RNAcode: Robust discrimination of coding and noncoding regions in comparative sequence data (original) (raw)
Abstract
With the availability of genome-wide transcription data and massive comparative sequencing, the discrimination of coding from noncoding RNAs and the assessment of coding potential in evolutionarily conserved regions arose as a core analysis task. Here we present RNAcode, a program to detect coding regions in multiple sequence alignments that is optimized for emerging applications not covered by current protein gene-finding software. Our algorithm combines information from nucleotide substitution and gap patterns in a unified framework and also deals with real-life issues such as alignment and sequencing errors. It uses an explicit statistical model with no machine learning component and can therefore be applied “out of the box,” without any training, to data from all domains of life. We describe the RNAcode method and apply it in combination with mass spectrometry experiments to predict and confirm seven novel short peptides in Escherichia coli and to analyze the coding potential of RNAs previously annotated as “noncoding.” RNAcode is open source software and available for all major platforms at http://wash.github.com/rnacode.
Keywords: coding sequence, comparative genomics, small peptides, transcriptome
INTRODUCTION
Distinguishing protein-coding from non-protein-coding sequence is the first and most crucial step in genome annotation. While the coding regions are subsequently investigated for properties of their protein products, a completely different toolkit is applied to the nucleic acid sequences of the noncoding regions. The quality of the analysis of coding potential therefore also affects the annotation of putative noncoding RNA (ncRNA) genes.
Discrimination between coding and noncoding regions poses technical as well as biological challenges not addressed by standard gene finders (Dinger et al. 2008). Ironically, investigators interested in noncoding RNAs hence have repeatedly implemented their own custom solutions to detect coding regions (see, e.g., Mourier et al. 2008; Shi et al. 2009). The tarsal-less gene in Drosophila melanogaster (also known as polished-rice in Trilobium) illustrates some of these challenges (Rosenberg and Desplan 2010). The transcript lacks a long open reading frame (ORF) and was originally annotated as noncoding RNA. Later it was found to produce several short, independently translated peptides of 11–32 amino acids (Galindo et al. 2007; Kondo et al. 2007) with a regulatory role in epidermal differentiation (Kondo et al. 2010). How many such short functional peptides may be hidden among RNAs remains an open question (Rosenberg and Desplan 2010).
The detection of protein-coding genes in genomic DNA data is a well-studied problem in computational biology (Burge and Karlin 1998). Using machine learning techniques, sophisticated models of genes have been built that can be used to annotate whole genomes (Brent 2008) and that have been constantly improved over the years (Flicek 2007; Brent 2008). Regular community meetings demonstrate a density of high-quality software not usually seen in other fields (Guigó et al. 2006; Coghlan et al. 2008). New types of high-throughput data, such as genome-wide transcription maps, massive comparative sequencing, and meta-genomics studies, however, have led to new challenges beyond classical gene finding. Many transcripts are found that do not overlap known or predicted genes (Carninci et al. 2005; The ENCODE Project Consortium 2007). Statistical methods are necessary to assess the coding potential of this “black matter” transcription (Frith et al. 2006). Similarly, comparative sequencing has revealed a plethora of evolutionarily conserved regions without other annotation (Siepel et al. 2005). A reliable analysis of the coding potential of these regions is an essential step preceding any downstream analysis.
Evolutionary analysis has previously proved useful for de novo detection of coding regions. Various algorithms have been developed to predict coding potential in pairwise alignments (Badger and Olsen 1999; Rivas and Eddy 2001; Mignone et al. 2003; Nekrutenko et al. 2003), and the power of multi-species comparison for the purpose of coding region prediction was demonstrated impressively in yeast (Kellis et al. 2003), human (Clamp et al. 2007), and more recently in 12 drosophilid genomes (Stark et al. 2007; Lin et al. 2008). There is no doubt that these types of analysis are powerful and useful additions to classical gene finders.
In this study, we introduce “RNAcode,” a program to detect protein-coding regions in multiple sequence alignments. The initial motivation was to use RNAcode in combination with the widely adopted structural RNA gene-finding program RNAz (Washietl et al. 2005). Similar in spirit to the program QRNA (Rivas and Eddy 2001), the goal is to produce more accurate annotations of ncRNAs by combining information from explicit models for structural RNAs and protein-coding RNAs. The direct identification of conserved regions as protein coding can reduce the number of false-positive ncRNA predictions, which is still the main problem in large-scale screens (Washietl et al. 2007).
More generally, RNAcode was designed to fill a specific gap in the current repertoire of comparative sequence analysis software. It provides the following features for which, to our knowledge, no other program is available: (1) RNAcode relies on evolutionary signatures only and is based on a direct statistical model. No machine learning or training is involved, and it can thus be applied in a generic way to data from all species. (2) It makes use of all evolutionary signatures that are known to be relevant rather than focusing on one particular feature. (3) It predicts local regions of high coding potential together with an estimate of statistical significance in the form of an intuitive _P_-value. (4) RNAcode deals with real-life issues such as sequencing and alignment errors. (5) It is provided as a robust, platform-independent, and easy-to-use C-implementation that is applicable to the analysis of selected regions and that can be integrated in annotation pipelines of larger scale.
ALGORITHM
Evolutionary changes in the nucleotide sequence of coding genes typically preserve the encoded protein. This type of negative (stabilizing) selection leads to frequent synonymous and conservative amino acid mutations, insertions/deletions preserving the reading frame, and the absence of premature stop codons. Our algorithm integrates this information in a unified scoring scheme. It takes as input a multiple nucleotide sequence alignment including a “reference” sequence, which is the one we wish to search for potential coding regions, and predicts local segments that show statistically significant protein-coding potential. Figure 1 shows an overview of the algorithm that is described in more detail in the following sections. First, we introduce a scoring scheme that acts on pairwise alignments and considers amino acid substitutions and gap patterns. Second, we describe how maximum scoring regions under this scheme can be computed for a multiple alignment by considering all pairwise combinations of a reference sequence to the other sequences in the alignment. Third, we indicate how assessment of the statistical significance of these regions can be performed.
FIGURE 1.

Overview of the RNAcode algorithm. First, a phylogenetic tree is estimated from the input alignment including a reference sequence (darker line) under a noncoding (neutral) nucleotide model. From this background model and a protein similarity matrix, a normalized substitution score is derived to evaluate observed mutations for evidence of negative selection. This substitution score and a gap scoring scheme are the basis for a dynamic programming (DP) algorithm to find local high-scoring coding segments. To estimate the statistical significance of these segments, a background score distribution is estimated from randomized alignments that are simulated along the same phylogenetic tree. The parameters of the extreme value distributed random scores are estimated and used to assign _P_-values to the observed segments in the native alignment.
Amino acid substitutions
Consider two aligned nucleotide triplets a and b that correspond to two potential codons. To see if they encode synonymous or biochemically similar amino acids, we can translate the triplets and use amino acid similarity matrices such as the widely used BLOSUM series of matrices (Henikoff and Henikoff 1992). Let Aa and Ab be the translated amino acids of the triplets a and b, respectively, and S(Aa,Ab) their BLOSUM score. In absolute terms, this score is of little value: Highly conserved nucleotide sequences will get high amino acid similarity scores upon translation even when noncoding.
We need to ask, therefore, what is the expected amino acid similarity score assuming that the two triplets evolve under some noncoding (neutral) nucleotide model. Deviations from this expectation will be evidence of coding potential. To this end, we estimate a phylogenetic tree for the input alignment using a maximum-likelihood method under the well-known HKY85 nucleotide substitution model (Hasegawa et al. 1985). Furthermore, we note that two aligned triplets can have zero, one, two, or three differing positions, i.e., they can have a Hamming distance h(a, b) ∈ {0,1,2,3}. It is straightforward to calculate the expected score for a given protein matrix, a parametrized HKY85 background model, and a given Hamming distance x:
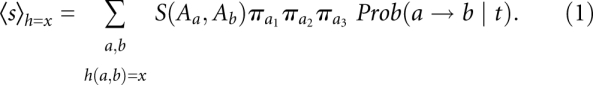
Here _a_1, _a_2, and _a_3 denote the first, second, and third nucleotide in triplet a; π is the stationary frequency in the HKY85 model; and Prob(a → b|t) is the probability that triplet a changes to b after some time t. The analytic expression for this probability is given by Hasegawa et al. (1985). The pairwise evolutionary distance t between two sequences is calculated as the sum of all branch lengths separating the two sequences in the estimated phylogenetic tree.
