Translocation of a gut pathobiont drives autoimmunity in mice and humans (original) (raw)
. Author manuscript; available in PMC: 2018 Sep 9.
Published in final edited form as: Science. 2018 Mar 9;359(6380):1156–1161. doi: 10.1126/science.aar7201
Abstract
Despite multiple associations between the microbiota and immune diseases, their role in autoimmunity is poorly understood. We found that translocation of a gut pathobiont, Enterococcus gallinarum, to the liver and other systemic tissues triggers autoimmune responses in a genetic background predisposing to autoimmunity. Antibiotic treatment prevented mortality in this model, suppressed growth of E. gallinarum in tissues, and eliminated pathogenic autoantibodies and Tcells. Hepatocyte–E. gallinarum cocultures induced autoimmune-promoting factors. Pathobiont translocation in monocolonized and autoimmune-prone mice induced autoantibodies and caused mortality, which could be prevented by an intramuscular vaccine targeting the pathobiont. _E. gallinarum_–specific DNA was recovered from liver biopsies of autoimmune patients, and cocultures with human hepatocytes replicated the murine findings; hence, similar processes apparently occur in susceptible humans. These discoveries show that a gut pathobiont can translocate and promote autoimmunity in genetically predisposed hosts.
The gut microbiota are implicated in the pathogenesis of multiple gut and systemic autoimmune diseases (1). The integrity of the gut barrier is essential to prevent the microbiota of a healthy individual from triggering adaptive immune responses (2–7). When intact commensals or pathogens escape the gut barrier, several defense mechanisms impede bacterial access to the systemic circulation. However, if these mechanisms fail, the mesenteric lymph nodes (MLNs) and liver represent further “firewalls” against commensal bacteria that escape the gut (2, 4, 7). Such mechanisms occur only during intestinal or vascular pathology, during chemotherapy, or in the absence of a functional innate immune system (2, 6, 8–11). Although recent studies show that gut commensals can reside within gastrointestinal-associated lymphoid tissues of unmanipulated, healthy hosts, it is unclear whether pathobiont translocation is involved in systemic autoimmunity (12).
In humans, systemic lupus erythematosus (SLE) is a multifactorial autoimmune disease that is associated with marked morbidity and mortality, especially in antiphospholipid antibody–positive patients. Besides the major histocompatibility complex locus, SLE is associated with several genetic risk loci for excessive signaling of RNA-sensing Toll-like receptor 7 (TLR7) and type I interferons (IFNs) (13). In normal animals, exogenous viral infections and retroviruses contribute to these immune responses and interact with the microbiota (14–19). In the specific pathogen-free (NZW × BXSB)F1 hybrid mouse, responses to endogenous retrovirus glycoprotein 70 (ERV gp70) have been shown to drive lupus kidney disease via TLR7 (20, 21). These mice eventually succumb to progressive autoimmune thrombi mediated by pathogenic antiphospholipid [β2-glycoprotein I (β2GPI)] antibodies. We asked whether, in animals predisposed to autoimmune responses, trans-location of gut commensals drives IFN and anti–double-stranded DNA (dsDNA), anti–ERV gp70, and anti-β2GPI responses. Consequently, we investigated whether pathological immune responses could be alleviated by therapeutic strategies such as antibiotic treatment or vaccination.
Mortality, lupus-related autoantibodies, and autoimmune manifestations were relieved in (NZW × BXSB)F1 hybrid mice after oral administration of vancomycin or ampicillin, implicating involvement of Gram-positive pathobionts in disease (Fig. 1, A to C, and fig. S1). In addition to anti-dsDNA and anti-RNA autoantibodies (Fig. 1, B and C), anti-β2GPI immunoglobulin G (IgG), hepatic and serum ERV gp70, and anti–ERV gp70 immune complexes (ICs) were all suppressed by vancomycin treatment (fig. S2, A to L). Uptake of orally fed fluorescein isothiocyanate (FITC)– dextran into the systemic circulation of (NZW × BXSB)F1 hybrid mice indicated grossly impaired gut barrier function relative to nonautoimmune C57BL/6 mice (Fig. 1D). At 16 weeks of age, we were able to detect marked bacterial growth in the mesenteric veins, MLNs, and liver, and 2 weeks later also in the spleen but not kidneys, which are affected by deposition of circulating immune complexes (Fig. 1, E to G, and fig. S2M). Translocation of microbiota was suppressed by vancomycin or ampicillin, both of which prevented mortality; neomycin was less effective at inhibiting translocation relative to vancomycin (Fig. 1, E to G).
