Two Fe-S clusters catalyse sulfur insertion by Radical-SAM methylthiotransferases (original) (raw)
. Author manuscript; available in PMC: 2014 Aug 1.
Published in final edited form as: Nat Chem Biol. 2013 Mar 31;9(5):333–338. doi: 10.1038/nchembio.1229
Abstract
How living organisms create carbon-sulfur bonds during biosynthesis of critical sulphur-containing compounds is still poorly understood. The methylthiotransferases MiaB and RimO catalyze sulfur insertion into tRNAs and ribosomal protein S12, respectively. Both belong to a sub-group of Radical-SAM enzymes that bear two [4Fe-4S] clusters. One cluster binds S-Adenosylmethionine and generates an Ado• radical via a well- established mechanism. However, the precise role of the second cluster is unclear. For some sulfur-inserting Radical-SAM enzymes, this cluster has been proposed to act as a sacrificial source of sulfur for the reaction. In this paper, we report parallel enzymological, spectroscopic and crystallographic investigations of RimO and MiaB, which provide the first evidence that these enzymes are true catalysts and support a new sulfation mechanism involving activation of an exogenous sulfur co-substrate at an exchangeable coordination site on the second cluster, which remains intact during the reaction.
Keywords: Enzymology, Radical-SAM, methylthiotransferase, Fe-S clusters, HYSCORE spectroscopy, X-ray crystallography
Living organisms depend on essential sulfur-containing cofactors such as biotin, lipoic acid, thiamine, and molybdopterin1. Their formation requires the introduction of sulfur atoms into metabolic precursors and the selective formation of C-S bonds. Similar chemistry is involved in site-specific enzymatic incorporation of sulfur atoms into biological macromolecules, which generates sulphated amino acids in proteins and thionucleosides in tRNAs. One such thionucleoside, 2-methylthio-N6- isopentenyl adenosine (ms2i6A), stabilizes tRNA interactions with mRNA and ribosomes, and defects in MiaB, the enzyme catalyzing the transformation of i6A to ms2i6A (Fig. 1a), lead to frame-shifting during protein translation2. Defects in CDKAL1, a paralogous enzyme catalyzing the conversion of N6-threonylcarbamoyl adenosine in tRNA to 2-methylthio-N6-threonylcarbamoyl adenosine (ms2t6A), inhibit insulin secretion and promotes diabetes in humans3. A related macromolecular sulfation reaction targeted at the translation apparatus is performed by RimO, another paralogous enzyme that catalyzes the conversion of aspartate to 2-methylthio-aspartate (msD) (Fig. 1b) in the S12 protein near the decoding center in prokaryotic ribosomes4,5. These enzymes, which all incorporate a methylthio group at a specific site on a macromolecular substrate, belong to the same family of methylthiotransferases (MTTases).
Figure 1. Reactions catalyzed by (a) MiaB and (b) RimO.
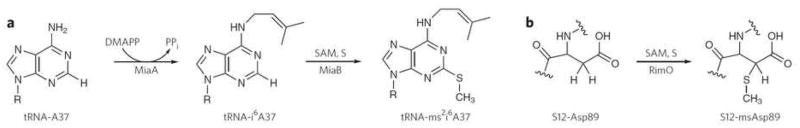
(a) MiaB catalyzes the insertion of a methylthio group on a modified A37 (i6A) of tRNAs reading codons beginning with U. DMAPP, dimethylallyldiphosphate; S, sulfur source. (b) RimO catalyzes the insertion of a methylthio group on the Asp89 residue of the protein S12 belonging to the small subunit of the bacterial ribosome.
Currently, there is limited understanding of the molecular mechanisms of MTTases or of the enzymes that perform the chemically challenging C-H to C-S bond conversion required to generate other sulphated biomolecules such as biotin and lipoic acid. Like many enzymes catalyzing biological sulfur-insertion reactions1, MiaB, CDKAL1, and RimO are iron-sulfur-containing Radical-SAM enzymes. This enzyme superfamily derives its name from the unique free-radical mechanism employed by its members, which all contain a catalytic [4Fe-4S] cluster (herein called the Radical-SAM cluster) chelated by three cysteines from a conserved CX3CX2C sequence6. The Radical-SAM cluster catalyzes the reductive cleavage of S-Adenosylmethionine (SAM) into the 5′-deoxyadenosyl radical (Ado•) that activates the substrate for transformation by abstracting a hydrogen atom from a specific C-H bond4. The sulfur inserting Radical-SAM enzymes have been shown to contain an additional iron-sulfur cluster (herein called cluster II), which is chelated by three additional conserved cysteines. The paralogous MTTases RimO, MiaB and CDKALl, all harbour two [4Fe-4S] clusters7–10, and they share a conserved domain architecture comprising an amino-terminal UPF0004 domain binding cluster II and a C-terminal TRAM (after TRM2, a family of uridine methylases, and MiaB) domain flanking the central Radical-SAM domain common to all enzymes in the Radical-SAM superfamily4 (Supplementary Results, Supplementary Fig. 1)11.
Studies of biotin synthase (BioB), from a different family of Radical-SAM- thiotransferases, suggest that the sulfur atom transferred to the substrate radical, following C-H hydrogen abstraction from the primary substrate by Ado•, is provided by a bridging sulfur in its second Fe-S cluster12. This mechanism implies loss of a sulfur atom from cluster II, and hence the partial disassembly and reconstitution of that cluster during each catalytic cycle13. This “sacrificial cluster” model based on experiments with biotin synthase has been assumed to apply to other families of Radical-SAM thiotransferases. Critical evaluation of this assumption has been impeded by the inability to establish in vitro conditions in which these enzymes turn over.
In this paper, we report parallel enzymological, spectroscopic and crystallographic investigations that substantially advance understanding of MTTases and the sulfation mechanism that they employ. We report the crystal structure of holo RimO from Thermotoga maritima (Tm_RimO)_, the first structure of an intact and fully functional MTTase. Moreover, we report the identification of in vitro reaction conditions in which _Tm_RimO and T. maritima MiaB (_Tm_MiaB) both catalyze multiple turnovers using sulfide, methylsulfide, selenide or methylselenide as co-substrates. Our data support a sulfation mechanism in which an exogenous sulfur co-substrate is activated at an exchangeable coordination site on cluster II, which remains intact during catalysis.
RESULTS
_Tm_MiaB catalyzes multiple turnovers of methylthiolation
Active preparations of _holo Tm_MiaB and _Tm_RimO containing two [4Fe-4S] clusters per monomer were obtained by reconstituting the clusters in corresponding apo proteins (Supplementary Fig. 2)8. Spectroscopic characterization of these enzymes is presented in Supplementary Figs. 3 and 4. Quantification of their Fe and S content consistently indicated an excess of sulfur atoms (12 ± 1 S vs. 8.5 ± 0.2 Fe per protomer of MiaB, 11.6 ± 0.8 S vs. 7.6 ± 0.2 Fe per protomer of RimO). Most of the excess S was in the S(0) redox state (2.5 ± 0.5 S(0) per MiaB protomer, 2 ± 1 S(0) per RimO protomer) (see Supplementary Results). Therefore, these in vitro reconstitution conditions supported not only assembly of Fe and S atoms into two [4Fe-4S] clusters, as previously demonstrated by Mössbauer spectroscopy8,10, but also the binding of additional sulfur to the proteins. On the basis of the X-ray crystal structure of _holo Tm_RimO prepared in this manner, presented below, we propose that the extra S atoms belong to a polysulfide moiety bound to the Fe-S clusters. This polysulfide species is likely formed during the reconstitution protocol and is expected, under the strongly reducing conditions, to be reduced to sulphide which is subsequently transferred to the substrate.
_Tm_MiaB activity was assayed in vitro in the presence of a physiological tRNA substrate by monitoring the formation of ms2i6A using HPLC (Fig. 2). Optimal conditions employed 0.5 μM enzyme at 65 °C in the presence of SAM, dithionite as a reducing agent, and a mixture of tRNAs prepared from a tRNA-Phe overexpressing E. coli strain carrying an inactivated miaB gene. Production of ms2i6A proceeded with an initial turnover number (TON) of 0.8 min−1 and reached a plateau after 6 min, generating 4.0 ± 1.0 moles of ms2i6A per mole of MiaB protomer (Fig. 2a). There was a striking correlation between the number of extra sulfur atoms retained by the reconstituted enzyme (i.e., in addition to those in the Fe-S clusters) and the maximal amount of ms2i6A produced under these conditions. This correlation suggests that these extra sulfur atoms are the ones incorporated into ms2i6A. A stoichiometric consumption of the substrate i6A and near-stoichiometric production of 5′-deoxyadenosine AdoH were observed during the reaction (AdoH/ms2i6A = 1.2), consistent with the activation of tRNA substrate by a SAM-derived Ado• radical (Fig. 2a)14. Consistent with SAM being the methyl donor15, formation of S-Adenosylhomocysteine (SAH) was also observed. However, an excess of SAH was observed with respect to ms2i6A (SAH/ms2i6A = 2–3.4- Fig. 2a). An analogous MiaB-dependent conversion of SAM to SAH occurs in the absence of the tRNA substrate if dithionite present (data not shown). The methyl acceptor for this uncoupled turnover of SAM is presently unknown, but ESI mass spectrometry analysis demonstrated that it is not the protein itself (data not shown). Further studies will be required to understand the mechanistic details of this uncoupled side-reaction.
Figure 2. Enzymological characterization of MiaB.