Put in simple terms, the score <_s_> is the average score over all possible pairs weighted by the probability of observing such a pair under our background assumption. We condition on the observed Hamming distance h(a, b) because this reduces the effect of implicit information on average amino acid frequencies contained in the BLOSUM matrix, and was found to give better results. We can use this expected score <_s_> to normalize our observed scores s arriving at the final protein-coding score σ for an aligned triplet:

To illustrate this with an example, consider the aligned triplets GAA and GAT. The triplets encode glutamic acid and aspartic acid, respectively, and score s = +3 in the BLOSUM62 matrix. Furthermore, assume that under some background model, the expected score for pairs with one difference is <_s_>h = 1 = −1. The overall score is thus σ = 3 − (−1) = +4. The positive score reflects the conservative mutation between the biochemically similar amino acids. A synonymous mutation usually gives the strongest support for negative selection. Since it also gives the highest scores in any protein matrix, there is no need to treat it differently from conservative mutations, and we can score both types of mutations using the same rules. Under this simple scoring scheme, the average triplet score in a coding alignment under negative selection will be positive, while in noncoding alignments, it will be 0 on average. We found that the HKY85 substitution model accurately models noncoding regions for this particular purpose (see the Results section).
Reading frames and gaps
It is straightforward to score an alignment that does not contain gaps. The alignment can simply be translated in all reading frames and the resulting triplets assigned a substitution score σ as described above. Real alignments, however, usually contain gaps. For the purpose of finding coding regions, gap patterns contribute valuable information (Kellis et al. 2004). Negative selection not only acts on the type of amino acid but also on the reading frame that is generally preserved when insertions/deletions occur. Our algorithm incorporates this information into the scoring scheme and, in addition, also deals with practical problems that occur in real-life data such as alignment and sequencing errors. Figure 2 shows some selected gap patterns to illustrate the basic principles. A more formal specification of the algorithm can be found in the Appendix.
FIGURE 2.

Examples of typical gap patterns and scoring paths in a pairwise alignment assumed to be coding. Nucleotides are shown as blocks, codons as three consecutive blocks of the same shading. (A) A gap of length three does not change the reading frame and in-frame-aligned codons are scored with the normalized substitution score σ. (B) A single gap destroys the reading frame but gets corrected downstream by another gap. The triplets that are out-of-phase because of this obvious alignment error are penalized by the two frameshift penalties Ω and ω. (C) A single gap that, in principle, destroys the reading frame is interpreted as a sequence error. Penalized by a high negative score Δ, this frameshift is ignored, and downstream codons are considered to be in-phase.
In real coding regions we will frequently encounter gap lengths that are multiples of three that do not break the coding frame (Fig. 2A). We treat this kind of gap neutrally and give it a score of 0. The aligned triplets before and after the gap are in the same phase and thus can be assigned a score of σ.
Any gap not a multiple of three will result in a frameshift and the sequences are out-of-phase. We assign a penalty score Ω < 0 for the frameshift event and each subsequent aligned triple that is out-of-phase receives an additional smaller penalty ω < 0. Changing the frame back is also penalized, again by Ω (Fig. 2B). The basic idea is that noncoding regions have many frameshifts, and long stretches in the same frame are rare. In contrast, coding regions should not have any frameshifts at all. In real data frameshifts can also be observed in coding regions because of alignment errors. However, they usually get reverted soon by another gap. Consequently, only relatively short regions are out-of-frame.
Gaps in coding regions that are not a multiple of three can also be the result of sequence errors. This is particularly problematic for low-coverage sequencing. In order not to miss substantial parts of true coding regions that appear to be out-of-frame because of a single sequence error, we allow change of the phase and penalize this event with a negative score Δ (Fig. 2C). Clearly, this event should be rare and hence the penalty must be high; the condition Δ < 2Ω must be met at least, or otherwise a sequence error event would always be chosen as a more favorable explanation than the frameshifting gaps in the optimization algorithm described below.
Stop codons
Under normal conditions, a reading frame cannot go beyond a stop codon. To reflect this in our algorithm, stop codons in the reference sequence get a score of −∞. We allow relaxation of this for stop codons in the other sequences because if they are of low quality, erroneous stop codons might be observed. These should not automatically destroy a potentially valid coding region but rather be penalized with a relatively large negative score.
Calculating the optimal score for a pairwise alignment
Using the scoring scheme introduced above, we need to find the interpretation of a given alignment as aligned codons in a particular reading frame, out-of-frame codons, and sequence errors that maximizes the score. This is achieved by a dynamic programming algorithm that is described in full detail in the Appendix.
Finding maximum scoring segments in a multiple alignment
To find regions of high coding potential in a multiple sequence alignment, we first consider the pairwise combinations of the reference sequence with each other sequence. In these pairwise alignments, we calculate the optimal score of each alignment block delimited by two columns i and j using the dynamic programming algorithm. Once the maximum scores have been found for each pairwise alignment, we take the average of all pairs and store the optimal scores for the blocks between any two columns i and j of the multiple alignment in a matrix Sij (for details, see Appendix). In this matrix, we identify maximal scoring segments, i.e., segments with a positive score that cannot be improved by elongating the segment in any direction. This approach is meaningful because in noncoding regions the average substitution score is ≈0 and gaps can only contribute negative scores.
Statistical evaluation
To assess the statistical significance of high scoring segments, we empirically estimate the score distribution of neutral alignments conditional on the phylogeny derived from the alignment under consideration. Again, we use the phylogenetic tree estimated under the HKY85 model as our null model. We simulate neutral alignments along this tree and calculate high-scoring segments in exactly the same way as for the native alignment. The score distribution follows an extreme value distribution, and we found that it is well approximated by the Gumbel variant with two free parameters (see the Results section). Fitting this distribution allows us to calculate a _P_-value for every high-scoring segment actually observed. This _P_-value expresses the probability that a segment with equal or higher score would be found in the given alignment by chance.
RESULTS
Classification accuracy
We tested RNAcode on six different comparative test sets. These test sets were created from genome-wide alignments (Blanchette et al. 2004; Schneider et al. 2006; Kuhn et al. 2009) typical of those that are widely used for comparative analysis today. The set consisted of alignments of E. coli with nine enterobacteria, Methanocaldococcus jannaschii with 10 methanogen Archaea, Saccharomyces cerevisiae with six other Saccharomyces strains, Drosophila melanogaster with 11 drosophilid species and three other insects, Caenorhabditis elegans with five other nematode species, and Homo sapiens aligned to 16 vertebrate genomes. From these alignments, we extracted both annotated coding regions/exons and randomly chosen regions without coding annotation. We then calculated the maximum coding potential score and its associated _P_-value for each alignment. We did not include explicit information on the reading direction, i.e., the coding regions were randomly either in forward or reverse complement direction and both directions were scored.
A typical score distribution (Fig. 3A) shows that random noncoding regions generally do not contain maximal scoring segments with scores higher than 15, whereas coding regions show a wide range of maximal scoring segments of much higher scores. The score efficiently discriminates coding and noncoding regions. Receiver operating curves (ROC) show the sensitivity and specificity of the classification at different score cutoffs (Fig. 3B). In general, we observe the area under the curves (AUC) of the ROCs to be close to 1, i.e., close to perfect discrimination. Usually, the high specificity range (Fig. 3B, insets) is of particular interest for large-scale analysis. At a false-positive rate of 0.05%, for example, we can detect ∼90% of coding regions in all six test sets.
FIGURE 3.

RNAcode results on comparative test sets from various species. (A) Score distributions of annotated coding regions and randomly chosen noncoding regions in the Drosophila test set. (B) ROC curves for all six test sets. The full curve for all ranges of sensitivity/specificity from 0 to 1 is shown in the main diagrams. (Insets) The high specificity rate with false positive rates from 0 to 0.1. (C) Score distribution of noncoding alignments. The same distribution of the Drosophila test set as shown in A is shown in more detail. The fitted Gumbel distribution is shown as dotted line. (Upper right diagram) Comparison of the calculated _P_-values (via simulation and fitting of the Gumbel distribution) to the empirical _P_-values, i.e., the actual observed frequencies in the test set.
Accuracy of _P_-value estimates
The fact that the amino acid similarity scores used in our scoring scheme are adjusted by the expected score under a neutral null model ensures that the RNAcode score is properly normalized with respect to base composition and sequence diversity (phylogeny). In other words, the RNAcode score is independent of sequence conservation and GC content. Unlike other abstract classifiers, it is therefore possible to interpret and compare scores in absolute terms. However, even more important is an accurate estimate of the statistical significance of a prediction. Similar to the well-known statistics of local alignments (e.g., BLAST), RNAcode scores follow an extreme value distribution (Fig. 3C). This allows us to calculate _P_-values (see the section “Statistical Evaluation”).
