Drug-Driven Phenotypic Convergence Supports Rational Treatment Strategies of Chronic Infections (original) (raw)
Summary
Chronic Pseudomonas aeruginosa infections evade antibiotic therapy and are associated with mortality in cystic fibrosis (CF) patients. We find that in vitro resistance evolution of P. aeruginosa toward clinically relevant antibiotics leads to phenotypic convergence toward distinct states. These states are associated with collateral sensitivity toward several antibiotic classes and encoded by mutations in antibiotic resistance genes, including transcriptional regulator nfxB. Longitudinal analysis of isolates from CF patients reveals similar and defined phenotypic states, which are associated with extinction of specific sub-lineages in patients. In-depth investigation of chronic P. aeruginosa populations in a CF patient during antibiotic therapy revealed dramatic genotypic and phenotypic convergence. Notably, fluoroquinolone-resistant subpopulations harboring nfxB mutations were eradicated by antibiotic therapy as predicted by our in vitro data. This study supports the hypothesis that antibiotic treatment of chronic infections can be optimized by targeting phenotypic states associated with specific mutations to improve treatment success in chronic infections.
Keywords: collateral sensitivity, drug resistance, Pseudomonas aeruginosa, cystic fibrosis, chronic infections, phenotypic convergence, nfxB, antibiotic treatment
Graphical Abstract
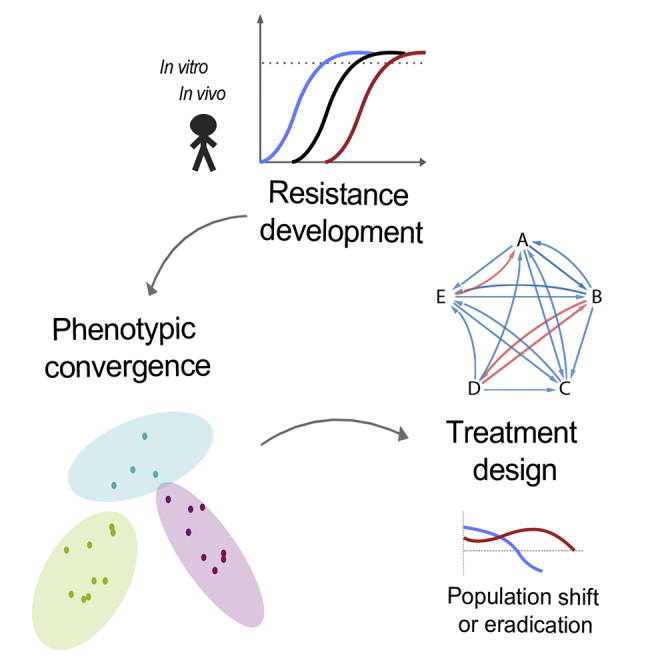
Highlights
- •
Collateral sensitivity can evolve from diverse genetic and phenotypic starting points - •
Collateral effects of resistance evolution converges to distinct phenotypic states - •
Genetic markers associated with convergent states were linked to nfxB mutations - •
nfxB mutants were eradicated in vivo from the lung of a CF patient during treatment
The evolution of antibiotic resistance of Pseudomonas infection in cystic fibrosis patients confers predictable sensitivities to other classes of antibiotics, suggesting new ways to optimize treatments for chronic infection.
Introduction
The emergence of drug-resistant bacteria coupled with a lack of novel structural classes of antibiotics have made antibiotic resistance one of the most eminent threats to global health (May, 2014, O’Neill, 2016). Therapeutic options and strategies are especially scarce for Gram-negative pathogens such as Pseudomonas aeruginosa (Boucher et al., 2013, Cabot et al., 2012). This versatile, opportunistic pathogen is a frequent cause of acute nosocomial infections as well as chronic infections in high-risk patient groups, such as those suffering from cystic fibrosis (CF) (Mesaros et al., 2007). CF is a recessive lethal genetic disorder among the Caucasian population that is caused by mutations in the CF transmembrane conductance regulator (CFTR) gene (Elborn et al., 2016). While intensive antibiotic treatment for the eradication of P. aeruginosa infections has been successful in young patients, eradication ultimately fails, leading to the chronic infections experienced by most adult CF patients (Folkesson et al., 2012, Gibson et al., 2003, Johansen et al., 2004). During chronic infection, antibiotic treatments can temporarily reduce airway infection and inflammation, thus extending the periods of stable disease status and maintained lung function (Fodor et al., 2012). Nevertheless, the ability of P. aeruginosa to sustain chronic infection and resist antibiotic treatment is associated with decline in lung function, respiratory failure, and death in CF patients (Hauser et al., 2011, Pittman et al., 2011, Taylor-Robinson et al., 2012).
The antibiotic resistance of P. aeruginosa is driven by several factors in CF patients, including the activation of chromosomally encoded resistance mechanisms, such as decreased production of the outer membrane porin, inducible chromosomal β-lactamase AmpC, and overexpression of several efflux systems (Lister et al., 2009, Marvig et al., 2015a). The main efflux pumps are tripartite systems consisting of a resistance nodulation cell division (RND) transporter, a membrane fusion protein (MFP), and an outer membrane factor (OMF). MexAB-OprM, MexCD-OprJ, MexEF-OprN, and MexXY-OprM are the main efflux pumps that expel functionally and structurally dissimilar antibiotics (Li et al., 2015).
When Escherichia coli and Staphylococcus aureus evolve resistance toward specific antibiotics, they also develop sensitivity toward other antibiotics (Baym et al., 2016, Imamovic and Sommer, 2013, Lázár et al., 2013, Munck et al., 2014, Rodriguez de Evgrafov et al., 2015). This observation led to the proposal of a new, rational drug treatment paradigm termed collateral sensitivity cycling, in which sequential drug treatments are designed to exploit collateral sensitivity resulting from resistance evolution (Imamovic and Sommer, 2013). Collateral sensitivity has also been demonstrated in cancer cell lines (Hall et al., 2009) and was recently successfully deployed for treatment of Ph(+) acute lymphoblastic leukemia in an animal model (Zhao et al., 2016).
Collateral sensitivity may be particularly useful for optimizing treatments of chronic infections since their nature and severity warrants and requires tailored treatment strategies. Chronic lung infections of CF patients caused by P. aeruginosa may be a useful clinical model to study the evolution of collateral sensitivity in response to antibiotic therapy. While a recent study reported a lack of collateral sensitivity in clinical isolates from CF patients (Jansen et al., 2016), the study did not investigate relative changes in strain susceptibility, and, thus, collateral sensitivity might be missed due to the lack of appropriate baseline controls. Moreover, if evolutionary tradeoffs, such as collateral sensitivity, do not occur in vivo, then ever-increasing resistance would be the consequence of decades of antibiotic exposure. Yet, previous phenotypic characterization of CF isolates did not observe such monotonic increase in antibiotic resistance over time (López-Causapé et al., 2013). Accordingly, we hypothesized that P. aeruginosa might evolve collateral sensitivity in response to antibiotic exposure both in vitro and in patients and that these vulnerabilities modulate population dynamics during antibiotic treatment.
Results
Complex Networks of Collateral Sensitivity and Collateral Resistance
To elucidate the collateral sensitivity network of drug-resistant strains of P. aeruginosa, PAO1 were experimentally evolved in media that resembled the chemical composition encountered in the lungs of CF patients (SCFM) (Palmer et al., 2007). Twenty-four clinically relevant antibiotics that included anti-pseudomonal antibiotics and other drugs were chosen from eight chemical classes affecting different targets in P. aeruginosa (Table 1). To exclude possible effects on resistance phenotypes from adaptation to novel growth conditions, we adapted the ancestral PA01 strain to SCFM for 10 days as a media control (WTE). At the last day of the adaptive evolution experiment, all lineages could grow in the media with antibiotic concentrations exceeding the clinical breakpoint defined by EUCAST for P. aeruginosa (Table 1; Figures S1A–S1F) (EUCAST, 2016). Collateral sensitivity or collateral resistance was defined as a decrease or increase in the MIC (minimal inhibitory concentration) of the antibiotic-resistant strain relative to the wild-type adapted to SCFM (WTE) (Figures S1G–S1I) (Imamovic and Sommer, 2013). To confirm the robustness of our susceptibility tests, we measured the significance of the fold increase or decrease in resistance relative to the WTE (see STAR Methods). We observed that collateral sensitivity toward ampicillin decreased by 8.5-fold for ciprofloxacin-resistant strain (p value 3.42e−21, t test). Increase in susceptibility for other antibiotics such as amikacin and colistin was 2.5- and 1.5-fold relative to the WTE; yet, in both cases, we observed that collateral sensitivity observed was statistically significant (p value 7.87e−55 and 2.07e−290, t test, respectively) (Figure S1J).
Table 1.
List of Antibiotics Used in the Study
| Antibiotic | Antibiotic Abbreviation | Class (sub-class) | Class Abbreviation | Target | EUCAST Breakpoints |
|---|---|---|---|---|---|
| Amikacin | AMI | aminoglycoside | A | protein synthesis, 30S | 16 |
| Gentamicin | GEN | aminoglycoside | A | protein synthesis, 30S | 4 |
| Tobramycin | TOB | aminoglycoside | A | protein synthesis, 30S | 4 |
| Ciprofloxacin | CIP | quinolone | Q | DNA gyrase | 1 |
| Levofloxacin | LEV | quinolone | Q | DNA gyrase | 2 |
| Ampicillin | AMP | β-lactam (penicillin) | B | cell wall | n.a. |
| Piperacillin | PIP | β-lactam (penicillin) | B | cell wall | 16 |
| Carbenicillin | CAR | β-lactam (penicillin) | B | cell wall | n.a. |
| Ticarcillin | TIC | β-lactam (penicillin) | B | cell wall | 16 |
| Aztreonam | AZE | β-lactam (monobactam) | B | cell wall | 16 |
| Cefepime | CFP | β-lactam (cephalosporin) | B | cell wall | 8 |
| Cefuroxime | CFX | β-lactam (cephalosporin) | B | cell wall | n.a. |
| Ceftazidime | CFZ | β-lactam (cephalosporin) | B | cell wall | 8 |
| Meropenem | MER | β-lactam (carbapenem) | B | cell wall | 8 |
| Imipenem | IMI | β-lactam (carbapenem) | B | cell wall | 8 |
| Minocycline | MIN | tetracycline | T | protein synthesis, 30S | n.a. |
| Doxycycline | DOX | tetracycline | T | protein synthesis, 30S | n.a. |
| Azithromycin | AZY | macrolide | M | protein synthesis, 50S | n.a. |
| Erythromycin | ERI | macrolide | M | protein synthesis, 50S | n.a. |
| Clarithromycin | CLA | macrolide | M | protein synthesis, 50S | n.a. |
| Colistin | COL | polymyxin | P | lipopolysaccharide | 4 |
| Fosfomycin | FOS | fosfomycin | F | cell wall biogenesis | n.a. |
| Rifampicin | RIF | rifamycin | R | RNA synthesis | n.a. |
| Trimethoprim/Sulfamethoxazole | TMS | antifolate | C | combination folic acid pathway/ synthesis of dihydrofolic acid | n.a. |
Figure S1.

