Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants (original) (raw)
Significance
Using high-throughput experiments, we determined the functional epigenomic landscape in pancreatic islet cells. Computational integration of these data along with similar data from the ENCODE project revealed the presence of large gene control elements across diverse cell types that we refer to as “stretch enhancers.” Stretch enhancers are cell type specific and are associated with increased expression of genes involved in cell-specific processes. We find that genetic variations associated with common disease are highly enriched in stretch enhancers; notably, stretch enhancers specific to pancreatic islets harbor variants linked to type 2 diabetes and related traits. We propose that stretch enhancers form as pluripotent cells differentiate into committed lineages, to program important cell-specific gene expression.
Abstract
Chromatin-based functional genomic analyses and genomewide association studies (GWASs) together implicate enhancers as critical elements influencing gene expression and risk for common diseases. Here, we performed systematic chromatin and transcriptome profiling in human pancreatic islets. Integrated analysis of islet data with those from nine cell types identified specific and significant enrichment of type 2 diabetes and related quantitative trait GWAS variants in islet enhancers. Our integrated chromatin maps reveal that most enhancers are short (median = 0.8 kb). Each cell type also contains a substantial number of more extended (≥3 kb) enhancers. Interestingly, these stretch enhancers are often tissue-specific and overlap locus control regions, suggesting that they are important chromatin regulatory beacons. Indeed, we show that (i) tissue specificity of enhancers and nearby gene expression increase with enhancer length; (ii) neighborhoods containing stretch enhancers are enriched for important cell type–specific genes; and (iii) GWAS variants associated with traits relevant to a particular cell type are more enriched in stretch enhancers compared with short enhancers. Reporter constructs containing stretch enhancer sequences exhibited tissue-specific activity in cell culture experiments and in transgenic mice. These results suggest that stretch enhancers are critical chromatin elements for coordinating cell type–specific regulatory programs and that sequence variation in stretch enhancers affects risk of major common human diseases.
High-throughput sequencing has been coupled to ChIP (ChIP-seq) and mRNA samples (RNA-seq) to survey the genomewide chromatin and transcription profiles in different cell types. Regulatory elements such as promoters, enhancers, insulators, transcribed, and repressed regions are marked by distinct patterns of histone modifications (1), including histone H3 lysine 27 acetylation (H3K27ac), H3K27 trimethylation (H3K27me3), H3K36me3, H3K4 monomethylation (H3K4me1), H3K4me3, and the CCCTC-binding factor (CTCF). Systematic chromatin state identification has recently emerged as a powerful technique to interpret and compare regulatory landscapes within and between cell types (2–7). Such methods use an unsupervised approach to identify recurrent combinations of histone modifications across the genome, thereby producing a map of representative chromatin states that are likely to be biologically relevant.
Results
Systematic Chromatin and Transcriptome Profiling in Human Islets.
To correlate chromatin features with the location of type 2 diabetes (T2D) genetic risk variants and with gene expression, we conducted high-throughput sequencing coupled to ChIP (ChIP-seq) and mRNA samples (RNA-seq) in human pancreatic islets, a cell type relevant to diabetes and to quantitative trait analysis of glucose and insulin levels (8). Using the ChromHMM algorithm (2), we uniformly integrated our islet ChIP-seq reads plus additional islet data sets (9) with those from nine Encyclopedia of DNA Elements (ENCODE) cell types to generate consistent chromatin state assignments across all 10 cell types. We anchored these assignments based on overlap with previously published chromatin states (2) in the nine ENCODE cell types to produce a consistent annotation of promoter, enhancer, insulator, transcribed, and repressed chromatin states (SI Appendix, Fig. S1). In parallel, we integrated our human islet RNA-seq data with ENCODE RNA-seq data, resulting in a unified set of chromatin state and mRNA maps for islets and the nine ENCODE cell types (Fig. 1_A_). After subsampling to normalize the amount of ChIP-seq reads, the fraction of the genome covered by select chromatin states remained relatively constant across any given cell type (Fig. 1_B_, Upper). However, we observed that additional read depth identified additional signal-enriched enhancer regions (SI Appendix, Fig. S2), a finding consistent with other studies (10, 11). Thus, in subsequent analyses, we used chromatin states identified using all reads (Fig. 1_B_, Lower) and note that the trends reported herein are consistently observed even when normalized read chromatin states are used. As shown in Fig. 1_A_, our integrative approach identified both common (e.g., POLD2 and YKT6) and cell-specific (e.g., GCK) chromatin state and expression patterns (Dataset S1).
