PrPC Controls via Protein Kinase A the Direction of Synaptic Plasticity in the Immature Hippocampus (original) (raw)
Abstract
The cellular form of prion protein PrPC is highly expressed in the brain, where it can be converted into its abnormally folded isoform PrPSc to cause neurodegenerative diseases. Its predominant synaptic localization suggests a crucial role in synaptic signaling. Interestingly, PrPC is developmentally regulated and its high expression in the immature brain could be instrumental in regulating neurogenesis and cell proliferation. Here, PrPC-deficient (_Prnp_0/0) mice were used to assess whether the prion protein is involved in synaptic plasticity processes in the neonatal hippocampus. To this aim, calcium transients associated with giant depolarizing potentials, a hallmark of developmental networks, were transiently paired with mossy fiber activation in such a way that the two events were coincident. While this procedure caused long-term potentiation (LTP) in wild-type (WT) animals, it caused long-term depression (LTD) in _Prnp_0/0 mice. Induction of LTP was postsynaptic and required the activation of cAMP-dependent protein kinase A (PKA) signaling. The induction of LTD was presynaptic and relied on G-protein-coupled GluK1 receptor and protein lipase C. In addition, at emerging CA3-CA1 synapses in WT mice, but not in _Prnp_0/0 mice, pairing Schaffer collateral stimulation with depolarization of CA1 principal cells induced LTP, known to be PKA dependent. Postsynaptic infusion of a constitutively active isoform of PKA catalytic subunit Cα into CA1 and CA3 principal cells in the hippocampus of _Prnp_0/0 mice caused a persistent synaptic facilitation that was occluded by subsequent pairing. These data suggest that PrPC plays a crucial role in regulating via PKA synaptic plasticity in the developing hippocampus.
Introduction
The cellular form of the prion protein PrPC is a conserved glycoprotein tethered to the cell membrane by a glycosylphosphatidylinositol anchor (Colby and Prusiner, 2011). The ubiquitous expression of PrPC in almost all tissues, most abundantly in the brain, suggests a key role for this protein in several cellular functions. Conversion of PrPC into its abnormally folded isoform PrPSc causes neurodegenerative disorders in mammals, including Creutzfeldt–Jakob disease in humans (Colby and Prusiner, 2011). Notwithstanding evidence supporting a central role for PrPC in the pathogenesis of transmissible spongiform encephalopathies, its normal physiological function is still unclear. PrPC-deficient mice (_Prnp_0/0) lack a consistent overt phenotype (Büeler et al., 1992), though they manifest a variety of rather minor deficits ranging from behavioral to electrophysiological and biochemical alterations (Steele et al., 2007). The predominant synaptic localization of PrPC suggests its function may be related to synaptic transmission and cell excitability. Previous in vitro studies on the hippocampus of _Prnp_0/0 mice have highlighted reduced GABAA-mediated synaptic inhibition (Collinge et al., 1994), altered glutamatergic excitation (Carleton et al., 2001), impaired long-term potentiation (LTP) (Collinge et al., 1994; Manson et al., 1995), and a reduced slow afterhyperpolarization associated with enhanced cell excitability (Colling et al., 1996; Mallucci et al., 2002; Fuhrmann et al., 2006).
Interestingly, PrPC is developmentally regulated (Manson et al., 1992; Salès et al., 2002; Benvegnù et al., 2010), and its high expression in the immature brain may account for the shorter incubation time of disease in younger animals (McKinley et al. 1989). Moreover, the high levels of the protein in late prenatal and early postnatal life are crucial for regulating neurogenesis and cell proliferation (Steele et al. 2006). In the hippocampus, developmental changes in PrPC immunoreactivity parallel those of mossy fiber (MF) terminals, the axons of granule cells in the dentate gyrus (Salès et al., 2002), which reach full maturation around postnatal day (P) 15–P20 (Amaral and Dent, 1981).
The immature hippocampus is characterized by network-driven giant depolarizing potentials (GDPs), which are generated by the synergistic action of glutamate and GABA, both of which are depolarizing and excitatory (Ben-Ari et al., 1989, 2007). Calcium transients associated with GDPs are thought to be instrumental for enhancing synaptic efficacy at emerging glutamatergic (Mohajerani et al., 2007) and GABAergic (Kasyanov et al., 2004) synapses.
Here, we tested the hypothesis that PrPC regulates synaptic plasticity processes at immature MF-CA3 synapses, which immediately after birth are mainly GABAergic (Safiulina et al., 2006). The rising phase of GDPs was used to trigger MF stimulation in such a way that synchronized network activity was coincident with MF activation. While in WT animals the pairing procedure produced a persistent increase in synaptic strength that depended on postsynaptic protein kinase A (PKA) signaling, in _Prnp_0/0 mice it caused a persistent weakening of synaptic efficacy that was presynaptic in origin and relied on G-protein-coupled GluK1 receptor and protein lipase C downstream to G-protein activation.
Materials and Methods
Animals.
All experiments were performed in accordance with the European Community Council Directive of November 24, 1986 (86/609EEC) and were approved by the local authority veterinary service. All efforts were made to minimize animal suffering and to reduce the number of animals used. Inbred FVB/N (Friend virus B-type susceptibility-NIH) wild-type and FVB _Prnp_0/0 mice were used in these experiments. The FVB _Prnp_0/0 mice were obtained from George A. Carlson, McLaughlin Research Institute, Great Falls, Montana, and were bred by backcrossing with the original _Prnp_0/0 mice at least 20 times.
Hippocampal slices.
Experiments were performed on hippocampal slices obtained from P3–P7-d-old mice as previously described (Le Magueresse et al., 2006). Briefly, animals were decapitated after being anesthetized with an intraperitoneal injection of urethane (2 g/kg). The brain was quickly removed from the skull and placed in ice-cold artificial CSF (ACSF) containing the following (in mm): 130 NaCl, 3.5 KCl, 1.2 NaH2PO4, 25 NaHCO3, 1.3 MgCl2, 2 CaCl2, 25 glucose, saturated with 95% O2 and 5% CO2, pH 7.3–7.4. Transverse hippocampal slices (400 μm thick) were cut with a vibratome and stored at room temperature in a holding bath containing the same solution as above. After a recovery period of at least 1 h, an individual slice was transferred to the recording chamber, where it was continuously superfused with oxygenated ACSF at a rate of 2–3 ml/min at 33–34°C.
Electrophysiological recordings.
