The ubiquitin proteasome system and myocardial ischemia (original) (raw)
Abstract
The ubiquitin proteasome system (UPS) has been the subject of intensive research over the past 20 years to define its role in normal physiology and in pathophysiology. Many of these studies have focused in on the cardiovascular system and have determined that the UPS becomes dysfunctional in several pathologies such as familial and idiopathic cardiomyopathies, atherosclerosis, and myocardial ischemia. This review presents a synopsis of the literature as it relates to the role of the UPS in myocardial ischemia. Studies have shown that the UPS is dysfunctional during myocardial ischemia, and recent studies have shed some light on possible mechanisms. Other studies have defined a role for the UPS in ischemic preconditioning which is best associated with myocardial ischemia and is thus presented here. Very recent studies have started to define roles for specific proteasome subunits and components of the ubiquitination machinery in various aspects of myocardial ischemia. Lastly, despite the evidence linking myocardial ischemia and proteasome dysfunction, there are continuing suggestions that proteasome inhibitors may be useful to mitigate ischemic injury. This review presents the rationale behind this and discusses both supportive and nonsupportive studies and presents possible future directions that may help in clarifying this controversy.
Keywords: ubiquitin proteasome system, myocardial ischemia, ischemic preconditioning, ubiquitin
this review article is part of a collection on Role of the Ubiquitin Proteosome System in Cardiac Disease. Other articles appearing in this collection, as well as a full archive of all Review collections, can be found online at http://ajpheart.physiology.org/.
Introduction
Since its discovery in 1979 (65), the ubiquitin proteasome system (UPS) has been the subject of intensive research and witnessed some exciting developments. Researchers have been investigating the UPS and defining its importance in normal physiology and in pathological conditions. Studies have described roles for the UPS in cell-cycle control (19, 64), cell signaling (22, 106), apoptosis (76, 154), immune response and antigen presentation (34, 85, 101), transcription regulation (59), protein quality control (157, 163, 164), and protein turnover under normal and pathological conditions (21, 61, 117, 145, 147). In regard to cardiovascular disease, the majority of studies suggest that at some point the UPS becomes dysfunctional in several pathologies including familial and idiopathic cardiomyopathies (17, 116, 139, 156), atherosclerosis (63, 151), and myocardial ischemia (11, 114). This review will focus in on the role of the UPS in myocardial ischemia. As a necessity, short descriptions of the structure, organization, and regulation of the UPS with a focus on the cardiac proteasome will be presented. These will be followed by a synopsis of the current knowledge of how the UPS is affected by myocardial ischemia and its potential roles in ischemic preconditioning (IPC). As part of each topic, any controversies or new hypotheses will be presented with a discussion of potential new directions.
Structure and Organization of the UPS
The four components of the UPS include the proteasome (in its various forms), ubiquitin, the ubiquitination machinery, and the deubiquitinases (DUBs).
The 20S proteasome.
The 20S proteasome is the central proteolytic structure of the UPS and is a barrel-shaped structure consisting of two pairs of rings each containing seven subunits (Fig. 1). The constitutive proteolytic activity resides in the inner two rings that contain the β-type subunits, designated β1 through β7. Three subunits are responsible for this activity: the β1 subunit which has “caspase-like” activity, the β2 subunit which has trypsin-like activity, and the β5 subunit which has chymotrypsin-like activity. These three subunits can be replaced by isoforms which are designated β1_i_, β2_i_, and β5_i_ (38). Earlier studies indicated that the isoforms were upregulated and incorporated into the 20S proteasome in response to exposure to γ-interferon primarily in immune cells. Replacement with these isoforms confers two additional proteolytic activities to the 20S proteasome: BrAAP, which is hydrolysis of peptide bonds after branched chain amino acids, and SNAAP, which is hydrolysis after small neutral amino acids. These changes favor the formation of peptides consistent with the major histocompatibility complex class I antigens; thus, proteasomes containing these isoforms are known as “immunoproteasomes” (102). However, this might be a misnomer because in depth proteomic analysis of the cardiac proteasome has also identified these isoforms in the constitutive proteasome in nonimmune cells. Once considered to be homogenous, in fact the β-subunit containing rings are quite heterogeneous and exist as a dynamic mixture of both constitutive and immunoform catalytic β-type subunits (33, 47). Sitting atop and aligned counter to the inner β-rings are the two outer rings containing the α-type subunits, designated α1 through α7 (Fig. 1). In the eukaryotic proteasome these subunits have no direct proteolytic activity, but subunits α1, α2, α3, α6, and α7 project into the openings at either end of the proteasome, sealing it to prevent access to the central chamber. When activated, the NH2-termini of these subunits retract and allow access of substrate into the proteolytic chamber (52). Molecular modeling studies have revealed that the 20S proteasome is arranged such that three chambers are present: two outer antechambers and one inner chamber containing the exposed active sites of the six catalytic subunits.
Fig. 1.
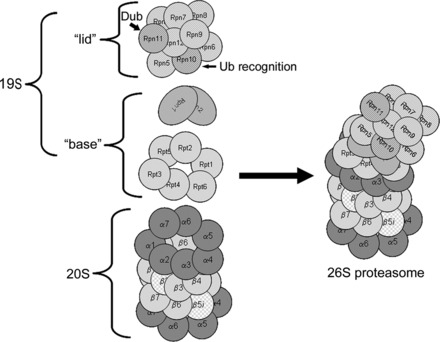
Proteasome structure. The 20S proteasome is a barrel-shaped structure composed of 4 rings, each consisting of 7 subunits. This 20S proteasome has immunoforms of the β2 (β2_i_) subunit in the upper β-ring and the β5 (β5_i_) subunit in the lower β-ring, illustrating the heterogeneity of this structure. Docked at one or both ends is the 19S regulatory particle. For the sake of clarity, the illustrated structure has only one 19S regulatory particle docked, known as the mushroom configuration and is actually 26S. Docking of two 19S regulatory particles at either end is called the dumbbell configuration and is actually 30S but is also commonly called the 26S proteasome. Rpn, regulatory particle non-ATPase subunit; Rpt, regulatory peptide triple A subunit; Ub, ubiquitin. Modified from Powell and Divald (110) with permission from Oxford University Press.
Different proteasome isoforms (zomes) and associated regulatory particles.
Proteasomes can be defined by the regulatory particles that sit at either end of the barrel. The process of mating of a regulatory particle at one or both ends of the 20S proteasome is called “docking.” There are at least three proteasome regulators described in eukaryotes that can be loosely classified according to whether or not they use ATP. Those that do not use ATP include the 11S activator ring and the PA200 activator. The ATP-utilizing activator is the 19S regulatory particle (135). The PA200 activator appears to be associated with DNA repair and genomic stability (8, 93, 149). Although subunits of this regulatory particle have been detected in heart (47), little else is known about its function. The following is a discussion of the 19S regulatory particle and associated ubiquitination machinery and the 11S activator ring.
The 19S regulatory particle and associated ubiquitination machinery.
The 19S regulatory particle is a large macromolecular structure containing an additional 18 subunits arranged in two distinct subcomplexes, the “base” and the “lid” (Fig. 1). The base contains six subunits, regulatory peptide triple A 1–6 (Rpt1 through Rpt6), that have ATPase activity and are arranged in a ring. Also included are the two largest of the regulatory particle non-ATPase subunits, Rpn1 and Rpn2. Some investigators include Rpn10, the ubiquitin recognition domain, as part of the base, and others do not. The base ATPase subunits use ATP to unfold the substrate and induce a conformational change in the α-subunits to open up the entrance channel to the catalytic chamber and thus activate the 20S core (121, 135). The Rpt2, Rpt5, and Rpn2 subunits play important roles in the attachment of the “base” to 20S proteasome α-rings and binding of the 19S particle “lid” to the “base” (26, 43, 49). The “lid” contains the remaining non-ATPase subunits, Rpn3 through Rpn12. As indicated, Rpn10 contains the main ubiquitin binding domain. Rpn11 is one of the intrinsic deubiquitinating enzymes. The functions of the remaining subunits are somewhat obscure. If just one 19S regulatory particle is docked at one end (mushroom configuration), the entire complex is known as the 26S proteasome. If two particles are docked, one at each end (dumbbell configuration), the complex is known as the 30S proteasome. The 19S regulatory particle confers selectivity for ubiquitin-conjugated proteins. The 26S or 30S proteasomes mediate the process known as ubiquitin-mediated degradation of proteins.
Ubiquitin-mediated degradation of proteins and associated ubiquitination machinery.
Over the past several years, there have been many advances in understanding the process of ubiquitination, and there are numerous excellent recent reviews on this topic, many cited throughout this section (12, 27, 50, 51, 68, 69, 81, 82, 126, 128, 130, 131, 150, 158). What follows is a brief description representing the currently accepted sequence of events that lead to the ubiquitination of a protein. The ubiquitination of a protein is an energy-requiring multistep E1-E2-E3 cascade that results in addition of ubiquitin to the ε-NH2 of a lysine residue (Fig. 2). Proteins can be either monoubiquitinated at a single substrate lysine, multiubiquitinated at several substrate lysines, or polyubiquitinated in which chains of ubiquitin are added to one or more substrate lysines. Ubiquitin itself contains seven lysines, all of which have the potential to be ubiquitinated or polyubiquitinated, although the most common appear to be Lys48 and Lys63. It is generally accepted that polyubiquitination through ubiquitins' Lys48 targets the substrate for proteasomal degradation. In the first step, a ubiquitin-activating enzyme (E1) binds ATP·Mg2+ and ubiquitin (or a ubiquitin-like protein) and then catalyzes ubiquitin COOH-terminus adenylation. This is followed by attacking of the catalytic E1-Cys on the ubiquitin-adenylate complex to form the activated E1-ubiquitin thioester complex. In humans, eight E1s that initiate ubiquitin or ubiquitin-like conjugation have been identified (50, 131). In the second step, the E1 transfers the activated ubiquitin (or ubiqutin-like protein) to a family of proteins that are called ubiquitin-conjugating enzymes (E2). These form a ubiquitin-E2 high-energetic conjugate through a highly conserved E2-Cys residue. The E2s play an essential role in conjugation and, by influencing which of ubiquitins' seven lysines is conjugated with the substrate, can also play a role in determining the ultimate fate of the substrate. There are at least 35 E2s that have been described in humans, and these are classified into one of four classes based on the presence of extensions to the NH2- or COOH-terminus of the catalytic core (150, 158). The third and final step is the selective interaction of the loaded E2 with a specific ubiquitin protein ligase (E3). E3s selectively recruit specific substrates and act as scaffolds, essentially bringing the activated ubiquitin into close proximity with the protein substrate. E3s represent the specificity of the UPS with over 600 having been identified at the time of this writing (27). E3s are generally classified into two basic groups: homologous to E6-AP COOH-terminus (HECT) and really interesting new gene (RING), which includes the U-box containing proteins, such as carboxyl terminus of Hsc70-interacting protein (CHIP). The basic differences between them is that HECT E3s use a conserved Cys residue to form a transient thioester linkage with the activated ubiquitin COOH- terminus before substrate ubiquitination. The RING E3s act as true scaffolds, binding both ubiquitin and substrate, providing an optimal conformation for conjugation of the ubiquitin COOH-terminus with the ε-amino group of one or more substrate lysines (69, 126, 128, 130, 175).