Fig. 1.
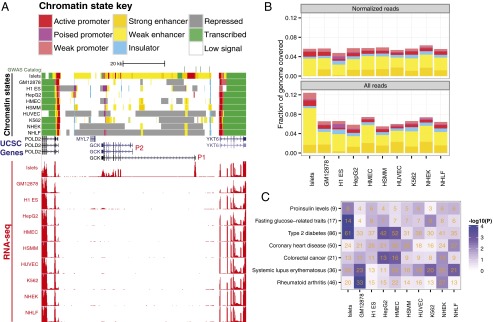
Systematic and simultaneous analysis of chromatin states and gene expression in human pancreatic islets and nine ENCODE cell types. (A) Chromatin states in and around the GCK locus. Human pancreatic islet chromatin states are similar to nine ENCODE cell types at commonly expressed flanking genes (POLD2 and YKT6) and unique at islet-specific expressed genes (GCK). (Upper) ChromHMM-defined chromatin states for each of 10 human cell types (islets; GM128278, lymphoblastoid cells; H1 ES, embryonic stem cells; HepG2, hepatocellular carcinoma; HMEC, mammary epithelial cells; HSMM, smooth muscle myoblasts; HUVEC, umbilical vein endothelial cells, K562, erythroleukemia cells, NHEK, keratinocytes; NHLF, lung fibroblasts). Chromatin state assignments are indicated in the key. (Lower) RNA-seq–based expression for each cell type is indicated in red and is measured in reads per million mapped reads (RPM) per base pair. Scale is from 0 to 2 for each cell type. Note the specific use of and expression from the β cell–specific upstream promoter of GCK (P1; red). SNPs associated with T2D and related quantitative traits are indicated in green in the GWAS catalog track and reside in our unique islet enhancers. All processed results are browsable and downloadable at http://research.nhgri.nih.gov/manuscripts/Collins/islet_chromatin/. (B) Chromatin state coverage is similar across cell types. Fraction of genome covered by each chromatin state is plotted. State assignment colors are as in A. Read depths were slightly higher for the islets, which were sequenced for comparison with the ENCODE data. (C) Islet enhancers show significant enrichment of GWAS SNPs for T2D and related quantitative traits. Positions of index and tightly linked (_r_2 ≥ 0.8) SNPs for different diseases or traits (y axis) were overlapped with those of enhancer states for each cell type (x axis). The number of SNP loci overlapping enhancer states in each cell type is indicated in orange. Blue shading indicates the significance of SNP locus enrichment relative to a null distribution (Materials and Methods). Notably, our analysis reproduced enrichment of lupus and rheumatoid arthritis SNPs in lymphoblastoid cell line enhancers and colorectal cancer SNPs in hepatocellular carcinoma cell line enhancers (2). The total number of GWAS loci for each trait is indicated in parentheses on the y axis.
Our 10 cell type chromatin state maps replicated previous disease- and trait-associated SNP enrichment in cell-specific enhancer states (2, 7), including rheumatoid arthritis in GM12878 and colorectal cancer in HepG2 (Fig. 1_C_). We discovered significant enrichment of T2D and related quantitative trait associated SNPs (glucose and proinsulin) in our newly identified islet enhancers (Fig. 1_C_). These results provide a basis for further understanding of the functional effects of common SNPs in common diseases. Interestingly, several linked SNPs (_r_2 ≥ 0.8) in an associated locus overlap enhancers more often than expected at random (SI Appendix, Figs. S3 and S4), hinting that multiple SNPs in enhancers may collude to alter gene expression and contribute to disease susceptibility.
Stretch Enhancer Links to Cell-Specific Transcriptional Programs.