To test the accuracy of this approach, we compared _P_-values calculated by this procedure to empirically determined _P_-values on a set of noncoding Drosophila alignments. To this end, we calculated the _P_-value for each alignment in the set and compared each to the proportion of alignments with better scores than the given one (Fig. 3C, inset). The excellent agreement of the _P_-values calculated by RNAcode and the actual observed frequencies confirms that the Gumbel distribution is an accurate approximation of the background scores. In addition, it also confirms that the HKY85 nucleotide substitution model and our simulation procedure accurately model real noncoding data.
Influence of parameter choice
The frameshift penalties in our algorithm are user-definable parameters. We found that the algorithm is relatively robust with respect to the particular choice of these parameters. Three different sets of parameters gave almost identical results (Supplemental Fig. 1). However, ignoring information from gap patterns altogether by setting all penalties to a neutral value of zero leads to a drop in classification performance. This shows that gap patterns do, indeed, hold relevant information for classification although most information is contained in the substitution score, a result that is consistent with previous reports (Lin et al. 2008).
Comparison to other comparative metrics
To further evaluate the performance of our new approach, we have created a more extensive data set that systematically covers alignments with varying numbers of sequences and different conservation levels (see Materials and Methods). On this data set, we have compared the RNAcode substitution score to two other commonly used metrics that are based on evolutionary signatures.
The ratio of nonsynonymous (dN) to synonymous substitutions (dS) gives information on the type of selection acting on a protein-coding sequence (Yang and Nielsen 2000). A low dN/dS ratio indicates negative selection, which was found to be a reliable way to detect coding regions in pairwise (Nekrutenko et al. 2003) and multiple alignments (Lin et al. 2008). The structure of the genetic code leads to a periodic pattern of evolutionary rates (Bofkin and Goldman 2007), another characteristic of protein-coding regions that was applied, for example, to assess the coding potential of unannotated transcripts in S. cerevisiae (David et al. 2006) and in human in the ENCODE pilot project (The ENCODE Project Consortium 2007).
We calculated the dN/dS ratio for all alignments in our data sets using a maximum likelihood method (Yang and Nielsen 2000). To quantify the substitution rate periodicity, we re-implemented a likelihood test described previously (Materials and Methods) (The ENCODE Project Consortium 2007). In essence, it compares a null model with equal rates for each nucleotide position to an alternative model allowing for a periodic pattern “…ABCABCABC…” of rates. It thus captures the periodicity of the codons without the need to explicitly determine the reading direction or frame.
We found that the RNAcode substitution score consistently outperforms the dN/dS ratio and the periodicity score (Fig. 4). The difference is particularly pronounced for alignments of low sequence conservation. These alignments presumably contain more conservative amino acid substitutions, which RNAcode—in contrast to the dN/dS ratio—can take advantage of. Interestingly, the fact that the dN/dS ratio and the periodicity score are calculated over a phylogenetic tree for the complete alignment does not lead to better performance than the RNAcode score, which is calculated from pairwise comparisons.
FIGURE 4.

Comparison of the RNAcode substitution score with other comparative metrics. The ROC curves show the classification performance of the dN/dS ratio, substitution rate variation, and the average substitution score σ used by RNAcode. Results are shown for alignments of length 30 from vertebrates, archaebacteria, yeasts, and drosophilid species grouped by the number of sequences in the alignment (N) and the mean pairwise sequence identity (MPI). The area under the ROC curve (AUC) as a measure for classification performance is shown for all methods and sets.
Influence of alignment properties
The performance of RNAcode depends on the evolutionary information contained in the alignment. The results shown in Figure 4 illustrate this dependency in terms of alignment size and sequence diversity. In the extreme case of pairwise alignments with very low sequence diversity (90%–100% mean pairwise sequence identity), the classification performance is relatively poor (AUC < 0.9). Adding more sequences (N = 4) and higher sequence diversity (identities below 90%) leads to much better performance (AUC ≈ 0.99). Adding even more sequences (N = 8) results in further improvement and almost perfect discrimination. We conclude that alignments with as few as four sequences that are <90% identical will give satisfactory results in practical applications of RNAcode.
The alignment method used might affect performance. All tests in this study were run on genome-wide alignments generated by MultiZ (Blanchette et al. 2004). We found that re-aligning with other commonly used alignment programs did not change our results (Supplemental Fig. 2).
Automatic annotation of Drosophila genome
The main purpose of RNAcode is to classify conserved regions of unknown function, to discriminate coding from noncoding transcripts, and to analyze the coding potential in non-standard genes (e.g., short ORFs or dual-function RNAs; see below for examples). RNAcode's algorithm is built on a direct statistical model that deliberately ignores any species-specific information and does not resort to machine learning. RNAcode is thus not optimized for the genome-wide annotation of protein-coding genes in well-known model organisms. However, to demonstrate that RNAcode is also efficient for this purpose and to study our algorithm in direct comparison to today's best gene finders, we automatically annotated chromosome 2L (≈23 Mb) of the D. melanogaster genome. We ran RNAcode with standard parameters and a _P_-value cutoff of 0.001 on MultiZ alignments available at the UCSC Genome Browser and compared the results to FlyBase (Drysdale and FlyBase Consortium 2008) annotation. Of the 10,535 annotated coding exons in FlyBase, 9245 overlapped (by at least one nucleotide) with an RNAcode prediction (sensitivity 87.8%). In total, RNAcode predicts 13,166 high-scoring coding regions with p < 0.001. Of these, 12,207 had overlap with one of the annotated exons, i.e., 959 were false positives (specificity: 92.7%). This result is surprisingly close to the currently best “full” gene finders. In the same overlap statistics, CONTRAST (Gross et al. 2007) achieves 91.0%/97.0% (sensitivity/specificity) and NSCAN (Gross and Brent 2006) 91.8%/97.2%. These algorithms can take advantage of species-specific features such as splice site signals, codon usage, exon length distributions, etc., information that is not available when studying non-model organisms or atypical genes (see below for examples). Our results show that evolutionary events alone hold a considerable amount of information and that RNAcode efficiently makes use of it.
Novel peptides in E. coli
The E. coli genome was one of the first completely sequenced genomes and is generally well annotated. However, even in this compact and extensively studied genome, the protein annotation is far from perfect. Protein gene annotation is largely based on compositional analysis and homology with known protein domains. The statistical power of these criteria is limited for small proteins. Standard gene-finding software is usually run with an arbitrary cutoff of 40–50 amino acids to avoid an excess of false positives and suffers from the lack of training data of verified short peptides.
Here, we attempted to produce a set of predictions based on evolutionary signatures only. We created alignments of the E. coli reference strain K12 MG1655 to 53 other completely sequenced enterobacteria strains including Erwinia, Enterobacter, and Yersinia (see Materials and Methods) (Supplemental Table 1). A screen of these alignments with RNAcode and a _P_-value cutoff of 0.05 resulted in 6542 high-scoring coding segments. We discarded all predictions that overlapped annotated proteins. For the remaining RNAcode predictions, we tried to identify a complete ORF (starting with AUG and ending in a stop codon) in the E. coli reference sequence (see Materials and Methods). This step is necessary because the boundaries of high-scoring segments usually do not correspond exactly to the ORF (a main problem here is the relatively short alignment blocks produced by MultiZ, which do not always cover an ORF over its full length). This procedure gave 35 potential new protein-coding genes between 11 and 73 amino acids in length (see Supplemental Table 2).
To assess the quality of these predictions, we first looked at the overall sensitivity of our screen on already annotated proteins. Of the 4267 RefSeq proteins, 3987 overlapped with a RNAcode prediction (sensitivity 93.4%). Hemm et al. (2008) revisited the annotation of small proteins in E. coli and found 18 novel examples using a combination of different bioinformatics and experimental methods. In a set of 18 new and 42 literature-curated proteins between 16 and 50 amino acids compiled by Hemm et al. (2008), 30 (50.0%) overlap with RNAcode predictions. These results show that our screen not only gives almost perfect results on typical E. coli proteins, but also recovers a substantial fraction of small proteins that are particularly difficult to detect. Moreover, our final list of 35 candidates for novel proteins is rather short and shows the high specificity in this screen.
For additional support, we compared our list of predicted candidates with publicly available transcriptome data (Tjaden et al. 2002; Cho et al. 2009). These data sets cover a broad range of experimental conditions and therefore reflect a comprehensive genome-wide transcription map of E. coli. Eight candidates (23%) overlap with regions that show clear evidence for transcription (Supplemental Table 2).