Selection for Resistance during Laboratory Evolution Experiments and Collateral Changes in Susceptibility Profiles, Related to Figure 1
(A–F) Increase in antibiotic resistance during adaptive evolution experiment in SCFM. Fold increase depicts the well from each day transfer was made in antibiotic gradient plate. Fold increase is calculated relative to the day 1.
(G–J) Resistance that alters the collateral sensitivity profiles of parallel evolved PAO1 strains. Three outcomes of resistance development were observed: (G) no change in susceptibility profiles relative to the WTE (black line), (H) collateral resistance or decrease in drug susceptibility relative to the WTE (red line) and (I) collateral sensitivity or increase in drug susceptibility relative to the WTE (blue line). For each strain, five replicates were performed to determine the drug susceptibility (±SD, error bars). (J) Significance test of the fold of difference in resistance between the WTE and ciprofloxacin resistant strain. Using the mean and standard deviation of the five replicates, growth inhibition of ciprofloxacin resistant strains toward three different antibiotics was calculated. _P_-value was determined using t test test with alternative hypothesis “greater” for the fold increase between one and 10 (by factor 0.5).
Given that the majority of resistant strains (75%) were collaterally sensitive to at least one antibiotic (Figure 1A; Table S1), we were able to construct a collateral sensitivity network for P. aeruginosa (Figures 1B and S2A). We simulated the number of collateral sensitivity cycles comprising: (1) all antibiotics in the study and (2) anti-pseudomonal antibiotics that have EUCAST-defined resistance breakpoints for P. aeruginosa (Table 1). For EUCAST-defined anti-pseudomonal antibiotics, we detected five collateral sensitivity cycles including two and three drugs (Figure 1C; Table S2). However, expanding the simulation for collateral sensitivity cycles to all antibiotics tested, the majority of antibiotics exhibiting collateral sensitivity (78%) could be employed in collateral sensitivity cycling. The number of simulated collateral sensitivity cycles including two and three drugs reached 18 and 51 cycles, respectively (Figures 1C and S2B).
Figure 1.

Consequence of Drug Resistance Evolution: Collateral Sensitivity and Collateral Resistance
(A) Heatmap represents quantification the collateral sensitivity profiles of the 24 evolved antibiotic-resistant PAO1 strains. Color coding represents the fold increase (red) or decrease (blue) in MIC value relative to the PAO1 strain evolved in SCFM media without antibiotics (WTE). For each strain, five replicates dose-response curves were performed to determine the drug susceptibility (Table S1). The order of the drugs and resistant strains was determined by hierarchical clustering using the similarity of normalized MIC values as the distance measure.
(B) Sub-network of collateral interactions among drugs commonly administered in treatments of CF patients. For collateral susceptibility networks, the directed path of each arrow represents the collateral sensitivity (blue) or collateral resistance (red) of an affected variable (drug-resistant strain) on the causal variable (drug). Collateral sensitivity cycling for two drugs would consist of alternating application of two drugs with collateral sensitivity (e.g., colistin and aztreonam) (full network for collateral sensitivity and resistance interaction depicted in Figures S2A and S2B).
(C) Number of possible collateral sensitivity cycles with anti-pseudomonal drugs (EUCAST) versus all drugs used in the study (Table S2).
See also Figures S1 and S2.
Figure S2.

Complex Networks of Interactions Based on the Collateral Susceptibility Profiles, Related to Figure 1
(A) Collateral sensitivity network. For collateral susceptibility networks, the directed path of each arrow represents the collateral sensitivity (blue) or collateral resistance (red) of an affected variable (drug-resistant strain) on the causal variable (drug). Antibiotic abbreviations are listed in Table 1.
(B) Number of collateral sensitivity cycles simulated for all drugs employed in the study. The cycles are based on PAO1 susceptibility profiles (Figure 1A).
Exploring the effects of exposure of drugs beyond typical anti-pseudomonal range is relevant for designing treatment strategies since the CF airways are frequently infected by complex microbiota, including potentially pathogenic bacteria such as Staphylococcus aureus, Haemophilus influenza, Burkholderia cepacia, or Stenotrophomonas maltophilia (Parkins and Floto, 2015, Willner et al., 2012). These bacteria may be treated with drugs toward which P. aeruginosa is considered intrinsically resistant or not used in treatment due to quick resistance development. Intriguingly, we observed changes in collateral susceptibility profiles for P. aeruginosa strains exposed to such drugs (Figure 1A), including trimethoprim-sulfamethoxazole (TMS) (Table 1), which was used in some centers for the treatment of S. aureus infections in CF patients (Gibson et al., 2003). The TMS-exposed PAO1 strain became collaterally sensitive toward aminoglycosides, polymyxin, and several β-lactam antibiotics. Simultaneously, the TMS-exposed PAO1 strain conferred resistance toward quinolone and tetracycline drugs (Figure 1A). These results suggest that, following treatment of S. aureus using TMS, P. aeruginosa treatment with aminoglycosides would be more effective than quinolones. This finding supports the hypothesis that the patient’s treatment history should be considered in order to exploit specific vulnerabilities of P. aeruginosa that result even from management of other pathogens.
Evolution of Drug Resistance Leads to Phenotypic Convergence toward Collateral States
To systematically elucidate the similarity of susceptibility phenotypes between the evolved strains, we computed Spearman correlation coefficients (ρ) for each pairwise comparison of their normalized susceptibility profiles (see STAR Methods). We detected 107 significant correlations (p < 0.05, a two-tailed significance test) between resistant strains. Interestingly, 67 (60%) pairwise comparisons had high positive correlation coefficients for strains resistant to drugs from different chemical classes suggesting convergent phenotypes (Figure S3A; Table S3). Among those strains were tetracycline- and macrolide- and quinolone-resistant strains. In contrast, 40 pairwise comparisons had a negative correlation between their susceptibility profiles (p < 0.05) suggesting orthogonal phenotypic states. Notably, negative correlations of susceptibility profiles were observed in 90% of cases between polymyxin- and β-lactam-resistant strains (Figure S3A). For instance, colistin- and ceftazidime-resistant strains had strong negatively correlated susceptibility (ρ = −0.74; p < 0.0001) (Figure S3A; Table S3). Interestingly, several strains resistant to β-lactams (aztreonam, carbenicillin, and ampicillin) also displayed reciprocal collateral sensitivity with colistin (Figure 1A).
Figure S3.

A Relationship between the Altered Susceptibilities of the Experimentally Evolved Resistant PAO1 and Collateral Sensitivity Profiles for DK2 Clinical Isolates, Related to Figure 2
(A) A Spearman correlation matrix for all pairwise susceptibility profiles of PAO1 drug-resistant strains. MIC values were normalized to WTE strain and log2 transformed. Upper right color panel is an indicator of the Spearman correlation coefficient (ρ). Circle size represents the strength of Spearman correlation’s coefficient (ρ.) Only statistically significant correlations are shown (p > 0.05, two-tailed test) (Table S3). Scatterplots represent a positive correlation between ciprofloxacin (CIP) and other strains resistant to four different chemical classes. The data displayed on the second scatterplot depict positive correlations (with aztreonam and two β-lactam resistant strains) and negative correlations (with aminoglycoside and polymyxin resistant strains). In addition, plots in yellow show strains for which no significant correlation was observed (p > 0.05, two-tailed test).
(B) Initial susceptibility levels for five DK2 isolates selected for the adaptive evolution experiment. Data are presented as the MIC fold change relative to the DK2 strain not exposed to antibiotics. (C) Collateral sensitivity and resistance during adaptive evolution for DK2 drug resistant strains. The MIC values were normalized to the baseline susceptibilities of each WT for evolved DK2 isolates (WTE) (Table S1).
To further examine the phenotypic states of the resistant strains, we reduced the dimensionality of the data using principal component analysis (PCA). This analysis revealed that resistant strains are divided into four groups that are positioned in different regions of PCA (Figure 2A), highlighting the convergence toward specific phenotypic states in response to antibiotic exposure. β-lactams, aminoglycoside, quinolone, and polymyxin antibiotics are commonly applied for the treatment of lung infections in CF patients. Notably, strains resistant to these antibiotic classes were positioned in different regions of PCA indicating that understanding the convergence toward drug-specific phenotypes for these drugs could inform treatment strategies. Interestingly, region II in the PCA plot included strains resistant to quinolones, macrolides, and tetracyclines (Figure 2A), highlighting that these different drug classes select for similar phenotypic states with strongly correlated susceptibility profiles (ρ = 0.84 – 0.97, p < 0.0001, Spearman correlation) (Figure S3A; Table S3).
Figure 2.

Drug Resistance Evolution Converges to Structural Phenotypic States
(A) Principal component analysis of the susceptibility profiles of evolved antibiotic resistance PAO1 strains reveals clustering of resistance phenotypes to specific phenotypic states. Principal component axes obtained from the PAO1 WTE normalized susceptibility data for the resistant PAO1 strains. Color coding depicts the drug classes listed in Table 1.
(B) Correlation analysis for PAO and DK2 susceptibility profiles. Exposure of different P. aeruginosa strains to a particular drug tends to increase the correlation between evolved antibiotic-resistant DK2 strains and PAO1 strains. Spearman’s correlation coefficients (ρ) summarize the pairwise correlative relationship between the altered susceptibility of drug resistant strains. Circle size represents the strength of ρ (Table S4).
(C and D) Shifts in phenotypic states toward specific group collateral states drug-resistant DK2 strains toward their corresponding resistant PAO1 strain. Space plot of two principal component axes obtained from the PAO1 WTE normalized and log2 transformed susceptibility data for DK2 WTE and DK2 ciprofloxacin-resistant strain (C) or aztreonam-resistant strain (D).
See also Figure S3.
Adaptive Evolution of Clinical Isolates Leads to Collateral Sensitivity
To explore further the phenotypic convergence in response to antibiotic exposure, we investigated whether antibiotic resistance evolution from different genetic starting points would lead to convergent evolution of their susceptibility profiles. We selected five clinical CF isolates from the DK2 clone type that share a common ancestor but have diverged during years of isolation in three different hosts (Marvig et al., 2013). We observed changes in susceptibility for all adapted clinical isolates, indicating that evolutionary trajectories toward collateral sensitivity can occur in divergent lineages (with diverse phenotypic and genotypic starting point) (Figure S3B). Importantly, several collateral sensitivity interactions remained preserved in the majority of strains tested (Figure S3C). For instance, resistance development for ciprofloxacin was consistently associated with collateral sensitivity toward aminoglycoside antibiotics in all different genetic backgrounds. In addition, the fold change relative to the ancestral strains was higher in clinical isolates than observed in PAO1 evolution. Notably, the clinical isolates evolved to azithromycin and ciprofloxacin resistance (173-1991-CIP and 173-1991-AZY) were 32-fold more sensitive to colistin antibiotics then the WT (Figure S3C; Table S1). Profound collateral sensitivity was also previously observed for E. coli clinical isolates (Imamovic and Sommer, 2013), indicating the potential for exploiting collateral sensitivity to optimize antibiotic regimens.
