NATURAL PRODUCTS: A CONTINUING SOURCE OF NOVEL DRUG LEADS (original) (raw)
. Author manuscript; available in PMC: 2014 Jun 1.
Published in final edited form as: Biochim Biophys Acta. 2013 Feb 18;1830(6):3670–3695. doi: 10.1016/j.bbagen.2013.02.008
Abstract
1. Background
Nature has been a source of medicinal products for millennia, with many useful drugs developed from plant sources. Following discovery of the penicillins, drug discovery from microbial sources occurred and diving techniques in the 1970s opened the seas. Combinatorial chemistry (late 1980s), shifted the focus of drug discovery efforts from Nature to the laboratory bench.
2. Scope of Review
This review traces natural products drug discovery, outlining important drugs from natural sources that revolutionized treatment of serious diseases. It is clear Nature will continue to be a major source of new structural leads, and effective drug development depends on multidisciplinary collaborations.
3. Major Conclusions
The explosion of genetic information led not only to novel screens, but the genetic techniques permitted the implementation of combinatorial biosynthetic technology and genome mining. The knowledge gained has allowed unknown molecules to be identified. These novel bioactive structures can be optimized by using combinatorial chemistry generating new drug candidates for many diseases.
4
General Significance: The advent of genetic techniques that permitted the isolation / expression of biosynthetic cassettes from microbes may well be the new frontier for natural products lead discovery. It is now apparent that biodiversity may be much greater in those organisms. The numbers of potential species involved in the microbial world are many orders of magnitude greater than those of plants and multi-celled animals. Coupling these numbers to the number of currently unexpressed biosynthetic clusters now identified (>10 per species) the potential of microbial diversity remains essentially untapped.
Keywords: Microbial diversity, synthesis, genomics, natural product drugs
1. Introduction
Throughout the ages humans have relied on Nature to cater for their basic needs, not the least of which are medicines for the treatment of a wide spectrum of diseases. Plants, in particular, have formed the basis of sophisticated traditional medicine systems, with the earliest records, dating from around 2600 BCE, documenting the uses of approximately 1000 plant-derived substances in Mesopotamia. These include oils of Cedrus species (cedar) and Cupressus sempevirens (cypress), Glycyrrhiza glabra (licorice), Commiphora species (myrrh), and Papaver somniferum (poppy juice), all of which are still used today for the treatment of ailments ranging from coughs and colds to parasitic infections and inflammation. Egyptian medicine dates from about 2900 B.C., but the best known record is the “Ebers Papyrus” dating from 1500 BCE, documenting over 700 drugs, mostly of plant origin [1]. The Chinese Materia Medica has been extensively documented over the centuries [2], with the first record dating from about 1100 B. C. (Wu Shi Er Bing Fang, containing 52 prescriptions), followed by works such as the Shennong Herbal (~100 B. C.; 365 drugs) and the Tang Herbal (659 A. D.; 850 drugs). Likewise, documentation of the Indian Ayurvedic system dates from before 1000 B. C. (Charaka; Sushruta and Samhitas with 341 and 516 drugs respectively) [3, 4].
The Greeks and Romans contributed substantially to the rational development of the use of herbal drugs in the ancient Western world. Dioscorides, a Greek physician (100 CE), accurately recorded the collection, storage, and use of medicinal herbs during his travels with Roman armies throughout the then “known world”, whilst Galen (130–200 CE.), a practitioner and teacher of pharmacy and medicine in Rome, is well known for his complex prescriptions and formulae used in compounding drugs. The Arabs, however, preserved much of the Greco-Roman expertise during the Dark and Middle Ages (5th to 12th centuries), and expanded it to include the use of their own resources, together with Chinese and Indian herbs unknown to the Greco-Roman world. A comprehensive review of the history of medicine may be found on the website of the National Library of Medicine (NLM), United States National Institutes of Health (NIH), at www.nlm.nih.gov/hmd/medieval/arabic.html.
2 The Role of Traditional Medicine and Plants in Drug Discovery
Plant-based systems continue to play an essential role in healthcare, and their use by different cultures has been extensively documented [5, 6]. The World Health Organization (WHO) estimated in 1985 that approximately 65% of the population of the world predominately relied on plant-derived traditional medicines for their primary health care, while plant products also play an important, though more indirect role in the health care systems of the remaining population who mainly reside in developed countries [7]. A survey of plant-derived pure compounds used as drugs in countries hosting WHO-Traditional Medicine Centers indicated that, of 122 compounds identified, 80% were used for the same or related ethnomedical purposes and were derived from only 94 plant species [7]. Some relevant examples are khellin, from Ammi visnaga (L) Lamk., which led to the development of chromolyn (in the form of sodium chromoglycate) as a bronchodilator; galegine, from Galega officinalis L., which was the model for the synthesis of metformin and other bisguanidine-type antidiabetic drugs [8]; and papaverine from Papaver somniferum which formed the basis for verapamil used in the treatment of hypertension (Fig. 1) [8]. The latter plant is better known as being the source of painkillers such as morphine and codeine [9], but probably the best example of ethnomedicine’s role in guiding drug discovery and development is that of the antimalarial drugs, particularly quinine and artemisinin.
Fig. 1.

Drugs based on traditional medicine leads (khellin, sodium chromoglycate, galegine, metformin, papaverine, verapamil)
Malaria remains one of the greatest health challenges confronting humankind, and the search for better drugs, both in terms of efficacy and cost, is a global health imperative. The isolation of the antimalarial drug, quinine (Fig. 2), from the bark of Cinchona species (e. g., C. officinalis), was reported in 1820 by the French pharmacists, Caventou and Pelletier [9]. The bark had long been used by indigenous groups in the Amazon region for the treatment of fevers, and was first introduced into Europe in the early 1600s for the treatment of malaria. Quinine formed the basis for the synthesis of the commonly used antimalarial drugs, chloroquine and mefloquine which largely replaced quinine in the mid 20th century, but with the emergence of resistance to both these drugs in many tropical regions, another plant long used in the treatment of fevers in Traditional Chinese Medicine (TCM), Artemisia annua (Quinhaosu), gained prominence [10].
Fig. 2.

Natural antimalarial agents and analogues
Quinine, chloroquine, mefloquine, artemisinin, OZ277, Dimeric analogue
This discovery in 1971 by Chinese scientists using data from ancient texts in Traditional Chinese Medicine provided an exciting new natural product lead compound, now known as artemisinin, which was subsequently reported from US-sourced Artemisia annua by investigators at the Walter Reed Army Institute of Research (WRAIR) in 1984 in an article in the Journal of Natural Products [11]. A fuller description of the importance of this drug class was given by the lead author in the WRAIR report in 1985 in an article in the journal Science [12]. In 2011, recognition of the importance of the initial finding was demonstrated by award of the Lasker prize to Dr. Y. Tu for her leadership in what was then known as Project 523 [13].
Artemisinin analogues are now used for the treatment of malaria in many countries [14]. There is still debate as to its actual mechanism of action (MOA) which may involve complexation with hemin by coordination of the unusual endoperoxide bridge with iron, which in turn interrupts the detoxification process used by the parasite and generates free radical species which can attack proteins in the parasite [15]. Or as suggested the same year, it might involve mitochondrial attack [16]. Many analogues of artemisinin have been prepared in attempts to improve its activity and utility [14], and two of the more promising of these are the totally synthetic analogue OZ277 (Fig. 2) [17], and the dimeric analogue (Fig. 2). Single doses of the latter compound were shown to cure malaria-infected mice, while corresponding treatments with artemisinin were much less effective [18]. It is also relevant that artemisinin and related compounds also have significant anti-tumor activity in vitro but again, MOAs are not known.
Other significant drugs developed from traditional medicinal plants include: the antihypertensive agent, reserpine, isolated from Rauwolfia serpentina used in Ayurvedic medicine for the treatment of snakebite and other ailments [3]; ephedrine, from Ephedra sinica (Ma Huang), a plant long used in traditional Chinese medicine, and the basis for the synthesis of the anti-asthma agents (beta agonists), salbutamol and salmetrol; and the muscle relaxant, tubocurarine, isolated from Chondrodendron and Curarea species used by indigenous groups in the Amazon as the basis for the arrow poison, curare [9]. An update on the use and requirements for further use of traditional medicines is very nicely covered by Cordell and Colvard [19] in a very recent review article, which should be read in conjunction with the earlier papers referred to above.
Plants have a long history of use in the treatment of cancer [20], though many of the claims for the efficacy of such treatment should be viewed with some skepticism because cancer, as a specific disease entity, is likely to be poorly defined in terms of folklore and traditional medicine [21]. Of the plant-derived anticancer drugs in clinical use, some of the best known are the so-called vinca alkaloids, vinblastine and vincristine (Fig. 3), isolated from the Madagascar periwinkle, Catharanthus roseus [22, 23], together with the two clinically-active agents, etoposide and teniposide (Fig. 3), which are semisynthetic derivatives of the natural product epipodophyllotoxin [24, 25].
Fig. 3.

Plant-derived anticancer agents
Vinblastine / vincristine, Etoposide, Paclitaxel, Taxotere, Cabazitaxel, Camptothecin / 9-NH2, 9-NO2 / Topotecan / Irinotecan / Belotecan, Maytansine
Paclitaxel (Taxol®; Fig. 3), the most exciting plant-derived anticancer drug discovered in recent years, occurs, along with several key precursors (the baccatins) in the leaves of various Taxus species [26, 27]. The discovery in 1979 of its mode of action through promotion of the assembly of tubulin into microtubules by Schiff and Horwitz and its report in 1980 [28], was a key milestone in the lengthy development process, and it was finally approved for clinical use against ovarian cancer in 1992 and against breast cancer in 1994. Since then it has become a blockbuster drug, with annual sales of over $1 billion, though nowadays, the drug is generic so the sales are spread amongst a significant number of companies. The success of paclitaxel spawned extensive studies on the synthesis of analogues, and the first one to be developed was the close chemical relative, docetaxel (Taxotere®; Fig. 3). The clinically-approved new albumin-bound formulation of paclitaxel known as Abraxane® offers some significant advantages compared with the original Cremophor formulation [29], and a nanoparticle formulation in cross-linked polymers with paclitaxel contained in the microspheres was approved in India in 2007 [30]. In 2010, the third taxane Cabazitaxel (Jevtana®; Fig 3) was approved in the USA and for readers who are interested they may consult recent reviews for information on the many new agents in development [27, 31].
Other recent additions to the armamentarium of plant-derived cancer chemotherapeutic agents are the three clinically-active agents, topotecan, irinotecan (CPT-11) and belotecan, plus their well-researched but not approved analogues 9-amino- and 9-nitro-camptothecin (Fig. 3), all of which were semi-synthetically derived from camptothecin, isolated from the Chinese ornamental tree, Camptotheca acuminata [32]. Camptothecin (as its sodium salt) was advanced to clinical trials by NCI in the 1970s, but was dropped because of severe bladder toxicity.
Very recently one of the first “plant-derived” tubulin interactive compounds to enter clinical trials, maytansine from the Ethiopian tree Maytenus serrata, was effectively granted a new lease of life as a slightly modified “warhead” on a monoclonal antibody. From the initial determination of its structure (Fig. 3) natural product chemists wondered if the compound was microbial in origin, due to its similarity to the “ansa” antibiotics such as the rifamycins. In 1977, scientists at Takeda Chemical Industries reported the structures of the bacterial products, the ansamitocins, which very closely resembled the maytansenoids. Later work on compounds isolated from the bacterium, subsequently renamed as Actinosynnema pretiosum, demonstrated that they were in fact identical to those isolated from other plant genera. For further information on the maytansenoids, the reader should consult the review by Kirchning et al., in 2008 [33] and / or the chapter by Yu et al., in 2012 [34] as these cover the chemistry and biosynthesis of these compounds.
As mentioned above, microbial “precursors” of maytansine, specifically DM-1 and DM-4 with suitable chemical linkages have been used as warheads linked to specific monoclonal antibodies directed against tumor-linked epitopes. A discussion of the utility of such linked materials was given by Senter in 2009 [35] and amplified the following year by both Alley [36] and Caravella [37]. An article specifically referring to the DM1-linked conjugates is the 2010 review by Lambert from Immunogen covering these constructs and their clinical efficacies [38]. This article should be read in conjunction with the 2011 paper by Kümler et al., covering the story of Trastuzumab emtansine, [39] the combination of Herceptin® with a specific linkage to DM1 that is cleaved by enzymes on uptake into the tumor. Currently this combination is in Phase III clinical trials against metastatic breast cancer, and a preliminary report from the Roche-sponsored EMILIA trial demonstrated significant extension of progression free survival compared to patients receiving lapatinib and capecitabine. Roche submitted an MMA to the EU and a BLA to the FDA before the end of 2012, with decisions expected in early 2013 from the FDA.
3. The Role of Marine Organisms in Drug Discovery
While marine organisms do not have a significant history of use in traditional medicine, the ancient Phoenicians employed a chemical secretion from marine molluscs to produce purple dyes for woolen cloth, and seaweeds have long been used to fertilize the soil. The world’s oceans, covering more than 70% of the earth’s surface, represent an enormous resource for the discovery of potential chemotherapeutic agents. Of the 33 animal phyla listed by Margulis and Schwartz [40] 32 are represented in aquatic environments, with fifteen being exclusively marine, seventeen in marine and non-marine (with five of these having more than 95% of their species only in marine environments), and only one, Onychophora, being exclusively non-marine. Before the development of reliable scuba diving techniques some 45–50 years ago, the collection of marine organisms was limited to those obtainable by skin diving. Subsequently, depths from approximately 3 meters to 40 meters became routinely attainable, and the marine environment has been increasingly explored as a source of novel bioactive agents. Deep water collections can be made by dredging or trawling, but these methods can suffer from disadvantages, such as environmental damage and nonselective sampling. These disadvantages can be partially overcome by use of manned submersibles or remotely operated vehicles (ROVs); however, the high cost of these forms of collecting precludes their extensive use in routine collection operations.
The systematic investigation of marine environments as sources of novel biologically active agents only began in earnest in the mid-1970s. During the decade from 1977–1987, about 2,500 new metabolites were reported from a variety of marine organisms and, to keep in perspective, the review article by Faulkner [41], covering just 1998, had over 840 novel structures described, and the latest in this series, published a few months ago, listed 1003 new compounds covering just publications in 2010 [42]. These studies have clearly demonstrated that the marine environment is a rich source of bioactive compounds, many of which belong to totally novel chemical classes not found in terrestrial sources.
While research has focused on the discovery of potential new anticancer agents [43, 44], the first marine derived product to gain approval as a drug was Ziconotide, a non-narcotic analgesic that is currently marketed as Prialt® [45]. This compound was isolated as a constituent of the venom, composed of combinatorial libraries of several hundred peptides that are injected by species of the cone snail genus Conus, to stun their prey prior to capture [46]. Ziconotide, though effective in its limited application, is not a drug that is taken easily as it has to be delivered via an intrathecal injection route. Recently however, Craik and co-workers in Australia have demonstrated that by taking structural cues from plant cyclotides, they can alter the pharmacological characteristics, producing oral analgesic activity in animal tests [47].
The complex alkaloid ecteinascidin 743 (Fig. 4) isolated from the colonial tunicate Ecteinascidia turbinata, which had been granted Orphan Drug designation in Europe and the USA under the name Yondelis®, was approved in September, 2007, by the EMEA for the treatment of soft tissue sarcomas (STS). Subsequently, it was approved in 2009 in the EU for treatment of relapsed ovarian cancer in conjunction with liposomal doxorubicin. It is also currently in a number of clinical trials ranging from Phase I to III in multiple countries for breast, prostate, liposarcoma and paediatric sarcomas. Although the initial clinical trials were performed with material from in sea and on land aquaculture of the producing organism, the subsequent development of a semisynthetic route from the microbial product cyanosafracin B (Fig. 4) has solved the issue of compound supply, always a problem with marine-sourced materials [48–50]. In a recent paper, Sherman’s group at the University of Michigan reported their isolation and identification of the putative producing NRPS gene cluster from a metagenomic analysis of Candidatus Endoecteinascidia frumentensis the putative producer of Et743 [51].
Fig. 4.
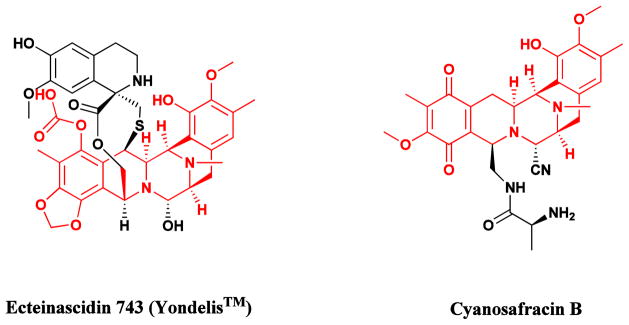
Ecteinascidin 743 (Yondelis®) and its semisynthetic precursor
Further examples of marine-derived anticancer agents are halichondrin B (Fig. 5) [52], a complex polyether isolated in miniscule yield from several sponge sources, and bryostatin 1 (Fig. 5) another complex macrolide originally isolated by Pettit and his collaborators from the bryozoan, Bugula neritina [53, 54]. The complex structure of halichondrin B appeared to make total synthesis impractical as a source for drug development, but fortunately total synthetic studies revealed that the right hand half of halichondrin B retained all or most of the potency of the parent compound. Large-scale synthesis of the analogue E7389 (Eribulin; Fig. 5) provided adequate supplies [55], and following initial trials in conjunction with the NCI and then further trials conducted by Eisai (the Eisai Research Institute in the USA and Eisai Inc., in other countries) and their collaborators, the compound was approved in November 2010 by the FDA for the treatment of refractory metastatic breast cancer. Details of the pivotal clinical trial leading to this approval have recently been published and can be consulted by interested readers [56]. As is customary with newly approved agents, it is currently in multiple clinical trials from Phase I to III against a variety of carcinomas in the USA, the EU and Japan.
Fig. 5.

