Occurrence, Biological Consequences, and Human Health Relevance of Oxidative Stress-Induced DNA Damage (original) (raw)
. Author manuscript; available in PMC: 2017 Dec 19.
Abstract
A variety of endogenous and exogenous agents can induce DNA damage and lead to genomic instability. Reactive oxygen species (ROS), an important class of DNA damaging agents, are constantly generated in cells as a consequence of endogenous metabolism, infection/inflammation, and/or exposure to environmental toxicants. A wide array of DNA lesions can be induced by ROS directly, including single-nucleobase lesions, tandem lesions, and hypochlorous acid (HOCl)/hypobromous acid (HOBr)-derived DNA adducts. ROS can also lead to lipid peroxidation, whose byproducts can also react with DNA to produce exocyclic DNA lesions. A combination of bioanalytical chemistry, synthetic organic chemistry, and molecular biology approaches have provided significant insights into the occurrence, repair, and biological consequences of oxidatively induced DNA lesions. The involvement of these lesions in the etiology of human diseases and aging was also investigated in the past several decades, suggesting that the oxidatively induced DNA adducts, especially bulky DNA lesions, may serve as biomarkers for exploring the role of oxidative stress in human diseases. The continuing development and improvement of LC-MS/MS coupled with the stable isotope-dilution method for DNA adduct quantification will further promote research about the clinical implications and diagnostic applications of oxidatively induced DNA adducts.
Graphical abstract
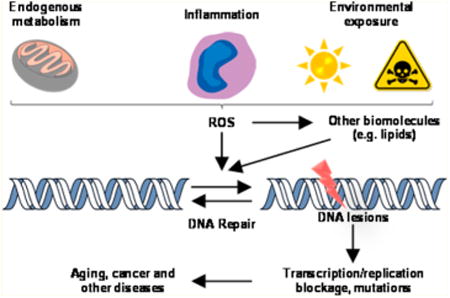
1. Oxidative Stress and ROS
The human genome is constantly exposed to a variety of endogenous and exogenous agents that can generate DNA damage, which may compromise genomic integrity.1 Reactive oxygen species (ROS) constitute an important class of DNA damaging agents, and they are continuously generated in cells as a consequence of endogenous metabolism and/or exposure to environmental toxicants.2 ROS encompass a variety of chemical species, e.g., superoxide anion radical (O2−•), hydrogen peroxide (H2O2), hydroxyl radical (•OH), and singlet oxygen (1O2). In this vein, mitochondrion is considered a major source of ROS production in cells, where electrons leaking from the electron transport chain during mitochondrial respiration can combine with molecular oxygen to generate O2−•, which can be subsequently converted to H2O2 by superoxide dismutase (SOD).3 H2O2, which diffuses freely in the cellular environment, may react with the reduced-state transition metal ions to give •OH via the Fenton-type reactions:4
Cu+/Fe2++H2O2→Cu2+/Fe3++•OH+OH−
Oxidation of biomolecules depends on the location of ROS production and the redox potential of the biomolecules. Some of the aforementioned ROS, such as O2−• and •OH, are extremely unstable, whereas others, like H2O2, are relatively long-lived.2,5,6
Infection and inflammation activate inflammatory cells, which induce and activate various oxidant-generating enzymes.7 Activated inflammatory cells produce O2−• through nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complexes or xanthine oxidase, and these cells release high concentrations of oxidant-generating enzymes such as myeloperoxidase and eosinophil peroxidase through degranulation.7–11 These enzymes produce high concentrations of reactive oxygen, nitrogen, and halogen species such as superoxide anion, nitric oxide, peroxynitrite, hydrogen peroxide, hypochlorous acid, and hypobromous acid at sites of inflammation.7,12 Although intended to neutralize invading pathogens, these reactive chemical species can result in collateral DNA damage of host cells.
Aside from damaging DNA directly, ROS may also lead to DNA damage indirectly, through reaction with lipids, proteins, and other cellular components to produce electrophilic species that can react with DNA.13,14 In particular, peroxidation of polyunsaturated fatty acids (PUFA) can give rise to a multitude of reactive aldehydes that can conjugate with DNA to yield DNA adducts.13,14 In this vein, DNA is susceptible to electrophilic attack because it contains many nucleophilic sites, including the _N_1, _N_2, _N_3, _N_7, and _O_6 of guanine; the _N_1, _N_3, _N_6, and _N_7 of adenine; the _O_2, _N_3, and _O_4 of thymine; and the _O_2, _N_3, and _N_4 of cytosine.15,16
In this review, we will discuss common types of oxidatively induced DNA lesions, including single-nucleobase lesions and tandem lesions that arise from direct ROS attack, as well as indirect ROS-induced DNA damage, such as those induced by inflammation and byproducts of lipid peroxidation. The emphasis is placed on their chemical mechanisms of formation, biological consequences, and human health relevance. In addition, we will discuss cellular replication and transcription studies of these lesions as well as their repair pathways and detection.
2. Chemistry of Oxidative Stress-Induced DNA Damage
2.1. Direct ROS-Induced DNA Lesions
2.1.1. Single-Nucleobase Lesions
•OH is highly reactive toward DNA; it can readily abstract a hydrogen atom from 2-deoxyribose or methyl group on nucleobases or be added to double bonds of purine and pyrimidine bases. Addition of hydroxyl radical to guanine leads to the formation of adduct radicals on the C4, C5, and C8 atoms, which have been previously reviewed.17 One-electron oxidation of the resulting C8-OH adduct radical gives rise to the formation of 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxo-dG, Figure 1), which was first reported in the 1980s.18 The C8-OH adduct radical of guanine can also undergo an intramolecular ring opening of its imidazole moiety by the cleavage of the C8–N7 bond, followed by one-electron reduction, to yield 2,6-diamino-4-hydroxy-5-formamidopyrimidine 2′-deoxynucleoside (Fapy-dG, Figure 1). Alternatively, one-electron reduction may also occur prior to the imidazole ring-opening process, generating 7-hydro-8-hydroxy-2′-deoxyguanosine, which subsequently undergoes ring-opening to form Fapy-dG.19,20 Similarly, the reaction between 2′-deoxyadenosine (dA) and hydroxyl radical gives 8-oxo-7,8-dihydro-2′-deoxyadenosine (8-oxo-dA)21 and 4,6-diamino-5-formamidopyrimidine 2′-deoxynucleoside (Fapy-dA).20 Apart from hydroxyl radical attack, singlet oxygen (1O2) may also oxidize dG to yield 8-oxo-dG. In this vein, 18O-labeled 8-oxo-dG could be detected in DNA isolated from cells incubated with water-soluble 18O-labeled nonionic 1,4-endoperoxide N,_N_′-di(2,3-dihydroxypropyl)-1,4-naphthalene-dipropanamide (DHPN18O2), whose thermal decomposition gives 18O-labeled singlet oxygen.22 The above-mentioned DNA adducts have already been detected at appreciable levels in vivo and summarized in previous reviews.23–28
Figure 1.
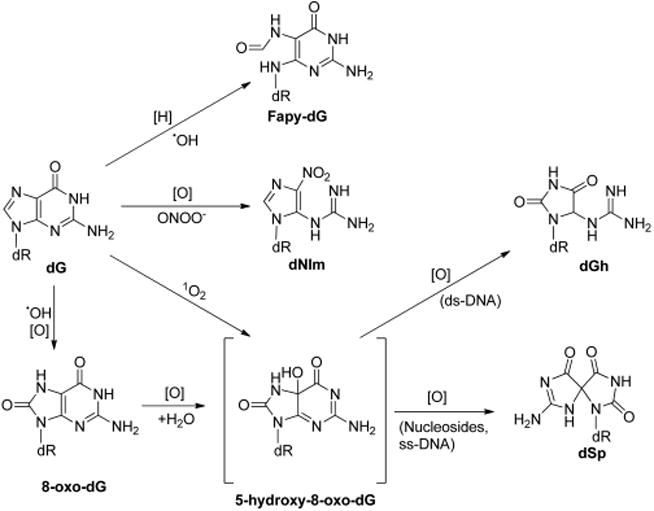
ROS-induced primary and secondary oxidation products of dG. [H] and [O] represent reduction and oxidation, respectively.
Guanine is the most easily oxidized among the four nucleobases in DNA, and a reduction potential of 1.29 V vs NHE was reported for guanosine.29 Compared to dG, 8-oxo-dG has an even lower reduction potential (0.74 V vs NHE).30 Indeed, it has been demonstrated that 8-oxo-dG can be readily oxidized by various oxidizing agents including Na2IrCl6, γ rays, peroxynitrite, Fenton-like reagents, hypochlorous acid (HOCl), etc.31–36 One-electron oxidation of 8-oxo-dG can give rise to a radical cation, which can undergo hydration, deprotonation, and another one-electron oxidation to produce 5-hydroxy-substituted derivative of 8-oxo-dG (5-OH-8-oxo-dG).31,34 Subsequent oxidation of 5-OH-8-oxo-dG leads to the formation of guanidinohydantoin 2′-deoxynucleoside (dGh), spiroiminodihydantoin 2′-deoxynucleoside (dSp), and various other oxidation products.31,34 The distributions of these products depend on the reaction context (e.g., nucleoside, single-stranded DNA, double-stranded DNA, and base pairing),37 oxidizing agents,32 and reaction conditions (e.g., pH and temperature).38 Generally speaking, the formation of dGh is favored in duplex DNA, while the formation of dSp is preferred in nucleosides, single-stranded DNA, and G-quadruplex DNA.39 dGh is the predominant product at acidic pH, while dSp is the major product at higher pH.40 Under physiological pH, especially within cells where nucleosides, ssDNA, and dsDNA are available, the formation of both dGh and dSp are feasible.
The pyrimidine bases are also prone to attack by free radicals.15 For instance, the hydroxyl radical can be added to the C5═C6 double bond of thymine and cytosine to yield C5-OH- and C6-OH-adduct radicals (Figure 2).17 The C5-OH- and C6-OH-adduct radicals can lead to the formation of 5,6-dihydroxy-5,6-dihydrothymidine (thymidine glycol) via different reaction pathways in the presence or absence of O2 (Figure 2), which was reviewed by Dizdaroglu and Jaruga41 and Teoule.42 Similar mechanisms may account for the formation of 5-methyl-2′-deoxycytidine (5-mdC) glycol, which can undergo deamination to yield thymidine glycol.43–47 The formation of 5-mdC glycol may be involved in the C → T transition mutations occurring at CpG dinucleotide sites, a type of mutation ubiquitously found in human cancers.48,49 Exposure to ionizing radiation50,51 and reaction with oxidizing agents, including KMnO4, OsO4,52 and Fenton reagents,47 can result in the formation of thymidine glycol in DNA.
Figure 2.
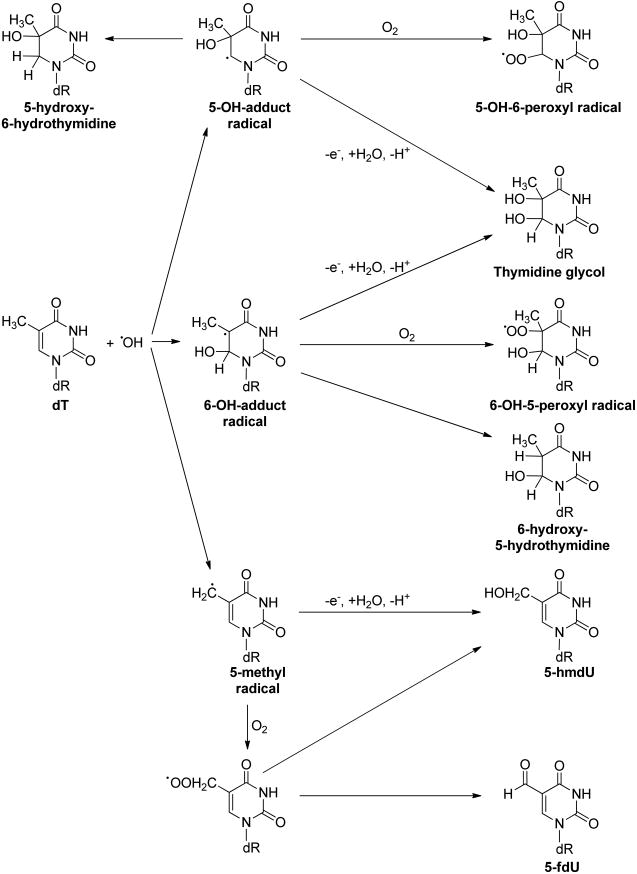
Pathways for hydroxyl radical-mediated oxidation of dT.
The hydroxyl radical can also abstract a hydrogen atom from the 5-methyl group of thymine and 5-methylcytosine to produce the 5-methyl radical of the two pyrimidine bases (Figures 2 and 3).17 In addition, the 5-methyl radical may also form from one-electron oxidation of the pyrimidine bases followed by deprotonation.53,54 The 5-methyl radical can be transformed to produce 5-hydroxymethyl-2′-deoxyuridine (5-hmdU) and 5-formyl-2′-deoxyuridine (5-fdU),41,42,55 5-hydroxymethyl-2′-deoxycytidine (5-hmdC), and 5-formyl-2′-deoxycytidine (5-fdC).53 Along this line, it was found recently that the oxidation of 5-mdC could also be catalyzed by Fe(II)- and 2-oxoglutarate (2-OG)-dependent ten-eleven translocation (TET) family dioxygenases.56–58 The resulting 5-hmdC, 5-fdC, and 5-carboxyl-2′-deoxycytidine (5-cadC) can be considered as epigenetic marks (Figure 3a).58–63 In addition, the removal of 5-fdC and 5-cadC by the base excision repair (BER) machinery is thought to play an important role in active cytosine demethylation in mammalian systems.60,64 Aside from being an oxidation product of thymidine, 5-hmdU may also arise from the deamination of 5-hydroxymethyl-2′-deoxycytidine (5-hmdC).65,66 In addition, Pfaffeneder et al.67 reported that TET enzymes could catalyze directly the formation of 5-hmdU from thymidine in the DNA of mouse embryonic stem cells (mESCs), and they also found that the deamination of 5-hmdC did not contribute significantly to the 5-hmdU level in mESCs (Figure 3b).67 5-fdU can be formed from thymidine upon exposure to ionizing radiation, one-electron photooxidation, and Fenton-type reactions, and its yield was similar or somewhat lower than that of 8-oxo-dG.55,68–71
Figure 3.
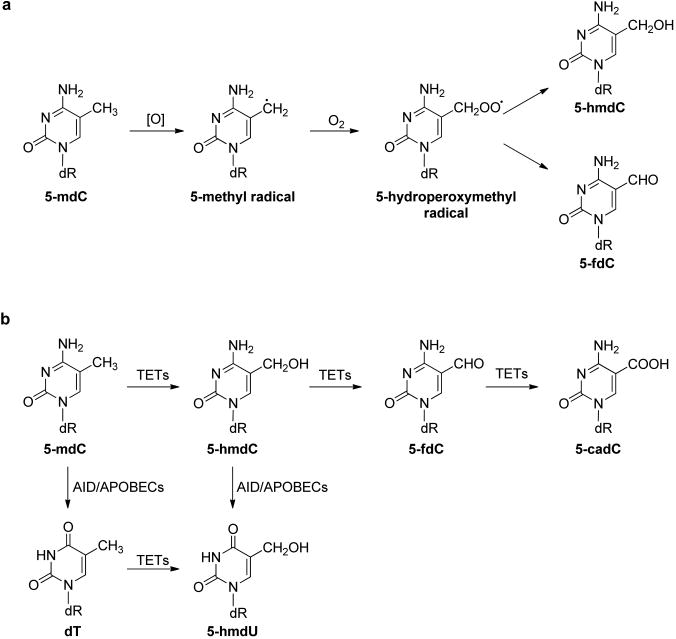
Oxidation pathways of 5-mdC. (a) ROS-induced oxidation of 5-mdC; (b) TET-mediated oxidation of 5-mdC.
2.1.2. Tandem Lesions
Apart from the above-mentioned single-nucleobase lesions, ROS may also induce the formation of bulky DNA lesions. In this context, exposure to ROS from a variety of experimental systems was found to induce CC → TT and mCG → TT tandem base substitutions, suggesting that ROS may induce the formation of intrastrand cross-link lesions.72–75 In addition, Randerath et al.,76,77 by using thin-layer chromatography (TLC) analysis of 32P-postlabeled DNA digestion products, demonstrated the existence of I (indigenous)-compounds, the bulky DNA modifications which increase markedly with aging in tissues of healthy laboratory animals and are derived from DNA-reactive intermediates arising from nutrient and oxygen metabolism. Especially, the type II I-compounds include several bulky DNA lesions, which are enhanced in kidney DNA of rodents treated with pro-oxidant carcinogen ferric nitrilotriacetate (Fe-NTA) and are identical to these lesions generated in DNA or oligodeoxyribonucleotides (ODNs) treated with Fenton reagents in vitro.76
Later, a modified 32P-postlabeling assay demonstrated that four type II I-compounds in mammalian tissue DNA are dinucleotides containing the bulky 5′S diastereomer of 8,5′-cyclo-2′-deoxyadenosine (cdA) as the 3′ nucleoside.78 The dinucleotides arise from the incomplete hydrolysis of the phosphodiester bond on the 5′ side of the modified nucleoside with the enzymes used in the 32P-postlabeling assay.78
The formation of purine cyclonucleosides (cPus) was proposed to arise from a single hydroxyl radical attack via a two-step mechanism (Figure 4).79,80 In this respect, the hydroxyl radical abstracts a hydrogen atom from the C5′ of 2-deoxyribose, yielding a carbon-centered radical, which attacks the C8 of adenine or guanine to form a new C–C bond. The resulting conjugate can lose an electron and a proton to give cdA and cdG. Molecular oxygen can inhibit this reaction by directly reacting with the C5′ radical, thereby preventing intramolecular cyclization.81,82 The above cyclization reaction yields two diastereomers at similar frequencies in calf thymus DNA exposed to ionizing radiation under anaerobic conditions.83 However, the 5′R diastereomers of cdA and cdG were induced in calf thymus DNA by Fenton-type reagents at markedly higher levels than the 5′S counterparts, with cdG being produced at a higher yield than cdA.84 Moreover, the cPus could be detected at appreciable levels in cells and animal tissues.23,85–91
Figure 4.
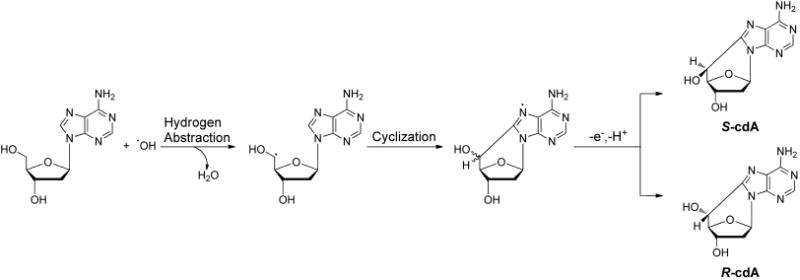
Proposed mechanism for the formation of 5′R_- and 5′_S diastereomers of 8,5′-cyclo-2′-deoxyadenosine.
The ROS-induced tandem DNA lesions with the adjacent nucleobases in the same DNA strand being covalently bonded were also investigated in the past few decades. Earlier studies by Box et al.92–95 showed the formation of intrastrand nucleobase–nucleobase cross-link lesions with guanine being covalently bonded with its adjacent thymine or cytosine when aqueous solutions of synthetic di- or tetranucleotides were exposed to X or γ rays under anaerobic conditions.
By introducing a photolabile precursor of the 5-methyl radical of thymine and 5-methylcytosine in synthetic dinucleoside monophosphates and ODNs, it was later found that the 5-methyl radical of the two pyrimidine bases can couple with the C8 position of its neighboring guanine and/or adenine to yield intrastrand cross-link lesions.96–99 Additionally, Zhang et al.100,101 revealed that an independently generated 5-hydroxy-5,6-dihydrothymidin-6-yl radical can conjugate with the C8 of guanine to give an intrastrand cross-link lesion in dinucleoside monophosphates and ODNs, though the formation of this lesion in duplex DNA was minimal. These studies provided important mechanistic insights into the ROS-induced formation of the intrastrand cross-link lesions. Furthermore, Zeng et al.102–104 and Hong et al.105 found that the UVB irradiation of duplex DNA containing a site-specifically inserted 5-bromocytosine or 5-bromouracil could give rise to efficient formation of intrastrand cross-link products with the C5 position of the pyrimidine base being covalently bonded with the C8 or _N_2 position of its neighboring guanine or with the C2, _N_6, and C8 position of its adjacent adenine. Together, these photochemical approaches offered a facile synthetic route for the generation of ODNs harboring site-specifically inserted and structurally defined intrastrand cross-link lesions, which are necessary for the characterizations of the repair of these lesions as well as their impact on DNA replication and transcription.
Further investigations were conducted about the formation of nucleobase–nucleobase intrastrand cross-links in vitro and in vivo. Along this line, Gu et al.106 showed that exposure of synthetic duplex DNA with γ rays under anaerobic conditions could give rise to the formation of the d(G[8–5]C) intrastrand cross-link. In addition, Zhang et al.98 observed that treatment of d(5mCG) with γ rays under anoxic conditions could lead to the formation of the d(5mC[5m-8]G) intrastrand cross-link. By using LC-MS/MS, Hong et al.71 further observed a dose dependent induction of d(G[8–5m]T) in calf thymus DNA upon treatment with the Fenton reagent, with a yield that is 2–3 orders of magnitude lower than that of common single-nucleobase lesions like 8-oxo-dG,5-hmdU and 5-fdU. Moreover, d(G[8–5]C) and d(G[8–5m]T) (Figure 5) could be detected in HeLa S3 cells upon exposure to γ rays, and the yields for these two lesions increase with the dose of γ rays.107 It was also observed that d(G[8–5m]5mC) (Figure 5)was formed at a higher yield than d(5mC[5m-8]G) in synthetic double-stranded DNA upon treatment with Fenton-type reagents.108 In addition to the above-mentioned intrastrand cross-links involving two adjacent nucleobases, Crean et al.109 demonstrated the induction of a nonadjacent intrastrand cross-link lesion between guanine and thymine bases separated by a cytosine in the single-stranded 5′-d(GpCpT)-3′ ODN exposed to a CO3•− radical. Similarly, the generation of nonadjacent and adjacent cross-link lesions between the C8 of guanine and the _N_3 of thymidine (d(G[8-_N_3]T)) (Figure 5) was observed in 5′-d(GpT)-3′ and 5′-d(GpCpT)-3′ ODN or calf thymus DNA treated with peroxynitrite/carbon dioxide/bicarbonate, in addition to the nitration/oxidation products of guanine such as 8-nitro-2′-deoxyguanosine (8-nitro-dG), 5-guanidino-4-nitroimidazole 2′-deoxynucleoside (dNIm), 8-oxo-dG, and dSp.110 Further study also demonstrated the formation of these two d(G[8-_N_3]T) lesions in HeLa cells upon one-electron oxidation initiated by intense nanosecond 266 nm laser irradiation.111
Figure 5.
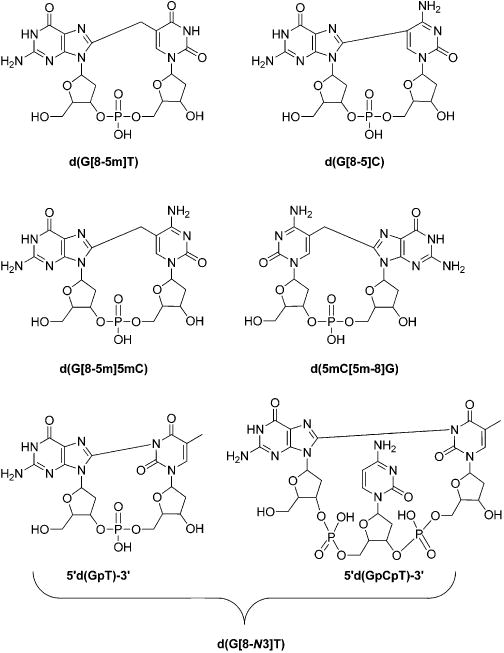
Structures of ROS-induced intrastrand cross-link lesions discussed in this review.
2.2. Inflammation-Induced DNA Damage
Chronic inflammation is an established risk factor for different types of cancers.112,113 Inflammatory responses protect human bodies from adverse effects inflicted by pathogens and damaged cells through the generation of reactive oxygen, nitrogen, and halogen species.12,114 These reactive chemical entities damage proteins and DNA of invaders as well as nearby healthy cells and tissues.12,112–114 The heme enzyme myeloperoxidase, which is secreted by activated neutrophils and monocytes, employs hydrogen peroxide (H2O2) and chloride ion as substrates to yield hypochlorous acid (HOCl) as the initial product (Figure 6).115,116 Likewise, eosinophil peroxidase, a structurally related heme protein released by activated eosinophils, preferentially oxidizes bromide to give hypobromous acid (HOBr, Figure 6).117 In addition, myeloperoxidase-induced production of HOCl is also involved in the formation of brominating species (Figure 6).117,118
Figure 6.
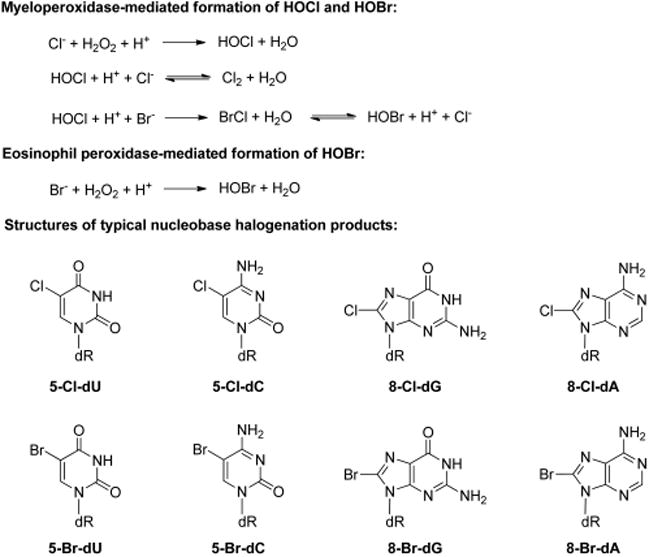
Inflammation-induced formation of hypochlorous acid (HOCl) and hypobromous acid (HOBR) and major nucleobase halogenation products. Myeloperoxidase can induce the formation of both HOCl and HOBr. The mechanism for the formation of HOBr by myeloperoxidase is different from that by eosinophil peroxidase.
