The B12-Radical SAM Enzyme PoyC Catalyzes Valine Cβ-Methylation during Polytheonamide Biosynthesis (original) (raw)
Abstract
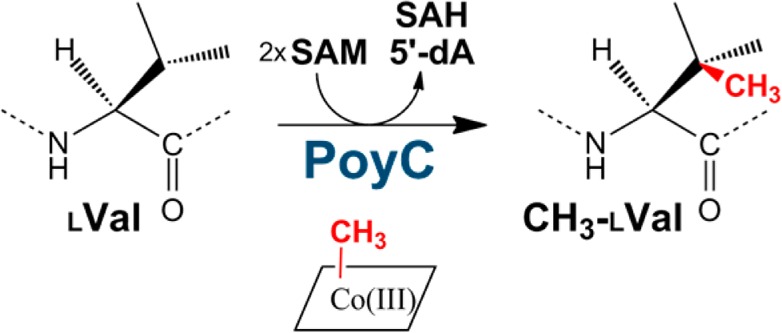
Genomic and metagenomic investigations have recently led to the delineation of a novel class of natural products called ribosomally synthesized and post-translationally modified peptides (RiPPs). RiPPs are ubiquitous among living organisms and include pharmaceutically relevant compounds such as antibiotics and toxins. A prominent example is polytheonamide A, which exhibits numerous post-translational modifications, some of which were unknown in ribosomal peptides until recently. Among these post-translational modifications, C-methylations have been proposed to be catalyzed by two putative radical _S_-adenosylmethionine (rSAM) enzymes, PoyB and PoyC. Here we report the in vitro activity of PoyC, the first B12-dependent rSAM enzyme catalyzing peptide Cβ-methylation. We show that PoyC catalyzes the formation of _S_-adenosylhomocysteine and 5′-deoxyadenosine and the transfer of a methyl group to l-valine residue. In addition, we demonstrate for the first time that B12-rSAM enzymes have a tightly bound MeCbl cofactor that during catalysis transfers a methyl group originating from _S_-adenosyl-l-methionine. Collectively, our results shed new light on polytheonamide biosynthesis and the large and emerging family of B12-rSAM enzymes.
B12-dependent radical _S_-adenosylmethionine (B12-rSAM) enzymes1,2 constitute one of the largest groups of enzymes within the rSAM enzyme superfamily, which contains more than 140 000 members.3,4 They have been identified in the biosynthetic pathways of many natural products, including several families of ribosomally synthesized and post-translationally modified peptides (RiPPs) such as thiostrepton,1,2,5 polytheonamides,6 and bottromycins.7 B12-rSAM enzymes are also involved in the biosynthesis of a wide range of antibiotics produced by Actinomycetes: carbapenem,8 chondrochloren,9 clorobiocin,10 fortimicin,11 fosfomycin,12 gentamicin,13 mitomycin,14 moenomycin,15 novobiocin,16 pactamycin,17 and l-phosphinothricin.18 However, their function often remains controversial, and the first in vitro studies have been reported only recently.2,18
B12-rSAM enzymes have been proposed to catalyze chemically challenging C- and P-methyl transfer reactions. Nevertheless, our understanding of their functions and catalytic mechanisms remains very limited. In an effort to gain insights into the unique chemistry of these enzymes and the biosynthesis of polytheonamides, we undertook a study of PoyB and PoyC, the two putative B12-rSAM enzymes encoded in the poy operon (Figure 1a).6
Figure 1.
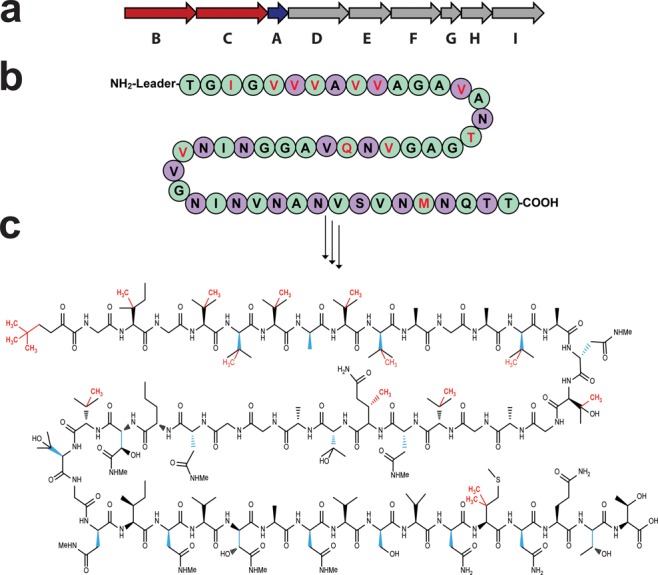
(a) Biosynthetic gene cluster of polytheonamide A. The two predicted B12-rSAM enzymes PoyB and PoyC (in red) and the precursor peptide PoyA (in blue) are indicated. (b) Sequence of PoyA showing the core peptide. Residues in red indicate C-methylated residues, while purple circles denote epimerized residues. (c) Structure of polytheonamide A. d-Configured residues are shown in blue, and methyl groups proposed to be inserted by PoyB and/or PoyC are shown in red.