Halichondrin B, Eribulin, Bryostatin 1
Bryostatin 1 demonstrated excellent anticancer activity, due at least in part, to its ability to interact with protein kinase C (PKC) isozymes, but compound supply has proved to be a major problem. Enough cGMP-grade material, however, was isolated from wild collections to supply material for clinical trials which indicated that the drug would be most effective in combination therapy, however, in spite of many clinical trials at both Phase I and Phase II, no effective drug regimen has yet been identified that would lead to further trials, due predominately to the severe side effects reported. The most recent clinical report highlighted these effects when bryostatin 1 was combined with cisplatin [57]. Currently there is a Phase II clinical trial (NCT00606164) approved for potential treatment of Alzheimer’s disease, but as yet no recruitment appears to have taken place, and there are three Phase II with bryostatin plus a cytotoxin or signal transduction inhibitor whose status is listed as unknown, though probably none of these are still underway and one Phase I trial with temsirolimus shown as recruiting listed in the clinical trials web site as of December 2012.
The bryostatins have been the target of many synthetic chemistry groups, with a focus being on the synthesis of simpler analogues possessing comparable or better activity, particularly related to binding to some of the target protein kinase C isozymes The result has been the preparation of nominally simpler compounds, particularly those from Wender and from Keck whose structures are given and in vitro activities discussed in the 2012 review by Newman [54].
4. The Role of Microorganisms in Drug Discovery
4.1. An Historical Perspective
The serendipitous discovery of penicillin from the filamentous fungus, Penicillium notatum, by Fleming in 1929, and the observation of the broad therapeutic use of this agent in the 1940s, ushered in a new era in medicine, “the Golden Age of Antibiotics”, and promoted the intensive investigation of Nature as a source of novel bioactive agents [58]. Microorganisms are a prolific source of structurally diverse bioactive metabolites and have yielded some of the most important products of the pharmaceutical industry. These include: antibacterial agents, such as the penicillins (from Penicillium species), cephalosporins (from Cephalosporium acremonium) (Fig. 6), aminoglycosides, tetracyclines and other polyketides of many structural types (from the Actinomycetales); immunosuppressive agents, such as the cyclosporins (from Trichoderma and Tolypocladium species) and rapamycin (from Streptomyces species) (Fig. 6); cholesterol lowering agents, such as mevastatin (compactin; from Penicillium species) and lovastatin (from Aspergillus species) (Fig. 6); and anthelmintics and antiparasitic drugs, such as the ivermectins (from Streptomyces species) [9].
Fig. 6.

Drugs from Microbes
Cephalosporins, cyclosporins, rapamycin, statins
4.2 Antitumor Antibiotics
Antitumor antibiotics are amongst the most important of the cancer chemotherapeutic agents, which include members of the anthracycline [59, 60], bleomycin [61, 62], mitomycin [63], the enediynes [64, 65], and the staurosporines [66]. The more recent aspects of the staurosporine story are covered in the 2012 update [67] and in two excellent reviews [68, 69]. All were isolated from various Streptomyces species. Examples of clinically useful agents from these compound families are the daunomycin-related agents, daunomycin itself, doxorubicin, idarubicin and epirubicin; the glycopeptidic bleomycins A2 and B2 (blenoxane®); the mitosanes such as mitomycin C; and the enediynes exemplified by the monoclonal antibody-linked calicheamicin conjugate, Mylotarg®. Although this compound has been withdrawn from clinical use, there are other similar compounds in clinical trials utilizing a similar warhead (Fig. 7).
Fig. 7.

Microbial-Derived Anticancer Agents
Daunomycin, bleomycin A2, mitomycin C, calicheamicin
In recent years, the epothilones isolated from myxobacteria have emerged as some of the most interesting natural product base structures being considered as agents for clinical trials in cancer chemotherapy. These macrolides, with a mechanism of action similar to that of paclitaxel (Taxol®), have been extensively studied, both from the biosynthetic and synthetic standpoints [70–74]. Classical fermentation and manipulation of the biosynthetic cluster have given significant quantities of epothilones A and B, and C and D, respectively (Fig. 8) [71], and the potential for the production of unnatural epothilones, whose properties may well be quite different from those previously reported, is being studied through a combination of precursor-directed biosynthesis coupled to chemical bond formation by standard techniques [71, 75].
Fig. 8.

Epothilone anticancer agents
Epothilones A–D, ixabepilone, sagopilone didehydroepothilone D, isoxazolefludelone
In addition, semi- and total syntheses are being performed by a number of groups, including variations on the macrolide ring. Thus the azaepothilone (ixabepilone or 16-azaepothilone B; Fig. 8) was approved for monotherapy against metastatic breast cancer by the FDA in 2007, and is currently in many trials from Phase I to Phase III against a variety of carcinomas. Although it reached Phase III trials with Novartis, epothilone B or patupilone was withdrawn from the Phase III level but it is currently in a number of Phase II trials under the auspices of Novartis, and recently, data from a recent Phase II trial was published demonstrating activity in prostate cancer [76]. Dehydelone (didehydroepothilone D) was in Phase II trials as late as 2008 (Fig. 8) [77] but is no longer listed as in trials.
Of the 8 examples of epothilone-based compounds currently listed as of December 2012 in the Thomson-Reuters Integrity™ database, only three are shown as being in current clinical trials aside from the approved ixabepilone. Thus epothilone B (patupilone, or EPO-906, referred to earlier) and the synthetically made sagopilone (ZK-EPO) are in current Phase II trials, though the changes in sagopilone are relatively modest it appears that the molecule is no longer an MDR substrate and is as active on resistant phenotypes as on their wild type precursors [78]. Finally isoxazolefludelone (Fig. 8) is now in Phase I trials at Memorial Sloan Kettering.
4.3 Glucan Synthase Inhibitors as Antifungal Agents: The Story of Cancidas®
In the late 1970s, one of the targets of new antifungal agents was the complex cell walls in organisms such as Candida albicans or Aspergillus species which were very significant infective agents in man, particularly in the emerging treatment of transplant patients who were on the early regimens of immunosuppressive therapy for treatment of transplants. Once the patient’s immune system was depressed, opportunistic infections, in particular Candida species became major problems. Since at that time, the major available treatments were either polyenes (amphotericin) or fluorinated cytosine, neither of which were particularly efficacious and had significant toxicity, scientists began to search for agents, almost all from fermentation of microbes, that had activity against the set of targets that were typical of fungi, the mannan, glucan or chitin syntheses in their cell walls.
The nikkomycins with an example being nikkomycin Z (Fig. 9) entered preclinical studies as potential chitin synthase inhibitors but languished for many years due to transport problems, however, fairly recently nikkomycin Z was reportedly under development as an orphan drug for coccidioiodomycoses (valley fever) [79].
Fig. 9.

Chitin and Glucan Inhibitors
Nikkomycin Z, Caspofungin, Anidulafungin, Micafungin
In the early to middle 1970s, and stretching into the early 1980s, came reports of lipopeptide antibiotics that had apparent activity against pathogenic fungi and appeared from early tracer methods, coupled to some experiments using microbes with genetically modified cell wall components (from use of classical methodologies, not recombinant DNA), to affect the metabolism related to the production of the glucans associated with cell wall production. These compounds fell into two relatively closely related chemical classes, the lipopeptide group known as echinocandins and a glycolipid group known as the papulacandins. There were a number of reviews published in the middle 1990s describing the different “classes” with examples being on the echinomycins by Debono and Gordee from Lilly [80], a shorter but more general one on the topic of new antifungal drugs by Georgopapadakou and Tkacz [81] followed by an extensive summation of their semisynthetic modifications published by Debono et al the next year [82], In 2006, a derivative from this series was launched by Pfizer (who assumed the compound on their acquisition of Vicuron) as anidulafungin (Fig. 9), though the Fujisawa (now Astellas) compound from the same general echinocandin class had been launched as micafungin (Fig. 9) in 2002.
However, the Merck efforts around the first molecule to be approved with this mechanism of action have a lot in common with the efforts around the development of taxol® and makes an interesting story as to the many paths that had to be followed, sometimes in series, sometimes in parallel in order to produce a successful drug, particularly as the work was performed in the pharmaceutical industry and not in a government / academic / industry consortium.
In 1985, a pneumocandin-producing microbe, subsequently named as Glarea lozoyensis was isolated from a water sample taken in the Lozoya River valley in Spain. The activity was discovered using a broad-based screen for cell wall active agents and reported in 1992 [83], together with information on yields of similar compounds from the fermentation the same year [84], where yields of 330 mg.l−1 for the A0 variant, plus 26 mg.l−1 for the B0 and 4 mg.l−1 for the D0 variant structures were reported. The same issue of the Journal of Antibiotics contained another very interesting report also from Merck that the A0 variant was also active against Pneumocystis carinii [85], thus linking parasitic and antifungal activity in the same molecule.
Bearing in mind these results and some demonstrating significant correlations between the MIC (minimum inhibitory concentration), MFC (minimum fungicidal concentration or concentration required to cause a 100 fold reduction in colony forming units of Candida) using a cleverly modified mouse model of kidney infection as an in vivo assay [86], plus other data, a decision was made to develop a modification of pneumocandin B0 as a candidate compound.
To cut a very long story short, a series of medium modifications, a search for strains that would produce more of the desired B0 starting material, very close linkages between medicinal chemists attempting to produce phosphate linked prodrugs, then altering one or more of the constituent amino acids in the ring structure, together with input from Merck geneticists studying Saccharomyces cerevisiae genetics, which materially aided in the identification of the corresponding genetic linkages in pathogenic Candida albicans and Aspergillus nidulans, led to the final identification of MK-0911 or L-743872 as the clinical candidate. An extremely thorough discussion and analysis, plus some entertaining side stories as to what was done both knowingly and unknowingly in adjacent Merck laboratories was published in 2001 by Kurtz and Rex [87]. This review in Advances in Protein Chemistry, an unusual journal in which to find the history of an antifungal drug development program, should be consulted for the interplay of chemistry, microbiology and pharmacology that led to the successful approval of the compound as Caspofungin (Fig. 9; Cancidas®) in 2001, the first of the glucan inhibitors to become an antifungal drug.
However, the story of molecules with this mechanism of action is still on-going, even with three clinical drugs approved in the early to middle 2000s. This is demonstrated by the publication over the last few years of some relevant reviews. In 2008, Mohr et al. [88], discussed all then current anti-fungal agents from a predominately clinical pharmacy perspective. In 2011, another clinical pharmacy based group, Pitman et al. [89], discussed the current and future prospects of the current drugs including the glucan inhibitors, and later that same year, a group of scientists who were intimately involved whilst at Merck, or who consulted with the earlier Merck scientists, published a very interesting perspective in Chemistry and Biology on the challenges associated with natural product-based antifungal drug discovery [90]. This article shows the new genetic techniques that were and are being employed in order to investigate materials that are still being used as potential sources of antimicrobial agents. This is a paper that deserves being read and reread by anyone who is in the field.
Finally, just to demonstrate that one cannot think that the mechanism(s) of action are well defined Ingham and Schneeburger [91] reported in a recent paper that caspofungin (Cancidas®) and anidulafungin reduce growth but not halt it when applied to Aspergillus fumigatus, with intact, halted but not lyzed cells showing rapid recovery when the drugs were removed. Thus designating a compound as fungistatic or fungicidal may be simplistic.
Thus as we said in the introduction to this section, the search for glucan inhibitors and in particular the work on caspofungin definitely is reminiscent of the story of the travails associated with the discovery and development of taxol®. In both cases, very significant amounts of time, changing sources of raw materials and significant chemistry and pharmacology was involved, but finally success was achieved.
5. Other Sources
Teprotide, isolated from the venom of the pit viper, Bothrops jaracaca, led to the design and synthesis of the ACE inhibitors, captopril and enalapril (Fig. 10) [9], used in the treatment of cardiovascular disease, while epibatidine, isolated from the skin of the poisonous frog, Epipedobates tricolor, has led to the development of a novel class of potential painkillers (Fig. 10) [92]. A further notable discovery was the isolation of exendin-4 from the venom of the Gila monster, Heloderma suspectum [93], that led to the development of the extenatide polypeptide, Byetta® (Fig. 10), as an injectable medicine used to improve glucose (blood sugar) control in adults with type 2 diabetes.
Fig. 10.

Drugs from amphibian, reptilian and human sources
Teprotide, captopril, epibatidine, Byetta®, liraglutide
In 2009, another peptide but this time a very close relative of human GLP-1 was approved for similar indications to extenatide in the treatment of type 2 diabetes in the EU, and then approved in Japan and the USA in 2010 under the generic name liraglutide (Fig. 10) [94]. Currently there are 20 other GLP-1 mimetics in clinical trials shown in the IntegrityTM database as of December 2012.
6. The Importance of Natural Products In Drug Discovery And Development
In Sections 2 to 5 we have listed a relatively small number of selected examples of some of the useful drugs discovered from a variety of natural sources. In our paper published in 2012 [95], we analyzed the sources of new drugs over the period 01/1981-12/2010, and classified these compounds as N (an unmodified natural product), NB (a natural product botanical), ND (a modified natural product), S (a synthetic compound with no natural product conception), S*, S*/NM (a synthetic compound with a natural product pharmacophore; /NM indicating competitive inhibition), and S/NM (a synthetic compound showing competitive inhibition of the natural product substrate). This analysis indicated that, while 66% of the 1073 small molecule, new chemical entities (NCEs) are “formally synthetic” due to the fact that they have an “S” in their coding, 17% correspond to synthetic molecules containing pharmacophores derived directly from natural products classified as S* and S*/NM. Furthermore, 14% are actually modeled on a natural product inhibitor of the molecular target of interest, or mimic (i.e., competitively inhibit) the endogenous substrate of the active site, such as ATP (S/NM). Thus, only 36% of the 1073 NCEs can be classified as truly synthetic (i.e., devoid of natural inspiration) in origin (S) (Fig. 11).
Fig. 11.
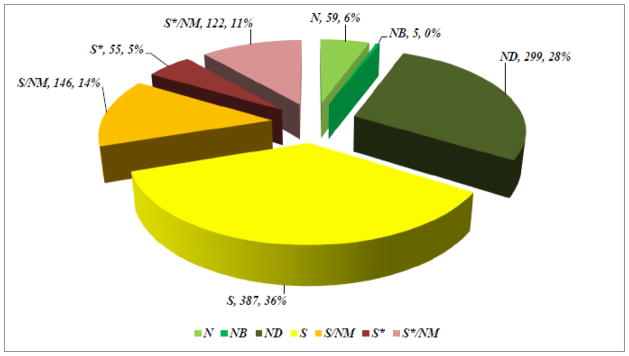
Sources of Drugs
Considering disease categories for the 1073 small molecules, 69% of anti-infectives (n = 195) which covers anti-bacterial, -fungal, -parasitic and –viral agents, are naturally derived or inspired (N; ND; S*; S*/NM; S/NM). In the cancer treatment area (n = 172) 75% are in these categories, with the figure being 59% if the S/NM category is excluded.
The steady decline in the output of the R&D programs of the pharmaceutical industry in recent years has been reflected in the figures shown in the 2012 review by the authors [95]. Inspection of the data shows that since the highpoints in the late 1980s (over 60 small moleculeNCEs / year) the trend line has been downwards, and over the 10 years from 2001 to 2010 the average was 23 NCEs per year, with a minimum of 18 and a maximum of 34 in this time period. Though various factors have been blamed for this downturn, it is significant that it has occurred during a period of declining interest in natural products on the part of major pharmaceutical companies, in favor of reliance on new chemical techniques such as combinatorial chemistry for generating molecular libraries.
The realization that the number of NCEs in drug development pipelines is declining may have led to the rekindling of interest in “rediscovering natural products” [96], as well as a mounting appreciation of the value of natural product-like models in “improving efficiency” in so-called diversity-oriented synthesis [97]. The urgent need for new pharmaceuticals for the treatment of cancer, HIV and other infectious diseases, as well as a host of other diseases, demands a vigorous exploration of all approaches to drug discovery, and it is clear that Nature has played, and will continue to play, a vital role in this process.
As stated by Berkowitz in 2003 commenting on natural products in Rouhi’s report [96], “We would not have the top-selling drug class today, the statins; the whole field of angiotensin antagonists and angiotensin-converting-enzyme inhibitors; the whole area of immuno-suppressives; nor most of the anticancer and antibacterial drugs. Imagine all of those drugs not being available to physicians or patients today.” Or, as was eloquently stated by Danishefsky in 2002, “a small collection of smart compounds may be more valuable than a much larger hodgepodge collection mindlessly assembled_” [98]. Six years ago he and a coauthor restated this theme in their review on the applications of total synthesis to problems in neurodegeneration: “_We close with the hope and expectation that enterprising and hearty organic chemists will not pass up the unique head start that natural products provide in the quest for new agents and new directions in medicinal discovery. We would chance to predict that even as the currently fashionable “telephone directory” mode of research is subjected to much overdue scrutiny and performance-based assessment, organic chemists in concert with biologists and even clinicians will be enjoying as well as exploiting the rich troves provided by Nature’s small molecules [99]. A comment that in the authors’ view is still absolutely relevant.
The complexity and molecular diversity of natural products are clearly evident from the foregoing discussion, and inspection of the structures shown in Figures 1 to 10 should be enough to convince any skeptic that few of them would have been discovered without the application of natural products chemistry.
Structural diversity, however, is not the only reason why natural products are of interest to drug development. An important additional feature is that they often possess highly selective and specific biological activities based on mechanisms of action. Two excellent examples are the HMG-CoA reductase inhibition exhibited by statins such as lovastatin (Fig. 6), and the tubulin-assembly promotion activity of paclitaxel (Fig. 3), neither of which would have been discovered without the natural product leads and investigation of their mechanisms of action. A further striking illustration of the influence of natural products involving the modulation of many of the enzymatic processes operative in cell cycle progression, may be found at the website of the Roscoff Biological Station (http://www.sb-roscoff.fr/usr3151.html), which covers diagrams originally published by Meijer [100] on natural products and the cell cycle, with a modified version shown below in Fig. 12.
Fig 12.