Electrophysiological experiments were performed mainly from CA3 pyramidal cells using the whole-cell configuration of the patch-clamp technique in current-clamp or voltage-clamp mode. Bipolar twisted NiCr-insulated electrodes localized into stratum granulosum of the dentate gyrus were used to evoke synaptic responses in CA3 pyramidal cells. We used minimal stimulation to activate only one or few presynaptic fibers. According to the technique described by Jonas et al. (1993) and Allen and Stevens (1994), the stimulation intensity was decreased until only a single axon was activated. This was achieved when the mean amplitude of the postsynaptic currents and failure probability remained constant over a range of stimulus intensities near the threshold for detecting a response. Further enhancing the stimulus intensity led to an abrupt increase in the mean peak amplitude of synaptic currents. This “all-or-none” behavior suggests that only a single granule cell was stimulated. When the stimulation intensity was turned down, the probability of failures in synaptic transmission was near 1. The latency and shape of individual synaptic responses remained constant over repeated stimuli. In most cases, paired stimuli were applied at 50 ms intervals.
Patch electrodes were pulled from borosilicate glass capillaries (Hilgenberg). They had a resistance of 4–6 MΩ when filled with an intracellular solution containing the following (in mm): 115 K-gluconate, 20 KCl, 10 disodium phosphocreatine, 10 HEPES, 4 MgATP, 0.3 GTP, 5 QX-314, pH 7.3. Recordings were made with a patch-clamp amplifier (Axopatch 1D; Molecular Devices). The stability of the patch was checked by repeatedly monitoring (every 5 min) the input and series resistance. Cells exhibiting >15% changes in series resistance were excluded from the analysis. During the first week of postnatal life, MF-CA3 responses are mainly GABAergic since they are insensitive to the AMPA receptor antagonist GYKI 52466 (Caiati et al., 2010), but they are readily and reversibly blocked by bicuculline (20 μm), picrotoxin (100 μm), or gabazine (2 μm; Walker et al., 2001; Safiulina et al., 2006). MF inputs were identified on the basis of their sensitivity to group III metabotropic glutamate receptor agonist 2-amino-4-phosphonobutyric acid (l-AP4, Gutiérrez et al., 2003; Kasyanov et al., 2004), their paired-pulse facilitation, and their short-term frequency-dependent facilitation (Safiulina et al., 2006). GABAA-mediated synaptic potentials or GABAA-mediated synaptic currents (GPSCs) were evoked at 0.05 Hz from a holding potential of −60 mV. After a control period of 5–10 min, the patch was switched from voltage-clamp to current-clamp mode and MF responses were paired with spontaneously occurring GDPs. “Pairing” consisted of triggering MF stimulation with the rising phases of GDPs during a 7–10 min period (Fig. 1A,B). The patch was subsequently switched back to voltage-clamp mode and synaptic currents were recorded as in control for an additional 20–40 min. In some experiments, recordings were performed with patch pipettes containing the calcium chelator 1,2-bis (2-aminophenoxy) ethane-N,N,N′,N′-tetraacetic acid (BAPTA, 20 mm; Sigma-Aldrich).
Figure 1.

Pairing GDPs with MF stimulation induces LTP in WT mice and LTD in _Prnp_0/0 mice. A, Diagram of the hippocampus showing a CA3 pyramidal neuron receiving synaptic input from the MF. The stimulating electrode (stim) was positioned in stratum granulosum of the dentate gyrus. B, GDPs recorded from a CA3 principal cell in current-clamp mode; the inset below represents the GDP (asterisk) on an expanded time scale. Note the absence of spikes riding on top of GDPs because of the block of the sodium channel with intracellular QX 314. The rising phase of GDPs (between the dashed lines) was used to trigger synaptic stimulation (stim). C, The mean amplitude of MF-GPSCs recorded in WT animals (n = 13) before and after pairing (arrow at time 0) is plotted versus time, in the absence or in the presence (bars) of l-AP4 and picrotoxin (PTX). Insets above the graphs represent individual traces of GPSCs evoked before and after pairing, after addition of l-AP4 and PTX. D, As in C but from _Prnp_0/0 mice (n = 15). In this and in the following figures, vertical bars refer to SEM.
Experiments aimed at assessing synaptic plasticity at glutamatergic CA3-CA1 synapses were performed from CA1 pyramidal cells using patch pipettes filled with a solution containing the following (in mm): 120 Cs-MeSO4, 20 KCl, 10 HEPES, 0.5 EGTA, 0.3 Na-GTP, and 4 Mg-ATP 4, pH 7.2 (osmolality, 275–280 mOsm).
Spontaneous miniature GABAergic currents (mGPSCs) were recorded from CA3 principal cells in the presence of DNQX (20 μm) and TTX (1 μm).
Drugs.
Drugs used were as follows: d-(-)-2-amino-5-phosphonopentaoic acid (d-AP5), 2-amino-4-phosphonobutyric acid (l-AP4), DNQX, (S)-1-(2-amino-2-carboxyethyl)-3-(2-carboxybenzyl)pyrimidine-2,4-dione (UBP 302), 1-[6-[[(17β)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1_H_-pyrrole-2,5-dione (U73122), picrotoxin, 6-imino-3-(4-methoxyphenyl)-1(6_H_)-pyridazinebutanoic acid hydrobromide (gabazine), PKA inhibitor 6-22 amide (PKI 6-22), PKA inhibitor 14-22 amide (PKI 14-22), and forskolin (all from Tocris Cookson); PKA catalytic subunits type Cα (Sigma-Aldrich). All drugs were dissolved in ACSF, except DNQX, picrotoxin, and forskolin, which were dissolved in dimethylsulphoxide (DMSO). The final concentration of DMSO in the bathing solution was 0.1%. At this concentration, DMSO alone did not modify the shape or the kinetics of synaptic currents. Drugs were applied in the bath via a three-way tap system by changing the superfusion solution to one differing only in its drug content. The ratio of flow rate to bath volume ensured complete exchange within 2 min.
Data analysis.
Data were acquired and digitized with an analog-to-digital converter (Digidata 1200, Molecular Devices) and stored on a computer hard disk. Acquisition and analysis of evoked responses were performed with Clampfit 9 (Molecular Devices). Data were sampled at 20 kHz and filtered with a cutoff frequency of 2 kHz. Mean GPSC amplitude was obtained by averaging successes and failures. The paired-pulse ratio (PPR) was calculated as the mean amplitude of the synaptic response evoked by the second stimulus over that evoked by the first one. The coefficient of variation (CV) of response amplitude was determined as the ratio between the SD and the mean.
Statistical analysis.
Statistical analysis was performed using Student's paired t test to compare conditions that allowed recordings from the same neuron before and after drug treatment. The unpaired t test was used to compare two independent groups and one-way ANOVA was used to compare several independent groups. When the ANOVA resulted in a significant general group effect, Dunnett's post hoc test was used to compare different groups versus the control group for single or multiple comparisons. A p value <0.05 was considered as statistically significant.