Inspection of islet chromatin states revealed an interesting feature: large, islet-specific enhancer domains were observed both within and near genes critical for islet function, such as GCK, KCNJ11/ABCC8, and INS (Fig. 1_A_; SI Appendix, Fig. S5). To determine whether this was a common feature, we investigated the length distribution of enhancer chromatin states (Fig. 2_A_). As expected, the majority of enhancers are small, with a median size of 0.8 kb. However, large enhancer states, although less common across the genome, occur more frequently than expected at random (Fig. 2_A_, Inset). Because of their exceptional length (top 10% of distribution), we refer to enhancer states ≥3 kb as stretch enhancers. These regions contain contiguous segments marked as enhancer states by ChromHMM, where there are no gaps in signal greater than or equal to 3 kb. Stretch enhancers were observed in all cell types (SI Appendix, Fig. S6), but their locations are different in and specific to each distinct cell type.
Fig. 2.
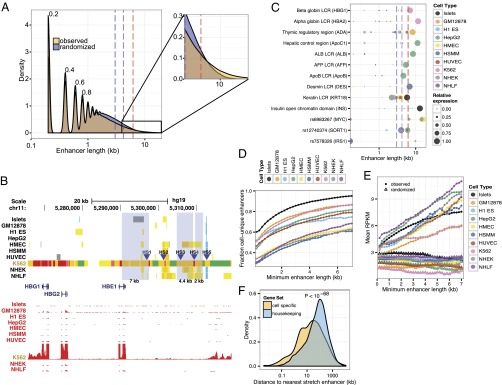
Stretch enhancer properties. (A) Enhancer states exhibit a range of length distribution. Density plot of observed (yellow) and random (blue) distribution of enhancer state lengths for all cell types combined. Distribution for each cell type is shown in SI Appendix, Fig. S6. Periodicity in the plot is a function of the 200-bp window of the ChromHMM algorithm. (Inset) Small but substantial enrichment for large enhancer states at the tail of the distribution. Enhancer sizes for the 90th (blue dashed line), 95th (purple dashed line), and 99th (red dashed line) percentile of the random distribution are indicated for reference. (B) Human β globin LCR contains multiple stretch enhancers. Chromatin states (Upper) and expression profiles (RNA-seq, red) near the β globin LCR show K562-specific chromatin states and robust HBG1/2 and HBE1 expression. Hypersensitive sites (HS) 1–3 and 5 (blue arrowheads) reside in K562-specific enhancer states (orange/yellow), two of which qualify as stretch enhancers. Chromatin states are color-coded as in Fig. 1_A_. (C) Celebrity enhancer regions overlap stretch enhancers. LCRs (15) and the INS/TH/IGF2 open chromatin domain (33) are contained in the top 10% of enhancer state size in relevant cell types (e.g., hepatic control region in hepatocellular carcinoma, thymic regulatory region in lymphoblastoid cell lines). Recently reported GWAS enhancer regions (18–21) also overlap stretch enhancers (see lower three rows). Colors of circles on the plot represent different cell types. Circle diameters indicate RNA-seq expression levels of the target gene relative to levels in the highest cell type, as indicated in the key. Dashed lines are the same as in A. (D) Cell type specificity of enhancer increases with length. Fraction of enhancers unique to a cell type is plotted against increasing enhancer length. (E) Nearby gene expression increases with enhancer length. Median RNA-seq expression (RPKM) of genes within 125 kb of enhancer states is plotted against increasing enhancer length. Filled circles denote observed mean expression. Empty triangles indicate mean expression levels from randomly assigned genes. (F) Cell-specific genes are close to stretch enhancers. The distance of cell-specific or housekeeping genes (Materials and Methods) to stretch enhancers in each cell type was measured and indicates that cell-specific genes are significantly closer (P < 10−68; Wilcoxon rank sum test) compared with housekeeping genes.