Resistance Evolution of Clinical Isolates Converges to Conserved Collateral States
To explore the link between the phenotypic changes of laboratory-evolved resistant PAO1 and clinical isolates, we determined the Spearman correlation coefficients for their resistance profiles. Overall, this analysis shows that exposure of different P. aeruginosa strains to a particular drug tends to increase the correlation between them (Figure 2B; Table S4). When considering selective antibiotic pressure in clinical isolates, all clinical isolates exposed to aztreonam and ciprofloxacin as well as 80% of azithromycin- and tobramycin-resistant DK2 strains increased the correlation of their susceptibility profiles to the respective antibiotic-resistant PAO1 strains (Figure 2B). For instance, the correlation coefficient for clinical isolate 173 evolved to ciprofloxacin changed from a negative ρ = −0.67 (p < 0.01) to a positive ρ = 0.65 (p < 0.01) (Table S4). The exception was the colistin-resistant strain, for which no changes in correlation ρ were linked to the colistin-resistant PAO1 strain, indicating that the difference with PAO1 might be reflected in the different genetic background between DK2 and PAO1 (Gutu et al., 2015). Using PCA we observed that the phenotypic states of the ciprofloxacin and aztreonam evolved clinical isolates shifted their corresponding resistant PAO1 strain in response to antibiotic resistance evolution (Figures 2C and 2D). Indeed, this analysis indicates that for some antibiotics, exposure and subsequent resistance evolution leads to convergence toward specific phenotypic states in diverse phenotypic and genotypic backgrounds.
Genetic Determinants Involved in Collateral Sensitivity and Resistance
To explore the genetic basis of phenotypic changes in susceptibility profiles, we sequenced the genomes of experimentally evolved strains. We observed that the impact of drug exposure on genome-wide evolutionary paths causing collateral resistance and sensitivity were associated with drug resistance genes also found to be undergoing selection in chronically infected CF patients (Marvig et al., 2013). Mutations in seven out of nine pathoadaptive, antibiotic resistance genes (fusA1, ampC, ampD, gyrA, gyrB, mexB, and pmrB) were observed in our adaptive evolution experiment (Figure S4A; Table S5). Uniformly, PAO1 strains resistant to quinolone, macrolide, and tetracyclines had mutations in the pathoadaptive gene nfxB (Marvig et al., 2015a, Marvig et al., 2015b) (Figure S4A). nfxB is a negative transcriptional regulator of MexCD-OprJ efflux (Poole et al., 1996). In addition, as observed for resistant PAO1 strains, we detected nfxB mutations in all five ciprofloxacin-resistant DK2 lineages and three out of five azithromycin-exposed lineages (Table S5).
Figure S4.

Mutational Events Leading to Drug Resistance for PAO1 and Changes in Proteome in Drug-Resistant Strains Sharing nfxB Mutation, Related to Figure 3
(A) Mutational events leading to drug resistance for PAO1 (Table S5). The order of the drugs and resistant strains was determined by hierarchical clustering using the shared mutation as a value for the distance measure. Antibiotic and class abbreviations are listed in Table 1.
(B) Changes in proteome in drug ciprofloxacin and azithromycin resistant strains harboring nfxB mutation. Fold change for specific proteins was calculated based on the level of the proteins in WTE sample. t test were preformed to determine the ratio of proteins with significantly altered abundance (95% confidence). Only proteins with at least 1.5-fold increase (_P_-value < 0.05, t test) were reported.
(C) MexCD-OprJ efflux is negatively regulated by NfxB repressor binding upstream of mexC gene.
(D) Mutation in nfxB lead affects repressor binding leading to expression of MexCD-OprJ efflux system. Expression of MexCD-OprJ efflux system resulted in increased abundance of MexC protein leading to collateral sensitivity and resistance.
To test whether nfxB mutations were indeed associated with collateral sensitivity and resistance, thereby leading to a selective disadvantage during exposure to particular drugs such amikacin or minocycline, we conducted a competition experiment between the PAO1 WT strain and a nfxB 119C > T mutant (azithromycin evolved PAO1). We evaluated the frequency of the nfxB mutation from each mix population exposed to amikacin and minocycline drugs for which the nfxB 119C > T mutant was collaterally sensitive or collaterally resistant, respectively. From these experiments, we observed that the strain carrying the nfxB mutation was selected against when treated with the collateral sensitivity drug amikacin (Figure 3A). As expected, the selective survival the nfxB 119C > T mutant was observed when the strains were exposed to the collateral resistance drug azithromycin (Figure 3B).
Figure 3.

Genetic Basis for Collateral Resistance and Collateral Sensitivity
(A) Competition experiment depicting the survival of WT over the resistant strain harboring the nfxB mutation when treated with collateral-sensitive antibiotic.
(B) The selective survival of resistant strain and eradication of the WT was observed in competition experiment when treated with collateral-resistant antibiotic
(C) MexC abundance in WTE, ciprofloxacin- and azithromycin-resistant strains. Data are presented as the means of three biological replicates and error bars represent SD. Significance levels indicate the p value of the t test (Table S6).
(D) nfxB mutations in populations at after gyrA mutation during adaptive evolution to ciprofloxacin.
(E) nfxB mutations at the end of the adaptive process for azithromycin exposed bacterial populations.
(F) Changes in susceptibility profiles for 173-2005 relative to ancestral clinical isolated (30-1979) and upon further resistance development to CIP and AZY (Table S1). Dashed lines mark the EUCAST clinical resistance breakpoints (Table 1).
See also Figure S4.
To further study the mechanisms associated with _nfxB_-induced collateral sensitivity, we conducted a proteomic characterization of the resistant strains evolved to azithromycin and ciprofloxacin resistance that harbored nfxB mutations. The MexC transporter protein, was the most upregulated protein in both strains (t ratio 4.2 for ciprofloxacin-resistant and 6.2 for azithromycin-resistant strains; p < 0.01) (Figures 3C and S4B). The increased abundance of the MexC protein suggested that the manipulation of drug efflux transporters could provide improved treatment effects by enhancing selective bacterial vulnerabilities. Importantly, nfxB is the only known regulator of MexCD-OprJ efflux (Poole et al., 1996) and mutations that lead to a truncated, incomplete, and non-functional NfxB protein would affect the repressor binding capacities (Figures S4C and S4D) suggesting that nfxB gene mutations could be a potential sequence-based biomarker indicating collateral sensitivity.
nfxB Mutations Accumulate Late during Resistance Evolution
A stepwise acquisition of the mutations that confer low-level resistance can lead to increased resistance to antibiotics (Solé et al., 2015) and, consequently, poorer treatment outcomes (Falagas et al., 2012). However, if such mutations also lead to collateral sensitivity, it may be possible to select against such resistance evolution through rational drug treatment. To assess this, ciprofloxacin- and azithromycin-treated populations were monitored for the presence of nfxB mutations at day 3, 6, and 8 during the adaptive evolution experiment. Day 8 was chosen because the treated populations reached maximal levels of resistance (Figure S1D). The first mutation detected in the ciprofloxacin-treated population was at day 6 in gyrA (248C > T) at a frequency of 12%, while no mutations were detected in nfxB (Figure 3D). Finally, at day 8, the gyrA mutation was present in 99.2% of the population, and three different nfxB were detected in the population at similar frequencies (Figure 3D). In populations exposed to azithromycin, four different mutations emerged, of which the mutation _nfxB_115A > C was detected in half of the total population at day 8 (Figure 3E). No other mutations were detected for the azithromycin-exposed strains (Table S5).
The tendency to fix nfxB mutations in populations at later stages of the adaptive evolution of ciprofloxacin resistance (after gyrA mutation) (Figure 3D) could suggest that strains harboring only a mutation in gyrA have the potential to acquire further mutations and change strain susceptibility profiles. To establish the association of collateral sensitivity with the nfxB mutations that emerged at the end of the adaptive process, we compared the PAO1 observations with selected the clinical isolate (173-2005). Strain 173-2005, which carried a mutation in the gyrA gene, also conferred elevated resistance to ciprofloxacin (Yang et al., 2011). Indeed, when the 173-2005 strain was exposed to ciprofloxacin for 10 days, the point mutation was detected in the nfxB gene (Table S5), and the MIC for ciprofloxacin increased further from 0.5 to 32 μg.mL−1 (Figure 3F). Consequently, resistant strains with altered nfxB gene also exhibited collateral sensitivity toward several different antibiotic classes (Figures 1A and S4A). This suggests that treatment strategies exploiting collateral sensitivity could also be applied to highly resistant strains to sensitize them for more effective treatment.
Drug Sensitivity Oscillations in Longitudinal P. aeruginosa Isolates
The current thinking on resistance evolution anticipates that pathogens causing chronic infections become increasingly resistant in response to antibiotic treatment. Yet, our findings of widespread collateral sensitivity interactions among clinically applied drugs would suggest that the resistance profiles of chronic infecting bacteria would fluctuate over time in response to different drug exposures. To test this hypothesis, we determined changes in drug susceptibility for longitudinally collected clinical isolates of the transmissible DK2 spanning a sampling period of over three decades, and corresponding to more than 200.000 generations from early and late chronic infection (Marvig et al., 2013, Yang et al., 2011). We selected isolates to represent particularly illustrative examples of population dynamics for the DK2 lineage with temporal and spatial heterogeneity (CF patients 173, 211, and 333). In addition, we also included the 30-1979 DK2 strain, which was previously determined to be the closest common ancestor of the DK2 lineage (Yang et al., 2011). For each longitudinal isolate, we determined changes in susceptibility profiles toward 22 drugs relative to the ancestral strain (30-1979) (Figure 4A; Table S1). In agreement with our hypothesis, we observed oscillatory dynamics in the resistance levels of the lineages sampled (Figure 4A). Importantly, we did not observe significant absolute increases in the resistance levels of isolates from patients across the decades of the longitudinal sampling. Indeed, the last longitudinal isolate analyzed for patient 333 exhibited increased susceptibly toward 14 drugs relative to the first isolate taken more than 15 years earlier (Figure 4A). Decreases in MICs were also observed for several antibiotics from the aminoglycoside, quinolone, and tetracycline drug classes in the two strains isolated in sub-lineage A (173-1991) and sub-lineage C (173-2005) (Figure 4A). Interestingly, the 173-1991 and 173-2005 isolates were the last isolates detected from each sub-lineage, suggesting that this reduction in MIC could be linked to their eradication.
Figure 4.

Oscillatory Dynamics in Susceptibility Profiles from P. aeruginosa Chronically Infected CF Patients
(A) Susceptibility profiles for clinical isolates obtained by longitudinal sampling of DK2 clinical isolates from three CF patients chronically infected by P. aeruginosa. For each strain, MIC values were determined toward 22 drugs. Drug susceptibility was determined based on the average values of five replicates. MIC values were normalized to the baseline susceptibilities of the immediate common ancestor of the DK2 isolates (isolate 30-1979) (Table S1).
(B) A heatmap of Spearman’s correlation coefficients (ρ) summarizes the pairwise correlative relationship between the altered susceptibilities of the experimentally evolved resistant PAO1 strains and DK2 isolates from CF patients. The upper-right color panel is an indicator of the Spearman correlation coefficient (ρ). Circle size represents the strength of ρ. Only statistically significant correlations are shown (p > 0.05, two-tailed test) (Table S6).