Fig. 2.
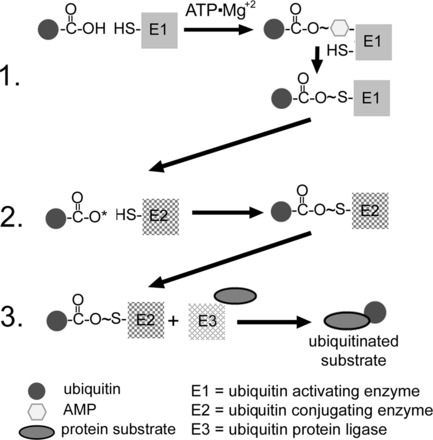
Ubiquitination of a protein. In step 1, a ubiquitin-activating enzyme (E1) binds ATP·Mg2+ and ubiquitin and then catalyzes ubiquitin COOH-terminus adenylation followed by attack of the catalytic E1-Cys on the ubiquitin-adenylate complex to form the activated E1-ubiquitin thioester complex. In step 2, ubiqutin is transferred to a ubiquitin-conjugating protein E2, producing a high-energy E2-ubiquitin thiol ester intermediate. In step 3, the intermediate is ligated to a protein substrate bound to a specific ubiquitin protein ligase E3. Modified from Powell and Divald (110) with permission from Oxford University Press.
Deubiquitinases.
DUBs are isopeptidases that belong to a superfamily of ∼80 ubiquitin-specific proteases. These maintain cellular ubiquitin homeostasis and counter the ubiquitination process by removing ubiquitin from substrates and by disassembling polyubiquitin chains. DUBs are classified into one of five structurally unrelated categories. Of these, the ubiquitin COOH-terminal hydrolases (UCHs), ubiquitin-specific proteases (USPs), ovarian tumour proteases (OTUs), and the Josephins are cysteine proteases. The JAB1/MPN/MOV34 family are zinc metalloproteases. DUBs have three main activities. The first is to generate free ubiquitin molecules from polyubiquitin expressed as a linear fusion protein. The second is to reverse ubiquitin signaling by removing ubiquitin chains from ubiquitinated substrates, which may lead to protein stabilization. If the protein is committed to degradation, the DUB can recycle the ubiquitin. Lastly, DUBs can trim ubiquitin chains and thus alter the form of ubiquitin modification to exchange one type of ubiquitin signal for another (i.e., receptor polyubiquitination degradation signal for monoubiquitination internalization signal). DUBS have considerable specificity for substrates and types of ubiquitin chains and are themselves subject to regulation by post-translational modifications, such as phosphorylation, and subcellular localization (12, 68, 82). With the exception of one study (23) that suggests that the DUB enzyme breast cancer-1 (BRCA-1)/BRCA-2-containing complex subunit 36 (BRCC36)-containing isopeptidase complex (BRISC), which deubiquitinates Lys63-linked polyubiquitin, has a protective role in myocardial ischemia-reperfusion injury, little else is known about the role of this class of ubiquitin associated proteins in the heart.
The 11S-activated proteasome.
The 11S-activated proteasome is a different proteasome configuration (“zomes”) that uses the 11S activator ring as its regulatory particle. The 11S-activated proteasome consists of a 20S proteasome that can be docked with one or two 11S activator rings or one 19S regulatory particle on one end and an 11S activator ring on the other end, called the hybrid proteasome. The 11S activator ring is a heterohexamer or heteroheptamer consisting of three protease activator 28α (PA28α) and three protease activator 28β (PA28β) subunits or three PA28α and four PA28β subunits, respectively, arranged in an alternating pattern (Fig. 3) (134, 170). When an 11S activator ring docks with a 20S proteasome, the carboxy-terminus tails of the PA28 subunits insert into pockets created by the α-subunits, resulting in a conformational shift that opens the access channel to a greater degree, enhancing substrate access to the catalytic chamber. This results in enhanced proteolytic capacity without altering catalytic subunit activity (138, 159). PA28 subunits are induced by γ-interferon, and despite the fact that knockdowns have very little effect, the 11S activator ring is thought to play a role in antigen presentation by favoring formation of peptides of specific size and composition. As mentioned above, the catalytic subunit immunoforms are also induced by γ-interferon and particularly in immune cells are often found in proteasomes in combination with the 11S activator ring. One form of the “immunoproteasome” is probably a hybrid containing immunoforms of the catalytic subunits in which the docking with 11S activator ring enables the hybrid to generate a high fraction of peptides that can serve as antigenic precursors (39, 51, 81, 100, 124). However, over the past few years it has become clear that 11S-activated proteasomes are involved in many cellular functions, particularly those involved in removal of oxidized and/or otherwise damaged proteins (34, 104, 105) and in clearing proteins damaged during inflammation (133). Indeed, we (113) have shown that 11S-activated proteasome may be upregulated in experimental hyperglycemia associated with increased oxidative stress. Moreover, recent studies indicate that forced expression of the PA28α subunits in cultured cardiomyocytes (87) and in heart (86) increases resistance to oxidative injury and ischemia-reperfusion injury, respectively.
Fig. 3.
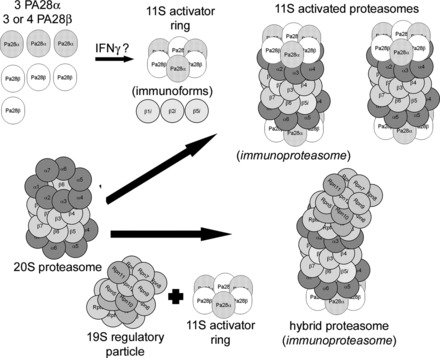
The 11S-activated proteasome and the related hybrid proteasome. 11S-activated proteasomes are formed when an 11S activator ring docks at one or both ends of a 20S proteasome. The 11S activator ring is composed of either 3 protease activator (PA) 28α and 3 PA28β subunits (shown) or 3 PA28α and 4 PA28β subunits and may be induced by interferon-γ. One form of an 11S-activated proteasome has been called the immunoproteasome and contains one or more of the immunoforms of the catalytic β-type subunits. Another form of an 11S-activated proteasome does not contain β-subunit immunoforms. Also shown is the hybrid proteasome which has a 11S activator ring docked at one end of the 20S proteasome and a 19S regulatory particle docked at the end that, if it contain β-subunit immunoforms (shown), has also been called the immunoproteasome. Modified from Powell and Divald (110) with permission from Oxford University Press.
Regulation of the Cardiac Proteasome
Proteasomes may be regulated by altering their composition (heterogeneity), post-translational modifications, and their associating partners. Dysfunction of the UPS in myocardial ischemia could very well be related to changes in any one of these regulatory elements or to the introduction of oxidative modifications so a brief discussion is relevant and important. The following discussion focuses on the cardiac proteasome but may be applicable to proteasomes found in other tissues.
Heterogeneity as a basis for regulation.
For many years, the prevailing hypothesis was that the two β-rings of the catalytic core of the 20S proteasome consisted of seven homologous β-subunits. The distribution of subunits β1, β2, and β5 were believed to be all constitutive or all replaced by the immunoform subunits β1_i_, β2_i_, and β5_i_. Several relatively recent studies have challenged this view and have provided novel evidence of heterogeneity within the β-rings (32, 45–47). Proteomic studies have demonstrated a high variability in cardiac proteasomes with regard to the distributions of the β-subunits. In addition to proteasomes having all constitutive or immunoform subunits, proteasomes may also contain one β-ring of constitutive subunits and one β-ring of immunoform subunits. Proteasomes may also be composed of a mixture of constitutive and immunoform subunits within each individual β-ring. Consistent with these varying structures, these subpopulations of 20S proteasomes, it is not surprising, exhibit varying degrees of proteolytic function and substrate specificity (32, 45, 166) (Fig. 4). In regard to the 19S regulatory particle, heterogeneity in the form of alternate splicing of the Rpn10 subunit (47) and two different populations of 19S regulatory particles in the murine heart (155) have also been reported. The presence of multiple proteasome subpopulations may regulate degradation of a particular protein or class of proteins, which in turn may alter the outcome of a particular disease.
Fig. 4.
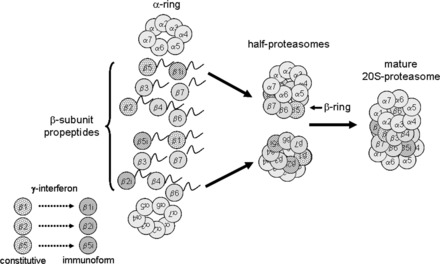
Regulation of 20S proteasomes by assembly of heterogeneous configurations. Under the influence of γ-interferon, select β-subunits are replaced by their immunoforms, which are directed by the complete α-rings to form half-proteasomes with specific configurations. In the example shown above, the top half-proteasome has constitutive β2 and β5 subunits and the β1 subunit has been replaced by the immunoform. The bottom half-proteasome has a constitutive β1 subunit, but the β2 and β5 subunits have been replaced by their immunoforms. These two distinctly different half-proteasomes mate to form a mature 20S proteasome that has a heterogeneous configuration. For the sake of clarity, the involvement of the proteasome assembly chaperones PAC1, PAC2, PAC3, and PAC4 and the proteasome maturation protein factor POMP are not depicted here. Modified from Powell (107) with the permission of Walters Kluwer Health.
Post-translational modification as a basis for regulation.
Proteasomes can be regulated by both reversible and irreversible post-translational modifications. Examples of reversible modifications include phosphorylation, ubiquitination, sumoylation, _O_-linked _N_-acetylglucosamination, and possibly some oxidative modifications. Irreversible modifications may include NH2-terminus acetylation and NH2-terminus myristoylation, as well as some oxidative modifications. For additional information, the reader is referred to an excellent review (132) on post-translational modification of the proteasome that has recently been published as part of the American Journal of Physiology-Heart Circulatory Physiology review collection on the “Role of the Ubiquitin Proteasome System in Cardiac Disease.”
Associating partners.
In general, the associating partner of the proteasome is often responsible for the post-translational modification discussed above. The best example of this is phosphorylation which can be mediated by several associating kinases, such as cAMP-dependent protein kinase A (PKA), calcium/calmodulin-dependent protein kinase II (CaMKII), and casein kinase II, which may phosphorylate multiple proteasome subunits including the α2, α3, α5, α7, β1, β2, β3, β5, β6, and β7 on the 20S proteasome and Rpt6 on the 19S. In general, phosphorylation of proteasome subunits tends to stabilize the proteasome and increase activity (10, 18, 31, 47, 90, 129, 141, 155, 167, 169, 176, 177). Phosphorylation of Rpt6, in particular, appears to enhance docking and stabilize the 26S (or 30S) proteasome (129, 169). The known exception to this is apoptosis signal-regulating kinase 1, which phosphorylates Rpt5, decreasing its ATPase activity and overall proteasome activity (148). In contrast, the associating serine threonine protein phosphatases PP2A and PP1γ dephosphorylate various proteasome subunits and, in general, decrease activity (47, 90, 176, 177). With regard to the 19S regulatory particle, a study has reported that heat shock protein 90 can associate with this regulatory particle and in doing so decreases functional potency (155). Inhibiting the associating heat shock protein 90 enhanced activity of the proteasome. We have proposed that the cardiac proteasome is actually part of and regulated by signal transduction pathways (107) (see Fig. 5), which is relevant to UPS dysfunction during myocardial ischemia as these pathways may become dysregulated.