To further substantiate our predictions, we used mass spectrometry (MS) as a direct experimental test for the existence of the novel peptides in E. coli cells. MS is particularly well suited to screen simultaneously for a large set of proteins without resorting to cloning or recombinant expression (Aebersold and Mann 2003). Many, but by no means all, proteins of an organism are expressed and detectable under the actual applied conditions by current MS-based proteomics. Detecting small peptides in complex protein mixtures is particularly challenging for various reasons. Compared to the overall protein expression level, short peptides often show low abundance, they are easily lost using standard proteomic protocols, and only a limited number of proteolytic peptides can be obtained (Klein et al. 2007). To meet these challenges, we developed a protocol that is specifically optimized for small proteins by avoiding sample loss by a simple extraction method and a combined purification and enrichment step using filtration (Müller et al. 2010; Materials and Methods). In order to improve the reliability of our results, we applied two different buffer systems for extractions, and for an improved coverage of peptides, we used two different proteases. This strategy led to an increased detection rate as well as to higher confidence in the hits by confirmation in independent experiments.
Using this protocol, we were able to identify 455 small molecular weight proteins (MW < 25 kDa) representing 27% of the 1672 known E. coli proteins below this size listed in the SWISS-PROT protein database (UniProt Consortium 2010). In a search against the list of 35 newly predicted proteins, we obtained evidence for the expression of seven candidates (20%) (Supplemental Table 3). For the rest of the candidates, we cannot distinguish whether they are false-positive RNAcode predictions or false negatives in the MS experiment. However, considering that the success rate of the MS experiments is roughly the same on known and predicted proteins (27% and 20%, respectively), we would expect a good fraction of our candidates to be true proteins not detectable by this particular growth conditions and MS approach.
Although it is not possible to give a conclusive statement on all predictions without additional experiments, compelling evidence from evolutionary analysis, transcriptomics data, and the MS experiments strongly suggests that several of the candidates are bona fide proteins. Figure 5 shows two examples in more detail. In both cases, RNAcode reported short but statistically highly significant (p ≈ 10−8 and p ≈ 10−6, respectively) signals between two well-annotated proteins. The loci overlap with transcribed regions as determined by Cho et al. (2009). In addition, our MS experiments detected several proteolytic fragments that can be assigned to these proteins.
FIGURE 5.

Examples of novel short proteins in Escherichia coli. Sequence, genomic context, the high-scoring RNAcode segment, and fragment ion mass spectra are shown. Genome browser screenshots were made at http://archaea.ucsc.edu (Schneider et al. 2006). Arrows within annotated elements indicate their reading direction. The shading of mutational patterns was directly produced by the RNAcode program. The full species names for the abbreviations can be found in Supplemental Table 1. The mass spectra are shown for two selected proteolytic peptides, which were scored with 80% probability and used in combination with the detection of additional peptides to confirm the expression of the candidates (for details, see Supplemental Table 3). The proteins shown in A and B correspond to candidates 28 and 19, respectively, listed in Supplemental Tables 2 and 3.
The coding potential of “noncoding” RNAs
In addition to assisting and complementing classical protein gene annotation strategies, a major area of application of RNAcode is the functional classification of individual conserved or transcribed regions. As an illustrative example, we analyzed the bacterial RNA C0343, which is listed in the Rfam database (Gardner et al. 2009) as noncoding RNA (ncRNA) of unknown function. The RNA originally detected by Tjaden et al. (2002) is also detected as transcript in the study of Cho et al. (2009) (Fig. 6). In our screen of the E. coli genome, we found a high-scoring coding segment with p ≈ 10−9 overlapping the C0343 ncRNA. The prediction corresponds to a potential ORF encoding 57 amino acids (Fig. 6A; candidate 8 in Supplemental Table 2). Analysis of the secondary structure using RNAz (Gruber et al. 2010) does not give any evidence for a functional RNA. Given the strong coding signal, we conclude that the “noncoding RNA” C0343 is, in fact, a small protein. This is also confirmed by our MS experiments that detected proteolytic fragments of this protein in E. coli cells (Supplemental Table 3).
FIGURE 6.

Examples of ambiguities between the coding and noncoding nature of three RNAs. (A) The RNA C0343 from E. coli is listed as a noncoding RNA in Rfam. However, it overlaps with an RNAcode-predicted coding segment. While there is no evidence for a RNA secondary structure according to the RNAz classification value, the highly significant RNAcode prediction and MS experiments suggest that C0343 is an mRNA and not an ncRNA. (B) RNAIII of Staphylococcus aureus (Rfam RF00503) contains a short ORF of a hemolysin gene. RNAcode predicts the open reading frame at the correct position, while RNAz clearly detects a structural signal. These results are consistent with the well-established dual nature of this molecule. (C) The Bacillus subtilis RNA SR1 is known to have function on the RNA level by targeting an mRNA. RNAcode detects a short ORF that was shown by Gimpel et al. (2010) to produce a small peptide and is thus another example of a dual-function RNA.
To test RNAcode on another example from Rfam, we analyzed RNAIII, an ncRNA known to regulate the expression of many genes in Staphylococcus aureus (Boisset et al. 2007). In addition to its role as regulatory RNA, the RNAIII transcript also contains an ORF coding for the 26-amino-acid-long delta-haemolysin gene (hld). We ran RNAcode with standard parameters on the Rfam seed alignment. It reports one high-scoring segment below a _P_-value cutoff of 0.05, which corresponds to the hld gene (Fig. 6B). The annotated alignment shows that the ORF is highly conserved with only few mutations. Nevertheless, these few mutations are sufficient to yield a statistically significant signal that allows RNAcode to locate the correct ORF. Again, we also ran RNAz on the alignment, which reports a conserved RNA secondary structure with a probability of 0.99. The combination of RNAcode and RNAz clearly shows the dual function of RNAIII. This example demonstrates how RNAcode can assist the classification of ncRNAs in particular for non-standard and ambiguous cases (Dinger et al. 2008).
As another example, we analyzed the SR1 RNA of Bacillus subtilis that was originally found by Licht et al. (2005) (Fig. 6C). Although the investigators noticed a potential short ORF in the transcript, the corresponding peptide could not be detected. Further experiments (Heidrich et al. 2006, 2007) clearly showed a function of SR1 in the arginine catabolism pathway by RNA/RNA interaction with the ahrC mRNA, thus confirming its nature as functional noncoding RNA. Using RNAcode, we found clear evolutionary evidence for a well-conserved small peptide deriving from SR1 (p ≈ 10−12), arguing for a role as dual-function RNA. Only recently, Gimpel et al. (2010) showed that the gapA operon is regulated by a short peptide encoded in SR1, which exactly corresponds to the high-scoring coding segment found by RNAcode (Fig. 6C).
Finally, we analyzed the tarsal-less gene mentioned in the Introduction (Galindo et al. 2007; Kondo et al. 2007). The small peptides produced by this unusually organized polycistronic gene were overlooked originally, and it was thought to be noncoding. Analysis using RNAcode predicts three significant high-scoring coding segments (_P_-values = 2.4 × 10−5, 5.5 × 10−5, 0.010) in this transcript, covering one known peptide and partially covering a second. Using a relaxed _P_-value cutoff, four of the five known peptides are identified (Supplemental Fig. 3).
Implementation and performance
RNAcode is implemented in ISO C. The program takes an alignment in either CLUSTAL W format or MAF format (popularized through the UCSC Genome Browser). It outputs relative coordinates and/or genomic coordinates of predicted coding regions, the raw score, and the _P_-value in either a human readable tabular format or as standard GTF annotation format. In addition, RNAcode offers an option to generate color annotations of the alignment. This kind of visualization helps to quickly identify mutational patterns, which allows visual discrimination between alignments of high and low coding potential. RNAcode produces publication-quality vector graphics in Postscript (EPS) format (see, e.g., Figs. 5, 6). To generate the color annotated images, it is not enough to know just the region and score of the high-scoring segments, but we also have to infer the state path that led to this prediction. Therefore, we have also implemented the backtracking step for the dynamic programming algorithm. In addition to the mutation patterns, this allows annotation of regions that are likely to be out-of-phase and the location of potential sequence errors inferred by the algorithm.
The dynamic programming algorithm used to score an alignment of N sequences with n columns requires 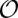 (N · _n_2) CPU time and memory. Large genomic alignments are therefore broken up into windows of several hundred nucleotides in length in practical applications (see Materials and Methods). There is nothing to be gained by feeding RNAcode with alignment windows that are longer than actual contiguous pieces of coding sequence.