Phenotypic Convergence of Laboratory-Evolved Strains and Clinical Isolates
To investigate potential associations between susceptibility profiles in the laboratory-evolved strains and clinical isolates, we calculated the Spearman correlation coefficients between their susceptibility dynamics of the clinical isolates and the collateral sensitivity and resistance profiles of the laboratory-evolved strains (Figure 4B). A Spearman covariance matrix revealed 38 significant correlations between clinical isolates and one or more of the resistant PAO1 strains (p < 0.05, a two-tailed significance test) (Figure 4B; Table S6). Notably, several clinical isolates with high positive correlation coefficients to resistant PAO1 strains exhibited increased susceptibility toward different antibiotic classes. Such changes in resistance dynamics were particularly pronounced for the last isolate from patient 333 (333-2007c). This strain had increased sensitivity toward drugs from the quinolone, β-lactam, fosfomycin, and rifampicin drug classes (Figure 4A) and had a strong positive correlation to the colistin-resistant PAO1 strain (ρ = 0.6; p < 0.01) (Figure 4B; Table S6). In addition, negative correlation between clinical isolates and one or more of the resistant PAO1 strains (p < 0.05) was also observed (Figure 4B). Between PAO1-resistant strains strong negative correlation coefficients were observed for antibiotics that had reciprocal collateral sensitivities (e.g., colistin and aztreonam) (Figure S3A) indicating incompatible drug resistance pathways.
Phenotypic Convergence during Antibiotic Therapy In Vivo
To assess whether collateral sensitivity might modulate population dynamics of a chronic P. aeruginosa population in the CF lung, we studied a chronically infected CF patient during a 2-week course of intensive antibiotic treatment. We performed a comprehensive phenotypic screen of 626 clinical isolates from sputum samples of a CF patient before (t1), during (t2), and at the end of intensive antibiotic therapy (t3) (see STAR Methods). Notably, the number of colonies that could be cultivated from the sputum sample t3 was significantly reduced compared to t1 and t2 (p < 0.01, one-way ANOVA) (Figure 5A), indicating effective antibiotic therapy for chronic CF lung infection. For all isolates recovered, we examined their phenotypic diversity based on antibiotic susceptibility for six antibiotics from the quinolone, β-lactam, aminoglycoside, and polymyxin classes (Figures 5B–5E; Table S7). The population before treatment (t1) exhibited the highest phenotypic diversity with regards to antibiotic susceptibility, which decreased significantly during the course of antibiotic treatment by 7-fold (p value = 6.125e−12, pairwise comparison of Euclidian distance using t test) (Figure 5B). Interestingly, the 275 isolates from the diverse population selected before intensive antibiotic treatment (t1) comprised both strains susceptible and strains resistant to all six antibiotics tested (according to EUCAST clinical breakpoints) (Figures 5C and S5A–S5F). For instance, different levels of colistin resistance were detected before treatment (e.g., isolates that were both 4-fold above and 7-fold below EUCAST clinical breakpoints for colistin were detected) (Figures 5C and S5A). Yet, all isolates from the end of antibiotic treatment (t3) exhibited MIC 4-fold below EUCAST clinical breakpoints for colistin (Figures 5E and S5A). Of note, collateral sensitivity toward colistin in laboratory evolution experiment was a consequence of resistance development to β-lactam and aminoglycoside antibiotics (Figure 1A), all of which were applied in the treatment of the studied CF patient. The observed selection for isolates susceptible to colistin suggest that this collateral sensitivity interaction is relevant when treating chronic lung infections in CF patients.
Figure 5.

Shift in Susceptibility Profiles during Intensive Antibiotic Treatment In Vivo
(A) Recovery of P. aeruginosa from sputum on selective media. Plating was done in five replicates for samples t1 and t2. For sample t3, 12 agar plates were used to recover P. aeruginosa. The line dots represent the average and error bars represent SD.
(B) Decrease in overall phenotypic diversity during the course of antibiotic treatment. Change in phenotypic diversity was calculated based on Euclidian distance between the normalized values for susceptibility profiles.
(C–E) Sensitivity profiles during antibiotic treatment of CF patient. Heatmap represents quantification of the drug response for single isolates obtained before (C), during (D), and at the end of treatment (E). Antibiotic and class abbreviations are listed in Table 1. Color coding represents the fold above (yellow) or below (blue) the isolate MIC value relative to the EUCAST clinical breakpoints (Table 1). Drug susceptibility was determined based on the average values of five replicates (Table S7). The order of isolates was determined by hierarchical clustering using the similarity of normalized MIC values as the distance measure.
(F–H) Space plot of two principal component axes obtained from the 626 clinical isolates normalized susceptibility data for the EUCAST resistance breakpoints. Color coding depicts the sampling time points before (F), during (G), and at the end of (H) treatment.
See also Figure S5.
Figure S5.

Phenotypic Convergences for Heterogeneous P. aeruginosa Population during Antibiotic Treatment, Related to Figure 5
(A–F) Distribution plots for susceptibility profiles of clinical isolates toward six clinically relevant antibiotics.
(G) PCA plot for antibiotic susceptibility of clinical isolates obtained before (t1), during (t2) and at the end (t3) of intensive antibiotic treatment of CF patient.
(H) PCA plot for quinolone resistant strains before (t1-b) and at the end of intensive antibiotic treatment (t3) for CF patient. For all panels, normalized MIC values were used as data input. MIC or the inhibitory concentration was defined as the lowest concentration of the drug that inhibited 90% of the growth of the strain tested. For each strain, five replicates were performed to determine the drug susceptibility. All MIC data were normalized to EUCAST resistant breakpoint values and log2 transformed.
Since the laboratory evolved strains converged toward specific phenotypic states (Figure 2A), we also applied PCA to characterize the phenotypic convergence of P. aeruginosa populations during in vivo treatment. Interestingly, we observed a striking phenotypic convergence of the characterized isolates during treatment (Figures 5F–5H and S5G). While isolates obtained before the intensive treatment were found in all four regions (I–IV) of the PCA plot (Figures 5F and S5H), the isolates at the end of the treatment converged to one region of PCA plot (region II) (Figure 5H) that represented a phenotypically uniform population highly susceptible to colistin (Figures 5E and S5A).
Antibiotic Therapy Selects for Genetically Distinct Subpopulations with Specific Phenotypes
Based on the observation that antibiotic treatment converges a phenotypically diverse population to a uniform antibiotic susceptibility phenotype (Figures 5F–5H), we decided to genetically characterize these populations. The majority of isolates sampled at t1 clustered in two subpopulations that were characterized by different levels of susceptibilities to fluoroquinolone antibiotics (Figure 5C). Accordingly, we separated the t1 population into quinolone susceptible (t1-a) and quinolone-resistant isolates (t1-b) based on the EUCAST breakpoints (Table 1). Given that both populations t1-b and t3 were characterized by a high level of quinolone resistance, we were interested whether population t3 was related to the quinolone-resistant subpopulation t1-b. We compared individual and shared mutations detected (Table S8) among different populations and found that 92.3% of the SNPs detected in population t3 were also observed in subpopulation t1-b (Figure 6A; Table S9). This finding suggested that population t3 and a subset of t1-b population originate from the same lineage. On the other hand, only 10 SNPs detected in t3 population were shared with quinolone susceptible populations t1-a and t2. Notably, both populations (t1-a and t2) that were more susceptible to quinolone drugs shared 98.6% of mutations (Figure 6A; Tables S8 and S9), which suggests that these populations originated from the same lineage. Emergence of the quinolone-resistant t3 population descendent from a small subset t1-b was observed after drug treatment was switched from ceftazidime to levofloxacin (Figures 5C–5E). The phylogenetic relatedness between the different populations determined by population sequencing was also supported by whole-genome sequencing of nine individual isolates selected from different subpopulations (Figure 6B; Table S8). We observed that isolates from t1-b-A and t3-J populations were DK2 type, while single isolates from t-1-a and t2 were different clone type (Marvig et al., 2015a).
Figure 6.

Population Switch during Antibiotic Treatment In Vivo
(A) Population sequencing of clinical isolates. Individual and shared mutations were plotted and used to evaluate population divergence during treatment. Variable percentage of shared mutations was observed among different subpopulations before (t1-a and t1-b), during (t2), and at the end of intensive treatment (t3) (Table S9). By calculating that majority mutations found in quinolone-resistant population t3 subpopulation are shared with t1-b, population divergence was estimated. Different color coding represent mutation shared by different subpopulations
(B) Genotype distance and susceptibility profiles among selected isolates. The order of isolates was determined by hierarchical clustering using the shared mutation as a value for the distance measure. Antibiotic and class abbreviations are listed in Table 1. Color coding represents the fold increase or decrease in MIC value relative to the EUCAST clinical breakpoints (Table 1). An average of five replicates were tested to determine the drug susceptibility (Table S1).
(C) nfxB mutation frequency in quinolone-resistant subpopulations t1-a and t1-b.
(D) Loss of resistant isolates harboring nfxB after exposure to β-lactam and aminoglycoside antibiotics during treatment of CF patient.
Selection against nfxB Mutants during Antibiotic Treatment in the CF Lung
Our in vitro work demonstrated that nfxB mutations lead to an overexpression of the MexC transporter (Figure 3C) leading to collateral sensitivity toward aminoglycosides, β-lactams, and colistin (Figure 1A). We showed that nfxB 119C > T mutants where outcompeted by WT during exposure to the aminoglycoside amikacin. Accordingly, we speculated that nfxB mutations present in the t1 population might also be eradicated during the intensive antibiotic therapy in the studied CF patient. Accordingly, we mapped the reads from population sequencing data from t1-a and t1-b to the nfxB gene to identify putative collateral sensitivity mutations. We found that 37.3% of t1-b subpopulation harbored a mutation in the nfxB gene (_nfxB_245G > T) (Figure 6C). Although the t1-b subpopulation had overlapping phenotypic and genotypic similarities with the population that emerged at the end of treatment (t3), none of the isolates in populations obtained at t1-a, during (t2), or at the end (t3) of antibiotic treatment harbored mutations in nfxB gene (Figure 6D). Loss of isolates with nfxB mutations followed after exposure to β-lactam and aminoglycoside antibiotics toward which resistant PAO1 and DK2 strains harboring nfxB mutations were collateral sensitive (Figures 1A and S4A). This observation suggests that the _nfxB_245G > T mutation is conferring collateral sensitivity in vivo in a similar manner as observed in lab evolved strains. Furthermore, this finding supports the notion that treatment of chronically infected CF patients could be individualized based on specific diagnostic markers that are associated with a collateral sensitivity to specific drugs.