HOCl and HOBr may lead to mutagenesis by damaging the nucleotide pool or reacting directly with DNA. In this vein, reactions of HOCl and HOBr with uracil produce 5-chlorouracil (5-ClU) and 5-bromouracil (5-BrU, Figure 6),117,118 respectively, which can be further converted to 5-chloro-2′-deoxyuridine (5-Cl-dU) and 5-bromo-2′-deoxyuridine (5-Br-dU) by thymidine phosphorylase.119,120 Both 5-Cl-dU and 5-Br-dU are dT analogues, and they can be converted to their corresponding nucleoside triphosphates and incorporated into DNA.121,122 In addition, bromination of 2′-deoxycytidine leads to the formation of 5-bromo-2′-deoxycytidine (5-Br-dC, Figure 6), which can undergo deamination before being incorporated into DNA as 5-Br-dU.117,118 Major products arising from the reaction of HOCl with DNA include 5-chloro-2′-deoxycytidine (5-Cl-dC), 5-Cl-dU, 8-chloro-2′-deoxyguanosine (8-Cl-dG), and 8-chloro-2′-deoxyadenosine (8-Cl-dA).118,123–127 Reaction of HOBr with DNA can lead to the formation of 8-bromo-2′-deoxyguanosine (8-Br-dG), 8-bromo-2′-deoxyadenosine (8-Br-dA), and 5-Br-dU (Figure 6).118,128–130 Among these halogenated nucleosides, 5-Cl-dC has been the most extensively studied and is considered a biomarker for chronic inflammation.131,132
During immune response, activated neutrophils and macrophages can also secrete other reactive chemical species, such as nitric oxide (•NO), which can further react with superoxide (O2−•), leading to the formation of peroxynitrite (ONOO−).12,133–136 Peroxynitrite is highly reactive toward DNA and may contribute to the cytotoxicity and carcinogenesis associated with excess generation of •NO and O2−• during chronic inflammation.137 ONOO− was found to react preferentially with dG, at a reaction rate that is at least 9 times higher than that of dA, dC, and dT in the nucleoside form.138 Along this line, previous in vitro experiments demonstrated that ONOO− induced the formation of dNIm (Figure 1) in synthesized ODNs and calf thymus DNA.110,139 Other in vitro studies also illustrated that the nitrogen dioxide radical (•NO2), produced from photolysis of nitrate with 308 nm nanosecond XeCl laser, could react with guanine neutral radicals (G(-H)•) in aqueous solution of ODNs and calf thymus DNA, leading to the formation of dNIm.140
2.3. DNA Damage Formed from Byproducts of Lipid Peroxidation
ROS can also attack biomolecules other than DNA. Specifically, the hydroxyl radical initiates the peroxidative degradation of lipids by abstracting a hydrogen atom from polyunsaturated fatty acids (PUFA).13,141–148 The resulting lipid radical (L•)is first converted to a lipid peroxyl radical (LOO•) in the presence of O2, leading to the formation of a lipid hydroperoxide (LOOH) via hydrogen atom abstraction, and finally to an alkoxyl radical (LO•) by the transition metal ion-catalyzed Fenton-type reaction. Further fragmentations of peroxyl and alkoxyl radicals give rise to reactive aldehydes, including malondialdehyde (MDA), acrolein, crotonaldehyde, 2-hexenal, 4-hydroxy-2-hexenal (HHE), 4-hydroxy-2-nonenal (HNE), 4-oxo-2-nonenal (ONE), 4-hydroperoxy-(2_E_)-nonenal (HPNE), 9,12-dioxo-(10_E_)-dodecenoic acid (DODE), 5,8-dioxo-(10_E_)-octenoic acid (DOOE), 2,4-decadienal (DDE), 4,5-epoxy-(2_E_)-decenal (EDE), etc. (Figure 7)13,14,148
Figure 7.
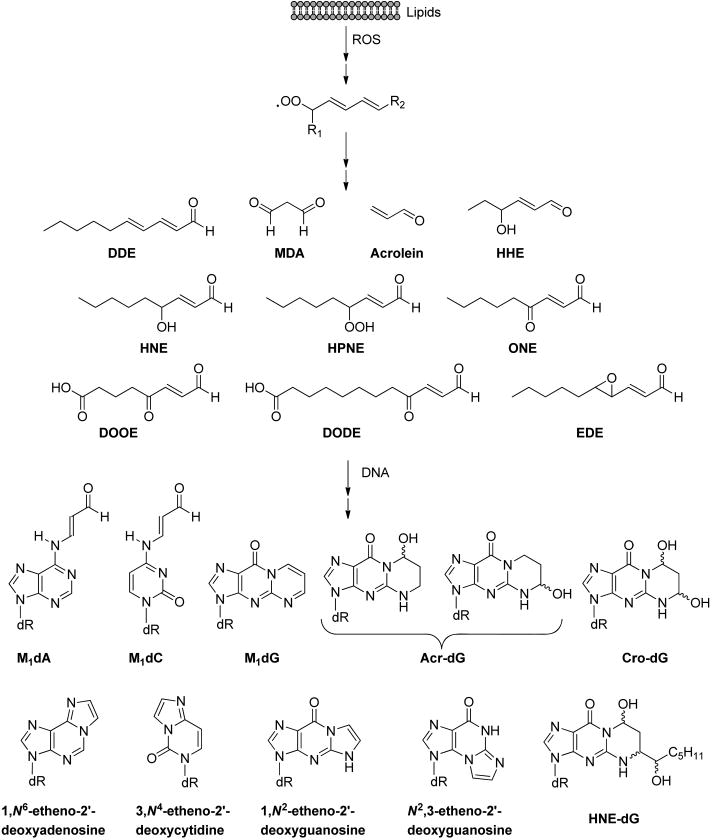
Structures of representative lipid peroxidation (LPO) byproducts and DNA adducts induced by these byproducts.
The aldehydes formed from lipid peroxidation can react with DNA to generate a variety of DNA adducts (Figure 7).13,14,148 In this vein, malondialdehyde reacts with guanine, adenine, and cytosine in DNA to form exocyclic pyrimido-[1,2-α_]purine-10(3_H)-one-2′-deoxyribose (M1dG), linear _N_6-(3-oxopropenyl)-2′-deoxyadenosine (M1dA), and _N_4-(3-oxopropenyl)-2′-deoxycytidine (M1dC), respectively (Figure 7).13 The α,_β_-unsaturated LPO products, such as acrolein, crotonaldehyde, and HNE, can lead to the formation of different diastereomers of exocyclic six-membered ring propano adducts in DNA with or without substituted alkyl side chains. The α,_β_-unsaturated aldehydes can undergo Michael addition with the _N_2-amino group of dG to give _N_2-(3-oxopropyl)-dG adducts, followed by cyclization of _N_1 with the aldehyde moiety to generate the corresponding exocyclic 1,_N_2-propano-dG products (Acr-dG, Cro-dG, and HNE-dG, etc.).149 This cyclization is reversible, and the ensuing release of the aldehyde functionality can induce the generation of DNA interstrand cross-links and DNA–protein cross-links.14,150,151 Along this line, it is worth noting that the unsubstituted Cro-dG can also arise from consecutive reactions of guanine with two molecules of acetaldehyde.152
Another type of LPO-induced DNA adduct, etheno adduct, possesses unsaturated five-membered exocyclic rings fused with heterocyclic nucleobases. Previous studies proposed a putative mechanism for the formation of etheno adducts from HNE-derived epoxide intermediate, where the α,_β_-unsaturated HNE can be converted to reactive intermediate 2,3-epoxy-4-hydroxynonanal by auto-oxidation or by oxidation with H2O2.143,144,153,154 The resulting reactive epoxy aldehyde can further react with DNA to yield the etheno adducts with or without substituted alkyl side chains, such as 1,_N_6-etheno-2′-deoxyadenosine (_ε_dA), 3,_N_4-etheno-2′-deoxycytidine (_ε_dC), 1,_N_2-etheno-2′-deoxyguanosine (1,_N_2-_ε_dG), and _N_2,3-etheno-2′-deoxyguanosine (_N_2,3-_ε_dG), etc.143,144,153,154 _N_2,3-_ε_dG can also be induced in DNA from exposure to carcinogen vinyl chloride via a similar epoxide initiation mechanism.155 Additionally, Lee et al.156,157 demonstrated possible HNE-independent pathways for the formation of 1,_N_2-_ε_dG initiated from either another LPO product EDE or HNE precursor HPNE, suggesting the existence of probable controversies concerning the mechanism for the formation of etheno DNA adducts in vivo. The above-mentioned DNA adducts emanating from products of LPO could be detected at appreciable levels in vivo.88,152,158–174
3. Repair and Biological Consequences of Oxidative Stress-Induced DNA Lesions
To minimize mutation induction and to maintain genome integrity, cells are equipped with multiple DNA repair systems to enable efficient removal of DNA lesions from the genome. Unrepaired DNA lesions may elicit cytotoxic and mutagenic effects by perturbing the accuracy and efficiency of DNA replication and transcription. Chemical synthesis of ODNs harboring site-specifically inserted and structurally defined DNA lesions, along with in vitro biochemical assay and shuttle vector-based cellular experiments, has provided important insights into how the oxidative stress-induced DNA lesions are repaired and how they compromise the flow of genetic information by inhibiting DNA replication and transcription and inducing mutations in these processes.175,176 Tables 1 and 2 summarize the key findings made from studies about how oxidative stress-induced DNA lesions perturb the efficiencies and fidelities of DNA replication and transcription in cells. Tables 3 and 4 provide information about the detection and repair mechanisms of oxidatively induced DNA modifications.
Table 1. Summary of Findings Made from Cellular Replication Studies of Oxidative Stress-Induced DNA Lesions.
| DNA lesions | assay system | bypass efficiency | mutation type (frequency) | refererces |
|---|---|---|---|---|
| 8-oxo-dG | M13 phage in E. coli | 50–90% | G → T (∼0.5–l%) | 179 |
| M13 phage in E. coli | G →T (0.72%) | 178 | ||
| 8-oxo-dA | double-stranded vector in NIH 3T3 cells | A → G + A → C (∼1% in total) | 180 | |
| Fapy-dG | single-stranded vector in COS-7 simian kidney cells | G → T (∼8–30%) | 181 | |
| G → C (∼2%) | ||||
| Fapy-dA | single-stranded vector in COS-7 cells | A → C (∼0.4%) | 181 | |
| dGh | single-stranded phage in wild-type AB1157 E. coli cells | ∼75 ± 5% | G → C (98%) | 191 |
| G → T (2%) | ||||
| single-stranded M13 phage in AB1157 E. coli cells (with MufY+ or Mut Y−) | ∼20% (MutY+) ∼30% (MutY−) | for MutY+ E. coli | 192 | |
| G → C (∼57%) | ||||
| G → T (∼40%) | ||||
| G → A (∼3%) | ||||
| no significant changes in mutation type or frequency were found in MutY− E. coli compared to MutY+ E. coli | ||||
| dSp | single-stranded M13 phage in wild-type AB1157 E. coli cells | stereoisomer 1: ∼9 ± 3% | stereoisomer 1: | 191 |
| G → C (72%) | ||||
| G → T (27%) | ||||
| stereoisomer 2: ∼9 ± 4% | stereoisomer 2: | |||
| G → C (57%) | ||||
| G → T (41%) | ||||
| single-stranded M13 phage in AB1157 E. coli cells (with MutY+ or MutY−) | MutY+ strain: <20% for both stereoisomers; MutY− strain: ∼30% for both stereoisomers | stereoisomer 1 (for MutY+ E. coli): G → C (∼19%) | 192 | |
| G → T (∼78%) | ||||
| G → A (∼1%) | ||||
| stereoisomer 2 (for MutY+ E. coli): | ||||
| G → C (∼48%) | ||||
| G → T (∼49%) | ||||
| G → A (∼3%) | ||||
| no significant changes in mutation type or frequency were found in MutY− E. coli compared to MutY+ E. coli | ||||
| thymidine glycol | single-stranded M13 phage in E. coli | lethal | not detectable | 202 |
| 5-hmdU | _ϕ_X-174am3 phage in E. coli spheroplasts | not detectable | 214 | |
| 5-fdU | double-stranded vectors in COS-7 simian kidney cells | not blocking | T → G + T → A (0.01–0.04% in total) | 226 |
| 5-hmdC | single-stranded M13 phage in E. coli cells | ∼90–110% | C → G + C → T + C → A (0.17–1.12% in total) | 240 |
| double-stranded vector in HEK-293T human kidney epithelial cells | ∼100% | not detectable | 242 | |
| 5-fdC | single-stranded M13 phage in E. coli cells | ∼90–110% | C → G + C → T + C → A (0.17–1.12% in total) | 240 |
| double-stranded vector in COS-7 cells | 39–90% | C → G + C → T + C → A (0.03–0.28% in total) | 241 | |
| double-stranded vector in HEK-293T cells | ∼70% | not detectable | 242 | |
| 5-cadC | single-stranded M13 phage in E. coli cells | ∼ 90–110% | C → G + C → T + C→A (0.17–1.12% in total) | 240 |
| double-stranded vector in HEK-293T cells | ∼70% | not detectable | 242 | |
| _S_-cdA | single-stranded M13 phage in E. coli | ∼10–31% | A → T (∼11%) | 253 |
| double-stranded vector in Pol _η_-deficient XP30RO cells and Pol η -complemented XP30RO cells | Pol η -deficient XP30RO cells: ∼3%; Pol η -complemented XP30RO cells: ∼5% | Pol _η_-deficient XP30RO cells: A →T (∼S%) Pol _η_-complemented XP30RO cells: A → T (∼9%) | 254 | |
| _S_-cdG | single-stranded plasmid in E. coli | <1% without SOS induction; <5.5% with SOS induction | G→A+G→T + deletion of 5′C (∼34% in total) | 252 |
| single-stranded M13 phage in E. coli | ∼4–11% | G→A (∼40% without SOS, ∼20% with SOS) | 253 | |
| double-stranded vector in Pol η -deficient XP30RO cells and Pol η -complemented XP30RO cells | Pol η -deficient XP30RO cells: ∼_2%;_ Pol η -complemented XP30RO cells: ∼4% | Pol _η_-deficient XP30RO cells: G→A (∼3%) | 254 | |
| G →T (∼27%); | ||||
| Pol _η_-complemented XP30RO cells: G→A (∼11%) | ||||
| G →T (∼32%) | ||||
| d(G[8-S]C) | single-stranded M13 phage in wild-type AB11S7 E. coli | 20% | G →T (8.7%) | 107 |
| G→C (1.2%) | ||||
| d(G[8-Sm]T) | single stranded pMS2 vector in E. coli (wild type and polymerase-deficient cells) | without SOS induction: 1.2–25%; with SOS induction: 3.1–35% | G→T (2.5% without SOS, 6.2% with SOS in wild-type cells) | 258 |
| 5-Cl-dC | single-stranded M13 phage in E. coli | 82–102% | C → T (∼5%) | 264 |
| dNIm | single stranded M13mp7L2 bacteriophage genome in AB1157 E. coli | without SOS induction: 7%; with SOS induction: 57% | G → C (8.9%) | 272 |
| G → A (19%) | ||||
| G → T (22%) | ||||
| εdA | single-stranded pMS2 vector in E. coli and COS-7 cells | in E. coli: very limited mutations; in COS-7 cells: | 275 | |
| A → G (63%) | ||||
| A → T (6%) | ||||
| A → C (1%) | ||||
| single-stranded pMS2 vector and double-stranded pSBK vector in HeLa and HCT116 cells | ssDNA in HeLa cells: | 277 | ||
| A → G (2%) | ||||
| A → T (8%) | ||||
| A → C (1%) | ||||
| dsDNA in HeLa cells (leading strand): | ||||
| A → G (2%) | ||||
| A → T (7%) | ||||
| A → C (5%) | ||||
| dsDNA in HCT116 cells (leading strand): | ||||
| A → G (5%) | ||||
| A → C (2%) | ||||
| dsDNA in HeLa cells (lagging strand): | ||||
| A → G (5%) | ||||
| A → T (4%) | ||||
| A → C (1%) | ||||
| double-stranded M13mp2S_Vori_L vectors in E. coli | A → C (1.6 X (10−4) | 276 | ||
| A → G (2.9 X (10−4) | ||||
| A → T (2.0 X 10−4) | ||||
| _ε_dC | single-stranded pMS2 vector in E. coli or COS-7 cells | uninduced E. coli cells: C → A + C → T (2% in total) | 278 | |
| SOS-induced cells: C → A + C → T (32% in total) | ||||
| COS-7 cells: C → A + C → T (81% in total) | ||||
| l,_N_2-_ε_dG | single-stranded M13MB19 phage in uvrA− E. coli | G → A (2.05%) | 279 | |
| G → T (0.74%) | ||||
| G → C (0.09%) | ||||
| _N_2,3-_ε_dG | single-stranded M13G*1 phage in E. coli | G → A (0.5%) | 280 | |
| _M_1G | doubled stranded M13MB102 phage in wild-type LM102 cells E. coli cells | 20% for the (–)-strand | G → A (0.35%) | 284 |
| G → T (0.4%) | ||||
| G → C (0.12%) | ||||
| single-stranded pS189 vector and double-stranded M13MB102-1 vector in E. coli and COS-7 cells | −1 or −2 frameshift in E. coli and COS-7 with reiterated (CpG)4 sequence (≤1% in total) G→A+G → T + G → C (≤2% in total) | 286 |
Table 2. Summary of Findings Made from Cellular Transcription Studies of Oxidative Stress-Induced DNA Lesions.
| DNA lesions | assay system | transcription bypass efficiency | mutagenic properties | refs |
|---|---|---|---|---|
| 8-oxo-dG | pBESTluc-fl luciferase reporter in E. coli | no detectable pausing or arrest | C → A (33%) | 184,185 |
| deletion of the first base (26%) | ||||
| pcDNA3.l(+) expression vector in mouse embryonic fibroblasts | Ogg+/+ cells: C → A + one-nucleotide deletion (∼2.6%) | 187 | ||
| _Ogg_−/− cells: C → A + one-nucleotide deletion: (∼10.8–13.9%) | ||||
| 5-hmdC | double-stranded vector in HEK293T cells | ∼90–100% | not detectable | 242 |
| 5-fdC | double-stranded vector in HEK293T cells | ∼69% | G → A (∼1%) | 242 |
| 5-cadC | double-stranded vector in HEK293T cells | ∼55% | G → A (∼1%) | 242 |
| S-cdA | double-stranded vector in SV40-transformed NER-deficient XP12BE cells | 5′A mutation (∼30%); multiple nucleotide deletion (−7, −13, and −21 nt deletions, ∼12.5%) | 255 | |
| double-stranded vectors in NER-proficient (GM00637) and NER-deficient (GM04429) human skin fibroblasts | increase with time, and up to 45% after 24 h in NER-proficient cells; no significant increase in bypass efficiency with time in NER-deficient cells | in NER-deficient cells: 5′A mutation (21%) | 250 | |
| _S_-cdG | double-stranded vectors in NER-proficient (GM00637) and NER-deficient (GM04429) human skin fibroblasts | increase with time, and up to 45% after 24 h in NER-proficient cells; no significant increase in bypass efficiency with time in NER-deficient cells | in NER-deficient cells: 5′A mutation (32%) | 250 |
Table 3. Levels of Oxidative Stress-Induced DNA Lesions in Cellular and Tissue DNA.
| DNA lesions | DNA sources | levels | refs |
|---|---|---|---|
| 8-oxo-G/8-oxo-dG | mouse | ∼2.5–4/106 nucleosides in liver; | 24,25 |
| ∼3–4/106 nucleosides in kidney; | |||
| ∼3/106 nucleosides in brain | |||
| mouse | ∼1–3/106 nucleosides in liver | 27,28 | |
| mouse | ∼ 1.5/106 nucleosides in brain; | 23,26 | |
| ∼3–4/106 nucleosides in liver; | |||
| ∼1–2/106 nucleosides in spleen | |||
| 8-oxD-A/8-oxo-dA | mouse | ∼0.3–0.5/106 nucleosides in brain; | 23,26 |
| ∼0.2–0.4/106 nucleosides in liver; | |||
| ∼0.4–0.7/106 nucleosides in spleen | |||
| Fapy-G/Fapy-dG | mouse | ∼0.6–1.2/106 nucleosides in liver; | 24,25 |
| ∼1–2/106 nucleosides in kidney; | |||
| ∼0.7–1.4/106 nucleosides in brain | |||
| mouse | ∼6–14/106 nucleosides in liver | 28 | |
| Fapy-A/Fapy-dA | mouse | ∼0.2–0.7/106 nucleosides in liver; | 24,25 |
| ∼0.2–1.2/106 nucleosides in kidney; | |||
| ∼0.2–0.6/106 nucleosides in brain | |||
| mouse | ∼1–3/106 nucleosides in liver | 27,28 | |
| dGh | mouse | 1.08–3.14/108 nucleosides in colon; | 131 |
| 3.09–7.99/108 nucleosides in liver | |||
| dSp | E. coli | ∼200–600/106 dG | 197 |
| mouse | 0.99–4.94/108 nucleosides in colon; | 131 | |
| 2.74–20.8/108 nucleosides in liver | |||
| thymine glycol/thymidine glycol | human urine | 3.1 pmol/μmol of creatinine | 293 |
| rat urine | 4.8–8.9 nmol/kg per day | 293 | |
| mouse urine | 8.62 nmol/kg per day | 294 | |
| monkey urine | 2.07 nmol/kg per day | 294 | |
| human white blood cells | 2.16 fmol/μg DNA (control); 2.83 fmol/μg DNA (ovarian cancer patients) | 301 | |
| 5-hmU/5-hmdU | HeLa cells | 3.0/106 nucleosides | 306 |
| WM-266–4 | 3.4/106 nucleosides | 306 | |
| human brain | 3.9/106 nucleosides | 306 | |
| mouse brain | 8.3/106 nucleosides | 306 | |
| mouse skin (red head) | 6.4/106 nucleosides | 306 | |
| mouse skin (albino) | 6.0/106 nucleosides | 306 | |
| LEA rat | 2.6/106 nucleosides in liver; | 85 | |
| 20.5/106 nucleosides in brain | |||
| LEC rat | 3.2–8.6/106 nucleosides in liver; | 85 | |
| 12–28.3/106 nucleosides in brain | |||
| human brain | ∼2–3/106 nucleosides (control); | 332 | |
| ∼3–6/106 nucleosides (XPA-deficient) | |||
| mouse (control and Ercc1_−/c)_ | for control, ∼6/106 nucleosides in liver, ∼1–2/106 nucleosides in kidney and ∼10/106 nucleosides in brain; | 332 | |
| For _Ercc1_−/c, -4-20/106 nucleosides in liver, ∼1–4/106 nucleosides in kidney and ∼5–10/106 nucleosides in brain | |||
| 5-fU/5-fdU | Hela-S3 cells (exposed with edrays) | formation rate: 0.022 lesion/106 nucleosides/Gy | 308 |
| LEA rat | 7/106 nucleosides in liver; | 85 | |
| 18.8/106 nucleosides in brain | |||
| LEC rat | 7.8–31.4/106 nucleosides in liver; | 85 | |
| 19.7–36.3/106 nucleosides in brain | |||
| human brain | ∼7–55/106 nucleosides (control); | 332 | |
| −10–45/106 nucleosides (XPA-deficient) | |||
| mouse (control and _Erccl_−/c) | for control, ∼10–25/106 nucleosides in liver, ∼5–20/106 nucleosides in kidney and ∼9–10/106 nucleosides in brain; | 332 | |
| for Erccl_−/_c, ∼10–95/106 nucleosides in liver, ∼10–35/106 nucleosides in kidney and ∼10–12/106 nucleosides in brain | |||
| 5-hmC/5-hmdC | mouse ES cells | 1300/106 C | 57 |
| LEA rat | 339/106 nucleosides in liver; | 88 | |
| 619/106 nucleosides in brain | |||
| LEC rat | 176/106 nucleosides in liver; | 88 | |
| 654/106 nucleosides in brain | |||
| human lung | 0.078–0.182% dG in normal lung; | 322 | |
| 0.033–0.096% dG in Stage-I lung squamous cell carcinoma (SCC) | |||
| human brain | 0.817–1.175% dG in normal brain; | 322 | |
| 0.028-0.753% dG in stage II/III astrocytomas | |||
| HeLa cells | 31.2/106 nucleosides | 306 | |
| WM-266–4 | 12.2/106 nucleosides | 306 | |
| mouse ES cells | 163/106 nucleosides | 306 | |
| human brain | 1550/106 nucleosides | 306 | |
| mouse brain | 560/106 nucleosides | 306 | |
| mouse skin (red head) | 277/106 nucleosides | 306 | |
| mouse skin (albino) | 217/106 nucleosides | 306 | |
| 5-fC/5-fdC | mouse ES cells | 20/106 dC | 57 |
| HeLa cells | 0.67/106 nucleosides | 306 | |
| WM-266–4 | 0.69/106 nucleosides | 306 | |
| mouse ES cells | 3.5/106 nucleosides | 306 | |
| human brain | 1.7/106 nucleosides | 306 | |
| mouse brain | 1.4/106 nucleosides | 306 | |
| mouse skin (red head) | 1.2/106 nucleosides | 306 | |
| mouse skin (albino) | 0.7/106 nucleosides | 306 | |
| 5-caC/5-cadC | mouse ES cells | 3/106 dC | 57 |
| HeLa cells | 0.27/106 nucleosides | 306 | |
| WM-266–4 | 0.29/106 nucleosides | 306 | |
| mouse ES cells | 0.83/106 nucleosides | 306 | |
| human brain | 0.15/106 nucleosides | 306 | |
| mouse brain | 0.12/106 nucleosides | 306 | |
| mouse skin (red head) | 0.21/106 nucleosides | 306 | |
| mouse skin (albino) | 0.19/106 nucleosides | 306 | |
| _S_-cdA | mouse | ∼0.1–0.2/106 nucleosides in brain; | 23 |
| ∼0.2–0.35/106 nucleosides in liver; | 26 | ||
| ∼0.15–0.2/106 nucleosides in spleen | |||
| LEA rat | 0.11/106 nucleosides in liver; | 85 | |
| 0.088/106 nucleosides in brain | |||
| LEC rat | 0.14–0.56/106 nucleosides in liver; | 85 | |
| 0.08–0.26/106 nucleosides in brain | |||
| mouse (control and _Csb_−/−) | for control, ∼0.05/106 nucleosides in brain, ∼0.02/106 nucleosides in kidney and ∼0.04/106 nucleosides in liver; | 87 | |
| for _Csb_−/−, ∼0.09/106 nucleosides in brain, ∼0.06/106 nucleosides in kidney and ∼0.08/106 nucleosides in liver | |||
| LEA rat | 1.2/106 nucleosides in liver; | 88 | |
| 1.54/106 nucleosides in brain | |||
| LEC rat | 2.68/106 nucleosides in liver; | 88 | |
| 1.41/106 nucleosides in brain | |||
| mouse (control and _Erccl_−/c) | for control, 0.21–0.42/106 nucleosides in liver, 0.26–1.25/106 nucleosides in kidney and 0.12–0.22/106 nucleosides in brain; | 86 | |
| for _Erccl_−/c, 0.96–4.09/106 nucleosides in livers, 0.19–1.81/106 nucleosides in kidney and 0.1–0.21/106 nucleosides in brain | |||
| mouse (albino and red head) | ∼0.15/106 nucleosides (albino); ∼0.35/106 nucleosides (red head); | 89 | |
| _R_-cdA | mouse | ∼0.025–0.035/106 nucleosides in brain; | 23 |
| ∼0.02–0.05/106 nucleosides in liver; | 26 | ||
| ∼0.04-0.05/106 nucleosides in spleen | |||
| LEA rat | 0.1/106 nucleosides in liver; | 85 | |
| 0.15/106 nucleosides in brain | |||
| LEC rat | 0.18–0.48/106 nucleosides in liver; | 85 | |
| 0.13–0.56/106 nucleosides in brain | |||
| mouse (control and Ercc1−/c) | for control, 0.17–0.95/106 nucleosides in liver, 0.2–0.65/106 nucleosides in kidney and 0.08–0.16/106 nucleosides in brain; | 86 | |
| for _Ercd_−/c, 2.54–8.37/106 nucleosides in liver, 0.27–0.72/106 nucleosides in kidney and 0.09–0.17/106 nucleosides in brain | |||
| mouse skin (albino and red head) | ∼0.15/106 nucleosides (albino); ∼0.3/106 nucleosides (red head); | 89 | |
| _S_-cdG | mouse | ∼2–3.5106 nucleosides in brains | 23 |
| ∼1–1.5/106 nucleosides in liver; | 26 | ||
| ∼2.5–3.5/106 nucleosides in spleen | |||
| LEA rat | 0.19/106 nucleosides in liver; | 85 | |
| 0.16/106 nucleosides in brain | |||
| LEC rat | 0.20–1.08/106 nucleosides in liver; | 85 | |
| 0.14–0.43/106 nucleosides in brain | |||
| LEA rat | 2.02/106 nucleosides in liver; | 88 | |
| 2.31/106 nucleosides in brain | |||
| LEC rat | 4.45/106 nucleosides in liver; | 88 | |
| 2.21/106 nucleosides in brain | |||
| mouse (control and _Ercc_−/c) | for control, 0.32–1.05/106 nucleosides in liver, 0.91–2.86/106 nucleosides in kidney and 0.37–0.53/106 nucleosides in brain; | 86 | |
| for _Ercd_−/c, 2.03–5.64/106 nucleosides in liver, 0.53–2.81/106 nucleosides in kidney and 0.25–0.66/106 nucleosides in brain | |||
| mouse skin (albino and red head) | ∼0.35/106 nucleosides (albino); ∼0.75/106 nucleosides (red head); | 89 | |
| _R_-cdG | mouse | ∼0.5–0.8/106 nucleosides in brain; | 23 |
| ∼0.5–0.55/106 nucleosides in liver; | 26 | ||
| ∼0.5–0.6/106 nucleosides in spleen | |||
| LEA rat | 0.13/106 nucleosides in liver; | 85 | |
| 0.14/106 nucleosides in brain | |||
| LEC rat | 0.16–0.54/106 nucleosides in liver; | 85 | |
| 0.13–0.52/106nucleosides in brain | |||
| mouse (control and _Erccl_−/c) | for control, 0.14–1.01/106 nucleosides in liver, 0.35–0.73/106 nucleosides in kidney and 0.11–0.17/106 nucleosides in brain; | 86 | |
| for _Ercrt_−/c, 2.43-7.31/106 nucleosides in liver, 0.28–0.73/106 nucleosides in kidney and 0.11–0.21/106 nucleosides in brain | |||
| mouse skin (albino and red head) | ∼0.15/106 nucleosides (albino); ∼0.3/106 nucleosides (red head); | 89 | |