Results
Basic synaptic transmission and network activity is unaffected in the hippocampus of Prnp0/0 mice
Whole-cell recordings from CA3 principal cells in P3–P7 hippocampal slices from WT (n = 30) and _Prnp_0/0 mice (n = 27) did not reveal any significant change in resting membrane potential (WT, −55.6 ± 1.55 mV; _Prnp_0/0, −52 ± 1.9 mV; p = 0.1), membrane capacitance (WT, 64 ± 4.8 pF; _Prnp_0/0, 57.4 ± 3.4 pF; p = 0.5), and input resistance (WT, 387 ± 27 MΩ; _Prnp_0/0, 382 ± 23 MΩ; p = 0.8) of the recorded cells. Both WT and _Prnp_0/0 mice exhibited similar spike half-width values (WT, 1.6 ± 0.04 ms; _Prnp_0/0, 1.6 ± 0.03 ms; p = 0.6) and responded to long depolarizing current pulses with repetitive and rapidly adapting firing (data not shown). In addition, they exhibited network-driven GDPs at the frequency of 0.056 ± 0.02 Hz in WT mice (n = 13) and 0.066 ± 0.02 Hz in _Prnp_0/0 mice (n = 15) (p > 0.05). Moreover, current responses underlying GDPs carried similar charge transfers as indicated by similar values of respective areas (WT, 30.7 ± 4.9 pAs−1; _Prnp_0/0, 43 ± 7.8 pAs−1; n = 12; p > 0.05; data not shown).
Minimal stimulation of granule cells in the dentate gyrus elicited in CA3 principal cells GPSCs of variable amplitude intermingled with synaptic failures. GPSCs exhibited a similar potency (the mean amplitude of successes without failures was 63.8 ± 8.1 pA in WT animals, n = 13, while in _Prnp_0/0 mice it was 60.5 ± 9.5 pA, n = 23 (p = 0.79, unpaired t test), but slightly different failure rates (WT mice, 0.37 ± 0.06; _Prnp_0/0, 0.57 ± 0.05; p = 0.01, unpaired t test). In accord with previous data (Safiulina et al., 2006; Sivakumaran et al., 2009; Caiati et al., 2010), GPSCs were of MF origin as they were sensitive to group III metabotropic glutamate receptor (mGluR) agonist l-AP4. This compound reduced the amplitude of GPSCs to 20.4 ± 3.4% and 18.3 ± 2.8% of controls (predrug values) in WT and _Prnp_0/0 mice, respectively (p > 0.05, paired t test, data not shown).
Pairing GDPs with MF stimulation induces LTP in WT mice and LTD in _Prnp_0/0 mice
After a control period of 10 min, a “pairing” procedure was used to change synaptic efficacy at MF-CA3 synapses (Kasyanov et al., 2004). This method consisted of switching the patch from voltage-clamp to current-clamp mode and of stimulating afferent fibers with the rising phase of GDPs (Fig. 1A,B). The mean number of GDPs recorded during the 7 min pairing was 18 ± 3 in WT mice and 20 ± 2 in _Prnp_0/0 mice (these values were not significantly different, p > 0.05; unpaired t test). After pairing, the patch was switched back to voltage-clamp mode and MF-GPSCs were recorded for additional 20–40 min. As illustrated in Figure 1C, the pairing procedure in WT animals produced a strong and persistent potentiation of MF-GPSCs (255 ± 48.2% of prepairing values, p < 0.001, paired t test, n = 13). This effect was associated with a significant increase in success rate (from 0.37 ± 0.06 to 0.7 ± 0.06, n = 13, p < 0.001; Fig. 2A), in the inverse square value of the coefficient of variation (CV−2) of response amplitude (from 1.09 ± 0.37 to 2.83 ± 0.97, n = 13, p < 0.001; Fig. 2A), as well as a significant reduction in paired-pulse ratio (from 2.7 ± 0.6 to 1.5 ± 0.35, n = 8, p < 0.05; Fig. 2A). This suggests an increased probability of GABA release.
Figure 2.

Presynaptic expression of LTP and LTD in WT and _Prnp_0/0 mice. A, B, Summary plot (columns) of successes, CV−2, PPR of MF-GPSCs measured in individual cells before pairing (Control, white) and after pairing (Pairing, black) in WT (A) and in _Prnp_0/0 animals (B). ***p < 0.001.
In _Prnp_0/0 mice, on the other hand, the pairing procedure always induced a persistent depression of GPSCs (30 min after pairing, the peak amplitude of GPSCs was reduced to 36.3 ± 5.6% of prepairing values, n = 15, p < 0.001, paired t test; Fig. 1D). This effect was mainly caused by a decrease in success rate (from 0.57 ± 0.06 to 0.21 ± 0.06, before and after pairing, respectively, n = 15, p < 0.001; Fig. 2B). As expected for a reduction in the probability of GABA release from MF terminals, the increased number of failures was associated with a significant increase in PPR (from 1.05 ± 0.1 to 2.1 ± 0.7, n = 7, p = 0.01; Fig. 2B) and a significant decrease in CV−2 (from 3.9 ± 1.3 to 2.2 ± 0.9, n = 15, p < 0.001; Fig. 2B).
In 6 of 15 cases, the pairing procedure led to synapse silencing (Fig. 3) (Voronin and Cherubini, 2004).
Figure 3.
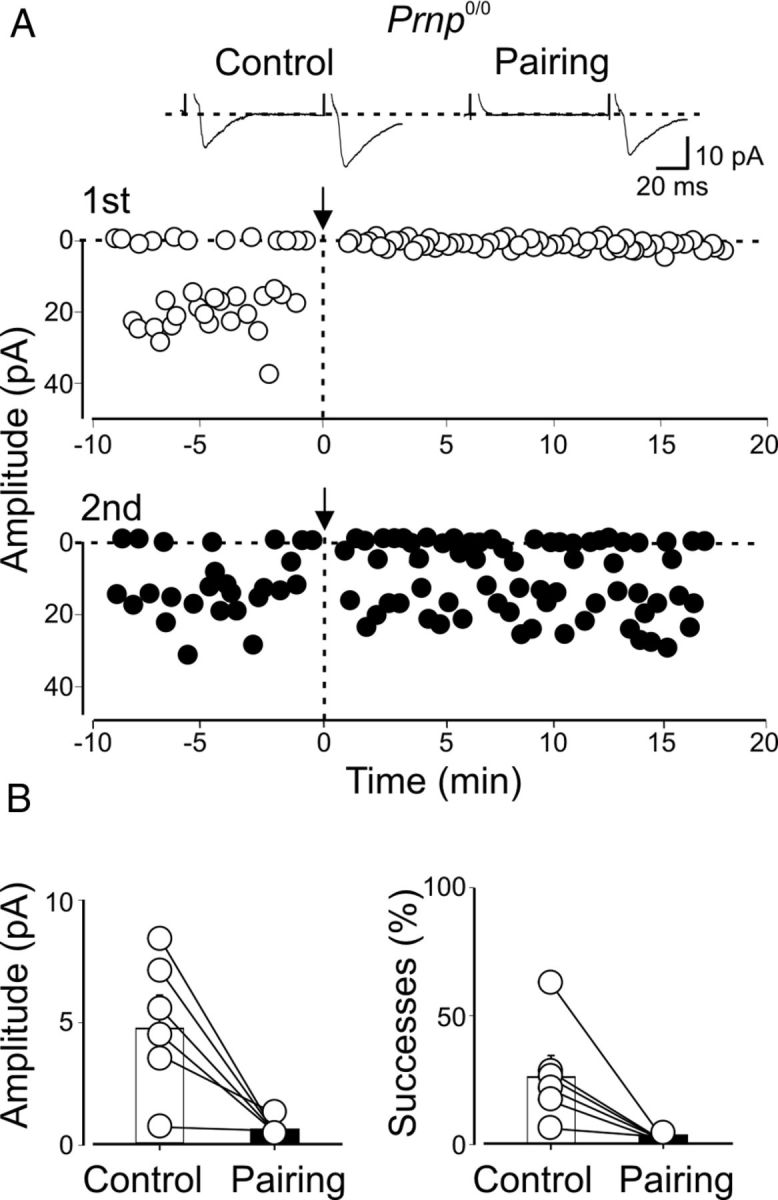
Pairing-induced synapse silencing in CA3 principal cells from _Prnp_0/0 mice. A, Amplitudes of synaptic responses (insets above graphs) evoked in a P4 CA3 principal cell by two stimuli (50 ms apart; open symbols, GPSCs evoked by first stimulus; closed symbols, GPSCs evoked by second stimulus) obtained before and after pairing (arrows at time 0) are plotted against time. B, Summary plots for six cells silenced after pairing. Amplitudes and successes measured in individual cells before pairing (Control, white columns) and after pairing (Pairing, black columns).