Discussion
Chronic lung infections caused by P. aeruginosa are challenging to treat due to the ability of this opportunistic pathogen to persist and develop resistance during treatment (Poole, 2011). In this study, we show that evolution of resistance was associated with up to 8- and 32-fold reductions in MICs toward other antibiotics for PAO1 and DK2, respectively. This level of collateral sensitivity could have substantial clinical impact for the management of chronic infections, in which patients are exposed to several rounds of antibiotics, or even lifelong antibiotic therapies, as is the case for CF patients (Johansen et al., 2004). Overall low MIC values within the susceptibility range are associated with better treatment outcomes and lower mortality rates (Falagas et al., 2012). For instance, a study on the impact of carbapenem MIC values on hospital mortalities revealed that for each 2-fold increase in the MIC value, the probability of death increased by 2-fold (Esterly et al., 2012). Accordingly, our findings indicate that treatment of chronic infections can be optimized through the rational deployment of drugs based on their collateral sensitivity and convergence toward distinct phenotypic states. For instance, our results suggest that application of ciprofloxacin results in subsequent resistance development and phenotypic convergence. Ciprofloxacin resistance could enhance the action of tobramycin, since the collateral sensitivity was observed in the resistant PAO1 and DK2 strains. Similarly, colistin action could be enhanced in bacteria that developed resistance toward ciprofloxacin or aztreonam. Collateral sensitivity observed toward colistin, an important drug for treatment of P. aeruginosa infections, additionally supports exploitation of collateral sensitivity to counter drug resistance.
Interestingly, we find that collateral sensitivity can evolve in response to exposure to drugs for which the organism is already resistant. This finding should be considered given the frequent polymicrobial infection of the CF airways. Indeed, P. aeruginosa may be exposed to drugs administered to target other organisms, such as S. aureus. We show that exposure to several chemical classes of drugs, used in CF patient therapy (tetracycline or macrolide) for other species than P. aeruginosa (Gibson et al., 2003), can modulate collateral sensitivity and resistance in P. aeruginosa. Indeed, we observed phenotypic convergences toward collateral states for DK2 strains treated with antibiotics regardless of initial resistance level. Thus, we hypothesize that by using commonly applied antibiotics according to a specific schedule, even highly resistant strains could be sensitized to enhance the treatment efficacy.
The genetic changes that are selected for during chronic infections by P. aeruginosa of the CF airways are being elucidated through recent clinical genome sequencing projects that have led to the identification of pathoadaptive genes (Marvig et al., 2013, Marvig et al., 2015a, Marvig et al., 2015b). Notably, there is a substantial overlap between the pathoadaptive genes that have been identified in longitudinal clinical sequencing studies and in our studies, suggesting that some pathoadaptive mutations are linked to collateral sensitivity. For instance, mutations in the nfxB that confers resistance to the quinolones, are frequently encountered in clinical isolates (Marvig et al., 2015a, Marvig et al., 2015b). We find that nfxB mutations are associated with collateral sensitivity driven by resistance evolution to several different drug classes and phenotypic convergence. nfxB mutants were eradicated from the lung of a chronically infected CF patient during treatment with aminoglycoside and beta-lactam drugs leading to the eradication of a quinolone-resistant subpopulation. These findings indicate that antibiotic treatment of chronic infections can be optimized by targeting specific mutations associated with collateral sensitivities and converged phenotypic states. Accordingly, we speculate that nfxB gene mutations or MexC protein abundance could be monitored in the clinic and potentially serve as a genomic or proteomic biomarker for collateral sensitivity.
Based on the analysis from in vivo evolved population, it seems likely that antibiotics could be grouped for cycling approaches to improve treatment success in chronic infections. Such treatment could be applied jointly to enhance the drug effect against susceptible isolates for treatment of infection against heterogeneous populations observed in chronically infected patients (Foweraker et al., 2009). Besides reducing the bacterial load, we also observed that such treatment lead to convergence to less heterogeneous populations with more uniform phenotype. We observed that, at the end of treatment, population was uniformly susceptible to colistin. This indicated that tailored treatment based on preserved sensitivity interactions could benefit infection management in treatment of CF patient infections and potentially lead to more complete eradication of the infecting population. Such treatment could be individualized based on specific genomic or proteomic biomarkers such as nfxB or MexC, which we have found to be linked to collateral sensitivity phenotypes. Future studies will likely identify additional genetic markers of collateral sensitivities, enabling improved and personalized treatment of chronically infected patients.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Pseudomonas aeruginosa PAO1 | Søren Molin lab | N/A |
| Pseudomonas aeruginosa DK2 | DK2 Collection (Marvig et al., 2013) | N/A |
| Biological Samples | ||
| Cystic fibrosis sputum | Helle Krogh Johansen | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Amikacin | Sigma-Aldrich | Cat No. A2324 |
| Gentamicin | Sigma-Aldrich | Cat No. G1264 |
| Tobramycin | Sigma-Aldrich | Cat No. T4014 |
| Ciprofloxacin | Sigma-Aldrich | Cat No. 17850 |
| Levofloxacin | Sigma-Aldrich | Cat No. 28266 |
| Ampicillin | Sigma-Aldrich | Cat No. A9518 |
| Piperacillin | Sigma-Aldrich | Cat No. P8396 |
| Carbenicillin | Sigma-Aldrich | Cat No. C1389 |
| Ticarcillin | Sigma-Aldrich | Cat No. T5639 |
| Aztreonam | Sigma-Aldrich | Cat No. A6848 |
| Cefepime | Sigma-Aldrich | Cat No. A3737 |
| Cefuroxime | Sigma-Aldrich | Cat No. C4417 |
| Ceftazidime | Sigma-Aldrich | Cat No. A6987 |
| Meropenem | Sigma-Aldrich | Cat No. M2574 |
| Imipenem | VWR | Cat No. ABCAAB141030-0 |
| Minocycline | Sigma-Aldrich | Cat No. M9511 |
| Doxycycline | Sigma-Aldrich | Cat No. D1822 |
| Azithromycin | Sigma-Aldrich | Cat No. 75199 |
| Erythromycin | Sigma-Aldrich | Cat No. E5389 |
| Clarithromycin | Sigma-Aldrich | Cat No. C9742 |
| Colistin | Sigma-Aldrich | Cat No. Y0000277 |
| Fosfomycin | Sigma-Aldrich | Cat No. 34089 |
| Rifampicin | Sigma-Aldrich | Cat No. R3501 |
| Trimethoprim | Sigma-Aldrich | Cat No. T7883 |
| Sulfamethoxazole | Sigma-Aldrich | Cat No. S7507 |
| Critical Commercial Assays | ||
| DNA Blood and Tissue Kit | QIAGEN | Cat No. 69504 |
| Nextera XT kit | Illumina | FC-131-1096 |
| TrueSeq Nano | Illumina | FC-121-4003 |
| Deposited Data | ||
| Raw genome sequence data | This study | PRJNA414086 |
| Raw proteomics data | This study | https://www.ebi.ac.uk/pride,ProteomeXchangePXD007972 |
| Antibiotic susceptibility data | This study | https://www.mendeley.com/sign-in/?routeTo=https%3A%2F%2Fapi.mendeley.com%2Foauth%2Fauthorize%3Fredirect_uri%3Dhttps%253A%252F%252Fdata.mendeley.com%252Fauth%252Fcallback%26scope%3Dall%26state%3D579590%26response_type%3Dcode%26client_id%3D1025 |
| Experimental Models: Organisms/Strains | ||
| Antibiotic resistant variants of PAO1 and DK2 | This study | N/A |
| Oligonucleotides | ||
| 2_mexD_nfxB-_up ACGCTGTTTCACCAGGGTAG | This study | N/A |
| 2_morA_nfxB-lp AGCATCAACAGGACCAGCAA | This study | N/A |
| Software and Algorithms | ||
| The collateral sensitivity drug cycle detection program | This study | https://github.com/MostafaEllabaan/DrugCyclesPrediction/blob/master/DrugCycleDetector.py |
| Cytoscape (3.1.0) | Shannon et al., 2003 | http://www.cytoscape.org/release_notes_3_1_0.html |
| R packages: PerformanceAnalytics, corrplot, FactoMineR, factoextra, ggdendro and ggplot2 | R software | https://www.r-project.org/ |
| PRISM 7.0a | GraphPad Software | https://www.graphpad.com/support/prism-7-updates/ |
| Progenesis QI for Proteomics versions 2.0 | Nonlinear Dynamics, A Waters Company | http://www.nonlinear.com/progenesis/qi-for-proteomics/v2.0/ |
| CLC Genomic Workbench 9.0.1. | CLC QIAGEN | https://www.qiagenbioinformatics.com |
| Other | ||
| Synthetic CF sputum | Palmer et al., 2007, Wong et al., 2012 | N/A |
Contact for Reagent and Resource Sharing
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Morten Otto Alexander Sommer (msom@bio.dtu.dk).
Experimental Model and Subject Details
Bacterial Strains and Growth Conditions
All experiments were performed in MHBII medium or Synthetic CF sputum (Palmer et al., 2007) supplemented with 5% mucin (Wong et al., 2012). PAO1 and DK2 drug-resistant phenotypes were selected as described below. Clinical isolates of the P. aeruginosa DK2 lineage were obtained from the Danish CF collection from patients 173, 211 and 333 (Marvig et al., 2013). The bacterial strains are described in Table S1.
CF Patient
Patient CF124 was 52 years old male suffering from cystic fibrosis genetic disorder. Patient was chronically infected with P.aeruginosa for 43 years at the collection date. Sputum sample was collected during routine diagnostic sampling from chronically infected CF patient. The local ethics committee at the Capital Region of Denmark Region Hovedstaden approved the use of the stored P. aeruginosa isolates: registration number H-4-2015-FSP. Patient gave informed consent.
At home, patient was receiving 600 mg each day of ceftizadime via inhalation. Patient was received to hospital during the period of acute exacerbation. First sputum sample was taken then (sample t1) and patient was treated with ceftazidime (2g, twice daily), azithromycin (250 mg, oral, once daily), meropenem (2 g, intravenously, twice daily) and tobramycin (400 mg, intravenously, once daily). On the day 7 of treatment, second sputum sample was taken (sample t2), and then the treatment was changes from ceftazidime to levofloxacin, twice daily (commercially available as Quinsar). Administration of other antibiotics remained the same following 7 days, at which point the third sputum sample was obtained (t3).
P.aeruginosa Isolation from Sputum Samples
Sputum samples were stored in 15%–20% glycerol at 80%. For each sampling point, 300 μL of stored sputum sample was plated onto large agar plates (area) containing selective P.aeruginosa media (Pseudomonas Isolation Agar, Sigma). Agar plates were incubated for 2 days (42 ± 2 h) at 37 °C. Single colonies were selected, grown in MHBII and stored at −80 °C for further phenotypic and genotypic characterization.
Adaptive Evolution Experiments
A single colony was grown overnight in synthetic CF sputum supplemented mucin (SCFM) media. Eight microliters of overnight culture were then inoculated into 1 mL of SCFM media containing the 2-fold dilution of antibiotics listed in Table 1. Populations were grown on an orbital shaker (180 rpm) at 37 °C in 24-well plates. After 22 hours, 8 μL of culture from the well with the highest concentration of antibiotics and showing minimal growth (as measured by an optical density of OD600 > 0.4) was transferred into 1 mL of fresh SCFM medium containing a 2-fold dilution of antibiotics. On day 8, the bacteria reached EUCAST levels of resistance. Thus, during day 9 and 10, antibiotic concentrations remained the same as on day 8. Final drug-treated cultures were frozen at −80 °C in 15% glycerol. The strains were then streaked on solid media (SCFM supplemented with 3.5% agar), and a single colony was selected, grown in liquid SCFM and stored at −80 °C for further phenotypic and genotypic characterization.