Fig. 5.
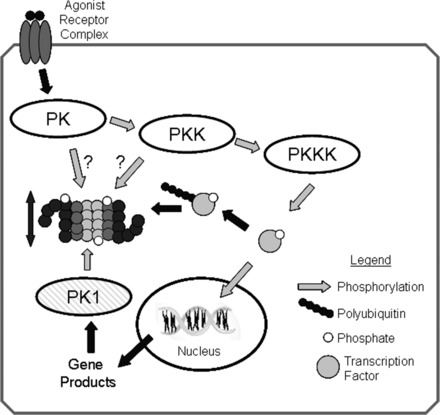
Regulation of proteasome activity by kinase cascades. In this scenario, binding of an agonist with a cell surface receptor initiates a phosphorylation cascade with an end effector protein kinase kinase kinase (PKKK) phosphorylating some transcription factor that can then enter the nucleus and interact with DNA resulting in gene transcription. Phosphorylation of signaling intermediates, in this case the transcription factor, is often a signal for ubiquitination and targeting to the 26S-proteasome for degradation. To amplify or dampen the effect of the signaling intermediate, any one of the protein kinases (PKs) within in the cascade might phosphorylate proteasome subunits, resulting in increased or decreased peptidase activity that would lessen or enhance availability of the signaling molecule accordingly. Another possibility would be for the gene product to activate a different protein kinase (PK1), which would phosphorylate proteasome resulting in activation and thus decreased availability of the transcription factor, essentially a form of feedback control. Reproduced from Powell (107) with the permission of Walters Kluwer Health.
UPS Dysfunction in Ischemia
Ischemia is reduced blood flow causing oxygen deprivation, reduced substrate supply, and decreased metabolite removal. Most studies have observed that the UPS becomes dysfunctional during myocardial ischemia (11, 114). For the purposes of this discussion, dysfunction means an impaired ability of the UPS to process ubiquitinated proteins. This is usually observed as diminished proteolytic activities in tissue extracts as measured by cleavage of small peptides linked to a fluorogen in combination with increases in total ubiquitinated or misfolded proteins (11, 114). In one study, this has been observed using a UPS reporter mouse expressing a green fluorescent protein fused with the CL1 degron sequence of amino acids (146). In theory, dysfunction of the UPS could involve any one of the components described earlier. While roles for some of the E3 ligases in ischemia have been proposed, they have not been shown to be dysfunctional. As this topic has been reviewed it will not be discussed here (125). The only component shown to be dysfunctional as a consequence of ischemia is the proteasome itself and this will be discussed in detail. Understanding how proteasome dysfunction occurs and how it effects myocardial ischemia is important as underscored by a recent study (146) which shows that even moderate knockdown (50%) of the β5 subunit is sufficient to worsen postischemic function and infarct size. The ensuing discussion starts with the cerebral proteasome since dysfunction was first described in this organ.
Cerebral ischemia.
Proteasome dysfunction, first described in 1992, was suggested by an increase in insoluble ubiquitin-conjugates in gerbil cortex and hippocampus following 5 min of transient forebrain ischemia (62). Formation of ubiquitin conjugates appears to be more a function of reperfusion rather than ischemia and may not be related to proteasome dysfunction (66). Activity of the proteasome has been measured in several models of cerebral ischemia in gerbil, rat, and mouse and in general was found to be decreased in several areas. The degree of dysfunction was generally found to be related to length of ischemia and was reversible with short durations of ischemia (2, 40, 66, 77, 79). Subsequent reports have suggested that proteasome dysfunction is secondary to oxidative damage (79) and disassembly of mature proteasome with deposition of subunits into protein aggregates (40). A very recent report (91) suggests that not only is cerebral proteasome dysfunctional but that a different form of proteasome containing immunoforms may predominate.
Myocardial ischemia.
Proteasome dysfunction in myocardial ischemia was first described by Bulteau et al. (11) in 2001 who reported that following 30 min of in vivo left anterior descending coronary artery occlusion, proteasomes exhibit decreased chymotryptic and caspase- and trypsin-like activities coupled with an increase in ubiquitinated proteins. However, following purification, only the decrease in trypsin-like activity was observed. This observation was confirmed in the isolated perfused heart in studies by Powell et al. (109, 114) who also demonstrate that ATP-dependent proteasome activity is preferentially affected. This is consistent with the suggestion that 26S proteasomal insufficiency can result in increased myocardial ubiquitinated proteins. A later study that examined turnover of signaling proteins known to be degraded by the UPS as a means of assessing proteasome function suggested that shorter periods of ischemia may result in selective proteasome dysfunction which may in turn affect turnover of specific proteins (54). In consideration of the presence of multiple proteasome subpopulations and regulation of the cardiac UPS as previously discussed, this is an intriguing observation.
Potential mechanisms involved in proteasome dysfunction.
Various mechanisms have been proposed but only two have received experimental support.
ISCHEMIA INDUCED ATP DEPLETION.
Docking of the 19S regulatory particle to the 20S proteasome, activation of the proteasome, and protein ubiquitination all require ATP (44, 67). Intracellular ATP levels are decreased during ischemia (137). One study has suggested that proteasome activities are regulated by intracellular ATP levels according to a biphasic relationship (increasing concentrations activate up to maxima and thereafter inhibit activity) (70). Therefore, it is reasonable to suggest that ATP depletion during ischemia could be partially responsible for decreased cardiac proteasome activity. In fact, two studies (3, 20) using two different animal models of ischemia, an in vivo canine model and an ex vivo isolated rat heart model, have suggested just this mechanism. Yet, when measuring lysate proteasome activity, ex vivo, adding back the ATP did not reverse the dysfunction. We have shown that lysate postischemic proteasome activity is still diminished following an addition of ATP at concentrations that evoke maximal activation (109). Another rather intriguing study suggests that some proteasomes may actually be activated by low ATP concentrations (41). While these studies may suggest that ATP depletion can be a contributing factor to proteasome dysfunction during ischemia, direct evidence is still lacking. What the ex vivo studies of proteasome activity in lysate do suggest is a defect in ATP utilization by postischemic proteasomes. This has led us and others to postulate that the subunits of the 20S proteasome and its regulatory particles may be targets for oxidative damage, certainly conceivable, given the numerous reports of oxidative stress and oxidized cytosolic, myofibrillar, and mitochondrial proteins in the postischemic heart (16, 80, 103, 111).
OXIDATIVE DAMAGE TO PROTEASOME AND/OR REGULATORY SUBUNITS.
Oxidative modifications to proteins may lead to unfolding and loss of function because of changes in secondary or tertiary structures. These modifications may also lead to exposure of hydrophobic patches and increased susceptibility to degradation (24, 25). Studies that have examined purified 20S and 26S proteasomes have shown that the 26S proteasome configuration is more vulnerable to oxidative damage (122). The initial report that cardiac proteasome was dysfunctional following ischemia suggested that several of the α-subunits of the 20S proteasome were 4-hydroxynonylated (11). However, it was not clear that the proteasome dysfunction was related to these modified subunits as it was later reported that concentrations of 4-hydroxynonenal in excess of 100 μM are required inactivate purified proteasome from rat hearts, suggesting relative resistance to this type of oxidative modification (37). We have developed evidence that postischemic ATP-dependent proteasome function may be preserved by pretreatment with α-tocotrienol, a vitamin E analog (Table 1) (unpublished data). Taking into consideration the effect of ischemia-reperfusion on ATP-dependent proteasome activity (20, 109) and the reported increased vulnerability of the 26S proteasome to oxidative inactivation (123) would lead one to suggest that perhaps the ATP-utilizing subunits of the 19S regulatory particle are subject to oxidative damage. In fact, the S6 ATPase subunit, Rpt3, has been reported to be sensitive to carbonylation in SH-SY5Y cells exposed to an oxidizing environment (75). This led us to examine the 19S regulatory particle for oxidative damage following myocardial ischemia and has identified significant carbonylation of subunits Rpt3 and Rpt5 of the 26S proteasome (29). These were the same subunits that appeared to be oxidized in explants from human hearts from patients with end-stage heart failure (116). These latter studies present the best evidence for a role of oxidative damage to proteasome subunits but do not provide cause and effect for a relationship between the dysfunction and the damage. This required an intervention, in this case IPC, which is described in a later section.
Table 1.
Pretreatment of hearts with tocotrienol-rich fraction preserves postischemic proteasome function
| Proteasome Chymotryptic Activity, %Preischemic | ||
|---|---|---|
| Treatment | 20S | 26S |
| PreIschemic | 100 ± 17 | 100 ± 15 |
| Ischemia (30 min) | 55 ± 10* | 54 ± 7* |
| + Reperfusion (120 min) | 78 ± 16 | 42 ± 9* |
| + Tocotrienol-rich fraction | 144 ± 22 | 152 ± 15 |
| + Reperfusion (120 min) | 102 ± 26 | 112 ± 45 |
The continuing controversy: can proteasome inhibitors improve outcomes following myocardial ischemia?
The use of proteasome inhibitors as a strategy to treat myocardial ischemia is an ongoing controversy that requires some discussion. Several studies have suggested that the use of proteasome inhibitors in myocardial ischemia is beneficial and have examined the proteasome inhibitor PS-519 (Millennium Pharmaceuticals). PS-519 is a lactacystin derivative originally tested and found to have some protective effects in experimental models of cerebral ischemia (7, 160–162). The rationale for testing this inhibitor is that proteasome inhibitors, in general, do have a well-documented anti-inflammatory effect (35) because of interference with UPS-mediated activation of the NF-κB pathways [reviewed in Krappmann and Scheidereit (84)]. The initial study (15) used a leukocyte-supplemented perfused heart preparation and did observe improved postischemic function when hearts were treated with PS-519. However, in the absence of the leukocytes, no effect of the inhibitor was observed. In a subsequent study (120) a single dose of PS-519 administered at time of reperfusion in an in vivo porcine model demonstrated reduced activation of myocardial NF-κB, release of creatine kinase and troponin I, and improved segmental function. However, a more recent study (98) in an in vivo murine model, while showing that PS-519 administered at time of reperfusion decreased infarct size, failed to have any effect on NF-κB activation. When administered 2 h postreperfusion, PS-519 had no effect at all, even though an IκK inhibitor was effective, thus suggesting that any protective effects may not be related to NF-κB signaling.
These types of studies have not been limited to PS-519. Pretreatment of isolated hearts with the proteasome inhibitor MG132 has been shown to improve posthypoxic function of excised isolated papillary muscles (136). However, this was after a delay of at least 30 min in the presence of increased heat shock proteins with no analysis of myocardial proteasome activity. This is in contrast to our studies (30, 114), which demonstrate that preischemic treatment of isolated rat hearts with MG132 results in a dose-dependent decrease in postischemic function and that the inhibitor lactacystin failed to have any effects on postischemic function even though preischemic proteasome activity was decreased by 40%. Other studies have examined bortezomib, a reversible nonselective proteasome inhibitor, in a canine model of left anterior descending coronary artery occlusion and have observed decreased arrhythmias and infarct size when the drug was administered 1 h preligation and then repeated 5 h postligation (168). A follow-up study showed similar effects when bortezomib was administered 1 h postligation (71). In both studies, the protective effect was associated with decreased loss of G protein receptor kinase 2 in the epicardial border zone and decreased proteasome activity in whole blood. Because proteasome activity was not determined in the corresponding epicardial border zone tissue, while an attractive hypothesis, it is unclear that the lack of change in the kinase was related to the inhibitory effect of bortezomib. Nonproteasome targets of bortezomib is an issue as a recent study indicates that this drug inhibits other proteolytic enzymes at potencies near that of the proteasome (1).