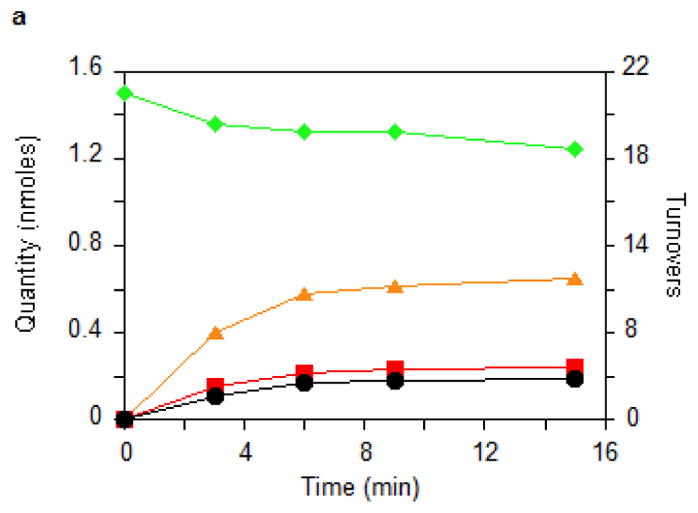
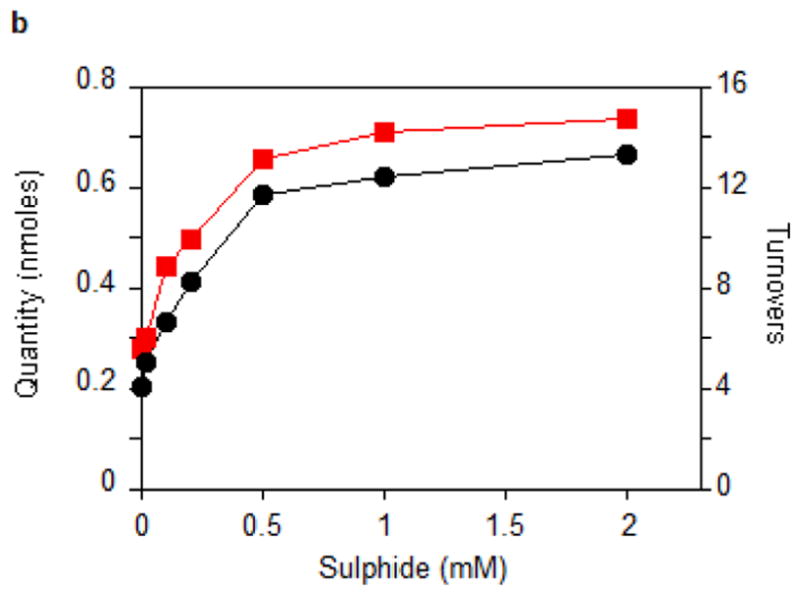
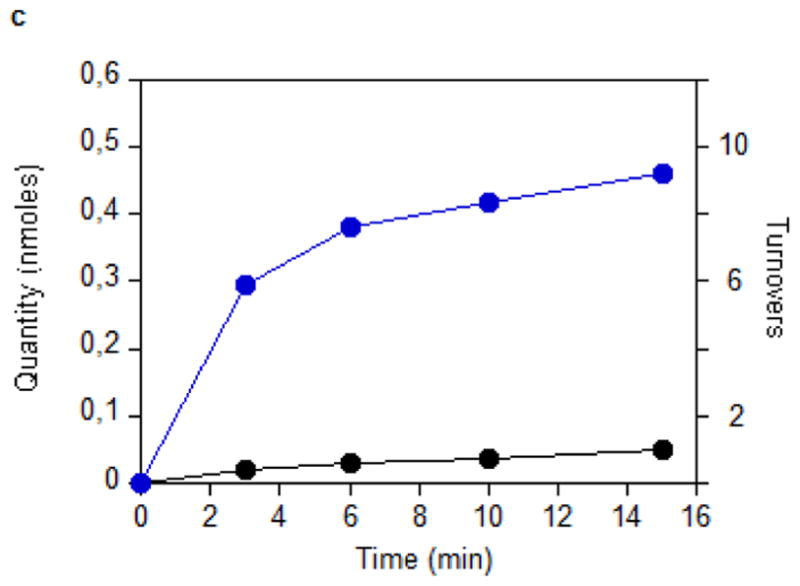

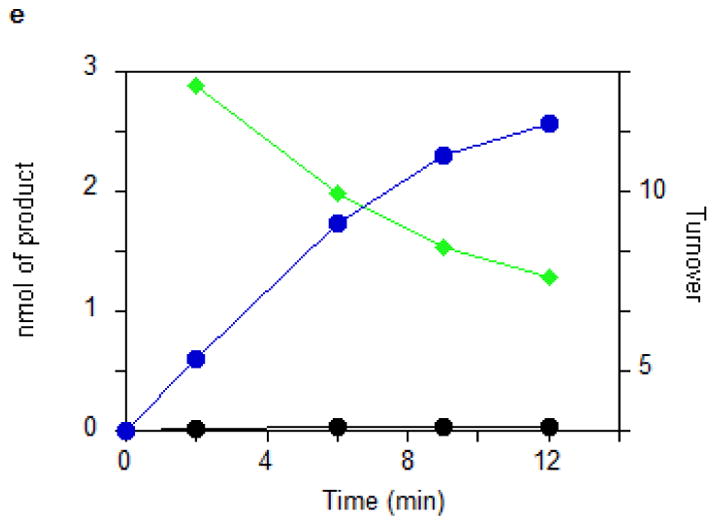
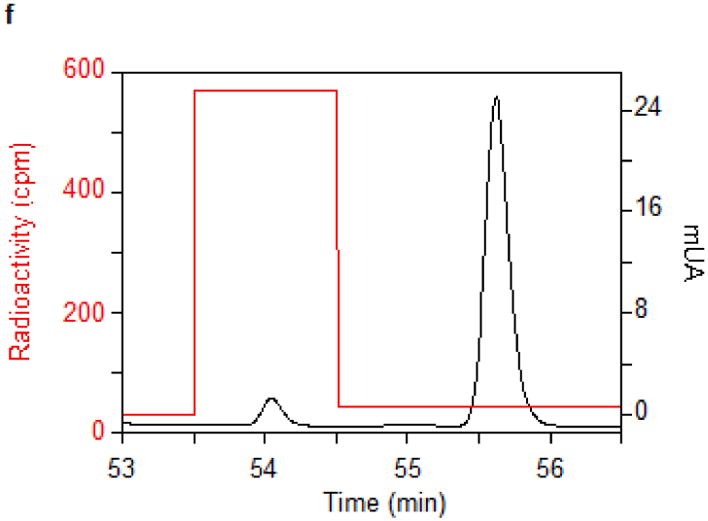
(a) Time courses for the disappearance of i6A (green ◆) and the formation of SAH (orange ▲), AdoH (red ■), and ms2i6A (black ●) in the reaction of MiaB (0.05 nmoles) with E. coli tRNA-Phe substrate. Turnover numbers refer to product formation. (b) The dependence of the MiaB-catalyzed production of ms2i6A (black ●) and AdoH (red ■) on the concentration of sulphide. The reaction time was 15 min. (c) Time course for the formation of mse2i6A (blue ●) and ms2i6A (black ●) when the assay is carried out in the presence of 1 mM methylselenide. Each point in panels a, b, and c represents the mean value from 5 experiments ± 10%. (d) HPLC analyses of the i6A/ms2i6A/mse2i6A region from the reactions depicted in panel b and c. The HPLC baseline from c has been shifted upwards for clarity (e) Time course for i6A disappearance (green ◆), formation of mse2i6A (blue ●) and ms2i6A (black ●), in the reaction of TmMiaB (0.2 nmol) with E. coli tRNA-Phe substrate in the presence of 500 μM Na2Se at 65°C. Turnover numbers refer to product formation. Each point in panel e represents the mean value from 3 experiments ± 5 % (f) Radioactive counting of the HPLC ms2i6A/mse2i6A region from a reaction analogous to that depicted in (c). Each point in panel f represents the mean value from 3 experiments ± 3%.
Figure 2b shows that the production of ms2i6A increased upon adding exogenous Na2S to the reaction mixture up to a concentration of 0.5 mM, allowing the enzyme to achieve 12 turnovers. This number varied between enzyme preparations, with the highest value observed being 21. At 0.5 mM sulphide, the initial enzyme TON was 1.1 min−1 (Fig. 2c). Figure 2c shows that the reaction stopped after 15–20 min of incubation time. These observations demonstrate that the enzyme can employ exogenous sulphide for catalytic methylthiolation. Catalytic methylselenide insertion by _Tm_MiaB also was observed by using 0.5 mM sodium selenide in place of sodium sulphide, with the reaction yielding almost exclusively 2-methylseleno-N6-isopentenyl adenosine (~12 mse2i6A per MiaB) and very little ms2i6A as monitored by HPLC (Fig. 2e).
To establish whether MiaB directly employs methylsulphide (CH3S−) or methylselenide (CH3Se−) as a co-substrate 1 mM CH3S− was provided in the assay resulting in ms2i6A production with a TON of 0.45 min−1 and a maximum of 6 ms2i6A per MiaB after 15 min, slightly less efficiently than using sulphide (data not shown). With 1 mM CH3SeNa, the enzyme generated mse2i6A, with a TON of 2 min−1 and producing 10 molecules of mse2i6A per MiaB molecule after 15 min (Fig. 2c). As shown by HPLC (Fig. 2d) ms2i6A was found to be a minor reaction product, indicating limited utilization of the MiaB-bound extra sulfur atoms under these conditions (Fig. 2c). Performing the assay using CH3Se− and radioactively labelled [14C-_methyl_]SAM yielded radioactivity exclusively in the minor ms2i6A peak but not in the major mse2i6A peak (Fig. 2f). These observations unambiguously demonstrate that CH3S− and CH3Se− behave as functional co-substrates of MiaB and are directly incorporated intact into the tRNA substrate.
Purified _Tm_RimO also turned over multiple times when assayed using similar reaction conditions (Supplementary Figs. 5 and 6). In this case the substrate is a synthetic peptide consisting of 20 residues flanking the target aspartate residue (D89) of ribosomal protein S12 from T. maritima 7. The physiological substrate, the S12 protein is unfortunately insoluble in vitro. With this peptide, _Tm_RimO turned over 3 times in the absence of sulphide or 5 times in the presence of sulphide or CH3Se−, producing msD or mseD respectively.