Method Details
Drugs
Drug working solutions were made from solid stock (Table 1). All drugs solutions were sterilized with 0.22 μm filters and stored at −20 °C until use.
Collateral Susceptibility Profiles and MIC Determination
Drug-resistant strains were streaked on Mueller-Hinton Agar (MHA) (Sigma) and incubated at 37 °C. The duration of incubation was variable for different strains. All PAO1 strains were incubated overnight (18 ± 2 h), while DK2 strains were incubated for 2 days (42 ± 2 h) due to slower growth. A single colony was selected and grown in Mueller-Hinton Broth II (MHBII) (Sigma). After overnight incubation (for the PAO1 strains) and 2-day incubation (for the DK2 strains) at 37 °C and 180 rpm, bacterial cultures were used for MIC testing. Approximately 1x105 cells per well were inoculated in 96-well plates. For each strain, 5 sets of experiments were performed to determine the drug susceptibility. The percentage of inhibition was calculated according to the following formula: 1 − [_A_600 drug/_A_600 untreated control]. The inhibitory concentration was defined as the lowest concentration of the drug that inhibited 90% of the growth of the strain tested (MIC) relative to the WTE.
Competition Experiments
Overnight cultures of WT and azithromycin-exposed PAO1 strain were inoculated 1:1 to approximately 5x105 cell/well into 96-well plates. Mixed cultures were treated with 2 μg.ml-1 amikacin or 4 μg.ml-1 minocycline in MHBII. Control samples were not treated with antibiotics. Samples were incubated at 37 °C for 18 h. All experiments were performed in triplicates. Specific strain abundance was calculated based on amplicon sequencing described below.
Amplicon sequencing
Three replicates done for each completion experiment were joined in one sample and genomic DNA of mixed population was prepared using the DNA Blood and Tissue Kit following manufacturer instructions (QIAGEN). Two μg of DNA was used as template for PCR reaction to amplify the nfxB region. PCR primers (2_mexD_nfxB-_up ACGCTGTTTCACCAGGGTAG and 2_morA_nfxB-lp AGCATCAACAGGACCAGCAA) were designed to amplify full nfxB gene (564 bp) and surrounding region (full length PCR product size = 3021 bp). Specific PCR product was selected on 1% agarose gel. Gel-purified PCR product was use as DNA template for library preparation using the NexteraXT kit (Illumina). Amplicon sequences of mixed populations were obtained on the MiSeq platform.
Whole-genome Sequencing
Genomic DNA was prepared using the DNA Blood and Tissue Kit following manufacturer instructions (QIAGEN). Libraries were prepared using the NexteraXT kit (Illumina) and TrueSeq Nano (Illumina). Genome sequences of resistant strains were obtained on the MiSeq platform with a coverage of > 50 fold.
Variant Detection
The sequences were trimmed to exclude low-quality reads and reads less than 75 bp. The remaining reads were then mapped against the P. aeruginosa PAO1 (GenBank Accession number NC_002516.2) or DK2 (GenBank Accession number CP003149) reference genomes using Genomic Workbench 9.0.1. (CLC Bio, QIAGEN). Parameters were as follows: mismatch gap 2, mismatch count 3, similarity fraction 0.9 and length fraction 0.5.
Mutation Detection in the Genomic Data
Point mutations or short InDels were identified based on Quality-based variant detection approaches implemented in Genomic Workbench 9.0.1. Single-nucleotide variant detection was carried out using a neighborhood quality standard algorithm with a minimum neighborhood quality score of 15, a maximum gap penalty of 2 and a minimum variant base of 10. The minimum variant frequency was set to 80%. For population sequencing, the minimum variant frequency was set to 10%. For each mutation detected in population, presence of mutation below 10% was verified by additional analysis allowing detection of known low frequency variant (1%–10%). Mutation detected for populations from CF sputum sample was filtered based on 5% frequencies and minimum 10 reads coverage. Variants were filtered using an additional WT PAO1 sequenced strain. The background mutations for WT PAO1 are listed in Tables S5.
Sample Preparation for Proteomic Analysis
P. aeruginosa overnight cultures were diluted 1:100 in 10 mL of MHBII. After 10 h of incubation at 37 °C and 180 rpm, cells were collected by centrifugation, snap frozen on dry ice and stored at −80 °C. Cell preparation and proteomic analysis were performed as previously described (Bonde et al., 2016). First, 100 μL of urea (8 M, 75 mM NaCl, 50 mM Tris-HCl, pH 8.2) was added to the cell pellets. After this, two 3-mm zirconium oxide beads (Glen Mills, NJ, USA) were added, and the cells were disrupted using a Mixer Mill (MM 400 Retsch, Haan, Germany) for 2 min at 25 Hz. Following 30 min at 4°C, the cells were again subjected to 2 min in the mixer mill, after which an additional 100 μL of the urea solution was added. Following another cycle of 30 min at 4°C followed by 2 min at 25 Hz, the samples were centrifuged 10 min, and 100 μL of supernatant was collected and diluted with 400 μL of 25 mM ammonium bicarbonate. Samples were concentrated to 100 μL using a 3 kDa cutoff filter. Samples containing 100 μg of proteins in 50 μL of solution were used for digestion. Before adding 1 ug/sample trypsin, 5 μL of 100 mM DTT was added, and samples were kept at 37°C for 45 min. Subsequently, 10 μL of 100 mM iodoacetamide was added, and samples were kept in the dark for 45 min. Tryptic digestion was carried out for 8 h, after which 10 μL of 10% TFA was added, and samples were StageTipped using C18 (Empore, 3M, USA) according to a previously described procedure (Rappsilber et al., 2007).
NanoUPLC-MSE Acquisition
For Nanoscale LC analysis of the trypsin-digested samples, a nanoACQUITY system (Waters, USA) equipped with a Symmetry C18 5-μm, 180 μm × 20 mm precolumn and a nanoACQUITY BEH130 C18 1.7-μm, 75 μm × 250 mm analytical reversed-phase column (Waters, USA) was used. For each sample, 1 μg of protein was trapped on the precolumn using mobile phase A, consisting of 0.1% formic acid in water with a flow rate of 8 μL min−1 for 4 min. Mobile phase B consisted of 0.1% formic acid in acetonitrile. A reversed-phase stepped gradient was used to separate peptides: i) from 6% to 14% acetonitrile in water over 28 min, ii) from 14% to 25% acetonitrile in water over 40 min, iii) from 25% to 38% acetonitrile in water over 15 min, iv) from 38% to 60% acetonitrile in water over 10 min, and v) from 60% to 99% acetonitrile in water over 20 min. Between each injection, a 30 min wash method was applied. Both methods used a constant column temperature of 35°C and a flow rate of 250 nL min−1.
The described gradient data were acquired using a Synapt G2 (Waters, Manchester, UK) Q-ToF instrument operated in positive mode using electrospray ionization with a NanoLock-spray source. Using the internal fluidics system of the mass spectrometer, leucine enkephalin was used as a lock mass. The lock mass channel was sampled every 60 s. For each injection, the mass spectrometer was operated in resolution mode, with continuum spectra being acquired. During acquisition, the mass spectrometer alternated between low- and high-energy modes using a scan time of 0.8 s for each mode over 50–2,000 Da. In the low-energy MS mode, data were collected at a constant collision energy of 4 eV. In the elevated-energy MS mode, the collision energy was increased from 15 to 40 eV.
Protein Identification
Protein identification and quantification were obtained using Progenesis QI for Proteomics version 2.0 and the P. aeruginosa UniProt proteome database (ID: UP 208964). Settings for the PLGS search engine were FDR 1%, tryptic peptides with one missed cleavage allowed, and carbamidomethylation of cysteine residues as fixed modification and oxidation of methionine residues as variable modification. For quantification, only unique peptides of the proteins of interest were used, enabling comparisons of protein abundance across the different samples (Bantscheff et al., 2007).
Quantification and Statistical Analysis
The inhibitory concentration was defined as the lowest drug concentration that prevented 90% growth (MIC or IC90). The percentage of growth inhibition was calculated according to the following formula: 1 − [_A_600 drug/_A_600 control]. Data for the MICs are presented as the means of 5 independent replicas (±SD). Heatmaps showing susceptibility profiles and correlations were generated in Excel (14.6.1). Hierarchical clustering was performed using the similarity of normalized MIC values as the distance measured in R software using “ggplot2” and “ggdendro” packages. To measure the significance of the fold of increase in resistance between WTE and resistant strains we calculated growth inhibition of resistant strain toward 3 different antibiotics. Based on these results, we approximated the distribution of growth inhibition using the mean and standard deviation of the five replicates. We then generated 3.000 of simulations using the growth inhibition distributions. The concentration associated with 90% growth inhibition is then extracted for both WTE and resistant strain from the results simulated for each antibiotic. For the two vectors (strains) associated, we employed t test with alternative hypothesis “greater” to test the fold of increase between 1 and 10 with increasing factor of 0.5. The p value associated with each fold of increase was reported.
Networks connecting collateral sensitivity or resistance were explored and visualized using Cytoscape (3.1.0) (Shannon et al., 2003). To detect collateral sensitivity drug cycles, we have developed a software system using Python 2.7.2 that reads the drugs collateral sensitivity profiles into a dictionary data structure which links the drug with its corresponding collaterally sensitive drugs. A cycle is found if the first drug deployed maps to a certain number of drugs that consequentially collaterally sensitive to each other and the first drug in the cycle is collaterally sensitive to the last one drug. To determine the correlations between the susceptibility profiles of resistant strains, the MIC values were normalized to the WTE and log2 transformed. A Spearman’s correlation was chosen due to improved handling of non-linear correlations. The packages “PerformanceAnalytics” and “corrplot” in R software were used for analysis of correlation plots, covariance matrix. The packages “FactoMineR,” “factoextra” were used for PCA analysis. The ggplot2 plotting system was used for data visualization.
Protein identification and quantification were obtained using Progenesis QI for Proteomics versions 2.0 with only unique peptides of the proteins of interest. To obtain a uniform normalization across all samples from different batches, 47 ribosomal proteins detected in each dataset were used to introduce the data normalization factors. Data are presented as the means of 3 independent replicas with the SE of difference. The abundance of specific proteins was calculated based on the levels of the proteins in the WTE samples. t tests were performed using PRISM 7.0a to determine the ratio of the proteins with significantly altered abundance (95% confidence). Only proteins with at least a 1.5-fold change relative to the WTE strains (p > 0.05, t test) and with 3 identified unique peptides were reported as the altered proteome in Figure S4B.
Phenotypic population diversity was calculated for each pair of isolates in the population. For this purpose, the Euclidian distance between the normalized value for susceptibility profiles of 6 antibiotics was calculated. This procedure formed a vector that includes all the pairwise distances between isolates in the population. The confidence level was determined using t test at 95% confidence level of the two vectors considering the alternative = “greater.” To specify the fold of increase, the diversity-declining population was multiplied by an increasing factor and compare with the diversity of population of interest.
Data and Software Availability
The collateral sensitivity drug cycle detection program is available at https://github.com/MostafaEllabaan/DrugCyclesPrediction/blob/master/DrugCycleDetector.py. All genome sequence data are available in Sequence Read Archive (SRA) under submission PRJNA414086. Raw proteomics data are available via ProteomeXchange with identifier PXD007972.