Whether a proteasome inhibitor has beneficial or detrimental effects is notoriously dose and time dependent (88, 96). Low nontoxic doses have been observed to upregulate several antioxidative enzymes and confer protection against H2O2-induced endothelial cell damage (95). In addition, sublethal doses of proteasome inhibitors may result in upregulation of proteasome subunits (94). It is quite possible that in the presence of mild UPS dysfunction, a low nontoxic dose of an inhibitor that upregulates antioxidant enzymes and subunits of the proteasome and is anti-inflammatory without effecting either UPS-mediated turnover of signaling proteins or protein quality control could be beneficial. From a clinical perspective, this is a tall order that requires specific doses and dosing intervals just not possible at this time with first generation proteasome inhibitors. Given the degree of proteasome dysfunction generally present during myocardial ischemia, there are serious concerns with the use of these inhibitors (108). This is best illustrated by the fact that the only clinically available proteasome inhibitors bortezomib (Velcade, Millenium Pharmaceuticals) and carfilzomib (Kyprolis, ONYX Pharmaceuticals) are indicated for treatment of multiple myeloma. While experience with carfilzomib is limited, bortezomib has been associated with several reports of “unexpected” cardiovascular toxicity, including heart failure and myocardial ischemia (36, 58, 144, 153). This raises the concern about nonselectivity of the first generation proteasome inhibitors. In this instance, nonselectivity refers to their ability to differentiate between constitutive subunits versus the immunoforms. As indicated earlier, proteasomes can be defined as to whether they contain the constitutive catalytic subunits β1, β2, or β5 or the immunoforms β1_i_, β2_i_, or β5_i_, originally observed to be upregulated in the presence of γ-interferon. The vast majority of the first generation inhibitors cannot discriminate between these subunits. Even the newest, carfilzomib, which is selective for the β5 and β5_i_ subunits, does not discriminate between them (174). The importance of subunit selectivity is best illustrated by a very recent study that demonstrates that selective cardiomyocyte-specific knockdown of the constitutive β5 subunit exacerbates myocardial ischemia-reperfusion injury (146). Does this mean that there is no role for proteasome inhibition as a strategy to decrease myocardial ischemia-reperfusion injury? No, rather, more selective inhibitors that selectively target the different subunits need to be developed. The recent literature suggests that it is a proteasome containing immunoforms, and specifically β1_i_ and β5_i_, that is responsible for activating the NF-κB inflammatory signaling pathways through processing of IκB (48, 152). ONYX-0914 is a second generation proteasome inhibitor that is 20–40 times more selective for the β5_i_ subunit (72, 99). This inhibitor has protective effects and blocks inflammatory cytokine production in models of experimental arthritis (99), lupus-induced nephritis (73), and experimental colitis (6). At the time of this writing, ONYX-0914 has not been examined in models of myocardial ischemia; thus it is unclear whether a strategy of specific inhibition of the immunoproteasome would be effective. Clearly, though, this is an area of research requiring additional development.
The UPS in IPC
IPC is an experimental technique that decreases vulnerability to ischemia-reperfusion injury. The current thinking is that IPC involves signaling changes that cause the inward mitochondrial ATP-sensitive K+ channel channels to open and prevent opening of the mitochondrial permeability transition pore (9, 53, 83). Several studies suggest that the UPS plays a role in IPC by facilitating some of the pre- and postischemic signaling changes (3, 20, 29). In order for this to occur, it is necessary that IPC preserve postischemic proteasome function in some way. Like the ischemia studies, the earliest investigations suggesting this possibility are found in the brain literature that examined IPC as a means of protection against cerebral ischemia. Most of these studies did not directly assess proteasome function but rather relied on decreased accumulation of proapoptotic proteins and protein aggregates in the postischemic brain as surrogate markers (89, 97, 115). One rather intriguing study suggests that myocardial IPC is prevented in a mouse deficient in the β1_i_ subunit (14). Perhaps the earliest study in the heart to suggest that postischemic function of the proteasome is protected to some extent by preconditioning is a study that used nicorandil to mimic IPC (114). All three of the aforementioned studies in the heart (3, 20, 29) agree that IPC is associated with improved proteasome peptidase activities and decreased accumulation of ubiquitinated or misfolded proteins. Where they differ is in the proposed mechanisms.
IPC preserves UPS function by preventing degradation of εPKC.
The heart has been reported to be protected from ischemic injury when the δ-isoform of protein kinase (δPKC), a prodeath kinase, is inhibited (74). As a result of IPC, the prosurvival kinase εPKC is activated and translocated to cardiac mitochondria (5). Churchill et al. (20) have shown that IPC alters the ratios of these two kinases and improves postischemic UPS function in turn favoring tissue survival. Considering that a proteasome inhibitor prevented the effects of IPC on postischemic function and the ratios of δPKC/εPKC, the authors proposed that the UPS regulates the ratio between these two kinases. This is an attractive hypothesis; however, IPC alters levels of many prosurvival and prodeath proteins thought to be regulated by the UPS, including PTEN (13), IκB (54), Bax (29), PKC, and Akt (protein kinase B) (146). Many diverse pathways are regulated by the UPS, and it is likely that UPS-mediated changes in δPKC account for only a portion of the protective effects of IPC.
IPC increases protein kinase A (PKA)-mediated activation of proteasome.
This theory is highly credible given the regulation of the cardiac proteasome by PKA (see Regulation of the Cardiac Proteasome). Proteasome peptidase activity is activated by PKA-induced phosphorylation of several subunits of the 20S proteasome and the 19S regulatory particle (169, 176). Evidence that PKA can enhance docking of 20S proteasomes and 19S regulatory particles was presented by Asai et al. (3) who suggest that assembly of intact 26S proteasome is enhanced by IPC-mediated transient postischemic increases in PKA, which would explain the higher peptidase activity observed in the immediate postischemic period. It is of interest that PKN (protein kinase C-related kinase 1) has also been shown to stimulate proteasome activity and possibly play a protective role in the heart during myocardial ischemia (142).
IPC decreases postischemic oxidation of 19S regulatory particle subunits.
As described above, the 19S regulatory particle subunit Rpt5 was observed to be susceptible to oxidative modifications during myocardial ischemia (29). In this study, IPC was used as an intervention to show cause and effect between changes in proteasome activity and 19S regulatory particle subunit oxidation, in this case decreased postischemic carbonylation of the Rpt5 subunit (29). The Rpt5 subunit has two roles: attachment of the “base” of the 19S regulatory particle to 20S proteasome α-rings (43) and binding of the 19S regulatory particle “lid” to the base (43). In consideration of these roles, it is conceivable that by decreasing oxidation of this subunit, IPC can improve the docking of these two complexes. However, this was not examined, and it is not clear that improved proteasome activity is related to decreased oxidation of this sole subunit. IPC has a high degree of complexity, and it is unlikely that a single mechanism accounts for the positive outcome of this procedure on postischemic proteasome function.
Roles for other Regulatory Particles in Myocardial Ischemia
The studies thus far have examined the role of ubiquitin-mediated degradation of proteins, or more specifically, dysfunction of 26S proteasome in myocardial ischemia. However, as described above, there are other forms of the proteasome. Recent studies have begun to define a role for the 11S-activated proteasome more related to protein quality control and removal of damaged proteins during myocardial ischemia.
The 11S-activated proteasome in myocardial ischemia.
As described above, if the 20S proteasome is docked with the 11S activator ring and does not contain immunoforms, it is called the 11S-activated proteasome. Studies have suggested this proteasome functions in the removal of oxidized proteins (104, 105). In consideration of the relationship between myocardial ischemia, oxidative stress, and protein damage (111, 112), as well as the reported upregulation of the 11S-activated proteasome in a related cardiomyopathy associated with oxidative stress (113), it is conceivable that the 11S-activated proteasome plays a role in removal of proteins oxidized during myocardial ischemia. We (30) have previously shown that the proteasome is responsible for the removal of proteins that have been oxidized during myocardial ischemia and that this appeared to occur in an ubiquitin-independent manner, suggesting involvement of a proteasome other than the 26S proteasome. A recent report that has shown that in cardiomyocytes, forced overexpression of the PA28α subunit of the 11S activator ring results in increased proteasome activity and an enhanced resistance to oxidative stress (87). Furthermore, it has been shown that postischemic function is improved and infarct size is decreased with overexpression of PA28α in a transgenic mouse model, providing strong evidence for a role of this zome in myocardial ischemia (86).
Autophagy and Ischemia
A major function of the UPS is protein quality control and the prevention of proteotoxic stress. However, this is not the only responsible system; thus this review would be incomplete without some discussion of autophagy, which is a parallel system also playing a major role in prevention of proteotoxic stress (55, 56). Discussion will be limited since there are excellent recent reviews on autophagy, in general, and in ischemia (4, 28, 55–57, 118, 143, 173). Also, whereas these two systems are related in function, their targets tend to be different with autophagy removing macromolecular structures and organelles. Unlike the UPS that tends to become dysfunctional during ischemia-reperfusion, most studies indicate that autophagic flux is increased during ischemia (60, 92, 127, 143, 165). In general, agents that enhance autophagy have a beneficial effect on ischemic injury (119, 127). It has been suggested that the enhanced flux is actually in response to the diminished capacity of the UPS in ischemic tissue and thus is an attempt to maintain cellular protein quality control. In fact, a recent study indicates that simply inhibiting the proteasome with an inhibitor is enough to result in upregulation of autophagy in cardiomyocytes (172) and that the protein p62/SQSTM1 may act as a proteotoxic sensor that connects the two systems (140, 171). This type of inverse relationship between UPS and autophagic flux is not limited to the ischemic myocardium but has also been observed in heart failure (78, 173).
Summary and Future Directions
The UPS has often been described as simply a means for the removal of unwanted, damaged, or otherwise unneeded proteins. Research over the past 20 years has shown there is much more to this concept. By removing proteins, some highly reactive, the UPS plays multiple critical roles in regulating many of the intracellular processes necessary for cell function and survival. The evidence to date is quite conclusive that the UPS is dysfunctional during myocardial ischemia-reperfusion, at least in experimental models. Given the absolute requirement for the UPS, it seemed reasonable that UPS dysfunction, particularly when a cell is being exposed to a stress such as ischemia, would be detrimental as it would result in defects in removal of highly reactive signaling molecules, for example, Bax (29) and proteins damaged by oxidative stress (30). Figure 6 presents a scheme describing the proposed functions of the 26S- and 11S-activated proteasomes and consequences of dysfunction. There have been multiple mechanisms suggested for the dysfunction, but the actual mechanism(s) is still not known, nor is it clear whether dysfunction is a primary or secondary process. As part of our discussion, we reviewed the evidence for and against the suggested use of proteasome inhibitors in treating myocardial ischemia. Part of this discussion suggests that perhaps new second generation proteasome inhibitors targeting specific catalytic subunit immunoforms might present a viable clinical option for decreasing the inflammatory response associated with myocardial ischemia, certainly an area requiring additional studies. Lastly, we discussed the potential role that the UPS might play in IPC and suggested that improved function of the UPS in the preconditioned heart might facilitate degradation of proapoptotic proteins in the postischemic period, although this story is also far from complete. The recent development of genetic models that either enhance or diminish proteasome function should help to shed some light on these issues as well as demonstrating the various roles of the different zomes and proteasome subunits in cardiac (patho)physiology. Hopefully, this review will spur research to expand our knowledge of how the UPS functions in the normal heart and how its dysfunction could lead to the worsening of cardiac pathologies.