(N · _n_2) CPU time and memory. Large genomic alignments are therefore broken up into windows of several hundred nucleotides in length in practical applications (see Materials and Methods). There is nothing to be gained by feeding RNAcode with alignment windows that are longer than actual contiguous pieces of coding sequence.
The analysis of 1 Mb of Drosophila MultiZ alignments with up to 12 species (10,426 alignment blocks) took 2 h and 6 min on a single Pentium 4 CPU running at 3.2 GHz. This includes calculation of _P_-values with 100 randomizations for all predictions. However, it is generally not of interest to calculate exact _P_-values for hits that are clearly not statistically significant. Therefore, we added an option to stop the sampling procedure as soon as too many of the randomizations score better than the original alignment (e.g., for 1000 randomizations and a significance level of p < 0.05, the sampling would stop after 50 random alignments with a better score than the native alignment). Depending on the density of coding regions in the input alignments, this simple heuristic can speed up the process considerably. Using this option, the 1 Mb of fly alignments could be scored in 1 h and 4 sec without any loss in sensitivity or specificity.
DISCUSSION
We have introduced RNAcode as a comparative genomics tool for the identification of protein-coding regions. Inspired by our own experiences in analysis of comparative sequence data in the context of ncRNA annotation, the design emphasized practicability and robustness and focused on the single task of discriminating protein-coding from noncoding regions. RNAcode therefore is not a gene-finder. By design, it neither uses nor predicts any features related to transcript structure such as splice sites, processing sites, or termination signals. Its direct statistical model is based on universal evolutionary signatures of coding sequence only. RNAcode is therefore a true ab initio approach that can be applied to data from all living species. In fact, it does not need any information on the source of its input data, facilitating, e.g., the application to meta-genomics data (Meyer et al. 2009; Shi et al. 2009).
We evaluated a variety of alternative possible metrics and algorithms, but found that pairwise BLOSUM-derived substitution scores together with the relatively simple gap scoring scheme presented was the most efficient solution. We were surprised that this algorithm also outperformed more sophisticated phylogenetic models acting on the whole tree. An exact dynamic programming scheme is used to determine high-scoring coding blocks in the input alignment in a way that is robust against sequence and alignment errors.
Although we do not include any species-specific features such as codon usage or splicing signals, the approach shows remarkable accuracy. Without any training or specifically optimizing the parameters, RNAcode could successfully discriminate between coding and noncoding regions in vertebrates, insects, nematodes, yeasts, bacteria, and even archaea that show a highly biased GC content. We also showed that it can reproduce accurately the current annotation in D. melanogaster and identified novel peptides in E. coli that have previously evaded annotation in this intensively studied organism. Case studies on individual examples of ncRNAs showed that RNAcode can help to identify mis-annotated ncRNAs and, in combination with RNAz, can identify dual-function RNAs.
The high discrimination performance in combination with accurate _P_-values, visualization, and the readily available open source implementation make RNAcode, we hope, an attractive and easy-to-use solution for many different applications in comparative genomics.
MATERIALS AND METHODS
Implementation details
To estimate the phylogenetic tree for the null model, we use a maximum likelihood implementation provided by PHYML (Guindon and Gascuel 2003). To simulate random alignments along this tree, we use code from Seq-Gen (Rambaut and Grassly 1997).
As a technical detail, we note that our simulation procedure does not simulate gap patterns. Instead, we simulate the alignments without gaps and introduce the original gap patterns afterward. The _P_-values for true coding regions are thus conservative because we use the coding gap pattern also for the background. There are algorithms to simulate the evolution of insertions and deletions. However, it is hard to estimate realistic parameters for these models, and thus we chose this conservative approach that has been successfully used in other applications (Goldman et al. 1998; Gesell and Washietl 2008).
We used the versions of the BLOSUM matrices that are provided with the EMBOSS package (Rice et al. 2000). The current implementation of RNAcode includes the EMBOSS62 and the EMBOSS90 matrices.
For fitting the extreme value parameters to the empirical score distributions, we used an implementation from Sean Eddy's HMMER package (http://hmmer.janelia.org).
Alignment data and benchmarks
Multiple sequence alignments were downloaded from the UCSC Genome Browser (http://genome.ucsc.edu; http://archaea.ucsc.edu). We used the following assemblies, alignments, reference annotations, and (if applicable) selected chromosomes, respectively: H. sapiens: hg18, multiz18, UCSC Genes, chr22; D. melanogaster: dm3, multiz15, FlyBase Genes (version 5.12), chr2L; C. elegans: ce6, multiz6, WormBase Genes (version WS190), chr5; S. cerevisiae: sacCer1, multiz7, SGD Genes (version from 01/30/2009), chr4; E. coli: eschColi_K12, multizEnterobacteria, GenBank RefSeq; M. jannaschii: methJann1, multizMethanococcus, GenBank RefSeq. All data from UCSC were downloaded around the middle of 2009.
To generate the positive test set of known exons, we first extracted alignment blocks corresponding to the annotated exons in the reference annotation. If an exon was covered by several blocks, these were merged. If the resulting alignment was longer than 200 columns, we only used the first 200 columns. As negative control, we selected a comparable number of random blocks that do not overlap annotated coding exons or repeats.
For the tests shown in Figure 4, we selected from the complete set of coding exons a balanced subset of alignments of varying window length (30 nt, 60 nt, 90 nt), varying number of sequences (N = 2, 4, 8), and mean pairwise identity (60%–100%). We discarded alignment windows that contained gaps and stop codons in any of the sequences so that they could be directly analyzed using PAML. It is unclear how to handle frameshifts and internal stop codons when calculating a phylogenetic model using PAML, which is not gene-finding software per se. By limiting the analysis to in-frame-aligned sense codons, we ensure a fair comparison to RNAcode that can take advantage of information in gap patterns and stop codons. To calculate the dN/dS ratio, we used the codeml program with the default codon model (“model 0”). The periodicity score is calculated as the log-likelihood ratio between two models. As the null model, we used an HKY nucleotide substitution model (“model 4” in PAML's baseml) with equal rates for each site. The alternative model considers three rate classes in a periodic pattern “…ABCABCABC…”. The maximum likelihood tree under this model was calculated using the partition model functions of baseml. We used the option “Mgene = 0” keeping all other parameters (κ and π) of the HKY model constant in all three rate classes. The results in Figure 4 are shown for length = 30; sets of length 60 and 90 show qualitatively similar results but saturate earlier to perfect discrimination (data not shown).
E. coli screen
For the screen of novel proteins in the E. coli genome, we generated multiple sequence alignments of our own because we noticed that the available alignments at UCSC missed many known coding regions. Moreover, we wanted to improve the evolutionary signal by adding additional species. We used the MultiZ alignment pipeline to align 54 species available from GenBank (Supplemental Table 1).
We then screened the alignments using the default parameters of RNAcode and a _P_-value cutoff of 0.05. This resulted in 20,528 high-scoring coding segments. This number is much higher than the actual number of ORFs mainly because the MultiZ alignments of such a high number of species fragmented the ORFs into relatively small blocks. We combined high-scoring coding segments if they were closer than 15 nt apart and in the same frame, yielding 6542 regions. We discarded all regions that overlapped with an annotated ORF, leaving 229 regions. For these regions, we inferred potential ORFs starting with an ATG and ending in a canonical stop codon. If we did not find an ORF within the RNAcode high-scoring segment, we extended the prediction by 51 nt upstream and downstream and repeated the search. We found 35 loci with a potential ORF (Supplemental Table 2).
Transcriptomics data
The analysis of Cho et al. (2009) represents a comprehensive transcription map for E. coli. The corresponding supplemental data were downloaded from http://systemsbiology.ucsd.edu/publication and the Gene Expression Omnibus web page http://www.ncbi.nlm.nih.gov/geo/. The data were converted into BED and WIG formatted files and loaded as custom tracks into the UCSC for visualization and comparison to the novel predicted proteins.
Mass spectrometry experiments
Cell growth
E. coli strain K12 cells were grown in LB medium to stationary phase. One liter of fresh medium was inoculated with 100 mL of a starter culture grown under the same conditions. Cells were collected by centrifugation (10 min, 8000_g_, 4°C).