Pairing-induced synaptic depression or silencing was not due to run down of synaptic responses. In fact, GPSCs evoked by a second stimulus (50 ms after the first one) were only slightly affected and, in the absence of pairing (just switching from voltage-clamp to current-clamp mode for 7 min), no changes in GPSC amplitude with time occurred (after 20 min, the amplitude of GPSCs was 108 ± 22% and 102 ± 12% of preswitching values in WT and _Prnp_0/0 mice, respectively; n = 5 for both; p > 0.05; data not shown).
In WT mice, LTP induction is postsynaptic, whereas in _Prnp_0/0 mice LTD is presynaptic
In agreement with earlier data (Kasyanov et al., 2004), loading postsynaptic neurons from WT animals with the calcium chelator BAPTA (20 mm) prevented pairing-induced synaptic potentiation and revealed synaptic depression (the mean amplitude of GPSCs was 47 ± 9% of prepairing values, n = 7, p < 0.001, paired t test; Fig. 4A). However, in _Prnp_0/0 mice, intracellular BAPTA did not affect pairing-induced synaptic depression (20 min after pairing, the peak amplitude of GPSCs was 41 ± 12% of prepairing values, n = 7, p < 0.05; Fig. 4B). In BAPTA, MF-GPSC depression did not significantly differ from that obtained in the absence of BAPTA (p = 0.6, unpaired t test). This suggests that in _Prnp_0/0 mice the induction of LTD is independent of postsynaptic calcium rise and possibly presynaptic in origin. To test this hypothesis in a new set of experiments, the conditioning protocol was applied to cells maintained in voltage-clamp mode (at −70 mV). This procedure prevents the depolarization of the postsynaptic cell and calcium influx through high-voltage-activated calcium channels and/or NMDA receptors (Malenka and Nicoll, 1999). In line with a presynaptic induction mechanism, this condition did not affect LTD in _Prnp_0/0 mice but it prevented synaptic potentiation and caused synaptic depression in WT mice (mean GPSC amplitude: WT mice, 44.6 ± 6.2% of prepairing values, n = 6, p = 0.007, paired t test; _Prnp_0/0 mice, 40.1 ± 6.5% of prepairing values, n = 9, p < 0.001, paired t test; Fig. 4C,D). The depressant effect observed in _Prnp_0/0 mice was not significantly different from that obtained when pairing was performed in current-clamp conditions (p = 0.6, unpaired t test).
Figure 4.

Loading the cells with BAPTA or maintaining them close to their resting values during pairing blocks LTP and reveals LTD in WT animals. A, C, Plot of mean MF-GPSCs amplitude from CA3 pyramidal cells loaded with BAPTA (n = 7; A) or voltage clamped at −70 mV (n = 6; C) before and after pairing (arrows at time 0) versus time. Insets above the graphs represent individual traces of GPSCs evoked before and after pairing. Note pairing-induced LTD instead of LTP. B, D, As in A and C but from _Prnp_0/0 mice (B, n = 7; D, n = 9).
As expected for a presynaptic type of action, LTD was associated with a significant reduction in success rate (WT mice, from 0.74 ± 0.06 to 0.42 ± 0.1, n = 7, p < 0.01; _Prnp_0/0 mice, from 0.67 ± 0.09 to 0.29 ± 0.1, n = 8, p < 0.01), a significant increase in PPR (WT mice, from 1.4 ± 0.1 to 2.1 ± 0.3, n = 5, p < 0.05; _Prnp_0/0 mice, from 1.1 ± 0.2 to 1.5 ± 0.3, n = 5, p < 0.05), and a marked decrease in CV−2 (WT mice, from 3 ± 0.67 to 2.3 ± 0.7, n = 7, p < 0.05; _Prnp_0/0 mice, from 3.3 ± 1.4 to 1.8 ± 1.2, n = 6, p < 0.01; Fig. 5).
Figure 5.

Presynaptic expression of pairing-induced LTD in CA3 pyramidal cells loaded with BAPTA. A, B, Summary plots of successes, CV−2, PPR of MF-GPSCs measured in individual cells before pairing (Control, white) and after pairing (Pairing, black) in WT (A, n = 7) and in _Prnp_0/0 animals (B, n = 7). *p < 0.05; **p < 0.01. As expected for a presynaptic type of action, pairing-induced LTD was associated with a significant reduction in success rate (WT mice, from 0.74 ± 0.06 to 0.42 ± 0.1, n = 7, p < 0.01; _Prnp_0/0 mice, from 0.67 ± 0.09 to 0.29 ± 0.1, n = 8, p < 0.01), a significant increase in PPR (WT mice, from 1.4 ± 0.1 to 2.1 ± 0.3, n = 5, p < 0.05; _Prnp_0/0 mice, from 1.1 ± 0.2 to 1.5 ± 0.3, n = 5, p < 0.05), and a marked decrease in CV−2 (WT mice, from 3 ± 0.67 to 2.3 ± 0.7, n = 7, p < 0.05; _Prnp_0/0 mice, 3.3 ± 1.4 to 1.8 ± 1.2, n = 6, p < 0.01).
In WT animals, synaptic depression can thus be revealed by either blocking postsynaptic calcium rise with BAPTA or by preventing the opening of voltage-gated calcium channels by clamping the membrane below the resting level.