Fig. 6.
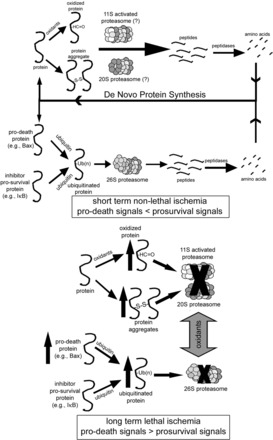
Potential roles for the ubiquitin proteasome system (UPS) and 11S-activated proteasome in short- and long-duration ischemia. In the nonischemic heart, oxidized, misfolded, and ubiquitinated proteins are degraded through both ubiquitin- and nonubiquitin-mediated pathways, recycling the constituent amino acids and maintaining a dynamic balance between prosurvival and prodeath signals. During an ischemic insult resulting in cell death or dysfunction, UPS function is inhibited leading to accumulation of oxidized and ubiquitinated proteins. In addition, a condition known as dysregulation may occur in which normal UPS-mediated degradation of prodeath proteins is depressed. Reproduced from Powell and Divald (110) with permission from Oxford University Press.
GRANTS
This work was supported by National Institute of Health Grant HL-68936 (to S. R. Powell).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
J.C. and S.R.P. prepared figures; J.C. and S.R.P. drafted manuscript; J.C. and S.R.P. approved final version of manuscript; S.R.P. edited and revised manuscript.
REFERENCES
- 1.Arastu-Kapur S, Anderl JL, Kraus M, Parlati F, Shenk KD, Lee SJ, Muchamuel T, Bennett MK, Driessen C, Ball AJ, Kirk CJ. Nonproteasomal targets of the proteasome inhibitors bortezomib and carfilzomib: a link to clinical adverse events. Clin Cancer Res 17: 2734–2743, 2011 [DOI] [PubMed] [Google Scholar]
- 2.Asai A, Tanahashi N, Qiu JH, Saito N, Chi S, Kawahara N, Tanaka K, Kirino T. Selective proteasomal dysfunction in the hippocampal CA1 region after transient forebrain ischemia. J Cereb Blood Flow Metab 22: 705–710, 2002 [DOI] [PubMed] [Google Scholar]
- 3.Asai M, Tsukamoto O, Minamino T, Asanuma H, Fujita M, Asano Y, Takahama H, Sasaki H, Higo S, Asakura M, Takashima S, Hori M, Kitakaze M. PKA rapidly enhances proteasome assembly and activity in in vivo canine hearts. J Mol Cell Cardiol 46: 452–462, 2009 [DOI] [PubMed] [Google Scholar]
- 4.Baines CP. How and when do myocytes die during ischemia and reperfusion: the late phase. J Cardiovasc Pharmacol Ther 16: 239–243, 2011 [DOI] [PubMed] [Google Scholar]
- 5.Baines CP, Song CX, Zheng YT, Wang GW, Zhang J, Wang OL, Guo Y, Bolli R, Cardwell EM, Ping P. Protein kinase Cepsilon interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circ Res 92: 873–880, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basler M, Dajee M, Moll C, Groettrup M, Kirk CJ. Prevention of experimental colitis by a selective inhibitor of the immunoproteasome. J Immunol 185: 634–641, 2010 [DOI] [PubMed] [Google Scholar]
- 7.Berti R, Williams AJ, Velarde LC, Moffett JR, Elliott PJ, Adams J, Yao C, Dave JR, Tortella FC. Effect of the proteasome inhibitor MLN519 on the expression of inflammatory molecules following middle cerebral artery occlusion and reperfusion in the rat. Neurotox Res 5: 505–514, 2003 [DOI] [PubMed] [Google Scholar]
- 8.Blickwedehl J, Agarwal M, Seong C, Pandita RK, Melendy T, Sung P, Pandita TK, Bangia N. Role for proteasome activator PA200 and postglutamyl proteasome activity in genomic stability. Proc Natl Acad Sci USA 105: 16165–16170, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bolli R. Preconditioning: a paradigm shift in the biology of myocardial ischemia. Am J Physiol Heart Circ Physiol 292: H19–H27, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bose S, Stratford FL, Broadfoot KI, Mason GG, Rivett AJ. Phosphorylation of 20S proteasome alpha subunit C8 (alpha7) stabilizes the 26S proteasome and plays a role in the regulation of proteasome complexes by gamma-interferon. Biochem J 378: 177–184, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bulteau AL, Lundberg KC, Humphries KM, Sadek HA, Szweda PA, Friguet B, Szweda LI. Oxidative modification and inactivation of the proteasome during coronary occlusion/reperfusion. J Biol Chem 276: 30057–30063, 2001 [DOI] [PubMed] [Google Scholar]
- 12.Burrows JF, Johnston JA. Regulation of cellular responses by deubiquitinating enzymes: an update. Front Biosci 17: 1184–1200, 2012 [DOI] [PubMed] [Google Scholar]
- 13.Cai Z, Semenza GL. PTEN activity is modulated during ischemia and reperfusion: involvement in the induction and decay of preconditioning. Circ Res 97: 1351–1359, 2005 [DOI] [PubMed] [Google Scholar]
- 14.Cai ZP, Shen Z, Van Kaer L, Becker LC. Ischemic preconditioning-induced cardioprotection is lost in mice with immunoproteasome subunit low molecular mass polypeptide-2 deficiency. FASEB J 22: 4248–4257, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campbell B, Adams J, Shin YK, Lefer AM. Cardioprotective effects of a novel proteasome inhibitor following ischemia and reperfusion in the isolated perfused rat heart. J Mol Cell Cardiol 31: 467–476, 1999 [DOI] [PubMed] [Google Scholar]
- 16.Canton M, Neverova I, Menabo R, Van Eyk J, Di Lisa F. Evidence of myofibrillar protein oxidation induced by postischemic reperfusion in isolated rat hearts. Am J Physiol Heart Circ Physiol 286: H870–H877, 2004 [DOI] [PubMed] [Google Scholar]
- 17.Carrier L, Schlossarek S, Willis MS, Eschenhagen T. The ubiquitin-proteasome system and nonsense-mediated mRNA decay in hypertrophic cardiomyopathy. Cardiovasc Res 85: 330–338, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Castano JG, Mahillo E, Arizti P, Arribas J. Phosphorylation of C8 and C9 subunits of the multicatalytic proteinase by casein kinase II and identification of the C8 phosphorylation sites by direct mutagenesis. Biochemistry 35: 3782–3789, 1996 [DOI] [PubMed] [Google Scholar]
- 19.Castro A, Bernis C, Vigneron S, Labbe JC, Lorca T. The anaphase-promoting complex: a key factor in the regulation of cell cycle. Oncogene 24: 314–325, 2005 [DOI] [PubMed] [Google Scholar]
- 20.Churchill EN, Ferreira JC, Brum PC, Szweda LI, Mochly-Rosen D. Ischaemic preconditioning improves proteasomal activity and increases the degradation of deltaPKC during reperfusion. Cardiovasc Res 85: 385–394, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ciechanover A. Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol 6: 79–87, 2005 [DOI] [PubMed] [Google Scholar]
- 22.Ciechanover A, Orian A, Schwartz AL. Ubiquitin-mediated proteolysis: biological regulation via destruction. Bioessays 22: 442–451, 2000 [DOI] [PubMed] [Google Scholar]
- 23.Cilenti L, Balakrishnan MP, Wang XL, Ambivero C, Sterlicchi M, del MF, Ma XL, Zervos AS. Regulation of Abro1/KIAA0157 during myocardial infarction and cell death reveals a novel cardioprotective mechanism for Lys63-specific deubiquitination. J Mol Cell Cardiol 50: 652–661, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davies KJ, Delsignore ME. Protein damage and degradation by oxygen radicals. III. Modification of secondary and tertiary structure. J Biol Chem 262: 9908–9913, 1987 [PubMed] [Google Scholar]
- 25.Davies KJ, Lin SW, Pacifici RE. Protein damage and degradation by oxygen radicals. IV. Degradation of denatured protein. J Biol Chem 262: 9914–9920, 1987 [PubMed] [Google Scholar]
- 26.DeMartino GN. Purification of PA700, the 19S regulatory complex of the 26S proteasome. Methods Enzymol 398: 295–306, 2005 [DOI] [PubMed] [Google Scholar]
- 27.Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem 78: 399–434, 2009 [DOI] [PubMed] [Google Scholar]
- 28.Ding WX, Yin XM. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy 4: 141–150, 2008 [DOI] [PubMed] [Google Scholar]
- 29.Divald A, Kivity S, Wang P, Hochhauser E, Roberts B, Teichberg S, Gomes AV, Powell SR. Myocardial ischemic preconditioning preserves postischemic function of the 26S proteasome through diminished oxidative damage to 19S regulatory particle subunits. Circ Res 106: 1829–1838, 2010 [DOI] [PubMed] [Google Scholar]
- 30.Divald A, Powell SR. Proteasome mediates removal of proteins oxidized during myocardial ischemia. Free Radic Biol Med 40: 156–164, 2006 [DOI] [PubMed] [Google Scholar]
- 31.Drews O, Tsukamoto O, Liem D, Streicher J, Wang Y, Ping P. Differential regulation of proteasome function in isoproterenol-induced cardiac hypertrophy. Circ Res 107: 1094–1101, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drews O, Wildgruber R, Zong C, Sukop U, Nissum M, Weber G, Gomes AV, Ping P. Mammalian proteasome subpopulations with distinct molecular compositions and proteolytic activities. Mol Cell Proteomics 6: 2021–2031, 2007 [DOI] [PubMed] [Google Scholar]
- 33.Drews O, Zong C, Ping P. Exploring proteasome complexes by proteomic approaches. Proteomics 7: 1047–1058, 2007 [DOI] [PubMed] [Google Scholar]
- 34.Ebstein F, Kloetzel PM, Kruger E, Seifert U. Emerging roles of immunoproteasomes beyond MHC class I antigen processing. Cell Mol Life Sci 69: 2543–2558, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Elliott PJ, Zollner TM, Boehncke WH. Proteasome inhibition: a new anti-inflammatory strategy. J Mol Med 81: 235–245, 2003 [DOI] [PubMed] [Google Scholar]
- 36.Enrico O, Gabriele B, Nadia C, Sara G, Daniele V, Giulia C, Antonio S, Mario P. Unexpected cardiotoxicity in haematological bortezomib treated patients. Br J Haematol 138: 396–397, 2007 [DOI] [PubMed] [Google Scholar]
- 37.Farout L, Mary J, Vinh J, Szweda LI, Friguet B. Inactivation of the proteasome by 4-hydroxy-2-nonenal is site specific and dependant on 20S proteasome subtypes. Arch Biochem Biophys 453: 135–142, 2006 [DOI] [PubMed] [Google Scholar]
- 38.Gaczynska M, Rock KL, Goldberg AL. Role of proteasomes in antigen presentation. Enzyme Protein 47: 354–369, 1993 [DOI] [PubMed] [Google Scholar]