Fig. 1. Effect of antibiotics on autoimmunity and E. gallinarum translocation to the liver.
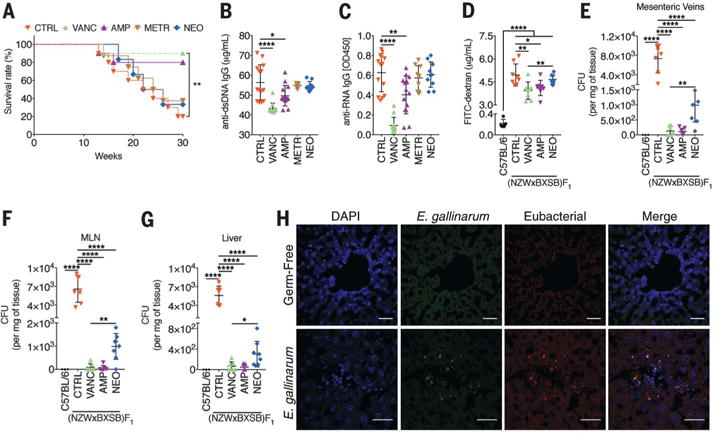
(A) Vancomycin (VANC), ampicillin (AMP), metronidazole (METR), neomycin (NEO), or control water (CTRL) were provided in the drinking water of (NZW × BXSB)F1 mice starting at 6 weeks of age (n = 15 each). Mice were followed for 30 weeks or until death from autoimmunity. (B and C) Serum anti-dsDNA (B) and anti-RNA (C) IgG at 16 weeks of age. (D) Serum levels of orally administered FITC-dextran as an indicator of gut barrier leakiness (n = 8 each). (E to G) Cultures of tissues from 16-week-old mice showed a selective growth of E. gallinarum in the mesenteric veins (E), MLN (F), and liver (G) (n = 7 each). (H) An _E. gallinarum_–specific FISH probe detects E. gallinarum in liver (scale bars, 30 μm) of _E. gallinarum_–monocolonized C57BL/6 mice 3 weeks after colonization in comparison to germ-free mice. One representative section from one mouse is shown from multiple sections with E. gallinarum signals within the tissues, representative of three mice in total. Data are presented as mean ± SD in (B) to (G); *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; log-rank test and Gehan-Breslow-Wilcoxon test in (A), analysis of variance (ANOVA) followed by Bonferroni multiple-comparisons test in (B) to (G).
Full-length 16_S_ rDNA sequencing of single colonies from aerobic and anaerobic MLN, liver, and spleen cultures detected Enterococcus gallinarum, a Gram-positive gut commensal of animals and humans, in the mesenteric veins of 82% of (NZW × BXSB)F1 hybrid mice. E. gallinarum was visualized in situ in MLNs and livers by fluorescence in situ hybridization (FISH) (Fig. 1H and fig. S2N). C57BL/6 control mice showed no systemic bacterial growth. Taxa identified by longitudinal fecal 16_S_ rDNA sequencing revealed that Enterococcus spp. were enriched only in some fecal samples (fig. S3, A to C, and fig. S4). Species-specific polymerase chain reaction (PCR) did not detect E. gallinarum DNA in stool samples from human or murine autoimmune hosts (fig. S5, A to D); however, fecal or mucosal tissue culture followed by species-specific PCR consistently revealed E. gallinarum in the feces and small intestine, as well as in the liver, of (NZW × BXSB)F1 mice (fig. S6, A to C). We also found E. gallinarum translocation to livers of (NZW × BXSB)F1 mice in two other animal facilities at Yale after transfer of newly weaned animals equilibrated to different microbiomes (fig. S6, D and E).
