Enhancer Identification through Comparative Genomics (original) (raw)
. Author manuscript; available in PMC: 2008 Feb 1.
Published in final edited form as: Semin Cell Dev Biol. 2007 Jan 5;18(1):140–152. doi: 10.1016/j.semcdb.2006.12.014
Abstract
With the availability of genomic sequence from numerous vertebrates, a paradigm shift has occurred in the identification of distant-acting gene regulatory elements. In contrast to traditional gene-centric studies in which investigators randomly scanned genomic fragments that flank genes of interest in functional assays, the modern approach begins electronically with publicly available comparative sequence datasets that provide investigators with prioritized lists of putative functional sequences based on their evolutionary conservation. However, although a large number of tools and resources are now available, application of comparative genomic approaches remains far from trivial. In particular, it requires users to dynamically consider the species and methods for comparison depending on the specific biological question under investigation. While there is currently no single general rule to this end, it is clear that when applied appropriately, comparative genomic approaches exponentially increase our power in generating biological hypotheses for subsequent experimental testing. It is anticipated that cardiac-related genes and the identification of their distant-acting transcriptional enhancers are particularly poised to benefit from these modern capabilities.
Keywords: cis-regulatory, comparative genomics, enhancer, review, transgenic
1. Introduction
One of the most intriguing features of biology is the identical DNA content across all cells within an organism and yet the ability of this genetic information to dictate the enormous cellular diversity within the body. Rather, cell type complexity arises predominantly from vast temporal and spatial differences in gene expression during development. The principal mechanism underlying this gene expression diversity across cell types is dynamic gene regulation induced by a variety of interacting transcription factors which are also encoded by our genome and subject to tight regulation [1–3]. Transcription factors recognize specific target sequences located within gene promoters and/or more distant acting _cis-_regulatory regions, and function to either enhance or repress a given gene’s cellular expression. Through this highly orchestrated process, higher organisms have been able to evolve beyond the limitations of unicellularity to create complex forms and functions, including the development of the cardiovascular system.
Insights into this complexity are beginning to emerge for the human genome with the availability of a complete genomic sequence template [4,5]. This starting point has led to the identification of the ~25,000 genes in the human genome, albeit work remains to be done in deciphering all of their functions. Gene identification was greatly facilitated by having access to protein sequence databases and “expressed sequence tags” where computational algorithms for gene identification could subsequently be built based upon knowledge gained from these experimental datasets. In contrast, the availability of the human genome sequence alone provided no additional clues as to the precise locations of distant-acting gene enhancers. Challenges included the large noncoding search space in the human genome (~98% of 3x109 bp), the small size and degenerate nature of transcription factor binding sites, and most importantly the lack of experimental training sets for computational methods to identify such sequences in a global manner. The recent determination of additional genome sequences from other vertebrates has proven to be powerful at identifying the location of candidate distant-acting _cis_-regulatory elements based on their evolutionary conservation across appropriately distanced species.
In this review, we describe the use of comparative genomics as an increasingly powerful strategy for sequence-based enhancer identification. In particular, we provide an overview of selected computational tools and resources that are useful for the identification of enhancers involved in development and/or specific gene function. We end by highlighting the challenges arising from the identification of large numbers of putative enhancers through comparative genomics and the need to develop high throughput functional assays to determine their spatiotemporal in vivo activity at a genomic scale.
2. Role of Noncoding Sequences in Development and Human Disease
Traditionally, most studies of the genetic networks underlying vertebrate development have focused on the proteins that are involved, since they are – compared to regulatory sequences – generally easier to identify and more readily accessible to a variety of experimental methods. However, these proteins are generally limited to functional activity only in tissues where they are expressed, thereby stressing the importance of understanding the intricacies of gene regulation to comprehend regulatory networks in their entirety. In this section, we provide a brief overview of insights gained from gene-centric in-depth studies. While the list of examples described here is by no means exhaustive, it illustrates some of the major properties and characteristics of distant-acting _cis-_regulatory elements and exemplifies their important role in vertebrate development and human disease.
2.1 Modularity of Transcriptional Regulation by Enhancers
A characteristic feature of enhancers is the modular mode by which they regulate gene expression. One of many insightful examples for these properties can be obtained by examination of the human apolipoprotein E (APOE) locus. At least six distinct sequence elements flanking this gene control different aspects of APOE expression. Namely, the enhancement of kidney expression has been ascribed to the promoter [6], while elements located downstream of the gene include two liver-specific enhancers [7,8], a skin enhancer [6,9], two multiple tissue enhancers directing gene expression to adipocytes, macrophages and brain astrocytes [9,10], and a distal brain-specific enhancer [11]. It is worth noting that each of these discrete elements are on the order of several hundred basepairs in length and are scattered across 42 kilobases. A second example where the modularity of transcriptional regulation has been experimentally studied in great detail is the cardiac homeobox gene Nkx2-5 (Csx). This gene is required for heart development [12] and series of deletions and transgenic reporter experiments were used to dissect both its proximal and distal regulatory regions [13–18]. These studies revealed that at least five distinct elements target Nkx2-5 gene expression to specific sub-regions of the developing heart as well as to non-cardiac tissues and it has been suggested that this regulatory complexity played a important role in the evolution of the multi-chambered mammalian heart [19]. Thus, modular transcriptional regulation appears to be a common mechanism of complex gene regulation and a number of gene-centric studies beyond the selected examples of APOE and Nkx2-5 have further supported the concept that the complex expression patterns of genes across tissues regularly arise from the combined activity of multiple elements.
2.2 Spatiotemporal Precision of Developmental Enhancers
Another remarkable feature of enhancers is the high spatiotemporal precision with which they regulate gene expression. One example of the tight restriction of the timing and tissue-specificity of enhancer activity during embryonic development is the Hoxd11 locus. Deletion of a single Hoxd11 regulatory element in mice delays expression of both Hoxd10 and Hoxd11 during somitogenesis, but at later stages normal expression of Hoxd10 and Hoxd11 is restored [20]. It is hypothesized that this partial gene expression rescue is mediated by complementary regulatory elements present in this region. Since only a subset of anatomical regions lack Hoxd11 expression temporally, this gene regulatory deletion results in vertebral patterning and specification defects but of lesser severity than complete Hoxd11 gene knockouts.
The Hoxd11 locus thus demonstrates how a single enhancer regulates a relatively subtle, yet functionally important spatiotemporal sub-aspect of the expression pattern of a key developmental gene. The general picture emerging from this and other similar gene-centric studies is that the high spatiotemporal precision of single enhancers – in combination with their modular mode of action – has allowed complex gene expression patterns to evolve. This is particularly the case for many developmentally important genes, whose expression patterns appear to be frequently the result of the orchestrated activity of several different enhancers with distinct spatiotemporal activity patterns. Importantly, these single elements tend to be more restricted in their tissue specificity than the mRNA expression patterns to which they contribute, providing researchers with reagents for tissue-specific targeting of gene expression.
2.3 Enhancers are Required for Vertebrate Development
Like mutations in the protein-coding portion of genes, deletions or mutations of regulatory elements can result in developmental defects, such as in the Hoxd11 locus (see section 2.2). Another example from the Hox gene family is the 200bp “early enhancer” (EE) of the Hoxc8 gene. Deletion of this enhancer results in delayed expression of the Hoxc8 protein and in skeletal defects that recapitulate aspects of the Hoxc8_−/_− phenotype [21], demonstrating that this regulatory element is required for normal embryogenesis. As a third example, deletion of three brain-specific enhancers of Otx2 [22,23] revealed that they are required for maintaining normal expression levels of Otx2 in the developing brain. While deletion of these enhancers did not result in obvious phenotypes, compound heterozygous embryos in which one Otx2 allele was null and the other allele was an Otx2 enhancer deletion displayed defects in brain development. These results support that while each of these elements is not absolutely required for viability, they play an important role in embryonic development through their coordinated and quantitative effects on gene expression.
Of note, defects resulting from deletion or mutation of regulatory elements are usually restricted to the tissue in which they drive expression. This property can be exploited to study gene functions that are otherwise difficult to assess experimentally. For example, the role of Hand2 in craniofacial development cannot be studied by targeted deletion of the gene itself because _Hand2_−/− embryos die from cardiac abnormalities before the differentiation of craniofacial features. However, deletion of a branchial arch-specific Hand2 enhancer in mice results in craniofacial defects including cleft palate and mandibular hypoplasia, demonstrating a role both for this enhancer and the Hand2 gene in craniofacial development [24]. These studies allowed for the dissection of the regulatory architecture of this locus through the separate assessment of the roles of this gene in cardiac and craniofacial development. Another important possibility arising from the identification of tissue-specific enhancers is the possibility to use them to drive the expression of Cre recombinase. Such constructs can be used to generate tissue-specific knockouts by introducing flanking LoxP sites to the gene of interest [25]. For example, the conditional Cre/Lox-mediated deletion of Mef2c using a myocardial-specific enhancer has been used to examine the role of Mef2c beyond developmental stages at which mice with a complete deletion of Mef2c die from cardiovascular defects [26]. Thus, even in cases where the deletion of an enhancer is insufficient to abolish gene expression in a particular tissue, the enhancer can be used to study the function of the respective gene in a tissue-specific manner.