We hypothesized that stretch enhancer chromatin states mark regulatory regions that govern robust, cell type–specific expression patterns. Locus control regions (LCRs) are classic examples of complex, cell type–specific regulatory regions. As shown in Fig. 2_B_, the β globin LCR (12, 13) is marked by multiple stretch enhancers, and the target gene HBG1 (13) is highly expressed specifically in the relevant K562 cell type. We extended this analysis to additional LCRs (14) for which 1 of the 10 cell types was an appropriate surrogate. Fig. 2_C_ demonstrates that additional LCR regions and other known enhancer domains that dictate strong and specific gene expression patterns are resoundingly marked as stretch enhancer states in the relevant cell types exhibiting robust target gene expression (specific locus views found in SI Appendix, Fig. S7, A–K and viewable at our interactive browser session http://research.nhgri.nih.gov/manuscripts/Collins/islet_chromatin/).
LCR analysis provided evidence that stretch enhancers overlap highly important and complex cell type–specific regulatory regions. We investigated, more generally, whether stretch enhancers are (i) more cell type specific than smaller enhancers and (ii) associated with robust expression of nearby genes. Indeed, cell type specificity of enhancers increases with length (Fig. 2_D_), as does expression of nearby genes (Fig. 2_E_; SI Appendix, Fig. S8). Furthermore, genes with cell-specific expression patterns are located significantly closer to stretch enhancers (P < 10−68; Wilcoxon rank sum test) compared with housekeeping genes (Fig. 2_F_; SI Appendix, Fig. S9). Longer enhancers tend to be more significantly enriched for overlapping insulator states in other cell types (SI Appendix, Fig. S10), suggesting that CTCF, the predominant DNA binding protein factor that distinguishes ChromHMM insulator states (SI Appendix, Fig. S1), can be co-opted in a cell type–specific fashion to expose important regulatory sequences in stretch enhancers.
The cohesin complex mediates inter- and intrachromosome interactions (15). A CTCF-independent role for cohesin in mediating enhancer-promoter interactions has been described (16). Additionally, dense clusters of transcription factor binding events were found to co-occur with cohesin binding (17). To test whether CTCF-independent cohesin binding sites are linked to stretch enhancers, we used ENCODE data to investigate the genomewide binding sites of the cohesin complex subunit RAD21 and compared this with our enhancer maps. We found that stretch enhancers, and not normal size enhancers, are highly enriched to overlap RAD21 binding sites in a cell-specific manner (SI Appendix, Fig. S11). Notably, H1 embryonic stem (H1 ES) cell enhancers are not enriched to overlap RAD21 regions (SI Appendix, Fig. S11), providing support for the concept that stretch enhancers are a mark of differentiated cell types.
Additionally, we demonstrate that three of three SNPs previously shown to alter long-range enhancer function [rs12740374/_SORT1_ (18) and rs6983267/MYC (19, 20)] or to reside in a long-range contact point [rs7578326/_IRS1_ (21)] overlap a stretch enhancer specifically in the relevant cell type(s) (SI Appendix, Fig. S7 I–K). We identified thousands of stretch enhancers across 10 cell types (SI Appendix, Table S1). Together, these results support the model that stretch enhancers are important chromatin-based regulatory regions in the genome that (i) direct cell-specific gene expression programs important to specialized cell type functions and (ii) may have their function altered by genetic variants, contributing to human disease.
To further investigate this model, we linked enhancers to nearby genes (Materials and Methods) and performed gene ontology (GO) term enrichment analyses (22), blinded to the identity of the gene expression patterns. As predicted, representative cell type specific GO terms are specifically and progressively enriched in relevant cell types as a function of enhancer length (Fig. 3_A_). For example, the GO term “regulation of B-cell proliferation” is specifically, significantly, and progressively enriched in genes nearby GM12878 stretch enhancers (Fig. 3_A_, Left). Similar trends are observed for terms “regulation of insulin secretion” in islets (Fig. 3_A_, Center) and “lipid localization” in HepG2 cells (Fig. 3_A_, Right). The GO corpus is hierarchically organized such that more specific terms near the bottom have higher information content than general terms near the top (23) (SI Appendix, Fig. S12). Accordingly, we use information content as a measure of GO term specificity. To generalize our GO term enrichment approach beyond the three examples above, we reasoned that if stretch enhancers encode more specific biological processes, they should be linked to higher specificity GO terms. Indeed, we observe a systematic and significant trend that genes near large enhancers are associated with higher specificity GO terms (Fig. 3 B and C; SI Appendix, Fig. S13), showing that specific biological processes (i.e., functions) correspond to stretch enhancer regions.