To test whether E. gallinarum induces proinflammatory pathways and alters gut barrier– related molecules in small intestinal tissue during translocation into internal organs, we performed RNA expression profiling of _E. gallinarum_– monocolonized C57BL/6 mice and compared these data with those from _Enterococcus faecalis_– and _Bacteroides thetaiotaomicron_–monocolonized mice (Fig. 2, A to J, and figs. S7 and S8). The presence of E. gallinarum down-regulated ileal molecules related to barrier function (e.g., occludin, claudins, Plvap, Axin2), the mucus layer (e.g., Mucin-2), and antimicrobial defense (e.g., Reg3b, Defa2) and up-regulated those related to inflammation (e.g., Cxcr2, AhR/Cyp1a1, Enpp3). Enpp3 is known to increase numbers of plasmacytoid dendritic cells (pDCs) (22), which are key cells contributing to the IFN signature in human SLE (13) and which were induced by E. gallinarum monocolonization (Fig. 2K).
Fig. 2. RNA expression profiling and plasmacytoid dendritic cell frequencies in small intestine from germ-free mice monocolonized with E. gallinarum, E. faecalis, or B. thetaiotaomicron.

Germ-free C57BL/6 mice were monocolonized with E. gallinarum (EG), E. faecalis (EF), or B. thetaiotaomicron (BT) for RNA-seq and fluorescence-activated cell sorting (FACS) analyses of the small intestine. (A) RNA-seq was performed with ileal tissue isolated from 14-week-old monocolonized mice (n = 3 each). Heat map shows transcripts differentially expressed in the ileum 8 hours after commensal delivery. (B to J) Reverse transcription quantitative PCR (RT-qPCR) analysis of ileum RNA (n = 6 each) as described in (A). (K) Plasmacytoid dendritic cell (pDC) and conventional dendritic cell (cDC) frequencies in the small intestinal lamina propria of 12-week-old germ-free mice (n = 4 each) were evaluated 3 weeks after monocolonization by FACS analysis. (L) Confocal imaging of gut tissues was performed as described in the supplementary materials. Localizations of TJ proteins are shown in green for occludin, JAM-A, claudin-3, and claudin-5. Images are representative of six different mice. DAPI, 4′,6-diamidino-2-phenylindole. Scale bars, 40 μm. Data are presented as mean ± SD in (B) to (K); *P < 0.05, **P < 0.01, ****P < 0.0001; ANOVA followed by Bonferroni multiple-comparisons test in (B) to (K).
We used confocal microscopy to visualize the gut epithelial, vascular, and lymphatic barrier molecule changes we detected by RNA. Intestinal epithelial and endothelial cells have tight junctions formed by occludin, zonula occludens–1 (ZO-1), cingulin, and junctional adhesion molecule– A (JAM-A) (23). These cells also have adherent junctions formed by vascular endothelial cadherin (VE-cadherin) and β-catenin. A loss of expression of these junctional proteins was seen in gnotobiotic C57BL/6 mice monocolonized with E. gallinarum, except for ZO-1 and VE-cadherin (Fig. 2L and figs. S9 to S11). Claudin-2, -3, and -5 are expressed in lymphatic endothelial tight junctions, but in _E. gallinarum_–monocolonized mice they were weakly expressed relative to germ-free controls, except for pore-forming claudin-2 (Fig. 2L and fig. S12).
Liver-resident E. gallinarum possibly induces hepatic overexpression of ERV gp70 that fuels anti-ERV immune complex formation and systemic autoimmunity (fig. S2). We found that (NZW × BXSB)F1–derived hepatocytes cocultured with E. gallinarum isolated from an (NZW × BXSB) F1 liver induced multiple autoimmune-promoting factors, including the autoantigens ERV gp70 and β2GPI, which were potently induced by E. gallinarum or E. gallinarum RNA (a potential TLR7/8 ligand) relative to E. faecalis (Fig. 3, A and B). In hepatocytes and dendritic cells, E. gallinarum RNA also induced expression of type I IFN and other proinflammatory cytokines (Fig. 3C and fig. S13).
Fig. 3. Induction of hepatic AhR by E. gallinarum, AhR antagonism in (NZW × BXSB)F1 mice, and Th17 and autoantibody induction in E. gallinarum monocolonized C57BL/ 6 mice.