Indeed, many enhancers do not cause an overt phenotype beyond changes in expression levels of the target gene when experimentally deleted in mice. Examples include tissue- or cell type-specific enhancers for Engrailed2 [27], Fgf4 [28], Gata1 [29] or MyoD [30]. An obvious explanation for the frequent absence of phenotypes in enhancer deletion experiments is that often only one aspect of a complex endogenous mRNA expression pattern is affected, while expression of the gene in other tissues or at other stages is maintained. This higher spatiotemporal restriction is therefore expected to result in generally milder effects than deletion of entire genes. A second explanation is functional redundancy, which might be more common among regulatory elements than it is among protein-coding genes. While being sufficient to drive expression in reporter assays, many enhancers could be dispensable for normal development and physiology because their function is complemented by other regulatory elements with similar tissue specificity. Such redundancy of regulatory elements has, for instance, been directly shown for the TCR-gamma locus, where a deletion of two enhancers results in severe reduction in gamma-delta-thymocytes, whereas single deletion of either element did not cause a major immunological phenotype [31]. Functional redundancy does not imply that these enhancers are functionally less important and that their deletion does not reduce reproductive fitness. Rather it indicates that many enhancers are involved in fine-tuning gene expression. These findings also raise the possibility that functional redundancies are a factor in the comparative studies described below, since they might result in reduced evolutionary conservation of such elements.
2.4 Enhancers Contribute to Human Disease
As a result of our limited knowledge about the location of most enhancers in the genome, the contribution of distant acting mutations to human disease has so far not been explored on a large scale. One of the few known examples is the limb-specific ZRS long-distance enhancer of Sonic hedgehog (SHH). This element is located at the extreme distance of one megabase from the gene it regulates, residing in the intron of a neighboring gene. Genetic lesions affecting this element cause polydactyly both in human individuals and in mutant mouse strains, demonstrating the crucial role of enhancers during mammalian development [32]. Elimination of the conserved intronic region in which this enhancer is embedded results in severe limb truncations in mice, strongly supporting human disease studies [33]. Even point mutations in this regulatory element cause human preaxial polydactyly [34], offering an explanation why many enhancers are highly constrained and therefore often conserved across long evolutionary distances. While hundreds of regulatory mutations contributing to human disease have been reported [35], most of them affect promoter regions whose precise location is known for many human genes. It is expected that with growing numbers of identified human enhancers it will become possible to target systematic screens increasingly for regulatory mutations in this distant-acting class of gene regulatory elements.
2.5 Challenges
The selected examples above highlight the important role of enhancers in development and disease. However, it must be emphasized that the vast majority of distant-acting regulatory sequences in the mammalian genome has so far not been experimentally characterized either in vitro or in vivo and their overall contribution to human disease remains unclear. Two major challenges have rendered large-scale studies of developmental enhancers difficult. First, the absence of suitable prediction methods continues to present a major obstacle for identifying the location of these elements, especially for those that act over long distances. Second, the limited number of known developmental enhancers has largely prevented prediction by computational analysis because no suitable training sets of enhancers characterized by standardized experimental methods have been available. In consequence, our understanding of the sequence features involved in enhancer function remains limited to gene-centric studies and single elements. In the next sections, we will describe recent efforts to tackle both of these problems. Namely, recently developed methods and computational tools for comparative genomics have significantly improved our ability to identify the location of putative enhancers in the human genome and provide a starting point for large-scale experimental characterization of enhancers.
3. Enhancer Identification by Comparative Genomic Strategies
Cross-species sequence comparisons were shown to be an efficient approach to identify putative functional regions in noncoding DNA even before whole genome sequences of humans and other vertebrates became available. Many variations on this theme have been presented, including variation of the species being compared and different comparison methods, yet they all rely on the same basic principle: that functionally relevant sequences are under purifying selection, whereas non-functional regions are subject to genetic drift and become increasingly different between species with increasing phylogenetic distance. As a result, functional sequences generally stand out as more “conserved” than non-functional sequences when genomic sequences of different species are compared. Sequence conservation between different species can thus be used to identify putative functional regions, and many of these will be _cis_-regulatory elements.
3.1 Pre-Genome-Scale Comparative Approaches
Bottom-up approaches provided the early foundation for the utility of cross-species comparisons for the identification of _cis-_regulatory elements in the genomic sequence of a gene of interest (for early examples, see references 36,37). In the absence of publicly available whole-genome sequence data and specialized computational tools for these purposes, this strategy usually included cloning and sequencing of orthologous noncoding sequences from two or more organisms, manual alignment and identification of conserved regions at the nucleotide level, often focusing on transcription factor binding sites. In reference to experimentally exploring these sequences through DNase footprinting, such approaches became known as “phylogenetic footprinting”.
Such gene-centric studies provided an important proof of principle, but the hypothesis that sequence conservation is a universal predictor of noncoding regulatory sequences was difficult to verify conclusively in the absence of sequence data for genome-wide comparisons. Thus, the prospect of genome-wide comparative identification of _cis_-regulatory regions was early recognized as an important motivation to sequence the genomes of the mouse and other vertebrates in addition to the human genome [38,39].
3.2 Using Genomic Data in Comparative Approaches
Even before sufficient sequence data for whole-genome comparisons became available, the merits of comparative approaches for enhancer identification were confirmed in studies that involved the sequencing of large genomic intervals. For example, Göttgens et al. [40] sequenced a 320kb interval of the stem cell leukemia (SCL) locus in human, mouse and chicken to identify regulatory candidate regions. A subset of these regions corresponded to known regulatory elements and functional testing of previously uncharacterized conservation peaks led to the discovery of a new neural enhancer in the SCL locus. In another study, Loots et al. [41] identified multiple noncoding elements regulating the human interleukin-4, -5, and -13 genes by sequencing and aligning one megabase of human chromosome 5 and the orthologous mouse genome region. These results lent further support to the notion that conservation of noncoding sequences can be used to predict functional regions including regulatory elements in genomic sequence data.
The publication of the mouse and the pufferfish genomes in 2002 marked the kick-off for genome-wide comparative approaches since they allowed for the first time systematic large-scale comparisons of the human with non-human vertebrate genomes [42,43]. Comparative analysis of the human and mouse genomes was particularly productive because their size is similar, 90% of these genomes are organized in syntenic blocks in which the respective order of genes is maintained, and in an initial analysis 40% of the two genomes were found to be alignable at the nucleotide level. Interestingly, while only ~1.5% of the human and mouse genome encode proteins, ~5% of these mammalian genomes were estimated to be under purifying selection, suggesting that much more than protein encoding functions are constrained within our genome [43]. However, a multitude of functions can potentially be embedded into non-protein-coding DNA, including activating and repressing regulatory binding sites, known and unknown functional RNA types, and structural chromatin features. Most of these cannot be reliably predicted by existing computational methods, therefore the functional relevance of constrained noncoding regions remained initially obscure.
Subsequent functional testing of such conserved regions revealed, however, that one of the predominant functions of constrained noncoding DNA seems in fact to be the tissue-specific spatial and temporal regulation of gene expression. One of the likely reasons for this is the large size of many enhancer sequences, conserved over hundreds of basepairs, which makes it possible to identify them through whole genome comparisons. In what follows, we provide an overview of comparative strategies that have so far been successfully used to find such _cis-_regulatory elements (for a more detailed discussion of general considerations regarding comparisons over different evolutionary distances, including the advantages and limitations of distant and close comparisons, see reference 44).
3.2.1 Deep Comparisons: Human-Fish
In the pre-genomic era, studies focusing on single genes suggested that distant evolutionary comparison could be useful to identify regulatory regions involved in core aspects of vertebrate development. For example over 10 years ago, Aparicio et al. [45] used comparisons between mouse and pufferfish (Takifugu rubripes) to identify functional regulatory elements in the Hoxb4 locus based on noncoding conservation. These and other results demonstrated that deep comparisons are an efficient tool for enhancer prediction, but genome-wide application was not possible at the time since none of these vertebrate genome sequences were available.
