Signaling Technology | Cat# 8242; RRID:AB 10859369 |
| anti-HO-1 | Cell Signaling Technology | Cat# 70081; RRID:AB 2799772 |
| anti-ASC for IF | Millipore | Cat# 04-147; RRID:AB 1977033 |
| Isotype Control antibody | Cell Signaling Technology | Cat# 3900S; RRID:AB 1550038 |
| anti-ASC (for IP) | Adipogen | Cat# AG-25B-0006; RRID:AB 2885200 |
| anti-NLRP12 (for IP) | Abcepta | Cat# AP14014a; RRID:AB 10893749 |
| anti-RIPK3 | Santa Cruz | Cat# sc-374639; RRID:AB 10332232 |
| anti-Myc | Cell Signaling Technology | Cat# 2276; RRID:AB 331783 |
| anti-NLRP3 | Adipogen | Cat# AG-20B-0014-C100; RRID:AB 2885199 |
| anti-FLAG | Cell Signaling Technology | Cat# 14793; RRID:AB 2572291 |
| anti-HA | Cell Signaling Technology | Cat# 2367; RRID:AB 10691311 |
| anti-human caspase-8 | Enzo Life Science | Cat# ALX-804-242; RRID:AB 2259459 |
| HRP-conjugated anti-GAPDH | Santa Cruz Biotechnology | Cat# sc-166574; RRID:AB 2107296 |
| HRP-conjugated anti-β-actin | Santa Cruz Biotechnology | Cat# sc-47778; RRID:AB 2714189 |
| HRP-conjugated secondary anti-rabbit | Jackson ImmunoResearch Laboratories | Cat# 111-035-047; RRID:AB 2337940 |
| HRP-conjugated secondary anti-mouse | Jackson ImmunoResearch Laboratories | Cat# 315-035-047; RRID:AB 2340068 |
| HRP-conjugated secondary anti-rat | Jackson ImmunoResearch Laboratories | Cat# 112-035-143; RRID:AB 2338138 |
| Alexa Fluor 488-conjugated anti-mouse IgG | Invitrogen | Cat# A11029; RRID:AB 2534088 |
| Biological Samples | ||
| PBMCs | This paper | N/A |
| HEK293T | ATCC | Cat# CRL-3216; RRID:CVCL 0063 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| IMDM | Thermo Fisher Scientific | Cat# 12440053 |
| RPMI 1640 | Corning | Cat# 10-040-CV |
| Fetal bovine serum | Biowest | Cat# S1620 |
| Non-essential amino acids | Thermo Fisher Scientific | Cat# 11140–050 |
| Penicillin and streptomycin | Thermo Fisher Scientific | Cat# 15070-063 |
| Lymphoprep™ | Stemcell Technologies | Cat# 07801/07811 |
| Human M-CSF | Peprotech | Cat# 300–25 |
| Human IFN-γ | Peprotech | Cat# 300–02 |
| DMEM | Gibco | Cat# 11995–065 |
| DPBS | Thermo Fisher Scientific | Cat# 14190–250 |
| Hemin | Sigma | Cat# H9039 |
| Hemin | Frontier Scientific | Cat# H651–9 |
| Pam3CSK4 | InvivoGen | Cat# tlrl-pms |
| Ultrapure LPS from E. coli 0111:B4 (in vitro) | InvivoGen | Cat# tlrl-3pelps |
| R848 | InvivoGen | Cat# tlrl-r848 |
| Poly(I:C) | InvivoGen | Cat# tlrl-picw |
| Monosodium urate (MSU) crystals | InvivoGen | Cat# tlrl-msu |
| Recombinant mouse S100A8/S100A9 heterodimer | bio-techne | Cat# 8916-S8–050 |
| Recombinant mouse HMGB1 | Abcam | Cat# ab255799 |
| TNF-α | Preprotech | Cat# 315-01A |
| Z-VAD(OMe)-FMK (zVAD) | Cayman Chemicals | Cat# 14463 |
| Z-VAD-FMK (for IP) | Selleckchem | Cat# S7023 |
| Necrostatin 2 racemate (Nec1) | Selleckchem | Cat# S8641 |
| N-acetyl L-cysteine (NAC) | Sigma | Cat# A9165 |
| Deferoxamine (DFO) | Cayman Chemicals | Cat# 14595 |
| Ferrostatin-1 (Fer1) | Cayman Chemicals | Cat# 17729 |
| Zinc (II) protoporphyrin IX | Sigma | Cat# 691550 |
| Propidium iodide (PI) | Life Technologies | Cat# P3566 |
| Paraformaldehyde | Electron Microscopy Sciences | Cat# 15710 |
| Triton X-100 | Sigma | Cat# T8787 |
| Normal goat serum | Vector Laboratories | Cat# S1000 |
| DAPI (4′,6-Diamidino-2-Phenylindole) | Biotium | Cat# 40043 |
| LPS (in vivo) | Sigma | Cat# L2630 |
| Phenylhydrazine | Sigma | Cat# 114715 |
| Protease inhibitor | Roche | Cat# 11697498001 |
| Phosphatase inhibitor | Roche | Cat# 04906837001 |
| Immobilon Forte western HRP substrate | Millipore | Cat# WBLUF0500 |
| SuperSignal™ West Femto Maximum Sensitivity Substrate | Thermo Fisher Scientific | Cat# 34096 |
| MitoSOX red dye | Thermo Fisher Scientific | Cat# M36008 |