Natural products and the cell cycle
The bioactivity of natural products stems from the hypothesis that essentially all natural products have some receptor-binding activity; the problem is to find which receptor a given natural product is binding to [101]. Experience shows that organisms often provide investigators with complex libraries of unique bioactive constituents, analogous to the libraries of crude synthetic products initially produced by combinatorial chemistry techniques. The natural products approach can thus be seen as complementary to the synthetic approach, each providing access to (initially) different lead structures. Indeed, combinatorial chemistry is an extremely powerful tool for the optimization of an active natural product structure, and the task of the natural products researcher is thus to select those initial lead compounds of pharmacological interest from the “natural combinatorial libraries” produced by extraction of organisms.
7. Classical Natural Sources: Untapped Potential
The exceptional complexity and molecular diversity of natural products has been highlighted in earlier sections, but even more remarkable is the fact that the surface of these unique natural resources has barely been scratched. Despite the intensive investigation of terrestrial flora, it is estimated that only 6% of the approximately 300,000 species (some estimates are as high as 500,000 species) of higher plants have been systematically investigated, pharmacologically, and only some 15% phytochemically [8, 102, 103]. The potential of the marine environment as a source of novel drugs remains virtually unexplored [43, 104], and until recently, the investigation of the marine environment had largely been restricted to tropical and subtropical regions; however, the exploration is being expanded to colder regions. The isolation of novel pyrido-pyrrolo-pyrimidine derivatives, the variolins (Fig. 13), from the Antarctic sponge, Kirkpatrickia varialosa, was reported in 1994 [105, 106], followed by their total synthesis in 2003 [107], while the isolation of a cytotoxic macrolide palmerolide A (Fig. 13) from an Antarctic tunicate was reported in 2006 [108], with total synthesis leading to a revision of the original structure the next year [109]. Recently, Wilson and Brimble published an excellent review on molecules from extreme environments, which included Antarctic sources. This should be consulted for further information on these compounds [110].
Fig. 13.
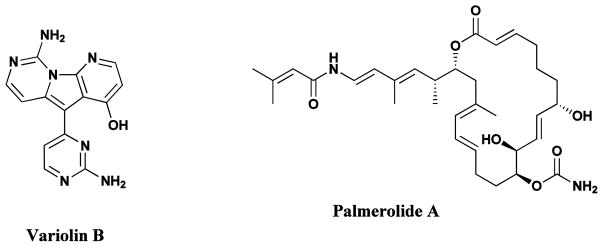
Natural Products from Antarctic sources
Variolins, Palmerolide
The selective and reproducible production of bioactive compounds has been induced through exposure of the roots of hydroponically grown plants to chemical elicitors [111], while feeding of seedlings with derivatives of selected biosynthetic precursors can lead to the production of non-natural analogs of the natural metabolites. Thus, the production of non-natural terpene indole alkaloids related to the vinca alkaloids has been reported through the feeding of seedlings of Catharanthus roseus with various tryptamine analogs [112]. Further work by the O’Connor group has demonstrated how genetic control of alkaloid synthesis in C. roseus can be modified by chemical and biological methods, thus opening up the possibility of novel agents via such manipulations [113–116]. In addition to these techniques, Gunatilaka’s group at Arizona have recently reported on a novel aeroponic system that produces unusual plant secondary metabolites, thus opening up yet another method of encouraging the expression of unrecognized biosynthetic clusters [117].
8. The Unexplored Potential of Microbial Diversity
Until recently, the inability to cultivate most naturally occurring microorganisms has severely limited the study of natural microbial ecosystems, and it has been estimated that much less than 1% of microorganisms seen microscopically have been cultivated. Yet, despite this limitation, the number of highly effective microbe-derived chemotherapeutic agents discovered and developed thus far has been impressive. Given the observation that “a handful of soil contain billions of microbial organisms” [118], and the assertion that “the workings of the biosphere depend absolutely on the activities of the microbial world” [119], the microbial universe clearly presents a vast untapped resource for drug discovery. In addition, substantial advances in the understanding of the gene clusters encoding multimodular enzymes involved in the biosynthesis of a multitude of microbial secondary metabolites, such as polyketide synthases (PKSs) and / or nonribosomal peptide synthetases (NRPSs), has enabled the sequencing and detailed analysis of the genomes of long-studied microbes such as Streptomyces avermitilis. These early studies revealed the presence of additional PKS and NRPS clusters resulting in the discovery of novel secondary metabolites not detected in standard fermentation and isolation processes [120]. Such genome mining has been used in the discovery of a novel peptide, coelichelin, from the soil bacterium, Streptomyces coelicolor [121], and this concept is further expanded on in the discussion in section 9.3.
8.1 Improved Culturing Procedures
Development of procedures for cultivating and identifying microorganisms is aiding microbiologists in their assessment of the earth’s full range of microbial diversity. For example, the use of “nutrient-sparse” media simulating the original natural environment, enabled the massive parallel cultivation of gel-encapsulated single cells (gel micro-droplets; GMDs) derived from microbes separated from environmental samples (sea water and soil) [122]. This has permitted “the simultaneous and relatively non-competitive growth of both slow- and fast-growing micro-organisms”, thereby preventing the overgrowth by fast-growing “microbial weeds”, and leading to the identification of previously undetected species (using 16S rRNA gene sequencing), as well as the culturing and scale-up cultivation of previously uncultivated microbes. Coupled with the initial report of the sequencing of the marine actinomycete, Salinispora tropica, where it was found that approximately 10 percent of the genome coded for potential secondary metabolites [123, 124], and the paper on cultivation of Gram-positive marine microbes by the same group [125], the potential for discovery of novel agents is immense. That this type of work and results is not limited to the prokaryotes is very well demonstrated by the recent review of Aspergillus genomic sequences and their potential in the production of previously unknown secondary metabolites by Keller’s group at Wisconsin [126].
8.2 Extraction of Environmental Samples (the Metagenome)
Despite improvement in culturing techniques, greater than 99% of microscopically observed microbes still defy culture. Extraction of nucleic acids (the metagenome) from environmental samples, however, permits the identification of uncultured microorganisms through the isolation and sequencing of ribosomal RNA or rDNA (genes encoding for rRNA). Samples from soils and seawater are currently being investigated [127–129], and whole-genome shotgun sequencing of environmental-pooled DNA obtained from water samples collected in the Sargasso Sea off the coast of Bermuda by the Venter group, indicated the presence of at least 1,800 genomic species which included 148 previously unknown bacterial phylotypes [128]. Venter et al., are examined microbial communities in water samples collected by the Sorcerer II Global Ocean Sampling (GOS) expedition, and their data predicted more than six million proteins, nearly twice the number of proteins present in the then current databases, and some of the predicted proteins bore no similarity to any currently known proteins, therefore representing new families [130]. These methods may be applied to other habitats, such as the microflora of insects and marine animals, and there was a report of an “Air Genome Project” launched in Manhattan in 2005 where samples of air were analyzed for content of DNA from bacteria, fungi and other microbes [131].
The cloning and understanding of the novel genes discovered through these processes, and the heterologous expression of gene clusters encoding the enzymes involved in biosynthetic pathways in viable host organisms, such as Escherichia coli, should permit the production of novel metabolites produced from as yet uncultured microbes. A 2009 review [129] and a very recent paper [132] demonstrated the potential for such techniques and further examples can be seen in the articles by Piel on recovery of clusters from marine invertebrate-related microbes [133, 134]. The production of the antibiotic**,** pantocin A (Fig. 14), from the bacterium, Pantoea agglomerans, is an example of such heterologous expression of genomic DNA [135]. In this case the production of pantocin A by the source microbe grown in liquid culture proved to be impractical due to low titers and the complexity of the mixture of metabolites produced. Expression of a genomic DNA library isolated from P. agglomerans in E. coli, however, provided access to reasonable quantities of the small molecule antibiotics of interest [135].
Fig 14.

Natural products from heterologous gene expression, extremophiles and endophytes
Pantocin, berkeleydione, berkeleytrione, ambuic acid, aspochalasins, terrequinone
8.3 Extremophiles
Extremophilic microbes (extremophiles) abound in “extreme habitats”. These include acidophiles (acidic sulfurous hot springs), alkalophiles (alkaline lakes), halophiles (salt lakes), piezo (baro)- and (hyper)thermophiles (deep-sea vents) [136–140], and psychrophiles (Arctic and Antarctic waters, alpine lakes).[141]. Early investigations generally centered on the isolation of thermophilic and hyperthermophilic enzymes (extremozymes) [142–146], but there is little doubt that these extreme environments can yield novel bioactive chemotypes.[110] Abandoned mine-waste disposal sites have yielded unusual acidophiles which thrive in the acidic, metal-rich, polluted environments which are generally toxic to most prokaryotic and eukaryotic organisms [147]. Thus the novel sesquiterpenoid and polyketide-terpenoid metabolites, berkeleydione and berkeleytrione (Fig. 14) showing activity against metalloproteinase-3 and caspase-1, activities relevant to cancer, Huntington’s disease and other diseases, have been isolated from Penicillium species found in the surface waters of Berkeley Pit Lake in Montana [148–151].
8.4 Endophytes
As indicated in Section 2, in relative terms, plants have been extensively studied as sources of bioactive metabolites, but the endophytic microbes which reside in the tissues between living plant cells have received little attention until fairly recently. Relationships between endophytes and their host plants may vary from symbiotic to opportunistic, and studies have revealed an interesting realm of novel chemistry [152–154]. Some of the new bioactive molecules discovered in the early 2000s included the novel wide-spectrum antibiotics, kakadumycins, isolated from an endophytic Streptomycete associated with the fern leafed grevillea (Grevillea pteridifolia) from the Northern Territory of Australia [155], ambuic acid (Fig. 14), an antifungal agent, which has been described from several isolates of Pestalotiopsis microspora found in many of the world’s rainforests [156, peptide antibiotics, the coronamycins, from a Streptomyces species associated with an epiphytic vine (Monastera species) found in the Peruvian Amazon, [Ezra, 2004 #94] and aspochalasins I, J, and K (Fig. 13) [157], isolated from endophytes of plants from the southwestern desert regions of the United States.
Recent reports of the isolation of important plant-derived anticancer drugs from endophytic fungi have served to focus attention on these sources. Examples are paclitaxel (Taxol®) from Taxomyces [158] and many Pestalotiopsis species [159], as well as camptothecin [160, 161], podophyllotoxin [162, 163], vinblastine [164], and vincristine [165, 166], all produced in relatively small amounts by endophytic fungi isolated from the producing plants. It has been demonstrated, however, that these compounds are not artifacts, and so the identification of the gene / gene products controlling metabolite production by these microbes could provide an entry into greatly increased production of key bioactive natural products.
The presence of potential gene products controlling metabolite production has been predicted in a recently reported genomic analysis of the fungus Aspergillus nidulans, which suggested not only the presence of clustered secondary metabolite genes having the potential to generate up to 27 polyketides, 14 nonribosomal peptides, one terpene, and two indole alkaloids, but also identified the potential controller of expression of these clusters [167]. This was demonstrated it by expressing terrequinone A (Fig. 14), a compound not previously reported from this species [167]. Similar predictions can be made from A. fumigatus and A. oryzae as a result of the analysis of the potential number of secondary metabolite clusters in these fungi [167]. Two reviews, both from the Keller group, expand on the genomic clusters and control of secondary metabolites in these organisms, one in 2007 [168] and the other in 2012 [126].
8.5 Marine Microbes
Deep ocean sediments are proving to be a valuable source of new actinomycete bacteria that are unique to the marine environment [125]. Use of a combination of culture and phylogenetic approaches has led to the description of the first truly marine actinomycete genus named Salinospora [125, 169], which was subsequently renamed as Salinispora, and its members are proving to be ubiquitous, being found in concentrations of up to 104 per mL in sediments on tropical ocean bottoms and in more shallow waters, as well as appearing on the surfaces of numerous marine plants and animals. On culturing using the appropriate selective isolation techniques, significant antibiotic and cytotoxic activity has been observed, and has resulted in the isolation of a potent cytotoxin, salinosporamide A (Fig. 15), a very potent proteasome inhibitor (IC50 = 1.3 nM) [170], currently in Phase I clinical trials. More recently, the isolation and cultivation of another new actinomycete genus, named Marinispora, has been reported, and novel macrolides called marinomycins have been isolated. Marinomycins A-D (Fig. 15) show potent activity against drug-resistant bacterial pathogens and some melanomas [171]. Further publications by the Fenical group on the novel and diverse chemistry of these new microbial genera include the isolation of potential chemopreventive agents, saliniketals A and B from Salinispora arenicola [172], while two new cyclic peptides, thalassospiramides A and B, possessing immunosuppressive activity have been isolated from a new member of the marine alpha-proteobacterium Thalassospira [173]. The potential for further development of antibacterial agents from marine sources, including isolated microbes was recently reviewed by Hughes and Fenical [174], with extension into other pharmacological areas in reviews by Waters et al., [175] and Pettit [176].
Fig. 15.

Examples of novel microbial natural products
Salinosporamide, marinomycins A-D, maytansine, pederin, onnamide, rhizoxin
8.6 Microbial Symbionts
Evidence is mounting indicating that many bioactive compounds isolated from various macro-organisms are actually metabolites synthesized by symbiotic bacteria [177, 178]. These include the anticancer compounds, the maytansanoids (Fig. 15), originally isolated from several plant genera of the Celastraceae family [34], and the pederin class (Fig. 15), isolated from beetles of genera Paederus and Paederidus as well as from several marine sponges [133, 177–180]. In addition, a range of antitumor agents isolated from marine organisms closely resemble bacterial metabolites [43, 181].
An interesting example of a complex symbiotic-pathogenic relationship involving a bacterium-fungus-plant interaction has been discovered in the case of rice seedling blight. The toxic metabolite, rhizoxin (Fig. 15), originally isolated from the contaminating Rhizopus fungus, has actually been found to be produced by an endo-symbiotic Burkholderia bacterial species [182–184]. Rhizoxin exhibits potent antitumor activity, but its further development as an anticancer drug has been precluded by toxicity problems. Thus, in addition to offering potentially new avenues for pest control, this unexpected finding has enabled the isolation of rhizoxin as well as rhizoxin analogs through the cultivation of the bacterium independently of the fungal host [185]. This may have significant implications in development of agents with improved pharmacological properties.
8.7 Combinatorial Biosynthesis
The substantial advances made in the understanding of the role of multifunctional polyketide synthase enzymes (PKSs) in bacterial aromatic polyketide biosynthesis have led to the identification of many such enzymes, together with their encoding genes [186–189]. The same applies to nonribosomal peptide synthases (NRPS) responsible for the biosynthesis of nonribosomal peptides (NRPs) [188]. The rapid developments in the analysis of microbial genomes has enabled the identification of a multitude of gene clusters encoding for polyketides, NRPs and hybrid polyketide-NRP metabolites, and have provided the tools for engineering the biosynthesis of novel “non-natural” natural products through gene shuffling, domain deletions and mutations [188, 190]. Results of the application of these combinatorial biosynthetic techniques to the production of novel analogs of anthracyclines, ansamitocins, epothilones, enediynes, and aminocoumarins have recently been reviewed by Van Lanen and Shen [191].
The efficient scale-up production of epothilone D (Section 4; Fig. 8) exemplifies the power of this technique. Epothilone D, the des-epoxy precursor of epothilone B, was the most active of the epothilone series isolated from the myxobacterium, Sorangium cellulosum, and entered clinical trials as a potential anticancer agent, reaching Phase II but has now been discontinued. The polyketide gene cluster producing epothilone B had been isolated and sequenced from two Sorangium cellulosum strains [192, 193], and the epoxidation of epothilone D to epothilone B was shown to be due to the last gene in the cluster, _epo_K, encoding a cytochrome P450. Heterologous expression of the gene cluster minus the _epo_K in Myxococcus xanthus resulted in large-scale production of crystalline epothilone D [194]. Further discussion on the integration of this technology into investigations of natural products is given in Section 9.3.
9. Natural Product Drug Discovery and Development: A Multidisciplinary Process
9.1 Total Synthesis
The total synthesis of complex natural products has long posed challenges to the top synthetic chemistry groups worldwide, and has led to dramatic advances in the field of organic chemistry, particularly at the turn of the 21st Century and onwards [195]. As eloquently stated by Nicolaou and his coauthors in 2000: “Today, natural product total synthesis is associated with prudent and tasteful selection of challenging and preferably biologically important target molecules; the discovery and invention of new synthetic strategies and technologies; and explorations in chemical biology through molecular design and mechanistic studies. Future strides in the field are likely to be aided by advances in the isolation and characterization of novel molecular targets from Nature, the availability of new reagents and synthetic methods, and information and automation technologies” [195].
In some instances, as noted in Section 7 regarding the cytotoxic macrolide palmerolide A (Fig. 13), total synthesis has led to a revision of the original published structure [109], with another notable example being that of the marine-derived antitumor compound, diazonamide A (Fig. 16) [109, 196].
Fig. 16.