A more recent study systematically exploited the remarkable potential of such distant vertebrate sequence comparisons to identify gene enhancers at the scale of larger genomic intervals [46]. In this work, the gene-sparse regions surrounding the human DACH locus were scanned for sequences that are not only highly conserved among mammals, but also had considerable sequence conservation in Xenopus as well as in pufferfish. Using an in vivo enhancer assay, these extremely conserved regions were found to be highly enriched for enhancers that drive tissue-specific gene transcription during embryogenesis. In fact, many of the conserved elements that are currently being tested in a large-scale transgenic in vivo screen in our laboratory (see section 4.3) were identified using human-fish conservation.
There are, however, several important limitations to distant comparative approaches. First, their high specificity is accompanied by moderate sensitivity. Depending on the alignment method, the comparative strategy, and the stringency of the applied filters, previously reported numbers of conserved non-coding elements identified by human-fish comparisons vary between 1,400 [47] and 5,700 [48]. Compared to estimates of the total number of protein-coding genes in the human genome [49], this is up to an order of magnitude lower, suggesting that many regulatory regions are missed by such distant comparisons. Second, to aggravate this problem, many elements with such extremely deep conservation occur in clusters around genes implicated in transcriptional regulation and development (trans-dev genes). For example, 85% of the 1,400 human-fish CNSs described by Woolfe et al. [47] are found in clusters of five or more elements. In total, only 165 distinct clusters were identified and 93% of these clusters are associated with trans-dev genes. In contrast, the majority of genes with other functions are not associated with any deeply conserved elements, despite modular regulation of gene expression in time and space. Third, extremely distant comparisons are expected to identify predominantly regulatory elements that are involved in molecular, developmental or physiological mechanisms that exist in both species under consideration, thereby explaining why they are anciently conserved. Human-fish comparisons would therefore be of limited utility for studies of enhancers that are involved in mammalian-specific developmental processes. As an example, we performed comparative analysis retrospectively on a subset of heart-specific _cis_-regulatory sequences originally identified through functional studies. These elements drive gene expression in the anterior heart field, a transient developmental structure, and heart regions derived from it [50]. The vast majority lacked conservation outside of mammals, which may be partially due to differences in heart development between mammals and non-mammalian vertebrates (Fig. 1B).
Fig. 1. Trade offs in comparative genomics of noncoding DNA based on different phylogenetic distances.
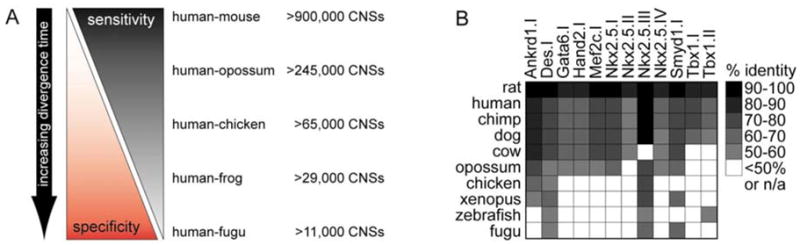
A) With simple definitions of CNSs, conservation depth can be used to calibrate specificity vs. sensitivity in comparative enhancer prediction. Closer sequence comparisons such as human-mouse provide a significant amount of noncoding conservation which provides strong sensitivity to identify known putative function, but at the cost of poor specificities. In contrast, human-fish comparison yields relatively little noncoding conservation and hence poor sensitivity to identify putative function, but with strong specificities for those conserved elements it does identify. B) Known heart enhancers lack deep sequence conservation. In this illustrative example, retrospective comparative analysis of twelve known heart-specific _cis_-regulatory elements in eleven vertebrate genomes reveals limited sensitivity of deep comparisons for detecting mammalian heart-specific enhancers (% identity refers to mouse as the base genome). Most of these elements are only minimally conserved beyond mammals and would have been missed by human-fish comparisons. These data indicate that biological context is an important factor for comparative-based approaches, though on occasion heart enhancers are anciently conserved to fish. For detailed description and experimental characterization of these elements, see references 13,15,18,51–57.
3.2.2 Extreme Conservation within Mammals
If conventional comparative criteria such as 70% identity over at least 100bp are used, human-rodent comparisons are of limited use for identification of enhancer elements. This is due to the fact that these two species share a relatively short divergence time since their last common ancestor which results in their high overall similarity even in nonfunctional genome regions. This results in the identification of an excess of elements as illustrated by the observation that ~40% of the human and mouse genome are alignable, yet only ~5% of the human genome are estimated to be under purifying selection [43]. In consequence, using human-mouse comparisons with relatively relaxed percent identity parameters for enhancer prediction is very sensitive, but results in a false-positive rate that is too high to be useful for most applications [58,59].
While an obvious solution is to seek more distant species for human genome comparison, this problem can be partially overcome by using more stringent conservation criteria in human-rodent comparisons alone. Human-rodent “ultraconserved” elements are one such class of extremely conserved human-rodent sequences and are defined as sequences of 200bp or more that are 100% identical between human, mouse and rat [60]. Thus, these sequences are at the extreme end of the conserved human-mouse continuum which is exemplified by there only being approximately 250 of such elements that do not overlap with protein-coding sequences in our genome. The function of these elements has not been exhaustively explored, but studies of single ultraconserved elements [46,61] as well as their genomic localization in clusters near key developmental genes [62] suggest that many of them may be long-range modulators of gene transcription.
While ultraconserved elements are highly likely to be enhancers or other functional elements, their value for large-scale prediction of enhancers is limited because they represent only a relatively small subset of the functionally conserved sequences in the human genome. Their low total number indicates a poor sensitivity, suggesting that many or most functional elements will be missed if ultraconservation alone is used to screen a genomic interval of interest. Moreover, because of the extreme conservation criteria of ultraconserved elements, most of them coincide with regions that are also conserved between human and fish. However, it has recently been suggested that statistically more rigorous methods than the original concept of ultraconservation might provide a way to extract larger populations with ultra-like constraints from human-rodent comparisons, increasing the sensitivity while maintaining the specificity associated with ultraconserved elements [48] (see section 4.1.2). Computational tools to exploit this concept are becoming increasingly available [48,63,64].
3.2.3 Comparison of Close Species: Phylogenetic Shadowing
For studying regulatory elements related to aspects of biology that are specific to humans or primates, but do not exist in more distant species such as rodents, distant comparisons will only be useful in cases where previously existing regulatory features have assumed a new function in the primate lineage. However, distant comparisons will miss elements that have evolved more recently and are possibly specific to the primate phylogenetic branch. On the other hand, comparison with other primates does not yield useful results when conventional sequence comparison is performed due to the relatively short period since the last common ancestor in the primate branch, e.g. ~25 million years for humans and Old World monkeys [65]. This is exemplified to a severe degree in comparisons of human and chimpanzee, which separated from their common ancestor ~7 million years ago. Between these two genomes ~99% of all nucleotides are conserved [66], rendering conventional comparative approaches useless because virtually all regions of the genome appear highly similar. This problem can be overcome using a “phylogenetic shadowing” approach [67]. In this method, the sequences of multiple, evolutionary close species such as humans, apes and monkeys are aligned. This depth of several species provides the nucleotide diversity that would otherwise be achieved through more distant pair-wise comparisons such as human-mouse. Moreover, this approach incorporates a molecular phylogenetic model to consider the phylogenetic relationships among the different species that are compared such that changes that occurred in a closely-related species are given more power than those in a more distantly-related species. Phylogenetic shadowing requires aligned sequences from multiple closely related species and has therefore so far only been used in the context of studies focusing on particular loci of interest [67,68]. However, this method will likely become increasingly used for the identification of regulatory elements as more and more closely-related genomes become available [69].
4. Tools and Resources for Comparative Genomics
A number of tools are available to identify conserved noncoding elements in genome sequences. In this section, we will provide an overview of computational approaches and web-based resources to interrogate and browse the human genome for such elements and retrieve their sequences for experimental studies. We also discuss approaches for experimental characterization of developmental enhancers and describe the Vista Enhancer Browser as a public database of experimentally validated enhancers. Relevant web addresses and references describing each of the listed resources are provided in table 1.
Tab. 1.