| EDTA-free protease inhibitor cocktail | Thermo Fisher Scientific | Cat# A32965 |
| Phosphatase inhibitor cocktail | Sigma | Cat# 4906845001 |
| TRIzol | Thermo Fisher Scientific | Cat# 15596026 |
| SYBR Green | Applied Biosystems | Cat# 4368706 |
| Critical Commercial Assays | ||
| BUN assay | HORIBA | Cat# A11A01641 |
| Creatinine assay | HORIBA | Cat# A11A01933 |
| Iron assay | HORIBA | Cat# A11A01637 |
| Heme assay kit | Sigma | Cat# MAK316 |
| Serum hemoglobin assay | Oxford Science | Cat# VetHemaChemRx |
| Protein A/G magnetic beads | Thermo Fisher Scientific | Cat# 88802 |
| Multiplex ELISA | Millipore | Cat# MCYTOMAG-70K |
| IL-18 ELISA | Invitrogen | Cat# BMS618-3 |
| EasySep Direct Human Monocyte isolation kit | Stemcell Technologies | Cat# 19669 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat# 4368814 |
| DeadEnd™ Colorimetric TUNEL System | Promega | Cat# PRG7130 |
| Neon™ Transfection System kit | Thermo Fisher Scientific | Cat# MPK5000 |
| Deposited Data | ||
| Gene expression in patients with malaria | Brito et al., 2022 | GSE136046 |
| Gene expression in patients with malaria | Idaghdour et al., 2012 | GSE34404 |
| Gene expression in patients with sickle cell disease | Lagresle-Peyrou et al., 2018 | GSE102881 |
| Gene expression in patients with sickle cell disease | Liu et al., 2021 | GSE168532 |
| Single cell transcriptomics dataset from patients with hemolytic diseases | Hua et al., 2019 | GSE133181 |
| Gene expression in patients with COVID-19 | Levy et al., 2021 | GSE171110 |
| Gene expression in patients with Crimean-Congo hemorrhagic fever virus infection | Neogi et al., 2022 | Sequence Read Archive: PRJNA680886 |
| Transcriptomic profile from macaques infected with Marburg virus | Connor et al., 2015 | GSE58287 |
| Gene expression in patients with influenza A infection, bacterial pneumonia or systemic inflammatory response | Parnell et al., 2012 | GSE40012 |
| Experimental Models: Organisms/Strains | ||
| _Myd88_−/− mice | Adachi et al., 1998 | N/A |
| _Irf1_−/− mice | Matsuyama et al., 1993 | N/A |
| _Ripk3_−/− mice | Newton et al., 2004 | N/A |
| _Ripk3_−/−_Casp8_−/− mice | Oberst et al., 2011 | N/A |
| Ripk3 −/− Casp8 −/− Casp1 −/− | This paper | N/A |
| _Casp7_−/− mice | Lakhani et al., 2006 | N/A |
| _Casp3_−/− mice | Zheng et al., 2000 | N/A |
| _Gsdmd_−/− mice | Karki et al., 2018 | N/A |
| _Gsdme_−/− mice | Skarnes et al., 2011 | N/A |
| _Gsdmd_−/−_Gsdme_−/− mice | Karki et al., 2021 | N/A |
| _Casp1_−/− mice | Man et al., 2016 | N/A |
| _Pycard_−/− mice | Van Gorp et al., 2016 | N/A |
| _Nlrp3_−/− mice | Kanneganti et al., 2006 | N/A |
| _Nlrp12_−/− mice | Zaki et al., 2011 | N/A |
| _Nlrp6_−/− mice | Chen et al., 2011 | N/A |
| _Tlr2_−/− mice | Takeuchi et al., 1999 | N/A |
| _Tlr4_−/− mice | Hoshino et al., 1999 | N/A |
| _Tlr2_−/−_Tlr4_−/− mice | This paper | N/A |
| Oligonucleotides | ||
| Non-targeting (control) siRNA | Horizon Discovery | Cat# D-001206-14-20 |
| Human-specific NLRP12 siRNA | Horizon Discovery | Cat# m015092-00-0005 |
| pcDNA3-Myc-hASC | Addgene | Cat# 73952 |
| pEGFP-C2_-hNLRP3_ | Addgene | Cat# 73955 |
| pcDNA3-hCASP8 | Addgene | Cat# 11817 |
| pcDNA3-HA-hRIPK3 | Addgene | Cat# 78804 |
| pcDNA3-_hNLRP12_-FLAG | Thermo Fisher Scientific | Cat# V790-20 |
| Software and Algorithms | ||
| GraphPad Prism 9.0 | GraphPad Software, Inc. | https://www.graphpad.com/ |
| R package v4.1.1 | The Comprehensive R Archive Network | https://cran.r-proiect.org |
| Complex Heatmap v2.8.0 | Gu et al., 2016 | N/A |
| AUCell v1.18.0 package | Bioconductor | https://bioconductor.org |
| Slidebook | Intelligent Imaging | https://www.intelligent-imaging.com |
| Halo v3.5 | Indica Labs | https://learn.indicalab.com |
Highlights.