Spectroscopic characterization of ligands to cluster II
Using HYSCORE spectroscopy we investigated the interactions of cluster II in MiaB with co-substrates. HYSCORE is a two-dimensional EPR technique that monitors nuclei interacting with an S=1/2 [4Fe-4S]+ system. To avoid interference from the Radical-SAM cluster, we used an inactive MiaB mutant (MiaB3C) in which the three cysteines (Cys150, Cys154 and Cys157) chelating the Radical-SAM cluster had been replaced by alanine, so that the protein only retains cluster II8. The HYSCORE data for MiaB3C demonstrate that cluster II binds exogenous ligands such as CH3Se− without being degraded. Upon CH3Se− addition, the EPR spectrum of reduced MiaB3C was slightly modified without loss of intensity (Supplementary Fig. 4a). CH377Se− addition resulted in a new feature in the HYSCORE spectrum (Figs. 3a and 3b), specifically a ridge perpendicular to the diagonal and centred on the nuclear frequency of 77Se (2.81 MHz for a static magnetic field of 3600 G). The shape and position of this feature is characteristic of the correlation pattern produced by a weak 77Se hyperfine coupling tensor A (|A| = 3.8 ± 0.5 MHz). This same feature appears in the HYSCORE spectra of reduced wild-type _Tm_MiaB and _Tm_RimO incubated with CH377Se− (Supplementary Figs. 7a and 7b).
Figure 3. HYSCORE spectra of variant MiaB3C.
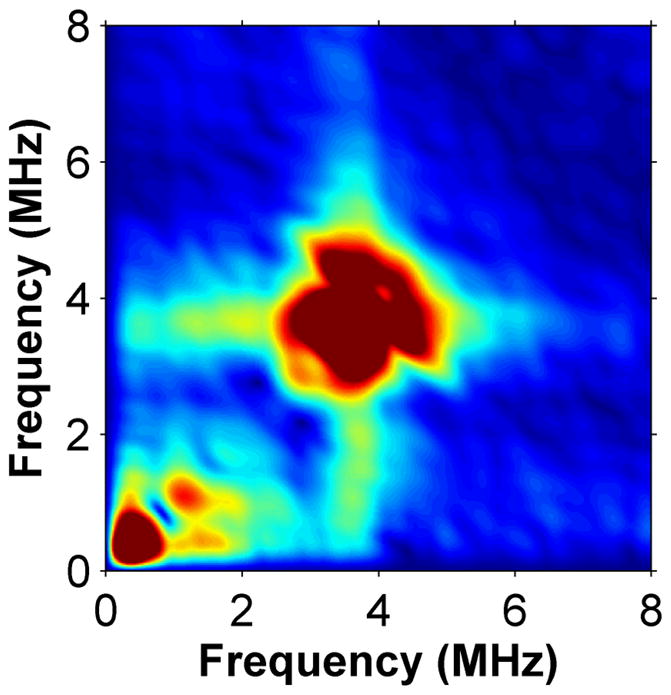
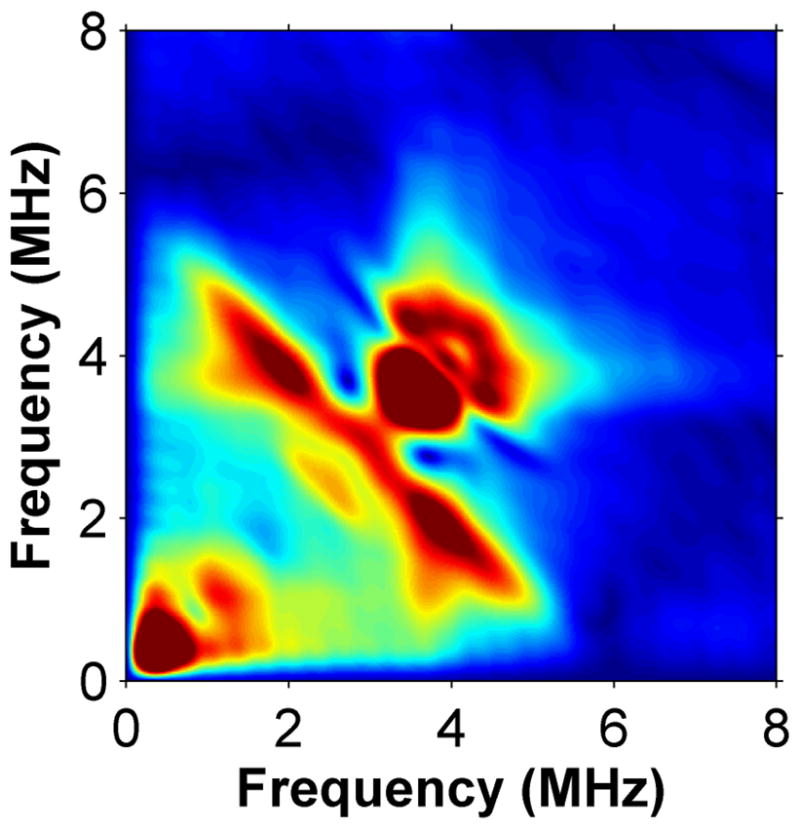
Low frequency region of the X-band HYSCORE spectra of MiaB3C without (panel a) and with (panel b) 77SeCH3Na.
The observed 77Se hyperfine coupling is due to adjacent magnetic iron atoms. To computationally estimate the magnitude of the coupling when 77Se is bound to the unligated Fe atom in a standard [4Fe-4S] cluster, and to exclude the possibility of CH3Se− replacing one of the internal ‘inorganic’ S ions in the cluster, we constructed two alternative atomic models and analyzed them using DFT methods (see Supplementary Note 1). In the first one, CH3Se− is linked externally to one of the iron atoms of an intact and unperturbed [4Fe-4S] cluster. This model was calculated using cluster II of the _holo_RimO crystal structure, by replacing the three cysteines by SCH2CH3 ligands and covalently attaching a CH3Se− ligand to the fourth iron ion (labelled #4). Because the electronic structure of the reduced (spin 1/2) cluster formally consists of two antiferromagnetically coupled pairs, a mixed-valence (spin 9/2) pair and a di-ferrous (spin 8/2) pair, there are six possible locations for the ferrous pair (labelled 12, 13, 14, 23, 24 and 34 in Supplementary Table 1). Each broken-symmetry state (Ms=1/2) mimicking the corresponding S=1/2 pure spin state was geometry-optimized (see Supplementary Note 1). We then computed the 77Se hyperfine coupling constant, A(Se) using standard spin-recoupling procedures. For the two energetically lowest electronic configurations (labelled 12 or 34), |A(Se)| was estimated at 6.7 and 4.2 MHz, respectively, close to the experimentally observed value in MiaB of |A| = 3.8 ± 0.5 MHz (Fig. 3b). Therefore, our spectroscopic studies are consistent with CH377Se− binding to the free coordination site of cluster II. To rule out the displacement of internal S atoms, a second in silico model was constructed, in which one of these internal S atoms in the cluster was replaced by CH3Se. The computational procedures outlined above now produce six alternative geometry-optimized broken-symmetry states. DFT calculations show that, in all of these states, the predicted lower limits for A(Se) are significantly larger (> 21 MHz) than the experimentally observed value in MiaB (Supplementary Table 2).
Further evidence for the stability of cluster II under enzyme assay conditions was obtained by exposing reconstituted MiaB3C (20 min at 65°C) to a 2000 fold excess of CH3S− both before and after reduction with dithionite. Assay of the amount of sulfur remaining bound to the enzyme demonstrated that no sulfur was released from the protein in either experiment (Supplementary Table 3).
The crystal structure of holo RimO_Tm_
_Holo Tm_RimO crystals with 44% solvent content grew in space group P212121 with two protomers in the asymmetric unit. The structure (Fig. 4 and Supplementary Fig. 8) was refined to working and free R-factors of 24.3% and 29.6%, respectively, at 3.3 Å resolution (Supplementary Table 4). The structures of roughly identical protomers (Supplementary Fig. 8) begin at residue 2 and extend past the native C-terminus to include two residues for subunit A from an engineered hexahistidine tag. Each protomer contains two [4Fe-4S] clusters with 3 cysteine ligands per cluster. Remarkably, a continuous chain of electron density was observed bridging the two clusters, which refined well when modeled as a covalently bonded pentasulfide chain (Figs. 4a, 4c, 4d and Supplementary Fig. 9). This observation supports the hypothesis that excess sulfur retained by _holo Tm_RimO and _Tm_MiaB after reconstitution (see above) is present in the form of a polysulfide moiety bound to the [4Fe-4S] clusters. The pentasulfide moiety bridges the two iron atoms in each cluster that have an open coordination site. Therefore, the crystal structure of _holo Tm_RimO provides further evidence for the binding of exogeneous sulfur atoms to cluster II, supporting the validity of the model inferred from the enzymological and spectroscopic experiments described above.
Figure 4. Crystal structure of holo TmRimO.