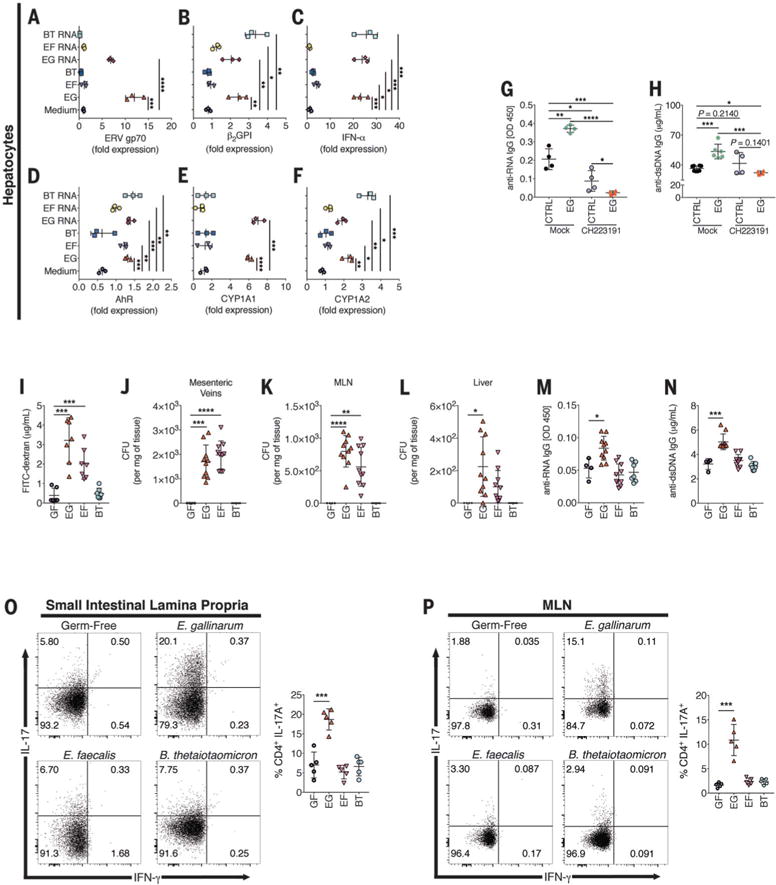
(A to F) E. gallinarum, E. faecalis, and B. thetaiotaomicron lysates or isolated RNA were cocultured with hepatocytes from 14-week-old (NZW × BXSB) F1 mice (n = 3 each), and expression of ERV gp70, the autoantigen β2GPI, IFN-α, and AhR was measured 6 hours later. (G and H) Serum anti-RNA (G) and anti-dsDNA (H) IgG of 16-week-old (NZWxBXSB)F1 mice (n = 4 each) gavaged with vehicle or EG and treated with AhR antagonist CH223191 or mock, as described in the supplementary materials. (I to L) C57BL/6 germ-free (GF) mice were monocolonized with E. gallinarum, E. faecalis, and B. thetaiotaomicron at 12 weeks of age (n = 6 to 10 mice) and evaluated 3 weeks later for integrity of the gut barrier with FITC-dextran (I), and for translocation to the mesenteric veins (J), MLNs (K), and liver (L). (M and N) C57BL/6 germ-free mice (n = 4 to 10 mice) were monocolonized as in (I), and anti-RNA (M) and anti-dsDNA (N) IgG autoantibodies were measured 8 weeks later. (O and P) Th17 cells and Th1 cell frequencies were determined by intracellular FACS analysis of IL-17A and IFN-g in small intestinal lamina propria (O) and MLN (P) CD45+ CD4+ CD44+ T cells from germ-free and monocolonized mice (n = 5 each). Data are presented as mean ± SD in (A) to (P); *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; ANOVA followed by Bonferroni multiple-comparisons test.