| d(G[8-5]C) | Hela-S3 cells (with y irradiation) | formation rate: ∼0.037 lesions/109 nucleosides per Gy | 106 |
| d(G[8-5m]T) | Hela-S3 cells (with y irradiation) | formation rate: 0.05 lesion/109 nucleosides/Gy | 107 |
| LEA rat | ∼0.005/106 nucleosides in liver; | 332 | |
| ∼0.02/106 nucleosides in brain | |||
| LEC rat | ∼0.01–0.04/106 nucleosides in liver; | 332 | |
| ∼0.01–0.08/106 nucleosides in brain | |||
| mouse liver (control and XPA-deficient) | ∼0.005/106 nucleosides (control); | 332 | |
| ∼0.01/106 nucleosides (XPA-deficient) | |||
| human brain (control and XPA-deficient) | ∼0.005–0.04/106 nucleosides (control); | 332 | |
| ∼0.015–0.07/106 nucleosides (XPA-deficient) | |||
| mouse (control and _Erccl_−/c) | for control, ∼0.007∼0.01/106 nucleosides in liver, ∼0.015–0.02/106 nucleosides in kidney and ∼0.001–0.005/106 nucleosides in brain; | 332 | |
| for _Erccl_−/c, ∼0.008–0.035/106 nucleosides in liver, ∼0.01–0.045/106 nucleosides in kidney and 0.001–0.007/106 nucleosides in brain | |||
| d(G[8-_N_3]T) | HeLa cells (with 266 nm laser pulse irradiation) | 0.21–1.19/106 nucleosides | 111 |
| 5–Cl-C/5-Cl-dC | mouse | 3.86–7.06/108 nt in colon; | 131 |
| 4.33–16.61/108 nt in liver | |||
| human leukocyte | 0.06–0.4/106 nucleosides | 127 | |
| human endothelial cell (treated with 300 _μ_M HOC1) | 40/106 dC | 343 | |
| human colon (from patients with inflammatory bowel disease) | 0.002–0.294/106 nucleobases | 132 | |
| mouse colon (_H. hepaticus-_infected) | 0.031–0.129/106 nucleobases | 132 | |
| 5–Br-C/5-Br-dC | human eosinophils | −60–225/106 cells | 117 |
| 5–C1-U/5-CU1U | rat | 20–80 pg/pouch | 341 |
| human atherosclerotic tissue | ∼0.1 pmol/g tissue (normal); ∼0.7 pmol/g tissue (atherosclerotic) | 342 | |
| 5–Br-U/5-Br-dU | human atherosclerotic tissue | ∼0.08 pmol/g tissue (normal); ∼0.18 pmol/g tissue (atherosclerotic) | 342 |
| M1G/M1dG | human | 0.5–1.2/106 nucleosides in liver; | 13 |
| 0.05–2.8/106 nucleosides in white cells; | |||
| 0.001–0.5/106 nucleosides in pancreas; 0.002–0.56/106 nucleosides in breast | |||
| human leukocyte | 64.9 fmol/mg DNA in smokers; | 171 | |
| 56.5 fmol/mg DNA in nonsmokers | |||
| rat liver | 0.52/106 nucleosides | 172 | |
| human colorectal mucosa | 0.43/106 nucleosides for men; 4.6/106 nucleosides for women | 174 | |
| ε_A/ε_dA | human asymptomatic colon epithelia | ∼0.025–0.065/106 nucleosides | 154 |
| rats exposed to 600 ppm vinyl chloride (4 h/day for 5 day) | 0.21/106 nucleosides in liver; | 155 | |
| 0.65/106 nucleosides in lung; | |||
| 0.04/106 nucleosides in kidney | |||
| human placenta | 2.3–2.5/106 nucleosides | 161 | |
| human | 0.282/106 nucleosides in placenta | 163 | |
| 0.162/106 nucleosides in leukocyte; | |||
| human saliva | 0.22–2.1/106 nucleosides | 165 | |
| LEC rat liver | ∼0.002–0.1/106 nucleosides | 170 | |
| LEA rat | 0.23/106 nucleosides in liver; 0.39/106 nucleosides in brain | 88 | |
| LEC rat | 0.3/106 nucleosides in liver; 0.44/106 nucleosides in brain | 88 | |
| 1, _N_2-_ε_G/l, _N_2-_ε_dG | human IMR-90 cells | ∼0.02/106 nucleosides | 152 |
| human urine | 95 pg/mL for smokers; | ||
| 50–68 pg/mL for nonsmokers | |||
| human | 0.085/106 nucleosides in placenta | 163 | |
| 0.086/106 nucleosides in leukocyte; | |||
| human saliva | 0.68–7.52/106 nucleosides | 165 | |
| Wistar rat | 2.47/108 dG in livers; | 168 | |
| 0.87/108 dG in lungs; | |||
| 2.96/108 dG in brains | |||
| LEA rat | 0.11/106 nucleosides in livers; 0.15/106 nucleosides in brains | 88 | |
| LEC rat | 0.16/106 nucleosides in livers; 0.13/106 nucleosides in brains | 88 | |
| _ε_C/_ε_dC | human asymptomatic colon epithelia | ∼0.015–0.035/106 nucleosides | 154 |
| rats exposed to 600 ppm vinyl chloride (4 h/day for 5 days) | 0.98/106 nucleosides in liver; | 155 | |
| 0.3/106 nucleosides in lung; | |||
| 0.29/106 nucleosides in kidney | |||
| human | 0.441/106 nucleosides in placenta | 163 | |
| 0.111/106 nucleosides in leukocyte; | |||
| human urine | 104–105 pg/mL | 164 | |
| human saliva | 0 –1.39/106 nucleosides | 165 | |
| human urine | 0.45 nM for smokers; | 166 | |
| 0.16 nM for nonsmokers | |||
| LEC rat liver | ∼0.035–0.25/106 nucleosides | 170 | |
| _N_2,3-_ε_G/_N_2,3-_ε_dG | rats exposed to 600 ppm vinyl chloride (4 h/day for 5 days) | 1.81/106 nucleosides in liver: | 155 |
| 0.21/106 nucleosides in lung; | |||
| 0.31/106 nucleosides in kidney | |||
| Acr-dG | human | 0.78/106 nucleosides in leukocyte; | 159 |
| 1.08/106 nucleosides in placenta | |||
| human saliva | 0.13/106 nucleosides | 165 | |
| human brain | 5.15/106 nucleosides (with Alzheimer's disease); | 336 | |
| 2.8/106 nucleosides (control) | |||
| Cro-dG | human IMR-90 cells | ∼0.05/106 nucleosides | 152 |
| human | 0.06/106 nucleosides in leukocyte; | 159 | |
| 0.26/106 nucleosides in placenta | |||
| human saliva | 0–0.485/106 nucleosides | 165 | |
| human | 14.57/109 dG in livers; 19.99/109 dG in lung | 167 | |
| Wistar rat | 4.61/108 dG in liver; | 168 | |
| 2.25/108 dG in lung; | |||
| 5.66/108 dG in brain |
Table 4. Repair Mechanisms of Oxidative Stress-Induced DNA Lesions.
| DNA lesions/modifications | repair pathways | refs |
|---|---|---|
| 8-oxo-dG | BER | 25,27,188–190 |
| 8-oxo-dA | BER | 25,27,188–190 |
| Fapy-dG | BER | 25,27,188–190 |
| Fapy-dA | BER | 25,27,188–190 |
| dGh | BER | 193,195–198 |
| dSp | BER | 193,195–198 |
| thymidine glycol | BER | 206,207,210 |
| NER | 211 | |
| 5-hmdU | BER | 188,219,220 |
| 5-fdU | BER | 228,230–236 |
| 5-fdC | BER | 65,220,246,247 |
| 5-cadC | BER | 65,220,246,247 |
| R/_S_-cdA | NER | 86,91,249,250 |
| R/_S_-cdG | NER | 86,91 |
| d(G[8-5]C) | NER | 257 |
| d(G[8-5m]T) | NER | 256,257,332 |
| d(G[8-_N_3]T) | NER | 259 |
| BER | 260 | |
| 8-Cl-dG | BER | 262 |
| 5-Cl-dU | BER | 263 |
| dNIm | BER | 199 |
| _ε_dA | AlkB/ALKBH | 281,282 |
| BER | 281 | |
| _ε_dC | AlkB/ALKBH | 281,282 |
| BER | 281 | |
| 1,_N_2-_ε_G/1,_N_2-_ε_dG | AlkB/ALKBH | 281,282 |
| BER | 281 | |
| _N_2,3-_ε_G/_N_2,3-_ε_dG | AlkB/ALKBH | 282 |
| 281 | ||
| BER | 281 | |
| M1dG | AlkB/ALKBH | 287 |
| NER | 14 | |
| Acr-dG | AlkB/ALKBH | 287 |
| NER | 14 | |
| Cro-dG | NER | 14 |
3.1. Direct ROS-Induced DNA Lesions
3.1.1. Single-Nucleobase Lesions
It has been illustrated that owing to the 8-oxo-dG:dA mispairing, replicative bypass of 8-oxo-dG leads to G → T transversion and that misincorporation of 8-oxodGTP formed in the nucleotide pool into DNA gives rise to A → C substitutions.177–179 8-oxo-dA induces A → G transitions and A → C transversions in mammalian cells.180 Fapy-dG can mispair with dA, which can induce G → T transversion (∼8–30% frequency).181,182 Fapy-dA is weakly mutagenic (0.4% frequency) and induces A → C transversion in vivo.181
In vitro experiments showed that 8-oxo-dG only slightly perturbed T7 RNA polymerase (T7RNAP)-mediated transcription, with bypass efficiency being up to 95%.183 E. coli RNA polymerase can efficiently bypass 8-oxo-dG in vivo, where the lesion induces C → A transversion and single-nucleotide deletion at frequencies of 33% and 26%, respectively.184,185 In addition, this lesion transiently paused transcription mediated by mammalian RNA polymerase II and led to a similar C → A transversion mutation,183,186 and Saxowsky et al.187 found that 8-oxo-dG induced one-nucleotide deletion and C → A transversion in transcripts in mouse embryonic fibroblasts (MEFs). Moreover, elevated levels of mutant transcripts were generated in the _Ogg1_−/− MEFs and _Csb_−/−_Ogg1_−/− MEFs, indicating the involvement of transcription-coupled repair and DNA glycosylase in the removal of 8-oxo-dG.187 The major known proteins and pathways for the repair of the aforementioned oxidatively induced nucleobase lesions were previously reviewed.188–190
A number of replication and repair studies have been conducted for dGh and dSp. These two lesions strongly block DNA polymerases, and once bypassed, they can be highly mutagenic and yield G → C and G → T transversions,191,192 and the frequencies of the dGh-induced mutations are pH-dependent.39 In addition, dGh and dSp adducts were found to be substrates for both BER and NER pathways. While neither lesion could be repaired by human hOGG1,193,194 both were found to be substrates for E. coli DNA glycosylases MutM, Nth and Nei,195–197 yeast yOGG1, yOGG2,193 murine NEIL1 and NEIL2,197 and human hNEIL1,198 where the repair mediated by hNEIL1 seems to be stereoselective.198 McKibbin et al.194 demonstrated the excision of dGh, dSp, and dSp-amine adducts by Nei, Fpg, NEIL1 (BER glycosylases), as well as the UvrABC system (bacterial NER pathway). It is worth noting that the authors demonstrated that bulky dSp-amine adducts (including dSp-Lys, dSp-GlcN, and dSp-GPRPGP) can be repaired by the BER pathway, indicating the presence of overlapping mechanisms for the removal of hydantoin and hydantoin-amine adducts.194 Shafirovich et al.199 also observed the formation of both BER and NER products when the dGh- and dSp-containing ODNs were treated with human cell extracts. The involvement of NER was further substantiated by the observation that the lack of XPA or XPC diminished the repair activity, whereas complementation of extracts of XPC-deficient cells with XPCRAD23B restored the NER activity.199 Comprehensive investigations into how BER and NER pathways are involved in the removal of dGh and dSp lesions in vivo will paint a more complete picture about the repair of these lesions.
Thymidine glycol effectively blocks DNA polymerases in vitro,200 while resulting in cell death in vivo.189,201 Although generally regarded as not highly mutagenic on its own,202 thymidine glycol was found to modulate the mutagenic properties of other closely placed DNA lesions. One example is that thymine glycol can form as part of clustered DNA damage with a neighboring 8-oxo-dG, where the presence of a neighboring thymidine glycol significantly increased the mutagenic potential of 8-oxo-dG.203 As noted elsewhere in this review, thymidine glycol can also arise from the deamination of 5-methyl-2′-deoxycytidine glycol;46,47 thus, the thymidine glycol/8-oxo-dG tandem lesion may be induced at methylated CpG sites upon ROS attack, thus contributing to CpG mutagenesis.47,48 When thymidine glycol is located opposite to an apurinic/apyrimidinic (AP) site, DNA double-strand breaks (DSBs) can be formed through a BER mechanism or at replication fork.204 When thymidine glycol is opposite to 8-oxo-dG, the DSB formation decreased, but mutation frequency of 8-oxo-dG increased compared to that found for 8-oxo-dG present as an isolated lesion.204 In addition, Almohaini and co-workers205 reported that the presence of a thymidine glycol at the first or second position from one 3′ terminus of a blunt-end DSB significantly impeded nonhomologous end-joining, while BER of thymidine glycol located at the fifth position from the blunt end interfered with the DSB ligation.
Thymidine glycol is mainly repaired via the BER pathway, and the lesion was found to be a substrate for a number of DNA glycosylases. These include endonucleases III (Endo III; Nth) and VIII (Endo VIII; Nei) in E. coli, yNTG1 (Ntg1) and yNTG2 (Ntg2) in S. cerevisiae, as well as NTH1, NEIL1, NEIL2, and NEIL3 in mammalian cells.189,201,206 In this context, it is worth noting that ADAR1-dependent adenosine-to-inosine editing of pre-mRNA of human NEIL1 gene yields a different form of NEIL1 protein with lysine 242 being converted to an arginine, and the edited form exhibits differential activity from its corresponding unedited form toward the removal of thymine glycol in duplex DNA.207 Both forms of NEIL1 can promote tautomerization of thymine glycol, thereby facilitating the recognition and removal of the lesion.208 Besides BER, some NER activity was also observed for the removal of thymidine glycol in E. coli and human systems,209–212 though the lesion is unlikely a substrate for the mismatch repair pathway in E. coli.213 5-hmdU formed from dT oxidation pairs with dA, while 5-hmdU produced by oxidation and deamination of 5-mdC pairs with dG. Earlier studies of 5-hmdU focused on its role as an oxidatively induced DNA lesion from dT and revealed that the modified nucleoside is weakly mutagenic,214 does not block DNA polymerases,215 pairs with dA in Watson–Crick geometry,216 and even replaces thymidine in bacteriophage DNA.217 The excision activity of 5-hmU in 5-hmU:A pair was much lower than that in 5-hmU:G pair by human cell extracts.218 5-hmU:G can be excised by TDG, SMUG1, MBD4,219–222 and with some weak activity by NEIL1.221 SMUG1 was also found to remove 5-hmU:A, albeit with lower activity.219 These attributes of 5-hmdU are in agreement with the recent hypothesis that this modified nucleoside may assume an epigenetic role.67,223 The occurrence, repair, and biological consequences of 5-hmdU were previously reviewed.189,224
5-fdU does not strongly block DNA polymerases.225 Although high-fidelity DNA polymerases can incorporate any of the four dNTPs opposite the lesion in vitro, 5-fdU is weakly mutagenic (0.01–0.04% mutation frequencies in double-stranded vectors), and it induces T → G and T → A transversions in simian COS-7 cells.226 5-fdU is predominantly repaired by the BER pathway. AlkA in E. coli227–229 and SMUG1 in mammalian cells230–232 are the major enzymes for the removal of 5-fdU from DNA. Nth, Fpg, and Nei in E. coli, SpNth1 in Schizosaccharomyces pombe (a homologue of E. coli endonuclease III), human Nth1 and Mbd4, and mouse Nth1 and Tdg can also excise 5-fdU from DNA.230,233–236 Similar to 5-hmU, 5-fdU in DNA exists as 5-fU:A (formed from A:T base pair) or 5-fU:G (formed from 5mC:G base pair). The former form can be removed by E. coli AlkA protein,227,228 and the latter may be repaired by AlkA protein and a MutHLS mismatch repair system (e.g., E. coli mismatch uracil DNA glycosylase, Mug),229,233 suggesting the involvement of the MMR pathway in the repair of 5-fdU. Meanwhile, repair initiated by SMUG1 can excise 5-fdU opposite any of the four nucleobases, with the highest activity toward 5-fU:C and 5-fU:T.232 The subsequent repair process will lead to T → G and T → A transversions, which are in agreement with the T → G and T → A transversions induced by this lesion in mammalian cells. In addition, KsgA was recently reported to remove the 5-fU:C mispair in E. coli, and mutation in ksgA resulted in increased spontaneous mutations in the mutM mutY and nth nei background.237 The repair of 5-fdU was also found to be modulated by a nearby apurinic/apyrimidinic (AP) site. When an AP site is located directly opposite 5-fdU, it is repaired through the long-patch BER pathway; by contrast, when the AP site is shifted it is primarily repaired by the short-patch BER pathway.238
Previous studies have provided significant insights about how the oxidized 5-mdC derivatives influence the efficiency and accuracy of DNA replication and transcription. An in vitro mutagenesis assay illustrated that, among the oxidized 5-mdC derivatives, only 5-fdC is marginally mutagenic, leading to 1–2% C → T transitions.239 5-hmdC, 5-fdC, and 5-cadC are slightly mutagenic in E. coli cells, where the C → T transition mutation occurs at frequencies of 0.17–1.12%.240 In this vein, 5-fdC was found to block DNA replication and lead to mutation in simian COS-7 cells, with bypass efficiencies and mutation frequencies being 39–90% and 0.03–0.28%, respectively.241 Moreover, Ji et al.242 demonstrated that, in HEK293T human embryonic kidney epithelial cells, 5-fdC and 5-cadC constituted modest blocks to DNA replication (with a 30% reduction in bypass efficiencies) without inducing detectable mutations in human cells, whereas replicative bypass of 5-hmdC is highly accurate and efficient.
An in vitro assay demonstrated that the yeast and mammalian RNA polymerase II (Pol II)-mediated polymerization rates and specificity constants for GTP incorporation against 5-fC and 5-caC were reduced significantly compared with those for unmodified C template, whereas no changes were observed for 5-mC and 5-hmC templates.243 Additionally, the substrate specificity was reduced by ∼30-fold for the 5-fC-containing template in comparison with the C template.243 Later, You et al.242 revealed that 5-fC and 5-caC displayed marginal mutagenic (∼0.7–1.7%) and modest inhibitory (31–50%) effects on transcription mediated by T7RNAP or human RNA polymerase II (hRNAPII) in vitro and in HEK293T cells. In addition, 5-hmC did not compromise appreciably the efficiency or accuracy of transcription in vitro or in HEK293T cells.242 The lack of pronounced deleterious effects of 5-hmC, 5-fC, and 5-caC on replication or transcription is in keeping with the potential roles of these oxidized 5-mC derivatives in epigenetic regulation.
Different from the passive DNA cytosine demethylation where 5-mdC is diluted during replication,244,245 it has been proposed that the TET-mediated oxidation of 5-mdC to 5-fdC and 5-cadC may play an important role in active cytosine demethylation in mammals, a process that results in the loss of 5-mdC independent of DNA replication. Along this line, the excision of 5-fdC and 5-cadC from DNA by thymine DNA glycosylase (TDG) and the following action through the BER pathway may result in the restoration with unmodified cytosine.59,246 It was found that genetic depletion or catalytic inactivation of TDG leads to embryonic lethality in mice,220,247 indicating the significant roles of TDG in maintaining epigenetic stability during embryonic development. An alternative active cytosine demethylation pathway was also proposed: The AID (activation-induced cytidine deaminase)/APOBEC (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like) proteins can deaminate 5-hmdC to give 5-hmdU (Figure 3b), which is then removed by TDG or SMUG1 (single-stranded-selective monofunctional uracil DNA glycosylase 1) and finally restored to unmodified cytosine through BER.65,220 Nevertheless, this mechanism is still controversial because the purified AID/APOBEC proteins exhibit no detectable activity toward 5-hmC deamination in vitro.248
3.1.2. Tandem Lesions
Multiple lines of evidence support that cdA and cdG are repaired by the NER pathway. First, cleavage products of cdA-containing duplex DNA emanating from NER activity in human cell nuclear extracts was observed, and depletion of XPA protein led to a pronounced decrease in such cleavage.249 By contrast, no correction of the lesion by direct damage reversal or base excision repair was detected.249 Second, genetic depletion of ERCC1 gave rise to elevated accumulation of cdA and cdG in mouse tissues.86,91 Third, a transcriptional bypass assay revealed that cdA and cdG are substrates for the transcription-coupled NER pathway in mammalian cells.250
cdA and cdG were found to be strong blockades to DNA replication, and replicative bypass of these lesions are highly mutagenic. With the use of DNA containing a site-specifically inserted cdA as template, both the 5′_R_- and 5′_S_-diastereomers of cdA were observed to block primer extension by replicative polymerases, including human DNA polymerase δ and T7 DNA polymerase.249,251 _S_-cdA and _S_-cdG were strongly blocking to DNA replication in E. coli and human cells and induced substantial frequencies of mutations at the lesion sites, where cdA induces A → T mutation, and cdG induces mainly G → A and G → T mutations.252–254 Additionally, Pol η, Pol ι, and Pol ζ, but not Pol κ, assume crucial roles in promoting replication across _S_-cdA and _S_-cdG in human cells,254 and Pol V plays a major role in bypassing these lesions in E. coli252,253
It was found that both _S_-cdA and _S_-cdG strongly inhibited transcription and induced transcriptional mutagenesis in vitro and in mammalian cells.250,255 Different from the observations made from replication studies, _S_-cdA and _S_-cdG primarily induce a 5′-A mutation during transcription in human cells, where the lesions direct human RNA polymerase II to misincorporate an adenosine nucleotide opposite the template base situated on the 5′ side of the lesions. Furthermore, when placed on the template strand of an actively transcribed gene, both _S_-cdA and _S_-cdG were primarily repaired by transcription-coupled NER pathways in mammalian cells.250
For the oxidatively induced intrastrand nucleobase-nucleobase cross-link lesions, d(G[8–5]C), d(G[8–5m]mC), and d(G[8–5m]T) could be recognized by E. coli UvrABC nuclease, suggesting the possible involvement of the NER pathway in the repair of these lesions in vivo.256,257 S. cerevisiae DNA polymerase η (Pol η) was able to bypass d(G[8–5]C) and d(G[8–5m]T), with the 5′ guanine portion of the lesion markedly reducing the efficiency and the fidelity of nucleotide incorporation.106,107 In line with in vitro replicative bypass studies, d(G[8–5]C) was found to block considerably DNA replication in E. coli cells, as reflected by a 20% bypass efficiency, and the lesion was significantly mutagenic in vivo, inducing G → T (8.7%) and G → C (1.2%) transversion mutations. In addition, Pol V was found to be responsible for the error-prone bypass of d(G[8–5]C).107 Similar observations were made for d(G[8–5m]T), where the guanine portion of the lesion induces G → T mutation, and among the three SOS-inducible DNA polymerases in E. coli, Pol V is the most efficient in bypassing the lesion, and it is required for most targeted G → T transversions.258 In vitro studies performed in HeLa cell extracts have demonstrated that the d(G[8-_N_3]T) lesions could be potential substrates of both NER and BER pathways.259,260 Another recent primer extension experiment revealed that the d(G[8-_N_3]T) lesions could strongly block the A-family BF polymerase from Bacillus stearothermophilus, Y-family polymerases Dpo4 from Sulfolobus sulfataricus P2 and human Pol κ, with bypass efficiencies being <1–2%, ∼8%, and 9–11%, respectively.261 In addition, the primer extension catalyzed by Pol η was also partially inhibited (with bypass efficiency being 28–45%) by the d(G[8-_N_3]T) cross-links, and more efficient bypass of nonadjacent d(G[8-_N_3]T) lesions in the GCT sequence context was observed than the adjacent counterpart in the GT sequence context.261,243
3.2. Inflammation-Induced DNA Damage
To date, not much is known about the repair of halogenated nucleobases. Notwithstanding the activity of the human MutT homologue (hMTH1) toward 8-Cl-dGTP,262 human 8-oxoguanine DNA glycosylase 1 (hOGG1), endonuclease VIII-like 1 (hNEIL1), alkyladenine DNA glycosylase (hAAG), E. coli formamidopyrimidine DNA glycosylase (FPG), or endonuclease V (EndoV) could not cleave 8-Cl-G when paired with a C in duplex DNA.130
5-Cl-dU can be incorporated into DNA as a thymidine analogue. It is more readily cleaved by hSMUG1 when paired with dG than dA.263 Relative to the T:G mispair, 5-Cl-U, when mispaired with G, is more efficiently repaired by TDG and hSMUG1.263 No specific repair pathway has yet been established for 5-Cl-C. On the other hand, DNA glycosylase MBD4-mediated excision repair of 5-Cl-U, 5-Br-U, and 5-Br-C within CpG and mCpG sites has been demonstrated.264,265 The lack of specific repair pathway may account for the accumulation of halogenated nucleobases in DNA.