Selected interactive genome browsing tools for the identification of vertebrate CNSs.
| Identification of conserved elements | Available at | URL | Based on alignment | Display/Download |
|---|---|---|---|---|
| Percent identity plot (PiP) [71,72] | Vista Genome Browser [73] | http://pipeline.lbl.gov | SLAGAN (pair-wise, glocal*) [74] | Percent identity curves; display and download of elements with adjustable threshold identity percentage |
| Dcode ECR Browser [75] | http://ecrbrowser.dcode.org | BLASTZ (pair-wise, local) [76] | Percent identity plots or curves; display and download of elements with adjustable threshold identity percentage | |
| PhastCons [64] | UCSC Genome Browser [77,78] | http://genome.ucsc.edu | MULTIZ (multiple, local) [79] | UCSC genome browser „Most Conserved“ track; download of elements with adjustable constraint threshold |
| Gumby [48] | Vista Genome Browser [73] | http://pipeline.lbl.gov | SLAGAN (pair-wise, glocal*) [74] | “RankVista” p-value bar plots; display and download of elements with adjustable threshold p-value |
| Vista Enhancer Browser | http://enhancer.lbl.gov | MLAGAN (multiple, global) [70] | Browsable list of human-mouse-rat CNSs; direct link to developmental enhancer assay results where available |
4.1 Identification of Candidate Regions at a Genomic Scale
Identification of conserved elements by comparison of genomes from different species is generally a two-step process. First, homologous regions of two or more different genomes are aligned at the nucleotide level, so that for each nucleotide position in the reference genome a best fit with the nucleotide at the respective position in the other genome(s) is determined. Second, based on this alignment, the different genomes are compared at the nucleotide level and statistical methods are used to identify regions where the sequence is more constrained (i.e. similar between the different organisms) than what would be expected for neutrally evolving DNA.
4.1.1 Aligning Genome Sequences
For the alignment step, a range of whole genome methods has been developed and several relevant programs are listed in table 1. These generally fall into two categories: local and global alignment approaches. Local methods compare relatively short intervals of genomic sequences with each other and return the best match between two genomes for each sub-region. However, because they do not take into account the region surrounding these matches, they can result in false hits, e.g. returning a paralogous sequence instead of the true ortholog. In contrast, global methods align entire syntenic regions and are less prone to return false-positive matches, but fail to recognize homologous regions that have been locally rearranged by translocations of inversions. Finally, “glocal” alignment [70] is a global alignment strategy that allows for local rearrangements, thereby eliminating some of the problems associated with local-only or global-only alignments.
While all three types of alignments have been successfully used for comparative identification of functional elements, it is important to keep in mind that they will often return slightly different results for a particular genomics region of interest. Thus, trial and error approaches are appropriate to maximize the likelihood of biological discovery.
4.1.2 Scoring Conservation in Aligned Genome Sequences
For defining highly conserved elements in aligned genomes, there is also a range of computational tools available. We focus here on a small subset of such tools that is of particular relevance for the identification of candidate enhancer sequences in the human genome by biomedical investigators (Fig. 2). The most straightforward way to identify highly constrained elements in genome alignments are pair-wise percent identity plots. When using local alignment methods such as BLASTZ [76], the length and percent identity of each aligned segment can be directly converted into a sequence plot [71] (Fig. 2A). Alternatively, for two globally aligned sequences, a sliding window of user-defined size (e.g. 100bp) is moved along the alignment and returns for each nucleotide position the percentage of identity within the window [72] (Fig. 2B). Conserved noncoding sequences (CNSs) are in both cases defined by a user-specified threshold, e.g. as regions exceeding 70% identity over at least 80bp.
Fig. 2. Sequence display of the same human genome region by various tools for comparative analysis.
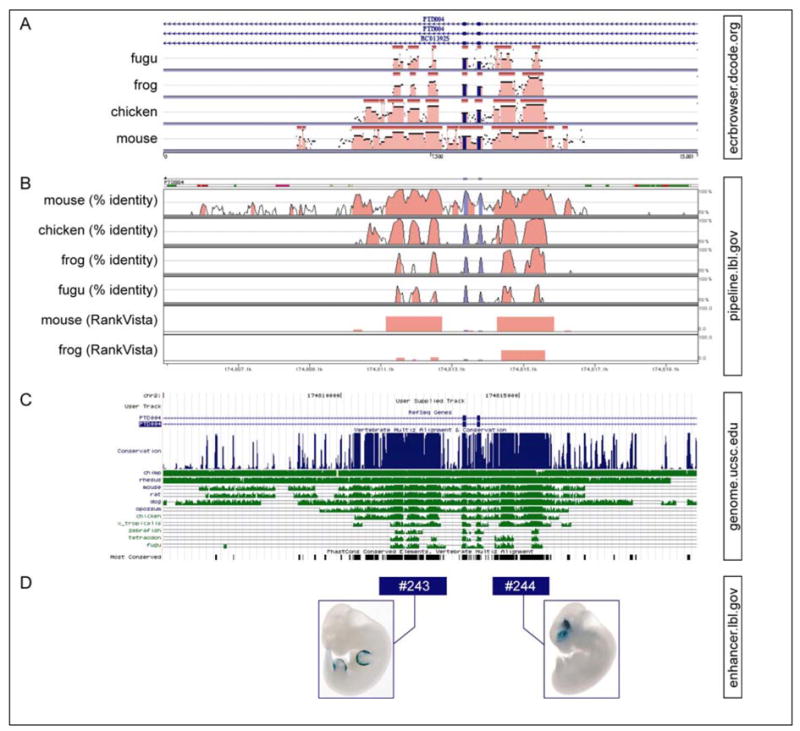
A 15kb region comprising two exons of the GTP-binding protein PTD004 is shown (chr2:174,805,000-174,820,000; hg17). A) Percent identity plots as displayed in the Dcode ECR browser. B) Percent identity tracks and RankVista tracks in the Vista Genome Browser. RankVista tracks are based on p-values of conserved elements determined by the Gumby algorithm. C) Conservation and PhastCons (“Most Conserved”) tracks in the UCSC genome browser. D) Experimental results for two CNSs in the Vista Enhancer Browser. See table 1 for relevant references.
Percent identity plots have been widely used because the concept is simple and readily implemented, but they have several important limitations. For example, they do not allow direct multi-species comparisons, but rather multiple species can be indirectly considered by aligning the pair-wise alignments to the same reference genome. Moreover, they do not take into account the evolutionary distance between the species that are being compared. When using the same threshold (e.g. 70% identity, ≥100bp), the choice of the species being compared can be used to roughly calibrate sensitivity versus specificity (Fig. 1A). For instance, CNSs identified by comparison of distant species such as human-fish are highly enriched in functional enhancers [46]. However, the relatively small number of such elements detected by this strategy indicates that it fails to capture many functional sequences (see section 3.2.1). In contrast, comparison of close species such as human-mouse identifies hundreds of thousands of elements and is thus more sensitive, but suffers from a high false-positive rate when such elements are tested for their tissue-specific enhancer activity in functional assays [58]. The problem of low specificity in percent-identity types of comparisons between close species can be partially alleviated by using more stringent threshold parameters. For example, human-mouse-rat “ultra”-conservation of 100% for ≥200bp [60] is similarly successful for enhancer identification as deep human-fish conservation (AV, LAP, unpublished observations), but is even less sensitive by an order of magnitude (see section 3.2.2).
Recently a new generation of advanced, mathematically and statistically rigorous tools have become available that allow direct multi-species (_n-_way) comparisons while also considering phylogenetic branch length and local neutral background substitution rates [48,64]. Importantly, these methods do not require a single pre-specified evolutionary distance [64] (Fig. 2C) and provide high specificity even in pair-wise comparisons of relatively close species such as human and mouse [48] (Fig 2B). Moreover, they use statistical tests to assign quantitative scores to elements, allowing a user to rank all elements within a given genomic interval according to the significance of their constraint. We have started to explore the relative value of these different comparative methods for prediction of tissue-specific enhancers by testing elements predicted by different methods in a transgenic reporter assay (see below), where we find that these more advanced comparative tools are indeed superior to simple percent identity plots in their ability to predict functional enhancers.
In order to browse the human or other vertebrate genomes for the presence of elements identified using the different methods described above, a variety of public resources is available online. We provide a list of such sites in table 1, limiting our selection to those resources that provide pre-aligned sequences and elements identified by the methods described above.
4.2 Experimental Validation of _cis_-Regulatory Elements
An array of experimental approaches is available to assess the potential for putative regulatory elements to influence the expression of genes. These include in vitro methods for determination of consensus binding sites of specific transcription factors, evaluation of potential accessibility of putative transcription factor binding sites (TFBSs) by DNase I hypersensitivity assays, electrophoretic mobility shift assays, and chromatin immunoprecipitation assays to determine the binding sites of a specific transcription factor within the genome. While this field has experienced considerable progress in the past, all of these methods, even when used in combination, are generally insufficient to successfully predict the location of a particular enhancer element or its tissue-specificity in an animal, prompting the need to validate and characterize putative enhancers in suitable in vivo assays.