- 39.Gaczynska M, Rock KL, Spies T, Goldberg AL. Peptidase activities of proteasomes are differentially regulated by the major histocompatibility complex-encoded genes for LMP2 and LMP7. Proc Natl Acad Sci USA 91: 9213–9217, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ge P, Luo Y, Liu CL, Hu B. Protein aggregation and proteasome dysfunction after brain ischemia. Stroke 38: 3230–3236, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Geng Q, Romero J, Saini V, Baker TA, Picken MM, Gamelli RL, Majetschak M. A subset of 26S proteasomes is activated at critically low ATP concentrations and contributes to myocardial injury during cold ischemia. Biochem Biophys Res Commun 390: 1136–1141, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gillette TG, Kumar B, Thompson D, Slaughter CA, DeMartino GN. Differential roles of the COOH termini of AAA subunits of PA700 (19 S regulator) in asymmetric assembly and activation of the 26 S proteasome. J Biol Chem 283: 31813–31822, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev 82: 373–428, 2002 [DOI] [PubMed] [Google Scholar]
- 45.Gomes AV, Young GW, Wang Y, Zong C, Eghbali M, Drews O, Lu H, Stefani E, Ping P. Contrasting proteome biology and functional heterogeneity of the 20 S proteasome complexes in mammalian tissues. Mol Cell Proteomics 8: 302–315, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gomes AV, Zong C, Edmondson RD, Berhane BT, Wang GW, Le S, Young G, Zhang J, Vondriska TM, Whitelegge JP, Jones RC, Joshua IG, Thyparambil S, Pantaleon D, Qiao J, Loo J, Ping P. The murine cardiac 26S proteasome: an organelle awaiting exploration. Ann NY Acad Sci 1047: 197–207, 2005 [DOI] [PubMed] [Google Scholar]
- 47.Gomes AV, Zong C, Edmondson RD, Li X, Stefani E, Zhang J, Jones RC, Thyparambil S, Wang GW, Qiao X, Bardag-Gorce F, Ping P. Mapping the murine cardiac 26S proteasome complexes. Circ Res 99: 362–371, 2006 [DOI] [PubMed] [Google Scholar]
- 48.Gong P, Canaan A, Wang B, Leventhal J, Snyder A, Nair V, Cohen CD, Kretzler M, D'Agati V, Weissman S, Ross MJ. The ubiquitin-like protein FAT10 mediates NF-kappaB activation. J Am Soc Nephrol 21: 316–326, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gorbea C, Taillandier D, Rechsteiner M. Assembly of the regulatory complex of the 26S proteasome. Mol Biol Rep 26: 15–19, 1999 [DOI] [PubMed] [Google Scholar]
- 50.Groettrup M, Pelzer C, Schmidtke G, Hofmann K. Activating the ubiquitin family: UBA6 challenges the field. Trends Biochem Sci 33: 230–237, 2008 [DOI] [PubMed] [Google Scholar]
- 51.Groettrup M, Soza A, Eggers M, Kuehn L, Dick TP, Schild H, Rammensee HG, Koszinowski UH, Kloetzel PM. A role for the proteasome regulator PA28alpha in antigen presentation. Nature 381: 166–168, 1996 [DOI] [PubMed] [Google Scholar]
- 52.Groll M, Huber R. Substrate access and processing by the 20S proteasome core particle. Int J Biochem Cell Biol 35: 606–616, 2003 [DOI] [PubMed] [Google Scholar]
- 53.Gross GJ, Fryer RM. Sarcolemmal versus mitochondrial ATP-sensitive K+ channels and myocardial preconditioning. Circ Res 84: 973–979, 1999 [DOI] [PubMed] [Google Scholar]
- 54.Gurusamy N, Goswami S, Malik G, Das DK. Oxidative injury induces selective rather than global inhibition of proteasomal activity. J Mol Cell Cardiol 44: 419–428, 2008 [DOI] [PubMed] [Google Scholar]
- 55.Gustafsson AB, Gottlieb RA. Eat your heart out: role of autophagy in myocardial ischemia/reperfusion. Autophagy 4: 416–421, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gustafsson AB, Gottlieb RA. Recycle or die: the role of autophagy in cardioprotection. J Mol Cell Cardiol 44: 654–661, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gustafsson AB, Gottlieb RA. Autophagy in ischemic heart disease. Circ Res 104: 150–158, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hacihanefioglu A, Tarkun P, Gonullu E. Acute severe cardiac failure in a myeloma patient due to proteasome inhibitor bortezomib. Int J Hematol 88: 219–222, 2008 [DOI] [PubMed] [Google Scholar]
- 59.Hammond-Martel I, Yu H, Affar el B. Roles of ubiquitin signaling in transcription regulation. Cell Signal 24: 410–421, 2012 [DOI] [PubMed] [Google Scholar]
- 60.Hariharan N, Zhai P, Sadoshima J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid Redox Signal 14: 2179–2190, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hasselgren PO, Fischer JE. Muscle cachexia: current concepts of intracellular mechanisms and molecular regulation. Ann Surg 233: 9–17, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hayashi T, Takada K, Matsuda M. Post-transient ischemia increase in ubiquitin conjugates in the early reperfusion. Neuroreport 3: 519–520, 1992 [DOI] [PubMed] [Google Scholar]
- 63.Herrmann J, Lerman LO, Lerman A. On to the road to degradation: atherosclerosis and the proteasome. Cardiovasc Res 85: 291–302, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hershko A. Roles of ubiquitin-mediated proteolysis in cell cycle control. Curr Opin Cell Biol 9: 788–799, 1997 [DOI] [PubMed] [Google Scholar]
- 65.Hershko A, Ciechanover A, Rose IA. Resolution of the ATP-dependent proteolytic system from reticulocytes: a component that interacts with ATP. Proc Natl Acad Sci USA 76: 3107–3110, 1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hochrainer K, Jackman K, Anrather J, Iadecola C. Reperfusion rather than ischemia drives the formation of ubiquitin aggregates after middle cerebral artery occlusion. Stroke 43: 2229–2235, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Horwitz AA, Navon A, Groll M, Smith DM, Reis C, Goldberg AL. ATP-induced structural transitions in PAN, the proteasome-regulatory ATPase complex in Archaea. J Biol Chem 282: 22921–22929, 2007 [DOI] [PubMed] [Google Scholar]
- 68.Hu HY. Editorial: protein ubiquitination and deubiquitination. Curr Protein Pept Sci 13: 413, 2012 [DOI] [PubMed] [Google Scholar]
- 69.Hua Z, Vierstra RD. The cullin-RING ubiquitin-protein ligases. Annu Rev Plant Biol 62: 299–334, 2011 [DOI] [PubMed] [Google Scholar]
- 70.Huang H, Zhang X, Li S, Liu N, Lian W, McDowell E, Zhou P, Zhao C, Guo H, Zhang C, Yang C, Wen G, Dong X, Lu L, Ma N, Dong W, Dou QP, Wang X, Liu J. Physiological levels of ATP negatively regulate proteasome function. Cell Res 20: 1372–1385, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang S, Patterson E, Yu X, Garrett MW, De AI, Kem DC. Proteasome inhibition 1 h following ischemia protects GRK2 and prevents malignant ventricular tachyarrhythmias and SCD in a model of myocardial infarction. Am J Physiol Heart Circ Physiol 294: H1298–H1303, 2008 [DOI] [PubMed] [Google Scholar]
- 72.Huber EM, Basler M, Schwab R, Heinemeyer W, Kirk CJ, Groettrup M, Groll M. Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell 148: 727–738, 2012 [DOI] [PubMed] [Google Scholar]
- 73.Ichikawa HT, Conley T, Muchamuel T, Jiang J, Lee S, Owen T, Barnard J, Nevarez S, Goldman BI, Kirk CJ, Looney RJ, Anolik JH. Beneficial effect of novel proteasome inhibitors in murine lupus via dual inhibition of type I interferon and autoantibody-secreting cells. Arthritis Rheum 64: 493–503, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Inagaki K, Chen L, Ikeno F, Lee FH, Imahashi K, Bouley DM, Rezaee M, Yock PG, Murphy E, Mochly-Rosen D. Inhibition of delta-protein kinase C protects against reperfusion injury of the ischemic heart in vivo. Circulation 108: 2304–2307, 2003 [DOI] [PubMed] [Google Scholar]
- 75.Ishii T, Sakurai T, Usami H, Uchida K. Oxidative modification of proteasome: identification of an oxidation-sensitive subunit in 26 s proteasome. Biochemistry 44: 13893–13901, 2005 [DOI] [PubMed] [Google Scholar]
- 76.Jesenberger V, Jentsch S. Deadly encounter: ubiquitin meets apoptosis. Nat Rev Mol Cell Biol 3: 112–121, 2002 [DOI] [PubMed] [Google Scholar]
- 77.Kamikubo T, Hayashi T. Changes in proteasome activity following transient ischemia. Neurochem Int 28: 209–212, 1996 [DOI] [PubMed] [Google Scholar]
- 78.Kassiotis C, Ballal K, Wellnitz K, Vela D, Gong M, Salazar R, Frazier OH, Taegtmeyer H. Markers of autophagy are downregulated in failing human heart after mechanical unloading. Circulation 120: S191–S197, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Keller JN, Huang FF, Zhu H, Yu J, Ho YS, Kindy TS. Oxidative stress-associated impairment of proteasome activity during ischemia-reperfusion injury. J Cereb Blood Flow Metab 20: 1467–1473, 2000 [DOI] [PubMed] [Google Scholar]
- 80.Khaliulin I, Schwalb H, Wang P, Houminer E, Grinberg L, Katzeff H, Borman JB, Powell SR. Preconditioning improves postischemic mitochondrial function and diminishes oxidation of mitochondrial proteins. Free Radic Biol Med 37: 1–9, 2004 [DOI] [PubMed] [Google Scholar]
- 81.Kloetzel PM, Soza A, Stohwasser R. The role of the proteasome system and the proteasome activator PA28 complex in the cellular immune response. Biol Chem 380: 293–297, 1999 [DOI] [PubMed] [Google Scholar]
- 82.Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol 10: 550–563, 2009 [DOI] [PubMed] [Google Scholar]
- 83.Korge P, Honda HM, Weiss JN. Protection of cardiac mitochondria by diazoxide and protein kinase C: implications for ischemic preconditioning. Proc Natl Acad Sci USA 99: 3312–3317, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Krappmann D, Scheidereit C. A pervasive role of ubiquitin conjugation in activation and termination of IkappaB kinase pathways. EMBO Rep 6: 321–326, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kruger E, Kloetzel PM. Immunoproteasomes at the interface of innate and adaptive immune responses: two faces of one enzyme. Curr Opin Immunol 24: 77–83, 2012 [DOI] [PubMed] [Google Scholar]
- 86.Li J, Horak KM, Su H, Sanbe A, Robbins J, Wang X. Enhancement of proteasomal function protects against cardiac proteinopathy and ischemia/reperfusion injury in mice. J Clin Invest 121: 3689–3700, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li J, Powell SR, Wang X. Enhancement of proteasome function by PA28alpha; overexpression protects against oxidative stress. FASEB J 25: 883–893, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lin KI, Baraban JM, Ratan RR. Inhibition versus induction of apoptosis by proteasome inhibitors depends on concentration. Cell Death Differ 5: 577–583, 1998 [DOI] [PubMed] [Google Scholar]