Tryptophan-derived indoles are bacterial ligands for the aryl hydrocarbon receptor (AhR), which activates the AhR-CYP1A1 pathway, a known innate antimicrobial defense mechanism and inducer of T helper 17 (Th17) cells (24–27) that we also found to be up-regulated (Fig. 3, D to F). Furthermore, whole-genome sequencing revealed that E. gallinarum encodes the shikimate pathway, which generates AhR ligands (fig. S14 and table S1) (26). We tested whether _E. gallinarum_–immune interactions, possibly mediated by the AhR pathway, induced Th17 and T follicular helper (Tfh) cells in vivo that are crucial for systemic autoantibody production (28, 29). Broad-spectrum antibiotic or vancomycin treatment in (NZW × BXSB)F1 mice reduced levels of Th17 cells, Tfh cells, and their cytokine signatures (fig. S15, A to C). Vancomycin had no direct influence on immune cells in vitro (fig. S15D). The in vivo effects of vancomycin on T cells correlated with reduced E. gallinarum translocation, less immunopathology, and fewer autoantibodies in these mice, as well as suppression of serum and hepatic levels of the ERV gp70 autoantigen and anti–ERV gp70 immune complexes (Fig. 1 and fig. S2). Administration of an AhR-selective antagonist abrogated the Th17- and autoantibody-inducing effects of E. gallinarum in (NZW × BXSB)F1 mice in vivo (Fig. 3, G and H, and fig. S16); this finding indicates that AhR signaling is involved in _E. gallinarum_–induced autoimmunity.
Because E. gallinarum has the potential for translocation in (NZW × BXSB)F1 mice but not in C57BL/6 mice under specific pathogen-free conditions, we tested translocation in germ-free C57BL/ 6 mice monocolonized with E. gallinarum. In the absence of competing microbiota, E. gallinarum induced barrier leakage, autoantibodies, and trans-location to mesenteric veins, MLNs, and livers of nonautoimmune C57BL/6 mice (Fig. 3, I to N). Autoantibody induction in C57BL/6 mice was not seen during monocolonization with other bacteria that either remain in the gut (B. thetaiotaomicron) or translocate to tissues (E. faecalis) (Fig. 3, I to N). Monocolonization with E. gallinarum induced Th17 cells in the small intestinal lamina propria and MLNs of C57BL/6 mice (Fig. 3, O and P), consistent with the finding that Th17 cells are abundant in (NZW × BXSB)F1 mice and were depleted after vancomycin treatment (fig. S15). Experiments with antibiotic-pretreated (NZW × BXSB)F1 hybrid mice gavaged with a trans-locating pathogen, Salmonella typhimurium, did not induce autoantibodies (fig. S17). By contrast, gavage of antibiotic-pretreated (NZW × BXSB)F1 hybrid mice with E. gallinarum caused systemic autoimmune pathology (fig. S17D and fig. S18, control group).
To deplete the pathobiont selectively, we developed an intramuscular vaccination strategy using heat-killed E. gallinarum (fig. S18A). Vaccination against E. gallinarum, but not against E. faecalis nor B. thetaiotaomicron, reduced levels of serum autoantibodies and prolonged survival in (NZW × BXSB)F1 mice (fig. S18, B to F). Vaccination also prevented translocation, as no growth of E. gallinarum was observed in internal organs (fig. S18G). Thus, pathobiont-specific treatment can abrogate host autoimmune processes without needing to suppress the immune system, which can lead to systemic adverse events in current clinical practice.
Longitudinal stool analyses from SLE patients revealed evidence for impaired gut barrier function with increased fecal albumin and calprotectin (Fig. 4, A and B). We thus tested for E. gallinarum translocation to human livers in patients with SLE and autoimmune hepatitis (AIH) who display serologic features of lupus, including antinuclear antibodies and anti-dsDNA IgG (table S2) (30). Liver biopsies from three SLE patients were positive for E. gallinarum (Fig. 4C); of six controls obtained from healthy liver transplant donors with normal liver histology, four were positive for the presence of other Enterococcus species but not E. gallinarum. Sterilely obtained human liver tissues from the same control patients, AIH patients, and cirrhosis patients [who are known to have grossly impaired gut barriers (31)] were subjected to 16_S_ rDNA sequencing; the results show that Enterococcus spp. predominated in diseased tissues (Fig. 4D). Note that the majority of AIH liver biopsies, but not the healthy control livers, were positive for E. gallinarum (Fig. 4E).
Fig. 4. Gut barrier function and E. gallinarum in liver biopsies of autoimmune patients with anti E. gallinarum serum reactivities.