Polytheonamides are the founding members of the so-called proteusins. They have been shown to contain 48 post-translational modifications, with 35 predicted to be catalyzed by three rSAM enzymes: PoyB, PoyC, and PoyD (Figure 1b,c). By an in vivo approach, it has been shown that PoyD is a novel type of rSAM enzyme generating d-configured amino acid residues.6 Although no experimental evidence was available, PoyB and PoyC were predicted to be responsible for the 13 Cβ-methylations and the formation of the unique N-terminal _tert_-butyl group (Figure 1c). Interestingly, recent characterization of the bottromycin biosynthetic cluster led to the identification of three B12-rSAM enzymes predicted to catalyze similar Cβ-methyl transfer reactions.7 Strikingly, in both operons the B12-rSAM enzymes do not contain the canonical rSAM motif but rather a Cx7Cx2C motif.
Our first attempts to express PoyB and PoyC in Escherichia coli proved to be largely unsuccessful, as reported for the majority of the B12-rSAM enzymes to date. Indeed, most B12-rSAM enzymes, including the P-methylase PhpK18 and the C-methylases Fom319 and GenK,13 had to be artificially refolded in the presence of cobalamin, sulfide, and iron. To isolate PoyB and PoyC, we adapted our expression conditions using the ethanolamine-M9 medium recently described for the expression of two B12-rSAM enzymes, TsrM20 and ThnK.8 Under these conditions, PoyB remained insoluble whereas PoyC was purified in a satisfactory yield (Figure 2a). The UV–vis spectrum of the aerobically purified enzyme exhibited a maximum absorption at ∼260/270 nm and two small shoulders at ∼420 and ∼464 nm (Figure 2b,c). The shoulder at 420 nm indicated the presence of an iron–sulfur center, while the latter was diagnostic of base-off alkylcobalamins.21,22
Figure 2.
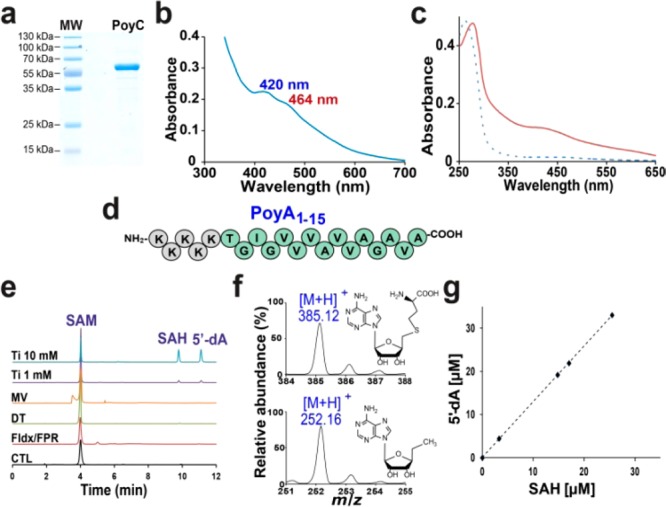
(a) Gel electrophoresis analysis of PoyC (SDS PAGE 12%). (b) UV–vis spectrum of as-isolated PoyC. (c) UV–vis spectra of PoyC before (dashed line) and after (solid line) iron–sulfur reconstitution. (d) Sequence of the PoyA1–15 peptide used as the substrate. (e) HPLC analysis of PoyC reactions after incubation under anaerobic conditions with SAM (2 mM), DTT (6 mM), and PoyA1–15 (1 mM) in the presence of various reductants, including sodium dithionite (DT), flavodoxin/flavodoxin reductase/NADPH (Fldx/FPR), methyl viologen/NADPH (MV), titanium citrate (Ti), or in the absence of reductant (CTL) (UV detection at 257 nm). See the Supporting Information (SI) for the experimental conditions. (f) MS analyses of SAH (upper trace) and 5′-dA (lower trace) produced by PoyC. (g) Correlation between SAH and 5′-dA produced by PoyC. Analyses were performed between 0 and 20 min as described in the SI.