- Cytosolic sensor NLRP12 drives lytic cell death in response to heme plus PAMPs/TNF
- NLRP12 forms a PANoptosome with the inflammasome as an integral component
- Caspase-8/RIPK3 drives PANoptosis downstream of the NLRP12-PANoptosome
- NLRP12 is upregulated during hemolytic disease and induces pathology
ACKNOWLEDGEMENTS
We thank all the members of the Kanneganti laboratory for their comments and suggestions during the development of this manuscript. We also thank R. Tweedell, PhD, and J. Gullett, PhD, for scientific editing and writing support. Work from our laboratory is supported by the US National Institutes of Health (AI101935, AI124346, AI160179, AR056296 and CA253095 to T.-D.K.) and the American Lebanese Syrian Associated Charities (to T.-D.K.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
DECLARATION OF INTERESTS
T.-D.K. previously served as a consultant for Pfizer.
St. Jude Children’s Research hospital filed a provisional patent application on methods for modulating NLRP12 described in this study, listing B.S. and T.-D.K. as inventors (serial no. 63/422,601).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Janeway CA Jr. (1989). Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol 54 Pt 1, 1–13. 10.1101/sqb.1989.054.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Medzhitov R, and Janeway CA Jr. (1997). Innate immunity: impact on the adaptive immune response. Curr Opin Immunol 9, 4–9. 10.1016/s0952-7915(97)80152-5. [DOI] [PubMed] [Google Scholar]
- 3.Janeway CA Jr. (2013). Pillars article: approaching the asymptote? Evolution and revolution in immunology. Cold spring harb symp quant biol. 1989. 54: 1–13. J Immunol 191, 4475–4487. [PubMed] [Google Scholar]
- 4.Kanneganti TD (2020). Intracellular innate immune receptors: Life inside the cell. Immunol Rev 297, 5–12. 10.1111/imr.12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harton JA, Linhoff MW, Zhang J, and Ting JP (2002). Cutting edge: CATERPILLER: a large family of mammalian genes containing CARD, pyrin, nucleotide-binding, and leucine-rich repeat domains. J Immunol 169, 4088–4093. [DOI] [PubMed] [Google Scholar]
- 6.Inohara N, and Nuñez G (2003). NODs: intracellular proteins involved in inflammation and apoptosis. Nat Rev Immunol 3, 371–382. 10.1038/nri1086. [DOI] [PubMed] [Google Scholar]
- 7.Zheng M K. R; Vogel P; Kanneganti TD (2020). Caspase-6 is a key regulator of innate immunity, inflammasome activation and host defense. Cell 181, 674–687.e613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Christgen S, Zheng M, Kesavardhana S, Karki R, Malireddi RKS, Banoth B, Place DE, Briard B, Sharma BR, Tuladhar S, Samir P, Burton A, Kanneganti T-D (2020). Identification of the PANoptosome: A molecular platform triggering pyroptosis, apoptosis, and necroptosis (PANoptosis). Front Cell Infect Microbiol 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee S, Karki R, Wang Y, Nguyen LN, Kalathur RC, and Kanneganti TD (2021). AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence. Nature. 10.1038/s41586-021-03875-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malireddi RKS, Kesavardhana S, Karki R, Kancharana B, Burton AR, and Kanneganti TD (2020). RIPK1 Distinctly Regulates Yersinia-Induced Inflammatory Cell Death, PANoptosis. Immunohorizons 4, 789–796. 10.4049/immunohorizons.2000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amarante-Mendes GP, Adjemian S, Branco LM, Zanetti LC, Weinlich R, and Bortoluci KR (2018). Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front Immunol 9, 2379. 10.3389/fimmu.2018.02379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matzinger P (1994). Tolerance, danger, and the extended family. Annu Rev Immunol 12, 991–1045. 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 13.Seong SY, and Matzinger P (2004). Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol 4, 469–478. 10.1038/nri1372. [DOI] [PubMed] [Google Scholar]
- 14.Medzhitov R (2008). Origin and physiological roles of inflammation. Nature 454, 428–435. 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 15.Gozzelino R, Jeney V, and Soares MP (2010). Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol 50, 323–354. 10.1146/annurev.pharmtox.010909.105600. [DOI] [PubMed] [Google Scholar]
- 16.Martins R, and Knapp S (2018). Heme and hemolysis in innate immunity: adding insult to injury. Curr Opin Immunol 50, 14–20. 10.1016/j.coi.2017.10.005. [DOI] [PubMed] [Google Scholar]