Fig. 3.

Stretch enhancers are linked to more specific GO terms. (A) Genes with cell type–specific functions occur near stretch enhancers. Cell type–specific GO terms exhibit progressive enrichment in relevant cell types with increasing enhancer length. Examples include regulation of B-cell proliferation in GM12878 (Left), regulation of insulin secretion in islets (Center), and lipid localization in HepG2 (Right). Each line color represents a different cell line as indicated in the key to Fig. 2. Size of the circle for each cell type indicates the statistical significance (Bonferroni-corrected P value; hypergeometric test) of GO term enrichment. (B) Mean term specificity of the top 10 enriched GO terms for each cell type at different minimum enhancer length thresholds. Note that specificity increases with enhancer length. (C) Term specificity of the top 10 enriched GO terms normalized to the mean term specificity of GO terms enriched in shuffled enhancers. For each cell type, we shuffled the genomic coordinates of enhancers 100 times along the same chromosome. For each shuffle, we assigned enhancers to nearby genes and calculated enriched GO terms. We computed the mean information content for each cell type by averaging the information content of the top 10 GO terms for each shuffle and enhancer size. Note that compared with random expectation, the increase of term specificity with enhancer length is even more pronounced (compare with B).
Stretch Enhancers Are Linked to Common Human Diseases.
Motivated by the enrichment of specific biological processes in stretch enhancers (Fig. 3; SI Appendix, Fig. S13) and cell type–specific enrichment of genomewide association study (GWAS) SNPs (Fig. 1_C_), we tested whether GWAS SNPs have a higher level of enrichment in stretch enhancers. Specifically, we asked if the observed cell type–specific enhancer SNP enrichments are disproportionately driven by stretch enhancers. Indeed, enhancer length-informed analysis uncovered such a trend (Fig. 4_A_; Materials and Methods). For example, rheumatoid arthritis GWAS SNPs are progressively more enriched in longer enhancers—specifically those of GM12878 (Fig. 4_A_). Enrichment was also observed for fasting glucose GWAS SNPs in islets (Fig. 4_B_). Specific examples of stretch enhancers in GM12878 associated with rheumatoid arthritis (Fig. 4_C_) and in islets associated with fasting glucose and T2D (Fig. 4_D_) demonstrate the large cell-specific enhancer landscape in these regions. Together, these results show that GWAS SNPs are enriched in stretch enhancers identified in cell types relevant to the associated disease or trait and suggest a specific and powerful biological role for these large enhancer elements in normal and disease states.
Fig. 4.

GWAS SNP enhancer enrichment signal is more pronounced in stretch enhancers. (A) Rheumatoid arthritis GWAS loci are progressively and specifically enriched in GM12878 stretch enhancers. Enrichment and significance is calculated using a permutation test (Materials and Methods). (B) Fasting glucose–related traits GWAS loci are progressively and specifically enriched in islet stretch enhancers. (C) Example of a rheumatoid arthritis GWAS SNP (rs615672) that overlaps a GM12878 stretch enhancer. (D) Example of a fasting glucose–related traits GWAS SNP (rs11071657) and a T2D GWAS SNP (rs7172432) that overlap islet stretch enhancers.
Functional Analysis of Stretch Enhancer DNA Sequences.
Finally, we sought to test whether chromatin-defined tissue-specific stretch enhancers demarcate important tissue-specific sequence elements. All enhancers were partitioned into different classes based on activity signatures across cell types (Materials and Methods). K-means clustering showed that 20 clusters were sufficient to partition the enhancers and identified cell type–specific groups for each of the 10 cell types considered (Fig. 5_A_). For example, enhancer cluster 17 is islet specific, whereas cluster 19 is K562 specific. Notably, these cell type–specific enhancer clusters are enriched for stretch enhancers (SI Appendix, Fig. S14). Although islet cluster 17 covers more genomic territory than other clusters (SI Appendix, Fig. S15), we surmise it is reflective of our increased sequencing depth and the mixture of several constituent cell types (as opposed to homogenous cell lines). Our analysis of these cell type–specific enhancer clusters shows GWAS SNP enrichment (SI Appendix, Fig. S16), supporting the concept that disease susceptibility is at least partially mediated by variation in cell type–specific enhancers (2, 6, 7, 24–26).