Protein preparation
Cells were resuspended in urea lysis buffer (40 mL, 8 M urea, 10 mM DTT, 1 M NaCl, 10 mM Tris/HCl at pH 8.0) (Klein et al. 2007) or acidic lysis buffer (40 mL, 0.1% TFA) (Dai et al. 1999) and disrupted using ultrasonication (5 min, 50% duty cycle, Branson Sonifier 250; Emerson, USA). Cell debris was removed by centrifugation (15 min, 10,000_g_, 4°C). High-molecular-weight proteins were depleted by centrifugation through a filter membrane (cutoff molecular weight 50 kDa, Pall Macrosep 50K; Pall Life Science, USA) (Harper et al. 2004). The flow-through was split into aliquots of 1200 μL. Where TFA was used for cell lysis, the samples were titrated to neutral pH by adding NH4HCO3 (final concentration 250 mM), and protein disulfide bonds were reduced by adding DTT (10 mM). Cysteine alkylation was conducted by adding 2-iodoacetamide (51.5 mM) and incubation for 45 min at room temperature in the dark.
Gel electrophoresis
Prior to protein separation by 1D gel electrophoresis, the proteins were desalted and concentrated by TCA precipitation (final concentration 20% [w/v]). The protein pellet was redissolved with SDS loading buffer (2% [w/v] SDS, 12% [w/v] glycerol, 120 mM 1,4-dithiothreitol, 0.0024% [w/v] bromophenol blue, 70 mM Tris/HCl) and adjusted to neutral pH by adding 10× cathode buffer solution (1 M Tris, 1 M Tricine, 1% [w/v] SDS at pH 8.25). Gel electrophoresis was performed according to Schägger (2006) (with slight modifications). In brief, a 20% T, 6% C separation gel combined with a 4% T, 3% C stacking was used. As protein marker, a prestained low-molecular-weight protein standard (molecular weight range 1.7 kDa–42 kDa, multicolor low-range protein ladder; Fermentas, Germany) was applied. For each cell lysis experiment, eight aliquots were used, of which two were stained with colloidal Coomassie, two were stored as a reserve, and four were used for further analysis. Nine gel slices per lane were excised between 1 and 25 kDa and used for in-gel digestion.
Protein digestion
The gel slices were washed twice with water for 10 min and once with NH4HCO3 (10 mM). The low-molecular-weight proteins were digested by adding modified porcine trypsin (100 ng; Sigma-Aldrich) or endoprotease AspN (100 ng; Sigma-Aldrich) in NH4HCO3 (10 mM, 30 μL volume). Digestion was performed overnight at 37°C. The supernatant and the solutions from two subsequent gel elution steps (first elution step 40% [v/v] acetonitril, second elution step 80% [v/v]) were collected and united. The samples were dried using vacuum centrifugation.
Mass spectrometry
For validation of the existence of the predicted protein by mass spectrometry, an unbiased bottom-up approach and a targeted analysis were applied. Peptides were reconstituted in 0.1% formic acid. Samples were injected by the autosampler and concentrated on a trapping column (nanoAcquity UPLC column, C18, 180 μm × 2 cm, 5 μm; Waters) with water containing 0.1% formic acid at flow rates of 15 μL/min. After 4 min, the peptides were eluted onto the separation column (nanoAcquity UPLC column, C18, 75 μm × 250 mm, 1.7 μm; Waters). Chromatography was performed with 0.1% formic acid in solvents A (100% water) and B (100% ACN). Peptides were eluted over 90 min with an 8%–40% solvent B gradient using a nano-HPLC system (nanoAcquity; Waters) coupled to an LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific). For an unbiased analysis, continuous scanning of eluted peptide ions was carried out between m/z 350 and 2000, automatically switching to CID-MS/MS mode upon detection of ions exceeding an intensity of 2000. For CID-MS/MS measurements, a dynamic precursor exclusion of 3 min was applied. For a targeted analysis, a scan range of m/z = 400–1800 was chosen. CID-MS/MS measurements were triggered if a precursor of a given inclusion list was measured with an error of <20 ppm. The inclusion lists contained all theoretically proteolytic peptides within a molecular weight range of 600 Da to 4000 Da of all predicted proteins considering methionine oxidation, cysteine carbamidomethylation, and up to one (for trypsin) or three (for AspN) proteolytic miscleavages.
Data analysis
Raw spectra were analyzed with ProteomeDiscoverer 1.0 software (Thermo Fisher Scientific, USA). Mascot (Perkins et al. 1999), Sequest (Yates et al. 1995), and X!Tandem (Craig and Beavis 2004) searches were conducted on a protein sequence database, which contains all sequences predicted by RNAcode (RNAcode database) as well as on an extended SWISS-PROT database containing protein sequences predicted by RNAcode and all validated proteins of Hemm et al. (2008). The searches were performed tolerating up to one proteolytic missed cleavage, a mass tolerance of 7 ppm for precursor ions, 0.5 Da for MS/MS product ions allowing for methionine oxidation (optional modification), and cysteine carbamidomethylation (fixed modification). Scaffold (version Scaffold_2_06_00; Proteome Software Inc.) was used to validate MS/MS-based peptide and protein identifications. Peptide identifications were accepted if they could be established at >50% probability as specified by the Peptide Prophet algorithm (Keller et al. 2002). Protein identifications were categorized to be unambiguously identified if they could be established at >99% probability and contained at least two identified peptides that had to achieve a score higher than 80%. Less stringent evidence for proteins was assigned if two peptides were observed with at least one peptide scored higher than 80% and the protein identification probability exceeds 90%. Protein probabilities were assigned by the Protein Prophet algorithm (Nesvizhskii et al. 2003). Additionally, the fragment spectra were checked manually.
Availability
RNAcode is open source software released under the GNU general public license version 3.0. The latest version is available at http://wash.github.com/rnacode.
The package includes a “Getting Started” guide that describes all steps involved in using RNAcode, including obtaining an alignment for analyses that start with a single sequence that is to be assessed for coding potential.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article. Additional data files can be downloaded from http://www.tbi.univie.ac.at/papers/SUPPLEMENTS/RNAcode.
ACKNOWLEDGMENTS
This work was supported in part by grants from the Wellcome Trust (grant 078968), the Deutsche Forschungsgemeinschaft (grant No. STA 850/7-1 under the auspices of SPP-1258, “Small Regulatory RNAs in Prokaryotes,” as well as the SFB Transregional Collaborative Research Centre 67: “Functional Biomaterials for Controlling Healing Processes in Bone and Skin—From Material Science to Clinical Application”), and the Austrian GEN-AU project “Noncoding RNA.” S.W. was supported by a GEN-AU mobility fellowship sponsored by the Bundesministerium für Wissenschaft und Forschung.
APPENDIX: DYNAMIC PROGRAMMING ALGORITHM
In the following, we formally describe the algorithms implemented in RNAcode. The core algorithm is a dynamic programming algorithm to find the optimal score for a pairwise alignment from all possible interpretations of the aligned sites as in-frame codons, out-of-frame codons, or sequence errors (cf. Fig. 2). The scores from pairwise alignments are then combined to find optimal scoring segments in a multiple alignment.
We start from a fixed multiple sequence alignment and assume that the first row is the reference sequence. The projected pairwise alignment of the reference sequence with sequence k is denoted by k. Now consider a position i in the reference sequence. It corresponds to a uniquely determined alignment column α(i), which, in turn, determines ik, the last position of sequence k that occurs in or before alignment column α(i).
Suppose i is a third codon position. Then the alignment block [α(i − 3) + 1, α(i)] corresponds to the (potential) codon ending in i. We define a score:
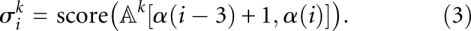
In the ungapped case, 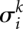 is the normalized BLOSUM score that was introduced in the main text. Let
is the normalized BLOSUM score that was introduced in the main text. Let 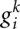 denote the number of gaps in sequence k in this block. We observe that sequences 1 (reference) and k stay in-frame if and only if
denote the number of gaps in sequence k in this block. We observe that sequences 1 (reference) and k stay in-frame if and only if 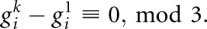 Otherwise, the two sequences change their phase within this interval. The local shift in frame between sequence k and the reference sequence is therefore:
Otherwise, the two sequences change their phase within this interval. The local shift in frame between sequence k and the reference sequence is therefore:
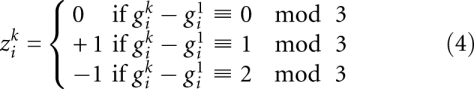
As discussed in the main text, alignment errors or sequence errors may destroy coherence between aligned codons and give 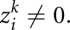 . Therefore, we introduce the penalties (negative scores) Ω for switching from in-frame to out-of-frame or back, as well as ω for every out-of-frame codon in between, and Δ for silently changing the phase and assuming subsequent codons are still in-frame (sequencing error). All penalties are negative; in particular,
. Therefore, we introduce the penalties (negative scores) Ω for switching from in-frame to out-of-frame or back, as well as ω for every out-of-frame codon in between, and Δ for silently changing the phase and assuming subsequent codons are still in-frame (sequencing error). All penalties are negative; in particular, 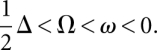 Furthermore, we set
Furthermore, we set 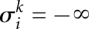 if
if 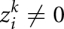 to mark the fact that we lose coherence of the frame and force the algorithm to select a frameshift or sequence error penalty and not a substitution score that would be meaningless for out-of-frame triples.
to mark the fact that we lose coherence of the frame and force the algorithm to select a frameshift or sequence error penalty and not a substitution score that would be meaningless for out-of-frame triples.