After anaerobic iron–sulfur reconstitution, the maximum absorption shifted to ∼280 nm and the absorption band at ∼420 nm increased (Figure 2c). Iron–sulfur determination indicated that PoyC contained 11.8 ± 0.3 mol of iron and 13.6 ± 0.8 mol of sulfide per polypeptide chain, supporting the presence of one to three [4Fe–4S] centers. Sequence alignment between PoyB and PoyC indicated the presence of 11 conserved cysteine residues (Supplementary Figure 1), which is compatible with the presence of an additional [4Fe–4S] center, although, as reported for ThnK8 and Fom3,19 excess iron and sulfide could not be ruled out.
Because PoyC is predicted to possess a RiPP precursor peptide recognition element,23 PoyA was the most suitable substrate. However, it has been shown that PoyA is not accessible by heterologous expression,6 hampering our understanding of polytheonamide biosynthesis. As shown in Figure 1c, most of the Cβ-methylated residues are located in the N-terminus region of polytheonamide A. We thus synthesized, as putative substrates, several peptides with a sequence corresponding to the first amino acid residues found in polytheonamide A. However, because of the intrinsic hydrophobic nature of these peptides (containing mainly valine and alanine residues), we obtained only low amounts of peptides, which were largely insoluble. To improve their solubility, we added a polylysine stretch upstream of the polytheonamide sequence. With this strategy, we obtained a synthetic peptide (PoyA1–15) that proved to be compatible with aqueous conditions and amenable to analysis by mass spectrometry (MS) (Figure 2d).
In order to preserve the cobalamin cofactor(s) present in PoyC, we incubated the anaerobically reconstituted enzyme in the dark in the presence of _S_-adenosyl-l-methionine (SAM), dithiothreitol (DTT), and various reducing systems. The reducing systems assayed were NADPH/flavodoxin/flavodoxin reductase (Fldx/FPR), sodium dithionite (DT), and methyl viologen/NADPH (MV) (Figure 2e). To date, MV has been reported to be the most efficient reducing system, successfully used with GenK13 and ThnK.8 Fldx/FPR proved to sustain activity but only at low levels for GenK. DT has been used with most of the B12-rSAM enzymes investigated to date,8,13,18,19,24 but it leads to poor activities, in line with its well-known deleterious effects on cobalamins and cobalamin-dependent enzymes.25 With PoyC, none of these reductants worked, as shown by the absence of the expected products, 5′-deoxyadenosine (5′-dA) and _S_-adenosylhomocysteine (SAH) (Figure 2e).
We then tried Ti(III) citrate (Ti), which has been used as a reducing agent for B12-dependent methionine synthase.26 In the presence of Ti, we monitored the formation of two products derived from SAM (Figure 2e), which were identified as SAH and 5′-dA, as demonstrated by their retention times, UV–vis spectra, and molecular weights of 385.1 and 252.1 Da, respectively (Figure 2f). Increasing the concentration of Ti led to an increase in the enzyme activity, further demonstrating the dependence of the reaction on an external electron donor. Kinetic analysis revealed a strict correlation between SAH production and 5′-dA production (Figure 2g) with _k_cat ∼9 min–1. A similar correlation between SAH and 5′-dA has been reported for GenK, which exhibited a lower in vitro activity with an estimated _k_cat of ∼48 min–1.
High-resolution LC–MS analysis showed that the PoyA1–15 peptide ([M + 2H]2+ = 962.129) was converted into a new peptide, PoyA1–15* ([M + 2H]2+ = 970.646), only in the presence of PoyC, methyl-deuterated SAM (CD3-SAM), and Ti (Figure 3a,b). This new peptide had a mass increment of 17.034 Da, which perfectly matched the replacement of one H atom by a CD3 group (theoretical mass shift [M + H]+ = 17.034). This result demonstrated that PoyC catalyzes a methyl transfer reaction from CD3-SAM to the PoyA1–15 peptide. Under our assay conditions, ∼6% methylated peptide was produced, hampering reliable kinetic analysis. However, as recently reported for ThnK,8 the presence of the substrate was not required for PoyC to catalyze the reductive cleavage of SAM.