Fig. 5.

Functional analysis of stretch enhancers. (A) K-means clustering all enhancers based on activity level (Materials and Methods) reveals 20 different enhancer clusters (y axis) of differing cell type specificity (x axis). Intensity of shading represents activity level. (B) Islet-specific enhancer cluster 17 sequences have significantly different enhancer activity compared with K562-specific enhancer cluster 19 sequences in relevant cell types. Significance is calculated using a Wilcoxon rank sum test. Relative luciferase activity is shown (Materials and Methods) and expressed in arbitrary units (a.u.). (C–F) Intragenic (C and D) and intergenic (E and F) human islet stretch enhancer sequences confer specific lacZ transgene expression in the pancreatic primordium of e11.5 mouse embryos. Whole mount (C and E) and histological analysis (D and F) of transgenic embryos expressing hsp-68 lacZ under the control of stretch enhancer sequences. Arrowhead indicates the specific, reproducible expression pattern observed. Numbers indicate the fraction of embryos exhibiting this pattern. (Scale bars in D and F, 100 μm.) dp, vp, dorsal or ventral pancreatic buds; st, stomach; li, liver; mg, midgut.
To determine whether DNA sequences underlying chromatin-defined stretch enhancers are sufficient to confer cell type–specific enhancer activity, we randomly selected and cloned islet-specific cluster 17 (n = 20) and K562-specific cluster 19 (n = 22) stretch enhancer sequences located at variable distances from the transcription start site (TSS; details in SI Appendix, Table S4). Transient transfection of luciferase reporter plasmids containing these sequences into MIN6 mouse insulinoma (pancreatic islet surrogate) and K562 cell lines confirmed that sequences underlying stretch enhancer chromatin states conveyed enhancer activity (Fig. 5_B_; SI Appendix, Fig. S17). Specifically, islet (cluster 17) sequences exhibited significantly higher enhancer activity in MIN6 compared with K562 (cluster 19) sequences of similar enhancer lengths (Fig. 5_B_; P = 0.008). In contrast, K562 (cluster 19) sequences show significantly higher activity in K562 (Fig. 5_B_; P = 0.004).
To determine whether islet stretch enhancer DNA sequences can direct tissue-specific expression patterns in a spatial and temporal manner in vivo, we tested sequences from intragenic (SI Appendix, Fig. S18; ABCC8) and intergenic (SI Appendix, Fig. S19; >164 kb from the nearest gene) islet-specific (cluster 17) stretch enhancers in transgenic mice. Both the intragenic (Fig. 5 C and D) and intergenic (Fig. 5 E and F) sequences conferred specific and reproducible pancreatic primordium expression patterns to a minimal promoter hsp-68-lacZ reporter transgene in stage e11.5 mouse embryos. Taken together, the in vitro luciferase and in vivo mouse reporter data show that DNA sequences underlying cell type–specific chromatin-defined stretch enhancers function as cell-specific transcriptional enhancers and are able to confer spatial and temporal gene expression cues in an intact organism.
Discussion
In this study, we created unified chromatin state (ChIP-seq) and transcriptome (RNA-seq) maps in human islets and nine ENCODE cell types and identified specific and robust enrichment of T2D, glucose, and insulin GWAS study SNPs in islet enhancer chromatin states (Fig. 1 A and B). We discovered that enhancer chromatin states exhibit a broad size distribution in all 10 cell types analyzed (Fig. 2_A_). As illustrated by their occurrence in and around LCRs (Fig. 2 B and C), we propose that stretch enhancers are critical chromatin elements for coordinating cell type–specific regulatory programs. Based on the enrichment of GWAS SNPs, we posit that sequence variation in stretch enhancers affects risk of major common human diseases.