- 17.Soares MP, and Bozza MT (2016). Red alert: labile heme is an alarmin. Curr Opin Immunol 38, 94–100. 10.1016/j.coi.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 18.Mogensen TH (2009). Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 22, 240–273, Table of Contents. 10.1128/cmr.00046-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fortes GB, Alves LS, de Oliveira R, Dutra FF, Rodrigues D, Fernandez PL, Souto-Padron T, De Rosa MJ, Kelliher M, Golenbock D, et al. (2012). Heme induces programmed necrosis on macrophages through autocrine TNF and ROS production. Blood 119, 2368–2375. 10.1182/blood-2011-08-375303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Q, Fu W, Yao J, Ji Z, Wang Y, Zhou Z, Yan J, and Li W (2014). Heme induces IL-1beta secretion through activating NLRP3 in kidney inflammation. Cell Biochem Biophys 69, 495–502. 10.1007/s12013-014-9823-9. [DOI] [PubMed] [Google Scholar]
- 21.Dutra FF, Alves LS, Rodrigues D, Fernandez PL, de Oliveira RB, Golenbock DT, Zamboni DS, and Bozza MT (2014). Hemolysis-induced lethality involves inflammasome activation by heme. Proc Natl Acad Sci U S A 111, E4110–4118. 10.1073/pnas.1405023111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, and Han J (2015). Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res 25, 1285–1298. 10.1038/cr.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671. 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 24.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, and Shao F (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Gao W, Shi X, Ding J, Liu W, He H, Wang K, and Shao F (2017). Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99–103. 10.1038/nature22393. [DOI] [PubMed] [Google Scholar]
- 26.Chen KW, Demarco B, Heilig R, Shkarina K, Boettcher A, Farady CJ, Pelczar P, and Broz P (2019). Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J 38. 10.15252/embj.2019101638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taabazuing CY, Okondo MC, and Bachovchin DA (2017). Pyroptosis and Apoptosis Pathways Engage in Bidirectional Crosstalk in Monocytes and Macrophages. Cell Chem Biol 24, 507–514 e504. 10.1016/j.chembiol.2017.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y, Karki R, Zheng M, Kancharana B, Lee S, Kesavardhana S, Hansen BS, Pruett-Miller SM, and Kanneganti TD (2021). Cutting Edge: Caspase-8 Is a Linchpin in Caspase-3 and Gasdermin D Activation to Control Cell Death, Cytokine Release, and Host Defense during Influenza A Virus Infection. J Immunol 207, 2411–2416. 10.4049/jimmunol.2100757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Idaghdour Y, Quinlan J, Goulet JP, Berghout J, Gbeha E, Bruat V, de Malliard T, Grenier JC, Gomez S, Gros P, et al. (2012). Evidence for additive and interaction effects of host genotype and infection in malaria. Proc Natl Acad Sci U S A 109, 16786–16793. 10.1073/pnas.1204945109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brito MAM, Baro B, Raiol TC, Ayllon-Hermida A, Safe IP, Deroost K, Figueiredo EFG, Costa AG, Armengol MDP, Sumoy L, et al. (2022). Morphological and Transcriptional Changes in Human Bone Marrow During Natural Plasmodium vivax Malaria Infections. J Infect Dis 225, 1274–1283. 10.1093/infdis/jiaa177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lagresle-Peyrou C, Lefrere F, Magrin E, Ribeil JA, Romano O, Weber L, Magnani A, Sadek H, Plantier C, Gabrion A, et al. (2018). Plerixafor enables safe, rapid, efficient mobilization of hematopoietic stem cells in sickle cell disease patients after exchange transfusion. Haematologica 103, 778–786. 10.3324/haematol.2017.184788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Y, Pal M, Bao W, Shi PA, Lobo CA, An X, Manwani D, Zhong H, and Yazdanbakhsh K (2021). Type I interferon is induced by hemolysis and drives antibody-mediated erythrophagocytosis in sickle cell disease. Blood 138, 1162–1171. 10.1182/blood.2021011629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seixas E, Gozzelino R, Chora A, Ferreira A, Silva G, Larsen R, Rebelo S, Penido C, Smith NR, Coutinho A, and Soares MP (2009). Heme oxygenase-1 affords protection against noncerebral forms of severe malaria. Proc Natl Acad Sci U S A 106, 15837–15842. 10.1073/pnas.0903419106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bradley JR (2008). TNF-mediated inflammatory disease. J Pathol 214, 149–160. 10.1002/path.2287. [DOI] [PubMed] [Google Scholar]