Products of Total Synthesis
Diazonamide, discodermolide, TZT-1027/auristatin PE, E-7974
Significant strides have been made in the synthesis and structural modification of drugs that are difficult to isolate in sufficient quantities for development. Adequate supply can be a serious limiting factor in the preclinical and clinical development of some naturally-derived drugs, and the focus of many top synthetic groups on devising economically feasible synthetic strategies is a very welcome development for both clinicians conducting clinical trials and patient populations. An excellent example is the marine-derived anticancer agent discodermolide (Fig. 16), where total synthesis provided sufficient quantities for thorough clinical trials, but unfortunately, these were terminated at the Phase I/II interface due to lack of objective responses and toxicity [197, 198].
The process of total synthesis can often lead to the identification of a sub-structural portion of the molecule bearing the essential features necessary for activity (the pharmacophore), and, in some instances, this has resulted in the synthesis of simpler analogs having similar or better activity than the natural product itself. One of the most notable examples is that of the marine derived antitumor agent, halichondrin B (Fig. 5; Section 3), where total synthetic studies revealed that the right hand half of the molecule retained all or most of the potency of the parent compound, and the analogue, E7389 (eribulin; Fig. 5), was approved by the FDA in 2010 [55].
The total synthesis of this compound by the group at the Eisai Research Institute in Massachusetts, USA is without question, the most ambitious production of a totally synthetic drug substance, first under good laboratory practice (GLP) and then under current good manufacturing practices (cGMP) ever performed. Around 200 derivatives were made before settling on the compound that ultimately became eribulin (Halaven®). Although not yet formally published, oral reports of the overcoming of problems inherent in the scale up process have been reported over the last few years, with the ultimate usage of multiple crystallization steps rather than chromatographic separations in the final commercial product.
Over the years, the Eisai Research Institute has continued its work with marine-sourced natural products, with known successes being the total synthesis of laulimalide and other simpler compounds such as E-7974 (Fig 16) which is based upon the modified tripeptide, hemiasterlin.
In some instances, clinical trials of the original natural product may fail, but totally synthetic analogues continue to be developed. Thus, while clinical trials of the marine-derived anticancer agents, dolastatin 10 and dolastatin 15, have been terminated, the synthetic analogue of dolastatin 10, TZT-1027 (auristatin PE or soblidotin; Fig. 16) was given a brand new “lease on life” by linking the peptide, with a slight modification, (auristatin F, Fig. 16), to specific antibodies using tumor specific scissionable linkages, with one antibody-drug complex (ADC) using auristatin F, brentuximab vedotin approved as Adcenris® by the FDA in 2011 for the treatment of lymphoma [199].
Similar success stories of syntheses of other complex agents from marine sources can be seen by inspection of the many compounds synthesized on “more than milligram quantities" by the groups of Paterson (at Cambridge University) working on peloruside [200], modifications of other marine and plant based agents, including dictyostatin [201] and more general aspects dealing with syntheses of polyketides [202]. To these examples one may also add the synthesis of spongiostatin 1 on a 1 gram scale by the Smith group at the University of Pennsylvania [203].
These are only a few of the examples that can be given of successes in the last few years where the use of very clever chemical strategies, coupled to novel reactions, in some cases, even the so-called “one-pot” synthesis, or the biomimetic syntheses with minimal protecting groups pioneered by Baran at Scripps [204].
9.2 Diversity-Oriented Synthesis, Privileged Structures and Combinatorial Chemistry
Early combinatorial work where an active natural product was utilized as the central synthetic scaffold, resulted in what can be described as the so-called parallel synthetic approach, giving large numbers of analogues for structure-activity studies, and is exemplified by the syntheses around the sarcodictyin (Fig. 17) scaffold [205]. The importance of natural products as leads for combinatorial synthesis (using any of the combinatorial methods) is further illustrated by the concept of “privileged structures” [206], and this approach has been successfully developed by several groups [207]. In one such case, a search of the literature yielded nearly four thousand 2,2-dimethyl-2H-benzopyran moieties (Fig. 17), with another 8000 structures identified through the inclusion of a slight modification of the search. Application of solid-phase synthetic methods led to the identification and subsequent optimization of a benzopyran with a cyanostilbene substitution (Fig. 17) that was effective against vancomycin-resistance bacteria [208–210].
Fig. 17.

Products of Diversity-Oriented and Parallel Synthesis and Privileged Structures
Dysidiolide, galanthamine, psammaplin, sarcodictyin, 2,2-dimethyl-2H-benzopyran, benzopyrans plus cyanostilbene substitution
While there were claims that combinatorial chemistry was generating new leads [211], the declining numbers of new NCEs [95] (Section 6) indicated that the use of de novo combinatorial chemistry approaches to drug discovery over the past decade have been disappointing, with some of the earlier libraries being described as “poorly designed, impractically large, and structurally simplistic” [211]. As stated in that article, “an initial emphasis on creating mixtures of very large numbers of compounds has largely given way in industry to a more measured approach based on arrays of fewer, well-characterized compounds” with “a particularly strong move toward the synthesis of complex natural-product-like compounds – molecules that bear a close structural resemblance to approved natural-product-based drugs”. The importance of the use of natural product-like scaffolds for generating meaningful combinatorial libraries has been further emphasized in a later article entitled “Rescuing Combichem. Diversity–oriented synthesis (DOS) aims to pick up where traditional combinatorial chemistry left off” [212]. In this article it was stated that “the natural product-like compounds produced in DOS have a much better shot at interacting with the desired molecular targets and exhibiting interesting biological activity”. A short discussion of the relative differences of the various chemical methods was given in the review by Cragg et al., in 2009 [213] with a very good discussion of the BIOS system presented by Waldmann in 2011 [214], which should be read in conjunction with his discussion of “chemical space with respect to natural products”, also published in 2011 [215].
The synthesis of natural-product-like libraries is exemplified by the work of the Schreiber group, who combined the simultaneous reaction of maximal combinations of sets of natural-product-like core structures (“latent intermediates”) with peripheral groups (“skeletal information elements”) in the synthesis of libraries of over 1000 compounds bearing significant structural and chiral diversity [216, 217]. Through detailed analyses of active natural product skeletons, relatively simple key precursor molecules may be identified which form the building blocks for use in combinatorial synthetic schemes, thereby enabling structure activity relationships to be probed. The generation of small libraries, built through the solid phase synthesis of molecules such as epothilone A (Fig. 8), dysidiolide (Fig. 17), galanthamine (Fig. 17) and psammaplin (Fig. 17), has been reviewed [207, 218–224]. Very recent aspects of syntheses around natural product structures can be seen by the work by the groups of Tan [225], and Quinn [226].
9.3 Interactions of Microbial Sources, Genomics and Synthetic Chemistry
The enormous unexplored potential of microbial diversity has been discussed in Section 8, and the strategy of genome mining was briefly mentioned in the introduction to that section. As a result of the rapid evolution of genomic sequencing and the ever dropping costs of performing such studies, the amount of genomic information is ever increasing, resulting in the potential for the expression of previously unrecognized metabolites, with an early (2005) review [227] and more recent reviews (2009) [129, 228] (2011) [229] and (2012) [230] covering general and more specific aspects of the influence of genomics on natural product research. These five reviews are only cited so the that reader can gain an idea of the “flavor” of the impact of these technological advances on natural product research, as a full discussion would be a book on the subject.
It has now become evident, initially through the pioneering work of Hopwood and Omura, that the genome of the Streptomycetes and by extension, Actinomycetes in general and as mentioned earlier, Aspergillus species contain large numbers of previously unrecognized secondary metabolite clusters. An excellent early example was the investigation of the genome of the vancomycin producer, Amycolatopsis orientalis (ATCC 43491), which resulted in the isolation of the novel antibiotic ECO-0501 (Fig.18). This compound was only found as a consequence of using the genomic sequence of the putative biosynthetic gene cluster to predict the molecular weight, and then looking for the molecule directly by HPLC-MS. The compound had a very similar biological profile to vancomycin but was masked by this compound in conventional bioassys [231]. Many more examples of the value of this type of investigation at that time, were provided in two subsequent reviews [232, 233], which should be read in conjunction with the later work reported by the groups of Hertweck [185], Keller [126], Moore [229], Piel [133] and Brady / Schmidt [129] to name but a few of the groups busy working in this field, all of which give up to date information on the manifold structures that can be found by expression of DNA whether directly from a microbe or from metagenomic operations..
Fig. 18.