Methods for in vivo testing of enhancer activities have been described for several vertebrate model organisms, including zebrafish and Xenopus [40,47]. In this article we will, however, focus on experimental approaches employing the mouse for determining the in vivo activity of candidate human enhancer sequences. Due to their shared phylogeny as mammals, the mouse is a suitable model for many aspects of human development, physiology, and disease. Importantly, mice are among the mammalian model organisms for which transgenic techniques have been available for many years, enabling the easy and efficient introduction of reporter constructs into the genome.
In order to study the in vivo properties of human enhancers, and in particular their ability to drive tissue-specific expression during embryonic development, we have recently set up a pipeline for testing of putative enhancers in transgenic mice (Fig. 3). We identify candidate elements by comparative criteria, such as human-fish comparison [46,80] or “ultra”-conservation between humans and rodents [60,61] (Fig. 2D). Then we assess the potential of such candidate regulatory regions experimentally in a transgenic mouse enhancer assay [81,82]. Candidate regions are PCR-amplified from human genomic DNA and cloned into a reporter vector in which they are fused to a minimal heat shock protein 68 promoter and a beta-galactosidase reporter gene. On its own, this vector does not drive beta-galactosidase gene expression in mammalian embryonic tissues [81,82], but when fused to a DNA fragment with gene enhancer properties, spatial and temporal patterns of expression can be robustly and reproducibly characterized. This construct is injected into one of the two pronuclei of fertilized mouse oocytes, where it integrates into the genomic DNA at a random position, usually in multiple copies. The oocytes are then implanted into pseudo-pregnant females, embryos are harvested at embryonic day 11.5 and stained for beta-galactosidase activity using X-Gal as a chromogenic substrate.
Fig. 3. Experimental design.
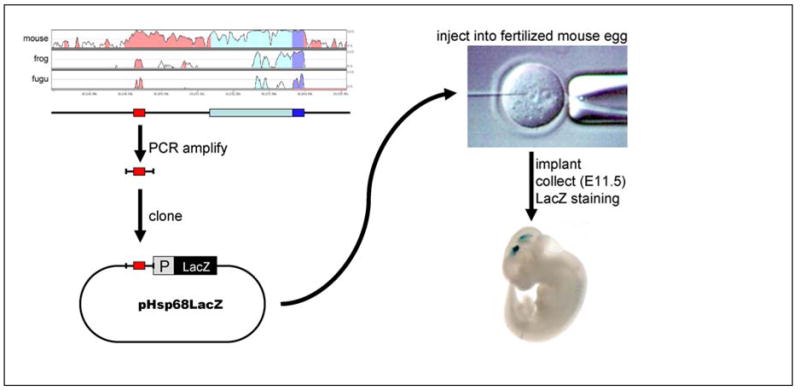
Identification (example alignment displayed as Vista track), cloning and transgenic testing of candidate enhancer sequences.
We chose this particular stage of development for analysis for several reasons. (1) Many human-fugu and ultra-conserved elements reside near genes that are expressed in early development [60,62]. (2) Whole embryo staining at this time-point enables the global identification of enhancer expression features without bias for particular tissues. (3) This is a key time-point during organogenesis at which most structures are present. Our preliminary studies of ~150 human-fugu elements indicate that this time-point is able to catch enhancer activities for >40% of the fragments tested, in contrast to moderately conserved human-rodent fragments where less than 5% of fragments behave as enhancers at this time-point [58]. Due to position effects that can alter in vivo enhancer characteristics as a result of the transgene integration site, we generate >5 independent transgenic animals per injection and require that at least 3 of these independent founders for each construct show reproducible spatial expression characteristics before assigning a conserved element an associated regulatory activity.
Compared with the generation of traditional BAC or YAC transgenic lines, use of this transient transgenic method results in a dramatically increased throughput that allows us to currently test 500 elements per year. This assay has previously been used in numerous gene-centric studies, where its reproducibility and high spatiotemporal resolution has provided valuable insights into the in vivo activities of single elements of interest. This increase in throughput allows application of this method at a genomic scale, without requiring guidance by their neighboring genes.
4.3 Enhancer Browser: Large-Scale Data Set of _in vivo-_Validated Enhancers
In order to make the results of our enhancer screen available to the scientific community, we have established a public database, the Vista Enhancer Browser, which is available at http://enhancer.lbl.gov (Fig. 4A). This browser houses two principal kinds of data: 1) experimental results from our in vivo screen and 2) a large collection of vertebrate noncoding sequences that are evolutionary conserved at varying distances.
Figure 4. Retrieving Data from the Vista Enhancer Browser.
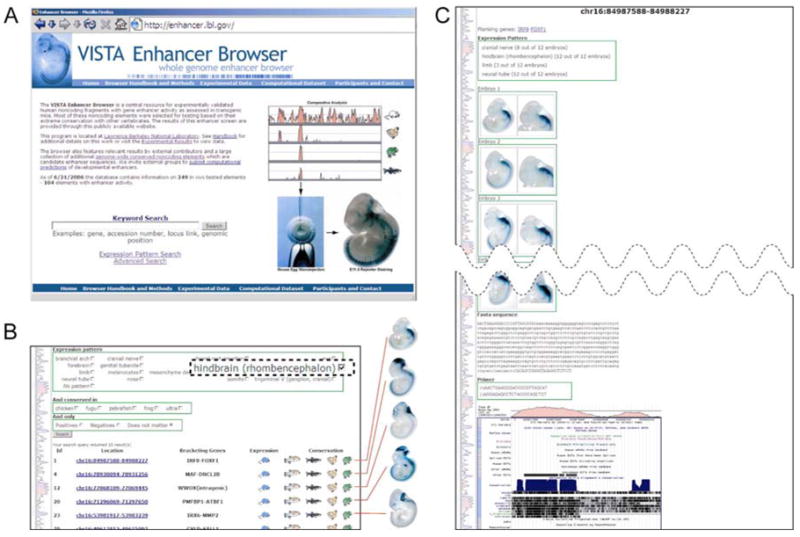
A) Entry page with basic query function. B) Advanced search page with query form for experimental data. The results of a search for enhancers with hindbrain expression are shown. Each row in the results table corresponds to one experimental data set. A representative embryo is shown for the first five data sets. C) Full data set display mode. Top: Coordinates of element, neighboring genes, anatomical description of expression patterns and pictures of representative embryos. Note that each embryo is an independent transgenic F0 animal. Overview pictures and magnified views of expression sites are provided; all images can be downloaded at high resolution. Bottom: Sequence of element, PCR primers used for cloning and conservation profile linked to UCSC genome browser.
4.3.1 Experimental Data
The experimental results of our transgenic in vivo screen constitute the core data set of the enhancer browser. Each tested fragment has an associated dataset (Fig. 4C) consisting of sequence-related information and the experimental results. Sequence-related information includes the genomic coordinates, names of neighboring genes, PCR primers used to amplify the element from human genomic DNA, and an overview of the conservation in various species. The results of the transgenic enhancer assay are provided both in the form of pictures of embryos with representative reporter gene activity and in anatomical annotation format. To be considered positive in our assay, an element has to drive reporter gene expression in the same anatomical structure in at least three independent transgenic embryos. Elements in which no such reproducibility is observed, although a sufficient number of transgenic embryos was generated (generally at least five transgenics confirmed by PCR genotyping) are reported as negative and no pictures of the embryos are shown. For positive elements, a selection of representative embryos is displayed. The images for each embryo can be retrieved as high-resolution files and are often supplemented by images at higher magnification or from more informative angles than the standard sagittal overview of the whole-mount specimen.
To enable searches of our data as well as bulk downloads, we annotate the tissue specificity of each positive enhancer identified using a list of anatomical terms that is largely consistent with existing standardized nomenclature [83]. We thus provide the ratio of X-gal stained-positive embryos versus all transgenic embryos separately for each structure (Fig. 4C). A text-based query function is available on the front page of the enhancer browser. Using this feature, the database can also be searched by genomic coordinates, gene names, accession number and Entrez Gene IDs. An additional comprehensive search tool is available for more advanced queries of the database. This includes searches for enhancers that are specific for a particular anatomical structure of interest (Fig. 4B) and/or restriction of the search to elements of a user-defined conservation depth (e.g. human-frog or human-fugu).
4.3.2 Computational Data Set
In addition to the experimental and external data, the enhancer browser also provides a genome-wide computationally generated set of more than 145,000 highly conserved elements for which no experimental data from the transgenic assay is available. These elements were identified using Gumby/RankVista with globally aligned human-mouse-rat sequences [48]. Only elements with a _p_-value <= 0.001 that do not overlap known mRNAs or spliced expressed sequence tags were considered for this data set. All of these elements were then checked for their conservation in chicken, frog, zebrafish and pufferfish to determine the conservation depth which is provided at the website. While we plan to test some subsets of this large collection of highly conserved elements in the future, the major purpose of this collection is to provide users with an easily accessible list of candidate regions for genomic intervals of interest for analysis in complementary computational and experimental approaches. Similar datasets can be obtained from other resources listed in table 1.