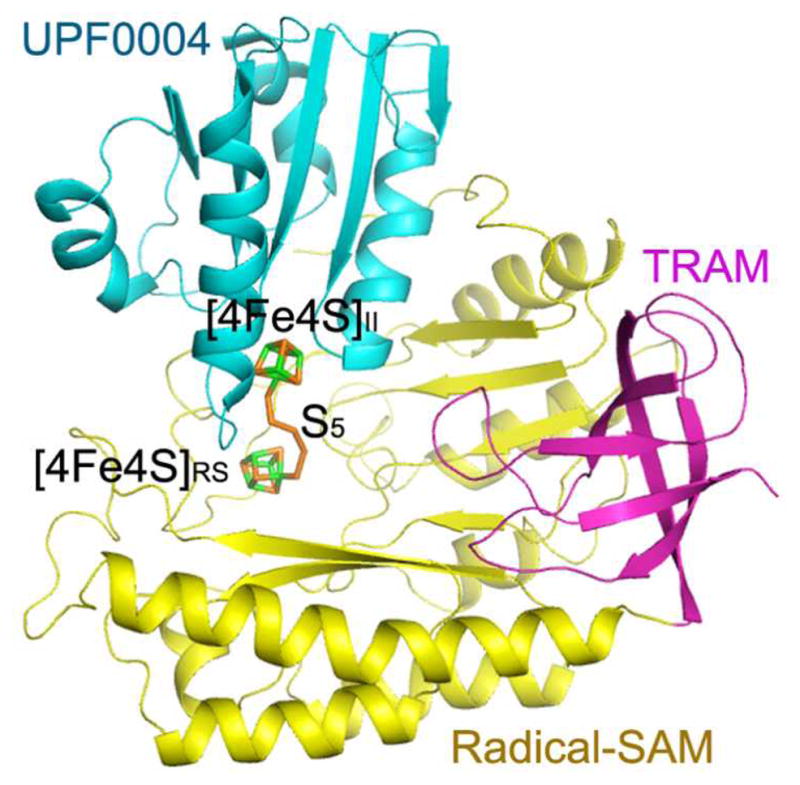
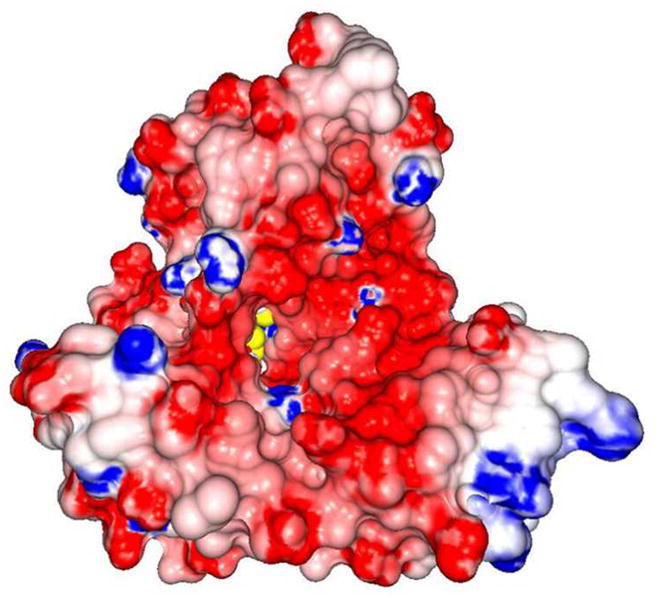

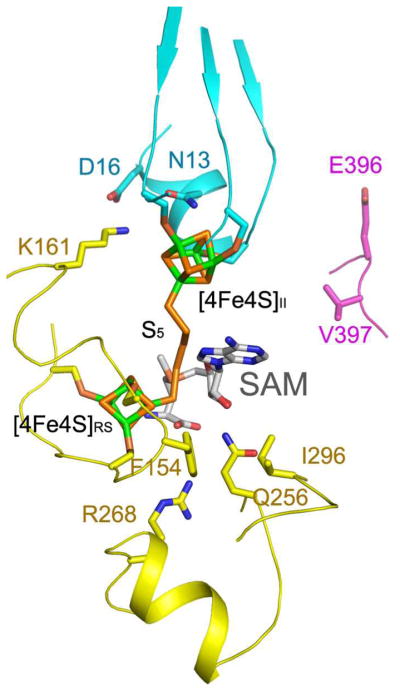
(a) Ribbon diagram showing the UPF0004, Radical-SAM, and TRAM domains in cyan, yellow, and magenta, respectively. The two [4Fe-4S] clusters and the bridging pentasulfide (S5), which connects the two clusters, are shown in stick representation, with iron and sulfur atoms colored green and orange, respectively. (b) The molecular surface is shown in the same orientation, color-coded according to its electrostatic potential. Fully saturated blue and red represent potentials of ±8 kT at 100 mM ionic strength31. The [4Fe-4S] clusters and bridging pentasulfide are shown in yellow. (c) Electron density from a simulated-annealing Fo-Fc omit map for the two [4Fe-4S] clusters and bridging pentasulfide, contoured at 1 σ (grey) and 5σ (blue) levels. (d) Active-site stereochemistry in protomer A. The [4Fe-4S] clusters and bridging pentasulfide are shown in stick representation, with iron and sulfur atoms colored green and orange, respectively. The six invariant cysteine residues that coordinate the clusters and other strongly conserved protein residues in MTTases are shown in stick representation, with carbon atoms colored according to the domain of origin and oxygen and nitrogen atoms colored red and blue, respectively. A molecule of SAM was docked into the active site based on structural alignment of the SAM-bound structure of the Radical-SAM enzyme MoaA (PDB id 1TV8); carbon atoms in SAM are colored gray. Some of the backbone of _Tm_RimO has been omitted in this image to improve clarity; see Supplementary Fig. 9 for a more detailed depiction of the active site.
The structures of the Radical-SAM and TRAM domains in _holo Tm_RimO closely match those of our previously published apo crystal structure lacking the N-terminal UPF0004 domain (root-mean-square deviation (rmsd) of 1 Å for 272 residues – Supplementary Fig. 10a10). The Radical-SAM domain is similar to that of other Radical-SAM enzymes16 and forms an incomplete or open TIM-barrel containing six parallel β-strands, each followed by an α-helix that packs parallel to the preceding β-strand on the outer surface of the open TIM-barrel (Fig. 3a). Following the fourth β-strand, there is an additional short but highly conserved α-helix (α8) that packs perpendicular to the β-sheet of the Radical-SAM domain (Fig. 3, and Supplementary Figs. 1 and 10); the loops immediately preceding and following this α-helix line the Radical-SAM active site. The TRAM domain in RimO, which contains five anti-parallel β-strands, docks on the surface of the Radical-SAM domain at the distal edge of its open TIM-barrel from its conserved [4Fe-4S] cluster (Fig. 3a). The relative locations of the Radical-SAM and TRAM domains in the new holo structure are shifted relative to one another by ~2.3 Å compared to the apo structure due to a rigid-body translation with minimal rotation (Supplementary Fig. 12a). The longest α-helix in the Radical-SAM domain, which is located at its C-terminus immediately preceding the TRAM domain, undergoes a similar rigid-body displacement and thus appears to move with the TRAM domain.
The Radical-SAM domain in RimO is most similar16 to those in two other Radical-SAM enzymes: oxygen-independent coproporphyringogen III (HemN -- PDB id 1OLT, Z-score of 13.7 and 5.2 Å rmsd for alignment of 244 Cα’s with 16% sequence identity) and molybdenum cofactor biosynthesis protein A (MoaA -- PDB id 1TV8, Z-score of 10.6 and 5.2 Å rmsd for alignment of 200 Cα’s with 12% sequence identity). RimO binds the Radical-SAM cluster at the same site and in equivalent geometry to those bound to the Radical-SAM domains in HemN and MoaA. Notably, the crystal structure of MoaA (Supplementary Fig. 13) shows the thiol group of a DTT directly bound to the second [4Fe-4S] cluster in that enzyme at the equivalent Fe atom to that ligating the pentasulfide bridge in the cluster II of RimO (although the second cluster in RimO is substantially closer to the Radical-SAM cluster than that in MoaA, which is 16 Å away).
Examination of the active site of RimO (Fig. 4d and Supplementary Fig. 11) shows relatively weak sequence conservation compared to other families of Radical-SAM enzymes, consistent with the trend observed in the superfamily. The weak inter-family conservation has been interpreted to indicate that direct interaction of the SAM cofactor with the [4Fe-4S] cluster is the dominant factor controlling generation of the reactive radical-SAM species and that the active-site structure has evolved primarily to control substrate-binding geometry and affinity17,18. Only residue F154 in RimO, which makes an edge-to-face interaction with the adenine moiety of SAM in existing ligand-bound stuctures, is broadly conserved in the Radical-SAM superfamily. Residues conserved in other MTTases include Asn13, Asp16, Lys161, Gln256, Arg268, and Ile296, some of which are likely to be involved in controlling the binding of cluster II or its interaction with exogenous sulfur species (Fig. 4d and Supplementary Fig. 11)
The UPF0004 domain binds to the opposite edge of the Radical-SAM domain from the TRAM domain and completes the active site of the enzyme (Figs. 4a and 4d), located at the bottom of an ~ 40 Å deep funnel with a highly acidic rim that is formed jointly by all three domains (Fig. 4b). The acidic character of the rim is consistent with binding of the highly basic ribosomal protein S12, the substrate of RimO, at this site. As expected, the three invariant cysteine residues in the UPF0004 domain ligate [4Fe-4S] cluster II.
Our crystal structure provides the first experimental data on the UPF0004 domain fold. The fold appears to comprise a five-stranded parallel β-sheet created by four α/β supersecondary motifs followed by a final β-strand. Cluster II is bound in a crevice between the first two β-strands at a so-called topological reversal point in the β-sheet19. While the electron density of our 3.3 Å structure is clearly defined for these first two α/β units, it becomes increasingly diffuse at greater distances from the active site. As a result, residues between positions 97–130 have poor electron density, from which residues 113–114 and 121–126 could not be assigned in the crystal structure. Analysis of backbone B-factors (Supplementary Figs. 8b and 8c) shows progressively greater mobility of the UPF0004 domain at greater distances from cluster II and its interface with the Radical-SAM domain, suggesting that it pivots around the interface. The UPF0004 domain is structurally similar to proteins in the CheY-related fold family20,21 which has a flavodoxin-like α/β fold. The strongest similarity is to bacterial signal-transduction proteins including the response regulator NarL (PDB id 1A04, Z-score of 6.5 and 2.9 Å rmsd for alignment of 94 Cα’s with 12% sequence identity – Supplementary Fig. 10b) and CheY (PDB id 3TMY, Z-score of 5.8 and 3.1 Å rmsd for alignment of 89 Cα’s with 16% sequence identity)22. Both proteins undergo conformational changes mediating signal-transduction processes23–25 upon phosphorylation of a residue in a loop corresponding to the one ligating cluster II in RimO. Therefore, substrate/product-responsive conformational changes in the UPF0004 domain may contribute to promoting efficient catalysis by RimO.