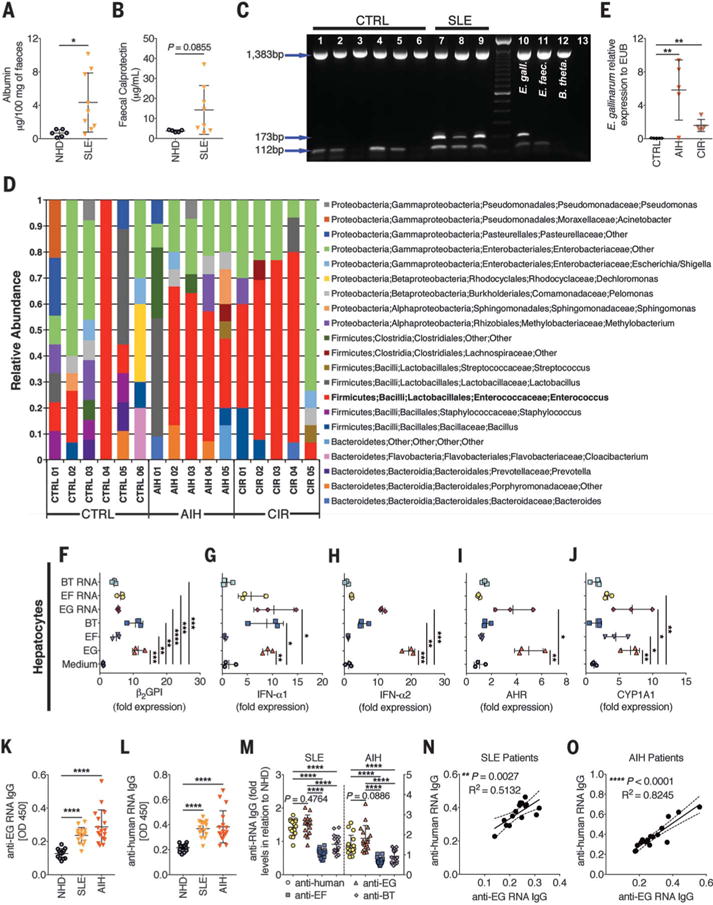
(A and B) Feces from patients with SLE (n = 9) were screened for increased albumin (A) and calprotectin (B) as signs of an impaired gut barrier. NHD, normal healthy donors (n = 9). (C) Multiplex PCR for eubacterial [1383 base pairs (bp)], Enterococcus genus (112 bp), and E. gallinarum (173 bp) DNA on sterilely obtained and processed liver biopsies from cadaveric liver transplant donors (CTRL) or SLE patients. Lanes 10 to 12, bacterial strains as indicated; lane 13, water. (D) 16_S_ rDNA sequencing of controls as in (C), autoimmune hepatitis (AIH) patients, and non-AIH cirrhosis (CIR) patients. (E) Liver biopsies from CTRL, AIH, and CIR patients (n = 5 to 6 each) were tested for E. gallinarum (EG) DNA by qPCR and normalized to any eubacterial (EUB) signal. (F to J) RT-qPCR of human primary hepatocytes (n = 3 each) stimulated with E. gallinarum, E. faecalis, or B. thetaiotaomicron as in Fig. 3. (K and L) SLE (n = 15) and AIH (n = 17) sera were screened for anti–E. gallinarum RNA IgG (K) and anti-human RNA IgG (L) by enzyme-linked immunosorbent assay (ELISA). (M) SLE and AIH serum IgG levels against human, E. gallinarum, E. faecalis, or B. thetaiotaomicron RNA normalized to NHD sera. (N and O) Correlation between anti–E. gallinarum RNA IgG and autoantibodies in SLE (N) and AIH (O) patients. Data are presented as mean ± SD; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; Student t test in (A) and (B), ANOVA followed by Bonferroni multiple-comparisons test in (E) to (M), and Pearson correlation in (N) and (O).
Similar to the effects seen with murine hepatocytes (Fig. 3, A to F), primary human hepatocytes from healthy livers stimulated with E. gallinarum induced β2GPI, type I interferon, and AHR/CYP1A1 (Fig. 4, F to J). Consistent with enhanced adaptive immune responses to E. gallinarum, the majority of SLE and AIH patients also showed increased serum antibody titers against E. gallinarum and particularly its RNA, which can act as a potential TLR7/8 stimulus and cross-reactive trigger (Fig. 4, K to O, and fig. S19). Comparison of anti– human RNA IgG autoantibody titers in SLE and AIH patients with anti–E. gallinarum RNA IgG showed that both were equally elevated, by contrast to anti–E. faecalis and anti–B. thetaiotaomicron RNA antibodies (Fig. 4, K to O).