In WT mice, pairing-induced LTP requires postsynaptic cAMP-dependent PKA activity
Interestingly, at immature glutamatergic Schaffer collateral-CA1 synapses, LTP requires the activation of postsynaptic cAMP-dependent PKA (Yasuda et al., 2003) for its induction. PKA activation is also necessary for spike time-dependent LTP at immature GABAergic MF-CA3 synapses (Sivakumaran et al., 2009), indicating that, in the immature hippocampus, PKA signaling is critically implicated in activity-dependent synaptic plasticity notwithstanding the nature of the neurotransmitter. To test whether postsynaptic PKA activity is responsible for pairing-induced LTP at MF-CA3 synapses in WT mice, we loaded the cell with the membrane-impermeable PKA inhibitor PKI 6-22 (1 μm). Under these conditions, the pairing procedure failed to induce LTP. Instead it revealed a persistent depression of MF-GPSCs (66 ± 9.8% of prepairing values, n = 11, p < 0.001, paired t test; Fig. 6).
Figure 6.
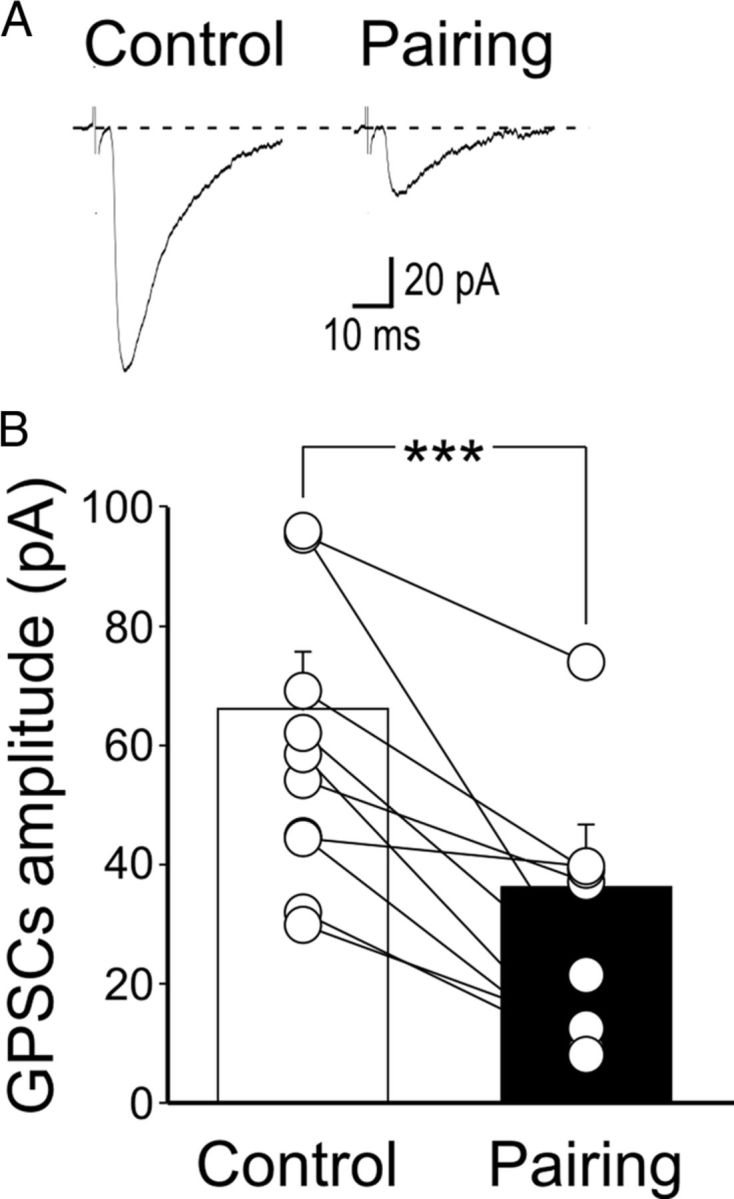
Inhibiting PKA in the postsynaptic neuron prevents pairing-induced LTP in WT mice. A, Sample traces of a P5 CA3 pyramidal cell (loaded with the membrane-impermeable PKA inhibitor PKI 6-22) obtained before pairing (Control) and 30 min after pairing. B, Summary plot of MF-GPSC amplitudes measured in individual cells loaded with PKI 6-22 before pairing (Control, white) and after pairing (Pairing, black). ***p < 0.001.
These data prove that, in WT animals, PKA activity is crucial for triggering LTP at immature MF-CA3 synapses. To examine whether a postsynaptic deficit of PKA activity accounts for the loss of LTP in _Prnp_0/0 mice, we tested forskolin, an activator of PKA, on spontaneous mGPSCs recorded from CA3 pyramidal cells. In WT animals, forskolin (50 μm) increased the frequency of mGPSCs only slightly and at a level that was not significant (138.7 ± 23%, n = 10, p > 0.05, paired t test; Fig. 7D). However, forskolin significantly increased mGPSC amplitude (128.4 ± 3.3% of control, n = 10, p < 0.001, paired _t_ test; Fig. 7_A_,_E_; Yasuda et al., 2003). The potentiating effects of forskolin on mGPSCs amplitude were prevented by loading the cell with the membrane-impermeable PKA inhibitor PKI 6-22 (1 μm; Fig. 7_B_,_E_). In _Prnp_0/0 mice, forskolin slightly increased the frequency of mGPSCs (124.7 ± 11.8% of control, _n_ = 8, _p_ > 0.05, paired t test; Fig. 7D), but failed to enhance their amplitude (92 ± 3.6% of control, n = 8, p > 0.05, paired t test; Fig. 7C,E).
Figure 7.

In WT but not in _Prnp_0/0 mice, forskolin enhances the amplitude of miniature GPSCs. A, Amplitude distribution of miniature GPSCs recorded in CA3 principal cells from WT animals under control conditions and during application of forskolin (n = 10). B, As in A but with the membrane-impermeable PKA inhibitor PKI 6-22 into the patch pipette (n = 8). C, As in A but from _Prnp_0/0 mice (n = 8). Note reduction of larger amplitude events with forskolin in B and C. Bin width, 5 pA. D, E, Summary plots of forskolin-induced increase in frequency (D) or in amplitude (E) of miniature GABAergic events in cells from WT animals (white columns), in cells from WT animals but loaded with PKI 6-22 (gray columns), or in cells from _Prnp_0/0 mice (black columns). ***p < 0.001 (respect to prepairing values, paired t test); values for WT plus PKI and _Prnp_0/0 versus WT were significantly different (p < 0.001, 1-way ANOVA).
These experiments provide evidence that PrPC is associated with the PKA signaling pathway at immature MF-CA3 synapses and this might account for the lack of pairing-induced LTP in PrPC-deficient mice.