A noteworthy feature of the active site of RimO is the close proximity of the two [4Fe-4S] clusters, which are only 8 Å apart and bridged by the bound pentasulfide moeity (Figs. 4a,4c,4d and Supplementary Fig. 9). In other Radical-SAM enzymes with two iron-sulfur clusters, the clusters are significantly farther apart (12 Å in BioB and 16 Å in MoaA)26,27. Superposition of the SAM-bound structure of MoaA provides a stereochemical model for SAM binding to the Radical-SAM cluster in RimO (Figs. 4d and Supplementary Fig. 11). This ligand can be accommodated in the canonical binding geometry without steric clash except for overlap with a portion of the bridging pentasulfide moiety bound to the Radical-SAM cluster (Figs. 4d and Supplementary Fig. 9). However, the two sulfur atoms in the pentasulfide moiety that are closest to cluster II do not overlap with the modeled SAM, indicating that the stereochemistry of the active site in RimO is compatible with the binding of exogenous sulfur to cluster II in the presence of SAM. This observation supports the enzymological and spectroscopic experiments reported above.
Manually docking the ribosome-bound conformation of the S12 protein into the ~40 Å deep active-site funnel of RimO positions the target Asp residue (Asp89 in the Thermotoga maritima S12 protein) adjacent to the two [4Fe-4S] clusters in RimO without any steric clash (Supplementary Fig. 12). However, the dimensions of the funnel are such that S12 will seal the active site of RimO, implying that the co-substrate SAM and possibly also the sulphide reactant must bind to the enzyme prior to the S12 substrate protein.
DISCUSSION
The biochemical and structural studies reported here demonstrate for the first time that the MTTases MiaB and RimO are true enzymes whose [4Fe-4S] clusters are not sacrificed as sulfur donors. The data support a catalytic mechanism involving activation of exogenous sulfur-containing co-substrates via binding to the free coordination site of cluster II. This mechanism stands in contrast to the proposed mechanism for the sulfating Radical-SAM-enzyme biotin synthase11, which involves a sulfur atom being extracted from its second iron-sulfur cluster for insertion into its substrate and hence the degradation of this cluster during turnover.
Indeed, EPR (Supplementary Fig. 4) and HYSCORE (Figs. 3a, 3b and Supplementary Fig. 7) spectroscopies unambiguously demonstrate that CH3Se− binds to an intact cluster II. The hypothesis that this coordination complex is a catalytically competent intermediate involved in the reaction is supported by a series of related enzymological observations. Most importantly, we show that CH3Se− or CH3S− can be used as co-substrates that are directly incorporated by MiaB (Figs. 2c, 2d and 2f) and RimO (Supplementary Figs. 5 and 6) into their macromolecular substrates. Both enzymes turn over multiple times using these co-substrates. Sulfide (Fig. 2b) and selenide (Fig. 2f) are also productive co-substrates, suggesting that these nucleophilic species can be methylated by SAM before incorporation into the substrates.
Additional evidence supporting our mechanistic model comes from the demonstration that excess sulfur retained following [4Fe-4S] clusters reconstitution can be mobilized for repeated methylthiolation reactions without addition of exogenous sulfur to enzyme reactions (Fig. 2a). This excess sulfur is observed in the crystal structure of _holo Tm_RimO in the form of a pentasulfide bridge between the two [4Fe-4S] clusters. Enzymatic activity of MTTases supported by excess sulfur retained during reconstitution (Fig. 2a) is inferred to derive from the reduction of this polysulfide species by dithionite which results in release of sulfide anions under the assay conditions. Importantly, the interaction of the pentasulfide bridge with cluster II in the crystal structure of _holo Tm_RimO demonstrates that cluster II has an accessible iron coordination site capable of ligating sulfur and simple stereochemical modeling shows that this site should remain accessible when SAM is bound to the Radical-SAM-cluster in the conserved geometry observed in other Radical-SAM enzymes26,27 (Figs. 4d and Supplementary Fig. 9). Therefore,, the crystal structure of _holo Tm_RimO provides another source of support for the hypothesis that cluster II activates sulfur-containing co-substrates, as inferred from our EPR, HYSCORE, and enzymological experiments. Notably, a similar mechanism has been proposed for the Radical-SAM enzyme MoaA, which has a second iron-sulfur cluster that interacts with a nitrogen atom on its GTP co-substrate, based on electron-nuclear double resonance spectroscopy 28.
Our mechanistic hypothesis is also supported by the observation that sulfur is not released from cluster II during incubation of the enzyme with excess CH3S− (Supplementary Table 3). This observation excludes nucleophilic substitution of bridging sulfide by CH3S−, a reaction for which there is notably not a chemical precedent29. Furthermore, a study of the reaction of synthetic [4Fe-4S] clusters with a strong methylating agent did not indicate methylation of bridging sulfur atoms30. The spectroscopic (Figs. 3a and 3b) and structural (Fig. 4) data presented above in this paper support instead a mechanism involving activation of an exogenous sulfur co-substrate at an open coordination site on cluster II in MiaB and RimO, which enables both enzymes to turnover repeatedly without degradation of their iron-sulfur clusters (Figs 2a–e).
In conclusion, our experimental data indicate that MTTases have evolved two distinct [4Fe-4S] clusters for binding and activation of two different co-substrates. The Radical-SAM cluster likely activates SAM to form the canonical Ado• radical, as proposed for all Radical-SAM enzymes, while cluster II serves to activate sulfide or methylsulfide, through a still undefined mechanism. Further investigation will be required to elucidate the exact chemical mechanism by which the Ado• radical and the [4Fe-4S]-SCH3 complex in the enzyme active site cooperate to convert a C-H bond into a C-SCH3 bond. An important issue to be addressed in such studies is whether there are differences in the mechanism used by RimO to modify an sp3-hybridized carbon compared to that used by MiaB to modify an aromatic carbon.
METHODS
Materials
Cysteine, 5′-deoxyadenosine (AdoH), S-adenosyl-L-homocysteine (SAH), 5′-methylthioadenosine (MTA), and dimethyldiselenide were purchased from Sigma. Sodium sulfide nonahydrate, selenium powder (200 mesh) and N-6-(delta-2-isopentenyl)adenosine hemihydrate (i6A) were from Acros. 77Se was a kind gift from Dr. J-M. Moulis, LCBM, CEA-Grenoble. SAM was synthesized and purified as described32. Sodium selenide was prepared by borohydride reduction of selenium powder and standardized with Pb(OAc)2 as described33. Sodium methyl-(77Se)selenide was prepared by borohydride reduction of the dimethyl di(77Se)-selenide prepared as follows34 : in a glove box, 7.5 mg (97 μmoles) of 77Se was mixed with 8.5 mg (212 μmoles) of powdered sodium hydroxide in 250 μL of anhydrous DMF. After 30 min. 15 μl of hydrazine hydrate (31 μmoles) in DMF were added and the reaction was allowed to proceed for 15 hours. To the dark brown solution, 6.5 μL of methyliodide (101μmoles) were added. The color rapidly changed to yellow and the reaction was continued for 5 hours. The reaction mixture was then diluted to 3 mL with H2O and loaded on top of a C18-SEP-PAK cartridge (Waters) equilibrated in water. The cartridge was washed with 5 mL water and the yellow diselenide eluted with 0.5 mL methanol. The concentration of the solution was determined using an extinction coefficient of 210 M−1cm−1 at 314 nm. Mass spectrometric analysis (Supplementary Fig. 13) gave only one molecular ion corresponding to the diselenide (m/z= 184) and fragments corresponding to loss of one and two methyl groups. The colorless reduced form was generated by reduction with an excess of NaBH4. After adding 0.2 M acetate pH : 4.0, the pH was adjusted to 8.0 with Tris 1M pH : 9.0. The construction of the triple C150/154/157A MiaB mutant, named MiaB3C, has been described previously8.
Protein, Fe, sulfide, and S(0) assays
Protein concentrations were determined by quantitative amino-acid analysis of the pure proteins that gave the following extinction coefficients (mM−1cm−1) at 280 nm: 50 for apo RimO, 65 for both wild-type MiaB and MiaB3C, 100 for _holo Tm_RimO, 95 for wild-type _holo Tm_MiaB, and 85 for holo MiaB3C. Iron concentrations were determined colorimetrically using bathophenanthroline disulfonate under reducing conditions as described35. Labile sulfide was determined according to a standard procedure36. S(0) was determined under anaerobic conditions in the following assay system37 : in a glove box, 100 μL samples containing the protein (0–8 nmoles) were incubated in 50 mM Tris-HC1 pH 8.8 with 30 mM NaCN for 30 min at 65°C. After cooling at room temperature, 50 μL zinc acetate (1%) were added and the samples were centrifuged at 13K for 10 min. Next, 50 μL ferric nitrate (0.75 M in 20% HNO3) were added to the supernatant and the absorbance of the resulting ferric thiocyanate was read at 460 nm against a reaction blank. The amount of S(0) per mole of monomer was calculated using the extinction coefficient ε460 = 3130 M−1.cm−1, which was established with standardized solutions of ferric thiocyanate.
Expression, purification and Fe-S cluster reconstitution into proteins
All expressions were conducted in LB medium at 37 °C in the E. coli BL21CodonPlusλ(DE3)-RIL38. All proteins were purified under aerobic conditions and contained sub-stoichiometric [4Fe-4S] clusters as judged by UV-visible absorption spectroscopy. Apoproteins were obtained by overnight exposure to EDTA (25 mM) under reducing conditions (10 mM sodium dithionite), followed by purification using a gel filtration column (G-25), which was equilibrated with 50 mM Tris-HCl buffer, pH 8, with 200 mM NaCl. The protein was washed and concentrated using a Centricon with a 30 kDa cut-off membrane.
The reconstitution of [4Fe-4S] clusters into the proteins was carried out using a 10-fold excess iron (Mohr’s salt), a 12-fold excess cysteine and a catalytic amount of cysteine desulfurase E. coli CSDA (2 μM). When the reaction was completed (A400/A278 = 0.30–32), the reaction mixture was loaded onto a NAP-25 desalting column, and the brown fractions were pooled, of which an aliquot (12 nmoles) was analysed by anaerobic FPLC on a Superdex-75 (analytic), which was already equilibrated with Tris-Cl 10 mM, NaCl 100 mM pH = 7.5 (Supplementary Fig. 2.). The chromatogram indicated the presence of significant amounts of multimeric aggregated protein forms. The bulk solution was then complemented with 10 mM DTT and heated for 1 h at 65°C. This solution was passed through a second NAP-25 column. The enzyme was subsequently concentrated (30–60 mg.mL−1) using micro-concentrators (Vivaspin 30 kDa), followed by addition of 15% (v/v) glycerol and stored as 25 μL aliquots at −80°C. An aliquot of the protein (12 nmoles) was subsequently analyzed on the Superdex-75 FPLC column (Supplementary Fig. 2), which showed that a more homogeneous protein in monomeric form was produced using this procedure.