- 89.Liu C, Chen S, Kamme F, Hu BR. Ischemic preconditioning prevents protein aggregation after transient cerebral ischemia. Neuroscience 134: 69–80, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lu H, Zong C, Wang Y, Young GW, Deng N, Souda P, Li X, Whitelegge J, Drews O, Yang PY, Ping P. Revealing the dynamics of the 20 S proteasome phosphoproteome: a combined CID and electron transfer dissociation approach. Mol Cell Proteomics 7: 2073–2089, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lu L, Wang H. Transient focal cerebral ischemia upregulates immunoproteasomal subunits. Cell Mol Neurobiol 32: 965–970, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Matsui Y, Kyoi S, Takagi H, Hsu CP, Hariharan N, Ago T, Vatner SF, Sadoshima J. Molecular mechanisms and physiological significance of autophagy during myocardial ischemia and reperfusion. Autophagy 4: 409–415, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McCullock S, Kinard T, McCullough L, Formosa T. blm3–1 is an allele of UBP3, a ubiquitin protease that appears to act during transcription of damaged DNA. J Mol Biol 363: 660–672, 2006 [DOI] [PubMed] [Google Scholar]
- 94.Meiners S, Heyken D, Weller A, Ludwig A, Stangl K, Kloetzel PM, Kruger E. Inhibition of proteasome activity induces concerted expression of proteasome genes and de novo formation of mammalian proteasomes. J Biol Chem 278: 21517–21525, 2003 [DOI] [PubMed] [Google Scholar]
- 95.Meiners S, Ludwig A, Lorenz M, Dreger H, Baumann G, Stangl V, Stangl K. Nontoxic proteasome inhibition activates a protective antioxidant defense response in endothelial cells. Free Radic Biol Med 40: 2232–2241, 2006 [DOI] [PubMed] [Google Scholar]
- 96.Meiners S, Ludwig A, Stangl V, Stangl K. Proteasome inhibitors: poisons and remedies. Med Res Rev 28: 309–327, 2008 [DOI] [PubMed] [Google Scholar]
- 97.Meller R, Cameron JA, Torrey DJ, Clayton CE, Ordonez AN, Henshall DC, Minami M, Schindler CK, Saugstad JA, Simon RP. Rapid degradation of Bim by the ubiquitin-proteasome pathway mediates short-term ischemic tolerance in cultured neurons. J Biol Chem 281: 7429–7436, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Moss NC, Tang RH, Willis M, Stansfield WE, Baldwin AS, Selzman CH. Inhibitory kappa B kinase-beta is a target for specific nuclear factor kappa B-mediated delayed cardioprotection. J Thorac Cardiovasc Surg 136: 1274–1279, 2008 [DOI] [PubMed] [Google Scholar]
- 99.Muchamuel T, Basler M, Aujay MA, Suzuki E, Kalim KW, Lauer C, Sylvain C, Ring ER, Shields J, Jiang J, Shwonek P, Parlati F, Demo SD, Bennett MK, Kirk CJ, Groettrup M. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat Med 15: 781–787, 2009 [DOI] [PubMed] [Google Scholar]
- 100.Murata S, Kawahara H, Tohma S, Yamamoto K, Kasahara M, Nabeshima Y, Tanaka K, Chiba T. Growth retardation in mice lacking the proteasome activator PA28gamma. J Biol Chem 274: 38211–38215, 1999 [DOI] [PubMed] [Google Scholar]
- 101.Neefjes J, Jongsma ML, Paul P, Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol 11: 823–836, 2011 [DOI] [PubMed] [Google Scholar]
- 102.Orlowski M, Wilk S. Catalytic activities of the 20 S proteasome, a multicatalytic proteinase complex. Arch Biochem Biophys 383: 1–16, 2000 [DOI] [PubMed] [Google Scholar]
- 103.Park Y, Kanekal S, Kehrer JP. Oxidative changes in hypoxic rat heart tissue. Am J Physiol Heart Circ Physiol 260: H1395–H1405, 1991 [DOI] [PubMed] [Google Scholar]
- 104.Pickering AM, Davies KJ. Differential roles of proteasome and immunoproteasome regulators Pa28alphabeta, Pa28gamma and Pa200 in the degradation of oxidized proteins. Arch Biochem Biophys 523: 181–190, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pickering AM, Koop AL, Teoh CY, Ermak G, Grune T, Davies KJ. The immunoproteasome, the 20S proteasome and the PA28alphabeta proteasome regulator are oxidative-stress-adaptive proteolytic complexes. Biochem J 432: 585–594, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Portbury AL, Ronnebaum SM, Zungu M, Patterson C, Willis MS. Back to your heart: ubiquitin proteasome system-regulated signal transduction. J Mol Cell Cardiol 52: 526–537, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Powell SR. The cardiac 26S proteasome: regulating the regulator. Circ Res 99: 342–345, 2006 [DOI] [PubMed] [Google Scholar]
- 108.Powell SR. Proteasome inhibitors in myocardial ischemia, some concerns. Ann Thorac Surg 85: 1503–1504, 2008 [DOI] [PubMed] [Google Scholar]
- 109.Powell SR, Davies KJA, Divald A. Optimal determination of heart tissue 26S Proteasome activity requires maximal stimulating concentrations of ATP. J Mol Cell Cardiol 42: 265–269, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Powell SR, Divald A. The ubiquitin-proteasome system in myocardial ischaemia and preconditioning. Cardiovasc Res 85: 303–311, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Powell SR, Gurzenda EM, Wahezi SE. Actin is oxidized during myocardial ischemia. Free Radic Biol Med 30: 1171–1176, 2001 [DOI] [PubMed] [Google Scholar]
- 112.Powell SR, Hall D. Use of salicylate as a probe for·OH formation in isolated ischemic rat hearts. Free Radic Biol Med 9: 133–141, 1990 [DOI] [PubMed] [Google Scholar]
- 113.Powell SR, Samuel SM, Wang P, Divald A, Thirunavukkarasu M, Koneru S, Wang X, Maulik N. Upregulation of myocardial 11S-activated proteasome in experimental hyperglycemia. J Mol Cell Cardiol 44: 618–621, 2008 [DOI] [PubMed] [Google Scholar]
- 114.Powell SR, Wang P, Katzeff HL, Shringarpure R, Teoh C, Khaliulin I, Das DK, Schwalb H. Oxidized and ubiquitinated proteins may predict recovery of postischemic cardiac function. Essential role of the proteasome. Antioxid Redox Signal 7: 538–535, 2005 [DOI] [PubMed] [Google Scholar]
- 115.Pradillo JM, Romera C, Hurtado O, Cardenas A, Moro MA, Leza JC, Davalos A, Castillo J, Lorenzo P, Lizasoain I. TNFR1 upregulation mediates tolerance after brain ischemic preconditioning. J Cereb Blood Flow Metab 25: 193–203, 2005 [DOI] [PubMed] [Google Scholar]
- 116.Predmore JM, Wang P, Davis F, Bartolone S, Westfall MV, Dyke DB, Pagani F, Powell SR, Day SM. Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation 21: 997–1004, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Price SR. Increased transcription of ubiquitin-proteasome system components: molecular responses associated with muscle atrophy. Int J Biochem Cell Biol 35: 617–628, 2003 [DOI] [PubMed] [Google Scholar]
- 118.Przyklenk K, Dong Y, Undyala VV, Whittaker P. Autophagy as a therapeutic target for ischaemia /reperfusion injury? Concepts, controversies, and challenges. Cardiovasc Res 94: 197–205, 2012 [DOI] [PubMed] [Google Scholar]
- 119.Przyklenk K, Undyala VV, Wider J, Sala-Mercado JA, Gottlieb RA, Mentzer RM., Jr Acute induction of autophagy as a novel strategy for cardioprotection: getting to the heart of the matter. Autophagy 7: 432–433, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pye J, Ardeshirpour F, McCain A, Bellinger DA, Merricks E, Adams J, Elliott PJ, Pien C, Fischer TH, Baldwin AS, Jr, Nichols TC. Proteasome inhibition ablates activation of NF-κB in myocardial reperfusion and reduces reperfusion injury. Am J Physiol Heart Circ Physiol 284: H919–H926, 2003 [DOI] [PubMed] [Google Scholar]
- 121.Rabl J, Smith DM, Yu Y, Chang SC, Goldberg AL, Cheng Y. Mechanism of gate opening in the 20S proteasome by the proteasomal ATPases. Mol Cell 30: 360–368, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Reinheckel T, Sitte N, Ullrich O, Kuckelkorn U, Davies KJ, Grune T. Comparative resistance of the 20S and 26S proteasome to oxidative stress. Biochem J 335: 637–642, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Reinheckel T, Ullrich O, Sitte N, Grune T. Differential impairment of 20S and 26S proteasome activities in human hematopoietic K562 cells during oxidative stress. Arch Biochem Biophys 377: 65–68, 2000 [DOI] [PubMed] [Google Scholar]
- 124.Rock KL, York IA, Saric T, Goldberg AL. Protein degradation and the generation of MHC class I-presented peptides. Adv Immunol 80: 1–70, 2002 [DOI] [PubMed] [Google Scholar]
- 125.Rodriguez JE, Schisler JC, Patterson C, Willis MS. Seek and destroy: the ubiquitin—-proteasome system in cardiac disease. Curr Hypertens Rep 11: 396–405, 2009 [DOI] [PubMed] [Google Scholar]
- 126.Rotin D, Kumar S. Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol 10: 398–409, 2009 [DOI] [PubMed] [Google Scholar]
- 127.Sala-Mercado JA, Wider J, Undyala VV, Jahania S, Yoo W, Mentzer RM, Jr, Gottlieb RA, Przyklenk K. Profound cardioprotection with chloramphenicol succinate in the swine model of myocardial ischemia-reperfusion injury. Circulation 122: S179–S184, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sarikas A, Hartmann T, Pan ZQ. The cullin protein family. Genome Biol 12: 220, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Satoh K, Sasajima H, Nyoumura KI, Yokosawa H, Sawada H. Assembly of the 26S proteasome is regulated by phosphorylation of the p45/Rpt6 ATPase subunit. Biochemistry 40: 314–319, 2001 [DOI] [PubMed] [Google Scholar]
- 130.Schulman BA. Twists and turns in ubiquitin-like protein conjugation cascades. Protein Sci 20: 1941–1954, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Schulman BA, Harper JW. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat Rev Mol Cell Biol 10: 319–331, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Scruggs SB, Zong NC, Wang D, Stefani E, Ping P. Post-translational modification of cardiac proteasomes: functional delineation enabled by proteomics. Am J Physiol Heart Circ Physiol 303: H9–H18, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Seifert U, Bialy LP, Ebstein F, Bech-Otschir D, Voigt A, Schröter F, Prozorovski T, Lange N, Steffen J, Rieger M, Kuckelkorn U, Aktas O, Kloetzel PM, Krüger E. Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell 142: 613–624, 2010 [DOI] [PubMed] [Google Scholar]