Figure 3.
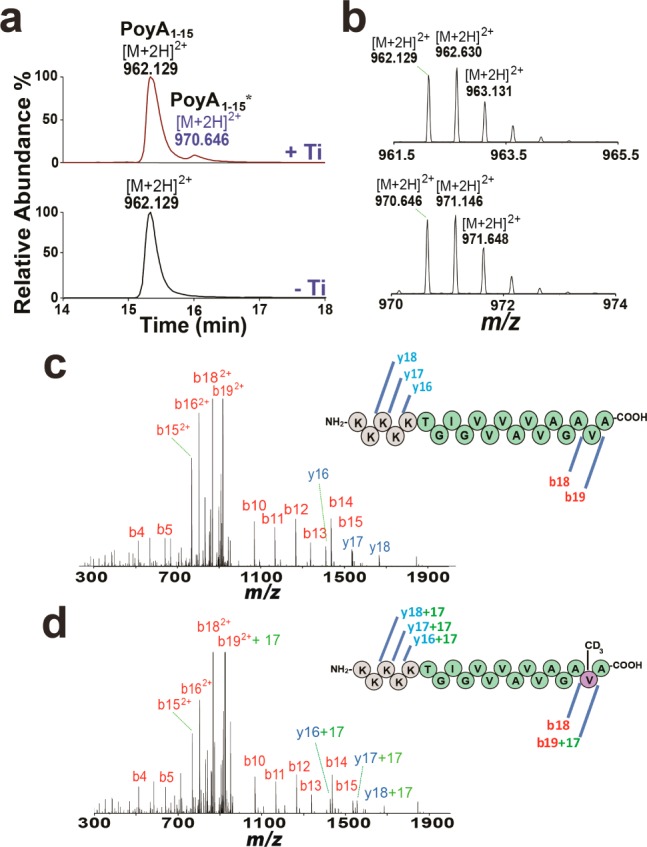
(a) LC–MS analysis of PoyA1–15 incubated with PoyC and CD3-SAM under anaerobic conditions in the presence (+Ti) or absence (−Ti) of titanium citrate. See the SI for the experimental conditions. (b) High-resolution LC–MS analyses of PoyA1–15 (upper trace) and PoyA1–15* (lower trace). (c, d) High-resolution LC–MS/MS analyses of (c) PoyA1–15 and (d) PoyA1–15*. See Supplementary Figure 2 for the full spectra.
LC–MS/MS analysis of the modified PoyA1–15* peptide revealed the positioning of the methyl group on the penultimate amino acid, l-Val14 (Figure 3c,d, Supplementary Figure 2, and Supplementary Tables 1 and 2). Interestingly, Val14 is one of the few C-methylated and epimerized residues found in native polytheonamide A (Figure 1b). Previous studies have shown that in vivo, heterologous coexpression of PoyA with PoyD6 leads to epimerization of amino acid residues, suggesting that epimerization is the first step in polytheonamide A biosynthesis. However, since E. coli is unable to produce de novo cobalamin, coexpressions of PoyA with PoyB or PoyC were inconclusive. To test whether PoyC acts prior to or after epimerization, we synthesized a peptide containing one d-Val at position 14 (PoyA1–15VD). As shown, introduction of one d-Val led to methyl transfer inhibition (Figure 4a). This result further supports the hypothesis that PoyC catalyzes methyl transfer only on l-amino acids. Consistent with this hypothesis, most of the C-methylated residues are l-configured in polytheonamide A (Figure 1b).
Figure 4.