Base-pairing energy of 5-Cl-C:G is only slightly lower than that of the C:G pair,266 suggesting that 5-Cl-C is likely a persistent DNA lesion. It was recently reported that all families of DNA polymerases predominantly decode 5-Cl-C as C in vitro; meanwhile, 5-Cl-C, when placed on single-stranded M13 plasmid and replicated in E. coli cells, induced 3–9% C → T transition, which is as mutagenic as 8-oxo-dG in similar assays.264 On the other hand, the 5-halogenated derivatives of cytosine may also perturb epigenetic signaling. In this vein, 5–Cl-C and 5–Br-C in a CpG sequence context can, similar to 5-mC, direct DNMT1-mediated maintenance DNA cytosine methylation and can bind to methyl-CpG-binding proteins.265,267,268 Such interactions may result in aberrant cytosine methylation and alteration of epigenetic signaling. Hence, 5-Cl-dC may contribute to inflammation-driven cancers through both epigenetic and mutagenic mechanisms.
The accumulation of 5-Cl-dU has been shown to cause mutations, such as T:A → C:G transition, and sister chromatid exchange.122,263,269,270 5-Cl-dU can be incorporated into DNA as a dT analogue, and it codes as a dT in an oligonucleotide template.263 Similar to 5-Cl-dU, 5-Br-dU is also a mutagenic analogue of thymidine, and it can mispair with guanine in DNA.117 Pols α, κ, and η were shown to incorporate predominantly a dG opposite 5-Br-dC, indicating that 5-Br-dC itself is not a mutagenic lesion. However, 5-Br-dC can be deaminated to 5-Br-dU and further lead to mutation.271 8-CldG is a mutagenic adduct; Sassa and co-workers130 showed that Pol α and Pol κ were slightly retarded at the 8-Cl-dG site, while Pol η readily bypassed the lesion. 8-Br-dG is a mutagenic lesion, and it may produce a broad spectrum of mutations at the site of inflammation. Pols α, κ, and η all incorporated the correct base opposite 8-Br-dA, indicating a low mutagenic potential of this lesion.271 Further investigations regarding the repair of halogenated nucleosides in DNA and the impact of these lesions on the efficiency and fidelity of DNA replication in mammalian cells are needed.
Recently, Shafirovich et al.199 revealed that dNIm was a substrate of the human BER pathway but was resistant to excision by the NER machinery when incubated with cell-free HeLa S3 cell extracts. Previous primer extension assays demonstrated that dNIm blocked significantly replication mediated by calf thymus polymerase α and E. coli polymerase I, but not human polymerase β.139 In addition, replicative bypass of dNIm by these two polymerases could induce G → T and G → C transversions.139 Along this line, by conducting cellular replication studies with the use of a single-stranded M13mp7L2 bacteriophage genome in E. coli AB1157 cells, Neeley et al.272 showed that dNIm strongly blocked DNA replication, with bypass efficiency being only (7.0 ± 1.6)% in uninduced wild-type cells. However, the bypass efficiency of dNIm markedly increased to (57 ± 1)% in SOS-induced cells. In wild-type AB1157 cells, dNIm induced (8.9 ± 0.5)% G → C mutations and roughly equal frequencies of G → A and G → T mutations, at (19 ± 2)% and (22 ± 3)%, respectively.272 Nonetheless, much lower frequencies of G → A and G → C mutations, at (13 ± 2)% and (2.5 ± 0.6)%, respectively, were found in SOS-induced cells.272 Later, Dimitri et al.273 found that dNIm displayed modest inhibitory effects, with bypass efficiency being (87 ± 5)% during transcription mediated by T7RNAP. However, dNIm strongly blocked transcription mediated by human RNA polymerase II (hRNAPII) in HeLa nuclear extract, with a bypass efficiency of (9 ± 5)%.273 Lesion bypass by T7RNAP induced base misinsertions and deletions opposite the dNIm (22% A, 13% –1 deletion, 7% > G and 1% U), while hRNAPII exhibited error-free nucleotide incorporation opposite the lesion.273
3.3. DNA Damage Formed from Byproducts of Lipid Peroxidation
Previous in vitro primer extension assays showed that both human Pol α and Pol β were primarily blocked by LPO-induced etheno DNA adduct _ε_dA with minimal extension.274 Pol η was capable of catalyzing a substantial amount of bypass across the lesion, where the polymerase incorporated all four nucleotides opposite _ε_dA with different preferences. Human Pol ι, a paralogue of Pol η, was blocked by _ε_dA with a very small amount of synthesis past _ε_dA, which results in insertion of dCMP and, to a much lesser extent, dTMP, opposite _ε_dA.274 The mutagenic potential of _ε_dA was also investigated using a single-stranded shuttle vector system in E. coli and in COS7 simian kidney cells. A nonmutagenic dTMP incorporation opposite _ε_dA was found as the nearly exclusive event in E. coli; the lesion is, however, highly mutagenic in COS7 cells, which leads to a very high frequency of A → G transition (63%), followed by A → T (6%) and A → C (1%) transversions.275 Different from what was observed in COS7 cells, _ε_dA induced all three possible base substitutions at similar frequencies (1.5–3% each) in HeLa human cervical cancer cells.276 Levine et al.277 also reported that, when placed on the leading strand, _ε_dA induces A → T (7%),A → C (5%), and A → G (2%) mutations in HeLa human cervical cancer cells and A → G (5%) and A → C (2%) mutations in HCT-116 human colorectal carcinoma cells. Similar to what was observed for _ε_dA, the mutagenic properties of _ε_dC were found to be strikingly different in E. coli and COS7 cells (2% in uninduced E. coli cells, 32% in SOS-induced E. coli cells, and 81% in COS7 cells).278 1,_N_2-_ε_dG is moderately mutagenic and directs the incorporation of the correct nucleotide (dCMP) in >80% of the replication events in E. coli.279 _N_2,3-_ε_dG specifically induces a very low frequency (0.5%) of G → A transition during DNA replication in E. coli.280 The above-mentioned etheno adducts can be repaired by multiple DNA repair pathways, including BER and AlkB/ALKBH family dioxygenases-mediated direct damage reversal, which were previously reviewed.281,282
The mutagenicity of malondialdehyde (MDA)-induced DNA adducts was measured in the lacZα forward mutation assay in E. coli.283 The most common type of mutations induced by MDA was base-pair substitution (76%), though frameshift mutations were detected in 16% of the induced mutants, and they comprised mainly single-nucleotide additions in runs of reiterated bases.283 Modified genomes containing a C opposite M1G resulted in roughly equal frequencies of G → A and G → T mutations with few G → C mutations. The (−)-strand was replicated only 20% of the time when M1dG was present. M1dG was also found to be a substrate for the NER pathway in E. coli.284 In addition, MDA-induced mutations, such as large insertions and deletions, were found after lesion-carrying shuttle vectors undergo replication in human cells.285 Furthermore, replication studies also demonstrated that M1dG can induce −1 and −2 frameshift mutations when positioned in a reiterated (CpG)4 sequence but not when positioned in a nonreiterated sequence in E. coli or COS-7 cells.286 Recently, Singh et al.287 revealed that AlkB could repair Acr-dG and M1dG in vitro, suggesting an important role for the AlkB family of dioxygenases in protecting against the deleterious biological consequences of acrolein- and MDA-induced DNA adducts in vivo.
For more detailed discussion about the mutagenic consequences, replication bypass and repair of DNA lesions induced by lipid peroxidation byproducts, the readers should consult a recent review by Minko et al.14
4. Implications of Oxidative Stress-Induced DNA Lesions in Human Diseases
The oxidative stress-induced DNA lesions may have significant impact on human health, including the natural processes of aging, neurodegeneration, and carcinogenesis. Recently, the development of LC-MS, coupled with the isotope-dilution method for the unambiguous identification and accurate quantification of multiple DNA lesions, has provided profound insights into the involvement of DNA lesions in different pathological conditions.15,16,288
4.1. Direct ROS-Induced DNA Lesions
4.1.1. Single-Nucleobase Lesions
Previous data from the investigation of Chinese and Japanese patients indicated that low BER activity arising from inactivating mutations of the NEIL1 gene may be involved in the pathogenesis of a subset of gastric cancers.289 In addition, elevated levels of Fapy-dA and Fapy-dG were observed in the liver, kidney, and brain tissues of _Neil1_−/− mice relative to the wild-type animals.24 These findings, along with the high incidence of pulmonary and hepatocellular tumors in _Nth1_−/−_Neil1_−/− mice, suggest the importance of DNA glycosylase NEIL1 in maintaining genomic stability.24
As discussed above, the dGh and dSp lesions are highly mutagenic.290 In the viewpoint that 8-oxo-dG is produced at high frequencies and that it is more readily oxidized than dG, the major oxidation products of 8-oxo-dG, i.e., dGh and dSp, may bear a significant impact on cellular functions. For example, the presence of dGh and dSp could influence the thermal stability and folding of the G-quadruplex,291 and dSp lesions could disturb the structure of duplex DNA and affect nucleosome positioning.292 dGh and dSp lesions have been detected in E. coli197 and in mice,131 though further studies are needed for systematically assessing their formation and repair in mammals.
Thymidine glycol has been suggested as a biomarker of oxidative stress and detected in urine samples of mammals.293–295 Thymidine glycol in DNA inhibits the nuclease P1-mediated hydrolysis of its neighboring 3′ phosphodiester bond,296–298 rendering the release of the lesion as a dinucleotide. The lesion-containing dinucleotide was thus utilized for the quantification of thymidine glycol in DNA using LC-MS/MS coupled with the stable isotope-dilution method.47,299,300 This method has been applied for probing oxidative stress in white blood cell DNA of ovarian cancer patients301 and BRCA mutation carriers302 as well as for examining the effect of smoking cessation303 and antioxidant usage304 on levels of oxidatively induced DNA damage. Rather than being highly mutagenic, thymidine glycol strongly inhibits DNA replication.203,305
As mentioned above, 5-hmdU can be produced from oxidative stress as well as epigenetic machinery (i.e., TET-mediated oxidation of thymidine). 5-hmdU has been detected in murine and human tissues,306 although the physiological implications are less well explored.
A number of studies have been conducted in measuring 5-fdU. Earlier quantification of 5-fdU in DNA was performed using GC-MS analysis of the modified nucleobase released from DNA with the use of formic acid or 70% (w/w) hydrogen fluoride in pyridine.68,69 Stable isotope dilution coupled with LC-MS/MS was later developed for the quantification of 5-fdU but with a relatively poor detection limit.307 Derivatization with Girard reagent T significantly improved the detection limit and has been applied for the detection of 5-fdU in DNA of HeLa-S3 cells308 and Trypanosoma brucei.309 5-fdU has also been detected in various tissues of LEA and LEC rats with LC-MS/MS/MS in the negative-ion mode.85 Higher levels of 5-fdU were observed in the liver of 3-month old LEC rats,85 which model Wilson disease, a disease characterized by hepatitis and hepatocellular carcinoma.310 Wilson's disease arises from mutations in ATP7B, which encodes a transporter protein required for hepatic excretion of copper ions.310 This results in the accumulation of copper ion and increased ROS production.311,312 This finding provides a vivid illustration of the role of oxidative DNA damage in transition metal-induced diseases such as Wilson's disease.
Global 5-hmC levels are lower in a variety of human cancers including breast, liver, lung, pancreatic, and prostate cancers than in normal tissues.245 One mechanism to explain this is that cancer-related gain-of-function mutations in IDH1 and IDH2 genes cause an increase in production of the oncometabolite (R)-2-hydroxyglutarate ((R)-2-HG) instead of production of the normal product 2-oxogluterate (2-OG). In addition, cancer-related mutations in two Krebs cycle genes, fumarate hydratase (FH) and succinate dehydrogenase (SDH), led to the accumulation of their substrates, fumarate and succinate, respectively. (R)-2-HG, succinate, and fumarate, which are structurally similar to 2-OG, act as competitive inhibitors of 2-OG-dependent TET activity, leading to diminished levels of 5-hmC in some tumors313–321 Additionally, Jin et al.322 reported pronounced depletion of 5-hmdC in multiple human cancers in an IDH mutation-independent manner, indicating the existence of alternative mechanism(s) involved in the loss of 5-hmdC. Together, the data suggest that 5-hmdC levels may serve as a useful molecular biomarker for cancer detection and diagnosis.322
It has been shown that 5-hmC, 5-fC, and 5-caC may serve as epigenetic marks in addition to being intermediates for active cytosine demethylation in mammals.62,63,88,323,324 In this vein, 5-hmC, 5-fC, and 5-caC are recognized by some specific cellular proteins critical for chromatin remodeling and transcriptional regulation.325–331 Thus, the homeostasis of these oxidized 5-mdC derivatives is crucial for maintaining normal cellular function, whereas the loss or aberrant accumulation of these epigenetic marks may lead to deleterious biological consequences and diseases. For instance, LC-MS/MS results revealed a significantly lower level of 5-hmdC in the liver of diseased LEC rats compared to that of control LEA rats, though no difference was found in the levels of 5-mdC.88 In vitro biochemical assays showed that Cu2+ ions could directly inhibit the activity of TET enzymes, suggesting that in LEC rats perturbation of 5-hmdC-mediated epigenetic signaling contributes to the etiology of Wilson's disease.88
4.1.2. Bulky Lesions
The implication of bulky DNA lesions, including cPus and the d(G[8–5m]T) intrastrand cross-link, were also investigated. Markedly elevated levels of 5′_R_- and 5′_S_-diastereomers of cdA and cdG were found in the liver of the LEC rat model of Wilson's disease85 Moreover, the levels of these lesions increased with age in the liver and brain of LEC rats, illustrating a correlation with disease progression.85 cPus also accumulate in the genomic DNA of wild-type mice with age, providing further evidence of the accumulation of endogenous lesions over the lifespan of mammals and a potential contribution of these lesions to aging.86 Mitra et al.89 detected greater levels of cPus lesions in the skin of a murine model of human melanoma. The increase in cPus levels is ultraviolet radiation-independent but pheomelanin pigment-dependent, suggesting that pigment-driven oxidative stress and the resultant oxidative DNA damage contribute to melanoma in humans even in the absence of UV exposure.89
Results from a similar LC-MS3 method revealed that the levels of d(G[8–5m]T) are elevated in tissues of LEC rats relative to LEA rats.332 Additionally, XPA-deficient human brain and mouse liver as well as various types of tissues of ERCC1-deficient mice contain higher levels of d(G[8–5m]T).332 In an earlier review, Brooks81 proposed that chemically stable bulky DNA lesions, such as cPus, may play a crucial role in neurodegeneration in XP patients.333 Since the brain is not directly exposed to sunlight, the accumulation of ROS-induced bulky DNA lesions in brains of XP patients may contribute to neuron loss in these patients. In support of this, in _Ercc1_−/Δ mice, with defective nucleotide excision repair of cPus and progressive neurodegeneration,334 the lesions accumulate more rapidly in the brain than in normal mice. However, as neurodegeneration progresses, the number of cPus decreases significantly in the brain, suggesting a direct causal role of oxidative DNA damage in the loss of neurons in the brain.86
A recent technique improvement by employing nanoflow liquid chromatography–nanoelectrospray ionization coupled with tandem mass spectrometry provided much better sensitivity in measuring cPus as well as LPO-induced _ε_dA and _ε_dG lesions in the liver and brain tissues of LEA and LEC rats.88 Simultaneous quantification of these two different types of lesions demonstrated a preferential accumulation of direct ROS-induced cPus in vivo.88 This result indicates that cPus may contribute to the etiology of oxidative stress-induced diseases to a greater extent than the etheno adducts arising from byproducts of lipid peroxidation.88
Earlier reviews discussed the application of LPO-induced DNA lesions as potential biomarkers for cancer risk assessment in humans with cancer-prone diseases such as chronic pancreatitis, ulcerative colitis, or Crohn's disease, as well as in patients with alcohol abuse-related chronic hepatitis, fatty liver, fibrosis, or cirrhosis.158,335 Results from an isotope dilution-capillary LC-MS/MS method revealed a significant increase in levels of Acr-dG in Alzheimer's disease (AD) patients compared to controls in DNA isolated from the hippocampus/parahippocampal gyrus.336 This, along with the existence of potential acrolein-derived DNA-peptide cross-links,149 provides evidence to support the correlation between acrolein-induced DNA damage and AD pathogenesis. The LPO-induced bulky exocyclic 1,_N_2-propano-dG adducts may play a significant role in driving congenital abnormalities, myelodysplasia, acute myeloid leukemia (AML), and certain solid tumors in patients suffering from Fanconi anemia (FA).337–339 FA is an autosomal recessive disease caused by deficiency in the repair of DNA interstrand cross-links. As discussed previously, the DNA interstrand cross-links generated from the ring-open form of 1,_N_2-propano-dG are likely substrates of the FA repair pathway, and if left unrepaired, these cross-links impede replication and transcription, causing chromosomal breaks and translocations that can drive disease in FA patients.340
4.2. Inflammation-Induced DNA Damage
Halogenated nucleobases are commonly associated with inflammation, and 5–Cl-C is often used as a biomarker for inflammation.131,132 Various halogenation products have been detected in mammalian cells, tissues, and fluids. Chlorinated DNA lesions such as 5-Cl-dC, 8-Cl-dG, 8-Cl-dA, and chlorinated ribonucleo-sides including 5-chlorocytidine (5-Cl-rC), 8-chloroguanosine (8-Cl-rG), and 8-chloroadenosine (8-Cl-rA) have been detected with an LC-MS/MS-based method in HOCl-treated cells and freshly isolated human white blood cells. 5-Cl-dC was the predominant DNA lesion, and 8-Cl-G and 5–Cl-C were present at higher levels in RNA than DNA.127 Jiang et al.341 found, from GC-MS analysis, increased levels of 5-Cl-dU in DNA from cells treated with HOCl, as well as in exudate fluid from carrageenan-induced inflammation sites, but not in the DNA from tissues at inflammation sites in rats, possibly due to the lack of proliferation in the isolated tissues.341 5-Cl-U has been detected in neutrophil-rich inflammatory sites118 as well as aortic tissue342 with isotope dilution coupled with GC-MS methods. The 5-Cl-U level in atherosclerotic aortic tissue was reported to be 10-fold higher than that in normal tissue, implying a potential role of halogenated nucleobases in atherogenesis. 8-Br-dG and 8-Cl-dG were quantitatively measured with stable isotope-dilution coupled with the LC-MS/MS method in rat liver, rat urine, and human urine samples, and elevated levels of 8-Br-dG and 8-Cl-dG were observed in urine samples of diabetic patients compared to those in healthy controls.129 Noyon et al.343 used LC-MS/MS for the quantification of 5-Cl-dC, 5-Cl-C, and 8-Cl-G and detected the presence of 5-Cl-C (1.0 ± 0.2 nM) in healthy human plasma. 5-Cl-C and 8-Cl-G could be detected in the cytoplasmic nucleotide pool and RNA, and 5-Cl-C is present in DNA from endothelial cells treated with HOCl.343
5. Conclusions and Perspectives
In this review, we summarized the chemical formation and biological consequences of a series of oxidatively induced DNA lesions. We also discussed the involvement of these lesions in the etiology of human diseases. Over the past several decades, much has been learned about the mechanisms of formation and repair of oxidative stress-induced DNA lesions. A number of shuttle vector-based studies have provided important molecule-level information about the degrees to which some of the oxidatively induced DNA lesions perturb the transmission of genetic information by inhibiting DNA replication and transcription as well as inducing mutations during these processes (Tables 1 and 2). Nevertheless, work remains to be done on assessing how other oxidatively induced DNA lesions, particularly the secondary oxidation products of dG (i.e., the dGhand dSp adducts) as well as the halogenated nucleosides induced by inflammatory processes, alter the efficiency and fidelity of DNA replication and transcription in mammalian cells.
The available data suggest that these mutagenic DNA adducts, especially the bulky DNA lesions such as exocyclic propano adducts, cPus, and nucleobase-nucleobase intrastrand cross-links, could be potential biomarkers for investigating the role of oxidative stress in human diseases. Along this line, future work must be done to further elucidate the relationship between the levels of oxidatively induced DNA lesions and the stages of disease (e.g., cancer), which will provide important knowledge for future diagnostic applications of DNA adduct measurements.
During the past two decades, the sensitivities of mass spectrometers for measuring DNA adducts have greatly improved, and the LC-MS coupled with the stable isotope-dilution method is considered the most powerful technique for the unequivocal identification and accurate quantification of oxidatively induced DNA lesions in cells and tissues. We envision that further development of the method for high-throughput and simultaneous quantification of multiple oxidatively induced DNA lesions is necessary. Adapting methods to the measurement of lesions in ever increasingly smaller quantities of genomic DNA will also improve translation of these techniques, for example, to tissue biopsies like bronchoalveolar lavage. Improvement of LC-MS instrument performance as well as sample preparation and cleanup are both needed. For instance, the use of small internal diameter columns coupled with nLC-nESI-MS systems has already facilitated the quantitative analysis of multiple oxidatively induced DNA lesions with the use of low microgram quantity of DNA.88,159,160,163,165 Recently, high-resolution Orbitrap mass spectrometers have been used for the detection of alkylated DNA lesions induced by carcinogens present in tobacco smoke.344,345 Owing to their accurate mass measurement capability and high sensitivity, we expect more applications of this type of instruments for the quantification of low levels of oxidatively generated DNA lesions in cellular and tissue samples in the future. The improvement in LC-MS-based quantification techniques will also enable systematic adductomics research,346–348 which may facilitate the discovery of novel oxidatively induced DNA lesions involved in the pathogenesis of human diseases.
Apart from the improvement in MS instrumentation, new sample preparation and cleanup techniques are also crucial for future high-throughput analysis of DNA lesions in vivo. For example, offline HPLC enrichment permits sensitive quantification of the 5′_R_- and 5′_S_- diastereomers of cdA and cdG.85,86,88,89 Although offline HPLC provides effective sample cleanup and removal of the excess amount of unmodified nucleosides in the DNA digestion mixture, it is relatively time-consuming and not conducive to rapid analysis. Because of the high separation ability, ultraperformance liquid chromatography (UPLC),349–351 nanoflow UPLC (nUPLC)-nESI coupled with high-sensitivity and high-resolution MS method may enable simultaneous and efficient quantification of multiple oxidatively induced DNA lesions in a DNA digestion mixture without offline HPLC enrichment.
Last, but not least, it is important to note that caution needs to be exerted during sample preparation (i.e., DNA extraction, enzymatic digestion of DNA, etc.) so that artificial generation of oxidatively induced DNA lesions can be minimized. In this vein, the levels of 8-oxo-dG in the DNA of pig liver or HeLa cells could vary by 2 orders of magnitude when the same samples were analyzed by a network of laboratories.352 Thus, extreme precautions should always be taken while measuring this and other direct ROS-induced DNA lesions as well as those DNA adducts arising from byproducts of lipid peroxidation. It is crucial that the final levels of DNA lesions measured reflect the levels of DNA adducts present in cellular and tissue DNA, not a combination of those present in initial DNA and artificially formed during sample preparation.
Acknowledgments
Funding: This work was supported by the National Institutes of Health (R01 CA101864 and P01 AG043376).
Abbreviations
Acr-dG
acrolein _N_2-(3-oxopropyl)-dG adduct
AD
Alzheimer's disease
AID/APOBEC proteins
activation-induced cytidine deaminase/apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like proteins
AML
acute myeloid leukemia
AP site
apurinic/apyrimidinic site
BER
base excision repair
cdA
8,5′-cyclo-2′-deoxyadenosine
Cro-dG
crotonaldehyde _N_2-(3-oxopropyl)-dG adduct
dA
2′-deoxyadenosine
dCMP
2′-deoxycytidine 5′-monophosphate
DDE
2,4-decadienal
dGh
guanidinohydantoin 2′-deoxynucleoside
dNIm
5-guanidino-4-nitroimidazole 2′-deoxynucleoside
DODE
9,12-dioxo-(10_E_)dodecenoic acid
DOOE
5,8-dioxo-(10_E_)-octenoic acid
DSB
double strand break
dSp
guanidinohydantoin 2′-deoxynucleoside
DHPN18O2
18O-labeled 1,4-endoperoxide of N,_N_′-di(2,3-dihydroxypropyl)-1,4-naphthalene-dipropanamide
EDE
4,5-epoxy-(2_E_)-decenal
EndoV
endonuclease V
FA
Fanconi anemia
Fapy-dA
4,6-diamino-5-formamidopyrimidine 2′-deoxynucleoside
Fapy-dG
2,6-diamino-4-hydroxy-5-formamidopyrimidine 2′-deoxynucleoside
Fe-NTA
ferric nitrilotriacetate
FH
fumarate hydratase
Fpg
formamidopyrimidine DNA glycosylase
G(-H)•
guanine neutral radicals
HHE
4-hydroxy-2-hexenal
hAAG
alkyladenine DNA glycosylase
hMTH1
human MutT homologue
hNEIL1
human endonuclease VIII-like 1
hOGG1
human 8-oxoguanine DNA glycosylase 1
HNE
4-hydroxy-2-nonenal
HNE-dG
HNE _N_2-(3-oxopropyl)-dG adduct
HPNE
4-hydroperoxy-(2_E_)nonenal
HOBr
hypobromous acid
HOCl
hypochlorous acid
H2O2
hydrogen peroxide
hRNAPII
human RNA polymerase II
M1dA
_N_6-(3-oxopropenyl)-2′-deoxyadenosine
M1dC
_N_4-(3-oxopropenyl)-2′-deoxycytidine
M1dG
pyrimido[1,2-α_]purine-10(3_H)-one-2′-deoxyribose
MDA
malondialde-hyde
MEFs
mouse embryonic fibroblasts
mESCs
mouse embryonic stem cells
_N_23-_ε_dG
_N_2,3-etheno-2′-deoxyguano-sine
NADPH
nicotinamide adenine dinucleotide phosphate
NER
nucleotide excision repair
ODN
oligodeoxyribonucleo-tide
ONE
4-oxo-2-nonenal
ONOO−
peroxynitrite
Pol η
DNA polymerase η
PUFA
polyunsaturated fatty acids
(R)-2-HG
(R)-2-hydroxyglutarate
ROS
reactive oxygen species
SDH
succinate dehydrogenase
SOD
superoxide dismutase
T7RNAP
T7 RNA polymerase
TDG
thymine DNA glycosylase
TET
ten-eleven translocation
thymidine glycol
5,6-dihydroxy-5,6-dihydro-2′-deoxythymidine
TLC
thin-layer chromatography
UPLC
ultraperformance liquid chromatography
1_N_2-_ε_dG
1,_N_2-etheno-2′-deoxyguanosine
2-OG
2-oxoglutarate
5-Br-dC
5-bromo-2′-deoxycytidine
5-Br-dU
5-bromo-2′-deoxyuridine
5-Br-U
5-bromouracil
5-cadC
5-carboxyl-2′-deoxycytidine
5-Cl-C
5-chlorocytosine
5-Cl-dC
5-chloro-2′-deoxycytidine
5-Cl-dU
5-chloro-2′-deoxyuridine
5-Cl-rC
5-chlorocytidine
5-Cl-U
5-chlorouracil
5-fdC
5-formyl-2′-deoxycytidine
5-fdU
5-formyl-2′-deoxyuridine
5-hmdC
5-hydroxymethyl-2′-deoxycytidine
5-hmdU
5-hydroxymethyl-2′-deoxyuridine
5-mdC
5-methyl-2′-deoxycytidine
5-OH-8-oxo-dG
5-hydroxy-substituted derivative of 8-oxo-dG
8-Br-dA
8-chloro-2′-deoxyadenosine
8-Br-dG
8-chloro-2′-deoxyadenosine
8-Cl-dA
8-chloro-2′-deoxyadenosine
8-Cl-dG
8-chloro-2′-deoxyguanosine
8-Cl-G
8-chloroguanine
8-Cl-rA
8-chloroadenosine
8-Cl-rG
8-chloroguanosine
8-nitro-dG
8-nitro-2′-deoxyguanosine
8-oxo-dA
8-oxo-7,8-dihydro-2′-deoxyadenosine
8-oxo-dG
8-oxo-7,8-dihydro-2′-deoxyguanosine
_ε_dA
1,_N_6-etheno-2′-deoxyadenosine
_ε_dC
3,_N_4-etheno-2′-deoxycytidine
Biographies
Biographies
Yang Yu received his B.S. (2009) and M.S. (2012) degrees in Biological Technology and Microbiology from Nankai University, China. He is currently a Ph.D. candidate in the Environmental Toxicology Graduate Program at the University of California Riverside under the supervision of Professor Yinsheng Wang. At the time of writing, Yang was close to completing his dissertation research project, which involves the development of mass spectrometric methods for the quantification of different types of structurally modified DNA and the application of these methods in assessing the biological implications of DNA adducts/modifications in various living cells/organisms.