The same procedure was utilized for the MiaB3C mutant except that lower excess of iron and cysteine (6 and 8 respectively) were used for reconstitution of the cluster.
tRNA substrate for MiaB
Overproduction of tRNAPhe was performed using the E. coli TX3346 _miaB_− strain lacking a functional miaB gene. The transformed cells with pTrc99-B-tRNAPhe were grown overnight in 5 ml of LB medium containing 100 μg of ampicillin/ml. This overnight culture was used to inoculate 5 liters of LB medium. When the culture reached 0.8 OD600, tRNAPhe expression was induced by adding isopropyl-1-thio-β-D-galactopyranoside to a final concentration of 0.5 mM followed by incubation for 15 h at 37 °C. Bacterial cells were harvested by centrifugation, resuspended in 200 mM Tris-Cl pH 8, and then extracted by an equal volume of phenol saturated with 200 mM Tris-Cl, pH 8. After 30 min of vigorous shaking at room temperature, the aqueous phase was collected by low speed centrifugation and extracted again under the same conditions. The small RNAs were precipitated with ethanol, resuspended in 50 ml of 500 mM Tris-Cl, pH 8.8, and incubated at 37 °C for 45 min in order to deacylate the extracted tRNAs. The solution was then neutralized by addition of 10 ml of 1 M sodium acetate, pH 5.1 and RNAs were precipitated with 3 volumes of cold ethanol. The total RNAs pellet was dissolved in 10 mM Tris-H3PO4, pH 6.3 containing 15% ethanol and 400 mM KCl, and the solution was applied to a Nucleobond column AX10000 (Clontech) equilibrated with the same buffer. The column was then washed extensively with the same buffer and tRNAs were eluted by increasing the concentration of KCl to 650 mM. tRNAs were precipitated with 0.7 volume of cold isopropyl alcohol for 1 h at 4 °C, washed with 70% ethanol, and dried. tRNAs were dissolved in water and stored at −20 °C in 250 mM NaCl and 3 volumes of ethanol. A final chromatography step on CHT-20 hydroxy-apatite column (BioRad) was performed as follows: a total of 10 mg (250 O.D.260 nm) of the material obtained after Nucleobond chromatography were loaded on a CHT-20 column equilibrated with 75% buffer A (10 mM potassium phosphate pH: 6.5) and 25% buffer B (0.5 M potassium phosphate pH: 6.5). The column was washed with 3 column volumes of this starting buffer then a linear gradient from 25 to 50% buffer B was applied (Supplementary Fig. 14). The 260 nm-absorbing materials were collected in 2 mL fractions and analysed by HPLC after overnight digestion with nuclease P1 and dephosphorylation by alkaline phosphatase. The purity of the tRNA-Phe fractions was estimated to be 65–85% by quantification of the amount of the i6A nucleoside with regard to the amount of digested tRNA, determined by its absorbance at 260 nm.
Peptide substrate for RimO
A 20-mer peptide LVRGGRVKDLPGVRYKIIRG was synthesized (98.3% pure, HPLC) by Proteogenics Strasbourg-France. The sequence of the peptide corresponds to residues 81–100 of the T. maritima S12 protein. The Asp residue (D) corresponds to D89, the site of modification by RimO.
In vitro enzyme assays
All assays were run under nitrogen inside a glove box (Jacomex, NT) containing less than 2 ppm O2. Activity assays were conducted at an enzyme concentration of 0.5 μM in a 100 μL volume in a buffer containing 150 μM SAM, 2.5 mM sodium dithionite, 0.1 M KCl, 25 mM Tris-HCl, pH 8. MiaB assays (Fig. 2a) contained 40–60 μg of tRNA-Phe-enriched (25 μM tRNA-Phe containing 15 μM i6A). RimO assays (Supplementary Figs. 5 and 6) contained 20 μM of peptide. Reactions were carried out at 65 °C and stopped by adding 5 μL of 3.5 M sodium formate, pH 4.3, before exposing them to air and analyzing them as described below.
Analysis of tRNA nucleoside composition by HPLC
The tRNAs-containing mixture was digested to nucleosides. The digested tRNAs (40–60 μg) were loaded onto a Zorbax SB-C-18 column connected to a Agilent-1100 HPLC system. A previously reported gradient profile39 was used to separate the different nucleosides and by-products of the reaction with the following retention times: SAH (22 min.), AdoH (30 min.), MTA (39 min), i6A (46 min.), ms2i6A (54 min.) and mse2i6A (57 min.). SAH, AdoH, MTA and i6A were quantified from standard curves established with the pure commercial compounds. ms2i6A was quantified as follows, using (_methyl_-14C)-SAM (Amersham) (specific activity = 160000 dpm.nmoles−1). In a kinetic experiment, aliquots (100 μL) were withdrawn at t = 0,5, 10, 20, 30 and 45 min. and processed as described above. HPLC ms2i6A peak was collected and counted. The amount of ms2i6A was correlated to the area under the peak at three wavelengths (245, 260 and 285 nm.) giving coherent results. The area at 260 nm was found to be linked to the amount of ms2i6A by the following equation: area260= 0.00152* nmoles (R= 0.9997). A correction factor of 1.3 was applied to this equation so as to estimate the amount of mse2i6A.
Analysis of modified peptide by mass spectrometry
The reaction mixtures were diluted 1000 times with a solution made with 5 % acetonitrile 0.1 % TFA, then washed and separated by a C18 chromatography before FT/MS measurement. The nano-LC-MS analysis was performed using an Ultimate 3000 followed by a LTQ-Orbitrap XL, Thermo Fischer Scientific. The LC method consisted in a 40-minute gradient at a flow rate of 300 nL/min using two solvents: A (5% acetonitrile and 0.1% formic acid in water) and B (80% acetonitrile and 0.08% formic acid in water). The LC system includes a 300 μm × 5 mm PepMap C18 precolumn and a 75 μm × 150 mm C18 column (PepMap C18 phase; Dionex). MS data were acquired using Xcalibur (Thermo Fischer Scientific) in a MS mode only using a 60000 Da resolution and a MS range of 380–1600 Da. For quantitative analyses, chromatograms corresponding to the mass of the 5, 4, and 3 charge states of the peptide, the methylthio and methyl seleno derivatives were extracted, from which the mass of the peptides present in the reaction mixture could be derived (Supplementary Figs. 5 and 6).
Sulfur exchange assays
MiaB3C (50 μM) was incubated either alone or in the presence of 0.1 M CH3SNa for 20 min at 65 °C in a final volume of 0.25 mL in the same buffer used for in vitro enzyme assays. The protein solutions were concentrated to 25 μL using micro-concentrators and diluted to 400 μL with the same buffer. This sequence was repeated four times and the resulting protein solutions analysed for their sulphide content (Supplementary Table 3). The same experiment was run in a buffer containing 500 μM sodium dithionite for reduction of MiaB3C (Supplementary Table 3).
Spectroscopic characterization of Fe-S centers
UV-visible absorption spectra were recorded in quartz cuvettes (optic path: 1 cm) under anaerobic conditions in a glove box on a XL-100 Uvikon spectrophotometer equipped with optical fibers. The UV-vis spectra of MiaB (a), MiaB3C (b) and RimO (c) are shown in Supplementary Fig. 3. X-band EPR spectra were measured on a Bruker ESP-300E EPR spectrometer operating with an ER-4116 dual mode cavity and an Oxford Instruments ESR-9 flow cryostat. The spectra were recorded at a temperature of 10 K at a frequency of 9.65 GHz using 25 μW power and 0.01 mT modulation. Resonances were quantified under non-saturating conditions by double integration against a standard containing 1 mM Cu-EDTA. X-band EPR spectra of reduced MiaB (Supplementary Fig. 4a) or RimO (Supplementary Fig. 4b) were recorded either alone or with a 10-fold excess of CH377SeNa. HYSCORE experiments were performed on a Bruker E-580 X band (frequency = 9.71 GHz) pulsed spectrometer with a Bruker ER4118X dielectric resonator and a continuous flow He cryostat (Oxford Instrument CF935) controlled by an Oxford Instrument temperature controller ITC 503. Experiments were performed at 10 K using the standard four-pulse sequence (π/2-t-π/2-t1-π-t2-π/2-echo) with a nominal pulse width of 16 ns for π/2 and of 32 ns for π pulses, a t value of 128 ns and a pulse repetition rate of 1 kHz. Unwanted echoes were removed by four-step phase cycling. A 128 x 128 dataset was recorded with times t1 and t2 incremented in 24 ns steps from an initial value of 200 ns. The background decay in both dimensions was subtracted using a linear fit followed by apodization with a Hamming window and zero-filling to 2048 points in each dimension. The 2D Fourier Transform magnitude spectrum was then calculated. Spectra were acquired at a magnetic field of 3600 G, corresponding to the g⊥ feature in CW EPR spectra.
DFT calculations
Electronic structures and subsequent hyperfine coupling constants were computed by the ADF2009 density functional code40. The standard Wilk, Vosko and Nusair functional41 was completed by Becke correction for the exchange42 and Perdew correction for the correlation43. It has been shown elsewhere44 that this exchange-correlation potential combination was suitable enough to correctly describe metal-ligand (i.e. iron-sulfur) covalency within Iron-Sulfur clusters. In addition, triple-zeta + two polarization functions were used for all atoms (TZ2P basis set in SCM-ADF nomenclature). Moreover, because we aimed at estimating hyperfine coupling constants, no frozen core approximation was set (i.e., all s atomic functions are polarizable).