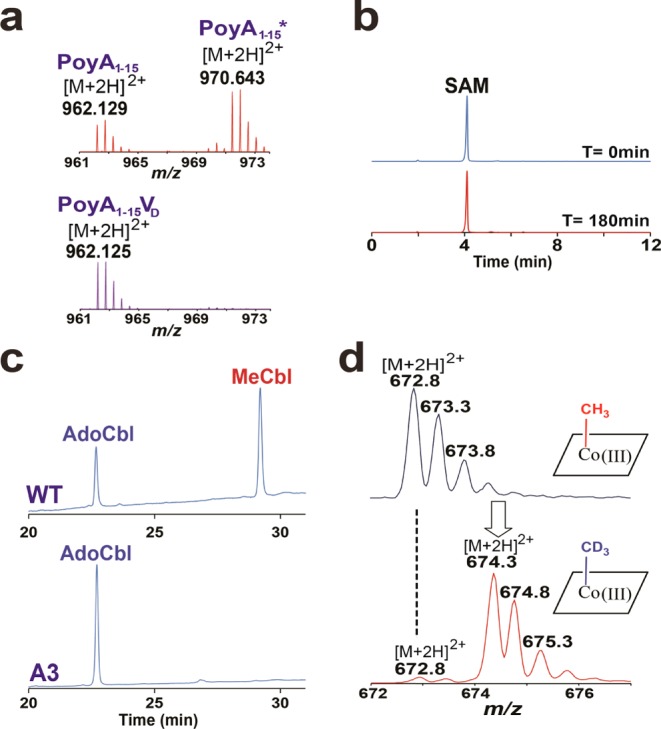
(a) LC–MS analyses of PoyA1–15 (upper trace) and PoyA1–15VD (lower trace) after incubation in the presence of PoyC. See the SI for the experimental conditions. (b) SAM cleavage assay performed with the PoyC Ax7Ax2A mutant (UV detection at 257 nm). Samples were assayed at T = 0 min (upper trace) and after 180 min of incubation with the mutant enzyme (lower trace). See the SI for the experimental conditions. (c) HPLC analysis of the cobalamin derivatives present in PoyC (WT) and the Ax7Ax2A mutant (A3) (UV detection at 278 nm). (d) LC–MS analyses of MeCbl present in PoyC at T = 0 h (upper trace) and after 1 h of incubation with its peptide substrate, Ti (10 mM) and CD3-SAM (2 mM) (lower trace).
PoyC possesses a predicted unusual rSAM motif: Cx7Cx2C. To probe its function, we generated the corresponding triple variant Ax7Ax2A (Supplementary Figure 3). As expected, this mutant was unable to produce 5′-dA and to transfer a methyl group to the substrate (Figure 4b). However, contrary to TsrM,1,2 this mutant was also impaired for SAH production, suggesting different functions for the [4Fe–4S] center in these two B12-rSAM enzymes.
Because PoyC is the first B12-rSAM enzyme isolated natively loaded with a B12 cofactor (i.e., without addition of cobalamin derivatives during enzyme refolding, purification or reaction), we aimed to characterize its B12 content. PoyC, carefully handled in the dark, was found to contain essentially two B12 derivatives identified as AdoCbl and MeCbl by their HPLC profiles (Figure 4c and Supplementary Figure 4), their UV–vis spectra and MS analyses (Supplementary Figure 5). PoyC proved to contain 0.27 ± 0.08 mol of MeCbl and 0.07 ± 0.02 mol of AdoCbl per polypeptide chain.
The apparent nonselectivity of PoyC for the upper axial ligand of cobalamin was surprising since the methionine synthase from E. coli was purified as a mixture of forms containing hydroxo- or methylcobalamin27 while the “corrinoid iron–sulfur protein” was isolated as a cob(II)alamin complex.28,29 It is well-known that when supplied with cobalamin derivatives, E. coli produces large amounts of AdoCbl,30 which was likely incorporated during the heterologous expression of PoyC. Interestingly, the Ax7Ax2A mutant also contained AdoCbl, but we did not detect MeCbl (Figure 4c). This result suggests a critical role of the [4Fe–4S] center in cobalamin methylation. It is also consistent with the fact that the Ax7Ax2A mutant is unable to produce SAH and thus to transfer the methyl group from SAM to cobalamin.
As shown, the AdoCbl/MeCbl content of the enzyme did not change during the reaction (Supplementary Figure 6). However, MeCbl was almost entirely converted into CD3-Cbl during the reaction (Figure 4d), establishing that the enzyme-bound cobalamin is recycled from SAM during catalysis.
Collectively, our results demonstrate that PoyC is a B12-rSAM enzyme with an unusual Cx7Cx2C radical SAM motif identified only in a putative rSAM enzyme31 and the predicted bottromycin methyltransferases.7 We purified PoyC with two bound alkylcobalamins: AdoCbl and MeCbl. On the basis of the UV–vis spectrum, it appears that these cobalamins have a base-off coordination, similar to what has recently been proposed for TsrM.20 Using Ti as the reducing system, we have also demonstrated that PoyC is a peptide C-methyltransferase catalyzing the reductive cleavage of SAM. Ti has been extensively used for the reactivation of methionine synthase26 and recently with a B12-dependent P-methyltransferase.24 However, in this latter case, no 5′-dA or SAH was reported.