5. Conclusions and Perspectives
While gene regulation studies were possible in the pre-genome era, they were exceedingly expensive and time-consuming. Distant enhancers flanking a gene of interest were usually painstakingly identified through historic deletion series in transgenic animals. These experiments occurred sequentially in a largely trial and error fashion until the minimum sequence necessary to drive a given expression pattern was identified. Retrospective comparative analysis reveals that many of these functionally identified fragments strongly overlap with highly conserved regions of the human genome. For example, the distal liver-specific enhancer of APOE, a protein that impacts cholesterol metabolism, cardiovascular and Alzheimer’s disease, was originally identified through such testing of many overlapping gene fragments in transgenic mice [6,7], but retrospective comparative analysis revealed that simple percent identity plot human-mouse comparisons would have readily identified this hepatic control region [84]. As is the case for numerous regulatory elements, had comparative data been available prior to beginning these experiments, hypotheses based on sequences under evolutionary constraint could have directly guided these studies from their inception.
Today, with this background experience, we are privileged to begin studies with computational sequence analysis followed by functional investigations. Such an approach can occur on a gene-by-gene basis or at a whole genome level of analysis. As a caveat, we should emphasize that comparative-based approaches are not without limitations. Some enhancers will lack conservation or may be missed by current computational tools, as illustrated in this article by the relatively weak conservation of many experimentally identified enhancers involved in heart development (Fig. 1B). While the thought of more vertebrate species genomic sequences is a daunting data management task, their availability will without doubt further improve our ability to know which species to compare to address which biological question and allow additional flexibility in the choice of organisms used in multi-species analyses.
Importantly, the possibility of deep alignments across a wide range of vertebrate taxa will also increasingly allow us to address the relation between noncoding sequences and phenotypic diversity. One paradigmatic example to this end was the analysis of the aforementioned Hoxc8 early enhancer in a panel of mammals that suggested that evolution of this enhancer contributed to the differences in axial morphology distinguishing baleen whales from other mammals [85]. While this study in the pre-genomic era relied on targeted sequencing of this regulatory element in a large number of species in the mammalian clade, the ever-growing number of available vertebrate sequences will increasingly allow for similar such studies at genomic scale.
The moderate-scale experimental testing of candidate enhancers through transgenic approaches such as that described here are expected to provide larger training sets for improved computational predictions of what activities conserved sequences are likely to contain. The first level of annotation in this area is occurring on the most highly (human-rodent "ultra") and deepest (human-fish) conserved elements in the human genome. These classes of conserved noncoding elements are enriched near genes active in early development and this is not universally applicable for all types of known enhancers. Rather, they will serve to demonstrate how one can go from comparative sequence data to their functional testing to using the resulting dataset to computationally predict additional such enhancer elements in the larger human genome. It is anticipated that through such an iterative process we will learn vital clues as to developmental enhancer function and that this knowledge will translate into a deeper understanding of the regulation of both developmental and non-developmental genes in vertebrates.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
L.A.P. was supported by grant HL066681, Berkeley-PGA, under the Programs for Genomic Applications, funded by National Heart, Lung, & Blood Institute, and HG003988 funded by National Human Genome Research Institute. Research was performed under Department of Energy Contract DE-AC02-05CH11231, University of California, E.O. Lawrence Berkeley National Laboratory. A.V. was supported by an American Heart Association postdoctoral fellowship.
References
- 1.Stathopoulos A, Levine M. Genomic regulatory networks and animal development. Dev Cell. 2005;9:449–62. doi: 10.1016/j.devcel.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 2.Levine M, Tjian R. Transcription regulation and animal diversity. Nature. 2003;424:147–51. doi: 10.1038/nature01763. [DOI] [PubMed] [Google Scholar]
- 3.Davidson EH. Genomic regulatory systems: Development and evolution. 1. San Diego: Academic Press; 2001. [Google Scholar]
- 4.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. The sequence of the human genome. Science. 2001;291:1304–51. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 5.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 6.Simonet WS, Bucay N, Pitas RE, Lauer SJ, Taylor JM. Multiple tissue-specific elements control the apolipoprotein e/c-i gene locus in transgenic mice. J Biol Chem. 1991;266:8651–4. [PubMed] [Google Scholar]
- 7.Simonet WS, Bucay N, Lauer SJ, Taylor JM. A far-downstream hepatocyte-specific control region directs expression of the linked human apolipoprotein e and c-i genes in transgenic mice. J Biol Chem. 1993;268:8221–9. [PubMed] [Google Scholar]
- 8.Allan CM, Walker D, Taylor JM. Evolutionary duplication of a hepatic control region in the human apolipoprotein e gene locus. Identification of a second region that confers high level and liver-specific expression of the human apolipoprotein e gene in transgenic mice. J Biol Chem. 1995;270:26278–81. doi: 10.1074/jbc.270.44.26278. [DOI] [PubMed] [Google Scholar]
- 9.Grehan S, Tse E, Taylor JM. Two distal downstream enhancers direct expression of the human apolipoprotein e gene to astrocytes in the brain. J Neurosci. 2001;21:812–22. doi: 10.1523/JNEUROSCI.21-03-00812.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shih SJ, Allan C, Grehan S, Tse E, Moran C, Taylor JM. Duplicated downstream enhancers control expression of the human apolipoprotein e gene in macrophages and adipose tissue. J Biol Chem. 2000;275:31567–72. doi: 10.1074/jbc.M005468200. [DOI] [PubMed] [Google Scholar]
- 11.Zheng P, Pennacchio LA, Le Goff W, Rubin EM, Smith JD. Identification of a novel enhancer of brain expression near the apoe gene cluster by comparative genomics. Biochim Biophys Acta. 2004;1676:41–50. doi: 10.1016/j.bbaexp.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 12.Lyons I, Parsons LM, Hartley L, Li R, Andrews JE, Robb L, et al. Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene nkx2-5. Genes Dev. 1995;9:1654–66. doi: 10.1101/gad.9.13.1654. [DOI] [PubMed] [Google Scholar]
- 13.Chi X, Chatterjee PK, Wilson W, 3rd, Zhang SX, Demayo FJ, Schwartz RJ. Complex cardiac nkx2-5 gene expression activated by noggin-sensitive enhancers followed by chamber-specific modules. Proc Natl Acad Sci U S A. 2005;102:13490–5. doi: 10.1073/pnas.0504295102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanaka M, Wechsler SB, Lee IW, Yamasaki N, Lawitts JA, Izumo S. Complex modular cis-acting elements regulate expression of the cardiac specifying homeobox gene csx/nkx2.5. Development. 1999;126:1439–50. doi: 10.1242/dev.126.7.1439. [DOI] [PubMed] [Google Scholar]
- 15.Searcy RD, Vincent EB, Liberatore CM, Yutzey KE. A gata-dependent nkx-2.5 regulatory element activates early cardiac gene expression in transgenic mice. Development. 1998;125:4461–70. doi: 10.1242/dev.125.22.4461. [DOI] [PubMed] [Google Scholar]
- 16.Reecy JM, Li X, Yamada M, DeMayo FJ, Newman CS, Harvey RP, et al. Identification of upstream regulatory regions in the heart-expressed homeobox gene nkx2-5. Development. 1999;126:839–49. doi: 10.1242/dev.126.4.839. [DOI] [PubMed] [Google Scholar]