- 134.Song X, von Kampen J, Slaughter CA, DeMartino GN. Relative functions of the alpha and beta subunits of the proteasome activator, PA28. J Biol Chem 272: 27994–28000, 1997 [DOI] [PubMed] [Google Scholar]
- 135.Stadtmueller BM, Hill CP. Proteasome activators. Mol Cell 41: 8–19, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Stangl K, Gunther C, Frank T, Lorenz M, Meiners S, Ropke T, Stelter L, Moobed M, Baumann G, Kloetzel PM, Stangl V. Inhibition of the ubiquitin-proteasome pathway induces differential heat-shock protein response in cardiomyocytes and renders early cardiac protection. Biochem Biophys Res Commun 291: 542–549, 2002 [DOI] [PubMed] [Google Scholar]
- 137.Stanley WC. Myocardial energy metabolism during ischemia and the mechanisms of metabolic therapies. J Cardiovasc Pharmacol Ther 9, Suppl 1: S31–S45, 2004 [DOI] [PubMed] [Google Scholar]
- 138.Stohwasser R, Salzmann U, Giesebrecht J, Kloetzel PM, Holzhutter HG. Kinetic evidences for facilitation of peptide channelling by the proteasome activator PA28. Eur J Biochem 267: 6221–6230, 2000 [DOI] [PubMed] [Google Scholar]
- 139.Su H, Wang X. The ubiquitin-proteasome system in cardiac proteinopathy: a quality control perspective. Cardiovasc Res 85: 253–262, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Su H, Wang X. p62 Stages an interplay between the ubiquitin-proteasome system and autophagy in the heart of defense against proteotoxic stress. Trends Cardiovasc Med 21: 224–228, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sucharov CC. Beta-adrenergic pathways in human heart failure. Expert Rev Cardiovasc Ther 5: 119–124, 2007 [DOI] [PubMed] [Google Scholar]
- 142.Takagi H, Hsu CP, Kajimoto K, Shao D, Yang Y, Maejima Y, Zhai P, Yehia G, Yamada C, Zablocki D, Sadoshima J. Activation of PKN mediates survival of cardiac myocytes in the heart during ischemia/reperfusion. Circ Res 107: 642–649, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Takagi H, Matsui Y, Sadoshima J. The role of autophagy in mediating cell survival and death during ischemia and reperfusion in the heart. Antioxid Redox Signal 9: 1373–1381, 2007 [DOI] [PubMed] [Google Scholar]
- 144.Takamatsu H, Yamashita T, Kotani T, Sawazaki A, Okumura H, Nakao S. Ischemic heart disease associated with bortezomib treatment combined with dexamethasone in a patient with multiple myeloma. Int J Hematol 91: 903–906, 2010 [DOI] [PubMed] [Google Scholar]
- 145.Taylor RG, Tassy C, Briand M, Robert N, Briand Y, Ouali A. Proteolytic activity of proteasome on myofibrillar structures. Mol Biol Rep 21: 71–73, 1995 [DOI] [PubMed] [Google Scholar]
- 146.Tian Z, Zheng H, Li J, Li YF, Su H, Wang X. Genetically induced moderate inhibition of the proteasome in cardiomyocytes exacerbates myocardial ischemia-teperfusion injury in mice. Circ Res 111: 532–542, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tisdale MJ. Pathogenesis of cancer cachexia. J Support Oncol 1: 159–168, 2003 [PubMed] [Google Scholar]
- 148.Um JW, Im E, Park J, Oh Y, Min B, Lee HJ, Yoon JB, Chung KC. ASK1 negatively regulates the 26 S proteasome. J Biol Chem 285: 36434–36446, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ustrell V, Hoffman L, Pratt G, Rechsteiner M. PA200, a nuclear proteasome activator involved in DNA repair. EMBO J 21: 3516–3525, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.van Wijk SJ, Timmers HT. The family of ubiquitin-conjugating enzymes (E2s): deciding between life and death of proteins. FASEB J 24: 981–993, 2010 [DOI] [PubMed] [Google Scholar]
- 151.Versari D, Herrmann J, Gossl M, Mannheim D, Sattler K, Meyer FB, Lerman LO, Lerman A. Dysregulation of the ubiquitin-proteasome system in human carotid atherosclerosis. Arterioscler Thromb Vasc Biol 26: 2132–2139, 2006 [DOI] [PubMed] [Google Scholar]
- 152.Visekruna A, Joeris T, Seidel D, Kroesen A, Loddenkemper C, Zeitz M, Kaufmann SH, Schmidt-Ullrich R, Steinhoff U. Proteasome-mediated degradation of IkappaBalpha and processing of p105 in Crohn disease and ulcerative colitis. J Clin Invest 116: 3195–3203, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Voortman J, Giaccone G. Severe reversible cardiac failure after bortezomib treatment combined with chemotherapy in a non-small cell lung cancer patient: a case report. BMC Cancer 6: 129, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Vucic D, Dixit VM, Wertz IE. Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nat Rev Mol Cell Biol 12: 439–452, 2011 [DOI] [PubMed] [Google Scholar]
- 155.Wang D, Zong C, Koag MC, Wang Y, Drews O, Fang C, Scruggs SB, Ping P. Proteome dynamics and proteome function of cardiac 19S proteasomes. Mol Cell Proteomics 10: M110, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wang X, Li J, Zheng H, Su H, Powell SR. Proteasome functional insufficiency in cardiac pathogenesis. Am J Physiol Heart Circ Physiol 301: H2207–H2219, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wang X, Robbins J. Heart failure and protein quality control. Circ Res 99: 1315–1328, 2006 [DOI] [PubMed] [Google Scholar]
- 158.Wenzel DM, Stoll KE, Klevit RE. E2s: structurally economical and functionally replete. Biochem J 433: 31–42, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Whitby FG, Masters EI, Kramer L, Knowlton JR, Yao Y, Wang CC, Hill CP. Structural basis for the activation of 20S proteasomes by 11S regulators. Nature 408: 115–120, 2000 [DOI] [PubMed] [Google Scholar]
- 160.Williams AJ, Berti R, Dave JR, Elliot PJ, Adams J, Tortella FC. Delayed treatment of ischemia/reperfusion brain injury: extended therapeutic window with the proteosome inhibitor MLN519. Stroke 35: 1186–1191, 2004 [DOI] [PubMed] [Google Scholar]
- 161.Williams AJ, Hale SL, Moffett JR, Dave JR, Elliott PJ, Adams J, Tortella FC. Delayed treatment with MLN519 reduces infarction and associated neurologic deficit caused by focal ischemic brain injury in rats via antiinflammatory mechanisms involving nuclear factor-kappaB activation, gliosis, and leukocyte infiltration. J Cereb Blood Flow Metab 23: 75–87, 2003 [DOI] [PubMed] [Google Scholar]
- 162.Williams AJ, Myers TM, Cohn SI, Sharrow KM, Lu XC, Tortella FC. Recovery from ischemic brain injury in the rat following a 10 h delayed injection with MLN519. Pharmacol Biochem Behav 81: 182–189, 2005 [DOI] [PubMed] [Google Scholar]
- 163.Willis MS, Schisler JC, Portbury AL, Patterson C. Build it up-tear it down: protein quality control in the cardiac sarcomere. Cardiovasc Res 81: 439–448, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Willis MS, Townley-Tilson WH, Kang EY, Homeister JW, Patterson C. Sent to destroy: the ubiquitin proteasome system regulates cell signaling and protein quality control in cardiovascular development and disease. Circ Res 106: 463–478, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yan L, Sadoshima J, Vatner DE, Vatner SF. Autophagy in ischemic preconditioning and hibernating myocardium. Autophagy 5: 709–712, 2009 [DOI] [PubMed] [Google Scholar]
- 166.Young GW, Wang Y, Ping P. Understanding proteasome assembly and regulation: importance to cardiovascular medicine. Trends Cardiovasc Med 18: 93–98, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Young LH, Li J, Baron SJ, Russell RR. AMP-activated protein kinase: a key stress signaling pathway in the heart. Trends Cardiovasc Med 15: 110–118, 2005 [DOI] [PubMed] [Google Scholar]
- 168.Yu X, Huang S, Patterson E, Garrett MW, Kaufman KM, Metcalf JP, Zhu M, Dunn ST, Kem DC. Proteasome degradation of GRK2 during ischemia and ventricular tachyarrhythmias in a canine model of myocardial infarction. Am J Physiol Heart Circ Physiol 289: H1960–H1967, 2005 [DOI] [PubMed] [Google Scholar]
- 169.Zhang F, Hu Y, Huang P, Toleman CA, Paterson AJ, Kudlow JE. Proteasome function is regulated by cyclic AMP-dependent protein kinase through phosphorylation of Rpt6. J Biol Chem 282: 22460–22471, 2007 [DOI] [PubMed] [Google Scholar]
- 170.Zhang Z, Krutchinsky A, Endicott S, Realini C, Rechsteiner M, Standing KG. Proteasome activator 11S REG or PA28: recombinant REG alpha/REG beta hetero-oligomers are heptamers. Biochemistry 38: 5651–5658, 1999 [DOI] [PubMed] [Google Scholar]
- 171.Zheng Q, Su H, Ranek MJ, Wang X. Autophagy and p62 in cardiac proteinopathy. Circ Res 109: 296–308, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Zheng Q, Su H, Tian Z, Wang X. Proteasome malfunction activates macroautophagy in the heart. Am J Cardiovasc Dis 1: 214–226, 2011 [PMC free article] [PubMed] [Google Scholar]
- 173.Zheng Q, Wang X. Autophagy and the ubiquitin-proteasome system in cardiac dysfunction. Panminerva Med 52: 9–25, 2010 [PMC free article] [PubMed] [Google Scholar]
- 174.Zhou HJ, Aujay MA, Bennett MK, Dajee M, Demo SD, Fang Y, Ho MN, Jiang J, Kirk CJ, Laidig GJ, Lewis ER, Lu Y, Muchamuel T, Parlati F, Ring E, Shenk KD, Shields J, Shwonek PJ, Stanton T, Sun CM, Sylvain C, Woo TM, Yang J. Design and synthesis of an orally bioavailable and selective peptide epoxyketone proteasome inhibitor (PR-047). J Med Chem 52: 3028–3038, 2009 [DOI] [PubMed] [Google Scholar]
- 175.Zimmerman ES, Schulman BA, Zheng N. Structural assembly of cullin-RING ubiquitin ligase complexes. Curr Opin Struct Biol 20: 714–721, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zong C, Gomes AV, Drews O, Li X, Young GW, Berhane B, Qiao X, French SW, Bardag-Gorce F, Ping P. Regulation of murine cardiac 20S proteasomes: role of associating partners. Circ Res 99: 372–380, 2006 [DOI] [PubMed] [Google Scholar]
- 177.Zong C, Young GW, Wang Y, Lu H, Deng N, Drews O, Ping P. Two-dimensional electrophoresis-based characterization of post-translational modifications of mammalian 20S proteasome complexes. Proteomics 8: 5025–5037, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]