Having defined all possible states and the associated scores, we now describe a dynamic programming algorithm to calculate the optimal score for a pairwise alignment. Let 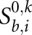 be the optimal score of the pairwise alignment _k_[α(b), α(i)] subject to the condition that i is a third codon position and sequence k ends in-frame, i.e., also with a third codon position. Analogously, we define
be the optimal score of the pairwise alignment _k_[α(b), α(i)] subject to the condition that i is a third codon position and sequence k ends in-frame, i.e., also with a third codon position. Analogously, we define 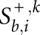 and
and 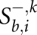 for those alignments where sequence k ends in the first and second codon position, respectively. Clearly, we initialize
for those alignments where sequence k ends in the first and second codon position, respectively. Clearly, we initialize 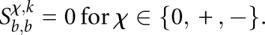
The entries in these matrices satisfy the following recursions:
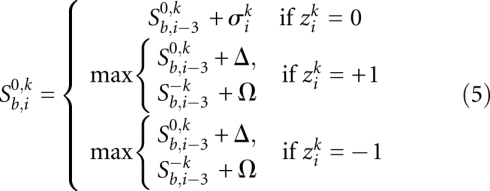
The expressions for the two out-of-frame scores are analogous. We show only one of them explicitly:
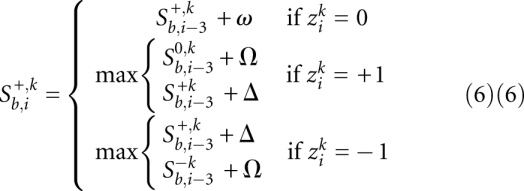
A state diagram corresponding to the above algorithm is shown in Figure 7. As presented here, the algorithm assumes that any sequence errors (penalized by Δ) occur in sequence k, not in the reference.
FIGURE 7.

Finite state automaton representing the scoring of pairwise alignments. The three states correspond to the relative phases of the sequences. Insertions and deletions with z ≠ 0 lead to local changes in-phase that are penalized by Ω. Extension in each of the two out-of-frame states S+ and S− is penalized by ω. In/dels interpreted as sequencing errors or true frameshifts are penalized by Δ.
Now we determine the optimal score Sbi of the multiple alignment [α(b), α(i)], subject to the condition that b is a first codon position and i is a third codon position.
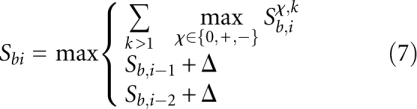
The second and third terms here correspond to frameshifts in the reference sequence.
It is easy now to determine the best scoring segment(s) of from the maximal entries in the matrix (Sbi). If we were to score only pairwise alignments, it would be possible to use a local alignment-like algorithm that does not keep track of the beginning of the segment, b. In the multiple alignment, however, the individual pairwise alignments are constrained by the requirement that a coding segment starts in the same column for all sequences, forcing us to keep track of b explicitly. The algorithm scales as (N · _n_2) in time and space, where n is the length of the reference sequence and N the number of rows in the alignment.
Footnotes
REFERENCES
- Aebersold R, Mann M 2003. Mass spectrometry-based proteomics. Nature 422: 198–207 [DOI] [PubMed] [Google Scholar]
- Badger JH, Olsen GJ 1999. CRITICA: Coding region identification tool invoking comparative analysis. Mol Biol Evol 16: 512–524 [DOI] [PubMed] [Google Scholar]
- Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AF, Roskin KM, Baertsch R, Rosenbloom K, Clawson H, Green ED, et al. 2004. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res 14: 708–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bofkin L, Goldman N 2007. Variation in evolutionary processes at different codon positions. Mol Biol Evol 24: 513–521 [DOI] [PubMed] [Google Scholar]
- Boisset S, Geissmann T, Huntzinger E, Fechter P, Bendridi N, Possedko M, Chevalier C, Helfer AC, Benito Y, Jacquier A, et al. 2007. Staphylococcus aureus RNAIII coordinately represses the synthesis of virulence factors and the transcription regulator Rot by an antisense mechanism. Genes Dev 21: 1353–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brent MR 2008. Steady progress and recent breakthroughs in the accuracy of automated genome annotation. Nat Rev Genet 9: 62–73 [DOI] [PubMed] [Google Scholar]
- Burge CB, Karlin S 1998. Finding the genes in genomic DNA. Curr Opin Struct Biol 8: 346–354 [DOI] [PubMed] [Google Scholar]
- Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, et al. 2005. The transcriptional landscape of the mammalian genome. Science 309: 1559–1563 [DOI] [PubMed] [Google Scholar]
- Cho BK, Zengler K, Qiu Y, Park YS, Knight EM, Barrett CL, Gao Y, Palsson BO 2009. The transcription unit architecture of the Escherichia coli genome. Nat Biotechnol 27: 1043–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clamp M, Fry B, Kamal M, Xie X, Cuff J, Lin MF, Kellis M, Lindblad-Toh K, Lander ES 2007. Distinguishing protein-coding and noncoding genes in the human genome. Proc Natl Acad Sci 104: 19428–19433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coghlan A, Fiedler TJ, McKay SJ, Flicek P, Harris TW, Blasiar D, nGASP Consortium, Stein LD 2008. nGASP—the nematode genome annotation assessment project. BMC Bioinformatics 9: 549 doi: 10.1186/1471-2105-9-549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig R, Beavis RC 2004. TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20: 1466–1467 [DOI] [PubMed] [Google Scholar]
- Dai Y, Li L, Roser DC, Long SR 1999. Detection and identification of low-mass peptides and proteins from solvent suspensions of Escherichia coli by high performance liquid chromatography fractionation and matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun Mass Spectrom 13: 73–78 [DOI] [PubMed] [Google Scholar]
- David L, Huber W, Granovskaia M, Toedling J, Palm CJ, Bofkin L, Jones T, Davis RW, Steinmetz LM 2006. A high-resolution map of transcription in the yeast genome. Proc Natl Acad Sci 103: 5320–5325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinger ME, Pang KC, Mercer TR, Mattick JS 2008. Differentiating protein-coding and noncoding RNA: Challenges and ambiguities. PLoS Comput Biol 4: e1000176 doi: 10.1371/journal.pcbi.1000176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drysdale R; FlyBase Consortium 2008. FlyBase: A database for the Drosophila research community. Methods Mol Biol 420: 45–59 [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447: 799–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P 2007. Gene prediction: compare and CONTRAST. Genome Biol 8: 233 doi: 10.1186/gb-2007-8-12-233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frith MC, Bailey TL, Kasukawa T, Mignone F, Kummerfeld SK, Madera M, Sunkara S, Furuno M, Bult CJ, Quackenbush J, et al. 2006. Discrimination of non-protein-coding transcripts from protein-coding mRNA. RNA Biol 3: 40–48 [DOI] [PubMed] [Google Scholar]
- Galindo MI, Pueyo JI, Fouix S, Bishop SA, Couso JP 2007. Peptides encoded by short ORFs control development and define a new eukaryotic gene family. PLoS Biol 5: e106 doi: 10.1371/journal.pbio.0050106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner PP, Daub J, Tate JG, Nawrocki EP, Kolbe DL, Lindgreen S, Wilkinson AC, Finn RD, Griffiths-Jones S, Eddy SR, et al. 2009. Rfam: updates to the RNA families database. Nucleic Acids Res 37: D136–D140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesell T, Washietl S 2008. Dinucleotide controlled null models for comparative RNA gene prediction. BMC Bioinformatics 9: 248 doi: 10.1186/1471-2105-9-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimpel M, Heidrich N, Mäder U, Krügel H, Brantl S 2010. A dual-function sRNA from B. subtilis: SR1 acts as a peptide encoding mRNA on the gapA operon. Mol Microbiol 76: 990–1009 [DOI] [PubMed] [Google Scholar]
- Goldman N, Thorne JL, Jones DT 1998. Assessing the impact of secondary structure and solvent accessibility on protein evolution. Genetics 149: 445–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross SS, Brent MR 2006. Using multiple alignments to improve gene prediction. J Comput Biol 13: 379–393 [DOI] [PubMed] [Google Scholar]