- 17.Lien CL, McAnally J, Richardson JA, Olson EN. Cardiac-specific activity of an nkx2-5 enhancer requires an evolutionarily conserved smad binding site. Dev Biol. 2002;244:257–66. doi: 10.1006/dbio.2002.0603. [DOI] [PubMed] [Google Scholar]
- 18.Lien CL, Wu C, Mercer B, Webb R, Richardson JA, Olson EN. Control of early cardiac-specific transcription of nkx2-5 by a gata-dependent enhancer. Development. 1999;126:75–84. doi: 10.1242/dev.126.1.75. [DOI] [PubMed] [Google Scholar]
- 19.Schwartz RJ, Olson EN. Building the heart piece by piece: Modularity of cis-elements regulating nkx2-5 transcription. Development. 1999;126:4187–92. doi: 10.1242/dev.126.19.4187. [DOI] [PubMed] [Google Scholar]
- 20.Zakany J, Gerard M, Favier B, Duboule D. Deletion of a hoxd enhancer induces transcriptional heterochrony leading to transposition of the sacrum. Embo J. 1997;16:4393–402. doi: 10.1093/emboj/16.14.4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Juan AH, Ruddle FH. Enhancer timing of hox gene expression: Deletion of the endogenous hoxc8 early enhancer. Development. 2003;130:4823–34. doi: 10.1242/dev.00672. [DOI] [PubMed] [Google Scholar]
- 22.Kurokawa D, Kiyonari H, Nakayama R, Kimura-Yoshida C, Matsuo I, Aizawa S. Regulation of otx2 expression and its functions in mouse forebrain and midbrain. Development. 2004;131:3319–31. doi: 10.1242/dev.01220. [DOI] [PubMed] [Google Scholar]
- 23.Kurokawa D, Takasaki N, Kiyonari H, Nakayama R, Kimura-Yoshida C, Matsuo I, et al. Regulation of otx2 expression and its functions in mouse epiblast and anterior neuroectoderm. Development. 2004;131:3307–17. doi: 10.1242/dev.01219. [DOI] [PubMed] [Google Scholar]
- 24.Yanagisawa H, Clouthier DE, Richardson JA, Charite J, Olson EN. Targeted deletion of a branchial arch-specific enhancer reveals a role of dhand in craniofacial development. Development. 2003;130:1069–78. doi: 10.1242/dev.00337. [DOI] [PubMed] [Google Scholar]
- 25.Gu H, Marth JD, Orban PC, Mossmann H, Rajewsky K. Deletion of a DNA polymerase beta gene segment in t cells using cell type-specific gene targeting. Science. 1994;265:103–6. doi: 10.1126/science.8016642. [DOI] [PubMed] [Google Scholar]
- 26.Vong LH, Ragusa MJ, Schwarz JJ. Generation of conditional mef2cloxp/loxp mice for temporal- and tissue-specific analyses. Genesis. 2005;43:43–8. doi: 10.1002/gene.20152. [DOI] [PubMed] [Google Scholar]
- 27.Li Song D, Joyner AL. Two pax2/5/8-binding sites in engrailed2 are required for proper initiation of endogenous mid-hindbrain expression. Mech Dev. 2000;90:155–65. doi: 10.1016/s0925-4773(99)00242-7. [DOI] [PubMed] [Google Scholar]
- 28.Iwahori A, Fraidenraich D, Basilico C. A conserved enhancer element that drives fgf4 gene expression in the embryonic myotomes is synergistically activated by gata and bhlh proteins. Dev Biol. 2004;270:525–37. doi: 10.1016/j.ydbio.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 29.Guyot B, Valverde-Garduno V, Porcher C, Vyas P. Deletion of the major gata1 enhancer hs 1 does not affect eosinophil gata1 expression and eosinophil differentiation. Blood. 2004;104:89–91. doi: 10.1182/blood-2004-01-0108. [DOI] [PubMed] [Google Scholar]
- 30.Chen JC, Goldhamer DJ. The core enhancer is essential for proper timing of myod activation in limb buds and branchial arches. Dev Biol. 2004;265:502–12. doi: 10.1016/j.ydbio.2003.09.018. [DOI] [PubMed] [Google Scholar]
- 31.Xiong N, Kang C, Raulet DH. Redundant and unique roles of two enhancer elements in the tcrgamma locus in gene regulation and gammadelta t cell development. Immunity. 2002;16:453–63. doi: 10.1016/s1074-7613(02)00285-6. [DOI] [PubMed] [Google Scholar]
- 32.Lettice LA, Horikoshi T, Heaney SJ, van Baren MJ, van der Linde HC, Breedveld GJ, et al. Disruption of a long-range cis-acting regulator for shh causes preaxial polydactyly. Proc Natl Acad Sci U S A. 2002;99:7548–53. doi: 10.1073/pnas.112212199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sagai T, Hosoya M, Mizushina Y, Tamura M, Shiroishi T. Elimination of a long-range cis-regulatory module causes complete loss of limb-specific shh expression and truncation of the mouse limb. Development. 2005;132:797–803. doi: 10.1242/dev.01613. [DOI] [PubMed] [Google Scholar]
- 34.Lettice LA, Heaney SJ, Purdie LA, Li L, de Beer P, Oostra BA, et al. A long-range shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum Mol Genet. 2003;12:1725–35. doi: 10.1093/hmg/ddg180. [DOI] [PubMed] [Google Scholar]
- 35.Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, et al. Human gene mutation database (hgmd): 2003 update. Hum Mutat. 2003;21:577–81. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- 36.Gumucio DL, Heilstedt-Williamson H, Gray TA, Tarle SA, Shelton DA, Tagle DA, et al. Phylogenetic footprinting reveals a nuclear protein which binds to silencer sequences in the human gamma and epsilon globin genes. Mol Cell Biol. 1992;12:4919–29. doi: 10.1128/mcb.12.11.4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tagle DA, Koop BF, Goodman M, Slightom JL, Hess DL, Jones RT. Embryonic epsilon and gamma globin genes of a prosimian primate (galago crassicaudatus). Nucleotide and amino acid sequences, developmental regulation and phylogenetic footprints. J Mol Biol. 1988;203:439–55. doi: 10.1016/0022-2836(88)90011-3. [DOI] [PubMed] [Google Scholar]
- 38.Hardison RC, Oeltjen J, Miller W. Long human-mouse sequence alignments reveal novel regulatory elements: A reason to sequence the mouse genome. Genome Res. 1997;7:959–66. doi: 10.1101/gr.7.10.959. [DOI] [PubMed] [Google Scholar]
- 39.Elgar G, Sandford R, Aparicio S, Macrae A, Venkatesh B, Brenner S. Small is beautiful: Comparative genomics with the pufferfish (fugu rubripes) Trends Genet. 1996;12:145–50. doi: 10.1016/0168-9525(96)10018-4. [DOI] [PubMed] [Google Scholar]
- 40.Gottgens B, Barton LM, Gilbert JG, Bench AJ, Sanchez MJ, Bahn S, et al. Analysis of vertebrate scl loci identifies conserved enhancers. Nat Biotechnol. 2000;18:181–6. doi: 10.1038/72635. [DOI] [PubMed] [Google Scholar]
- 41.Loots GG, Locksley RM, Blankespoor CM, Wang ZE, Miller W, Rubin EM, et al. Identification of a coordinate regulator of interleukins 4, 13, and 5 by cross-species sequence comparisons. Science. 2000;288:136–40. doi: 10.1126/science.288.5463.136. [DOI] [PubMed] [Google Scholar]
- 42.Aparicio S, Chapman J, Stupka E, Putnam N, Chia JM, Dehal P, et al. Whole-genome shotgun assembly and analysis of the genome of fugu rubripes. Science. 2002;297:1301–10. doi: 10.1126/science.1072104. [DOI] [PubMed] [Google Scholar]
- 43.Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–62. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 44.Boffelli D, Nobrega MA, Rubin EM. Comparative genomics at the vertebrate extremes. Nat Rev Genet. 2004;5:456–65. doi: 10.1038/nrg1350. [DOI] [PubMed] [Google Scholar]
- 45.Aparicio S, Morrison A, Gould A, Gilthorpe J, Chaudhuri C, Rigby P, et al. Detecting conserved regulatory elements with the model genome of the japanese puffer fish, fugu rubripes. Proc Natl Acad Sci U S A. 1995;92:1684–8. doi: 10.1073/pnas.92.5.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nobrega MA, Ovcharenko I, Afzal V, Rubin EM. Scanning human gene deserts for long-range enhancers. Science. 2003;302:413. doi: 10.1126/science.1088328. [DOI] [PubMed] [Google Scholar]
- 47.Woolfe A, Goodson M, Goode DK, Snell P, McEwen GK, Vavouri T, et al. Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol. 2005;3:e7. doi: 10.1371/journal.pbio.0030007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prabhakar S, Poulin F, Shoukry MI, Afzal V, Rubin EM, Couronne O, et al. Close sequence comparisons are sufficient to identify human cis-regulatory elements. Genome Res. 2006;16:855–63. doi: 10.1101/gr.4717506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–45. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 50.Buckingham M, Meilhac S, Zaffran S. Building the mammalian heart from two sources of myocardial cells. Nat Rev Genet. 2005;6:826–35. doi: 10.1038/nrg1710. [DOI] [PubMed] [Google Scholar]