Yuxiang Cui obtained her B.S. degree in Chemistry at Nankai University and M.S. degree in Food Safety and Toxicology at the University of Hong Kong. She joined the Environmental Toxicology Graduate Program at the University of California Riverside in 2014 and is currently a Ph.D. candidate under the guidance of Professor Yinsheng Wang. Her research projects mainly focus on the development and application of LC-MS methods for the quantitative measurement of DNA adducts.
Laura Niedernhofer is an Associate Professor in the Department of Metabolism and Aging at The Scripps Research Institute in Florida. She has a Bachelor of Sciences in Chemistry from Duke University and completed the Medical Scientist Training Program at Vanderbilt University, earning her Ph.D. in Biochemistry. She followed this with postdoctoral training in mouse genetics at the Erasmus Medical Center in Rotterdam, The Netherlands. Her area of expertise is DNA damage and repair. Dr. Niedernhofer's current research program is focused on discovering the mechanism by which DNA damage contributes to aging and age-related diseases using genetic and pharmacologic approaches.
Yinsheng Wang obtained his Ph.D. in Chemistry from Washington University in St. Louis in 2001. He is currently a professor of Chemistry and the Director for the Environmental Toxicology Graduate Program at the University of California Riverside. Yinsheng's research concentrates on the use of mass spectrometry, along with molecular biology and/or synthetic organic chemistry, to understand the occurrence and biological consequences of DNA adducts and post-translational modifications of proteins.
Footnotes
Notes: The authors declare no competing financial interest.
Special Issue: Mass Spectrometry and Emerging Technologies for Biomarker Discovery in the Assessment of Human Health and Disease
References
- 1.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 2.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 3.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 4.Stohs SJ, Bagchi D. Oxidative mechanisms in the toxicity of metal ions. Free Radical Biol Med. 1995;18:321–336. doi: 10.1016/0891-5849(94)00159-h. [DOI] [PubMed] [Google Scholar]
- 5.De Bont R, van Larebeke N. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis. 2004;19:169–185. doi: 10.1093/mutage/geh025. [DOI] [PubMed] [Google Scholar]
- 6.Evans MD, Dizdaroglu M, Cooke MS. Oxidative DNA damage and disease: induction, repair and significance. Mutat Res Rev Mutat Res. 2004;567:1–61. doi: 10.1016/j.mrrev.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 7.Ohshima H, Tatemichi M, Sawa T. Chemical basis of inflammation-induced carcinogenesis. Arch Biochem Biophys. 2003;417:3–11. doi: 10.1016/s0003-9861(03)00283-2. [DOI] [PubMed] [Google Scholar]
- 8.Winterbourn CC, Kettle AJ. Redox reactions and microbial killing in the neutrophil phagosome. Antioxid Redox Signaling. 2013;18:642–660. doi: 10.1089/ars.2012.4827. [DOI] [PubMed] [Google Scholar]
- 9.Rayner BS, Love DT, Hawkins CL. Comparative reactivity of myeloperoxidase-derived oxidants with mammalian cells. Free Radical Biol Med. 2014;71:240–255. doi: 10.1016/j.freeradbiomed.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 10.Panday A, Sahoo MK, Osorio D, Batra S. NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cell Mol Immunol. 2015;12:5–23. doi: 10.1038/cmi.2014.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Valko M, Izakovic M, Mazur M, Rhodes CJ, Telser J. Role of oxygen radicals in DNA damage and cancer incidence. Mol Cell Biochem. 2004;266:37–56. doi: 10.1023/b:mcbi.0000049134.69131.89. [DOI] [PubMed] [Google Scholar]
- 12.Lonkar P, Dedon PC. Reactive species and DNA damage in chronic inflammation: reconciling chemical mechanisms and biological fates. Int J Cancer. 2011;128:1999–2009. doi: 10.1002/ijc.25815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marnett LJ. Lipid peroxidation—DNA damage by malondialdehyde. Mutat Res Fundam Mol Mech Mutagen. 1999;424:83–95. doi: 10.1016/s0027-5107(99)00010-x. [DOI] [PubMed] [Google Scholar]
- 14.Minko IG, Kozekov ID, Harris TM, Rizzo CJ, Lloyd RS, Stone MP. Chemistry and biology of DNA containing 1,N2-deoxyguanosine adducts of the α,β-unsaturated aldehydes acrolein, crotonaldehyde, and 4-hydroxynonenal. Chem Res Toxicol. 2009;22:759–778. doi: 10.1021/tx9000489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu S, Wang Y. Mass spectrometry for the assessment of the occurrence and biological consequences of DNA adducts. Chem Soc Rev. 2015;44:7829–7854. doi: 10.1039/c5cs00316d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tretyakova N, Villalta PW, Kotapati S. Mass spectrometry of structurally modified DNA. Chem Rev. 2013;113:2395–2436. doi: 10.1021/cr300391r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.von Sonntag C. The Chemical Basis of Radiation Biology. Taylor & Francis; London: 1987. [Google Scholar]
- 18.Kasai H, Nishimura S. Hydroxylation of deoxyguanosine at the C-8 position by ascorbic acid and other reducing agents. Nucleic Acids Res. 1984;12:2137–2145. doi: 10.1093/nar/12.4.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steenken S. Purine bases, nucleosides, and nucleotides: aqueous solution redox chemistry and transformation reactions of their radical cations and e− and OH adducts. Chem Rev. 1989;89:503–520. [Google Scholar]
- 20.Dizdaroglu M, Kirkali G, Jaruga P. Formamidopyrimidines in DNA: Mechanisms of formation, repair, and biological effects. Free Radical Biol Med. 2008;45:1610–1621. doi: 10.1016/j.freeradbiomed.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 21.Cho BP, Evans FE. Structure of oxidatively damaged nucleic acid adducts 3 Tautomerism, ionization and protonation of 8-hydroxyadenosine studied by 15N NMR spectroscopy. Nucleic Acids Res. 1991;19:1041–1046. doi: 10.1093/nar/19.5.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ravanat JL, Di Mascio P, Martinez GR, Medeiros MHG, Cadet J. Singlet oxygen induces oxidation of cellular DNA. J Biol Chem. 2000;275:40601–40604. doi: 10.1074/jbc.M006681200. [DOI] [PubMed] [Google Scholar]
- 23.Egler RA, Fernandes E, Rothermund K, Sereika S, de Souza-Pinto N, Jaruga P, Dizdaroglu M, Prochownik EV. Regulation of reactive oxygen species, DNA damage, and c-Myc function by peroxiredoxin 1. Oncogene. 2005;24:8038–8050. doi: 10.1038/sj.onc.1208821. [DOI] [PubMed] [Google Scholar]
- 24.Chan MK, Ocampo-Hafalla MT, Vartanian V, Jaruga P, Kirkali G, Koenig KL, Brown S, Lloyd RS, Dizdaroglu M, Teebor GW. Targeted deletion of the genes encoding NTH1 and NEIL1 DNA N-glycosylases reveals the existence of novel carcinogenic oxidative damage to DNA. DNA Repair. 2009;8:786–794. doi: 10.1016/j.dnarep.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dizdaroglu M. Oxidatively induced DNA damage: Mechanisms, repair and disease. Cancer Lett. 2012;327:26–47. doi: 10.1016/j.canlet.2012.01.016. [DOI] [PubMed] [Google Scholar]
- 26.Dizdaroglu M, Coskun E, Jaruga P. Measurement of oxidatively induced DNA damage and its repair, by mass spectrometric techniques. Free Radical Res. 2015;49:525–548. doi: 10.3109/10715762.2015.1014814. [DOI] [PubMed] [Google Scholar]
- 27.Dizdaroglu M. Oxidatively induced DNA damage and its repair in cancer. Mutat Res Rev Mutat Res. 2015;763:212–245. doi: 10.1016/j.mrrev.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 28.Hu J, de Souza-Pinto NC, Haraguchi K, Hogue BA, Jaruga P, Greenberg MM, Dizdaroglu M, Bohr VA. Repair of formamidopyrimidines in DNA involves different glycosylases: role of the OGG1, NTH1, and NEIL1 enzymes. J Biol Chem. 2005;280:40544–40551. doi: 10.1074/jbc.M508772200. [DOI] [PubMed] [Google Scholar]
- 29.Steenken S, Jovanovic SV. How easily oxidizable is DNA? One-electron reduction potentials of adenosine and guanosine radicals in aqueous solution. J Am Chem Soc. 1997;119:617–618. [Google Scholar]
- 30.Steenken S, Jovanovic SV, Bietti M, Bernhard K. The trap depth (in DNA) of 8-oxo-7,8-dihydro-2′-deoxyguanosine as derived from electron-transfer equilibria in aqueous solution. J Am Chem Soc. 2000;122:2373–2374. [Google Scholar]
- 31.Luo WC, Muller JG, Rachlin EM, Burrows CJ. Characterization of hydantoin products from one-electron oxidation of 8-oxo-7,8-dihydroguanosine in a nucleoside model. Chem Res Toxicol. 2001;14:927–938. doi: 10.1021/tx010072j. [DOI] [PubMed] [Google Scholar]
- 32.Cui L, Ye WJ, Prestwich EG, Wishnok JS, Taghizadeh K, Dedon PC, Tannenbaum SR. Comparative analysis of four oxidized guanine lesions from reactions of DNA with peroxynitrite, singlet oxygen, and γ-radiation. Chem Res Toxicol. 2013;26:195–202. doi: 10.1021/tx300294d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Niles JC, Wishnok JS, Tannenbaum SR. Spiroiminodihydantoin is the major product of the 8-oxo-7,8-dihydroguanosine reaction with peroxynitrite in the presence of thiols and guanosine photooxidation by methylene blue. Org Lett. 2001;3:963–966. [PubMed] [Google Scholar]
- 34.Niles JC, Wishnok JS, Tannenbaum SR. Spiroiminodihydantoin and guanidinohydantoin are the dominant products of 8-oxoguanosine oxidation at low fluxes of peroxynitrite: Mechanistic studies with 18O. Chem Res Toxicol. 2004;17:1510–1519. doi: 10.1021/tx0400048. [DOI] [PubMed] [Google Scholar]
- 35.Fleming AM, Muller JG, Ji IS, Burrows CJ. Characterization of 2′-deoxyguanosine oxidation products observed in the Fenton-like system Cu(II)/H2O2/reductant in nucleoside and oligodeoxynucleotide contexts. Org Biomol Chem. 2011;9:3338–3348. doi: 10.1039/c1ob05112a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suzuki T, Masuda M, Friesen MD, Ohshima H. Formation of spiroiminodihydantoin nucleoside by reaction of 8-oxo-7,8-dihydro-2′-deoxyguanosine with hypochlorous acid or a myeloperoxidase-H2O2-Cl− system. Chem Res Toxicol. 2001;14:1163–1169. doi: 10.1021/tx010024z. [DOI] [PubMed] [Google Scholar]
- 37.Fleming AM, Burrows CJ. G-quadruplex folds of the human telomere sequence alter the site reactivity and reaction pathway of guanine oxidation compared to duplex DNA. Chem Res Toxicol. 2013;26:593–607. doi: 10.1021/tx400028y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ye Y, Muller JG, Luo WC, Mayne CL, Shallop AJ, Jones RA, Burrows CJ. Formation of 13C-, 15N-, and 18O-labeled guanidinohydantoin from guanosine oxidation with singlet oxygen. Implications for structure and mechanism. J Am Chem Soc. 2003;125:13926–13927. doi: 10.1021/ja0378660. [DOI] [PubMed] [Google Scholar]
- 39.Zhu J, Fleming AM, Orendt AM, Burrows CJ. pH-Dependent equilibrium between 5-guanidinohydantoin and iminoallantoin affects nucleotide insertion opposite the DNA lesion. J Org Chem. 2016;81:351–359. doi: 10.1021/acs.joc.5b02180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luo WC, Muller JG, Burrows CJ. The pH-dependent role of superoxide in riboflavin-catalyzed photooxidation of 8-oxo-7,8-dihydroguanosine. Org Lett. 2001;3:2801–2804. doi: 10.1021/ol0161763. [DOI] [PubMed] [Google Scholar]
- 41.Dizdaroglu M, Jaruga P. Mechanisms of free radical-induced damage to DNA. Free Radical Res. 2012;46:382–419. doi: 10.3109/10715762.2011.653969. [DOI] [PubMed] [Google Scholar]
- 42.Teoule R. Radiation-induced DNA damage and its repair. Int J Radiat Biol Relat Stud Phys Chem Med. 1987;51:573–589. doi: 10.1080/09553008414552111. [DOI] [PubMed] [Google Scholar]
- 43.Castro GD, Stamato CJ, Castro JA. 5-Methylcytosine attack by free radicals arising from bromotrichloro-methane in a model system: structures of reaction products. Free Radical Biol Med. 1994;17:419–428. doi: 10.1016/0891-5849(94)90168-6. [DOI] [PubMed] [Google Scholar]
- 44.Zuo SJ, Boorstein RJ, Teebor GW. Oxidative damage to 5-methylcytosine in DNA. Nucleic Acids Res. 1995;23:3239–3243. doi: 10.1093/nar/23.16.3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bienvenu C, Cadet J. Synthesis and kinetic study of the deamination of the cis diastereomers of 5,6-dihydroxy-5,6-dihydro-5-methyl-2′-deoxycytidine. J Org Chem. 1996;61:2632–2637. doi: 10.1021/jo951900e. [DOI] [PubMed] [Google Scholar]
- 46.Tremblay S, Douki T, Cadet J, Wagner JR. 2′-Deoxycytidine glycols, a missing link in the free radical-mediated oxidation of DNA. J Biol Chem. 1999;274:20833–20838. doi: 10.1074/jbc.274.30.20833. [DOI] [PubMed] [Google Scholar]
- 47.Cao HC, Jiang Y, Wang YS. Kinetics of deamination and Cu(II)/H2O2/Ascorbate-induced formation of 5-methylcytosine glycol at CpG sites in duplex DNA. Nucleic Acids Res. 2009;37:6635–6643. doi: 10.1093/nar/gkp615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pfeifer GP. p53 mutational spectra and the role of methylated CpG sequences. Mutat Res Fundam Mol Mech Mutagen. 2000;450:155–166. doi: 10.1016/s0027-5107(00)00022-1. [DOI] [PubMed] [Google Scholar]
- 49.Kanwal R, Gupta S. Epigenetic modifications in cancer. Clin Genet. 2012;81:303–311. doi: 10.1111/j.1399-0004.2011.01809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Teoule R, Bonicel A, Bert C, Cadet J, Polverel M. Identification of radioproducts resulting from breakage of thymine moiety by gamm-irradiation of Escherichia coli DNA in an aerated aqueous-solution. Radiat Res. 1974;57:46–58. [PubMed] [Google Scholar]
- 51.Teoule R, Bert C, Bonicel A. Thymine fragment damage retained in the DNA polynucleotide chain after gamma irradiation in aerated solutions. II. Radiat Res. 1977;72:190–200. [PubMed] [Google Scholar]
- 52.Frenkel K, Goldstein MS, Duker NJ, Teebor GW. Identification of the cis-thymine glycol moiety in oxidized deoxyribonucleic acid. Biochemistry. 1981;20:750–754. doi: 10.1021/bi00507a014. [DOI] [PubMed] [Google Scholar]
- 53.Bienvenu C, Wagner JR, Cadet J. Photosensitized oxidation of 5-methyl-2′-deoxycytidine by 2-methyl-1,4-naphthoquinone: Characterization of 5-(hydroperoxymethyl)-2′-deoxycytidine and stable methyl group oxidation products. J Am Chem Soc. 1996;118:11406–11411. [Google Scholar]
- 54.Wagner JR, Cadet J. Oxidation reactions of cytosine DNA components by hydroxyl radical and one-electron oxidants in aerated aqueous solutions. Acc Chem Res. 2010;43:564–571. doi: 10.1021/ar9002637. [DOI] [PubMed] [Google Scholar]
- 55.Kasai H, Iida A, Yamaizumi Z, Nishimura S, Tanooka H. 5-Formyldeoxyuridine: a new type of DNA damage induced by ionizing-radiation and its mutagenicity to Salmonella strain TA102. Mutat Res Lett. 1990;243:249–253. doi: 10.1016/0165-7992(90)90139-b. [DOI] [PubMed] [Google Scholar]
- 56.Tahiliani M, Koh KP, Shen YH, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.He YF, Li BZ, Li Z, Liu P, Wang Y, Tang QY, Ding JP, Jia YY, Chen ZC, Li L, Sun Y, Li XX, Dai Q, Song CX, Zhang KL, He C, Xu GL. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pastor WA, Pape UJ, Huang Y, Henderson HR, Lister R, Ko M, McLoughlin EM, Brudno Y, Mahapatra S, Kapranov P, Tahiliani M, Daley GQ, Liu XS, Ecker JR, Milos PM, Agarwal S, Rao A. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473:394–397. doi: 10.1038/nature10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu H, D'Alessio AC, Ito S, Wang ZB, Cui KR, Zhao KJ, Sun YE, Zhang Y. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev. 2011;25:679–684. doi: 10.1101/gad.2036011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Raiber EA, Beraldi D, Ficz G, Burgess HE, Branco MR, Murat P, Oxley D, Booth MJ, Reik W, Balasubramanian S. Genome-wide distribution of 5-formylcytosine in embryonic stem cells is associated with transcription and depends on thymine DNA glycosylase. Genome Biol. 2012;13:R69. doi: 10.1186/gb-2012-13-8-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shen L, Wu H, Diep D, Yamaguchi S, D'Alessio AC, Fung HL, Zhang K, Zhang Y. Genome-wide analysis reveals TET- and TDG-dependent 5-methylcytosine oxidation dynamics. Cell. 2013;153:692–706. doi: 10.1016/j.cell.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Song CX, Szulwach KE, Dai Q, Fu Y, Mao SQ, Lin L, Street C, Li YJ, Poidevin M, Wu H, Gao J, Liu P, Li L, Xu GL, Jin P, He C. Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell. 2013;153:678–691. doi: 10.1016/j.cell.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lu XY, Zhao BS, He C. TET family proteins: oxidation activity, interacting molecules, and functions in diseases. Chem Rev. 2015;115:2225–2239. doi: 10.1021/cr500470n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guo JU, Su YJ, Zhong C, Ming GL, Song HJ. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Branco MR, Ficz G, Reik W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat Rev Genet. 2012;13:7–13. doi: 10.1038/nrg3080. [DOI] [PubMed] [Google Scholar]
- 67.Pfaffeneder T, Spada F, Wagner M, Brandmayr C, Laube SK, Eisen D, Truss M, Steinbacher J, Hackner B, Kotljarova O, Schuermann D, Michalakis S, Kosmatchev O, Schiesser S, Steigenberger B, Raddaoui N, Kashiwazaki G, Muller U, Spruijt CG, Vermeulen M, Leonhardt H, Schar P, Muller M, Carell T. Tet oxidizes thymine to 5-hydroxymethyluracil in mouse embryonic stem cell DNA. Nat Chem Biol. 2014;10:574–581. doi: 10.1038/nchembio.1532. [DOI] [PubMed] [Google Scholar]
- 68.Bjelland S, Eide L, Time RW, Stote R, Eftedal I, Volden G, Seeberg E. Oxidation of thymine to 5-formyluracil in DNA: mechanisms of formation, structural implications, and base excision by human cell free extracts. Biochemistry. 1995;34:14758–14764. doi: 10.1021/bi00045a017. [DOI] [PubMed] [Google Scholar]
- 69.Douki T, Delatour T, Paganon F, Cadet J. Measurement of oxidative damage at pyrimidine bases in gamma-irradiated DNA. Chem Res Toxicol. 1996;9:1145–1151. doi: 10.1021/tx960095b. [DOI] [PubMed] [Google Scholar]
- 70.MurataKamiya N, Kamiya H, Muraoka M, Kaji H, Kasai H. Comparison of oxidation products from DNA components by γ-irradiation and Fenton-type reactions. J Radiat Res. 1997;38:121–131. doi: 10.1269/jrr.38.121. [DOI] [PubMed] [Google Scholar]
- 71.Hong H, Cao H, Wang Y, Wang Y. Identification and quantification of a guanine–thymine intrastrand cross-link lesion induced by Cu(II)/H2O2/ascorbate. Chem Res Toxicol. 2006;19:614–621. doi: 10.1021/tx060025x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Feig DI, Reid TM, Loeb LA. Reactive oxygen species in tumorigenesis. Cancer Res. 1994;54:1890s–1894s. [PubMed] [Google Scholar]
- 73.Lee DH, O'Connor TR, Pfeifer GP. Oxidative DNA damage induced by copper and hydrogen peroxide promotes CGàTT tandem mutations at methylated CpG dinucleotides in nucleotide excision repair-deficient cells. Nucleic Acids Res. 2002;30:3566–3573. doi: 10.1093/nar/gkf478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Reid TM, Loeb LA. Tandem double CCàTT mutations are produced by reactive oxygen species. Proc Natl Acad Sci U S A. 1993;90:3904–3907. doi: 10.1073/pnas.90.9.3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Newcomb TG, Allen KJ, Tkeshelashvili L, Loeb LA. Detection of tandem CCàTT mutations induced by oxygen radicals using mutation-specific PCR. Mutat Res Fundam Mol Mech Mutagen. 1999;427:21–30. doi: 10.1016/S0027-5107(99)00075-5. [DOI] [PubMed] [Google Scholar]
- 76.Randerath K, Randerath E, Zhou GD, Li D. Bulky endogenous DNA modifications (I-compounds)—possible structural origins and functional implications. Mutat Res Fundam Mol Mech Mutagen. 1999;424:183–194. doi: 10.1016/s0027-5107(99)00018-4. [DOI] [PubMed] [Google Scholar]
- 77.Randerath K, Reddy MV, Disher RM. Age- and tissue-related DNA modifications in untreated rats: detection by 32Ppostlabeling assay and possible significance for spontaneous tumor induction and aging. Carcinogenesis. 1986;7:1615–1617. doi: 10.1093/carcin/7.9.1615. [DOI] [PubMed] [Google Scholar]
- 78.Randerath K, Zhou GD, Somers RL, Robbins JH, Brooks PJ. A 32P-postlabeling assay for the oxidative DNA lesion 8,5′-cyclo-2′-deoxyadenosine in mammalian tissues: Evidence that four type II I-compounds are dinucleotides containing the lesion in the 3′ nucleotide. J Biol Chem. 2001;276:36051–36057. doi: 10.1074/jbc.M105472200. [DOI] [PubMed] [Google Scholar]
- 79.Jaruga P, Dizdaroglu M. 8,5′-Cyclopurine-2′-deoxynucleosides in DNA: mechanisms of formation, measurement, repair and biological effects. DNA Repair. 2008;7:1413–1425. doi: 10.1016/j.dnarep.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 80.Chatgilialoglu C, Ferreri C, Terzidis MA. Purine 5′,8-cyclonucleoside lesions: chemistry and biology. Chem Soc Rev. 2011;40:1368–1382. doi: 10.1039/c0cs00061b. [DOI] [PubMed] [Google Scholar]
- 81.Brooks PJ. The case for 8,5′-cyclopurine-2′-deoxynucleosides as endogenous DNA lesions that cause neuro-degeneration in xeroderma pigmentosum. Neuroscience. 2007;145:1407–1417. doi: 10.1016/j.neuroscience.2006.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dizdaroglu M. Free-radical-induced formation of an 8,5′-cyclo-2′-deoxyguanosine moiety in deoxyribonucleic acid. Biochem J. 1986;238:247–254. doi: 10.1042/bj2380247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dirksen ML, Blakely WF, Holwitt E, Dizdaroglu M. Effect of DNA conformation on the hydroxyl radical-induced formation of 8,5′-cyclopurine 2′-deoxyribonucleoside residues in DNA. Int J Radiat Biol. 1988;54:195–204. doi: 10.1080/09553008814551631. [DOI] [PubMed] [Google Scholar]
- 84.Guerrero CR, Wang J, Wang Y. Induction of 8,5′-cyclo-2′-deoxyadenosine and 8,5′-cyclo-2′-deoxyguanosine in isolated DNA by Fenton-type reagents. Chem Res Toxicol. 2013;26:1361–1366. doi: 10.1021/tx400221w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang J, Yuan B, Guerrero C, Bahde R, Gupta S, Wang Y. Quantification of oxidative DNA lesions in tissues of Long-Evans Cinnamon rats by capillary high-performance liquid chromatography–tandem mass spectrometry coupled with stable isotope-dilution method. Anal Chem. 2011;83:2201–2209. doi: 10.1021/ac103099s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang J, Clauson CL, Robbins PD, Niedernhofer LJ, Wang Y. The oxidative DNA lesions 8,5′-cyclopurines accumulate with aging in a tissue-specific manner. Aging Cell. 2012;11:714–716. doi: 10.1111/j.1474-9726.2012.00828.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kirkali G, de Souza-Pinto NC, Jaruga P, Bohr VA, Dizdaroglu M. Accumulation of (5′S)-8,5′-cyclo-2′-deoxyadenosine in organs of Cockayne syndrome complementation group B gene knockout mice. DNA Repair. 2009;8:274–278. doi: 10.1016/j.dnarep.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yu Y, Guerrero CR, Liu S, Amato NJ, Sharma Y, Gupta S, Wang Y. Comprehensive assessment of oxidatively induced modifications of DNA in a rat model of human Wilson's disease. Mol Cell Proteomics. 2016;15:810–817. doi: 10.1074/mcp.M115.052696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mitra D, Luo X, Morgan A, Wang J, Hoang MP, Lo J, Guerrero CR, Lennerz JK, Mihm MC, Wargo JA, Robinson KC, Devi SP, Vanover JC, D'Orazio JA, McMahon M, Bosenberg MW, Haigis KM, Haber DA, Wang Y, Fisher DE. An ultraviolet-radiation-independent pathway to melanoma carcinogenesis in the red hair/fair skin background. Nature. 2012;491:449–453. doi: 10.1038/nature11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shaked H, Hofseth LJ, Chumanevich A, Chumanevich AA, Wang J, Wang Y, Taniguchi K, Guma M, Shenouda S, Clevers H, Harris CC, Karin M. Chronic epithelial NF-κB activation accelerates APC loss and intestinal tumor initiation through iNOS up-regulation. Proc Natl Acad Sci U S A. 2012;109:14007–14012. doi: 10.1073/pnas.1211509109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tilstra JS, Robinson AR, Wang J, Gregg SQ, Clauson CL, Reay DP, Nasto LA, St Croix CM, Usas A, Vo N, Huard J, Clemens PR, Stolz DB, Guttridge DC, Watkins SC, Garinis GA, Wang Y, Niedernhofer LJ, Robbins PD. NF-κB inhibition delays DNA damage–induced senescence and aging in mice. J Clin Invest. 2012;122:2601–2612. doi: 10.1172/JCI45785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Box HC, Budzinski EE, Dawidzik JB, Gobey JS, Freund HG. Free radical-induced tandem base damage in DNA oligomers. Free Radical Biol Med. 1997;23:1021–1030. doi: 10.1016/s0891-5849(97)00166-4. [DOI] [PubMed] [Google Scholar]