- 35.Vladimer GI, Weng D, Paquette SW, Vanaja SK, Rathinam VA, Aune MH, Conlon JE, Burbage JJ, Proulx MK, Liu Q, et al. (2012). The NLRP12 inflammasome recognizes Yersinia pestis. Immunity 37, 96–107. 10.1016/j.immuni.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ataide MA, Andrade WA, Zamboni DS, Wang D, Souza Mdo C, Franklin BS, Elian S, Martins FS, Pereira D, Reed G, et al. (2014). Malaria-induced NLRP12/NLRP3-dependent caspase-1 activation mediates inflammation and hypersensitivity to bacterial superinfection. PLoS Pathog 10, e1003885. 10.1371/journal.ppat.1003885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Allen IC, Wilson JE, Schneider M, Lich JD, Roberts RA, Arthur JC, Woodford RM, Davis BK, Uronis JM, Herfarth HH, et al. (2012). NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-κB signaling. Immunity 36, 742–754. 10.1016/j.immuni.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zaki MH, Man SM, Vogel P, Lamkanfi M, and Kanneganti TD (2014). Salmonella exploits NLRP12-dependent innate immune signaling to suppress host defenses during infection. Proc Natl Acad Sci U S A 111, 385–390. 10.1073/pnas.13176431111317643111 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lukens JR, Gurung P, Shaw PJ, Barr MJ, Zaki MH, Brown SA, Vogel P, Chi H, and Kanneganti TD (2015). The NLRP12 Sensor Negatively Regulates Autoinflammatory Disease by Modulating Interleukin-4 Production in T Cells. Immunity 42, 654–664. 10.1016/j.immuni.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zaki MH, Vogel P, Malireddi RK, Body-Malapel M, Anand PK, Bertin J, Green DR, Lamkanfi M, and Kanneganti TD (2011). The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell 20, 649–660. 10.1016/j.ccr.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hua P, Roy N, de la Fuente J, Wang G, Thongjuea S, Clark K, Roy A, Psaila B, Ashley N, Harrington Y, et al. (2019). Single-cell analysis of bone marrow-derived CD34+ cells from children with sickle cell disease and thalassemia. Blood 134, 2111–2115. 10.1182/blood.2019002301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lazarian G, Quinquenel A, Bellal M, Siavellis J, Jacquy C, Re D, Merabet F, Mekinian A, Braun T, Damaj G, et al. (2020). Autoimmune haemolytic anaemia associated with COVID-19 infection. Br J Haematol 190, 29–31. 10.1111/bjh.16794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.AbouYabis AN, and Bell GT (2021). Hemolytic Anemia Complicating COVID-19 Infection. J Hematol 10, 221–227. 10.14740/jh906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sato SB, Kawasaki K, and Ohnishi S (1983). Hemolytic activity of influenza virus hemagglutinin glycoproteins activated in mildly acidic environments. Proc Natl Acad Sci U S A 80, 3153–3157. 10.1073/pnas.80.11.3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang RT, Rott R, and Klenk HD (1981). Influenza viruses cause hemolysis and fusion of cells. Virology 110, 243–247. 10.1016/0042-6822(81)90030-1. [DOI] [PubMed] [Google Scholar]
- 46.Khan FY, and A.y M. (2009). Mycoplasma pneumoniae associated with severe autoimmune hemolytic anemia: case report and literature review. Braz J Infect Dis 13, 77–79. 10.1590/s1413-86702009000100018. [DOI] [PubMed] [Google Scholar]
- 47.Wang JL, Ho MY, and Shen EY (2004). Mycoplasma pneumoniae infection associated with hemolytic anemia--report of one case. Acta Paediatr Taiwan 45, 293–295. [PubMed] [Google Scholar]
- 48.Larsen R, Gozzelino R, Jeney V, Tokaji L, Bozza FA, Japiassu AM, Bonaparte D, Cavalcante MM, Chora A, Ferreira A, et al. (2010). A central role for free heme in the pathogenesis of severe sepsis. Sci Transl Med 2, 51ra71. 10.1126/scitranslmed.3001118. [DOI] [PubMed] [Google Scholar]
- 49.Meinders AJ, and Dijkstra I (2014). Massive hemolysis and erythrophagocytosis in severe sepsis. Blood 124, 841. 10.1182/blood-2014-04-565663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brightbill HD, Libraty DH, Krutzik SR, Yang RB, Belisle JT, Bleharski JR, Maitland M, Norgard MV, Plevy SE, Smale ST, et al. (1999). Host defense mechanisms triggered by microbial lipoproteins through toll-like receptors. Science 285, 732–736. 10.1126/science.285.5428.732. [DOI] [PubMed] [Google Scholar]
- 51.Aliprantis AO, Yang RB, Mark MR, Suggett S, Devaux B, Radolf JD, Klimpel GR, Godowski P, and Zychlinsky A (1999). Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science 285, 736–739. 10.1126/science.285.5428.736. [DOI] [PubMed] [Google Scholar]