New compounds from a variety of approaches
ECO 0501, chivosazol, platensimycin, platencin, phomallenic acid C, lucensimycin A, (−)-adamantaplatensimycin
As mentioned earlier in the discussion of the epothilones, the myxobacteria have now yielded to genomic analyses, and the identification and utilization of ChiR, the gene controlling production of chivosazol, an extremely potent eukaryotic antibiotic, has been reported [234]. This paper also deals with the major problem in secondary metabolite expression, whether in homologous or heterologous hosts, which is the identification and application of the transcriptional control mechanisms involved. An article published very recently demonstrates how one may go from a hypothetical intermediate in a myxobacterium to a potential drug candidate utilizing biosynthetic and chemical information obtained from myxobacterial genomic information plus clever chemistry [235].
It is also becoming evident that searching older sources for novel agents by utilization of new screens, frequently involving the use of genetically modified organisms or cell lines, and then coupling these to new synthetic methods, is leading to the discovery and development of very interesting bioactive compounds. In the field of antibiotics from microbial sources, the last few years have seen a veritable explosion of novelty in compounds and screens. In 2006, microbial and chemical groups at Merck demonstrated that, by screening their older microbial extract libraries against novel screens utilizing antisense technologies, three entirely new chemical structural classes were identified. The first two, utilizing a screen for FabF/H inhibitors (coding for the β-ketoacyl carrier protein synthase I/II) yielded platensimycin (Fig. 18) from S. platensis [236], platencin (Fig. 17) [237] and phomallenic acid C (Fig. 18) from a Phoma species [238]. The third, also utilizing antisense technologies but this time directed against ribosomal protein synthesis, specifically ribosomal protein S4, led to the identification of the lucensimycin A (Fig. 18) from S. lucensis [239].
Within a year of the publication of the structure of platensimycin, both the racemic [240] and asymmetric [241] syntheses of this molecule had been published, and the synthesis of the adamantyl derivative (Fig. 18) was also reported, substituting the more accessible adamantyl substituent in place of the parent caged ketolide [242]. When resolved, the (minus;)-adamanta-platensimycin (Fig. 18) exhibited comparable activity to (minus;)-platensimycin against methicillin resistant Staphylococcus aureus and vancomycin resistant Enterococcus faecium, with the other enantiomers being inactive. This clearly demonstrates that there are synthetic chemists eager to synthesize novel molecules isolated from a natural source, and ready to modify these structures in ways that will permit scale-up. Recently, the Merck group published a much fuller report on these agents and this should be consulted for the full details of these very interesting agents [243].
The examples given in this section are merely a small portion of the immense amount of information that is currently available in the literature. It is hoped that this information will lead to novel and efficient methods for generating new agents from many natural sources. With this goal in mind, appropriate combinations of microbial fermentation, combinatorial biosynthesis or total chemical syntheses will make original naturally-occurring compounds and their derivatives or analogues available to assist in the generation of new medicinal or other agents for use in the human, veterinary and agricultural arenas.
10. Potential Problems with Natural Product Drugs
10.1 The Rio Convention
In May of 1992, the Nairobi Conference led directly to the opening of what has become known as the Convention on Biodiversity (CBD) which was opened for signature at the Earth Summit in Rio, and by the end of the first year, it had been signed by 168 nations. The history can be found at the following URL: http://www.cbd.int/history/
Although the CBD was signed by the US President, it has never been ratified by the US Senate, so the USA is one of the very few nations that are not a voting member of the Conference of Parties (COP) though it can attend these regular meetings as an observer. However, what this lack of ratification has done is to make it difficult for US-government funded organizations to obtain permission to collect materials from a Sovereign Nation’s territories. Effectively, each investigator has to establish a specific agreement that covers the rights and expectations of countries in whose lands and waters he or she wishes to operate. This is made even more difficult by the lack of specific infrastructure in a large number of countries (legal and intra-country difficulties) plus at times, an unrealistic expectation of what the odds are against finding a drug from collections.
Luckily for the collections organized by the National Cancer Institute (NCI) from the late 1980s, the NCI had developed what is now known as the NCI’s Letter of Collection and had signed the first agreement with the Government of the Malagasy Republic in 1990, two years before RIO. This document effectively contained all of the requirements of the original CBD with one exception, a royalty statement. This lack of such a clause was due to the US Code which effectively states that the US government may not “encumber a future invention”. Since under almost all conditions, the act of collection is not an inventive process, no royalty could be pre-assigned as it would “encumber a future invention”. However, this was overcome by stating that in the event that a patent was obtained from the collections (dealing now with discoveries made by US government personnel), the licensee of the patent must produce an agreement within one year with the country of origin, that covers their expectations as to suitable recompense, etc.
This has worked quite successfully, and is the basis for a very large number of similar agreements made by US government-funded investigators with a variety of countries. US-based organizations such as universities can establish agreements that have royalty clauses, even if funded by the US government; it is only US government organizations such as NCI that have this proscription.
Needless to say, this can lead to a variety of problems, and the authors of this review have a fund of stories about situations that have occurred over the years. Further information on the NIH’s methodologies, particularly in the marine environment, has recently been published in the Handbook of Marine Natural Products [244] and from a slightly different perspective, by the same authors in a recent Natural Product Reports review [245].
10.2 Screening Methods for Natural Products
With the advent of the initial high throughput screening systems (HTS) in the early 1990s, and the current iteration of >250,000 samples per day per assay, using robots, advanced data capture, 1536 well plate formats (meaning ~2 to 5 μl per well), isolated target proteins (enzymes, receptors etc., cloned from a variety of sources) and data mining techniques, the perception was that natural products other than as pure compounds, were passé. Perhaps the major, but unspoken reason was that at that particular time, the requirements in terms of time required to purify the bioactive(s) from crude natural product extracts, and then identify the compound(s), was significantly in excess of the life-time of the assay. To this one had to add the costs associated with maintenance of the necessary scientific and technical infrastructure related to natural products collections and curation of raw material samples and extracts.
There are examples of the lack of productivity of HTS screens using isolated proteins (thought to be essential components of the cellular target) using massive libraries of synthetic compounds in the literature, with perhaps the most telling being the report from GSK investigators on the lack of success when searching for antibiotics using HTS and isolated proteins [246]. In this report, over 70 different target proteins considered essential to the growth of microbes were evaluated over a 7 year period with no success. We should point out however, that a somewhat different view was expressed by Macarron et al., (also from GSK) when looking at different disease targets [247]. On the other hand, the utility of phenotypic cell-based screens in the antibiotic area is aptly demonstrated by the work reported in section 9.3 above where Merck scientists using their microbial extract collection identified novel agents against new targets using whole cell techniques.
A point that is often not taken into account in HTS / isolated protein screening is the “loss in formal activity” when one moves from in vitro to in cellulo screening systems. The usual rule of thumb is that a “hit” in the HTS format, is a 100 to a 1000 fold less “active” when tested in a suitable cell-line model. Thus a potential method of overcoming such a problem is to use phenotypic cell assays in a semi HTS mode with natural product extracts. Obviously, depending upon the individual assay design, crude extracts might have to be “cleaned up” (removal of promiscuous materials such as tannins, or pre-fractionated) prior to such cell-based, or even isolated protein HTS screening. This type of pre-fractionation / pretreatment of natural product extracts is being done in both academic and pharmaceutical company laboratories but reports at this time are usually oral presentations without too much details as to hit rates, losses etc. There are some reports of high content cell based screening that may well be suitable for partially purified NP extracts are in the literature, with an example being the review by Lang et al., in 2006 [248], and many advances in instrumentation have been made since that publication.
Thus the future of natural product extract screening is probably in the utilization of a combination of techniques that may include pre-fractionation, HTS using “designer” cell lines that have particular aspects of the cell’s metabolic machinery adapted to give a signal when inhibited or activated, coupled to microliter volumes of extract. The advent of modern hyphenated chemical analytical methodologies then enables an “active” hit to be rapidly assessed as to novelty on 50 – 100 micrograms of material, even in a semi-purified form. What one might call “modernized phenotypic screening”. There is irony in the fact that these situations, and potential methodologies that could help in screening of extracts, were covered in a review article published by one of the authors in 1989, including a set of assay screening rules that are still valid today [249].
10.3 The “Valley of Death”
This is a term that has a variety of meanings but in this review, we are using it to mean the “gap in availability / access to funding” that occurs when a small company and / or an academic investigator identifies a candidate that they consider a potential drug, It is at this point, that the full realization of what has to be accomplished; preclinical pharmacology and toxicology, preparation of cGMP quality active ingredients and the necessary regulatory filings, causes a search for a partner with “experience and deep pockets”. In general, the funding to that initial point has been via venture capitalists (VCs), US Government Small Business Innovation Research (SBIR) grants or contracts and conventional hypothesis-driven governmental grants.
In the early days of “biotechnology” meaning the middle 1980s to early 1990s, such funding was often available, predominately from VCs, but in the last seven plus years, sources of finance for preclinical development and early clinical development have dried up. Adding to the difficulties was the realization that the few large pharmaceutical companies remaining “in play” now want to see activity in at least a Phase I clinical trial and preferably at a Phase II level, before any significant discussions as to funding or even outright purchase, commence.
There is a perception that the costs of developing a natural product-sourced drug are significantly greater than those of a synthetic agent. To some extent, the initial costs might well be higher (meaning the sourcing and purification of enough material to commence preclinical work at the GLP level), but if the initial compound was found from a relatively small number of candidates rather than from massive (1 million or greater synthetic compounds) high throughput screens then the costs of the relative screening techniques need to be considered.
However, with the advent of the advances in modern synthetic chemistry techniques (exemplified in Section 9.1 above), it is now feasible to consider synthesis of the base molecule. Such syntheses are often performed using strategies that permit rapid combinatorial modification as the ADME (absorption, distribution, metabolism and excretion) properties of the original compound, and subsequent modifications are explored to determine the optimal preclinical candidate. Estimates of numbers of structural modifications of the base structure in order to optimize ADME properties range from <10 to >100 in oral presentations but since most such modifications are performed in pharmaceutical houses, no reliable published data are available
An example of where a discovery made in academia, then moved to a small company set up by the investigator and his university, that has been successful in overcoming the “Valley of Death” and only slightly modifying the natural product derivative for stability under ADME conditions, is the very recent approval (July 2012) of the proteasome inhibitor carfilzomib (Fig. 19; Kyprolis®) based upon the bacterial natural product epoxomicin (Fig. 19). Crews at Yale synthesized epoxomicin, a molecule that had been discovered by Bristol-Banyu from a bacterial source and then dropped by Bristol Myers due to not knowing the target. The Crews’ group discovered that it was a proteasome inhibitor inhibiting the chymotrypsin-like part of the 20 S proteasome, modified it slightly to produce what was then known as YU-101 and set up a small company (Proteolix) with the help of Yale University [250]. Subsequently Proteolix scientists made around a 100 derivatives but ended up improving the base YU-101 by addition of a morpholino end-group thus helping with the ADME properties (mainly water solubility and distribution from an oral perspective) [251]. Proteolix was acquired by Onyx (thus overcoming their “Valley of Death”) and the FDA approved the compound in July 2012 on the basis of responses from a Phase IIb clinical trial against multiple myeloma (ClinicalTrials.gov Identifier NCT00511238). Trials are ongoing in the US, Europe and Japan to extend the spectrum of treatments for this compound. We should add that this is the exception rather than the rule, but it does show that with luck and an excellent compound, it is possible.
Fig. 19.
Carfilzomib, Epoxomicin
11. Summary
The preceding sections have provided a very brief impression of the importance of natural products, both as pharmaceutical agents and/or as leads to bioactive molecules. With the emergence of novel screening systems related to the explosion of genetic information accelerating, the need to rapidly identify effective, novel lead structures is a vital necessity. We believe that a very significant portion of these leads will continue to be natural product derived. It should be remembered that Mother Nature has had three billion years to refine her chemistry and we are only now scratching the surface in exploring Nature’s molecular diversity!
To date, the relative ease of access to plants has resulted in the discovery of a majority of plant-derived materials as far as sources are concerned, with microbial sources being especially important in the antibiotic area. Recent work, however, suggests that marine organisms, and perhaps a group of organisms that have not had very much published on them until recently, the marine-sourced fungi [252], will play an increasingly important role in the future, especially given the impressive advances in the power of organic synthesis to address the supply problems inherent with this source material. In the future, with the advent of genetic techniques that permit the isolation and expression of biosynthetic cassettes, microbes and their marine invertebrate hosts [253] may well be the new frontier for natural products lead discovery, though plant endophytes also offer an exciting new resource. An interesting and very recent review by Janos Berdy [254] should also be read on the past, current and future aspects of antibiotic discovery and development, as it covers from a slightly different perspective, a significant number of the points alluded to in this article, but from the perspective of over 50 years of work in microbial natural products.
Coupling these novel sources to revamped phenotypic screens that utilize high-content imaging systems and that can be run in microliter volumes, hopefully will enable investigators to rapidly assess the activity of individual agents and their potential for future development. This will also require the expertize of talented synthetic chemists who have the ability to “tailor” structures from Nature in order to optimize their ADME properties for future drug use.
Highlights.
- We have given a history of natural products as drugs.
- We have discussed the reasons for the decreased interest in NPs in the Pharmaceutical industry.
- We have shown that the use of genomic techniques has allowed the recognition of new microbial sources of structures
- We demonstrate that the new frontier will be the interplay of genomics, chemistry and controlled biosynthesis.
- We demonstrate that biodiversity is in the microbial realm.
Footnotes
Note: This chapter reflects the opinions of the authors, not necessarily those of the US Government
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Borchardt JK. The beginnings of drug therapy: Ancient mesopotamian medicine. Drug News Perspect. 2002;15:187–192. doi: 10.1358/dnp.2002.15.3.840015. [DOI] [PubMed] [Google Scholar]
- 2.Huang KC. The pharmacology of chinese herbs. 2. CRC Press; Boca Raton, FL: 1999. [Google Scholar]
- 3.Kapoor LD. Crc handbook of ayurvedic medicinal plants. CRC Press; Boca Raton, FL: 1990. [Google Scholar]
- 4.Dev S. Ancient-modern concordance in ayurvedic plants: Some examples. Environ Health Persp. 1999;107:783–789. doi: 10.1289/ehp.99107783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moerman DE. Medicinal plants of native america. University of Michigan Museum of Anthropology; Ann Arbor, MI: 1986. [Google Scholar]
- 6.Johnson T. Crc ethnobotany desk reference. CRC Press; Boca Raton, FL: 1999. [Google Scholar]
- 7.Farnsworth NR, Akerele RO, Bingel AS, Soejarto DD, Guo Z. Medicinal plants in therapy. Bull World Health Org. 1985;63:965–981. [PMC free article] [PubMed] [Google Scholar]
- 8.Fabricant DS, Farnsworth NR. The value of plants used in traditional medicine for drug discovery. Environ Health Perspect. 2001;109(supplement):69–75. doi: 10.1289/ehp.01109s169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buss AD, Waigh RD. Natural products as leads for new pharmaceuticals. In: Wolff ME, editor. Burger’s medicinal chemistry and drug discovery. Principles and practice. Vol. 1. John Wiley & Sons, Inc; New York, NY: 1995. pp. 983–1033. [Google Scholar]