As the journey from pluripotency to terminal cell types traverses a series of commitments, bivalent promoters resolve to a monovalent active or repressed status (27, 28). The NIH Epigenome Roadmap recently determined that large heterochromatin domains are established to restrict committed cells from performing off-target functions (28). Correspondingly, we note that H1 ES cells contain fewer stretch enhancers (SI Appendix, Table S1 and Fig. S6) and that they occur further away from cell-specific genes compared with other cell types (SI Appendix, Fig. S9), suggesting that lineage commitment may also be accompanied by increases in number and size of stretch enhancers nearer to cell-specific genes.
We propose that stretch enhancers may serve as molecular runways or beacons to focus activity and attract tissue-specific transcription factors necessary to assemble productive transcriptional activation complexes. In support of the molecular runway hypothesis, the broad and high H3K27ac signal observed at stretch enhancer regions would serve to neutralize the positive charge on the histone lysine residues of multiple nucleosomes, thereby weakening their interaction with the negatively charged DNA backbone. This relaxation could result in DNA regions that are more exposed to transcription factors. Likewise, transcription factors have intrinsically disordered positively charged tails that enable efficient target site searching (29). Thus, the lengthening of enhancers could serve as a molecular runway that facilitates effective localization of transcription factors in the 3D nuclear compartment. More specifically, stretch enhancers could function as a nuclear beacon that attracts transcription factors, which can then scan the underlying DNA for a target binding sequence. Notably, this hypothesis does not distinguish whether stretch enhancers are contiguous concatenations of multiple independent regulatory sequences or one large functional unit.
While this manuscript was in preparation, two reports (30, 31) described the presence of large enhancers in mouse cells and human cancer cell lines associated with highly expressed cell identity genes and densely occupied by the Mediator coactivator complex. The human data reported here expand on and complement those data with direct evidence for the presence of chromatin-defined stretch enhancers in multiple human cell types, demonstrate colocalization of stretch enhancers with human disease risk variants, and reveal tissue-specific function of stretch enhancers in expression vector experiments.
Continued efforts to identify stretch enhancers in additional cell types will be an important objective. Because these enhancers exhibit high cell type specificity, we propose they represent important regulatory features essential for understanding cellular functions, disease susceptibility, and potential opportunities for therapeutic intervention (32).
Materials and Methods
A detailed description of experimental and computational analyses is provided in the SI Appendix. Briefly, we conducted ChIP-seq and RNA-seq in human islets and integrated these data with other cell types. Function of stretch enhancer sequences were investigated using luciferase reporter assays in the MIN6 and K562 cell lines or lacZ transgenic experiments in mouse embryos.
Supplementary Material
Supporting Information
Acknowledgments
We thank the Finland United States Investigation of NIDDM Genetics Consortium and National Institutes of Health (NIH) Chromatin community for valuable critiques and discussion. We thank Lori Bonnycastle, Laura Elnitski, Andrea Ramirez, Mario Morken, and Adam Woolfe for helpful comments. Human islets were obtained from organ donors through the ICR Basic Science Islet Distribution Program (Universities of Illinois, Washington, Miami, and Alabama at Birmingham) and the National Disease Research Interchange. We thank the reviewers for excellent feedback and suggestions. This work was supported by Postdoctoral Research Associate (PRAT) fellowship (to S.C.J.P.); Grants 1ZIAHG000024 (to F.S.C.), K99DK092251 (to M.L.S.), K99DK099240 (to S.C.J.P.), R01HL64658 (to B.L.B.), and R01DE019118 (to B.L.B.). A.V. and L.A.P. are supported by Grant R01HG003988 and conducted research at the E.O. Lawrence Berkeley National Laboratory, performed under Department of Energy Contract DEAC02-05CH11231, University of California.
Footnotes
The authors declare no conflict of interest.
A complete list of the NISC Comparative Sequencing Program can be found in the Supporting Information.
Data deposition: The sequence reported in this paper has been deposited in the Gene Expression Omnibus (GEO) database (accession no. GSE51312).