In agreement with our results, it is reasonable to assume that PoyC catalyzes the reductive cleavage of SAM into the 5′-dA radical. This radical species likely abstracts a _C_β-H atom,32 generating a carbon-centered radical intermediate. Currently, how the methyl group is attached and what are the physiological oxidation states of cobalamin remain open questions. The homolytic cleavage of MeCbl and the formation of a methyl radical intermediate, as elegantly reported in methyl-coenzyme M reductase,33 would provide a reasonable solution to attach the methyl group to the carbon-centered radical. Cob(II)alamin, after reduction to cob(I)alamin, could react with SAM to regenerate MeCbl. We have previously shown that TsrM catalyzes methyl transfer to sp2 carbon atoms2 and rigorously demonstrated that TsrM, contrary to PoyC, does not catalyze the reductive cleavage of SAM or abstracts a substrate H atom.1,2 It thus appears that, depending on the hybridization of the carbon atom (sp2 vs sp3), B12-rSAM enzymes have evolved different mechanisms requiring or not requiring the formation of 5′-dA radical.
Finally, our data show that PoyC catalyzes methyl transfer only on l-amino acid residues. This result indicates that methylation occurs prior epimerization and suggests that PoyB is likely responsible for the synthesis of the N-terminal _tert_-butyl group in polytheonamide A, although other roles cannot be ruled out yet.
Acknowledgments
This work was supported by the European Research Council (Consolidator Grant 617053 to O.B.). High-resolution MS analyses were performed on the INRA PAPPSO proteomics platform.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b06697.
- Experimental procedures and supplementary figures (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Benjdia A.; Pierre S.; Gherasim C.; Guillot A.; Carmona M.; Amara P.; Banerjee R.; Berteau O. Nat. Commun. 2015, 6, 8377. 10.1038/ncomms9377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierre S.; Guillot A.; Benjdia A.; Sandstrom C.; Langella P.; Berteau O. Nat. Chem. Biol. 2012, 8 (12), 957–9. 10.1038/nchembio.1091. [DOI] [PubMed] [Google Scholar]
- Broderick J. B.; Duffus B. R.; Duschene K. S.; Shepard E. M. Chem. Rev. 2014, 114 (8), 4229–317. 10.1021/cr4004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjdia A.; Berteau O. Biochem. Soc. Trans. 2016, 44 (1), 109–15. 10.1042/BST20150191. [DOI] [PubMed] [Google Scholar]
- Kelly W. L.; Pan L.; Li C. J. Am. Chem. Soc. 2009, 131 (12), 4327–34. 10.1021/ja807890a. [DOI] [PubMed] [Google Scholar]
- Freeman M. F.; Gurgui C.; Helf M. J.; Morinaka B. I.; Uria A. R.; Oldham N. J.; Sahl H. G.; Matsunaga S.; Piel J. Science 2012, 338 (6105), 387–90. 10.1126/science.1226121. [DOI] [PubMed] [Google Scholar]
- Huo L.; Rachid S.; Stadler M.; Wenzel S. C.; Muller R. Chem. Biol. 2012, 19 (10), 1278–87. 10.1016/j.chembiol.2012.08.013. [DOI] [PubMed] [Google Scholar]
- Marous D. R.; Lloyd E. P.; Buller A. R.; Moshos K. A.; Grove T. L.; Blaszczyk A. J.; Booker S. J.; Townsend C. A. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (33), 10354–8. 10.1073/pnas.1508615112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachid S.; Scharfe M.; Blocker H.; Weissman K. J.; Muller R. Chem. Biol. 2009, 16 (1), 70–81. 10.1016/j.chembiol.2008.11.005. [DOI] [PubMed] [Google Scholar]
- Westrich L.; Heide L.; Li S. M. ChemBioChem 2003, 4 (8), 768–73. 10.1002/cbic.200300609. [DOI] [PubMed] [Google Scholar]
- Dairi T.; Ohta T.; Hashimoto E.; Hasegawa M. Mol. Gen Genet 1992, 236 (1), 39–48. 10.1007/BF00279641. [DOI] [PubMed] [Google Scholar]