- 51.Kuo H, Chen J, Ruiz-Lozano P, Zou Y, Nemer M, Chien KR. Control of segmental expression of the cardiac-restricted ankyrin repeat protein gene by distinct regulatory pathways in murine cardiogenesis. Development. 1999;126:4223–34. doi: 10.1242/dev.126.19.4223. [DOI] [PubMed] [Google Scholar]
- 52.Kuisk IR, Li H, Tran D, Capetanaki Y. A single mef2 site governs desmin transcription in both heart and skeletal muscle during mouse embryogenesis. Dev Biol. 1996;174:1–13. doi: 10.1006/dbio.1996.0046. [DOI] [PubMed] [Google Scholar]
- 53.Molkentin JD, Antos C, Mercer B, Taigen T, Miano JM, Olson EN. Direct activation of a gata6 cardiac enhancer by nkx2.5: Evidence for a reinforcing regulatory network of nkx2.5 and gata transcription factors in the developing heart. Dev Biol. 2000;217:301–9. doi: 10.1006/dbio.1999.9544. [DOI] [PubMed] [Google Scholar]
- 54.McFadden DG, Charite J, Richardson JA, Srivastava D, Firulli AB, Olson EN. A gata-dependent right ventricular enhancer controls dhand transcription in the developing heart. Development. 2000;127:5331–41. doi: 10.1242/dev.127.24.5331. [DOI] [PubMed] [Google Scholar]
- 55.Dodou E, Verzi MP, Anderson JP, Xu SM, Black BL. Mef2c is a direct transcriptional target of isl1 and gata factors in the anterior heart field during mouse embryonic development. Development. 2004;131:3931–42. doi: 10.1242/dev.01256. [DOI] [PubMed] [Google Scholar]
- 56.Phan D, Rasmussen TL, Nakagawa O, McAnally J, Gottlieb PD, Tucker PW, et al. Bop, a regulator of right ventricular heart development, is a direct transcriptional target of mef2c in the developing heart. Development. 2005;132:2669–78. doi: 10.1242/dev.01849. [DOI] [PubMed] [Google Scholar]
- 57.Hu T, Yamagishi H, Maeda J, McAnally J, Yamagishi C, Srivastava D. Tbx1 regulates fibroblast growth factors in the anterior heart field through a reinforcing autoregulatory loop involving forkhead transcription factors. Development. 2004;131:5491–502. doi: 10.1242/dev.01399. [DOI] [PubMed] [Google Scholar]
- 58.Nobrega MA, Zhu Y, Plajzer-Frick I, Afzal V, Rubin EM. Megabase deletions of gene deserts result in viable mice. Nature. 2004;431:988–93. doi: 10.1038/nature03022. [DOI] [PubMed] [Google Scholar]
- 59.Pennacchio LA. Insights from human/mouse genome comparisons. Mamm Genome. 2003;14:429–36. doi: 10.1007/s00335-002-4001-1. [DOI] [PubMed] [Google Scholar]
- 60.Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS, et al. Ultraconserved elements in the human genome. Science. 2004;304:1321–5. doi: 10.1126/science.1098119. [DOI] [PubMed] [Google Scholar]
- 61.Poulin F, Nobrega MA, Plajzer-Frick I, Holt A, Afzal V, Rubin EM, et al. In vivo characterization of a vertebrate ultraconserved enhancer. Genomics. 2005;85:774–81. doi: 10.1016/j.ygeno.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 62.Sandelin A, Bailey P, Bruce S, Engstrom PG, Klos JM, Wasserman WW, et al. Arrays of ultraconserved non-coding regions span the loci of key developmental genes in vertebrate genomes. BMC Genomics. 2004;5:99. doi: 10.1186/1471-2164-5-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cooper GM, Stone EA, Asimenos G, Green ED, Batzoglou S, Sidow A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–13. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–50. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Goodman M. The genomic record of humankind's evolutionary roots. Am J Hum Genet. 1999;64:31–9. doi: 10.1086/302218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- 67.Boffelli D, McAuliffe J, Ovcharenko D, Lewis KD, Ovcharenko I, Pachter L, et al. Phylogenetic shadowing of primate sequences to find functional regions of the human genome. Science. 2003;299:1391–4. doi: 10.1126/science.1081331. [DOI] [PubMed] [Google Scholar]
- 68.Clark VJ, Cox NJ, Hammond M, Hanis CL, Di Rienzo A. Haplotype structure and phylogenetic shadowing of a hypervariable region in the capn10 gene. Hum Genet. 2005;117:258–66. doi: 10.1007/s00439-005-1305-0. [DOI] [PubMed] [Google Scholar]
- 69.Margulies EH, Vinson JP, Miller W, Jaffe DB, Lindblad-Toh K, Chang JL, et al. An initial strategy for the systematic identification of functional elements in the human genome by low-redundancy comparative sequencing. Proc Natl Acad Sci U S A. 2005;102:4795–800. doi: 10.1073/pnas.0409882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brudno M, Do CB, Cooper GM, Kim MF, Davydov E, Green ED, et al. Lagan and multi-lagan: Efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 2003;13:721–31. doi: 10.1101/gr.926603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schwartz S, Zhang Z, Frazer KA, Smit A, Riemer C, Bouck J, et al. Pipmaker--a web server for aligning two genomic DNA sequences. Genome Res. 2000;10:577–86. doi: 10.1101/gr.10.4.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dubchak I, Brudno M, Loots GG, Pachter L, Mayor C, Rubin EM, et al. Active conservation of noncoding sequences revealed by three-way species comparisons. Genome Res. 2000;10:1304–6. doi: 10.1101/gr.142200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. Vista: Computational tools for comparative genomics. Nucleic Acids Res. 2004;32:W273–9. doi: 10.1093/nar/gkh458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brudno M, Malde S, Poliakov A, Do CB, Couronne O, Dubchak I, et al. Glocal alignment: Finding rearrangements during alignment. Bioinformatics. 2003;19 (Suppl 1):i54–62. doi: 10.1093/bioinformatics/btg1005. [DOI] [PubMed] [Google Scholar]
- 75.Loots GG, Ovcharenko I. Dcode. Org anthology of comparative genomic tools. Nucleic Acids Res. 2005;33:W56–64. doi: 10.1093/nar/gki355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schwartz S, Kent WJ, Smit A, Zhang Z, Baertsch R, Hardison RC, et al. Human-mouse alignments with blastz. Genome Res. 2003;13:103–7. doi: 10.1101/gr.809403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bejerano G, Siepel AC, Kent WJ, Haussler D. Computational screening of conserved genomic DNA in search of functional noncoding elements. Nat Methods. 2005;2:535–45. doi: 10.1038/nmeth0705-535. [DOI] [PubMed] [Google Scholar]
- 78.Hinrichs AS, Karolchik D, Baertsch R, Barber GP, Bejerano G, Clawson H, et al. The ucsc genome browser database: Update 2006. Nucleic Acids Res. 2006;34:D590–8. doi: 10.1093/nar/gkj144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AF, Roskin KM, et al. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004;14:708–15. doi: 10.1101/gr.1933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ahituv N, Rubin EM, Nobrega MA. Exploiting human--fish genome comparisons for deciphering gene regulation. Hum Mol Genet. 2004;13(Spec No 2):R261–6. doi: 10.1093/hmg/ddh229. [DOI] [PubMed] [Google Scholar]
- 81.Kothary R, Clapoff S, Brown A, Campbell R, Peterson A, Rossant J. A transgene containing lacz inserted into the dystonia locus is expressed in neural tube. Nature. 1988;335:435–7. doi: 10.1038/335435a0. [DOI] [PubMed] [Google Scholar]
- 82.Kothary R, Clapoff S, Darling S, Perry MD, Moran LA, Rossant J. Inducible expression of an hsp68-lacz hybrid gene in transgenic mice. Development. 1989;105:707–14. doi: 10.1242/dev.105.4.707. [DOI] [PubMed] [Google Scholar]
- 83.Bard JL, Kaufman MH, Dubreuil C, Brune RM, Burger A, Baldock RA, et al. An internet-accessible database of mouse developmental anatomy based on a systematic nomenclature. Mech Dev. 1998;74:111–20. doi: 10.1016/s0925-4773(98)00069-0. [DOI] [PubMed] [Google Scholar]
- 84.Pennacchio LA, Rubin EM. Genomic strategies to identify mammalian regulatory sequences. Nat Rev Genet. 2001;2:100–9. doi: 10.1038/35052548. [DOI] [PubMed] [Google Scholar]
- 85.Shashikant CS, Kim CB, Borbely MA, Wang WC, Ruddle FH. Comparative studies on mammalian hoxc8 early enhancer sequence reveal a baleen whale-specific deletion of a cis-acting element. Proc Natl Acad Sci U S A. 1998;95:15446–51. doi: 10.1073/pnas.95.26.15446. [DOI] [PMC free article] [PubMed] [Google Scholar]