- 93.Box HC, Budzinski EE, Dawidzik JB, Wallace JC, Iijima H. Tandem lesions and other products in X-irradiated DNA oligomers. Radiat Res. 1998;149:433–439. [PubMed] [Google Scholar]
- 94.Box HC, Budzinski EE, Dawidzik JD, Wallace JC, Evans MS, Gobey JS. Radiation-induced formation of a crosslink between base moieties of deoxyguanosine and thymidine in deoxygenated solutions of d(CpGpTpA) Radiat Res. 1996;145:641–643. [PubMed] [Google Scholar]
- 95.Budzinski EE, Dawidzik JB, Rajecki MJ, Wallace JC, Schroder EA, Box HC. Isolation and characterization of the products of anoxic irradiation of d(CpGpTpA) Int J Radiat Biol. 1997;71:327–336. doi: 10.1080/095530097144210. [DOI] [PubMed] [Google Scholar]
- 96.Bellon S, Ravanat JL, Gasparutto D, Cadet J. Cross-Linked thymine-purine base tandem lesions: Synthesis, characterization, and measurement in γ-Irradiated Isolated DNA. Chem Res Toxicol. 2002;15:598–606. doi: 10.1021/tx015594d. [DOI] [PubMed] [Google Scholar]
- 97.Romieu A, Bellon S, Gasparutto D, Cadet J. Synthesis and UV photolysis of oligodeoxynucleotides that contain 5-(phenylthiomethyl)-2′-deoxyuridine: a specific photolabile precursor of 5-(2′-deoxyuridilyl)methyl radical. Org Lett. 2000;2:1085–1088. doi: 10.1021/ol005643y. [DOI] [PubMed] [Google Scholar]
- 98.Zhang Q, Wang Y. Independent generation of 5-(2′-deoxycytidinyl)methyl radical and the formation of a novel cross-link lesion between 5-methylcytosine and guanine. J Am Chem Soc. 2003;125:12795–12802. doi: 10.1021/ja034866r. [DOI] [PubMed] [Google Scholar]
- 99.Zhang Q, Wang Y. Generation of 5-(2′-deoxycytidyl)methyl radical and the formation of intrastrand cross-link lesions in oligodeoxyribonucleotides. Nucleic Acids Res. 2005;33:1593–1603. doi: 10.1093/nar/gki301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang Q, Wang Y. Independent generation of the 5-hydroxy-5,6-dihydrothymidin-6-yl radical and its reactivity in dinucleoside monophosphates. J Am Chem Soc. 2004;126:13287–13297. doi: 10.1021/ja048492t. [DOI] [PubMed] [Google Scholar]
- 101.Zhang Q, Wang Y. The reactivity of the 5-hydroxy-5,6-dihydrothymidin-6-yl radical in oligodeoxyribonucleotides. Chem Res Toxicol. 2005;18:1897–1906. doi: 10.1021/tx050195u. [DOI] [PubMed] [Google Scholar]
- 102.Zeng Y, Wang Y. Facile formation of an intrastrand cross-link lesion between cytosine and guanine upon pyrex-filtered UV light irradiation of d(BrCG) and duplex DNA containing 5-bromocytosine. J Am Chem Soc. 2004;126:6552–6553. doi: 10.1021/ja049760q. [DOI] [PubMed] [Google Scholar]
- 103.Zeng Y, Wang Y. Sequence-dependent formation of intrastrand crosslink products from the UVB irradiation of duplex DNA containing a 5-bromo-2′-deoxyuridine or 5-bromo-2′-deoxycytidine. Nucleic Acids Res. 2006;34:6521–6529. doi: 10.1093/nar/gkl892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zeng Y, Wang Y. UVB-induced formation of intrastrand cross-link products of DNA in MCF-7 cells treated with 5-bromo-2′-deoxyuridine. Biochemistry. 2007;46:8189–8195. doi: 10.1021/bi700431q. [DOI] [PubMed] [Google Scholar]
- 105.Hong H, Wang Y. Formation of intrastrand cross-link products between cytosine and adenine from UV irradiation of d(BrCA) and duplex DNA containing a 5-bromocytosine. J Am Chem Soc. 2005;127:13969–13977. doi: 10.1021/ja0531677. [DOI] [PubMed] [Google Scholar]
- 106.Gu C, Wang Y. LC-MS/MS identification and yeast polymerase η bypass of a novel γ-irradiation-induced intrastrand cross-link lesion G[8–5]C. Biochemistry. 2004;43:6745–6750. doi: 10.1021/bi0497749. [DOI] [PubMed] [Google Scholar]
- 107.Jiang Y, Hong H, Cao H, Wang Y. In vivo formation and in vitro replication of a guanine-thymine intrastrand cross-link lesion. Biochemistry. 2007;46:12757–12763. doi: 10.1021/bi7012195. [DOI] [PubMed] [Google Scholar]
- 108.Hong H, Cao H, Wang Y. Formation and genotoxicity of a guanine–cytosine intrastrand cross-link lesion in vivo. Nucleic Acids Res. 2007;35:7118–7127. doi: 10.1093/nar/gkm851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Crean C, Uvaydov Y, Geacintov NE, Shafirovich V. Oxidation of single-stranded oligonucleotides by carbonate radical anions: generating intrastrand cross-links between guanine and thymine bases separated by cytosines. Nucleic Acids Res. 2008;36:742–755. doi: 10.1093/nar/gkm1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yun BH, Geacintov NE, Shafirovich V. Generation of Guanine–Thymidine Cross-Links in DNA by Peroxynitrite/Carbon Dioxide. Chem Res Toxicol. 2011;24:1144–1152. doi: 10.1021/tx200139c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Madugundu GS, Wagner JR, Cadet J, Kropachev K, Yun BH, Geacintov NE, Shafirovich V. Generation of Guanine–Thymine Cross-Links in Human Cells by One-Electron Oxidation Mechanisms. Chem Res Toxicol. 2013;26:1031–1033. doi: 10.1021/tx400158g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mangerich A, Dedon PC, Fox JG, Tannenbaum SR, Wogan GN. Chemistry meets biology in colitis-associated carcinogenesis. Free Radical Res. 2013;47:958–986. doi: 10.3109/10715762.2013.832239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Halliwell B. Phagocyte-derived reactive species: salvation or suicide? Trends Biochem Sci. 2006;31:509–515. doi: 10.1016/j.tibs.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 115.Henderson JP, Byun J, Heinecke JW. Molecular chlorine generated by the myeloperoxidase-hydrogen peroxide-chloride system of phagocytes produces 5-chlorocytosine in bacterial RNA. J Biol Chem. 1999;274:33440–33448. doi: 10.1074/jbc.274.47.33440. [DOI] [PubMed] [Google Scholar]
- 116.Klebanoff SJ. Myeloperoxidase: friend and foe. J Leukocyte Biol. 2005;77:598–625. doi: 10.1189/jlb.1204697. [DOI] [PubMed] [Google Scholar]
- 117.Henderson JP, Byun J, Mueller DM, Heinecke JW. The eosinophil peroxidase-hydrogen peroxide-bromide system of human eosinophils generates 5-bromouracil, a mutagenic thymine analogue. Biochemistry. 2001;40:2052–2059. doi: 10.1021/bi002015f. [DOI] [PubMed] [Google Scholar]
- 118.Henderson JP, Byun J, Takeshita J, Heinecke JW. Phagocytes produce 5-chlorouracil and 5-bromouracil, two mutagenic products of myeloperoxidase, in human inflammatory tissue. J Biol Chem. 2003;278:23522–23528. doi: 10.1074/jbc.M303928200. [DOI] [PubMed] [Google Scholar]
- 119.Gotto AM, Belkhode ML, Touster O. Stimulatory effects of inosine and deoxyinosine on the incorporation of uracil-2-14C, 5-fluorouracil-2-14C, and 5-bromouracil-2-14C into nucleic acids by Ehrlich ascites tumor cells in vitro. Cancer Res. 1969;29:807–811. [PubMed] [Google Scholar]
- 120.Pal BC, Cumming RB, Walton MF, Preston RJ. Environmental pollutant 5-chlorouracil is incorporated in mouse liver and testes DNA. Mutat Res Lett. 1981;91:395–401. doi: 10.1016/0165-7992(81)90021-x. [DOI] [PubMed] [Google Scholar]
- 121.Morris SM. The genetic toxicology of 5-bromodeoxyuridine in mammalian cells. Mutat Res Rev Genet Toxicol. 1991;258:161–188. doi: 10.1016/0165-1110(91)90007-i. [DOI] [PubMed] [Google Scholar]
- 122.Morris SM. The genetic toxicology of 5-fluoropyrimidines and 5-chlorouracil. Mutat Res Rev Genet Toxicol. 1993;297:39–51. doi: 10.1016/0165-1110(93)90006-9. [DOI] [PubMed] [Google Scholar]
- 123.Whiteman M, Jenner A, Halliwell B. Hypochlorous acid-induced base modifications in isolated calf thymus DNA. Chem Res Toxicol. 1997;10:1240–1246. doi: 10.1021/tx970086i. [DOI] [PubMed] [Google Scholar]
- 124.Winterbourn CC, Kettle AJ. Biomarkers of myeloperoxidase-derived hypochlorous acid. Free Radical Biol Med. 2000;29:403–409. doi: 10.1016/s0891-5849(00)00204-5. [DOI] [PubMed] [Google Scholar]
- 125.Kawai Y, Morinaga H, Kondo H, Miyoshi N, Nakamura Y, Uchida K, Osawa T. Endogenous formation of novel halogenated 2′-deoxycytidine - Hypohalous acid-mediated DNA modification at the site of inflammation. J Biol Chem. 2004;279:51241–51249. doi: 10.1074/jbc.M408210200. [DOI] [PubMed] [Google Scholar]
- 126.Kang JI, Sowers LC. Examination of hypochlorous acid-induced damage to cytosine residues in a CpG dinucleotide in DNA. Chem Res Toxicol. 2008;21:1211–1218. doi: 10.1021/tx800037h. [DOI] [PubMed] [Google Scholar]
- 127.Badouard C, Masuda M, Nishino H, Cadet J, Favier A, Ravanat JL. Detection of chlorinated DNA and RNA nucleosides by HPLC coupled to tandem mass spectrometry as potential biomarkers of inflammation. J Chromatogr B: Anal Technol Biomed Life Sci. 2005;827:26–31. doi: 10.1016/j.jchromb.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 128.Shen ZZ, Mitra SN, Wu WJ, Chen YH, Yang YW, Qin J, Hazen SL. Eosinophil peroxidase catalyzes bromination of free nucleosides and double-stranded DNA. Biochemistry. 2001;40:2041–2051. doi: 10.1021/bi001961t. [DOI] [PubMed] [Google Scholar]
- 129.Asahi T, Kondo H, Masuda M, Nishino H, Aratani Y, Naito Y, Yoshikawa T, Hisaka S, Kato Y, Osawa T. Chemical and immunochemical detection of 8-halogenated deoxyguanosines at early stage inflammation. J Biol Chem. 2010;285:9282–9291. doi: 10.1074/jbc.M109.054213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sassa A, Kamoshita N, Matsuda T, Ishii Y, Kuraoka I, Nohmi T, Ohta T, Honma M, Yasui M. Miscoding properties of 8-chloro-2′-deoxyguanosine, a hypochlorous acid-induced DNA adduct, catalysed by human DNA polymerases. Mutagenesis. 2013;28:81–88. doi: 10.1093/mutage/ges056. [DOI] [PubMed] [Google Scholar]
- 131.Mangerich A, Knutson CG, Parry NM, Muthupalani S, Ye WJ, Prestwich E, Cui L, McFaline JL, Mobley M, Ge ZM, Taghizadeh K, Wishnok JS, Wogan GN, Fox JG, Tannenbaum SR, Dedon PC. Infection-induced colitis in mice causes dynamic and tissue-specific changes in stress response and DNA damage leading to colon cancer. Proc Natl Acad Sci U S A. 2012;109:E1820–E1829. doi: 10.1073/pnas.1207829109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Knutson CG, Mangerich A, Zeng Y, Raczynski AR, Liberman RG, Kang P, Ye WJ, Prestwich EG, Lu K, Wishnok JS, Korzenik JR, Wogan GN, Fox JG, Dedon PC, Tannenbaum SR. Chemical and cytokine features of innate immunity characterize serum and tissue profiles in inflammatory bowel disease. Proc Natl Acad Sci U S A. 2013;110:E2332–E2341. doi: 10.1073/pnas.1222669110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dedon PC, Tannenbaum SR. Reactive nitrogen species in the chemical biology of inflammation. Arch Biochem Biophys. 2004;423:12–22. doi: 10.1016/j.abb.2003.12.017. [DOI] [PubMed] [Google Scholar]
- 134.Xia Y, Zweier JL. Superoxide and peroxynitrite generation from inducible nitric oxide synthase in macrophages. Proc Natl Acad Sci U S A. 1997;94:6954–6958. doi: 10.1073/pnas.94.13.6954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Carreras MC, Pargament GA, Catz SD, Poderoso JJ, Boveris A. Kinetics of nitric oxide and hydrogen peroxide production and formation of peroxynitrite during the respiratory burst of human neutrophils. FEBS Lett. 1994;341:65–68. doi: 10.1016/0014-5793(94)80241-6. [DOI] [PubMed] [Google Scholar]
- 136.Huie RE, Padmaja S. The Reaction of no With Superoxide. Free Radical Res Commun. 1993;18:195–199. doi: 10.3109/10715769309145868. [DOI] [PubMed] [Google Scholar]
- 137.Juedes MJ, Wogan GN. Peroxynitrite-induced mutation spectra of pSP189 following replication in bacteria and in human cells. Mutat Res Fundam Mol Mech Mutagen. 1996;349:51–61. doi: 10.1016/0027-5107(95)00152-2. [DOI] [PubMed] [Google Scholar]
- 138.Burney S, Niles JC, Dedon PC, Tannenbaum SR. DNA Damage in Deoxynucleosides and Oligonucleotides Treated with Peroxynitrite. Chem Res Toxicol. 1999;12:513–520. doi: 10.1021/tx980254m. [DOI] [PubMed] [Google Scholar]
- 139.Gu F, Stillwell WG, Wishnok JS, Shallop AJ, Jones RA, Tannenbaum SR. Peroxynitrite-Induced Reactions of Synthetic Oligo 2′-Deoxynucleotides and DNA Containing Guanine: Formation and Stability of a 5-Guanidino-4-nitroimidazole Lesion. Biochemistry. 2002;41:7508–7518. doi: 10.1021/bi020148q. [DOI] [PubMed] [Google Scholar]
- 140.Misiaszek R, Crean C, Geacintov NE, Shafirovich V. Combination of Nitrogen Dioxide Radicals with 8-Oxo-7,8-dihydroguanine and Guanine Radicals in DNA: Oxidation and Nitration End-Products. J Am Chem Soc. 2005;127:2191–2200. doi: 10.1021/ja044390r. [DOI] [PubMed] [Google Scholar]
- 141.Girotti AW. Mechanisms of lipid peroxidation. J Free Radicals Biol Med. 1985;1:87–95. doi: 10.1016/0748-5514(85)90011-x. [DOI] [PubMed] [Google Scholar]
- 142.Halliwell B, Chirico S. Lipid peroxidation: its mechanism, measurement, and significance. Am J Clin Nutr. 1993;57:715S–724S. doi: 10.1093/ajcn/57.5.715S. [DOI] [PubMed] [Google Scholar]
- 143.Burcham PC. Genotoxic lipid peroxidation products: their DNA damaging properties and role in formation of endogenous DNA adducts. Mutagenesis. 1998;13:287–305. doi: 10.1093/mutage/13.3.287. [DOI] [PubMed] [Google Scholar]
- 144.Chung FL, Chen HJC, Nath RG. Lipid peroxidation as a potential endogenous source for the formation of exocyclic DNA adducts. Carcinogenesis. 1996;17:2105–2111. doi: 10.1093/carcin/17.10.2105. [DOI] [PubMed] [Google Scholar]
- 145.Nair J, Barbin A, Velic I, Bartsch H. Etheno DNA-base adducts from endogenous reactive species. Mutat Res Fundam Mol Mech Mutagen. 1999;424:59–69. doi: 10.1016/s0027-5107(99)00008-1. [DOI] [PubMed] [Google Scholar]
- 146.Winczura A, Zdżalik D, Tudek B. Damage of DNA and proteins by major lipid peroxidation products in genome stability. Free Radical Res. 2012;46:442–459. doi: 10.3109/10715762.2012.658516. [DOI] [PubMed] [Google Scholar]
- 147.Prado FM, Oliveira MCB, Miyamoto S, Martinez GR, Medeiros MHG, Ronsein GE, Di Mascio P. Thymine hydroperoxide as a potential source of singlet molecular oxygen in DNA. Free Radical Biol Med. 2009;47:401–409. doi: 10.1016/j.freeradbiomed.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 148.Blair IA. DNA adducts with lipid peroxidation products. J Biol Chem. 2008;283:15545–15549. doi: 10.1074/jbc.R700051200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Minko IG, Kozekov ID, Kozekova A, Harris TM, Rizzo CJ, Lloyd RS. Mutagenic potential of DNA–peptide crosslinks mediated by acrolein-derived DNA adducts. Mutat Res Fundam Mol Mech Mutagen. 2008;637:161–172. doi: 10.1016/j.mrfmmm.2007.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Stone MP, Cho YJ, Huang H, Kim HY, Kozekov ID, Kozekova A, Wang H, Minko IG, Lloyd RS, Harris TM, Rizzo CJ. Interstrand DNA cross-links induced by α,β-unsaturated aldehydes derived from lipid peroxidation and environmental sources. Acc Chem Res. 2008;41:793–804. doi: 10.1021/ar700246x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kozekov ID, Nechev LV, Moseley MS, Harris CM, Rizzo CJ, Stone MP, Harris TM. DNA interchain cross-links formed by acrolein and crotonaldehyde. J Am Chem Soc. 2003;125:50–61. doi: 10.1021/ja020778f. [DOI] [PubMed] [Google Scholar]
- 152.Garcia CCM, Angeli JPF, Freitas FP, Gomes OF, de Oliveira TF, Loureiro APM, Di Mascio P, Medeiros MHG. [13C2]-acetaldehyde promotes unequivocal formation of 1,N2-Propano-2′-deoxyguanosine in human cells. J Am Chem Soc. 2011;133:9140–9143. doi: 10.1021/ja2004686. [DOI] [PubMed] [Google Scholar]
- 153.el Ghissassi F, Barbin A, Nair J, Bartsch H. Formation of 1,N6-ethenoadenine and 3,N4-ethenocytosine by lipid peroxidation products and nucleic acid bases. Chem Res Toxicol. 1995;8:278–283. doi: 10.1021/tx00044a013. [DOI] [PubMed] [Google Scholar]
- 154.Bartsch H, Nair J, Owen RW. Exocyclic DNA adducts as oxidative stress markers in colon carcinogenesis: potential role of lipid peroxidation, dietary fat and antioxidants. Biol Chem. 2002;383:915–921. doi: 10.1515/BC.2002.098. [DOI] [PubMed] [Google Scholar]
- 155.Swenberg JA, Fedtke N, Ciroussel F, Barbin A, Bartsch H. Etheno adducts formed in DNA of vinyl chloride-exposed rats are highly persistent in liver. Carcinogenesis. 1992;13:727–729. doi: 10.1093/carcin/13.4.727. [DOI] [PubMed] [Google Scholar]
- 156.Lee SH, Arora JA, Oe T, Blair IA. 4-Hydroperoxy-2-nonenal-Induced Formation of 1,N2-Etheno-2′-deoxyguanosine Adducts. Chem Res Toxicol. 2005;18:780–786. doi: 10.1021/tx0497088. [DOI] [PubMed] [Google Scholar]
- 157.Lee SH, Oe T, Blair IA. 4,5-Epoxy-2(E)-decenal-Induced Formation of 1,N6-Etheno-2′-deoxyadenosine and 1,N2-Etheno-2′-deoxyguanosine Adducts. Chem Res Toxicol. 2002;15:300–304. doi: 10.1021/tx010147j. [DOI] [PubMed] [Google Scholar]
- 158.Nair U, Bartsch H, Nair J. Lipid peroxidation-induced DNA damage in cancer-prone inflammatory diseases: A review of published adduct types and levels in humans. Free Radical Biol Med. 2007;43:1109–1120. doi: 10.1016/j.freeradbiomed.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 159.Chen HJ, Lin WP. Simultaneous quantification of 1,N2-propano-2′-deoxyguanosine adducts derived from acrolein and crotonaldehyde in human placenta and leukocytes by isotope dilution nanoflow LC nanospray ionization tandem mass spectrometry. Anal Chem. 2009;81:9812–9818. doi: 10.1021/ac9019472. [DOI] [PubMed] [Google Scholar]
- 160.Chen HJC. Analysis of DNA adducts in human samples: Acrolein-derived exocyclic DNA adducts as an example. Mol Nutr Food Res. 2011;55:1391–1400. doi: 10.1002/mnfr.201100185. [DOI] [PubMed] [Google Scholar]
- 161.Chen HJC, Chiang LC, Tseng MC, Zhang LL, Ni J, Chung FL. Detection and quantification of 1,N6-ethenoadenine in human placental DNA by mass spectrometry. Chem Res Toxicol. 1999;12:1119–1126. doi: 10.1021/tx990074s. [DOI] [PubMed] [Google Scholar]
- 162.Chen HJC, Chiu WL. Association between cigarette smoking and urinary excretion of 1,N2-ethenoguanine measured by isotope dilution liquid chromatography-electrospray ionization/tandem mass spectrometry. Chem Res Toxicol. 2005;18:1593–1599. doi: 10.1021/tx050145p. [DOI] [PubMed] [Google Scholar]
- 163.Chen HJC, Lin GJ, Lin WP. Simultaneous quantification of three lipid peroxidation-derived etheno adducts in human DNA by stable isotope dilution nanoflow liquid chromatography nanospray ionization tandem mass spectrometry. Anal Chem. 2010;82:4486–4493. doi: 10.1021/ac100391f. [DOI] [PubMed] [Google Scholar]
- 164.Chen HJC, Lin TC, Hong CL, Chiang LC. Analysis of 3,N4-ethenocytosine in DNA and in human urine by isotope dilution gas chromatography/negative ion chemical ionization/mass spectrometry. Chem Res Toxicol. 2001;14:1612–1619. doi: 10.1021/tx015551x. [DOI] [PubMed] [Google Scholar]
- 165.Chen HJC, Lin WP. Quantitative analysis of multiple exocyclic DNA adducts in human salivary DNA by stable isotope dilution nanoflow liquid chromatography-nanospray ionization tandem mass spectrometry. Anal Chem. 2011;83:8543–8551. doi: 10.1021/ac201874d. [DOI] [PubMed] [Google Scholar]
- 166.Chen HJC, Wu CF, Hong CL, Chang CM. Urinary excretion of 3,N4-etheno-2′-deoxycytidine in humans as a biomarker of oxidative stress: association with cigarette smoking. Chem Res Toxicol. 2004;17:896–903. doi: 10.1021/tx0342013. [DOI] [PubMed] [Google Scholar]
- 167.Zhang S, Villalta PW, Wang M, Hecht SS. Analysis of crotonaldehyde- and acetaldehyde-derived 1,N2-propanodeoxyguanosine adducts in DNA from human tissues using liquid chromatography electrospray ionization tandem mass spectrometry. Chem Res Toxicol. 2006;19:1386–1392. doi: 10.1021/tx060154d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Garcia CCM, Freitas FP, Di Mascio P, Medeiros MHG. Ultrasensitive simultaneous quantification of 1,N2-etheno-2′-deoxyguanosine and 1,N2-propano-2′-deoxyguanosine in DNA by an online liquid chromatography–electrospray tandem mass spectrometry assay. Chem Res Toxicol. 2010;23:1245–1255. doi: 10.1021/tx1001018. [DOI] [PubMed] [Google Scholar]
- 169.Loureiro APM, Marques SA, Garcia CCM, Di Mascio P, Medeiros MHG. Development of an on-line liquid chromatography-electrospray tandem mass spectrometry assay to quantitatively determine 1,N2-Etheno-2′-deoxyguanosine in DNA. Chem Res Toxicol. 2002;15:1302–1308. doi: 10.1021/tx025554p. [DOI] [PubMed] [Google Scholar]
- 170.Nair J, Strand S, Frank N, Knauft J, Wesch H, Galle PR, Bartsch H. Apoptosis and age-dependant induction of nuclear and mitochondrial etheno-DNA adducts in Long-Evans Cinnamon (LEC) rats: enhanced DNA damage by dietary curcumin upon copper accumulation. Carcinogenesis. 2005;26:1307–1315. doi: 10.1093/carcin/bgi073. [DOI] [PubMed] [Google Scholar]
- 171.Ma B, Villalta PW, Balbo S, Stepanov I. Analysis of a malondialdehyde–deoxyguanosine adduct in human leukocyte DNA by liquid chromatography nanoelectrospray–high-resolution tandem mass spectrometry. Chem Res Toxicol. 2014;27:1829–1836. doi: 10.1021/tx5002699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Chaudhary AK, Nokubo M, Marnett LJ, Blair IA. Analysis of the malondialdehyde-2′-deoxyguanosine adduct in rat liver DNA by gas chromatography/electron capture negative chemical ionization mass spectrometry. Biol Mass Spectrom. 1994;23:457–464. doi: 10.1002/bms.1200230802. [DOI] [PubMed] [Google Scholar]
- 173.Zhang Y, Chen SY, Hsu T, Santella RM. Immunohistochemical detection of malondialdehyde–DNA adducts in human oral mucosa cells. Carcinogenesis. 2002;23:207–211. doi: 10.1093/carcin/23.1.207. [DOI] [PubMed] [Google Scholar]
- 174.Leuratti C, Watson MA, Deag EJ, Welch A, Singh R, Gottschalg E, Marnett LJ, Atkin W, Day NE, Shuker DEG, Bingham SA. Detection of malondialdehyde DNA adducts in human colorectal mucosa: relationship with diet and the presence of adenomas. Cancer Epidemiol Biomarkers Prev. 2002;11:267–273. [PubMed] [Google Scholar]
- 175.Delaney JC, Essigmann JM. Biological properties of single chemical-DNA adducts: a twenty year perspective. Chem Res Toxicol. 2008;21:232–252. doi: 10.1021/tx700292a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.You C, Wang Y. Mass spectrometry-based quantitative strategies for assessing the biological consequences and repair of DNA adducts. Acc Chem Res. 2016;49:205–213. doi: 10.1021/acs.accounts.5b00437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991;349:431–434. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- 178.Cheng KC, Cahill DS, Kasai H, Nishimura S, Loeb LA. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G——T and A——C substitutions. J Biol Chem. 1992;267:166–172. [PubMed] [Google Scholar]
- 179.Wood ML, Dizdaroglu M, Gajewski E, Essigmann JM. Mechanistic studies of ionizing radiation and oxidative mutagenesis: genetic effects of a single 8-hydroxyguanine (7-hydro-8-oxoguanine) residue inserted at a unique site in a viral genome. Biochemistry. 1990;29:7024–7032. doi: 10.1021/bi00482a011. [DOI] [PubMed] [Google Scholar]