Acknowledgments
We thank Rasmus Lykke Marvig, Lars Jelsbak, Mari Cristina Rodriguez de Evgrafov, and Eugene Flecher for discussion and suggestions; Daniel Simon, Ulla Rydahl Johansen, and Elio Rossi for help with the storage of sputum samples; and Anna Koza for work on library preparations for population sequencing. We acknowledge the use of Pseudomonas Genome Database web resource (http://www.pseudomonas.com/). This research was funded by the EU H2020 ERC-20104-STG LimitMDR (638902) and the Danish Council for Independent Research Sapere Aude Program DFF 4004-00213. H.K.J. was funded by a clinical research stipend from The Novo Nordisk Foundation, Rigshospitalet Rammebevilling 2015–17, and RegionH rammebevilling R144-A5287. M.O.A.S. acknowledges additional funding from the Novo Nordisk Foundation and The Lundbeck Foundation.
Author Contributions
Conceptualization, L.I. and M.O.A.S.; Methodology and Project Administration, L.I.; Investigations, L.I., A.M.D.M., and L.C.; Software, M.M.H.E., T.W., and L.I.; Writing – Original Draft, L.I. and M.O.A.S.; Writing – Reviewing and Editing, L.I. and M.O.A.S.; Supervision, L.I. and M.O.A.S.; Resources, H.K.J., S.M., and M.O.A.S.; Funding Acquisition, M.O.A.S.
Declaration of Interests
The authors declare no competing interests.
Published: January 4, 2018
Footnotes
Contributor Information
Lejla Imamovic, Email: lejim@bio.dtu.dk.
Morten Otto Alexander Sommer, Email: msom@bio.dtu.dk.
Supplemental Information
Table S1. Susceptibility Profiles, Related to Figures 1, 2, 4, and 6
MIC values were determined in MHBII based on five replicates. Concentration of antibiotics is given in μg.ml-1. MIC values were determined in two-fold broth dilution. Concentration that inhibited 90% strain growth was determined by using five sets of replicates. Concentration of antibiotic is given in μg.ml-1.
Table S2. Collateral Sensitivity Cycles, Related to Figure 1
All drugs were included in the analysis. No restriction was given to the number of cycles. For 24 drugs, max 11-drug cycle was discovered. Overall 5698 collateral sensitivity cycles were discovered with max 11 antibiotics in the cycle. In the table below only collateral sensitivity cycles with two and three drugs were listed. Antipseudomonal antibiotics were considered antibiotics for which EUCAST breakpoints (Table 1) were defined. Ten out of 13 antipseudomonal antibiotics exhibited collateral sensitivity. Based on that collateral sensitivity cycles were determined for this group. Five collateral sensitivity cycles were identified with antipseudomonal (labeled with ∗ in the table) and a maximum possible drug in the cycle was three.
Table S3. Correlation between Collateral Susceptibilities of PAO1 Drug-Resistant Strains, Related to Figure 2
Red shading indicates measurements that had positive and blue indicates negative Spearman correlation coefficient (ρ)
Table S4. Correlation between Collateral Susceptibilities of PAO1- and DK2-Resistant Strains, Related to Figure 4
All MIC data were normalized to PAO1 WT values and log2 transformed. Red shading indicates measurements that had positive and blue indicates negative Spearman correlation coefficient (ρ). Spearman ρ was calculated for each pair of variables for strains DK2 and PAO1 (A–E). Values in the row below ρ indicate _P_-values (a two-tailed significance test).
Table S5. Genetic Changes in PAO1- and DK2-Resistant Strains during Laboratory Evolution Experiment, Related to Figure 3
Details on genetic variants detected in PAO1 and DK2 resistant strains. For single isolates, variants had minimal allele frequency of 75%. For population sequencing during resistance development toward ciprofloxacin and azithromycin, the variants had minimal allele frequency (Frq.) of 10%. For each mutation detected in population, presence of mutation below 10% was verified by additional analysis allowing detection of known low frequency variant (1-10%). SNV – Single Nucleotide Variant; MNV – Multi Nucleotide Variant.
Table S6. Correlation between Collateral Susceptibilities of Experimentally Evolved Resistant Strains and DK2 Clinical Isolates, Related to Figure 4
All MIC data were normalized to PAO1 WTE values and log2 transformed. Red shading indicates measurements that had positive and blue indicates negative Spearman correlation coefficient (ρ). Spearman ρ was calculated for each pair of variables for strains DK2 and PAO1 (A-E). Values in the row below ρ indicate _P_-values.
Table S7. Antibiotic Susceptibility for Clinical Isolates from Sputum Sample, Related to Figure 5
MIC values for clinical isolates. MIC or the inhibitory concentration was defined as the lowest concentration of the drug that inhibited 90% of the growth of the strain tested. For each strain, five replicates were performed to determine the drug susceptibility. The MIC values were normalized to EUCAST breakpoints (Table 1) and log2 transformed.
Table S8. Sequencing of P. aeruginosa Single Isolates and Populations from Sputum Samples, Related to Figure 6
Details on genetic variants detected in subset of population during intensive antibiotic treatment of CF patient. The variants had minimal allele frequency (Frq.) of 5%. To call a mutation in the population, minimal read coverage was set to 10. The genetic variants detected in single isolates during treatment had minimal allele frequency of 75%. SNV – Single Nucleotide Variant; MNV – Multi Nucleotide Variant.
Table S9. Mutation Overlap between Subpopulations during In Vivo Antibiotic Treatment, Related to Figure 6
Mutation overlap in subset of population during intensive antibiotic treatment of CF patient (Table S8).
References
- Bantscheff M., Schirle M., Sweetman G., Rick J., Kuster B. Quantitative mass spectrometry in proteomics: A critical review. Anal. Bioanal. Chem. 2007;389:1017–1031. doi: 10.1007/s00216-007-1486-6. [DOI] [PubMed] [Google Scholar]
- Baym M., Stone L.K., Kishony R. Multidrug evolutionary strategies to reverse antibiotic resistance. Science. 2016;351:aad3292. doi: 10.1126/science.aad3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonde M.T., Pedersen M., Klausen M.S., Jensen S.I., Wulff T., Harrison S., Nielsen A.T., Herrgård M.J., Sommer M.O.A. Predictable tuning of protein expression in bacteria. Nat. Methods. 2016;13:233–236. doi: 10.1038/nmeth.3727. [DOI] [PubMed] [Google Scholar]
- Boucher H.W., Talbot G.H., Benjamin D.K., Jr., Bradley J., Guidos R.J., Jones R.N., Murray B.E., Bonomo R.A., Gilbert D., Infectious Diseases Society of America 10 x ’20 Progress—Development of new drugs active against gram-negative bacilli: An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2013;56:1685–1694. doi: 10.1093/cid/cit152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabot G., Ocampo-Sosa A.A., Domínguez M.A., Gago J.F., Juan C., Tubau F., Rodríguez C., Moyà B., Peña C., Martínez-Martínez L., Oliver A., Spanish Network for Research in Infectious Diseases (REIPI) Genetic markers of widespread extensively drug-resistant Pseudomonas aeruginosa high-risk clones. Antimicrob. Agents Chemother. 2012;56:6349–6357. doi: 10.1128/AAC.01388-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elborn J.S., Ramsey B.W., Boyle M.P., Konstan M.W., Huang X., Marigowda G., Waltz D., Wainwright C.E., VX-809 TRAFFIC and TRANSPORT study groups Efficacy and safety of lumacaftor/ivacaftor combination therapy in patients with cystic fibrosis homozygous for Phe508del CFTR by pulmonary function subgroup: a pooled analysis. Lancet Respir. Med. 2016;4:617–626. doi: 10.1016/S2213-2600(16)30121-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esterly J.S., Wagner J., McLaughlin M.M., Postelnick M.J., Qi C., Scheetz M.H. Evaluation of clinical outcomes in patients with bloodstream infections due to Gram-negative bacteria according to carbapenem MIC stratification. Antimicrob. Agents Chemother. 2012;56:4885–4890. doi: 10.1128/AAC.06365-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EUCAST 2016 The European Committee on Antimicrobial Susceptibility Testing. MIC Clinical breakpoints version 6.0. http://www.eucast.org/.