- 10.Wongsrichanalai C, Pickard AL, Wernsdorfer WH, Meshnick SR. Epidemiology of drug-resistant malaria. Lancet Infect Dis. 2002;2:209–218. doi: 10.1016/s1473-3099(02)00239-6. [DOI] [PubMed] [Google Scholar]
- 11.Klayman DL, Lin AJ, Acton N, Scovill JP, Hoch JM, Milhous WK, Theoharides AD. Isolation of artemisinin (qinghaosu) from artemisia annua growing in the United States. J Nat Prod. 1985;47:715–717. doi: 10.1021/np50034a027. [DOI] [PubMed] [Google Scholar]
- 12.Klayman DL. Qinghaosu (artemisinin): An antimalarial drug from China. Science. 1985;228:1049–1055. doi: 10.1126/science.3887571. [DOI] [PubMed] [Google Scholar]
- 13.Miller LH, Su X. Atermisinin: Discovery from the Chinese herbal garden. Cell. 2011;146:855–858. doi: 10.1016/j.cell.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Neill PM, Posner GH. A medicinal chemistry perspective on artemisinin and related endoperoxides. J Med Chem. 2004;47:2945–2964. doi: 10.1021/jm030571c. [DOI] [PubMed] [Google Scholar]
- 15.O’Neill PM, Barton VE, Ward SA. The molecular mechanism of action of artemisinin - the debate continues. Molecules. 2010;15:1705–1721. doi: 10.3390/molecules15031705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang J, Huang L, Li J, Fan Q, Long Y, Li Y, Zhou B. Artemisinin directly targets malarial mitochondria through its specific mitochondrial activation. PLoS One. 2010;5:e9582. doi: 10.1371/journal.pone.0009582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vennerstrom JL, Arbe-Barnes S, Brun R, Charman SA, Chiu FCK, Chollet J, Dong Y, Dorn A, Hunziker D, Matile H, McIntosh K, Padmanilayam M, Santo Tomas J, Scheurer C, Scorneaux B, Tang Y, Urwyler H, Sergio W, Charman WN. Identification of an antimalarial synthetic trioxolane drug development candidate. Nature. 2004;430:900–904. doi: 10.1038/nature02779. [DOI] [PubMed] [Google Scholar]
- 18.Posner GH, Paik IH, Chang W, Borstnik K, Sinishtaq S, Rosenthal AS, Shapiro TA. Malaria-infected mice are cured by a single dose of novel artemisinin derivatives. J Med Chem. 2007;50:2516–2519. doi: 10.1021/jm070149m. [DOI] [PubMed] [Google Scholar]
- 19.Cordell GA, Colvard MD. Natural products and traditional medicine: Turning on a paradigm. J Nat Prod. 2012;75:514–525. doi: 10.1021/np200803m. [DOI] [PubMed] [Google Scholar]
- 20.Hartwell JL. Plants used against cancer. Quarterman; Lawrence, MA: 1982. [Google Scholar]
- 21.Cragg GM, Boyd MR, Cardellina JH, II, Newman DJ, Snader KM, McCloud TG. Ethnobotany and drug discovery: The experience of the us national cancer institute. In: Chadwick DJ, Marsh J, editors. Ethnobotany and the search for new drugs, ciba foundation symposium. Vol. 185. John Wiley & Sons, Inc; New York, NY: 1994. pp. 178–196. [DOI] [PubMed] [Google Scholar]
- 22.Gueritte F, Fahy J. The vinca alkaloids. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer agents from natural products. Taylor and Francis; Boca Raton, FL: 2005. pp. 123–135. [Google Scholar]
- 23.Roussi F, Gueritte F, Fahy J. The vinca alkaloids. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer agents from natural products. 2. Taylor and Francis; Boca Raton, FL: 2012. pp. 177–198. [Google Scholar]
- 24.Lee K-H, Xiao Z. Podophyllotoxin and analogs. In: Cragg GMK, DGI, Newman DJ, editors. Anticancer agents from natural products. Taylor and Francis; Boca Raton, Fl: 2005. pp. 71–87. [Google Scholar]
- 25.Lee K-H, Xiao Z. Podophyllotoxin and analogs. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer agents from natural products. 2. Taylor and Francis; Boca Raton, FL: 2012. pp. 95–122. [Google Scholar]
- 26.Kingston DGI. Taxol and its analogs. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer agents from natural products. Taylor and Francis; Boca Raton, FL: 2005. pp. 89–122. [Google Scholar]
- 27.Kingston DGI. Taxol and its analogs. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer agents from natural products. 2. Taylor and Francis; Boca Raton, Fl: 2012. pp. 123–175. [Google Scholar]
- 28.Schiff PB, Horwitz SB. Taxol stabilizes microtubules in mouse fibroblast cells. Proc Nat Acad Sci USA. 1980;77:1561–1565. doi: 10.1073/pnas.77.3.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Green MR, Manikhas GM, Orlov S, Afanasyev B, Makhson AM, Bhar P, Hawkins MJ. Abraxane, a novel cremophor-free, albumin-bound particle form of paclitaxel for the treatment of advanced non-small-cell lung cancer. Ann Oncol. 2006;17:1263–1268. doi: 10.1093/annonc/mdl104. [DOI] [PubMed] [Google Scholar]
- 30.Burman AC, Mukherjee R, Khattar D, Kumar M, Bala H, Shrivastava RK. Patent EP 1216042. Formulations of paclitaxel entrapped into nanoparticles of polymeric micelles. 2000 Jun 12;
- 31.Kingston DGI, Newman DJ. Taxoids: Cancer-fighting compounds from nature. Curr Opin Drug Disc Devel. 2007;10:130–144. [PubMed] [Google Scholar]
- 32.Rahier NJ, Thomas CJ, Hecht SM. Camptothecin and its analogs. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer agents from natural products. Taylor and Francis; Boca Raton, FL: 2005. pp. 5–21. [Google Scholar]
- 33.Kirschning A, Harmrolfs K, Knobloch T. The chemistry and biology of the maytansinoid antitumor agents. C R Chimie. 2008;11:1523–1543. [Google Scholar]
- 34.Yu J-W, Floss HG, Cragg GM, Newman DJ. Ansamitocins (maytansenoids) In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer agents from natural products. 2. Taylor and Francis; Boca Raton, FL: 2012. pp. 407–427. [Google Scholar]
- 35.Senter PD. Potent antibody drug conjugates for cancer therapy. Curr Opin Chem Biol. 2009;13:235–244. doi: 10.1016/j.cbpa.2009.03.023. [DOI] [PubMed] [Google Scholar]
- 36.Alley SC, Okeley NM, Senter PD. Antibody-drug conjugates: Targeted drug delivery for cancer. Curr Opin Chem Biol. 2010;14:529–537. doi: 10.1016/j.cbpa.2010.06.170. [DOI] [PubMed] [Google Scholar]
- 37.Caravella J, Lugovskoy A. Design of next-generation protein therapeutics. Curr Opin Chem Biol. 2010;14:520–528. doi: 10.1016/j.cbpa.2010.06.175. [DOI] [PubMed] [Google Scholar]
- 38.Lambert JM. Antibody-maytansinoid conjugates: A new strategy for the trreatment of cancer. Drugs Fut. 2010;35:471–480. [Google Scholar]
- 39.Kümler I, Ehlers Mortensen C, Nielsen DL. Trastuzumab emtansine: Tumor-activated prodrug (tap) immunoconjugate oncolytic. Drugs Fut. 2011;36:825–834. [Google Scholar]
- 40.Margulis L, Schwartz KV. Five kingdoms, an illustrated guide to the phyla of life on earth. 2. W. H. Freeman & Co; New York: 1988. [Google Scholar]
- 41.Faulkner DJ. Marine natural products. Nat Prod Rep. 2000;17:7–55. doi: 10.1039/a809395d. [DOI] [PubMed] [Google Scholar]
- 42.Blunt JW, Copp BR, Keyzers RA, Munro MHG, Prinsep MR. Marine natural products. Nat Prod Rep. 2012;29:144–222. doi: 10.1039/c2np00090c. [DOI] [PubMed] [Google Scholar]
- 43.Newman DJ, Cragg GM. Marine natural products and related compounds in clinical and advanced preclinical trials. J Nat Prod. 2004;67:1216–1238. doi: 10.1021/np040031y. [DOI] [PubMed] [Google Scholar]
- 44.Gerwick WH, Moore BS. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem Biol. 2012;19:85–98. doi: 10.1016/j.chembiol.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wallace MS. Ziconotide: A new nonopioid intrathecal analgesic for the treatment of chronic pain. Expert Rev Neurother. 2006;6:1423–1428. doi: 10.1586/14737175.6.10.1423. [DOI] [PubMed] [Google Scholar]
- 46.Bulaj G, Buczek O, Goodsell I, Jiminez EC, Kranski J, Nielsen JS, Garrett JE, Olivera BM. Efficient oxidative folding of conotoxins and the radiation of venomous cone snails. Proc Natl Acad Sci USA. 2003;100:14562–14568. doi: 10.1073/pnas.2335845100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Clark RJ, Akcan M, Kaas Q, Daly NL, Craik DJ. Cyclization of conotoxins to improve their biopharmaceutical properties. Toxicon. 2012;59:446–455. doi: 10.1016/j.toxicon.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 48.Henriquez R, Faircloth G, Cuevas C. Ecteinascidin 743 (et-743); yondelistm, aplidin and kahalahide f. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer agents from natural products. 2005. pp. 215–240. [Google Scholar]
- 49.Cuevas C, Francesch A. Development of yondelis® (trabectedin, et-743). A semisynthetic process solves the supply problem. Nat Prod Rep. 2009;26:322–337. doi: 10.1039/b808331m. [DOI] [PubMed] [Google Scholar]
- 50.Cuevas C, Francesch A, Galmarini CM, Aviles P, Munt S. Ecteinascidin-743 (yondelis(r)), aplidine(r), and irvalec(r) In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer agents from natural products. 2. Taylor and Francis; Boca Raton, FL: 2012. pp. 291–316. [Google Scholar]
- 51.Rath CM, Janto B, Earl J, Ahmed A, Hu FZ, Hiller L, Dahlgren M, Kreft R, Yu F, Wolff JJ, Kweon HK, Christiansen MA, Håkansson K, Williams RM, Ehrlich GD, Sherman DH. Meta-omic characterization of the marine invertebrate microbial consortium that produces the chemotherapeutic natural product et-743. ACS Chem Biol. 2011;6:1244–1256. doi: 10.1021/cb200244t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu MJ, Kishi Y, Littlefield BA. Discovery of e7389, a fully synthetic macrocyclic ketone analog of halichondrin b. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer agents from natural products. Taylor and Francis; Boca Raton, FL: 2005. pp. 241–265. [Google Scholar]
- 53.Newman DJ. The bryostatins. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer agents from natural products. Taylor and Francis; Boca Raton, FL: 2005. pp. 137–150. [Google Scholar]
- 54.Newman DJ. The bryostatins. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer agents from natural products. 2. Taylor and Francis; Boca Raton, Fl: 2012. pp. 199–218. [Google Scholar]
- 55.Yu MJ, Zheng W, Seletsky BM, Littlefield BA, Kishi Y. Case history: Discovery of eribulin (halaventm), a halichondrin b analogue that prolongs overall survival in patients with metastatic breast cancer. In: Macor JA, editor. Ann Rept Med Chem. Vol. 46. Academic Press; Amsterdam: 2011. pp. 227–241. [Google Scholar]
- 56.Cortes J, O’Shaughnessy J, Loesch D, Blum JL, Vahdat LT, Petrakova K, Chollet P, Manikas A, Diéras V, Delozier T, Vladimirov V, Cardoso F, Koh H, Bougnoux P, Dutcus CE, Seegobin S, Mir D, Meneses N, Wanders J, Twelves C. Eribulin monotherapy versus treatment of physician’s choice in patients with metastatic breast cancer (embrace): A phase 3 open-label randomised study. Lancet. 2011;377:914–923. doi: 10.1016/S0140-6736(11)60070-6. [DOI] [PubMed] [Google Scholar]
- 57.Morgan RJ, Jr, Leong L, Chow W, Gandara D, Frankel P, Garcia A, Lenz HJ, Doroshow JH. Phase II trial of bryostatin-1 in combination with cisplatin in patients with recurrent or persistent epithelial ovarian cancer: A California cancer consortium study. Invest New Drugs. 2012;30:723–728. doi: 10.1007/s10637-010-9557-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scriabine A. Discovery and development of major drugs currently in use. In: Landau R, Achilladelis B, Scriabine A, editors. Pharmaceutical innovation: Revolutionizing human health. Chemical Heritage Press; Philadelphia: 1999. pp. 148–270. [Google Scholar]
- 59.Arcamone FM. Anthracyclines. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer agents from natural products. Taylor and Francis; Boca Raton, FL: 2005. pp. 299–320. [Google Scholar]
- 60.Arcamone FM. Anthracyclines. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer agents from natural products. 2. Taylor and Francis; Boca Raton, FL: 2012. pp. 383–405. [Google Scholar]
- 61.Hecht SM. Bleomycin group antitumor agents. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer agents from natural products. Taylor and Francis; Boca Raton, FL: 2005. pp. 357–381. [Google Scholar]
- 62.Hecht SM. Bleomycin group antitumor agents. In: Cragg GM, Kingston DGI, Newman DJ, editors. Antitmor agents from natural products. Taylor and Francis; Boca Raton, Fl: 2012. pp. 451–478. [Google Scholar]
- 63.Remers WA. The mitomycins. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer agents from natural products. Taylor and Francis; Boca Raton, FL: 2005. pp. 475–497. [Google Scholar]
- 64.Hamann PR, Upeslacis J, Borders DB. Enediynes. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer agents from natural products. 2005. pp. 451–474. [Google Scholar]
- 65.Hamann PR, Upeslacis J, Borders DB. Enediynes. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer agents from natural products. 2. Taylor and Francis; Boca Raton, FL: 2012. pp. 575–619. [Google Scholar]
- 66.Prudhomme M. Staurosporines and structurally related indolocarbazoles as antitumor agents. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer agents from natural products. Taylor and Francis; Boca Raton: 2005. pp. 499–517. [Google Scholar]
- 67.Prudhomme M. Staurosporines and structurally related indolocarbazoles as antitumor agents. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer agents from natural products. 2. Taylor and Francis; Boca Raton, FL: 2012. pp. 647–669. [Google Scholar]
- 68.Knolker H-J, Reddy KR. Chemistry and biology of the carbazole alkaloids. In: Cordell GA, editor. The alkaloids, chemistry and biology. Vol. 65. Academic Press; London: 2008. pp. 1–430. [DOI] [PubMed] [Google Scholar]
- 69.Nakano H, Omura S. Chemical biology of natural indolocarbazole products: 30 years since the discovery of staurosporine. J Antibiot. 2009;62:17–26. doi: 10.1038/ja.2008.4. [DOI] [PubMed] [Google Scholar]
- 70.Hofle G, Reichenbach H. Epothilone, a myxobacterial metabolite with promising antitumor activity. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer agents from natural products. Taylor and Francis; Boca Raton, FL: 2005. pp. 413–450. [Google Scholar]
- 71.Hofle G, Reichenbach H. Epothilone, a myxobacterial metabolite with promising antitumor activity. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer agents from natural products. Taylor and Francis; Boca Raton, FL: 2012. pp. 513–573. [Google Scholar]
- 72.Wilson RM, Danishefsky SJ. Small molecule natural products in the discovery of therapeutic agents: The synthesis connection. J Org Chem. 2006;71:8329–8351. doi: 10.1021/jo0610053. [DOI] [PubMed] [Google Scholar]
- 73.Altmann KH, Gertsch J. Anticancer drugs from nature-natural products as a unique source of new microtubule-stabilizing agents. Nat Prod Rep. 2007;24:327–357. doi: 10.1039/b515619j. [DOI] [PubMed] [Google Scholar]
- 74.Altmann KH, Pfeifer B, Arseniyadis S, Pratt BA, Nicolaou KC. The chemistry and biology of epothilones - the wheel keeps turning. ChemMedChem. 2007;2:396–423. doi: 10.1002/cmdc.200600206. [DOI] [PubMed] [Google Scholar]
- 75.Tse ML, Watts RE, Khosla C. Substrate tolerance of module 6 of the epothilone synthase. Biochemistry. 2007;46:3385–3393. doi: 10.1021/bi0616448. [DOI] [PubMed] [Google Scholar]
- 76.Chi KN, Beardsley E, Eigl BJ, Venner P, Hotte SJ, Winquist E, Ko YJ, Sridhar SS, Weber D, Saad F. A phase 2 study of patupilone in patients with metastatic castration-resistant prostate cancer previously treated with docetaxel: Canadian urologic oncology group study p07a. Ann Oncol. 2012;23:53–58. doi: 10.1093/annonc/mdr336. [DOI] [PubMed] [Google Scholar]
- 77.McMeekin S, Patel R, Verschraegen C, Celano P, Burke J, 2nd, Plaxe S, Ghatage P, Giurescu M, Stredder C, Wang Y, Schmelter T. Phase i/ii study of sagopilone (zk-epo) plus carboplatin in women with recurrent platinum-sensitive ovarian cancer. Br J Cancer. 2012;106:707–706. doi: 10.1038/bjc.2011.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Klar U, Buchmann B, Schwede W, Skuballa W, Hoffman J, Lichtner RB. Total synthesis and antitumor activity of zk-epo: The first fully synthetic epothilone in clinical development. Angew Chem Int Ed. 2006:45. doi: 10.1002/anie.200602785. [DOI] [PubMed] [Google Scholar]
- 79.Nix DE, Swezey RR, Hector R, Galgiani JN. Pharmacokinetics of nikkomycin z after single rising oral doses. Antimicrob Agents Chemother. 2009;53:2517–2521. doi: 10.1128/AAC.01609-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Debono M, Gordee RS. Antibiotics that inhibit fungal cell wall development. Annu Rev Microbiol. 1994;48:471–497. doi: 10.1146/annurev.mi.48.100194.002351. [DOI] [PubMed] [Google Scholar]
- 81.Georgopapadakou NH, Tkacz JS. The fungal cell wall as a drug target. Trends Microbiol. 1995;3:98–104. doi: 10.1016/s0966-842x(00)88890-3. [DOI] [PubMed] [Google Scholar]
- 82.Debono M, Turner WW, LaGrandeur L, Burkhardt FJ, Nissen JS, Nichols KK, Rodriguez MJ, Zweifel MJ, Zeckner DJ, Gordee RS, Tang J, Parr TR., Jr Semisynthetic chemical modification of the antifungal lipopeptide echinocandin b (ecb): Structure-activity studies of the lipophilic and geometric parameters of polyarylated acyl analogs of ecb. J Med Chem. 1995;38:3271. doi: 10.1021/jm00017a012. [DOI] [PubMed] [Google Scholar]
- 83.Schwartz RE, Sesin DF, Joshua H, Wilson KE, Kempf AJ, Goklen KA, Kuehner D, Gailliot P, Gleason C, White R, Inamine E, Bills G, Salmon P, Zitano L. Pneumocandins from Zalerion arboricola I. Discovery and isolation. J Antibiot. 1992;45:1853–1866. doi: 10.7164/antibiotics.45.1853. [DOI] [PubMed] [Google Scholar]
- 84.Masurekar PS, Fountoulakis JM, Hallada TC, Sosa MS, Kaplan L. Pneumocandins from Zalerion arboricola II. Modification of product spectrum by mutation and medium manipulation. J Antibiot. 1992;45:1867–1874. doi: 10.7164/antibiotics.45.1867. [DOI] [PubMed] [Google Scholar]
- 85.Schmatz DM, Abruzzo G, Powles MA, McFadden DC, Balkovec JM, Black RM, Nollstadt K, Bartizal K. Pneumocandins from Zalerion arboricola IV. Biological evaluation of natural and semisynthetic pneumocandins for activity against Pneumocystis carinii and Candida species. J Antibiot. 1992;45:1886–1891. doi: 10.7164/antibiotics.45.1886. [DOI] [PubMed] [Google Scholar]
- 86.Bartizal K, Abruzzo G, Trainor C, Krupa D, Nollstadt K, Schmatz D, Schwartz R, Hammond M, Balkovec J, Vanmiddlesworth F. In vitro antifungal activities and in vivo efficacies of 1,3-beta-d-glucan synthesis inhibitors l-671,329, l-646,991, tetrahydroechinocandin b, and l-687,781, a papulacandin. Antimicrob Agents Chemother. 1992;36:1648–1657. doi: 10.1128/aac.36.8.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kurtz MB, Rex JH. Glucan synthase inhibitors as antifungal agents. Adv Protein Chem. 2001;36:423–475. doi: 10.1016/s0065-3233(01)56011-8. [DOI] [PubMed] [Google Scholar]
- 88.Mohr J, Johnson M, Cooper T, Lewis JS, II, Ostrosky-Zeicher L. Current options in antifungal pharmacotherapy. Pharmacotherapy. 2008;28:614–645. doi: 10.1592/phco.28.5.614. [DOI] [PubMed] [Google Scholar]
- 89.Pitman SK, Drew RH, Perfect JR. Addressing current medical needs in invasive fungal infection prevention and treatment with new antifungal agents, strategies and formulations. Exp Opin Emerging Drugs. 2011;16:559–586. doi: 10.1517/14728214.2011.607811. [DOI] [PubMed] [Google Scholar]