- 52.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, et al. (1998). Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282, 2085–2088. 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 53.Qureshi ST, Lariviere L, Leveque G, Clermont S, Moore KJ, Gros P, and Malo D (1999). Endotoxin-tolerant mice have mutations in Toll-like receptor 4 (Tlr4). J Exp Med 189, 615–625. 10.1084/jem.189.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, Alves LS, Oliveira MF, Oliveira PL, Graca-Souza AV, and Bozza MT (2007). Characterization of heme as activator of Toll-like receptor 4. J Biol Chem 282, 20221–20229. 10.1074/jbc.M610737200. [DOI] [PubMed] [Google Scholar]
- 55.Min H, Choi B, Jang YH, Cho IH, and Lee SJ (2017). Heme molecule functions as an endogenous agonist of astrocyte TLR2 to contribute to secondary brain damage after intracerebral hemorrhage. Mol Brain 10, 27. 10.1186/s13041-017-0305-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deguine J, and Barton GM (2014). MyD88: a central player in innate immune signaling. F1000Prime Rep 6, 97. 10.12703/P6-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen ST, Chen L, Lin DS, Chen SY, Tsao YP, Guo H, Li FJ, Tseng WT, Tam JW, Chao CW, et al. (2019). NLRP12 Regulates Anti-viral RIG-I Activation via Interaction with TRIM25. Cell Host Microbe 25, 602–616 e607. 10.1016/j.chom.2019.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Senaldi G, Shaklee CL, Guo J, Martin L, Boone T, Mak TW, and Ulich TR (1999). Protection against the mortality associated with disease models mediated by TNF and IFN-gamma in mice lacking IFN regulatory factor-1. J Immunol 163, 6820–6826. [PubMed] [Google Scholar]
- 59.Tan RS, Feng C, Asano Y, and Kara AU (1999). Altered immune response of interferon regulatory factor 1-deficient mice against Plasmodium berghei blood-stage malaria infection. Infect Immun 67, 2277–2283. 10.1128/IAI.67.5.2277-2283.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pamplona A, Ferreira A, Balla J, Jeney V, Balla G, Epiphanio S, Chora A, Rodrigues CD, Gregoire IP, Cunha-Rodrigues M, et al. (2007). Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat Med 13, 703–710. 10.1038/nm1586. [DOI] [PubMed] [Google Scholar]
- 61.Ezerina D, Takano Y, Hanaoka K, Urano Y, and Dick TP (2018). N-Acetyl Cysteine Functions as a Fast-Acting Antioxidant by Triggering Intracellular H(2)S and Sulfane Sulfur Production. Cell Chem Biol 25, 447–459 e444. 10.1016/j.chembiol.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Beutler E (1969). Drug-induced hemolytic anemia. Pharmacol Rev 21, 73–103. [PubMed] [Google Scholar]
- 63.Ramos S, Carlos AR, Sundaram B, Jeney V, Ribeiro A, Gozzelino R, Bank C, Gjini E, Braza F, Martins R, et al. (2019). Renal control of disease tolerance to malaria. Proc Natl Acad Sci U S A 116, 5681–5686. 10.1073/pnas.1822024116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vermeulen Windsant IC, Snoeijs MG, Hanssen SJ, Altintas S, Heijmans JH, Koeppel TA, Schurink GW, Buurman WA, and Jacobs MJ (2010). Hemolysis is associated with acute kidney injury during major aortic surgery. Kidney Int 77, 913–920. 10.1038/ki.2010.24. [DOI] [PubMed] [Google Scholar]
- 65.Borghini S, Tassi S, Chiesa S, Caroli F, Carta S, Caorsi R, Fiore M, Delfino L, Lasigliè D, Ferraris C, et al. (2011). Clinical presentation and pathogenesis of cold-induced autoinflammatory disease in a family with recurrence of an NLRP12 mutation. Arthritis Rheum 63, 830–839. 10.1002/art.30170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jéru I, Duquesnoy P Fau - Fernandes-Alnemri T, Fernandes-Alnemri T Fau - Cochet E, Cochet E Fau - Yu JW, Yu Jw Fau - Lackmy-Port-Lis M, Lackmy-Port-Lis M Fau - Grimprel E, Grimprel E Fau - Landman-Parker J, Landman-Parker J Fau - Hentgen V, Hentgen V Fau - Marlin S, Marlin S Fau - McElreavey K, et al. (2008). Mutations in NALP12 cause hereditary periodic fever syndromes. Proc Natl Acad Sci U S A 105, 1614–1619. 10.1073/pnas.0708616105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, and Tschopp J (2007). Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ 14, 1583–1589. 10.1038/sj.cdd.4402195. [DOI] [PubMed] [Google Scholar]
- 68.Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, and Núñez G (2013). K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153. 10.1016/j.immuni.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Groß CJ, Mishra R, Schneider KS, Médard G, Wettmarshausen J, Dittlein DC, Shi H, Gorka O, Koenig PA, Fromm S, et al. (2016). K(+) Efflux-Independent NLRP3 Inflammasome Activation by Small Molecules Targeting Mitochondria. Immunity 45, 761–773. 10.1016/j.immuni.2016.08.010. [DOI] [PubMed] [Google Scholar]