- Falagas M.E., Tansarli G.S., Rafailidis P.I., Kapaskelis A., Vardakas K.Z. Impact of antibiotic MIC on infection outcome in patients with susceptible Gram-negative bacteria: a systematic review and meta-analysis. Antimicrob. Agents Chemother. 2012;56:4214–4222. doi: 10.1128/AAC.00663-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodor A.A., Klem E.R., Gilpin D.F., Elborn J.S., Boucher R.C., Tunney M.M., Wolfgang M.C. The adult cystic fibrosis airway microbiota is stable over time and infection type, and highly resilient to antibiotic treatment of exacerbations. PLoS ONE. 2012;7:e45001. doi: 10.1371/journal.pone.0045001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkesson A., Jelsbak L., Yang L., Johansen H.K., Ciofu O., Høiby N., Molin S. Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: an evolutionary perspective. Nat. Rev. Microbiol. 2012;10:841–851. doi: 10.1038/nrmicro2907. [DOI] [PubMed] [Google Scholar]
- Foweraker J.E., Laughton C.R., Brown D.F., Bilton D. Comparison of methods to test antibiotic combinations against heterogeneous populations of multiresistant Pseudomonas aeruginosa from patients with acute infective exacerbations in cystic fibrosis. Antimicrob. Agents Chemother. 2009;53:4809–4815. doi: 10.1128/AAC.00269-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson R.L., Burns J.L., Ramsey B.W. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2003;168:918–951. doi: 10.1164/rccm.200304-505SO. [DOI] [PubMed] [Google Scholar]
- Gutu A.D., Rodgers N.S., Park J., Moskowitz S.M. Pseudomonas aeruginosa high-level resistance to polymyxins and other antimicrobial peptides requires cprA, a gene that is disrupted in the PAO1 strain. Antimicrob. Agents Chemother. 2015;59:5377–5387. doi: 10.1128/AAC.00904-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall M.D., Handley M.D., Gottesman M.M. Is resistance useless? Multidrug resistance and collateral sensitivity. Trends Pharmacol. Sci. 2009;30:546–556. doi: 10.1016/j.tips.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser A.R., Jain M., Bar-Meir M., McColley S.A. Clinical significance of microbial infection and adaptation in cystic fibrosis. Clin. Microbiol. Rev. 2011;24:29–70. doi: 10.1128/CMR.00036-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamovic L., Sommer M.O.A. Use of collateral sensitivity networks to design drug cycling protocols that avoid resistance development. Sci. Transl. Med. 2013;5:204ra132. doi: 10.1126/scitranslmed.3006609. [DOI] [PubMed] [Google Scholar]
- Jansen G., Mahrt N., Tueffers L., Barbosa C., Harjes M., Adolph G., Friedrichs A., Kreinz-Weinreich A., Rosenstiel P., Schulenburg H. Association between clinical antibiotic resistance and susceptibility of Pseudomonas in the cystic fibrosis lung. Evol. Med. Public Heal. 2016;2016:182–194. doi: 10.1093/emph/eow016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen H.K., Nørregaard L., Gøtzsche P.C., Pressler T., Koch C., Høiby N. Antibody response to Pseudomonas aeruginosa in cystic fibrosis patients: A marker of therapeutic success?--A 30-year cohort study of survival in Danish CF patients after onset of chronic P. aeruginosa lung infection. Pediatr. Pulmonol. 2004;37:427–432. doi: 10.1002/ppul.10457. [DOI] [PubMed] [Google Scholar]
- Lázár V., Pal Singh G., Spohn R., Nagy I., Horváth B., Hrtyan M., Busa-Fekete R., Bogos B., Méhi O., Csörgő B. Bacterial evolution of antibiotic hypersensitivity. Mol. Syst. Biol. 2013;9:700. doi: 10.1038/msb.2013.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.-Z., Plésiat P., Nikaido H. The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin. Microbiol. Rev. 2015;28:337–418. doi: 10.1128/CMR.00117-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister P.D., Wolter D.J., Hanson N.D. Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin. Microbiol. Rev. 2009;22:582–610. doi: 10.1128/CMR.00040-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Causapé C., Rojo-Molinero E., Mulet X., Cabot G., Moyà B., Figuerola J., Togores B., Pérez J.L., Oliver A. Clonal dissemination, emergence of mutator lineages and antibiotic resistance evolution in Pseudomonas aeruginosa cystic fibrosis chronic lung infection. PLoS ONE. 2013;8:e71001. doi: 10.1371/journal.pone.0071001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvig R.L., Johansen H.K., Molin S., Jelsbak L. Genome analysis of a transmissible lineage of pseudomonas aeruginosa reveals pathoadaptive mutations and distinct evolutionary paths of hypermutators. PLoS Genet. 2013;9:e1003741. doi: 10.1371/journal.pgen.1003741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvig R.L., Sommer L.M., Molin S., Johansen H.K. Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat. Genet. 2015;47:57–64. doi: 10.1038/ng.3148. [DOI] [PubMed] [Google Scholar]
- Marvig R.L., Dolce D., Sommer L.M., Petersen B., Ciofu O., Campana S., Molin S., Taccetti G., Johansen H.K. Within-host microevolution of Pseudomonas aeruginosa in Italian cystic fibrosis patients. BMC Microbiol. 2015;15:218. doi: 10.1186/s12866-015-0563-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May M. Drug development: Time for teamwork. Nature. 2014;509:S4–S5. doi: 10.1038/509S4a. [DOI] [PubMed] [Google Scholar]
- Mesaros N., Nordmann P., Plésiat P., Roussel-Delvallez M., Van Eldere J., Glupczynski Y., Van Laethem Y., Jacobs F., Lebecque P., Malfroot A. Pseudomonas aeruginosa: resistance and therapeutic options at the turn of the new millennium. Clin. Microbiol. Infect. 2007;13:560–578. doi: 10.1111/j.1469-0691.2007.01681.x. [DOI] [PubMed] [Google Scholar]
- Munck C., Gumpert H.K., Wallin A.I.N., Wang H.H., Sommer M.O.A. Prediction of resistance development against drug combinations by collateral responses to component drugs. Sci. Transl. Med. 2014;6:262ra156. doi: 10.1126/scitranslmed.3009940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill (2016). Tackling drug-resistant infections globally: Final report and recommendations. https://amr-review.org/sites/default/files/160518_Final%20paper_with%20cover.pdf.
- Palmer K.L., Aye L.M., Whiteley M. Nutritional cues control Pseudomonas aeruginosa multicellular behavior in cystic fibrosis sputum. J. Bacteriol. 2007;189:8079–8087. doi: 10.1128/JB.01138-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkins M.D., Floto R.A. Emerging bacterial pathogens and changing concepts of bacterial pathogenesis in cystic fibrosis. J. Cyst. Fibros. 2015;14:293–304. doi: 10.1016/j.jcf.2015.03.012. [DOI] [PubMed] [Google Scholar]
- Pittman J.E., Calloway E.H., Kiser M., Yeatts J., Davis S.D., Drumm M.L., Schechter M.S., Leigh M.W., Emond M., Van Rie A., Knowles M.R. Age of Pseudomonas aeruginosa acquisition and subsequent severity of cystic fibrosis lung disease. Pediatr. Pulmonol. 2011;46:497–504. doi: 10.1002/ppul.21397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole K. Pseudomonas aeruginosa: resistance to the max. Front. Microbiol. 2011;2:65. doi: 10.3389/fmicb.2011.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole K., Gotoh N., Tsujimoto H., Zhao Q., Wada A., Yamasaki T., Neshat S., Yamagishi J., Li X.Z., Nishino T. Overexpression of the mexC-mexD-oprJ efflux operon in nfxB-type multidrug-resistant strains of Pseudomonas aeruginosa. Mol. Microbiol. 1996;21:713–724. doi: 10.1046/j.1365-2958.1996.281397.x. [DOI] [PubMed] [Google Scholar]
- Rappsilber J., Mann M., Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007;2:1896–1906. doi: 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- Rodriguez de Evgrafov M., Gumpert H., Munck C., Thomsen T.T., Sommer M.O.A. Collateral resistance and sensitivity modulate evolution of high-level resistance to drug combination treatment in Staphylococcus aureus. Mol. Biol. Evol. 2015;32:1175–1185. doi: 10.1093/molbev/msv006. [DOI] [PubMed] [Google Scholar]
- Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solé M., Fàbrega A., Cobos-Trigueros N., Zamorano L., Ferrer-Navarro M., Ballesté-Delpierre C., Reustle A., Castro P., Nicolás J.M., Oliver A. In vivo evolution of resistance of Pseudomonas aeruginosa strains isolated from patients admitted to an intensive care unit: mechanisms of resistance and antimicrobial exposure. J. Antimicrob. Chemother. 2015;70:3004–3013. doi: 10.1093/jac/dkv228. [DOI] [PubMed] [Google Scholar]
- Taylor-Robinson D., Whitehead M., Diderichsen F., Olesen H.V., Pressler T., Smyth R.L., Diggle P. Understanding the natural progression in %FEV1 decline in patients with cystic fibrosis: a longitudinal study. Thorax. 2012;67:860–866. doi: 10.1136/thoraxjnl-2011-200953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willner D., Haynes M.R., Furlan M., Schmieder R., Lim Y.W., Rainey P.B., Rohwer F., Conrad D. Spatial distribution of microbial communities in the cystic fibrosis lung. ISME J. 2012;6:471–474. doi: 10.1038/ismej.2011.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong A., Rodrigue N., Kassen R. Genomics of adaptation during experimental evolution of the opportunistic pathogen Pseudomonas aeruginosa. PLoS Genet. 2012;8:e1002928. doi: 10.1371/journal.pgen.1002928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Jelsbak L., Marvig R.L., Damkiær S., Workman C.T., Rau M.H., Hansen S.K., Folkesson A., Johansen H.K., Ciofu O. Evolutionary dynamics of bacteria in a human host environment. Proc. Natl. Acad. Sci. USA. 2011;108:7481–7486. doi: 10.1073/pnas.1018249108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B., Sedlak J.C., Srinivas R., Creixell P., Pritchard J.R., Tidor B., Lauffenburger D.A., Hemann M.T. Exploiting temporal collateral sensitivity in tumor clonal evolution. Cell. 2016;165:234–246. doi: 10.1016/j.cell.2016.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Susceptibility Profiles, Related to Figures 1, 2, 4, and 6
MIC values were determined in MHBII based on five replicates. Concentration of antibiotics is given in μg.ml-1. MIC values were determined in two-fold broth dilution. Concentration that inhibited 90% strain growth was determined by using five sets of replicates. Concentration of antibiotic is given in μg.ml-1.
Table S2. Collateral Sensitivity Cycles, Related to Figure 1
All drugs were included in the analysis. No restriction was given to the number of cycles. For 24 drugs, max 11-drug cycle was discovered. Overall 5698 collateral sensitivity cycles were discovered with max 11 antibiotics in the cycle. In the table below only collateral sensitivity cycles with two and three drugs were listed. Antipseudomonal antibiotics were considered antibiotics for which EUCAST breakpoints (Table 1) were defined. Ten out of 13 antipseudomonal antibiotics exhibited collateral sensitivity. Based on that collateral sensitivity cycles were determined for this group. Five collateral sensitivity cycles were identified with antipseudomonal (labeled with ∗ in the table) and a maximum possible drug in the cycle was three.
Table S3. Correlation between Collateral Susceptibilities of PAO1 Drug-Resistant Strains, Related to Figure 2
Red shading indicates measurements that had positive and blue indicates negative Spearman correlation coefficient (ρ)
Table S4. Correlation between Collateral Susceptibilities of PAO1- and DK2-Resistant Strains, Related to Figure 4
All MIC data were normalized to PAO1 WT values and log2 transformed. Red shading indicates measurements that had positive and blue indicates negative Spearman correlation coefficient (ρ). Spearman ρ was calculated for each pair of variables for strains DK2 and PAO1 (A–E). Values in the row below ρ indicate _P_-values (a two-tailed significance test).
Table S5. Genetic Changes in PAO1- and DK2-Resistant Strains during Laboratory Evolution Experiment, Related to Figure 3
Details on genetic variants detected in PAO1 and DK2 resistant strains. For single isolates, variants had minimal allele frequency of 75%. For population sequencing during resistance development toward ciprofloxacin and azithromycin, the variants had minimal allele frequency (Frq.) of 10%. For each mutation detected in population, presence of mutation below 10% was verified by additional analysis allowing detection of known low frequency variant (1-10%). SNV – Single Nucleotide Variant; MNV – Multi Nucleotide Variant.
Table S6. Correlation between Collateral Susceptibilities of Experimentally Evolved Resistant Strains and DK2 Clinical Isolates, Related to Figure 4
All MIC data were normalized to PAO1 WTE values and log2 transformed. Red shading indicates measurements that had positive and blue indicates negative Spearman correlation coefficient (ρ). Spearman ρ was calculated for each pair of variables for strains DK2 and PAO1 (A-E). Values in the row below ρ indicate _P_-values.
Table S7. Antibiotic Susceptibility for Clinical Isolates from Sputum Sample, Related to Figure 5
MIC values for clinical isolates. MIC or the inhibitory concentration was defined as the lowest concentration of the drug that inhibited 90% of the growth of the strain tested. For each strain, five replicates were performed to determine the drug susceptibility. The MIC values were normalized to EUCAST breakpoints (Table 1) and log2 transformed.
Table S8. Sequencing of P. aeruginosa Single Isolates and Populations from Sputum Samples, Related to Figure 6
Details on genetic variants detected in subset of population during intensive antibiotic treatment of CF patient. The variants had minimal allele frequency (Frq.) of 5%. To call a mutation in the population, minimal read coverage was set to 10. The genetic variants detected in single isolates during treatment had minimal allele frequency of 75%. SNV – Single Nucleotide Variant; MNV – Multi Nucleotide Variant.
Table S9. Mutation Overlap between Subpopulations during In Vivo Antibiotic Treatment, Related to Figure 6
Mutation overlap in subset of population during intensive antibiotic treatment of CF patient (Table S8).