- 180.Kamiya H, Miura H, Murata-Kamiya N, Ishikawa H, Sakaguchi T, Inoue H, Sasaki T, Masutanl C, Hanaoka F, Nishimura S, Ohtsuka E. 8-Hydroxyadenine (7, 8-dihydro-8-oxoadenine) induces misincorporation in in vitro DNA synthesis and mutations in NIH 3T3 cells. Nucleic Acids Res. 1995;23:2893–2899. doi: 10.1093/nar/23.15.2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kalam MA, Haraguchi K, Chandani S, Loechler EL, Moriya M, Greenberg MM, Basu AK. Genetic effects of oxidative DNA damages: comparative mutagenesis of the imidazole ring-opened formamidopyrimidines (Fapy lesions) and 8-oxo-purines in simian kidney cells. Nucleic Acids Res. 2006;34:2845. doi: 10.1093/nar/gkl099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wiederholt CJ, Greenberg MM. Fapy·dG instructs Klenow exo− to misincorporate deoxyadenosine. J Am Chem Soc. 2002;124:7278–7279. doi: 10.1021/ja026522r. [DOI] [PubMed] [Google Scholar]
- 183.Tornaletti S, Maeda LS, Kolodner RD, Hanawalt PC. Effect of 8-oxoguanine on transcription elongation by T7 RNA polymerase and mammalian RNA polymerase II. DNA Repair. 2004;3:483–494. doi: 10.1016/j.dnarep.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 184.Viswanathan A, Doetsch PW. Effects of nonbulky DNA base damages on Escherichia coli RNA polymerase-mediated elongation and promoter clearance. J Biol Chem. 1998;273:21276–21281. doi: 10.1074/jbc.273.33.21276. [DOI] [PubMed] [Google Scholar]
- 185.Brégeon D, Doddridge ZA, You HJ, Weiss B, Doetsch PW. Transcriptional mutagenesis induced by uracil and 8-oxoguanine in Escherichia coli. Mol Cell. 2003;12:959–970. doi: 10.1016/s1097-2765(03)00360-5. [DOI] [PubMed] [Google Scholar]
- 186.Kuraoka I, Endou M, Yamaguchi Y, Wada T, Handa H, Tanaka K. Effects of endogenous DNA base lesions on transcription elongation by mammalian RNA polymerase II: Implications for transcription-coupled DNA repair and transcriptional mutagenesis. J Biol Chem. 2003;278:7294–7299. doi: 10.1074/jbc.M208102200. [DOI] [PubMed] [Google Scholar]
- 187.Saxowsky TT, Meadows KL, Klungland A, Doetsch PW. 8-Oxoguanine-mediated transcriptional mutagenesis causes Ras activation in mammalian cells. Proc Natl Acad Sci U S A. 2008;105:18877–18882. doi: 10.1073/pnas.0806464105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 2003;17:1195–1214. doi: 10.1096/fj.02-0752rev. [DOI] [PubMed] [Google Scholar]
- 189.Bjelland S, Seeberg E. Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat Res Fundam Mol Mech Mutagen. 2003;531:37–80. doi: 10.1016/j.mrfmmm.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 190.Wallace SS. Biological consequences of free radical-damaged DNA bases. Free Radical Biol Med. 2002;33:1–14. doi: 10.1016/s0891-5849(02)00827-4. [DOI] [PubMed] [Google Scholar]
- 191.Henderson PT, Delaney JC, Muller JG, Neeley WL, Tannenbaum SR, Burrows CJ, Essigmann JM. The hydantoin lesions formed from oxidation of 7,8-dihydro-8-oxoguanine are potent sources of replication errors in vivo. Biochemistry. 2003;42:9257–9262. doi: 10.1021/bi0347252. [DOI] [PubMed] [Google Scholar]
- 192.Delaney S, Neeley WL, Delaney JC, Essigmann JM. The substrate specificity of MutY for hyperoxidized guanine lesions in vivo. Biochemistry. 2007;46:1448–1455. doi: 10.1021/bi061174h. [DOI] [PubMed] [Google Scholar]
- 193.Leipold MD, Workman H, Muller JG, Burrows CJ, David SS. Recognition and removal of oxidized guanines in duplex DNA by the base excision repair enzymes hOGG1, yOGG1, and yOGG2. Biochemistry. 2003;42:11373–11381. doi: 10.1021/bi034951b. [DOI] [PubMed] [Google Scholar]
- 194.McKibbin PL, Fleming AM, Towheed MA, Van Houten B, Burrows CJ, David SS. Repair of hydantoin lesions and their amine adducts in DNA by base and nucleotide excision repair. J Am Chem Soc. 2013;135:13851–13861. doi: 10.1021/ja4059469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Hazra TK, Muller JG, Manuel RC, Burrows CJ, Lloyd RS, Mitra S. Repair of hydantoins, one electron oxidation product of 8-oxoguanine, by DNA glycosylases of Escherichia coli. Nucleic Acids Res. 2001;29:1967–1974. doi: 10.1093/nar/29.9.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Leipold MD, Muller JG, Burrows CJ, David SS. Removal of hydantoin products of 8-oxoguanine oxidation by the Escherichia coli DNA repair enzyme, FPG. Biochemistry. 2000;39:14984–14992. doi: 10.1021/bi0017982. [DOI] [PubMed] [Google Scholar]
- 197.Hailer MK, Slade PG, Martin BD, Sugden KD. Nei deficient Escherichia coli are sensitive to chromate and accumulate the oxidized guanine lesion spiroiminodihydantoin. Chem Res Toxicol. 2005;18:1378–1383. doi: 10.1021/tx0501379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Krishnamurthy N, Zhao XB, Burrows CJ, David SS. Superior removal of hydantoin lesions relative to other oxidized bases by the human DNA glycosylase hNEIL1. Biochemistry. 2008;47:7137–7146. doi: 10.1021/bi800160s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Shafirovich V, Kropachev K, Anderson T, Liu Z, Kolbanovskiy M, Martin BD, Sugden K, Shim Y, Chen XJ, Min JH, Geacintov NE. Base and nucleotide excision repair of oxidatively generated guanine lesions in DNA. J Biol Chem. 2016;291:5309–5319. doi: 10.1074/jbc.M115.693218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Aller P, Rould MA, Hogg M, Wallace SS, Doublie S. A structural rationale for stalling of a replicative DNA polymerase at the most common oxidative thymine lesion, thymine glycol. Proc Natl Acad Sci U S A. 2007;104:814–818. doi: 10.1073/pnas.0606648104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Dolinnaya NG, Kubareva EA, Romanova EA, Trikin RM, Oretskaya TS. Thymidine glycol: the effect on DNA molecular structure and enzymatic processing. Biochimie. 2013;95:134–147. doi: 10.1016/j.biochi.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 202.Hayes RC, Petrullo LA, Huang H, Wallace SS, Leclerc JE. Oxidative damage in DNA:lack of mutagenicity by thymine glycol lesions. J Mol Biol. 1988;201:239–246. doi: 10.1016/0022-2836(88)90135-0. [DOI] [PubMed] [Google Scholar]
- 203.Yuan BF, Jiang Y, Wang YS, Wang YS. Efficient formation of the tandem thymine glycol/8-oxo-7,8-dihydroguanine lesion in isolated DNA and the mutagenic and cytotoxic properties of the tandem lesions in Escherichia coli cells. Chem Res Toxicol. 2010;23:11–19. doi: 10.1021/tx9004264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Bellon S, Shikazono N, Cunniffe S, Lomax M, O'Neill P. Processing of thymine glycol in a clustered DNA damage site: mutagenic or cytotoxic. Nucleic Acids Res. 2009;37:4430–4440. doi: 10.1093/nar/gkp422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Almohaini M, Chalasani L, Bafail D, Akopiants K, Zhou T, Yannone SM, Ramsden DA, Hartman MCT, Povirk LF. Nonhomologous end joining of complex DNA double-strand breaks with proximal thymine glycol and interplay with base excision repair. DNA Repair. 2016;41:16–26. doi: 10.1016/j.dnarep.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Miller H, Fernandes AS, Zaika E, McTigue MM, Torres MC, Wente M, Iden CR, Grollman AP. Stereoselective excision of thymine glycol from oxidatively damaged DNA. Nucleic Acids Res. 2004;32:338–345. doi: 10.1093/nar/gkh190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Yeo JC, Goodman RA, Schirle NT, David SS, Beal PA. RNA editing changes the lesion specificity for the DNA repair enzyme NEIL1. Proc Natl Acad Sci U S A. 2010;107:20715–20719. doi: 10.1073/pnas.1009231107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Zhu CX, Lu LM, Zhang J, Yue ZW, Song JH, Zong S, Liu MH, Stovicek O, Gao YQ, Yi CQ. Tautomerization-dependent recognition and excision of oxidation damage in base-excision DNA repair. Proc Natl Acad Sci U S A. 2016;113:7792–7797. doi: 10.1073/pnas.1604591113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Lin JJ, Sancar A. A new mechanism for repairing oxidative damage to DNA: (A)BC excinuclease removes AP sites and thymine glycols from DNA. Biochemistry. 1989;28:7979–7984. doi: 10.1021/bi00446a002. [DOI] [PubMed] [Google Scholar]
- 210.Kow YW, Wallace SS, Vanhouten B. UvrABC nuclease complex repairs thymine glycol, an oxidative DNA base damage. Mutat Res DNA Repair. 1990;235:147–156. doi: 10.1016/0921-8777(90)90068-g. [DOI] [PubMed] [Google Scholar]
- 211.Reardon JT, Bessho T, Kung HC, Bolton PH, Sancar A. In vitro repair of oxidative DNA damage by human nucleotide excision repair system: Possible explanation for neuro-degeneration in Xeroderma pigmentosum patients. Proc Natl Acad Sci U S A. 1997;94:9463–9468. doi: 10.1073/pnas.94.17.9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Brown KL, Roginskaya M, Zou Y, Altamirano A, Basu AK, Stone MP. Binding of the human nucleotide excision repair proteins XPA and XPC/HR23B to the 5R-thymine glycol lesion and structure of the cis-(5R,6S) thymine glycol epimer in the 5′-GTgG-3′ sequence: destabilization of two base pairs at the lesion site. Nucleic Acids Res. 2010;38:428–440. doi: 10.1093/nar/gkp844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Perevozchikova SA, Trikin RM, Heinze RJ, Romanova EA, Oretskaya TS, Friedhoff P, Kubareva EA. Is thymidine glycol containing DNA a substrate of E. coli DNA mismatch repair system? PLoS One. 2014;9:e104963. doi: 10.1371/journal.pone.0104963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Levy DD, Teebor GW. Site directed substitution of 5-hydroxymethyluracil for thymine in replicating phi-X-174am3 DNA via synthesis of 5-hydroxymethyl-2′-deoxyuridine-5′-triphosphate. Nucleic Acids Res. 1991;19:3337–3343. doi: 10.1093/nar/19.12.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Herrala AM, Vilpo JA. Template-primer activity of 5-(hydroxymethyl)uracil-containing DNA for prokaryotic and eukaryotic DNA and RNA polymerases. Biochemistry. 1989;28:8274–8277. doi: 10.1021/bi00447a003. [DOI] [PubMed] [Google Scholar]
- 216.Mellac S, Fazakerley GV, Sowers LC. Structures of base pairs with 5-(hydroxymethyl)-2′-deoxyuridine in DNA determined by NMR spectroscopy. Biochemistry. 1993;32:7779–7786. doi: 10.1021/bi00081a025. [DOI] [PubMed] [Google Scholar]
- 217.Kallen RG, Marmur J, Simon M. Occurrence of a new pyrimidine base replacing thymine in a bacteriophage DNA: 5-hydroxymethyl uracil. J Mol Biol. 1962;5:248–250. doi: 10.1016/s0022-2836(62)80087-4. [DOI] [PubMed] [Google Scholar]
- 218.Rusmintratip V, Sowers LC. An unexpectedly high excision capacity for mispaired 5-hydroxymethyluracil in human cell extracts. Proc Natl Acad Sci U S A. 2000;97:14183–14187. doi: 10.1073/pnas.97.26.14183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Wibley JEA, Waters TR, Haushalter K, Verdine GL, Pearl LH. Structure and specificity of the vertebrate anti-mutator uracil-DNA glycosylase SMUG1. Mol Cell. 2003;11:1647–1659. doi: 10.1016/s1097-2765(03)00235-1. [DOI] [PubMed] [Google Scholar]
- 220.Cortellino S, Xu JF, Sannai M, Moore R, Caretti E, Cigliano A, Le Coz M, Devarajan K, Wessels A, Soprano D, Abramowitz LK, Bartolomei MS, Rambow F, Bassi MR, Bruno T, Fanciulli M, Renner C, Klein-Szanto AJ, Matsumoto Y, Kobi D, Davidson I, Alberti C, Larue L, Bellacosa A. Thymine DNA glycosylase Is essential for active DNA demethylation by linked deamination-base excision repair. Cell. 2011;146:67–79. doi: 10.1016/j.cell.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Morera S, Grin I, Vigouroux A, Couve S, Henriot V, Saparbaev M, Ishchenko AA. Biochemical and structural characterization of the glycosylase domain of MBD4 bound to thymine and 5-hydroxymethyuracil-containing DNA. Nucleic Acids Res. 2012;40:9917–9926. doi: 10.1093/nar/gks714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Hashimoto H, Hong S, Bhagwat AS, Zhang X, Cheng XD. Excision of 5-hydroxymethyluracil and 5-carboxylcytosine by the thymine DNA glycosylase domain: its structural basis and implications for active DNA demethylation. Nucleic Acids Res. 2012;40:10203–10214. doi: 10.1093/nar/gks845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Zarakowska E, Gackowski D, Foksinski M, Olinski R. Are 8-oxoguanine (8-oxoGua) and 5-hydroxymethyluracil (5-hmUra) oxidatively damaged DNA bases or transcription (epigenetic) marks? Mutat Res Genet Toxicol Environ Mutagen. 2014:764–765. 58–63. doi: 10.1016/j.mrgentox.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 224.Olinski R, Starczak M, Gackowski D. Enigmatic 5-hydroxymethyluracil: Oxidatively modified base, epigenetic mark or both? Mutat Res Rev Mutat Res. 2016;767:59–66. doi: 10.1016/j.mrrev.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 225.Klungland A, Paulsen R, Rolseth V, Yamada Y, Ueno Y, Wiik P, Matsuda A, Seeberg E, Bjelland S. 5-Formyluracil and its nucleoside derivatives confer toxicity and mutagenicity to mammalian cells by interfering with normal RNA and DNA metabolism. Toxicol Lett. 2001;119:71–78. doi: 10.1016/s0378-4274(00)00308-8. [DOI] [PubMed] [Google Scholar]
- 226.Kamiya H, Murata-Kamiya N, Karino N, Ueno Y, Matsuda A, Kasai H. Induction of TàG and TaÀ transversions by 5-formyluracil in mammalian cells. Mutat Res Genet Toxicol Environ Mutagen. 2002;513:213–222. doi: 10.1016/s1383-5718(01)00312-6. [DOI] [PubMed] [Google Scholar]
- 227.Bjelland S, Birkeland NK, Benneche T, Volden G, Seeberg E. DNA glycosylase activities for thymine residues oxidized in the methyl-troup are functions of the AlkA enzyme in Escherichia coli. J Biol Chem. 1994;269:30489–30495. [PubMed] [Google Scholar]
- 228.Masaoka A, Terato H, Kobayashi M, Honsho A, Ohyama Y, Ide H. Enzymatic repair of 5-formyluracil I. Excision of 5-formyluracil site-specifically incorporated into oligonucleotide substrates by AlkA protein (Escherichia coli 3-methyladenine DNA glycosylase II) J Biol Chem. 1999;274:25136–25143. doi: 10.1074/jbc.274.35.25136. [DOI] [PubMed] [Google Scholar]
- 229.Terato H, Masaoka A, Kobayashi M, Fukushima S, Ohyama Y, Yoshida M, Ide H. Enzymatic repair of 5-formyluracil II. Mismatch formation between 5-formyluracil and guanine during DNA replication and its recognition by two proteins involved in base excision repair (AlkA) and mismatch repair (MutS) J Biol Chem. 1999;274:25144–25150. doi: 10.1074/jbc.274.35.25144. [DOI] [PubMed] [Google Scholar]
- 230.Matsubara M, Masaoka A, Tanaka T, Miyano T, Kato N, Terato H, Ohyama Y, Iwai S, Ide H. Mammalian 5-formyluracil-DNA glycosylase. 1. Identification and characterization of a novel activity that releases 5-formyluracil from DNA. Biochemistry. 2003;42:4993–5002. doi: 10.1021/bi027322v. [DOI] [PubMed] [Google Scholar]
- 231.Masaoka A, Matsubara M, Hasegawa R, Tanaka T, Kurisu S, Terato H, Ohyama Y, Karino N, Matsuda A, Ide H. Mammalian 5-formyluracil-DNA glycosylase. 2. role of SMUG1 uracilDNA glycosylase in repair of 5-formyluracil and other oxidized and deaminated base lesions. Biochemistry. 2003;42:5003–5012. doi: 10.1021/bi0273213. [DOI] [PubMed] [Google Scholar]
- 232.Knaevelsrud I, Slupphaug G, Leiros I, Matsuda A, Ruoff P, Bjelland S. Opposite-base dependent excision of 5-formyluracil from DNA by hSMUG1. Int J Radiat Biol. 2009;85:413–420. doi: 10.1080/09553000902818915. [DOI] [PubMed] [Google Scholar]
- 233.Liu PF, Burdzy A, Sowers LC. Repair of the mutagenic DNA oxidation product, 5-formyluracil. DNA Repair. 2003;2:199–210. doi: 10.1016/s1568-7864(02)00198-2. [DOI] [PubMed] [Google Scholar]
- 234.Zhang QM, Miyabe I, Matsumoto Y, Kino K, Sugiyama H, Yonei S. Identification of repair enzymes for 5-formyluracil in DNA - Nth, Nei, and MutM proteins of Escherichia coli. J Biol Chem. 2000;275:35471–35477. doi: 10.1074/jbc.M006125200. [DOI] [PubMed] [Google Scholar]
- 235.Miyabe I, Zhang QM, Kino K, Sugiyama H, Takao M, Yasui A, Yonei S. Identification of 5-formyluracil DNA glycosylase activity of human hNTH1 protein. Nucleic Acids Res. 2002;30:3443–3448. doi: 10.1093/nar/gkf460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Yonekura S, Nakamura N, Doi T, Sugiyama H, Yamamoto K, Yonei S, Zhang Q. Recombinant Schizosaccharomyces pombe Nth1 protein exhibits DNA glycosylase activities for 8-oxo-7,8-dihydroguanine and thymine residues oxidized in the methyl group. J Radiat Res. 2007;48:417–424. doi: 10.1269/jrr.07042. [DOI] [PubMed] [Google Scholar]
- 237.Zhang-Akiyama QM, Morinaga H, Kikuchi M, Yonekura SI, Sugiyama H, Yamamoto K, Yonei S. KsgA, a 16S rRNA adenine methyltransferase, has a novel DNA glycosylase/AP lyase activity to prevent mutations in Escherichia coli. Nucleic Acids Res. 2009;37:2116–2125. doi: 10.1093/nar/gkp057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Belousova EA, Vasil'eva IA, Moor NA, Zatsepin TS, Oretskaya TS, Lavrik OI. Clustered DNA lesions containing 5-formyluracil and AP Site: repair via the BER system. PLoS One. 2013;8:e68576. doi: 10.1371/journal.pone.0068576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Münzel M, Lischke U, Stathis D, Pfaffeneder T, Gnerlich FA, Deiml CA, Koch SC, Karaghiosoff K, Carell T. Improved synthesis and mutagenicity of oligonucleotides containing 5-hydroxymethylcytosine, 5-formylcytosine and 5-carboxylcytosine. Chem - Eur J. 2011;17:13782–13788. doi: 10.1002/chem.201102782. [DOI] [PubMed] [Google Scholar]
- 240.Xing XW, Liu YL, Vargas M, Wang Y, Feng YQ, Zhou X, Yuan BF. Mutagenic and cytotoxic properties of oxidation products of 5-methylcytosine revealed by next-generation sequencing. PLoS One. 2013;8:e72993. doi: 10.1371/journal.pone.0072993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Kamiya H, Tsuchiya H, Karino N, Ueno Y, Matsuda A, Harashima H. Mutagenicity of 5-formylcytosine, an oxidation product of 5-methylcytosine, in DNA in mammalian cells. J Biochem. 2002;132:551–555. doi: 10.1093/oxfordjournals.jbchem.a003256. [DOI] [PubMed] [Google Scholar]
- 242.Ji D, You C, Wang P, Wang Y. Effects of Tet-Induced oxidation products of 5-methylcytosine on DNA replication in mammalian cells. Chem Res Toxicol. 2014;27:1304–1309. doi: 10.1021/tx500169u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Kellinger MW, Song CX, Chong J, Lu XY, He C, Wang D. 5-formylcytosine and 5-carboxylcytosine reduce the rate and substrate specificity of RNA polymerase II transcription. Nat Struct Mol Biol. 2012;19:831–833. doi: 10.1038/nsmb.2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Zheng G, Fu Y, He C. Nucleic acid oxidation in DNA damage repair and epigenetics. Chem Rev. 2014;114:4602–4620. doi: 10.1021/cr400432d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Lu X, Zhao BS, He C. TET family proteins: Oxidation activity, interacting molecules, and functions in diseases. Chem Rev. 2015;115:2225–2239. doi: 10.1021/cr500470n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Maiti A, Drohat AC. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J Biol Chem. 2011;286:35334–35338. doi: 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Cortazar D, Kunz C, Selfridge J, Lettieri T, Saito Y, MacDougall E, Wirz A, Schuermann D, Jacobs AL, Siegrist F, Steinacher R, Jiricny J, Bird A, Schar P. Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature. 2011;470:419–423. doi: 10.1038/nature09672. [DOI] [PubMed] [Google Scholar]
- 248.Nabel CS, Jia H, Ye Y, Shen L, Goldschmidt HL, Stivers JT, Zhang Y, Kohli RM. AID/APOBEC deaminases disfavor modified cytosines implicated in DNA demethylation. Nat Chem Biol. 2012;8:751–758. doi: 10.1038/nchembio.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Kuraoka I, Bender C, Romieu A, Cadet J, Wood RD, Lindahl T. Removal of oxygen free-radical-induced 5′,8-purine cyclodeoxynucleosides from DNA by the nucleotide excision-repair pathway in human cells. Proc Natl Acad Sci U S A. 2000;97:3832–3837. doi: 10.1073/pnas.070471597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.You C, Dai X, Yuan B, Wang J, Wang J, Brooks PJ, Niedernhofer LJ, Wang Y. A quantitative assay for assessing the effects of DNA lesions on transcription. Nat Chem Biol. 2012;8:817–822. doi: 10.1038/nchembio.1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Kuraoka I, Robins P, Masutani C, Hanaoka F, Gasparutto D, Cadet J, Wood RD, Lindahl T. Oxygen free radical damage to DNA: translesion synthesis by human DNA polymerase η and resistance to exonuclease action at cyclopurine deoxynucleoside residues. J Biol Chem. 2001;276:49283–49288. doi: 10.1074/jbc.M107779200. [DOI] [PubMed] [Google Scholar]
- 252.Jasti VP, Das RS, Hilton BA, Weerasooriya S, Zou Y, Basu AK. 5′S)-8,5′-cyclo-2′-deoxyguanosine is a strong block to replication, a potent pol V-dependent mutagenic lesion, and is inefficiently repaired in Escherichia coli. Biochemistry. 2011;50:3862–3865. doi: 10.1021/bi2004944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Yuan B, Wang J, Cao H, Sun R, Wang Y. High-throughput analysis of the mutagenic and cytotoxic properties of DNA lesions by next-generation sequencing. Nucleic Acids Res. 2011;39:5945–5954. doi: 10.1093/nar/gkr159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.You C, Swanson AL, Dai X, Yuan B, Wang J, Wang Y. Translesion synthesis of 8,5′-cyclopurine-2′-deoxynucleo-sides by DNA polymerases η, ι, and ζ. J Biol Chem. 2013;288:28548–28556. doi: 10.1074/jbc.M113.480459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Marietta C, Brooks PJ. Transcriptional bypass of bulky DNA lesions causes new mutant RNA transcripts in human cells. EMBO Rep. 2007;8:388–393. doi: 10.1038/sj.embor.7400932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Yang Z, Colis LC, Basu AK, Zou Y. Recognition and incision of gamma-radiation-induced cross-linked γ-thymine tandem lesion G[8,5-Me]T by UvrABC nuclease. Chem Res Toxicol. 2005;18:1339–1346. doi: 10.1021/tx050147+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Gu C, Zhang Q, Yang Z, Wang Y, Zou Y, Wang Y. Recognition and incision of oxidative intrastrand cross-link lesions by UvrABC nuclease. Biochemistry. 2006;45:10739–10746. doi: 10.1021/bi060423z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Raychaudhury P, Basu AK. Genetic requirement for mutagenesis of the G[8,5-Me]T cross-link in Escherichia coli: DNA polymerases IV and V compete for error-prone bypass. Biochemistry. 2011;50:2330–2338. doi: 10.1021/bi102064z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Ding S, Kropachev K, Cai Y, Kolbanovskiy M, Durandina SA, Liu Z, Shafirovich V, Broyde S, Geacintov NE. Structural, energetic and dynamic properties of guanine(C8)– thymine(N3) cross-links in DNA provide insights on susceptibility to nucleotide excision repair. Nucleic Acids Res. 2012;40:2506–2517. doi: 10.1093/nar/gkr1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Talhaoui I, Shafirovich V, Liu Z, Saint-Pierre C, Akishev Z, Matkarimov BT, Gasparutto D, Geacintov NE, Saparbaev M. Oxidatively Generated Guanine(C8)-Thymine(N3) Intrastrand Cross-links in Double-stranded DNA Are Repaired by Base Excision Repair Pathways. J Biol Chem. 2015;290:14610–14617. doi: 10.1074/jbc.M115.647487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Lee YA, Lee YC, Geacintov NE, Shafirovich V. Translesion synthesis past guanine(C8)-thymine(N3) intra-strand cross-links catalyzed by selected A- and Y-family polymerases. Mol BioSyst. 2016;12:1892–1900. doi: 10.1039/c6mb00160b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Fujikawa K, Yakushiji H, Nakabeppu Y, Suzuki T, Masuda M, Ohshima H, Kasai H. 8-chloro-dGTP, a hypochlorous acid-modified nucleotide, is hydrolyzed by hMTH1, the human MutT homolog. FEBS Lett. 2002;512:149–151. doi: 10.1016/s0014-5793(02)02240-8. [DOI] [PubMed] [Google Scholar]