- Gross SS, Do CB, Sirota M, Batzoglou S 2007. CONTRAST: a discriminative, phylogeny-free approach to multiple informant de novo gene prediction. Genome Biol 8: R269 doi: 10.1186/gb-2007-8-12-r269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber AR, Findeiß S, Washietl S, Hofacker IL, Stadler PF 2010. RNAz 2.0: Improved noncoding RNA detection. Pac Symp Biocomput 15: 69–79 [PubMed] [Google Scholar]
- Guigó R, Flicek P, Abril JF, Reymond A, Lagarde J, Denoeud F, Antonarakis S, Ashburner M, Bajic VB, Birney E, et al. 2006. EGASP: the human ENCODE genome annotation assessment project. Genome Biol 7: S2.1–S31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Gascuel O 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52: 696–704 [DOI] [PubMed] [Google Scholar]
- Harper RG, Workman SR, Schuetzner S, Timperman AT, Sutton JN 2004. Low-molecular-weight human serum proteome using ultrafiltration, isoelectric focusing, and mass spectrometry. Electrophoresis 25: 1299–1306 [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Kishino H, Yano T 1985. Dating of the human–ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22: 160–174 [DOI] [PubMed] [Google Scholar]
- Heidrich N, Chinali A, Gerth U, Brantl S 2006. The small untranslated RNA SR1 from the Bacillus subtilis genome is involved in the regulation of arginine catabolism. Mol Microbiol 62: 520–536 [DOI] [PubMed] [Google Scholar]
- Heidrich N, Moll I, Brantl S 2007. In vitro analysis of the interaction between the small RNA SR1 and its primary target ahrC mRNA. Nucleic Acids Res 35: 4331–4346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemm MR, Paul BJ, Schneider TD, Storz G, Rudd KE 2008. Small membrane proteins found by comparative genomics and ribosome binding site models. Mol Microbiol 70: 1487–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Henikoff JG 1992. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci 89: 10915–10919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A, Nesvizhskii AI, Kolker E, Aebersold R 2002. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem 74: 5383–5392 [DOI] [PubMed] [Google Scholar]
- Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES 2003. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature 423: 241–254 [DOI] [PubMed] [Google Scholar]
- Kellis M, Patterson N, Birren B, Berger B, Lander ES 2004. Methods in comparative genomics: genome correspondence, gene identification and regulatory motif discovery. J Comput Biol 11: 319–355 [DOI] [PubMed] [Google Scholar]
- Klein C, Aivaliotis M, Olsen JV, Falb M, Besir H, Scheffer B, Bisle B, Tebbe A, Konstantinidis K, Siedler F, et al. 2007. The low molecular weight proteome of Halobacterium salinarum. J Proteome Res 6: 1510–1518 [DOI] [PubMed] [Google Scholar]
- Kondo T, Hashimoto Y, Kato K, Inagaki S, Hayashi S, Kageyama Y 2007. Small peptide regulators of actin-based cell morphogenesis encoded by a polycistronic mRNA. Nat Cell Biol 9: 660–665 [DOI] [PubMed] [Google Scholar]
- Kondo T, Plaza S, Zanet J, Benrabah E, Valenti P, Hashimoto Y, Kobayashi S, Payre F, Kageyama Y 2010. Small peptides switch the transcriptional activity of shavenbaby during Drosophila embryogenesis. Science 329: 336–339 [DOI] [PubMed] [Google Scholar]
- Kuhn RM, Karolchik D, Zweig AS, Wang T, Smith KE, Rosenbloom KR, Rhead B, Raney BJ, Pohl A, Pheasant M, et al. 2009. The UCSC Genome Browser database: Update 2009. Nucleic Acids Res 37: D755–D761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licht A, Preis S, Brantl S 2005. Implication of CcpN in the regulation of a novel untranslated RNA (SR1) in Bacillus subtilis. Mol Microbiol 58: 189–206 [DOI] [PubMed] [Google Scholar]
- Lin MF, Deoras AN, Rasmussen MD, Kellis M 2008. Performance and scalability of discriminative metrics for comparative gene identification in 12 Drosophila genomes. PLoS Comput Biol 4: e1000067 doi: 10.1371/journal.pcbi.1000067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MM, Ames TD, Smith DP, Weinberg Z, Schwalbach MS, Giovannoni SJ, Breaker RR 2009. Identification of candidate structured RNAs in the marine organism ‘Candidatus Pelagibacter ubique’. BMC Genomics 10: 268 doi: 10.1186/1471-2164-10-268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignone F, Grillo G, Liuni S, Pesole G 2003. Computational identification of protein coding potential of conserved sequence tags through cross-species evolutionary analysis. Nucleic Acids Res 31: 4639–4645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourier T, Carret C, Kyes S, Christodoulou Z, Gardner PP, Jeffares DC, Pinches R, Barrell B, Berriman M, Griffiths-Jones S, et al. 2008. Genome-wide discovery and verification of novel structured RNAs in Plasmodium falciparum. Genome Res 18: 281–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller SA, Kohajda T, Findeiß S, Stadler PF, Washietl S, Kellis M, von Bergen M, Kalkhof S 2010. Optimization of parameters for coverage of low molecular weight proteins. Anal Bioanal Chem 398: 2867–2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekrutenko A, Chung WY, Li WH 2003. ETOPE: Evolutionary test of predicted exons. Nucleic Acids Res 31: 3564–3567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii AI, Keller A, Kolker E, Aebersold R 2003. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem 75: 4646–4658 [DOI] [PubMed] [Google Scholar]
- Perkins DN, Pappin DJ, Creasy DM, Cottrell JS 1999. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20: 3551–3567 [DOI] [PubMed] [Google Scholar]
- Rambaut A, Grassly NC 1997. Seq-Gen: an application for the Monte Carlo simulation of DNA sequence evolution along phylogenetic trees. Comput Appl Biosci 13: 235–238 [DOI] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16: 276–277 [DOI] [PubMed] [Google Scholar]
- Rivas E, Eddy SR 2001. Noncoding RNA gene detection using comparative sequence analysis. BMC Bioinformatics 2: 8 doi: 10.1186/1471-2105-2-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg MI, Desplan C 2010. Molecular biology. Hiding in plain sight. Science 329: 284–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schägger H 2006. Tricine-SDS-PAGE. Nat Protoc 1: 16–22 [DOI] [PubMed] [Google Scholar]
- Schneider KL, Pollard KS, Baertsch R, Pohl A, Lowe TM 2006. The UCSC archaeal genome browser. Nucleic Acids Res 34: D407–D410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Tyson GW, DeLong EF 2009. Metatranscriptomics reveals unique microbial small RNAs in the ocean's water column. Nature 459: 266–269 [DOI] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15: 1034–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark A, Lin MF, Kheradpour P, Pedersen JS, Parts L, Carlson JW, Crosby MA, Rasmussen MD, Roy S, Deoras AN, et al. 2007. Discovery of functional elements in 12 Drosophila genomes using evolutionary signatures. Nature 450: 219–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjaden B, Saxena RM, Stolyar S, Haynor DR, Kolker E, Rosenow C 2002. Transcriptome analysis of Escherichia coli using high-density oligonucleotide probe arrays. Nucleic Acids Res 30: 3732–3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium 2010. The universal protein resource (UniProt) in 2010. Nucleic Acids Res 38: D142–D148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washietl S, Hofacker IL, Stadler PF 2005. Fast and reliable prediction of noncoding RNAs. Proc Natl Acad Sci 102: 2454–2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washietl S, Pedersen JS, Korbel JO, Stocsits C, Gruber AR, Hackermüller J, Hertel J, Lindemeyer M, Reiche K, Tanzer A, et al. 2007. Structured RNAs in the encode selected regions of the human genome. Genome Res 17: 852–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Nielsen R 2000. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol Biol Evol 17: 32–43 [DOI] [PubMed] [Google Scholar]
- Yates JR III, Eng JK, McCormack AL, Schieltz D 1995. Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal Chem 67: 1426–1436 [DOI] [PubMed] [Google Scholar]