To validate the hypothesis that the lack of postsynaptic PKA activity is responsible for the loss of LTP in _Prnp_0/0 mice, additional experiments were performed using a patch pipette containing the constitutively active PKA catalytic subunits Cα to mimic PKA activation (Duffy and Nguyen, 2003). In these conditions, MF-GPSCs started to increase in amplitude immediately after breaking the membrane, and attained a steady-state value in 15 ± 2 min (after 20 min, the peak amplitude of GPSCs was 149 ± 3% of the values obtained during the first 3 min, n = 9, p < 0.001, paired t test; Fig. 8).
Figure 8.

Postsynaptic application of the catalytic subunit of PKA (Cα) induces MF-GPSC facilitation in the hippocampus of _Prnp_0/0 mice. Infusion of the constitutively active isoform of PKA catalytic subunit Cα into CA3 pyramidal neurons via the patch pipette progressively enhanced the amplitude of MF-GPSCs, which reached a steady state after ∼10 min. Subsequent pairing (at the time 0, arrow) did not modify GPSC amplitude, suggesting occlusion. While Cα was applied to nine CA3 pyramidal neurons, pairing was performed only on six of nine cells. Insets, Sample traces from an individual experiment taken at the time indicated.
At this point we switched from voltage-clamp to current-clamp mode and we used a pairing protocol that consisted of stimulating MFs with the rising phase of GDPs. This procedure failed to induce LTD. Instead, MF-GPSCs remained potentiated, suggesting occlusion (15 min after pairing, the peak amplitude of MF-GPSCs was 155 ± 21%, a value not significantly different from that obtained immediately before pairing, p = 0.9, paired t test).
Loss of PKA-dependent LTP at Schaffer collateral-CA1 synapses in the hippocampus of _Prnp_0/0 mice
To further assess whether the lack of postsynaptic PKA activity may account for the loss of LTP in _Prnp_0/0 mice, a pairing procedure was applied to immature CA1 principal cells known to critically depend on postsynaptic PKA signaling for LTP (Yasuda et al., 2003). Pairing repetitive stimulation (50 stimuli at 1 Hz) of Schaffer collateral with depolarization of CA1 principal cells to 0 mV (Yasuda et al. 2003) caused in P3–P7 WT animals a persistent increase in synaptic efficacy (30 min after pairing, the mean EPSC amplitude was 143.6 ± 8.9% of controls, n = 6, p = 0.01, paired t test; Fig. 9A).
Figure 9.

Lack of PKA-dependent LTP at Schaffer collateral-CA1 synapses in the hippocampus of _Prnp_0/0 mice. A, The mean EPSC amplitude recorded before and after pairing (arrow at time 0) is plotted against time in WT (open symbols; n = 6) and in _Prnp_0/0 mice (closed symbols; n = 14). Insets above the graph represent sample traces of Schaffer collateral-CA1 EPSCs obtained in WT and _Prnp_0/0 mice before (left) and after pairing (right). B, Blocking postsynaptic PKA with the membrane-impermeable PKA inhibitor PKI 6-22 prevents LTP induction. Each column represents pairing-induced changes of EPSC amplitude (normalized to prepairing values) in CA1 pyramidal cells from WT animals (WT, white, n = 6), from WT animals in the presence of PKI 6-22 (+PKI, gray, n = 5), and from _Prnp_0/0 mice (_Prnp_0/0, black, n = 14). Data obtained from WT animals in the presence of PKI 6-22 and from _Prnp_0/0 versus WT were significantly different (p < 0.001, 1-way ANOVA). C, Infusion of the constitutively active isoform of PKA catalytic subunit Cα into CA1 pyramidal cells (n = 7) via the patch pipette progressively increased the amplitude of EPSCs evoked by Schaffer collateral stimulation. The amplitude reached a peak after ∼10 min. The pairing protocol (applied at time 0, arrow) failed to modify EPSC amplitude, suggesting occlusion. Insets, Sample traces from an individual experiment taken at the time indicated.
This effect was prevented when the postsynaptic cell was loaded with the membrane-impermeable PKA inhibitor PKI 6-22 (1 μm; after pairing, the mean EPSC amplitude was 88 ± 8% of prepairing values, n = 5, p > 0.05, paired t test; Fig. 9B). In contrast, in _Prnp_0/0 mice, the same protocol failed to induce LTP (the mean EPSC amplitude was 85.2 ± 4.6% of control, n = 14, p < 0.05, paired t test; Fig. 9). In 5 of 14 cases, included in the summary graph of Figure 9, pairing produced a synaptic depression of 33% of controls, n = 5, p = 0.003, paired t test). However, similarly to GPSCs at MF-CA3 synapses, infusion of the constitutively active PKA catalytic subunit Cα induced an increase in amplitude of EPSCs evoked in CA1 principal cells by Schaffer collateral stimulation. Amplitude reached a steady state in 11 ± 2 min (after 20 min, the peak amplitude of EPSCs was 141 ± 2% of the values obtained during the first 3 min, n = 7, p = 0.01, paired t test; Fig. 9C). At this stage, the pairing procedure failed to further potentiate EPSCs, suggesting occlusion (15 min after pairing, the peak amplitude of EPSCs was 143 ± 2%, a value not significantly different from that obtained immediately before pairing, p = 0.07, paired t test; Fig. 9C).
Together, these data further indicate that, in PrPC-deficient mice, the loss of PKA activity is responsible for the lack of pairing-induced LTP at both MF-CA3 synapses and Schaffer collateral-CA1 synapses.
In _Prnp_0/0 mice, pairing-induced LTD requires the activation of presynaptic GluK1 receptors and phospholipase C
While the present experiments clearly show that PKA-dependent LTP in _Prnp_0/0 mice is lost not only at MF-CA3 synapses but also at Schaffer collateral-CA1 synapses, the experiments do not clarify the mechanisms responsible for pairing-induced LTD. The PKA signaling pathway was not involved in pairing-induced LTD because application of the membrane-permeable PKA inhibitor PKI 14-22 (1 μm) in _Prnp_0/0 mice failed to prevent LTD (GPSC amplitude was 42.2 ± 8.2% of controls, n = 5, p = 0.043, paired t test; data not shown).
Among presynaptic receptors known to depress GABA release from immature MF terminals, high-affinity GluK1 receptors play a crucial role (Caiati et al., 2010). We hypothesized that these receptors could be activated during the pairing procedure by a large amount of glutamate released from network-driven GDPs. In the following experiments from _Prnp_0/0 mice, pairing was thus performed in the presence of the GluK1 antagonist UBP 302 (10 μm). As shown in Figure 10, this compound fully prevented pairing-induced synaptic depression (on average the peak amplitude of GPSCs was 91.8 ± 4.5% of prepairing values, n = 9, p > 0.05, paired t test). This value significantly differed from that obtained in the absence of UBP 302 (p < 0.001, 1-way ANOVA), indicating that GluK1 receptors are indeed responsible for LTD.