Two different molecular models were considered. The first, an intact cluster model, was generated from the crystal structure of _holo Tm_RimO (Fig. 4 and Supplementary Table 4) by replacing the cysteinyl ligands with SCH2CH3 thiolate ligands and adding a SeCH3 ligand to the unliganded iron site 4. In its reduced state (S=1/2), the [4Fe-4S] cluster is made of a mixed-valence pair (of spin +9/2) antiferromagnetically coupled to a ferrous pair of spin −8/2 resulting into the S=1/2 ground state. There are therefore six possible electronic (and spin) arrangements among the four iron atoms. As the spin-coupled S=1/2 state is not directly accessible through DFT mono-determinantal codes, we relied on the computation of spin-uncoupled broken-symmetry states for which the magnetic quantum number Ms=1/2 is constrained while preserving local iron high spins45. Geometrical optimization was conducted of each of the six possible Ms=1/2 broken symmetry states, labeled BSij where “ij” refers to the ferrous pair (i.e., ij = 12, 13, 14, 23, 24 and 34). The second was generated by inserting a SeCH3 moiety into the [4Fe-4S] core, thus replacing one of the internal ‘inorganic’ S ions, resulting in a molecular model with structure [4Fe-3S(SeCH3)](SCH2CH3)4. In this model, the Se ion is linked to the iron ions 2, 3 and 4. Again, geometrical optimization was conducted of each of the six possible Ms=1/2 broken symmetry states, labeled BSij where “ij” refers to the ferrous pair (i.e., ij = 12, 13, 14, 23, 24 and 34).
Protein crystallization
Production of RimO (TM1862) protein from Thermotoga maritima (_Tm_RimO) for crystallization was carried out as part of the high-throughput protein production process of the Northeast Structural Genomics Consortium (NESG)46. The TM1862 protein corresponds to NESG target VR77. The full-length TM1862 gene from Thermotoga maritima (strain DSM8) was cloned into a pET21d (Novagen) derivative, generating plasmid pVR77-21. The recombinant protein contains eight non-native residues (LEHHHHHH) at the C-terminus. The construct was verified by standard DNA sequence analysis. Escherichia coli BL21 (DE3) pMGK cells, a rare codon enhanced strain, were transformed with pVR77-21. A single isolate was cultured in MJ9 minimal media47 supplemented with selenomethionine, lysine, phenylalanine, threonine, isoleucine, leucine and valine for the production of selenomethionine-labeled TM186248. Initial growth was carried out at 37 °C until the OD600 of the culture reached 0.6–0.8. The incubation temperature was then decreased to 17 °C and protein expression was induced by the addition of IPTG (isopropyl-β-D-thiogalactopyranoside) at a final concentration of 1 mM. Following overnight incubation, the cells were harvested by centrifugation.
Selenomethionyl TM1862 was purified by standard methods. Cell pellets were resuspended in lysis buffer (50 mM NaH2PO4 (pH 8.0), 300 mM NaCl, 10 mM imidazole and 5 mM β-mercaptoethanol) and disrupted by sonication. The resulting lysate was clarified by centrifugation at 26,000 × g for 45 min at 4 °C. The supernatant was loaded onto a Ni-NTA column (Qiagen) and eluted in lysis buffer containing 250 mM imidazole. Fractions containing partially purified TM1862 were pooled and buffer conditions providing mono-disperse samples were optimized by analytical gel filtration detected by static light scattering, as described elsewhere46. Preparative gel filtration (Superdex 75, GE Healthcare) was then performed using a buffer containing 10 mM Tris, pH 7.5, 100 mM NaCl, 5 mM DTT, and 0.02% NaN3. The purified TM1862 protein was concentrated to 8–20 mg/ml, flash frozen in aliquots, and used for crystallization screening. Sample purity (>95%) and molecular weight were verified by SDS-PAGE and MALDI-TOF mass spectrometry, respectively. The yield of the purified TM1862 protein was approximately 36 mg/L.
Prior to the protein crystallization, the _apo Tm_RimO was treated with 5 mM DTT for 30 minutes, followed by its reconstitution with 10 equivalents Fe2+ and S2− in a COY anaerobic glove box whose oxygen level was kept below 2 ppm. The resulting _holo Tm_RimO protein was crystallized at 23° C using microbatch method. 2 μL of _holo Tm_RimO were mixed with 2 μL precipitation cocktail consisting of 100 mM CAPS, pH 10.0, 40% PEG 4000, and 100 mM sodium thiosulphate. The RimO crystals appeared after three weeks and grew to full size in four weeks and were directly flash-frozen in liquid propane. While crystals were consistently obtained from five different _holo Tm_RimO protein preparations with stock concentrations ranging from 8–20 mg/mL, only two crystals were obtained that were suitable for diffraction data to be collected at sufficient resolution for structure determination. Although, both crystals were highly mosaic, one diffracted X-ray to a resolution 4 Å, whereas the other, which yielded the structure reported in this paper, diffracted to 3.3 Å. These two crystals were obtained when the excess S2− and Fe2+ ions were not removed from the protein solution prior to its crystallization. In contrast, all crystals that were grown in the absence of the aforementioned excess ions did not diffract X-ray beyond 5 Å. The _holo Tm_RimO crystals belong to space group _P_212121 with cell parameters of _a_=59.72 Å, _b_=86.95 Å, _c_=172.79 Å. The _holo Tm_RimO crystals contain two protomers forming a pseudo-dimer per asymmetric unit.
Crystal structure determination and refinement
A single-wavelength anomalous diffraction (SAD) was collected for each crystal maintained at 100 K on beamline X4C at the National Synchrotron Light Source (NSLS). Data collected at the peak absorption wavelength of selenium were integrated and scaled using the HKL package49 (Supplementary Table 4). The crystal structure of _holo Tm_RimO was determined by the Molecular Replacement method using the Phaser crystallographic software The structure of the truncated _apo Tm_RimO, comprising Radical-SAM and TRAM domains (PDB id: 2QGQ), was used as a search model for structure determination of the _holo Tm_RimO. Subsequently, the N-domain of _holo Tm_RimO (TM1862) was manually built with the program XtalView50 and refined by DEN-assisted refinement procedure implemented in CNS 1.351. Non-crystallographic symmetry restraint was applied at all stages of the refinement for most of UPF0004 and entire Radical-SAM and TRAM domains. The data processing and refinement statistics are summarized in Supplementary Table 4. The structure _holo Tm_RimO has been deposited into Protein Data Bank (PDB id: 4JC0).
Supplementary Material
Supplementary Text and Figures
Acknowledgments
We thank R. Abramowitz, and J. Schwanof for assistance with synchrotron data collection, B. Gibney for advice on Fe-S reconstitution for crystallization, and R. Breslow for discussion of reaction mechanism. We thank Drs. O. Hamelin for GC-MS analysis and J-M Moulis for providing 77Se. This work was supported by NIH Protein Structure Initiative grants U54-GM074958 and U54-GM094597 to the Northeast Structural Genomics Consortium (www.nesg.org), a GIS-CNRS fellowship to SA, ANR Blanc-2010 grant INSERAD, and Région Rhône-Alpes grant CIBLE 2008-2011.
Footnotes
Author contributions. E.M., SA, M.A., and M.F., designed the biochemical and enzymological experiments, which were conducted by E.M. and S.A.. J-M.M. and S.G. designed and conducted the EPR, HYSCORE, and DFT experiments. S.K-J conducted the HPLC-MS experiments. T.B.A., R.X., J.F.H., J. S., and G.T.M. designed the target-selection and protein purification/crystallization pipeline of the Northeast Structural Genomics Consortium, which purified a wide variety of MTTases for this project. F.F. with advice from J.F.H. developed reconstitution methods for crystallization, which was performed by M.H. F.F. solved and refined related crystal structures. M.F., E.M., J.F.H., F.F., M.A., and G.T.M. interpreted the results, while M.F., E.M., J.F.H., and F.F. wrote the manuscript.
Competing financial interests. The authors declare no competing financial interests.