- 70.Chen J, and Chen ZJ (2018). PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 564, 71–76. 10.1038/s41586-018-0761-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, and Horng T (2012). Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci U S A 109, 11282–11287. 10.1073/pnas.1117765109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee GS, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, Germain RN, Kastner DL, and Chae JJ (2012). The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 492, 123–127. 10.1038/nature11588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xu H, Shi J, Gao H, Liu Y, Yang Z, Shao F, and Dong N (2019). The N-end rule ubiquitin ligase UBR2 mediates NLRP1B inflammasome activation by anthrax lethal toxin. EMBO J. 10.15252/embj.2019101996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chui AJ, Okondo MC, Rao SD, Gai K, Griswold AR, Johnson DC, Ball DP, Taabazuing CY, Orth EL, Vittimberga BA, and Bachovchin DA (2019). N-terminal degradation activates the NLRP1B inflammasome. Science 364, 82–85. 10.1126/science.aau1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sandstrom A, Mitchell PS, Goers L, Mu EW, Lesser CF, and Vance RE (2019). Functional degradation: A mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science 364. 10.1126/science.aau1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gai K, Okondo MC, Rao SD, Chui AJ, Ball DP, Johnson DC, and Bachovchin DA (2019). DPP8/9 inhibitors are universal activators of functional NLRP1 alleles. Cell Death Dis 10, 587. 10.1038/s41419-019-1817-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ball DP, Tsamouri LP, Wang AE, Huang HC, Warren CD, Wang Q, Edmondson IH, Griswold AR, Rao SD, Johnson DC, and Bachovchin DA (2022). Oxidized thioredoxin-1 restrains the NLRP1 inflammasome. Sci Immunol 7, eabm7200. 10.1126/sciimmunol.abm7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hollingsworth LR, Sharif H, Griswold AR, Fontana P, Mintseris J, Dagbay KB, Paulo JA, Gygi SP, Bachovchin DA, and Wu H (2021). DPP9 sequesters the C terminus of NLRP1 to repress inflammasome activation. Nature 592, 778–783. 10.1038/s41586-021-03350-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jenster LM, Lange KE, Normann S, vom Hemdt A, Wuerth JD, Schiffelers LDJ, Tesfamariam YM, Gohr FN, Klein L, Kaltheuner IH, et al. (2023). P38 kinases mediate NLRP1 inflammasome activation after ribotoxic stress response and virus infection. J Exp Med 220. 10.1084/jem.20220837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Man SM, Karki R, Sasai M, Place DE, Kesavardhana S, Temirov J, Frase S, Zhu Q, Malireddi RKS, Kuriakose T, et al. (2016). IRGB10 Liberates Bacterial Ligands for Sensing by the AIM2 and Caspase-11-NLRP3 Inflammasomes. Cell 167, 382–396 e317. 10.1016/j.cell.2016.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zheng TS, Hunot S, Kuida K, Momoi T, Srinivasan A, Nicholson DW, Lazebnik Y, and Flavell RA (2000). Deficiency in caspase-9 or caspase-3 induces compensatory caspase activation. Nat Med 6, 1241–1247. 10.1038/81343. [DOI] [PubMed] [Google Scholar]
- 82.Lakhani SA, Masud A, Kuida K, Porter GA Jr., Booth CJ, Mehal WZ, Inayat I, and Flavell RA (2006). Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science 311, 847–851. 10.1126/science.1115035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Newton K, Sun X, and Dixit VM (2004). Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol 24, 1464–1469. 10.1128/MCB.24.4.1464-1469.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Van Gorp H, Saavedra PH, de Vasconcelos NM, Van Opdenbosch N, Vande Walle L, Matusiak M, Prencipe G, Insalaco A, Van Hauwermeiren F, Demon D, et al. (2016). Familial Mediterranean fever mutations lift the obligatory requirement for microtubules in Pyrin inflammasome activation. Proc Natl Acad Sci U S A 113, 14384–14389. 10.1073/pnas.1613156113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kanneganti TD, Ozoren N, Body-Malapel M, Amer A, Park JH, Franchi L, Whitfield J, Barchet W, Colonna M, Vandenabeele P, et al. (2006). Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 440, 233–236. 10.1038/nature04517. [DOI] [PubMed] [Google Scholar]