Our findings show that the Gram-positive gut pathobiont E. gallinarum translocates, as a result of gut barrier breakdown, into systemic organs in autoimmune-prone hosts to drive autoimmune pathogenesis (fig. S20). Translocating bacteria may not only skew T helper cell differentiation but may also directly act on colonized tissues, such as the liver, to induce autoantigens, ERV proteins, cytokines, and other autoimmune-promoting factors. If the complexity of host tissue–microbiota interactions is considered in chronic autoimmunity, it may offer new therapeutic avenues for these debilitating and potentially lethal diseases.
Supplementary Material
SOM
Acknowledgments
We thank all patients enrolled under the IRB protocols 1408014402 and 1602017150 (ClinicalTrials.gov identifier NCT02394964) who have participated in this study, as well as K. DeFrancesco and I. Matos for patient recruitment at the Yale Center for Clinical Investigation, which is supported by CTSA grant UL1 RR024139 from the National Center for Research Resources, the National Center for Advancing Translational Science, and the NIH Roadmap for Medical Research. We also thank D. Assis, J. Boyer, and the Yale Liver Center for contributions of liver biopsy material; J. Galan for provision of S. typhimurium; J. Sterpka for technical assistance; O. Pagovich for initial antibiotics experiments; J. Weinstein for assistance with Tfh studies; N. Palm for use of an anaerobic chamber; M. Tokuyama and A. Iwasaki for contributing ERV anti-gp70 antibodies for ERV and ERV IC ELISAs; and E. Meffre, J. Craft, and G. Eberl for critically reading the manuscript. S.M.V. and M.A.K. are inventors on a patent application filed by Yale University related to the use of antibiotics and commensal vaccination to treat autoimmunity (U.S. Provisional Patent Application 62/448,510). Supported by NIH grants K08AI095318, R01AI118855, T32AI07019, and T32DK007017-39; Yale Rheumatic Diseases Research Core (NIH grant P30 AR053495); the Yale Liver Center (NIH grant P30 DK34989); Women’s Health Research at Yale; the O’Brien Center at Yale (NIH grant P30DK079310); the Arthritis National Research Foundation; the Arthritis Foundation; and the Lupus Research Institute. Author contributions: S.M.V. designed, performed, and analyzed all murine, gnotobiotic, and human experiments; M.H., D.Z.R., N.K., F.R.C.C., E.T., T.G., and C.K. assisted in murine experiments including gnotobiotics; S.M.V., F.R.C.C., and D.Z.R. performed translocation and barrier function experiments; V.K., C.D., and T.G. were involved in consenting, recruiting, and sampling human study subjects; V.K., A.B., and D.J. obtained all liver biopsy samples; S.M.V. processed and analyzed data related to human material; S.M.V., V.K., C.D., A.B., and D.J. performed histopathologic studies and scoring; S.M.V. and C.D. performed FISH, immunofluorescence staining, and confocal microscopy; S.M.V., V.K., and W.R. performed 16_S_ rDNA sequencing, data processing, and analysis; A.L.G. assisted in microbiologic studies and bioinformatics related to 16_S_ rDNA sequencing; S.M.V. and S.S.M. processed and analyzed the RNA sequencing data; S.S.M. and J.R.K. assembled the bacterial genome; S.M.V. and S.S.M. analyzed the genome; S.M.V. produced the chemical structures drawing and biochemical pathway; S.M.V. and M.A.K. wrote the manuscript with input from all authors; M.A.K. conceived the study and supervised the project; and S.M.V. and M.A.K. participated in design and interpretation of all experiments. All data to understand and assess the conclusions of this research are available in the main text and supplementary materials, as well as via the following repositories: Whole-genome shotgun sequences have been deposited at DDBJ/ENA/GenBank under accession number PPHK00000000. The version described in this paper is version PPHK01000000. RNA-seq and 16_S_ rRNA sequences have been deposited with the European Bioinformatics Institute under accession numbers PRJEB24586 and PRJEB24587, respectively.