References
- 1.Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12(1):7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
- 2.Ernst J, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473(7345):43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ernst J, Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat Biotechnol. 2010;28(8):817–825. doi: 10.1038/nbt.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ernst J, Kellis M. ChromHMM: Automating chromatin-state discovery and characterization. Nat Methods. 2012;9(3):215–216. doi: 10.1038/nmeth.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoffman MM, et al. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat Methods. 2012;9(5):473–476. doi: 10.1038/nmeth.1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoffman MM, et al. Integrative annotation of chromatin elements from ENCODE data. Nucleic Acids Res. 2013;41(2):827–841. doi: 10.1093/nar/gks1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bernstein BE, et al. ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Visscher PM, Brown MA, McCarthy MI, Yang J. Five years of GWAS discovery. Am J Hum Genet. 2012;90(1):7–24. doi: 10.1016/j.ajhg.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stitzel ML, et al. NISC Comparative Sequencing Program Global epigenomic analysis of primary human pancreatic islets provides insights into type 2 diabetes susceptibility loci. Cell Metab. 2010;12(5):443–455. doi: 10.1016/j.cmet.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium (2011) A user’s guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol 9:e1001046. [DOI] [PMC free article] [PubMed]
- 11.Landt SG, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22(9):1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grosveld F, van Assendelft GB, Greaves DR, Kollias G. Position-independent, high-level expression of the human beta-globin gene in transgenic mice. Cell. 1987;51(6):975–985. doi: 10.1016/0092-8674(87)90584-8. [DOI] [PubMed] [Google Scholar]
- 13.Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature. 2012;489(7414):109–113. doi: 10.1038/nature11279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Q, Peterson KR, Fang X, Stamatoyannopoulos G. Locus control regions. Blood. 2002;100(9):3077–3086. doi: 10.1182/blood-2002-04-1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dorsett D, Merkenschlager M. Cohesin at active genes: A unifying theme for cohesin and gene expression from model organisms to humans. Curr Opin Cell Biol. 2013;25(3):327–333. doi: 10.1016/j.ceb.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmidt D, et al. A CTCF-independent role for cohesin in tissue-specific transcription. Genome Res. 2010;20(5):578–588. doi: 10.1101/gr.100479.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan J, et al. Transcription factor binding in human cells occurs in dense clusters formed around cohesin anchor sites. Cell. 2013;154(4):801–813. doi: 10.1016/j.cell.2013.07.034. [DOI] [PubMed] [Google Scholar]
- 18.Musunuru K, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466(7307):714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tuupanen S, et al. The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nat Genet. 2009;41(8):885–890. doi: 10.1038/ng.406. [DOI] [PubMed] [Google Scholar]
- 20.Sur IK, et al. Mice lacking a Myc enhancer that includes human SNP rs6983267 are resistant to intestinal tumors. Science. 2012;338(6112):1360–1363. doi: 10.1126/science.1228606. [DOI] [PubMed] [Google Scholar]
- 21.Li G, et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012;148(1-2):84–98. doi: 10.1016/j.cell.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boyle EI, et al. GO:TermFinder—Open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics. 2004;20(18):3710–3715. doi: 10.1093/bioinformatics/bth456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mistry M, Pavlidis P. Gene Ontology term overlap as a measure of gene functional similarity. BMC Bioinformatics. 2008;9:327. doi: 10.1186/1471-2105-9-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maurano MT, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337(6099):1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trynka G, et al. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat Genet. 2013;45(2):124–130. doi: 10.1038/ng.2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ward LD, Kellis M. Interpreting noncoding genetic variation in complex traits and human disease. Nat Biotechnol. 2012;30(11):1095–1106. doi: 10.1038/nbt.2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bernstein BE, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125(2):315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 28.Zhu J, et al. Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell. 2013;152(3):642–654. doi: 10.1016/j.cell.2012.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vuzman D, Levy Y. DNA search efficiency is modulated by charge composition and distribution in the intrinsically disordered tail. Proc Natl Acad Sci USA. 2010;107(49):21004–21009. doi: 10.1073/pnas.1011775107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whyte WA, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153(2):307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lovén J, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153(2):320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith RP, Lam ET, Markova S, Yee SW, Ahituv N. Pharmacogene regulatory elements: From discovery to applications. Genome Med. 2012;4(5):45. doi: 10.1186/gm344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mutskov V, Felsenfeld G. The human insulin gene is part of a large open chromatin domain specific for human islets. Proc Natl Acad Sci USA. 2009;106(41):17419–17424. doi: 10.1073/pnas.0909288106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information