- 86.Chen GY, Liu M, Wang F, Bertin J, and Núñez G (2011). A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J Immunol 186, 7187–7194. 10.4049/jimmunol.1100412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Matsuyama T, Kimura T, Kitagawa M, Pfeffer K, Kawakami T, Watanabe N, Kundig TM, Amakawa R, Kishihara K, Wakeham A, and et al. (1993). Targeted disruption of IRF-1 or IRF-2 results in abnormal type I IFN gene induction and aberrant lymphocyte development. Cell 75, 83–97. [PubMed] [Google Scholar]
- 88.Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, and Green DR (2011). Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367. 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, and Akira S (1999). Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 11, 443–451. 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 90.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, and Akira S (1999). Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol 162, 3749–3752. [PubMed] [Google Scholar]
- 91.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, and Akira S (1998). Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 9, 143–150. 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 92.Karki R, Lee E, Place D, Samir P, Mavuluri J, Sharma BR, Balakrishnan A, Malireddi RKS, Geiger R, Zhu Q, et al. (2018). IRF8 Regulates Transcription of Naips for NLRC4 Inflammasome Activation. Cell 173, 920–933 e913. 10.1016/j.cell.2018.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, Mujica AO, Thomas M, Harrow J, Cox T, et al. (2011). A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474, 337–342. 10.1038/nature10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Karki R, Sundaram B, Sharma BR, Lee S, Malireddi RKS, Nguyen LN, Christgen S, Zheng M, Wang Y, Samir P, et al. (2021). ADAR1 restricts ZBP1-mediated immune response and PANoptosis to promote tumorigenesis. Cell Rep 37, 109858. 10.1016/j.celrep.2021.109858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Connor JH, Yen J, Caballero IS, Garamszegi S, Malhotra S, Lin K, Hensley L, and Goff AJ (2015). Transcriptional Profiling of the Immune Response to Marburg Virus Infection. J Virol 89, 9865–9874. 10.1128/JVI.01142-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Parnell GP, McLean AS, Booth DR, Armstrong NJ, Nalos M, Huang SJ, Manak J, Tang W, Tam OY, Chan S, and Tang BM (2012). A distinct influenza infection signature in the blood transcriptome of patients with severe community-acquired pneumonia. Crit Care 16, R157. 10.1186/cc11477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Levy Y, Wiedemann A, Hejblum BP, Durand M, Lefebvre C, Surenaud M, Lacabaratz C, Perreau M, Foucat E, Dechenaud M, et al. (2021). CD177, a specific marker of neutrophil activation, is associated with coronavirus disease 2019 severity and death. iScience 24, 102711. 10.1016/j.isci.2021.102711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Neogi U, Elaldi N, Appelberg S, Ambikan A, Kennedy E, Dowall S, Bagci BK, Gupta S, Rodriguez JE, Svensson-Akusjarvi S, et al. (2022). Multi-omics insights into host-viral response and pathogenesis in Crimean-Congo hemorrhagic fever viruses for novel therapeutic target. Elife 11. 10.7554/eLife.76071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Benjamini Y, and Hochberg Y (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol 57, 289–300. [Google Scholar]
- 101.Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 102.Hettmansperger TP, and McKean JW (2010). Robust Nonparametric Statistical Methods. 10.1201/b10451. [DOI] [Google Scholar]
- 103.Rossi M, Delbauve S, Wespes E, Roumeguere T, Leo O, Flamand V, Le Moine A, and Hougardy JM (2018). Dual effect of hemin on renal ischemia-reperfusion injury. Biochem Biophys Res Commun 503, 2820–2825. 10.1016/j.bbrc.2018.08.046. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1
Data Availability Statement
- The datasets generated and analyzed during the current study are contained within the manuscript and accompanying extended data figures and tables, and publicly available datasets analyzed can be found in the Gene Expression Omnibus database (GSE133181, GSE34404, GSE136046, GSE102881, GSE168532, GSE58287, GSE40012, GSE171110). Data for patients with CCHFV was obtained from Sequence Read Archive (Accession: PRJNA680886).
- This paper does not report original code.
- Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.