Footnotes
REFERENCES AND NOTES
- 1.Ruff WE, Kriegel MA. Trends Mol Med. 2015;21:233–244. doi: 10.1016/j.molmed.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balmer ML, et al. Sci Transl Med. 2014;6:237ra66. doi: 10.1126/scitranslmed.3008618. [DOI] [PubMed] [Google Scholar]
- 3.Hand TW, et al. Science. 2012;337:1553–1556. doi: 10.1126/science.1220961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Macpherson AJ, Uhr T. Science. 2004;303:1662–1665. doi: 10.1126/science.1091334. [DOI] [PubMed] [Google Scholar]
- 5.Sandler NG, Douek DC. Nat Rev Microbiol. 2012;10:655–666. doi: 10.1038/nrmicro2848. [DOI] [PubMed] [Google Scholar]
- 6.Slack E, et al. Science. 2009;325:617–620. doi: 10.1126/science.1172747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spadoni I, et al. Science. 2015;350:830–834. doi: 10.1126/science.aad0135. [DOI] [PubMed] [Google Scholar]
- 8.Costa FR, et al. J Exp Med. 2016;213:1223–1239. doi: 10.1084/jem.20150744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sonnenberg GF, et al. Science. 2012;336:1321–1325. doi: 10.1126/science.1222551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stanley D, et al. Nat Med. 2016;22:1277–1284. doi: 10.1038/nm.4194. [DOI] [PubMed] [Google Scholar]
- 11.Viaud S, et al. Science. 2013;342:971–976. doi: 10.1126/science.1240537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fung TC, et al. Immunity. 2016;44:634–646. doi: 10.1016/j.immuni.2016.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crow MK. J Immunol. 2014;192:5459–5468. doi: 10.4049/jimmunol.1002795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abt MC, et al. Immunity. 2012;37:158–170. doi: 10.1016/j.immuni.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ichinohe T, et al. Proc Natl Acad Sci USA. 2011;108:5354–5359. doi: 10.1073/pnas.1019378108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kane M, et al. Science. 2011;334:245–249. doi: 10.1126/science.1210718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuss SK, et al. Science. 2011;334:249–252. doi: 10.1126/science.1211057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vieira SM, Pagovich OE, Kriegel MA. Lupus. 2014;23:518–526. doi: 10.1177/0961203313501401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Young GR, et al. Nature. 2012;491:774–778. doi: 10.1038/nature11599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baudino L, Yoshinobu K, Morito N, Santiago-Raber ML, Izui S. Autoimmun Rev. 2010;10:27–34. doi: 10.1016/j.autrev.2010.07.012. [DOI] [PubMed] [Google Scholar]
- 21.Tabata N, et al. J Virol. 2000;74:4116–4126. doi: 10.1128/jvi.74.9.4116-4126.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furuta Y, et al. PLOS ONE. 2017;12:e0172509. doi: 10.1371/journal.pone.0172509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luissint AC, Parkos CA, Nusrat A. Gastroenterology. 2016;151:616–632. doi: 10.1053/j.gastro.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moura-Alves P, et al. Nature. 2014;512:387–392. doi: 10.1038/nature13684. [DOI] [PubMed] [Google Scholar]
- 25.Schiering C, et al. Nature. 2017;542:242–245. doi: 10.1038/nature21080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stockinger B, Di Meglio P, Gialitakis M, Duarte JH. Annu Rev Immunol. 2014;32:403–432. doi: 10.1146/annurev-immunol-032713-120245. [DOI] [PubMed] [Google Scholar]
- 27.Veldhoen M, et al. Nature. 2008;453:106–109. doi: 10.1038/nature06881. [DOI] [PubMed] [Google Scholar]
- 28.Campisi L, et al. Nat Immunol. 2016;17:1084–1092. doi: 10.1038/ni.3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Craft JE. Nat Rev Rheumatol. 2012;8:337–347. doi: 10.1038/nrrheum.2012.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fallatah HI, Akbar HO. Autoimmune Dis. 2012;2012:312817. doi: 10.1155/2012/312817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cirera I, et al. J Hepatol. 2001;34:32–37. doi: 10.1016/s0168-8278(00)00013-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SOM