References
- 1.Fontecave M, Ollagnier-de-Choudens S, Mulliez E. Biological radical sulfur insertion reactions. Chemical Reviews. 2003;103:2149–2166. doi: 10.1021/cr020427j. [DOI] [PubMed] [Google Scholar]
- 2.Jenner L, Demeshkina N, Yusupova G, Yusupov M. Structural rearrangements of the ribosome at the tRNA proofreading step. Nature Structural & Molecular Biology. 2010;17:1072–1076. doi: 10.1038/nsmb.1880. [DOI] [PubMed] [Google Scholar]
- 3.Wei FY, et al. Deficit of tRNA(Lys) modification by Cdkal1 causes the development of type 2 diabetes in mice. Journal of Clinical Investigation. 2011;121:3598–3608. doi: 10.1172/JCI58056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Atta M, et al. S-Adenosylmethionine-dependent radical-based modification of biological macromolecules. Current Opinion in Structural Biology. 2010;20:684–692. doi: 10.1016/j.sbi.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 5.Kowalak JA, Walsh KA. beta-methylthio-aspartic acid: Identification of a novel posttranslational modification in ribosomal protein S12 from Escherichia coli. Protein Science. 1996;5:1625–1632. doi: 10.1002/pro.5560050816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, Miller NE. Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucleic Acids Research. 2001;29:1097–1106. doi: 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee KH, et al. Characterization of RimO, a New Member of the Methylthiotransferase Subclass of the Radical SAM Superfamily. Biochemistry. 2009;48:10162–10174. doi: 10.1021/bi900939w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hernandez HL, et al. MiaB, a bifunctional radical-S-adenosylmethionine enzyme involved in the thiolation and methylation of tRNA, contains two essential [4Fe-4S] clusters. Biochemistry. 2007;46:5140–5147. doi: 10.1021/bi7000449. [DOI] [PubMed] [Google Scholar]
- 9.Arragain S, et al. Identification of Eukaryotic and Prokaryotic Methylthiotransferase for Biosynthesis of 2-Methylthio-N-6-threonylcarbamoyladenosine in tRNA. Journal of Biological Chemistry. 2010;285:28425–28433. doi: 10.1074/jbc.M110.106831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arragain S, et al. Post-translational Modification of Ribosomal Proteins. Structural and functional characterization of RimO from Thermotoga maritima, a Radical S-Adenosylmethionine methylthiotransferase. Journal of Biological Chemistry. 2010;285:5792–5801. doi: 10.1074/jbc.M109.065516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anantharaman V, Koonin EV, Aravind L. TRAM, a predicted RNA-binding domain, common to tRNA uracil methylation and adenine thiolation enzymes. Fems Microbiology Letters. 2001;197:215–21. doi: 10.1111/j.1574-6968.2001.tb10606.x. [DOI] [PubMed] [Google Scholar]
- 12.Booker SJ, Cicchillo RM, Grove TL. Self-sacrifice in radical S-adenosylmethionine proteins. Current Opinion in Chemical Biology. 2007;11:543–552. doi: 10.1016/j.cbpa.2007.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ugulava NB, Sacanell CJ, Jarrett JT. Spectroscopic changes during a single turnover of biotin synthase: Destruction of a [2Fe-2S] cluster accompanies sulfur insertion. Biochemistry. 2001;40:8352–8358. doi: 10.1021/bi010463x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frey PA, Hegeman AD, Ruzicka FJ. The radical SAM superfamily. Critical Reviews in Biochemistry and Molecular Biology. 2008;43:63–88. doi: 10.1080/10409230701829169. [DOI] [PubMed] [Google Scholar]
- 15.Pierrel F, Douki T, Fontecave M, Atta M. MiaB protein is a bifunctional radical-S-adenosylmethionine enzyme involved in thiolation and methylation of tRNA. Journal of Biological Chemistry. 2004;279:47555–47563. doi: 10.1074/jbc.M408562200. [DOI] [PubMed] [Google Scholar]
- 16.Vey JL, Drennan CL. Structural Insights into Radical Generation by the Radical SAM Superfamily. Chemical Reviews. 2011;111:2487–2506. doi: 10.1021/cr9002616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duschene KS, Veneziano SE, Silver SC, Broderick JB. Control of radical chemistry in the AdoMet radical enzymes. Current Opinion in Chemical Biology. 2009;13:74–83. doi: 10.1016/j.cbpa.2009.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Q, Liu W. Complex Biotransformations Catalyzed by Radical S-Adenosylmethionine Enzymes. Journal of Biological Chemistry. 2011;286:30245–30252. doi: 10.1074/jbc.R111.272690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Branden C, Tooze J. Introduction to Protein Structure. Garland Publishing; NY, NY: 1999. [Google Scholar]
- 20.Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 38:W545–9. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andreeva A, et al. Data growth and its impact on the SCOP database: new developments. Nucleic Acids Research. 2008;36:D419–D425. doi: 10.1093/nar/gkm993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baikalov I, et al. Structure of the Escherichia coli response regulator NarL. Biochemistry. 1996;35:11053–11061. doi: 10.1021/bi960919o. [DOI] [PubMed] [Google Scholar]
- 23.Schnell R, Agren D, Schneider G. 1.9 angstrom structure of the signal receiver domain of the putative response regulator NarL from Mycobacterium tuberculosis. Acta Crystallographica Section F-Structural Biology and Crystallization Communications. 2008;64:1096–1100. doi: 10.1107/S1744309108035203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maris AE, et al. Dimerization allows DNA target site recognition by the NarL response regulator. Nature Structural Biology. 2002;9:771–778. doi: 10.1038/nsb845. [DOI] [PubMed] [Google Scholar]
- 25.Porter SL, Wadhams GH, Armitage JP. Signal processing in complex chemotaxis pathways. Nature Reviews Microbiology. 2011;9:153–165. doi: 10.1038/nrmicro2505. [DOI] [PubMed] [Google Scholar]
- 26.Berkovitch F, Nicolet Y, Wan JT, Jarrett JT, Drennan CL. Crystal structure of biotin synthase, an S-adenosylmethionine-dependent radical enzyme. Science. 2004;303:76–79. doi: 10.1126/science.1088493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanzelmann P, Schindelin H. Crystal structure of the S-adenosylmethionine-dependent enzyme MoaA and its implications for molybdenum cofactor deficiency in humans. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:12870–12875. doi: 10.1073/pnas.0404624101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lees NS, et al. ENDOR Spectroscopy Shows That Guanine N1 Binds to [4Fe-4S] Cluster II of the S-Adenosylmethionine-Dependent Enzyme MoaA: Mechanistic Implications. Journal of the American Chemical Society. 2009;131:9184–9185. doi: 10.1021/ja903978u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng B, Chen XD, Zheng SL, Holm RH. Selenium as a Structural Surrogate of Sulfur: Template-Assisted Assembly of Five Types of Tungsten-Iron-Sulfur/Selenium Clusters and the Structural Fate of Chalcogenide Reactants. Journal of the American Chemical Society. 2012;134:6479–6490. doi: 10.1021/ja3010539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilker JJ, Lippard SJ. Methylation of iron-sulfur complexes by trimethyl phosphate. Inorganic Chemistry. 1999;38:3569–3574. doi: 10.1021/ic9808899. [DOI] [PubMed] [Google Scholar]
- 31.Petrey D, Honig B. GRASP2: visualization, surface properties, and electrostatics of macromolecular structures and sequences. Methods Enzymol. 2003;374:492–509. doi: 10.1016/S0076-6879(03)74021-X. [DOI] [PubMed] [Google Scholar]
- 32.Iwig DF, Booker SJ. Insight into the polar reactivity of the onium chalcogen analogues of S-adenosyl-L-methionine. Biochemistry. 2004;43:13496–13509. doi: 10.1021/bi048693+. [DOI] [PubMed] [Google Scholar]
- 33.Bui BTS, Mattioli TA, Florentin D, Bolbach G, Marquet A. Escherichia coli biotin synthase produces selenobiotin. Further evidence of the involvement of the [2Fe-2S](2+) cluster in the sulfur insertion step. Biochemistry. 2006;45:3824–3834. doi: 10.1021/bi052388m. [DOI] [PubMed] [Google Scholar]
- 34.Syper L, Mlochowski J. The Convenient Syntheses of Organoselenium Reagents. Synthesis-Stuttgart. 1984:439–442. [Google Scholar]
- 35.Fish WW. Rapid Colorimetric Micromethod for the Quantitation of Complexed Iron in Biological Samples. Methods in Enzymology. 1988;158:357–364. doi: 10.1016/0076-6879(88)58067-9. [DOI] [PubMed] [Google Scholar]
- 36.Beinert H. Semi-Micro Methods for Analysis of Labile Sulfide and of Labile Sulfide Plus Sulfane Sulfur in Unusually Stable Iron Sulfur Proteins. Analytical Biochemistry. 1983;131:373–378. doi: 10.1016/0003-2697(83)90186-0. [DOI] [PubMed] [Google Scholar]
- 37.Then J, Truper HG. Sulfide Oxidation in Ectothiorhodospira-Abdelmalekii - Evidence for the Catalytic Role of Cytochrome-C-551. Archives of Microbiology. 1983;135:254–258. [Google Scholar]
- 38.Pierrel F, Hernandez HL, Johnson MK, Fontecave M, Atta M. MiaB protein from Thermotoga maritima - Characterization of an extremely thermophilic tRNA-methylthiotransferase. Journal of Biological Chemistry. 2003;278:29515–29524. doi: 10.1074/jbc.M301518200. [DOI] [PubMed] [Google Scholar]
- 39.Gehrke CW, Kuo KC. Ribonucleoside Analysis by Reversed-Phase High-Performance Liquid-Chromatography. Journal of Chromatography. 1989;471:3–36. doi: 10.1016/s0021-9673(00)94152-9. [DOI] [PubMed] [Google Scholar]
- 40.Velde GT, Baerends EJ. Numerical-Integration for Polyatomic Systems. Journal of Computational Physics. 1992;99:84–98. [Google Scholar]
- 41.Vosko SH, Wilk L, Nusair M. Accurate Spin-Dependent Electron Liquid Correlation Energies for Local Spin-Density Calculations - a Critical Analysis. Canadian Journal of Physics. 1980;58:1200–1211. [Google Scholar]
- 42.Becke AD. Density-Functional Exchange-Energy Approximation with Correct Asymptotic-Behavior. Physical Review A. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 43.Perdew JP. Density-Functional Approximation for the Correlation-Energy of the Inhomogeneous Electron-Gas. Physical Review B. 1986;33:8822–8824. doi: 10.1103/physrevb.33.8822. [DOI] [PubMed] [Google Scholar]
- 44.Szilagyi RK, Winslow MA. On the accuracy of density functional theory for iron - Sulfur clusters. Journal of Computational Chemistry. 2006;27:1385–1397. doi: 10.1002/jcc.20449. [DOI] [PubMed] [Google Scholar]
- 45.Noodleman L, Peng CY, Case DA, Mouesca JM. Orbital Interactions, Electron Delocalization and Spin Coupling in Iron-Sulfur Clusters. Coordination Chemistry Reviews. 1995;144:199–244. [Google Scholar]
- 46.Acton TB, et al. Robotic cloning and Protein Production Platform of the Northeast Structural Genomics Consortium. Nuclear Magnetic Resonance of Biological Macromolecules, Part C. 2005;394:210-+. doi: 10.1016/S0076-6879(05)94008-1. [DOI] [PubMed] [Google Scholar]
- 47.Jansson M, et al. High-level production of uniformly N-15- and C-13-enriched fusion proteins in Escherichia coli. Journal of Biomolecular NMR. 1996;7:131–141. doi: 10.1007/BF00203823. [DOI] [PubMed] [Google Scholar]
- 48.Doublie S, et al. Crystallization and preliminary X-ray analysis of the 9 kDa protein of the mouse signal recognition particle and the selenomethionyl-SRP9. FEBS Letters. 1996;384:219–221. doi: 10.1016/0014-5793(96)00316-x. [DOI] [PubMed] [Google Scholar]
- 49.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography, Pt A. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 50.McRee DE. XtalView Xfit - A versatile program for manipulating atomic coordinates and electron density. Journal of Structural Biology. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 51.Brunger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Text and Figures