Figure 10.

In _Prnp_0/0 mice, at MF-CA3 synapses, pairing-induced LTD is prevented by blocking G-protein-coupled GluK1 receptors or phospholipase C downstream to G-protein activation. A, The mean amplitude of MF-GPSCs recorded in _Prnp_0/0 mice (n = 9) in the presence of the GluK1 antagonist UBP 302 before and after pairing (arrow at time 0) is plotted versus time. Insets above the graph represent individual traces of MF-GPSCs evoked before (Control) and after pairing. B, Each column represents the mean MF-GPSC amplitude obtained 30 min after pairing and normalized to prepairing values in controls (white, n = 15), in the presence of UBP 302 (black, n = 9), and in the presence of PLC inhibitor U73122 (gray, n = 8). Data obtained in the presence of UBP 302 and U73122 versus controls were significantly different (p < 0.001; 1-way ANOVA).
In the absence of pairing, UBP 302 only slightly enhanced the amplitude of MF-GPSCs (to 121.7 ± 9.8% of controls) without reaching a significant level (p > 0.05). This suggests that under resting conditions the amount of glutamate present in the extracellular medium is not sufficient to tonically activate presynaptic GluK1 receptors. However, in agreement with a previous study on rats (Caiati et al., 2010), GluK1 receptors are present on MF terminals of _Prnp_0/0 mice since the GluK1 agonist (RS)-2-amino-3-(3-hydoxy-5-tert-butylisoxazol-4-yl) popanoic acid (ATPA) (1 μm) caused a reduction of MF-GPSC of 41.5 ± 7.2% compared to controls (n = 5; p < 0.05; data not shown).
In the developing hippocampus, endogenously released glutamate depresses MF-GPSCs via presynaptic Gi/o-protein-coupled high-affinity kainate receptors (Caiati et al., 2010). The most common signaling pathway, downstream to G-protein activation, involves phospholipase C (PLC) and intracellular calcium (Lerma, 2006; Rodríguez-Moreno and Sihra, 2007). Therefore, in the following experiments using _Prnp_0/0 mice, we tested whether the PLC inhibitor U73122 is able to block pairing-induced LTD. As shown in Figure 10B, U73122 (10 μm) prevented pairing-induced synaptic depression (after pairing, the mean GPSC amplitude was 89 ± 8.7% of prepairing values, n = 8, p > 0.05, paired t test). Data obtained in the presence of U73122 were significantly different from controls (p < 0.001, 1-way ANOVA). Pairing-induced synaptic weakening was not mediated by other receptors known to depress transmitter release, such as GABAB receptors (Safiulina and Cherubini, 2009), nicotine receptors, muscarinic receptors, acetylcholine receptors (Maggi et al., 2004), purinergic P2Y receptors (Zhang et al., 2003; Safiulina et al., 2005), adenosine receptors, and mGluRs (Scanziani et al., 1997), because blocking these receptors with a mixture containing selective antagonists did not modify LTD (Fig. 11). In addition, pairing-induced synaptic depression was still present when NMDA receptors were blocked with d-AP5 (50 μm), indicating that this form of synaptic plasticity is independent of NMDA receptor activation (Fig. 11).
Figure 11.

At MF-CA3 synapses in slices from _Prnp_0/0 mice, LTD results from the direct effect of pairing on GluK1 receptors. Each column represents the mean MF-GPSC amplitude obtained 30 min after pairing and normalized to prepairing values in control samples (white, n = 15), in the presence of different receptor antagonists (Antag, gray, n = 6), and in the presence of d-AP5 (black, n = 7). Receptor antagonists comprise the following: 3 μm CGP 52432, 1 μm atropine, 50 μm dihydro-β-erythroidine, 10 μm 8-cyclopentyl-1,3-dipropylxanthine, 50 μm pyridoxal-phosphate-6-azophenyl-2-disulphonic acid, and 100 μm LY341495 to block GABAB, muscarinic, nicotinic, adenosine, purinergic P2Y, and mGluR receptors, respectively. Data obtained in the presence of different receptor antagonists and d-AP5 were not significantly different from controls: p = 0.73 and 0.44, respectively (1-way ANOVA).
Discussion
In this study we describe a previously unknown effect of PrPC, namely its ability to control, at early developmental stages, the direction of synaptic plasticity in the hippocampus. In particular, we show that pairing GDP-associated calcium transients with stimulation of granule cells in the dentate gyrus triggers synaptic potentiation in WT animals and synaptic depression in _Prnp_0/0 mice. As for other forms of synaptic plasticity, calcium entering the postsynaptic neuron through voltage-dependent calcium channels opening by the depolarizing action of GABA during GDPs (Ben-Ari et al., 2007) is responsible for LTP. Hence, in agreement with a previous report (Kasyanov et al., 2004), LTP induction was clearly postsynaptic as it could be prevented by loading the postsynaptic cell with BAPTA. Interestingly, in the presence of BAPTA, the pairing procedure unveiled LTD, suggesting the two phenomena are tightly correlated (Kasyanov et al., 2004; Sivakumaran et al., 2009). While the induction of LTP was clearly postsynaptic, its expression was presynaptic as indicated by pairing-induced decrease in failure rate and paired-pulse facilitation, as well as by the increase in CV−2, all traditional indices of presynaptic changes in release probability (Zucker, 1989). In _Prnp_0/0 mice, on the other hand, the pairing procedure failed to induce synaptic potentiation, yet it caused synaptic depression, which in some cases led to synapse silencing. LTD was unaffected by loading the postsynaptic cell with BAPTA, indicating a possible presynaptic origin. This was further supported by the observation that LTD was not affected when the pairing protocol was delivered to cells kept close to their resting membrane potential to prevent membrane depolarization. However, this procedure revealed LTD in WT animals, indicating that also in this case LTD was presynaptic. Like LTP, LTD is also likely to involve network-driven GDPs, which, being synchronous over the entire hippocampus, may act as coincident detectors of signals occurring at both postsynaptic and presynaptic sites.
Similarly to our results, theta burst stimulation of cerebellar MFs was unable to induce LTP in granule cells in slices from juvenile (but not adult) PrP0/0 mice (Zurich-I; Büeler et al., 1992), a process probably related to impaired motor control and reinforcement (Prestori et al., 2008). However, while in the cerebellum the lack of LTP probably depended on the reduced cell excitability that prevented granule cells from firing, in our case lack of LTP was independent of any apparent change in passive membrane properties, neuronal spiking, and synaptic or networking activity.