- Woodyer R. D.; Li G.; Zhao H.; van der Donk W. A. Chem. Commun. (Cambridge, U. K.) 2007, 4, 359–61. 10.1039/B614678C. [DOI] [PubMed] [Google Scholar]
- Kim H. J.; McCarty R. M.; Ogasawara Y.; Liu Y. N.; Mansoorabadi S. O.; Levieux J.; Liu H. W. J. Am. Chem. Soc. 2013, 135, 8093–6. 10.1021/ja312641f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y.; Varoglu M.; Sherman D. H. Chem. Biol. 1999, 6 (4), 251–63. 10.1016/S1074-5521(99)80040-4. [DOI] [PubMed] [Google Scholar]
- Ostash B.; Walker S. Nat. Prod. Rep. 2010, 27 (11), 1594–617. 10.1039/c001461n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffensky M.; Muhlenweg A.; Wang Z. X.; Li S. M.; Heide L. Antimicrob. Agents Chemother. 2000, 44 (5), 1214–22. 10.1128/AAC.44.5.1214-1222.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo F.; Kasama Y.; Hirayama T.; Eguchi T. J. Antibiot. 2007, 60 (8), 492–503. 10.1038/ja.2007.63. [DOI] [PubMed] [Google Scholar]
- Werner W. J.; Allen K. D.; Hu K.; Helms G. L.; Chen B. S.; Wang S. C. Biochemistry 2011, 50 (42), 8986–8. 10.1021/bi201220r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen K. D.; Wang S. C. Arch. Biochem. Biophys. 2014, 543, 67–73. 10.1016/j.abb.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaszczyk A. J.; Silakov A.; Zhang B.; Maiocco S. J.; Lanz N. D.; Kelly W. L.; Elliott S. J.; Krebs C.; Booker S. J. J. Am. Chem. Soc. 2016, 138 (10), 3416–26. 10.1021/jacs.5b12592. [DOI] [PubMed] [Google Scholar]
- Kim J.; Gherasim C.; Banerjee R. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (38), 14551–4. 10.1073/pnas.0805989105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R. V.; Frasca V.; Ballou D. P.; Matthews R. G. Biochemistry 1990, 29 (50), 11101–9. 10.1021/bi00502a013. [DOI] [PubMed] [Google Scholar]
- Burkhart B. J.; Hudson G. A.; Dunbar K. L.; Mitchell D. A. Nat. Chem. Biol. 2015, 11 (8), 564–70. 10.1038/nchembio.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen K. D.; Wang S. C. Biochim. Biophys. Acta, Proteins Proteomics 2014, 1844 (12), 2135–2144. 10.1016/j.bbapap.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews R. G.; Koutmos M.; Datta S. Curr. Opin. Struct. Biol. 2008, 18 (6), 658–66. 10.1016/j.sbi.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z.; Chakraborty S.; Banerjee R. J. Biol. Chem. 1995, 270 (33), 19246–9. 10.1074/jbc.270.33.19246. [DOI] [PubMed] [Google Scholar]
- Banerjee R. V.; Johnston N. L.; Sobeski J. K.; Datta P.; Matthews R. G. J. Biol. Chem. 1989, 264 (23), 13888–95. [PubMed] [Google Scholar]
- Menon S.; Ragsdale S. W. Biochemistry 1998, 37 (16), 5689–98. 10.1021/bi9727996. [DOI] [PubMed] [Google Scholar]
- Kung Y.; Ando N.; Doukov T. I.; Blasiak L. C.; Bender G.; Seravalli J.; Ragsdale S. W.; Drennan C. L. Nature 2012, 484 (7393), 265–9. 10.1038/nature10916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escalante-Semerena J. C.; Warren M. J. EcoSal Plus 2008, 10.1128/ecosalplus.3.6.3.8. [DOI] [PubMed] [Google Scholar]
- Hiratsuka T.; Suzuki H.; Kariya R.; Seo T.; Minami A.; Oikawa H. Angew. Chem., Int. Ed. 2014, 53 (21), 5423–6. 10.1002/anie.201402623. [DOI] [PubMed] [Google Scholar]
- Benjdia A.; Leprince J.; Sandstrom C.; Vaudry H.; Berteau O. J. Am. Chem. Soc. 2009, 131 (24), 8348–9. 10.1021/ja901571p. [DOI] [PubMed] [Google Scholar]
- Wongnate T.; Sliwa D.; Ginovska B.; Smith D.; Wolf M. W.; Lehnert N.; Raugei S.; Ragsdale S. W. Science 2016, 352 (6288), 953–8. 10.1126/science.aaf0616. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.