- 263.Kim CH, Darwanto A, Theruvathu JA, Herring JL, Sowers LC. Polymerase incorporation and miscoding properties of 5-chlorouracil. Chem Res Toxicol. 2010;23:740–748. doi: 10.1021/tx900302j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Fedeles BI, Freudenthal BD, Yau E, Singh V, Chang SC, Li DY, Delaney JC, Wilson SH, Essigmann JM. Intrinsic mutagenic properties of 5-chlorocytosine: A mechanistic connection between chronic inflammation and cancer. Proc Natl Acad Sci U S A. 2015;112:E4571–E4580. doi: 10.1073/pnas.1507709112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Valinluck V, Liu P, Kang JI, Burdzy A, Sowers LC. 5-Halogenated pyrimidine lesions within a CpG sequence context mimic 5-methylcytosine by enhancing the binding of the methyl-CpG-binding domain of methyl-CpG-binding protein 2 (MeCP2) Nucleic Acids Res. 2005;33:3057–3064. doi: 10.1093/nar/gki612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Theruvathu JA, Yin YW, Pettitt BM, Sowers LC. Comparison of the structural and dynamic effects of 5-methylcytosine and 5-chlorocytosine in a CpG dinucleotide sequence. Biochemistry. 2013;52:8590–8598. doi: 10.1021/bi400980c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Valinluck V, Wu W, Liu PF, Neidigh JW, Sowers LC. Impact of cytosine 5-halogens on the interaction of DNA with restriction endonucleases and methyltransferase. Chem Res Toxicol. 2006;19:556–562. doi: 10.1021/tx050341w. [DOI] [PubMed] [Google Scholar]
- 268.Valinluck V, Sowers LC. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 2007;67:946–950. doi: 10.1158/0008-5472.CAN-06-3123. [DOI] [PubMed] [Google Scholar]
- 269.Mazrimas JA, Stetka DG. Direct evidence for the role of incorporated BUdR in the induction of sister chromatid exchanges. Exp Cell Res. 1978;117:23–30. doi: 10.1016/0014-4827(78)90423-8. [DOI] [PubMed] [Google Scholar]
- 270.Heartlein MW, O'Neill JP, Pal BC, Preston RJ. The induction of specific-locus mutations and sister-chromatid exchanges by 5-bromo- and 5-chloro-deoxyuridine. Mutat Res Fundam Mol Mech Mutagen. 1982;92:411–416. doi: 10.1016/0027-5107(82)90239-1. [DOI] [PubMed] [Google Scholar]
- 271.Sassa A, Ohta T, Nohmi T, Honma M, Yasui M. Mutational specificities of brominated DNA adducts catalyzed by human DNA polymerases. J Mol Biol. 2011;406:679–686. doi: 10.1016/j.jmb.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 272.Neeley WL, Delaney JC, Henderson PT, Essigmann JM. In vivo Bypass Efficiencies and Mutational Signatures of the Guanine Oxidation Products 2-Aminoimidazolone and 5-Guanidino-4-nitroimidazole. J Biol Chem. 2004;279:43568–43573. doi: 10.1074/jbc.M407117200. [DOI] [PubMed] [Google Scholar]
- 273.Dimitri A, Jia L, Shafirovich V, Geacintov NE, Broyde S, Scicchitano DA. Transcription of DNA containing the 5-guanidino-4-nitroimidazole lesion by human RNA polymerase II and bacteriophage T7 RNA polymerase. DNA Repair. 2008;7:1276–1288. doi: 10.1016/j.dnarep.2008.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Hang B, Chenna A, Guliaev AB, Singer B. Miscoding properties of 1,N6-ethanoadenine, a DNA adduct derived from reaction with the antitumor agent 1,3-bis(2-chloroethyl)-1-nitrosourea. Mutat Res Fundam Mol Mech Mutagen. 2003;531:191–203. doi: 10.1016/j.mrfmmm.2003.07.006. [DOI] [PubMed] [Google Scholar]
- 275.Pandya GA, Moriya M. 1,N6-Ethenodeoxyadenosine, a DNA adduct highly mutagenic in mammalian cells. Biochemistry. 1996;35:11487–11492. doi: 10.1021/bi960170h. [DOI] [PubMed] [Google Scholar]
- 276.Tolentino JH, Burke TJ, Mukhopadhyay S, McGregor WG, Basu AK. Inhibition of DNA replication fork progression and mutagenic potential of 1,N-6-ethenoadenine and 8-oxoguanine in human cell extracts. Nucleic Acids Res. 2008;36:1300–1308. doi: 10.1093/nar/gkm1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Levine RL, Yang IY, Hossain M, Pandya GA, Grollman AP, Moriya M. Mutagenesis induced by a single 1,N6-ethenodeoxyadenosine adduct in human cells. Cancer Res. 2000;60:4098–4104. [PubMed] [Google Scholar]
- 278.Moriya M, Zhang W, Johnson F, Grollman AP. Mutagenic potency of exocyclic DNA adducts: marked differences between Escherichia coli and simian kidney cells. Proc Natl Acad Sci U S A. 1994;91:11899–11903. doi: 10.1073/pnas.91.25.11899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Langouët S, Mican AN, Müller M, Fink SP, Marnett LJ, Muhle SA, Guengerich FP. Misincorporation of nucleotides opposite five-membered exocyclic ring guanine derivatives by Escherichia coli polymerases in vitro and in vivo:1,N2-ethenoguanine, 5,6,7,9-tetrahydro-9-oxoimidazo[1,2-a]purine, and 5,6,7,9-tetrahydro-7-hydroxy-9-oxoimidazo[1,2-a]purine. Biochemistry. 1998;37:5184–5193. doi: 10.1021/bi972327r. [DOI] [PubMed] [Google Scholar]
- 280.Cheng KC, Preston BD, Cahill DS, Dosanjh MK, Singer B, Loeb LA. The vinyl chloride DNA derivative N2,3-ethenoguanine produces GaÀ transitions in Escherichia coli. Proc Natl Acad Sci U S A. 1991;88:9974–9978. doi: 10.1073/pnas.88.22.9974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Gros L, Ishchenko AA, Saparbaev M. Enzymology of repair of etheno-adducts. Mutat Res Fundam Mol Mech Mutagen. 2003;531:219–229. doi: 10.1016/j.mrfmmm.2003.07.008. [DOI] [PubMed] [Google Scholar]
- 282.Zdżalik D, Domańska A, Prorok P, Kosicki K, van den Born E, Falnes PØ, Rizzo CJ, Guengerich FP, Tudek B. Differential repair of etheno-DNA adducts by bacterial and human AlkB proteins. DNA Repair. 2015;30:1–10. doi: 10.1016/j.dnarep.2015.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Benamira M, Johnson K, Chaudhary A, Bruner K, Tibbetts C, Marnett LJ. Induction of mutations by replication of malondialdehyde modified M13 DNA in Escherichia coil: determination of the extent of DNA modification, genetic requirements for mutagenesis, and types of mutations induced. Carcinogenesis. 1995;16:93–99. doi: 10.1093/carcin/16.1.93. [DOI] [PubMed] [Google Scholar]
- 284.Fink SP, Reddy GR, Marnett LJ. Mutagenicity in Escherichia coli of the major DNA adduct derived from the endogenous mutagen malondialdehyde. Proc Natl Acad Sci U S A. 1997;94:8652–8657. doi: 10.1073/pnas.94.16.8652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Niedernhofer LJ, Daniels JS, Rouzer CA, Greene RE, Marnett LJ. Malondialdehyde, a product of lipid peroxidation, is mutagenic in human cells. J Biol Chem. 2003;278:31426–31433. doi: 10.1074/jbc.M212549200. [DOI] [PubMed] [Google Scholar]
- 286.VanderVeen LA, Hashim MF, Shyr Y, Marnett LJ. Induction of frameshift and base pair substitution mutations by the major DNA adduct of the endogenous carcinogen malondialde-hyde. Proc Natl Acad Sci U S A. 2003;100:14247–14252. doi: 10.1073/pnas.2332176100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Singh V, Fedeles BI, Li D, Delaney JC, Kozekov ID, Kozekova A, Marnett LJ, Rizzo CJ, Essigmann JM. Mechanism of repair of acrolein- and malondialdehyde-derived exocyclic guanine adducts by the α-ketoglutarate/Fe(II) dioxygenase AlkB. Chem Res Toxicol. 2014;27:1619–1631. doi: 10.1021/tx5002817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Tretyakova N, Goggin M, Sangaraju D, Janis G. Quantitation of DNA adducts by stable isotope dilution mass spectrometry. Chem Res Toxicol. 2012;25:2007–2035. doi: 10.1021/tx3002548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Shinmura K, Tao H, Goto M, Igarashi H, Taniguchi T, Maekawa M, Takezaki T, Sugimura H. Inactivating mutations of the human base excision repair gene NEIL1 in gastric cancer. Carcinogenesis. 2004;25:2311–2317. doi: 10.1093/carcin/bgh267. [DOI] [PubMed] [Google Scholar]
- 290.Delaney S, Delaney JC, Essigmann JM. Chemical-biological fingerprinting: Probing the properties of DNA lesions-formed by peroxynitrite. Chem Res Toxicol. 2007;20:1718–1729. doi: 10.1021/tx700273u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Zhou J, Fleming AM, Averill AM, Burrows CJ, Wallace SS. The NEIL glycosylases remove oxidized guanine lesions from telomeric and promoter quadruplex DNA structures. Nucleic Acids Res. 2015;43:4039–4054. doi: 10.1093/nar/gkv252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Norabuena EM, Williams SB, Klureza MA, Goehring LJ, Gruessner B, Radhakrishnan ML, Jamieson ER, Nunez ME. Effect of the spiroiminodihydantoin lesion on nucleosome stability and positioning. Biochemistry. 2016;55:2411–2421. doi: 10.1021/acs.biochem.6b00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Cathcart R, Schwiers E, Saul RL, Ames BN. Thymine glycol and thymidine glycol in human and rat urine: a possible assay for oxidative DNA damage. Proc Natl Acad Sci U S A. 1984;81:5633–5637. doi: 10.1073/pnas.81.18.5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Adelman R, Saul RL, Ames BN. Oxidative damage to DNA: relation to species metabolic rate and life span. Proc Natl Acad Sci U S A. 1988;85:2706–2708. doi: 10.1073/pnas.85.8.2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Cao EH, Wang JJ. Oxidative damage to DNA: levels of thymine glycol and thymidine glycol in neoplastic human urines. Carcinogenesis. 1993;14:1359–1362. doi: 10.1093/carcin/14.7.1359. [DOI] [PubMed] [Google Scholar]
- 296.Wang Y. Bulky DNA lesions induced by reactive oxygen species. Chem Res Toxicol. 2008;21:276–281. doi: 10.1021/tx700411g. [DOI] [PubMed] [Google Scholar]
- 297.Box HC, Budzinski EE, Evans MS, French JB, Maccubbin AE. The differential lysis of phosphoester bonds by nuclease P1. Biochim Biophys Acta, Protein Struct Mol Enzymol. 1993;1161:291–294. doi: 10.1016/0167-4838(93)90227-i. [DOI] [PubMed] [Google Scholar]
- 298.Falcone JM, Box HC. Selective hydrolysis of damaged DNA by nuclease P1. Biochim Biophys Acta, Protein Struct Mol Enzymol. 1997;1337:267–275. doi: 10.1016/s0167-4838(96)00172-0. [DOI] [PubMed] [Google Scholar]
- 299.Bailey DT, DeFedericis HCC, Greene KF, Iijima H, Budzinski EE, Patrzyc HB, Dawidzik JB, Box HC. A novel approach to DNA damage assessments: Measurement of the thymine glycol lesion. Radiat Res. 2006;165:438–444. doi: 10.1667/rr3534.1. [DOI] [PubMed] [Google Scholar]
- 300.Greene KE, Budzinski EE, Iijima H, Dawidzik JB, DeFedericis HC, Patrzyc HB, Evans MS, Bailey DT, Freund HG, Box HC. Assessment of DNA damage at the dimer level: Measurement of the formamide lesion. Radiat Res. 2007;167:146–151. doi: 10.1667/rr0693.1. [DOI] [PubMed] [Google Scholar]
- 301.Iijima H, Patrzyc HB, Budzinski EE, Freund HG, Dawidzik JB, Rodabaugh KJ, Box HC. A study of pyrimidine base damage in relation to oxidative stress and cancer. Br J Cancer. 2009;101:452–456. doi: 10.1038/sj.bjc.6605176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Budzinski EE, Patrzyc HB, Dawidzik JB, Freund HG, Frederick P, Godoy HE, Voian NC, Odunsi K, Box HC. Pyrimidine base damage is increased in women with BRCA mutations. Cancer Lett. 2013;338:267–270. doi: 10.1016/j.canlet.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Box HC, O'Connor RJ, Patrzyc HB, Iijima H, Dawidzik JB, Freund HG, Budzinski EE, Cummings KM, Mahoney MC. Reduction in oxidatively generated DNA damage following smoking cessation. Tob Induced Dis. 2011;9:5. doi: 10.1186/1617-9625-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Box HC, Patrzyc HB, Budzinski EE, Dawidzik JB, Freund HG, Zeitouni NC, Mahoney MC. Profiling oxidative DNA damage: Effects of antioxidants. Cancer Sci. 2012;103:2002–2006. doi: 10.1111/j.1349-7006.2012.02391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.McNulty JM, Jerkovic B, Bolton PH, Basu AK. Replication inhibition and miscoding properties of DNA templates containing a site-specific cis-thymine glycol or urea residue. Chem Res Toxicol. 1998;11:666–673. doi: 10.1021/tx970225w. [DOI] [PubMed] [Google Scholar]
- 306.Liu S, Wang J, Su Y, Guerrero C, Zeng Y, Mitra D, Brooks PJ, Fisher DE, Song H, Wang Y. Quantitative assessment of Tet-induced oxidation products of 5-methylcytosine in cellular and tissue DNA. Nucleic Acids Res. 2013;41:6421–6429. doi: 10.1093/nar/gkt360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Frelon S, Douki T, Ravanat JL, Pouget JP, Tornabene C, Cadet J. High-performance liquid chromatography-tandem mass spectrometry measurement of radiation-induced base damage to isolated and cellular DNA. Chem Res Toxicol. 2000;13:1002–1010. doi: 10.1021/tx000085h. [DOI] [PubMed] [Google Scholar]
- 308.Hong HH, Wang YS. Derivatization with Girard reagent T combined with LC-MS/MS for the sensitive detection of 5-formyl-2′-deoxyuridine in cellular DNA. Anal Chem. 2007;79:322–326. doi: 10.1021/ac061465w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Bullard W, da Rosa-Spiegler JL, Liu S, Wang YS, Sabatini R. Identification of the glucosyltransferase that converts hydroxymethyluracil to base J in the trypanosomatid genome. J Biol Chem. 2014;289:20273–20282. doi: 10.1074/jbc.M114.579821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Wu J, Forbes JR, Chen HS, Cox DW. The LEC rat has a deletion in the copper transporting ATPase gene homologous to the Wilson disease gene. Nat Genet. 1994;7:541–545. doi: 10.1038/ng0894-541. [DOI] [PubMed] [Google Scholar]
- 311.Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML. Wilson's disease. Lancet. 2007;369:397–408. doi: 10.1016/S0140-6736(07)60196-2. [DOI] [PubMed] [Google Scholar]
- 312.Roberts EA, Schilsky ML. Diagnosis and treatment of Wilson disease: An update. Hepatology. 2008;47:2089–2111. doi: 10.1002/hep.22261. [DOI] [PubMed] [Google Scholar]
- 313.Killian JK, Kim SY, Miettinen M, Smith C, Merino M, Tsokos M, Quezado M, Smith WI, Jahromi MS, Xekouki P, Szarek E, Walker RL, Lasota J, Raffeld M, Klotzle B, Wang Z, Jones L, Zhu Y, Wang Y, Waterfall JJ, O'Sullivan MJ, Bibikova M, Pacak K, Stratakis C, Janeway KA, Schiffman JD, Fan JB, Helman L, Meltzer PS. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discovery. 2013;3:648–657. doi: 10.1158/2159-8290.CD-13-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, Peeters JK, Liu W, Choe SE, Fantin VR, Paietta E, Löwenberg B, Licht JD, Godley LA, Delwel R, Valk PJM, Thompson CB, Levine RL, Melnick A. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Yang H, Lin H, Xu H, Zhang L, Cheng L, Wen B, Shou J, Guan K, Xiong Y, Ye D. TET-catalyzed 5-methylcytosine hydroxylation is dynamically regulated by metabolites. Cell Res. 2014;24:1017–1020. doi: 10.1038/cr.2014.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Ye D, Xiong Y, Guan KL. The mechanisms of IDH mutations in tumorigenesis. Cell Res. 2012;22:1102–1104. doi: 10.1038/cr.2012.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE, Liau LM, Rabinowitz JD, Cantley LC, Thompson CB, Vander Heiden MG, Su SM. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, Rabinowitz JD, Carroll M, Su SM, Sharp KA, Levine RL, Thompson CB. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, Liu L, Liu Y, Yang C, Xu Y, Zhao S, Ye D, Xiong Y, Guan KL. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26:1326–1338. doi: 10.1101/gad.191056.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature. 2012;491:364–373. doi: 10.1038/nature11706. [DOI] [PubMed] [Google Scholar]
- 321.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, Liu Lx, Jiang Wq, Liu J, Zhang Jy, Wang B, Frye S, Zhang Y, Xu Yh, Lei Qy, Guan KL, Zhao Sm, Xiong Y. Oncometabolite 2-hydroxyglutarate is acompetitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Jin SG, Jiang Y, Qiu R, Rauch TA, Wang Y, Schackert G, Krex D, Lu Q, Pfeifer GP. 5-hydroxymethylcytosine is strongly depleted in human cancers but its levels do not correlate with IDH1 mutations. Cancer Res. 2011;71:7360–7365. doi: 10.1158/0008-5472.CAN-11-2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Shen L, Song CX, He C, Zhang Y. Mechanism and function of oxidative reversal of DNA and RNA methylation. Annu Rev Biochem. 2014;83:585–614. doi: 10.1146/annurev-biochem-060713-035513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Song CX, He C. Potential functional roles of DNA demethylation intermediates. Trends Biochem Sci. 2013;38:480–484. doi: 10.1016/j.tibs.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Spruijt CG, Gnerlich F, Smits Arne H, Pfaffeneder T, Jansen Pascal WTC, Bauer C, Münzel M, Wagner M, Müller M, Khan F, Eberl HC, Mensinga A, Brinkman Arie B, Lephikov K, Müller U, Walter J, Boelens R, van Ingen H, Leonhardt H, Carell T, Vermeulen M. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152:1146–1159. doi: 10.1016/j.cell.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 326.Mellén M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151:1417–1430. doi: 10.1016/j.cell.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Yildirim O, Li R, Hung JH, Chen Poshen B, Dong X, Ee LS, Weng Z, Rando Oliver J, Fazzio TG. Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell. 2011;147:1498–1510. doi: 10.1016/j.cell.2011.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Frauer C, Hoffmann T, Bultmann S, Casa V, Cardoso MC, Antes I, Leonhardt H. Recognition of 5-hydroxymethylcytosine by the Uhrf1 SRA domain. PLoS One. 2011;6:e21306. doi: 10.1371/journal.pone.0021306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Iurlaro M, Ficz G, Oxley D, Raiber EA, Bachman M, Booth MJ, Andrews S, Balasubramanian S, Reik W. A screen for hydroxymethylcytosine and formylcytosine binding proteins suggests functions in transcription and chromatin regulation. Genome Biol. 2013;14:R119. doi: 10.1186/gb-2013-14-10-r119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Sun Z, Dai N, Borgaro JG, Quimby A, Sun D, Correâ Ivan R, Jr, Zheng Y, Zhu Z, Guan S. A sensitive approach to map genome-wide 5-hydroxymethylcytosine and 5-formylcytosine at single-base resolution. Mol Cell. 2015;57:750–761. doi: 10.1016/j.molcel.2014.12.035. [DOI] [PubMed] [Google Scholar]
- 331.Jin SG, Zhang ZM, Dunwell Thomas L, Harter Matthew R, Wu X, Johnson J, Li Z, Liu J, Szabó Piroska E, Lu Q, Xu Gl, Song J, Pfeifer GP. Tet3 reads 5-carboxylcytosine through its CXXC domain and is a potential guardian against neurodegeneration. Cell Rep. 2016;14:493–505. doi: 10.1016/j.celrep.2015.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Wang J, Cao H, You C, Yuan B, Bahde R, Gupta S, Nishigori C, Niedernhofer LJ, Brooks PJ, Wang Y. Endogenous formation and repair of oxidatively induced G[8–5m]T intrastrand cross-link lesion. Nucleic Acids Res. 2012;40:7368–7374. doi: 10.1093/nar/gks357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Kraemer KH, Patronas NJ, Schiffmann R, Brooks BP, Tamura D, DiGiovanna JJ. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: A complex genotype–phenotype relationship. Neuroscience. 2007;145:1388–1396. doi: 10.1016/j.neuroscience.2006.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Borgesius NZ, de Waard MC, van der Pluijm I, Omrani A, Zondag GC, van der Horst GT, Melton DW, Hoeijmakers JH, Jaarsma D, Elgersma Y. Accelerated age-related cognitive decline and neurodegeneration, caused by deficient DNA repair. J Neurosci. 2011;31:12543–12553. doi: 10.1523/JNEUROSCI.1589-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Bartsch H, Nair UJ. Cancer and Inflammation Mechanisms. John Wiley & Sons, Inc; New York: 2014. Lipid peroxidation-derived DNA adducts and the role in inflammation-related carcinogenesis; pp. 61–74. [Google Scholar]
- 336.Liu X, Lovell MA, Lynn BC. Development of a method for quantification of acrolein–deoxyguanosine adducts in DNA using isotope dilution-capillary LC/MS/MS and its application to human brain tissue. Anal Chem. 2005;77:5982–5989. doi: 10.1021/ac050624t. [DOI] [PubMed] [Google Scholar]
- 337.Moldovan GL, D'Andrea AD. How the fanconi anemia pathway guards the genome. Annu Rev Genet. 2009;43:223–249. doi: 10.1146/annurev-genet-102108-134222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.D'Andrea AD. Susceptibility pathways in Fanconi's anemia and breast cancer. N Engl J Med. 2010;362:1909–1919. doi: 10.1056/NEJMra0809889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Kottemann MC, Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature. 2013;493:356–363. doi: 10.1038/nature11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Tischkowitz MD, Hodgson SV. Fanconi anaemia. J Med Genet. 2003;40:1–10. doi: 10.1136/jmg.40.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Jiang Q, Blount BC, Ames BN. 5-chlorouracil, a marker of DNA damage from hypochlorous acid during inflammation - A gas chromatography-mass spectrometry assay. J Biol Chem. 2003;278:32834–32840. doi: 10.1074/jbc.M304021200. [DOI] [PubMed] [Google Scholar]
- 342.Takeshita J, Byun J, Nhan TQ, Pritchard DK, Pennathur S, Schwartz SM, Chait A, Heinecke JW. Myeloperoxidase generates 5-chlorouracil in human athero-sclerotic tissue - A potential pathway for somatic mutagenesis by macrophages. J Biol Chem. 2006;281:3096–3104. doi: 10.1074/jbc.M509236200. [DOI] [PubMed] [Google Scholar]
- 343.Noyon C, Delporte C, Dufour D, Cortese M, Rousseau A, Poelvoorde P, Neve J, Vanhamme L, Boudjeltia KZ, Roumeguere T, Van Antwerpen P. Validation of a sensitive LC/MSMS method for chloronucleoside analysis in biological matrixes and its applications. Talanta. 2016;154:322–328. doi: 10.1016/j.talanta.2016.03.087. [DOI] [PubMed] [Google Scholar]
- 344.Balbo S, Villalta PW, Hecht SS. Quantitation of 7-ethylguanine in leukocyte DNA from smokers and nonsmokers by liquid chromatography-nanoelectrospray-high resolution tandem mass spectrometry. Chem Res Toxicol. 2011;24:1729–1734. doi: 10.1021/tx200262d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Zhao L, Balbo S, Wang M, Upadhyaya P, Khariwala SS, Villalta PW, Hecht SS. Quantitation of pyridyloxobutyl-DNA adducts in tissues of rats treated chronically with (R)- or (S)-N′-nitrosonornicotine (NNN) in a carcinogenicity study. Chem Res Toxicol. 2013;26:1526–1535. doi: 10.1021/tx400235x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Bessette EE, Goodenough AK, Langouët S, Yasa I, Kozekov ID, Spivack SD, Turesky RJ. Screening for DNA Adducts by data-dependent constant neutral loss-triple stage mass spectrometry with a linear quadrupole ion trap mass spectrometer. Anal Chem. 2009;81:809–819. doi: 10.1021/ac802096p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Balbo S, Hecht SS, Upadhyaya P, Villalta PW. Application of a high-resolution mass-spectrometry-based DNA adductomics approach for identification of DNA adducts in complex mixtures. Anal Chem. 2014;86:1744–1752. doi: 10.1021/ac403565m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Balbo S, Turesky RJ, Villalta PW. DNA Adductomics. Chem Res Toxicol. 2014;27:356–366. doi: 10.1021/tx4004352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Zhao YY, Wu SP, Liu S, Zhang Y, Lin RC. Ultra-performance liquid chromatography–mass spectrometry as a sensitive and powerful technology in lipidomic applications. Chem Biol Interact. 2014;220:181–192. doi: 10.1016/j.cbi.2014.06.029. [DOI] [PubMed] [Google Scholar]
- 350.Guo J, Yun BH, Upadhyaya P, Yao L, Krishnamachari S, Rosenquist TA, Grollman AP, Turesky RJ. Multiclass carcinogenic DNA adduct quantification in formalin-fixed paraffin-embedded tissues by ultraperformance liquid chromatography–tandem mass spectrometry. Anal Chem. 2016;88:4780–4787. doi: 10.1021/acs.analchem.6b00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Li H, Cui S, Wang S, Jiang X, Zhang S, Zhang R, Fu PP, Sun X. Ultrasensitive UPLC–MS/MS method for analysis of etheno-DNA adducts in human white blood cells. Free Radical Res. 2015;49:1049–1054. doi: 10.3109/10715762.2015.1006213. [DOI] [PubMed] [Google Scholar]
- 352.ESCODD (European Standards Committee on Oxidative DNA Damage. Comparative analysis of baseline 8-oxo-7,8-dihydroguanosine in mammalian cell DNA, by different methods in different laboratories: an approach to consensus. Carcinogenesis. 2002;23:2129–2133. doi: 10.1093/carcin/23.12.2129. [DOI] [PubMed] [Google Scholar]