- 90.Roemer T, Xu D, Singh SB, Parish CA, Harris G, Wang H, Davies JE, Bills GF. Confronting the challenges of natural product-based antifungal discovery. Chem Biol. 2011;18:148–164. doi: 10.1016/j.chembiol.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 91.Ingham CJ, Schneeburger PM. Microcolony imaging of Aspergillus fumigatus treated with echinocandins reveals both fungistatic and fungicidal activities. PLoS one. 2012;7:e35478. doi: 10.1371/journal.pone.0035478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Daly JW, Spande TF, Garrafo HM. Alkaloids from amphibian skins. A tabulation of over eight hundred compounds. J Nat Prod. 2005;68:1556–1575. doi: 10.1021/np0580560. [DOI] [PubMed] [Google Scholar]
- 93.Eng J, Kleinman WA, Singh L, Singh G, Raufman JP. Isolation and characterization of exendin-4, an exendin-3 analogue, from heloderma suspecturn venom. J Biol Chem. 1992;267:7402–7405. [PubMed] [Google Scholar]
- 94.Gallwitz B. Liraglutide. Drugs Fut. 2008;33:13–20. [Google Scholar]
- 95.Newman DJ, Cragg GM. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod. 2012;75:311–338. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rouhi M. Rediscovering natural products. Chem Eng News. 2003;81(41):77–91. [Google Scholar]
- 97.Borman S. Improving efficiency. Chem Eng News. 2006;84(25):56–78. [Google Scholar]
- 98.Borman S. Organic lab sparks drug discovery. Chem Eng News. 2002;80(40):23–24. [Google Scholar]
- 99.Wilson RM, Danishefsky SJ. Applications of total synthesis to problems in neurodegeneration: Fascinating chemistry along the way. Acc Chem Res. 2006;39:539–549. doi: 10.1021/ar068018n. [DOI] [PubMed] [Google Scholar]
- 100.Meijer L. Le cycle de division cellulaire et sa regulation. Oncologie. 2003;5:311–326. [Google Scholar]
- 101.Cragg GM, Newman DJ. Natural product sources of drugs: Plants, microbes, marine organisms and animals. In: Kennewell PD, Triggle D, Taylor J, editors. Comprehensive medicinal chemistry ii. Vol. 1. Elsevier; Oxford: 2006. pp. 355–403. [Google Scholar]
- 102.Balandrin MF, Kinghorn AD, Farnsworth NR. Plant-derived natural products in drug discovery and development. In: Kinghorn AD, Balandrin MF, editors. Human medicinal agents from plants. Vol. 534. Amer. Chem. Soc; Washington, DC: 1993. pp. 2–12. [Google Scholar]
- 103.Raskin I, Ribnicky DM, Komarnytsky S, Ilic N, Poulev A, Borisjuk N, Brinker A, Moreno DA, Ripoll C, Yakoby N, O’Neal JM, Cornwell T, Pastor I, Fridlander B. Plants and human health in the twenty-first century. Trends Biotech. 2002;20:522–531. doi: 10.1016/s0167-7799(02)02080-2. [DOI] [PubMed] [Google Scholar]
- 104.Newman DJ, Hill RT. New drugs from marine microbes: The tide is turning. J Ind Microbiol Biotechnol. 2006;33:539–544. doi: 10.1007/s10295-006-0115-2. [DOI] [PubMed] [Google Scholar]
- 105.Perry NB, Ettouati L, Litaudon M, Blunt JW, Munro MHG. Alkaloids from the Antarctic sponge Kirkpatrickia varialosa. : Part 1: Variolin B, a new antitumour and antiviral compound. Tetrahedron. 1994;50:3987–3992. [Google Scholar]
- 106.Trimurtulu G, Faulkner DJ, Perry NB, Ettouati L, Litaudon M, Blunt JW, Munro MHG, Jameson GB. Alkaloids from the Antarctic sponge Kirkpatrickia varialosa. Part 2: Variolin a and n(3′)-methyl tetrahydrovariolin B. Tetrahedron. 1994;50:3993–4000. [Google Scholar]
- 107.Ahaidar A, Fernández D, Danelón G, Cuevas C, Manzanares I, Albericio F, Joule JA, Álvarez M. Total syntheses of variolin B and deoxyvariolin B. J Org Chem. 2003;68:10020–10029. doi: 10.1021/jo035332b. [DOI] [PubMed] [Google Scholar]
- 108.Diyabalanage T, Amsler CD, McClintock JB, Baker BJ. Palmerolide a, a cytotoxic macrolide from the Antarctic tunicate Synoicum adareanum. J Am Chem Soc. 2006;128:5630–5631. doi: 10.1021/ja0588508. [DOI] [PubMed] [Google Scholar]
- 109.Nicolaou KC, GR, Sun Y-P, Banerji B, Chen DYK. Synthesis of the originally proposed and revised structures of palmerolide A. Angew Chem Int Ed. 2007;46:5896–5900. doi: 10.1002/anie.200702243. [DOI] [PubMed] [Google Scholar]
- 110.Wilson ZE, Brimble MA. Molecules derived from the extremes of life. Nat Prod Rep. 2009;26:44–71. doi: 10.1039/b800164m. [DOI] [PubMed] [Google Scholar]
- 111.Poulev A, O’Neal JM, Logendra S, Pouleva RB, Timeva V, Garvey AS, Gleba D, Jenkins IS, Halpern BT, Kneer R, Cragg GM, Raskin I. Elicitation a new window into plant chemodiversity and phytochemical drug discovery. J Med Chem. 2003:2542–2547. doi: 10.1021/jm020359t. [DOI] [PubMed] [Google Scholar]
- 112.McCoy E, O’Connor SE. Directed biosynthesis of alkaloid analogs in the medicinal plant Catharanthus roseus. J Am Chem Soc. 2006;128:14276–14277. doi: 10.1021/ja066787w. [DOI] [PubMed] [Google Scholar]
- 113.Maresh JJ, Giddings LA, Friedrich A, Loris EA, Panjikar S, Trout BL, Stöckigt J, Peters B, O’Connor SE. Strictosidine synthase: Mechanism of a Pictet-Spengler catalyzing enzyme. J Am Chem Soc. 2008;130:710–723. doi: 10.1021/ja077190z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.McCoy E, O’Connor SE. Natural products from plant cell cultures. Prog Drug Res. 2008;65:330–370. [PubMed] [Google Scholar]
- 115.Giddings LA, Liscombe DK, Hamilton JP, Childs KL, DellaPenna D, Buell CR, O’Connor SE. A stereoselective hydroxylation step of alkaloid biosynthesis by a unique cytochrome p450 in Catharanthus roseus. J Biol Chem. 2011;286:16751–16757. doi: 10.1074/jbc.M111.225383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Liscombe DK, O’Connor SE. A virus-induced gene silencing approach to understanding alkaloid metabolism in Catharanthus roseus. Phytochem. 2011;72:1969–1977. doi: 10.1016/j.phytochem.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Xu YM, Gao S, Bunting DP, Gunatilaka AAL. Unusual withanolides from aeroponically grown Withania somnifera. Phytochem. 2011;72:518–522. doi: 10.1016/j.phytochem.2010.12.020. [DOI] [PubMed] [Google Scholar]
- 118.Pace NR. A molecular view of microbial diversity and the biosphere. Science. 1997;276:734–740. doi: 10.1126/science.276.5313.734. [DOI] [PubMed] [Google Scholar]
- 119.Madigan MT, Martinko JM, Parker JB. Biology of microorganisms. 8. Prentice-Hall; Upper Saddle River, NJ: 1996. [Google Scholar]
- 120.McAlpine JB, Bachmann BO, Piraee M, Tremblay S, Alarco AM, Zazopoulos E, Farnet CM. Microbial genomics as a guide to drug discovery and structural elucidation: Eco-02301, a novel antifungal agent, as an example. J Nat Prod. 2005;68:493–496. doi: 10.1021/np0401664. [DOI] [PubMed] [Google Scholar]
- 121.Lautru S, Deeth RJ, Bailey L, Challis GM. Discovery of a new peptide natural product by Streptomyces coelicolor genome mining. Nature Chem Biol. 2005;1:265–269. doi: 10.1038/nchembio731. [DOI] [PubMed] [Google Scholar]
- 122.Zengler K, Toledo G, Rappe M, Elkins J, Mathur EJ, Short JM, Keller M. Cultivating the uncultured. Proc Nat Acad Sci USA. 2002;99:15681–15686. doi: 10.1073/pnas.252630999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Udwary DW, Zeigler L, Asolkar RN, Singan V, Lapidus A, Fenical W, Jensen PR, Moore BS. Genome sequencing reveals comples secondary metabolome in the marine actinomycete Salinispora tropica. Proc Natl Acad Sci USA. 2007;104:10376–10381. doi: 10.1073/pnas.0700962104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nett M, Ikeda H, Moore BS. Genomic basis for natural product biosynthetic diversity in the actinomycetes. Nat Prod Rep. 2009;26:1362–1384. doi: 10.1039/b817069j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gontang EA, Fenical W, Jensen PR. Phylogenetic diversity of Gram-positive bacteria cultured from marine sediments. Appl Environ Microbiol. 2007;73:3272–3282. doi: 10.1128/AEM.02811-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sanchez JF, Somoza AD, Keller NP, Wang CCC. Advances in Aspergillus secondary metabolite research in the post-genomic era. Nat Prod Rep. 2012;29:351–371. doi: 10.1039/c2np00084a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rondon MR, August PR, Bettermann AD, Brady SF, Grossman TH, Liles MR, Loiacono KA, Lynch BA, MacNeil IA, Minor C, Tiong CL, Gilman M, Osburne MS, Clardy J, Handelsman JM, Goodman RM. Cloning the soil metagenome: A strategy for accessing the genetic and functional diversity of uncultured microorganisms. App Environ Microbiol. 2000;66:2541–2547. doi: 10.1128/aem.66.6.2541-2547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Venter JC, Remington K, Heidelberg JF, Halpern AL, Rusch D, Eisen JA, Wu D, Paulsen I, Nelson KE, Nelson W, Fouts DE, Levy S, Knap AH, Lomas MW, Nealson K, White O, Peterson J, Hoffman J, Parsons R, Baden-Tillson H, Pfannkoch C, Rogers YH, Smith HO. Environmental genome shotgun sequencing of the sargasso sea. Science. 2004;304:66–74. doi: 10.1126/science.1093857. [DOI] [PubMed] [Google Scholar]
- 129.Brady SF, Simmons L, Kim JH, Schmidt EW. Metagenomic approaches to natural products from free-living and symbiotic organisms. Nat Prod Rep. 2009;26:1488–1503. doi: 10.1039/b817078a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yooseph S, Sutton G, Rusch DB, Halpern AL, Williamson SJ, Remington K, Eisen JA, Heidelberg KB, Manning G, Li W, Jaroszewski L, Cieplak P, Miller CS, Li H, Mashiyama ST, Joachimiak MP, van Belle C, Chandonia JM, Soergel DA, Zhai Y, Natarajan K, Lee S, Raphael BJ, Bafna B, Friedman R, Brenner SE, Godzik A, Eisenberg D, Dixon JE, Taylor SS, Strausberg RL, Frazier M, Craig Venter J. The sorcerer ii global ocean sampling expedition: Expanding the universe of protein families. PLoS Biology. 2007;5:e16. doi: 10.1371/journal.pbio.0050016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Holden C. Life in the air. Science. 2005;307:1558. [Google Scholar]
- 132.Crawford J, Clardy J. Microbial genome mining answers longstanding biosynthetic questions. Proc Nat Acad Sci USA. 2012;109:7589–7590. doi: 10.1073/pnas.1205361109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Piel J. Metabolites from symbiotic bacteria. Nat Prod Rep. 2009;26:338–362. doi: 10.1039/b703499g. [DOI] [PubMed] [Google Scholar]
- 134.Zimmermann K, Engeser M, Blunt JW, Munro MHG, Piel J. Pederin-type pathways of uncultivated bacterial symbionts: Analysis of o-methyltransferases and generation of a biosynthetic hybrid. J Am Chem Soc. 2009;131:2780–2781. doi: 10.1021/ja808889k. [DOI] [PubMed] [Google Scholar]
- 135.Jin M, Liu L, Wright SAI, Beer SV, Clardy J. Structural and functional analysis of pantocin A: An antibiotic from Pantoea agglomerans discovered by heterologous expression of cloned genes. Angew Chem Int Ed. 2003;42:2898–2901. doi: 10.1002/anie.200351053. [DOI] [PubMed] [Google Scholar]
- 136.Abe F, Horikoshi K. The biotechnological potential of piezophiles. Trends Biotechnol. 2001;19:102–108. doi: 10.1016/s0167-7799(00)01539-0. [DOI] [PubMed] [Google Scholar]
- 137.Amato A. Microbes live near undersea CO2 lake. Chem Eng News. 2006;84(38):14. [Google Scholar]
- 138.Persidis A. Extremophiles. Nature Biotechnol. 1998;16:593–594. doi: 10.1038/nbt0698-593. [DOI] [PubMed] [Google Scholar]
- 139.Rossi M, Ciaramella M, Cannio R, Pisani FM, Moracci M, Bartolucci S. Extremophiles 2002. J Bacteriol. 2003;185:3683–3689. doi: 10.1128/JB.185.13.3683-3689.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Short PL. New Zealand plays to its strengths. Chem Eng News. 2007;85(4):20–21. [Google Scholar]
- 141.Cavicchioli R, Siddiqui KS, Andrews D, Sowers KR. Low-temperature extremophiles and their applications. Curr Opin Biotechnol. 2002;13:253–261. doi: 10.1016/s0958-1669(02)00317-8. [DOI] [PubMed] [Google Scholar]
- 142.Schiraldi C, De Rosa M. The production of biocatalysts and biomolecules from extremophiles. Trends Biotechnol. 2002;20:515–521. doi: 10.1016/s0167-7799(02)02073-5. [DOI] [PubMed] [Google Scholar]
- 143.van den Burg B. Extremophiles as a source for novel enzymes. Curr Opin Microbiol. 2003;6:213–218. doi: 10.1016/s1369-5274(03)00060-2. [DOI] [PubMed] [Google Scholar]
- 144.Gomes J, Steiner W. The biocatalytic potential of extremophiles and extremozymes. Food Technol Biotechnol. 2004;42:223–235. [Google Scholar]
- 145.Hoyoux A, Blaise V, Collins T, D’Amico S, Gratia E, Huston AL, Marx JC, Sonan G, Zeng YX, Feller G, Gerday C. Extreme catalysts from low-temperature environments. J Biosci Bioeng. 2004;98:317–330. doi: 10.1016/S1389-1723(04)00290-7. [DOI] [PubMed] [Google Scholar]
- 146.Wiegel J, Kevbrin VV. Alkalithermophiles. Biochem Soc Trans. 2004;32:193–198. doi: 10.1042/bst0320193. [DOI] [PubMed] [Google Scholar]
- 147.Johnson DB, Hallberg KB. The microbiology of acidic mine waters. Res Microbiol. 2003;154:466–473. doi: 10.1016/S0923-2508(03)00114-1. [DOI] [PubMed] [Google Scholar]
- 148.Stierle AA, Stierle DB, Kemp K. Novel sesquiterpenoid matrix metalloproteinase-3 inhibitors from an acid mine waste extremophile. J Nat Prod. 2004;67:1392–1395. doi: 10.1021/np049975d. [DOI] [PubMed] [Google Scholar]
- 149.Stierle DB, Stierle AA, Hobbs D, Stokken J, Clardy J. Berkeleydione and berkeleytrione, new bioactive metabolites from an acid mine organism. Org Lett. 2004;6:1049–1052. doi: 10.1021/ol049852k. [DOI] [PubMed] [Google Scholar]
- 150.Stierle AA, Stierle DB. Bioprospecting in the Berkeley pit: Bioactive metabolites from acid mine waste extremophiles. Studies Nat Prod Chem. 2005;32:1123–1175. [Google Scholar]
- 151.Stierle DB, Stierle AA, Patacini B, McIntyre K, Girtsman T, Bolstad E. Berkeleyones and related meroterpenes from a deep water acid mine waste fungus that inhibit the production of interleukin 1-β from induced inflammasomes. J Nat Prod. 2011;74:2273–2277. doi: 10.1021/np2003066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Tan RX, Zou WX. Endophytes: A rich source of functional metabolites. Nat Prod Rep. 2001;18:448–459. doi: 10.1039/b100918o. [DOI] [PubMed] [Google Scholar]
- 153.Strobel G, Daisy B, Castillo U, Harper J. Natural products from endophytic microorganisms. J Nat Prod. 2004;67:257–268. doi: 10.1021/np030397v. [DOI] [PubMed] [Google Scholar]
- 154.Gunatilaka AAL. Natural products from plant-associated microorganisms: Distribution, structural diversity, bioactivity, and implications of their occurrence. J Nat Prod. 2006;69:509–526. doi: 10.1021/np058128n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Castillo U, Harper JK, Strobel GA, Sears J, Alesi K, Ford E, Lin J, Hunter M, Maranta M, Ge H, Yaver D, Jensen JB, Porter H, Robinson R, Millar D, Hess WM, Condron M, Teplow D. Kakadumycins, novel antibiotics from streptomyces sp. Nrrl 30566, an endophyte of Grevillea pteridifolia. FEMS Microbiol Lett. 2003;224:183–190. doi: 10.1016/S0378-1097(03)00426-9. [DOI] [PubMed] [Google Scholar]
- 156.Li JY, Harper JK, Grant DM, Tombe BO, Bashyal B, Hess WM, Strobel GA. Ambuic acid, a highly functionalized cyclohexenone with antifungal activity from Pestalotiopsis spp. and Monochaetia sp. Phytochem. 2001;56:463–468. doi: 10.1016/s0031-9422(00)00408-8. [DOI] [PubMed] [Google Scholar]
- 157.Zhou G-X, Wijeratne EMK, Bigelow D, Pierson LS, III, VanEtten HD, Gunatilaka AAL. Aspochalasins i, j, and k: Three new cytotoxic cytochalasans of Aspergillus flavipes from the rhizosphere of Ericameria laricifolia of the sonoran desert. J Nat Prod. 2004;67:328–332. doi: 10.1021/np030353m. [DOI] [PubMed] [Google Scholar]
- 158.Stierle A, Strobel G, Stierle D. Taxol and taxane production by Taxomyces andreanae, an endophytic fungus of pacific yew. Science. 1993;260:214–216. doi: 10.1126/science.8097061. [DOI] [PubMed] [Google Scholar]
- 159.Li JY, Sidhu RS, Bollon A, Strobel GA. Stimulation of taxol production in liquid cultures of Pestalotiopsis microspora. Mycolog Res. 1998;102:461–464. [Google Scholar]
- 160.Puri SC, Verma V, Amna T, Qazi GN, Spiteller M. An endophytic fungus from Nothapodytes foetida that produces camptothecin. J Nat Prod. 2005;68:1717–1719. doi: 10.1021/np0502802. [DOI] [PubMed] [Google Scholar]
- 161.Amna T, Puri SC, Verma V, Sharma JP, Khajuria RK, Musarrat J, Spiteller M, Qazi GN. Bioreactor studies on the endophytic fungus Entrophosphora infrequens for the production of an anticancer alkaloid camptothecin. Can J Microbiol. 2006;52:189–196. doi: 10.1139/w05-122. [DOI] [PubMed] [Google Scholar]
- 162.Eyberger AL, Dondapati R, Porter JR. Endophyte fungal isolates from Podophyllum peltatum produce podophyllotoxin. J Nat Prod. 2006;69:1121–1124. doi: 10.1021/np060174f. [DOI] [PubMed] [Google Scholar]
- 163.Puri SC, Nazir A, Chawla R, Arora R, Riyaz-ul-Hasan S, Amna T, Ahmed B, Verma V, Singh S, Sagar R, Sharma A, Kumar R, Sharma RK, Qazi GN. The endophytic fungus Trametes hirsuta as a novel alternative source of podophyllotoxin and related tetralin lignans. J Biotech. 2006;122:494–510. doi: 10.1016/j.jbiotec.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 164.Guo B, Li H, Zhang L. Isolation of a fungus producing vinblastine. J Yunnan Univ. 1998;20:214–215. [Google Scholar]
- 165.Zhang LQ, Guo B, Li H, Zeng S, Shao H, Gu S, Wei R. Preliminary study on the isolation of endophytic fungus of Catharanthus roseus and its fermentation to produce products of therapeutic value. Zhong Cao Yao (Chinese Tradit Herb Drugs) 2000;31:805–807. [Google Scholar]
- 166.Yang X, Zhang L, Guo B, Guo S. Preliminary study of a vincristine-producing endophytic fungus isolated from leaves of Catharanthus roseus. Zhong Cao Yao (Chinese Tradit Herb Drugs) 2004;35:79–81. [Google Scholar]
- 167.Bok JW, Hoffmeister D, Maggio-Hall LA, Murillo R, Glasner JD, Keller NP. Genomic mining for Aspergillus natural products. Chem Biol. 2006;13:31–37. doi: 10.1016/j.chembiol.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 168.Hoffmeister D, Keller NP. Natural products of filamentous fungi: Enzymes, genes, and their regulation. Nat Prod Rep. 2007;24:393–416. doi: 10.1039/b603084j. [DOI] [PubMed] [Google Scholar]
- 169.Mincer TJ, Jensen PR, Kauffman CA, Fenical W. Widespread and persistent populations of a major new marine Actinomycete taxon in ocean sediments. Appl Environ Microbiol. 2002;68:5005–5011. doi: 10.1128/AEM.68.10.5005-5011.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Feling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR, Fenical W. Salinosporamide A: A highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus salinospora. Angew Chem Int Ed. 2003;42:355–357. doi: 10.1002/anie.200390115. [DOI] [PubMed] [Google Scholar]
- 171.Kwon HC, Kauffman CA, Jensen PR, Fenical W. Marinomycins A-D, antitumor-antibiotics of a new structure class from a marine actinomycete of the recently discovered genus “Marinispora”. J Am Chem Soc. 2006;128:1622–1632. doi: 10.1021/ja0558948. [DOI] [PubMed] [Google Scholar]
- 172.Williams PG, Asolkar RN, Kondratyuk T, Pezzuto JM, Jensen PR, Fenical W. Saliniketals A and B, bicyclic polyketides from the marine actinomycete Salinispora arenicola. J Nat Prod. 2007;70:83–88. doi: 10.1021/np0604580. [DOI] [PubMed] [Google Scholar]
- 173.Oh DC, Strangman WK, Kauffman CA, Jensen PR, Fenical W. Thalassospiramides A and B, immunosuppressive peptides from teh marine bacterium Thalassopira sp. Org Lett. 2007;9:1525–1528. doi: 10.1021/ol070294u. [DOI] [PubMed] [Google Scholar]
- 174.Hughes CC, Fenical W. Antibacterials from the sea. Chem Eur J. 2010;16:12512–12525. doi: 10.1002/chem.201001279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Waters AL, Hill RT, Place AR, Hamann MT. The expanding role of marine microbes in pharmaceutical development. Curr Opin Biotech. 2010;21:780–786. doi: 10.1016/j.copbio.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Pettit RK. Culturability and secondary metabolite diversity of extreme microbes: Expanding contribution of deep sea and deep-sea vent microbes to natural product discovery. Mar Biotech. 2011;13:1–11. doi: 10.1007/s10126-010-9294-y. [DOI] [PubMed] [Google Scholar]
- 177.Piel J. Metabolites from symbiotic bacteria. Nat Prod Rep. 2004;21:519–538. doi: 10.1039/b310175b. [DOI] [PubMed] [Google Scholar]
- 178.Piel J, Butzke D, Fusetani N, Hui D, Platzer M, Wen G, Matsunaga S. Exploring the chemistry of uncultivated bacterial symbionts: Antitumor polyketides of the pederin family. J Nat Prod. 2005;68:472–479. doi: 10.1021/np049612d. [DOI] [PubMed] [Google Scholar]
- 179.Piel J, Hofer I, Hui D. Evidence for a symbiosis island involved in horizontal acquisition of pederin biosynthetic capabilities by the bacterial symbiont of Paederus fuscipes beetles. J Bacteriol. 2004;186:1280–1286. doi: 10.1128/JB.186.5.1280-1286.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Piel J, Hui D, Wen G, Butzke D, Platzer M, Fusetani N, Matsunaga S. Antitumor polyketide biosynthesis by an uncultivated bacterial symbiont of the marine sponge Theonella swinhoei. Proc Nat Acad Sci USA. 2004;101:16222–16227. doi: 10.1073/pnas.0405976101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Radjasa OK, Vaske YM, Navarro G, Vervoort HC, Tenney K, Linington RG, Crews P. Highlights of marine invertebrate-derived biosynthetic products: Their biomedical potential and possible production by microbial associants. Bioorg Med Chem. 2011;19:6658–6674. doi: 10.1016/j.bmc.2011.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Partida-Martinez LP, Hertweck C. Pathogenic fungus harbours endosymbiotic bacteria for toxin production. Nature. 2005;437:884–888. doi: 10.1038/nature03997. [DOI] [PubMed] [Google Scholar]
- 183.Partida-Martinez LP, De Looss CF, Ishida K, Ishida M, Roth M, Buder K, Hertweck C. Rhizonin, the first mycotoxin isolated from the zygomycota, is not a fungal metabolite but is produced by bacterial endosymbionts. App Environ Microbiol. 2007;73:793–797. doi: 10.1128/AEM.01784-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Partida-Martinez LP, Groth I, Schmitt I, Richter W, Roth M, Hertweck C. Burkholderia rhizoxinica sp. nov. and Burkholderia endofungorum sp. nov., bacterial endosymbionts of the plant-pathogenic fungus Rhizopus microsporous. Int J Sys Evol Microbiol. 2007;57:2583–2590. doi: 10.1099/ijs.0.64660-0. [DOI] [PubMed] [Google Scholar]
- 185.Lackner G, Möbius N, Scherlach K, Partida-Martinez LP, Winkler R, Schmitt I, Hertweck C. Global distribution and evolution of a toxinogenic Burkholderia-Rhizopus symbiosis. App Environ Microbiol. 2009;75:2982–2986. doi: 10.1128/AEM.01765-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Khosla C. Natural product biosynthesis: A new interface between enzymology and medicine. J Org Chem. 2000;65:8127–8133. doi: 10.1021/jo000849y. [DOI] [PubMed] [Google Scholar]
- 187.Staunton J, Weissman KJ. Polyketide biosynthesis: A millennium review. Nat Prod Rep. 2001;18:380–416. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]
- 188.Walsh CT. Polyketide and nonribosomal peptide antibiotics: Modularity and versatility. Science. 2004;303:1805–1810. doi: 10.1126/science.1094318. [DOI] [PubMed] [Google Scholar]
- 189.Walsh CT. The chemical versatility of natural-product assembly lines. Acc Chem Res. 2007;41:4–10. doi: 10.1021/ar7000414. [DOI] [PubMed] [Google Scholar]
- 190.Clardy J, Walsh CT. Lessons from natural molecules. Nature. 2004;432:829–837. doi: 10.1038/nature03194. [DOI] [PubMed] [Google Scholar]
- 191.Van Lanen SG, Shen B. Combinatorial biosynthesis of anticancer natural products. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer agents from natural products. 2. Taylor and Francis; Boca Raton, FL: 2012. pp. 671–698. [Google Scholar]
- 192.Julien B, Shah S, Ziemann R, Goldman R, Katz L, Khosla C. Isolation and characterization of the epothilone biosynthetic gene cluster from Sorangium cellulosum. Gene. 2000;249:153–160. doi: 10.1016/s0378-1119(00)00149-9. [DOI] [PubMed] [Google Scholar]
- 193.Molnar I, Schupp T, Ono M, Zirkle RE, Milnamow M, Nowak-Thompson B, Engel N, Toupet C, Stratmann A, Cyr DD, Gorlach J, Mayo JM, Hu A, Goff S, Schmid J, Ligon JM. The biosynthetic gene cluster for the microtubule-stabilizing agents epothilones A and B from Sorangium cellulosum so ce90. Chem Biol. 2000;7:97–109. doi: 10.1016/s1074-5521(00)00075-2. [DOI] [PubMed] [Google Scholar]
- 194.Lau J, Frykman S, Regentin R, Ou S, Tsuruta H, Licari P. Optimizing the heterologous production of epothilone D in Myxococcus xanthus. Biotechnol Bioeng. 2002;78:280–288. doi: 10.1002/bit.10202. [DOI] [PubMed] [Google Scholar]
- 195.Nicolaou KC, Vourloumis D, Winssinger N, Baran PS. The art and science of total synthesis at the dawn of the twenty-first century. Angew Chem Int Ed. 2000;39:44–122. [PubMed] [Google Scholar]
- 196.Li J, Burgett AWG, Esser L, Amezcua C, Harran PG. Total synthesis of nominal diazonamides - part 2: On the true structure and origin of natural isolates. Angew Chem Int Ed. 2001;40:4770–4773. doi: 10.1002/1521-3773(20011217)40:24<4770::aid-anie4770>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 197.Freemantle M. Scaled-up synthesis of discodermolide. Chem & Eng News. 2004;82(9):33–35. [Google Scholar]
- 198.Mickel SJ, Niederer D, Daeffler R, Osmani A, Kuesters E, Schmid E, Schaer K, Gamboni R, Chen WC, Loeser E, Kinder FR, Jr, Konigsberger K, Prasad K, Ramsey TM, Repic O, Wang RM, Florence G, Lyothier I, Paterson I. Large-scale synthesis of the anti-cancer marine natural product (+)-discodermolide. Part 5: Linkage of fragments c1–6 and c7–24 and finale. Org Process Res Dev. 2004;8:122–130. [Google Scholar]
- 199.Copeland A, Younes A. Brentuximab vedotin. Drugs Fut. 2010;35:797–801. [Google Scholar]
- 200.Singh AJ, Razzak M, Teesdale-Spittle P, Gaitanos TN, Wilmes A, Paterson I, Goodman JM, Miller JH, Northcote PT. Structure-activity studies of the pelorusides: New congeners and semi-synthetic analogues. Org Biomol Chem. 2011;9:4456–4466. doi: 10.1039/c0ob01127d. [DOI] [PubMed] [Google Scholar]
- 201.Paterson I, Naylor GJ, Gardner NM, Guzmán E, Wright AE. Total synthesis and biological evaluation of a series of macrocyclic hybrids and analogues of the antimitotic natural products dictyostatin, discodermolide, and taxol. Chemistry - An Asian Journal. 2011;6:459–473. doi: 10.1002/asia.201000541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Dalby SM, Paterson I. Synthesis of polyketide natural products and analogs as promising anticancer agents. Curr Opin Drug Discov Develop. 2010;13:777–794. [PubMed] [Google Scholar]
- 203.Smith AB, III, Sfouggatakis C, Risatti CA, Sperry JB, Zhu W, Doughty VA, Tomioka T, Gotchev DB, Bennett CS, Sakamoto S, Atasoylu O, Shirakami S, Bauer D, Takeuchi M, Koyanagi J, Sakamoto Y. Spongipyran synthetic studies. Evolution of a scalable total synthesis of (+)-spongistatin 1. Tetrahedron. 2009;65:6489–6509. doi: 10.1016/j.tet.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Seiple IB, Su S, Young IS, Lewis CA, Yamaguchi J, Baran PS. Total synthesis of palau’amine. Angew Chem Int Ed. 2010;49:1095–1098. doi: 10.1002/anie.200907112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Nicolaou KC, Kim S, Pfefferkorn JA, Xu J, Oshima T, Hosokawa S, Li T. Synthesis and biological activity of sarcodictyins. Angew Chem Int Ed. 1998;37:1418–1421. doi: 10.1002/(SICI)1521-3773(19980605)37:10<1418::AID-ANIE1418>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 206.Evans BE, Rittle KE, Bock MG, DiPardo RM, Fredinger RM, Whitter WL, Lundell GF, Veber DF, Anderson PS, Chang RSL, Lotti VJ, Cerino DJ, Chen TB, Kling P, Kunkel KA, Springer JP, Hirshfield J. Methods for drug discovery: Development of potent, selective, orally effective cholecystokinin antagonists. J Med Chem. 1988:2235–2246. doi: 10.1021/jm00120a002. [DOI] [PubMed] [Google Scholar]
- 207.Koch MA, Waldmann H. Protein structure similarity clustering and natural product structure as guiding principles in drug discovery. Drug Discov Today. 2005;10:471–483. doi: 10.1016/S1359-6446(05)03419-7. [DOI] [PubMed] [Google Scholar]
- 208.Nicolaou KC, Pfefferkorn JA, Roecker AJ, Cao GQ, Barluenga S, Mitchell HJ. Natural product-like combinatorial libraries based on privileged structures. 1. General principles and solid-phase synthesis of benzopyrans. J Am Chem Soc. 2000;122:9939–9953. [Google Scholar]
- 209.Nicolaou KC, Pfefferkorn JA, Barluenga S, Mitchell HJ, Roecker AJ, Cao GQ. Natural product-like combinatorial libraries based on privileged structures. 3. The “libraries from libraries” principle for diversity enhancement of benzopyran libraries. J Am Chem Soc. 2000;122:9968–9976. [Google Scholar]
- 210.Nicolaou KC, Pfefferkorn JA, Mitchell HJ, Roecker AJ, Barluenga S, Cao GQ, Affleck RL, Lillig JE. Natural product-like combinatorial libraries based on privileged structures. 2. Construction of a 10 000-membered benzopyran library by directed split-and-pool chemistry using nanokans and optical encoding. J Am Chem Soc. 2000;122:9954–9967. [Google Scholar]
- 211.Borman S. The many faces of combinatorial chemistry. Chem Eng News. 2003;81(43):45–56. [Google Scholar]
- 212.Borman S. Rescuing combichem. Diversity-oriented synthesis (dos) aims to pick up where traditional combinatorial chemistry left off. Chem Eng News. 2004;82(40):32–40. [Google Scholar]
- 213.Cragg GM, Grothaus PG, Newman DJ. Impact of natural products on developing new anti-cancer agents. Chem Rev. 2009;109:3012–3043. doi: 10.1021/cr900019j. [DOI] [PubMed] [Google Scholar]
- 214.Wetzel S, Bon RS, Kumar K, Waldmann H. Biology-oriented synthesis. Angew Chem Int Ed. 2011;50:10800–10826. doi: 10.1002/anie.201007004. [DOI] [PubMed] [Google Scholar]
- 215.Eberhardt L, Kumar K, Waldmann H. Exploring and exploiting biologically relevant chemical space. Curr Drug Targ. 2011;12:1531–1546. doi: 10.2174/138945011798109482. [DOI] [PubMed] [Google Scholar]
- 216.Burke MD, Berger EM, Schreiber SL. Generating diverse skeletons of small molecules combinatorially. Science. 2003;302:613–618. doi: 10.1126/science.1089946. [DOI] [PubMed] [Google Scholar]
- 217.Burke MD, Schreiber SL. A planning strategy for diversity-oriented synthesis. Angew Chem Int Ed. 2004;43:46–58. doi: 10.1002/anie.200300626. [DOI] [PubMed] [Google Scholar]
- 218.Breinbauer R, Manger M, Scheck M, Waldmann H. Natural product guided compound library development. Curr Med Chem. 2002;9:2129–2145. doi: 10.2174/0929867023368773. [DOI] [PubMed] [Google Scholar]
- 219.Breinbauer R, Vetter IR, Waldmann H. From protein domains to drug candidates natural products as guiding principles in the design and synthesis of compound libraries. Angew Chem Int Ed. 2002;41:2878–2890. doi: 10.1002/1521-3773(20020816)41:16<2878::AID-ANIE2878>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 220.Brohm D, Metzger S, Bhargava A, Muller O, Lieb F, Waldmann H. Natural products are biologically validated starting points in structural space for compound library development: Solid-phase synthesis of dysidiolide-derived phosphatase inhibitors. Angew Chem Int Ed. 2002;41:307–311. doi: 10.1002/1521-3773(20020118)41:2<307::aid-anie307>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 221.Ganesan A. Natural products as a hunting ground for combinatorial chemistry. Curr Opin Biotech. 2004;15:584–590. doi: 10.1016/j.copbio.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 222.Stahl P, Kissau L, Mazitschek R, Giannis A, Waldmann H. Natural product derived receptor tyrosine kinase inhibitors: Identification of igf1r, tie-2, and vegfr-3 inhibitors. Angew Chem Int Ed. 2002;41:1174–1178. doi: 10.1002/1521-3773(20020402)41:7<1174::aid-anie1174>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 223.Thutewohl M, Kissau L, Popkirova B, Karaguni IM, Nowak T, Bate M, Kuhlmann J, Muller O, Waldmann H. Solid-phase synthesis and biological evaluation of a pepticinnamin E library. Angew Chem Int Ed. 2002;41:3616–3620. doi: 10.1002/1521-3773(20021004)41:19<3616::AID-ANIE3616>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 224.Ganesan A. The impact of natural products upon modern drug discovery. Curr Opin Chem Biol. 2008;12:306–317. doi: 10.1016/j.cbpa.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 225.Kopp F, Stratton CF, Akella LB, Tan DS. A diversity-oriented synthesis approach to macrocycles via oxidative ring expansion. Nature Chem Biol. 2012;8:358–365. doi: 10.1038/nchembio.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Camp D, Davis RA, Campitelli M, Ebdon J, Quinn RJ. Drug-like properties: Guiding principles for the design of natural product libraries. J Nat Prod. 2012;75:72–81. doi: 10.1021/np200687v. [DOI] [PubMed] [Google Scholar]
- 227.Bode HB, Muller R. The impact of bacterial genomics on natural product research. Angew Chem Int Ed. 2005;44:6828–6846. doi: 10.1002/anie.200501080. [DOI] [PubMed] [Google Scholar]
- 228.Yadav G, Gokhale RS, Mohanty D. Towards prediction of metabolic products of polyketide synthases: An in silico analysis. PLoS Comp Biol. 2009;5:e1000351. doi: 10.1371/journal.pcbi.1000351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Lane AL, Moore BS. A sea of biosynthesis: Marine natural products meet the molecular age. Nat Prod Rep. 2011;28:411–428. doi: 10.1039/c0np90032j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Akey DL, Gehret JJ, Khare D, Smith JL. Insights from the sea: Strucutral biology of marine polyketide synthases. Nat Prod Rep. 2012 doi: 10.1039/c2np20016c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Banskota AH, Mcalpine JB, Sørensen D, Ibrahim A, Aouidate M, Piraee M, Alarco AM, FCM, Zazopoulos E. Genomic analyses lead to novel secondary metabolites. Part 3. Eco-0501, a novel antibacterial of a new class. J Antibiot. 2006;59:533–542. doi: 10.1038/ja.2006.74. [DOI] [PubMed] [Google Scholar]
- 232.Clardy J, Fischbach MA, Walsh CT. New antibiotics from bacterial natural products. Nature Biotech. 2006;24:1541–1550. doi: 10.1038/nbt1266. [DOI] [PubMed] [Google Scholar]
- 233.Lam KS. New aspects of natural products in drug discovery. TRENDS Microbiol. 2007;15:279–289. doi: 10.1016/j.tim.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 234.Rachid S, Gerth K, Kochems I, Muller R. Deciphering regulatory mechanisms for secondary metabolite production in the myxobacterium Sorangium cellulosum so ce56. Mol Microbiol. 2007;63:1783–1796. doi: 10.1111/j.1365-2958.2007.05627.x. [DOI] [PubMed] [Google Scholar]
- 235.Herrmann J, Elnakady YA, Wiedmann RM, Ullrich A, Rohde M, Kazmaier U, Vollmar AM, Müller R. Pretubulysin: From hypothetical biosynthetic intermediate to potential lead in tumor therapy. PLoS ONE. 2012;7:e37416. doi: 10.1371/journal.pone.0037416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Wang J, Soisson SM, Young K, Shoop W, Kodali S, Galgoci A, Painter R, Parthasarathy G, Tang YS, Cummings R, Ha S, Dorso K, Motyl M, Jayasuriya H, Ondeyka J, Herath K, Zhang C, Hernandez L, Allocco J, Basilio A, Tormo JR, Genilloud O, Vicente F, Pelaez F, Colwell L, Lee SH, Michael B, Felcetto T, Gill C, Silver LL, Hermes JD, Bartizal K, Barrett J, Schmatz D, Becker JW, Cully D, Singh SB. Platensimycin is a selective fabf inhibitor with potent antibiotic properties. Nature. 2006;441:358–361. doi: 10.1038/nature04784. [DOI] [PubMed] [Google Scholar]
- 237.Jayasuriya H, Herath KB, Zhang C, Zink DL, Basilio A, Genilloud O, Diez MT, Vicente F, Gonzalez I, Salazar O, Pelaez F, Cummings R, Ha S, Wang J, Singh SB. Isolation and structure of platencin: A fabh and fabf dual inhibitor with potent broad-spectrum antibiotic activity. Angew Chem Int Ed. 2007:4684–4688. doi: 10.1002/anie.200701058. [DOI] [PubMed] [Google Scholar]
- 238.Young K, Jayasuriya H, Ondeyka JG, Herath K, Zhang C, Kodali S, Galgoci A, Painter R, Brown-Driver V, Yamamoto R, Silver LL, Zheng Y, Ventura JI, Sigmund J, Ha S, Basilio A, Vincente F, Tormo JR, Pelaez F, Youngman P, Cully D, Barett JF, Schmatz D, Singh SB, Wang J. Discovery of fabh/fabf inhibitors from natural products. Antimicrob Ag Chemotherap. 2006;50:519–526. doi: 10.1128/AAC.50.2.519-526.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Singh SB, Zink DL, Huber J, Genilloud O, Salazar O, Diez MT, Basilio A, Vincente F, Byrne KM. Discovery of lucensimycins A and B from Streptomyces lucensis ma7349 using an antisense strategy. Org Lett. 2006;8:5449–5452. doi: 10.1021/ol062041r. [DOI] [PubMed] [Google Scholar]
- 240.Nicolaou KC, Li D, Edmonds DJ. Total synthesis of platensimycin. Angew Chem Int Ed. 2006;45:7086–7090. doi: 10.1002/anie.200603892. [DOI] [PubMed] [Google Scholar]
- 241.Nicolaou KC, Edmonds DJ, Li A, Tria GS. Asymmetric total syntheses of platensimycin. Angew Chem Int Ed. 2007;46:3942–3945. doi: 10.1002/anie.200700586. [DOI] [PubMed] [Google Scholar]
- 242.Nicolaou KC, Lister T, Denton RM, Montero A, Edmonds DJ. Adamanta-platensimycin: A bioactive analogue of platensimycin. Angew Chem Int Ed. 2007;46:4712–4714. doi: 10.1002/anie.200701548. [DOI] [PubMed] [Google Scholar]
- 243.Zhang C, Ondeyka J, Herath K, Jayasuriya H, Guan Z, Zink DL, Dietrich L, Burgess B, Ha SN, Wang J, Singh SB. Platensimycin and platencin congeners from Streptomyces platensis. J Nat Prod. 2011;74:329–340. doi: 10.1021/np100635f. [DOI] [PubMed] [Google Scholar]
- 244.Cragg GM, Katz F, Newman DJ, Rosenthal J. Legal and ethical issues involving marine biodiscovery and development. In: Fattorusso E, Gerwick WH, Taglialatela-Scafati O, editors. Handbook of marine natural products. Vol. 2. Springer Science; Dordrecht: 2012. pp. 1316–1342. [Google Scholar]
- 245.Cragg GM, Katz F, Newman DJ, Rosenthal J. The impact of the united nations convention on biological diversity on natural products research. Nat Prod Rep. 2012;29:1407–1423. doi: 10.1039/c2np20091k. [DOI] [PubMed] [Google Scholar]
- 246.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nature Rev Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 247.Macarron R, Banks MN, Bojanic D, Burns DJ, Cirovic DA, Garyantes T, Green DVS, Hertzberg RP, Janzen WP, Paslay JW, Schopfer U, Sittampalam GS. Impact of high-throughput screening in biomedical research. Nature Rev Drug Discov. 2011;10:188–195. doi: 10.1038/nrd3368. [DOI] [PubMed] [Google Scholar]
- 248.Lang P, Yeow K, Nichols A, Scheer A. Cellular imaging in drug discovery. Nature Rev Drug Discov. 2006;5:343–356. doi: 10.1038/nrd2008. [DOI] [PubMed] [Google Scholar]
- 249.Suffness M, Newman DJ, Snader KM. Discovery and development of antineoplastic agents from natural sources. In: Scheuer PJ, editor. Bioorganic marine chemistry. Vol. 3. Springer-Verlag; Berlin: 1989. pp. 132–168. [Google Scholar]
- 250.Sin N, Kim KB, Elofsson M, Meng L, Auth H, Kwok BHB, Crews CM. Total synthesis of the potent proteasome inhibitor epoximicin: A useful tool for understanding proteosome biology. Bioorg Med Chem Lett. 1999;9:2283–2288. doi: 10.1016/s0960-894x(99)00376-5. [DOI] [PubMed] [Google Scholar]
- 251.Davies S, Pandian R, Bolos J, Castaner R. Carfilzomib. Drugs Fut. 2009;34:708–716. [Google Scholar]
- 252.Rateb ME, Ebel R. Secondary metabolites of fungi from marine habitats. Nat Prod Rep. 2011;28:290–344. doi: 10.1039/c0np00061b. [DOI] [PubMed] [Google Scholar]
- 253.Imhoff JF, Labes A, Wiese J. Bio-mining the microbial treasures of the ocean: New natural products. Biotech Adv. 2011;29:468–482. doi: 10.1016/j.biotechadv.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 254.Berdy J. Thoughts and facts about antibiotics: Where we are now and where we are heading. J Antibiot. 2012;65:385–395. doi: 10.1038/ja.2012.27. [DOI] [PubMed] [Google Scholar]