We also found that in WT animals, LTP required the activation of cAMP-dependent PKA since pairing-induced synaptic potentiation was lost when the postsynaptic cell was loaded with the membrane-impermeable PKA inhibitor PKI 6-22. This compound was also able to block the potentiating effect of the adenylate cyclase activator forskolin on the amplitude of miniature GABAergic currents, further indicating that the PKA signaling pathway is crucial for modifying synaptic strength, possibly through phosphorylation of GABAA receptors (Cherubini and Conti, 2001). Note that while in neonates forskolin acts mainly at postsynaptic sites (Yasuda et al., 2003), in juvenile animals it affects mainly presynaptic sites by increasing GABA release (Sciancalepore and Cherubini, 1995). Postsynaptic PKA activity has been involved in LTP induced in the immature hippocampus when MF stimulation is paired with back-propagating action potentials (Sivakumaran et al., 2009). As for spike time-dependent plasticity, cAMP-dependent PKA activity is likely also in the present case to play an essential role in regulating intracellular calcium levels and second messenger cascades (Obrietan and van den Pol, 1997).
Interestingly, PKA together with ERK1/2 signaling pathways are controlled by PrPC, thus contributing to memory consolidation (Coitinho et al., 2006). The loss of pairing-induced potentiation of MF-GPSCs, together with the loss of sensitivity of miniature GABAergic currents to forskolin in _Prnp_0/0 mice, provides evidence that the prion protein controls synaptic activity by interacting directly or indirectly with postsynaptic PKA activity. The contribution of PrPC to the regulation of hippocampal synaptic plasticity through the PKA signaling pathway was further confirmed by the observation that in _Prnp_0/0 mice, stimulation of Schaffer collateral failed to induce LTP, known to depend on postsynaptic PKA activation (Yasuda et al., 2003). Further support in favor of the hypothesis that the loss of postsynaptic PKA activity prevents pairing-induced LTP in PrPC-deficient mice is the observation that intracellular infusion of the constitutively active PKA catalytic subunit Cα is able to potentiate synaptic events at both MF-CA3 and Schaffer collateral-CA1 synapses, an effect that can be occluded by a subsequent pairing protocol. It should be stressed that, in the developing brain, PrPC is particularly abundant in plastic regions, including the hippocampus where, acting as a growth factor, it contributes to neurogenesis and differentiation via different transduction pathways, including cAMP-dependent PKA (Kanaani et al., 2005; Steele et al., 2006).
If the loss of postsynaptic PKA activity is responsible for the lack of LTP in _Prnp_0/0 mice, what determines LTD at these synapses? Several lines of evidence suggest that this form of synaptic plasticity is presynaptic in origin. Early in postnatal development, MFs are endowed with several neurotransmitter receptors, including GABAB receptors (Safiulina and Cherubini, 2009), GluK1 receptors (Caiati et al., 2010), mGluRs (Scanziani et al., 1997), nAChRs, mAChRs (Maggi et al., 2004), adenosine, and purinergic P2Y receptors (Zhang et al., 2003; Safiulina et al., 2005) known to downregulate GABA release. Among these, high-affinity GluK1 receptors, which are highly expressed in the immature hippocampus mainly at presynaptic sites, play a crucial role (Lauri et al., 2005; Caiati et al., 2010; present data with ATPA). Indeed, blocking these receptors with a selective antagonist fully prevented LTD induction in _Prnp_0/0 mice. This effect was not mediated by other signaling molecules known to inhibit GABA release since the pairing protocol was still able to cause LTD in the presence of various receptor antagonists. LTD was also NMDA independent as it could be triggered in the presence of d-AP5.
In a previous study on rats, we demonstrated that GluK1 receptors localized on MF terminals are activated by a high level of glutamate in the extracellular space, probably maintained by a less efficient glutamate transport system and a poorly developed diffusional barrier (Caiati et al., 2010). Compared with previously reported data (Caiati et al., 2010), the amount of “ambient” glutamate in the extracellular milieu of _Prnp_0/0 mice was probably lower because the GluK1 antagonist only slightly enhanced the amplitude of MF-GPSCs. Although we have not ascertained the reason for this apparent discrepancy, it is likely that during network-driven bursts, extracellular glutamate rises to a level sufficient to activate high-affinity G-coupled GluK1 receptors present on MF terminals, which are known to exert a ionotropic and metabotropic type of action (Lerma, 2006). In _Prnp_0/0 mice, the effect of GluK1 on the persistent depression of GABA release from MF terminals depended on a metabotropic type of action as LTD was prevented by blocking PLC downstream of G-protein activation. The blocking effect of the PLC inhibitor on spike time-dependent depression observed in rats (Caiati et al., 2010) is in line with the present observations. In _Prnp_0/0 mice, however, blocking GluK1 did not reveal LTP as in rats (Caiati et al., 2010) because of the loss of postsynaptic PKA signaling.
We cannot exclude the possibility that the loss of postsynaptic PKA activity in Prnp0/0 mice is due to genetic manipulations that may alter the biochemical signaling at synapses. However, electrophysiological experiments did not unveil any difference in basic synaptic transmission and/or correlated network activity between WT and Prnp0/0 animals, making this possibility unlikely. In addition, a recent analysis of the effects of Prnp disruption on the whole transcriptome has shown only minor modifications of the gene expression profile (Benvegnù et al., 2011). It is worth mentioning that the genes coding for the proteins involved in the PKA signaling do not localize in the proximity of Prnp, thus ruling out the possibility that the alteration of this pathway may be due to the genetic manipulation of Prnp. Altogether, these observations suggest that the lack of expression of PrPC is the cause of activity-dependent modifications found in _Prnp_0/0 and highlight the “gating” role PrPC-dependent PKA signaling in regulating synaptic plasticity and information processing in the developing hippocampus.
Footnotes
This work was supported by a grant from the Ministero Istruzione Università e Ricerca to E.C. (Grant MIUR-PRIN 2009) and by a grant from the European Union (FP7/2007-2013 N. 222887) to G.L. We are grateful to L. Gasperini for participating in some experiments and to Erica Sarnataro for editing support.
The authors declare no competing financial interests.
References
- Allen C, Stevens CF. An evaluation of causes for unreliability of synaptic transmission. Proc Natl Acad Sci U S A. 1994;91:10380–10383. doi: 10.1073/pnas.91.22.10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral DG, Dent JA. Development of the mossy fibers of the dentate gyrus: I. A light and electron microscopic study of the mossy fibers and their expansions. J Comp Neurol. 1981;195:51–86. doi: 10.1002/cne.901950106. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Cherubini E, Corradetti R, Gaiarsa JL. Giant synaptic potentials in immature rat CA3 hippocampal neurones. J Physiol. 1989;416:303–325. doi: 10.1113/jphysiol.1989.sp017762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Gaiarsa JL, Tyzio R, Khazipov R. GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev. 2007;87:1215–1284. doi: 10.1152/physrev.00017.2006. [DOI] [PubMed] [Google Scholar]
- Benvegnù S, Poggiolini I, Legname G. Neurodevelopmental expression and localization of the cellular prion protein in the central nervous system of the mouse. J Comp Neurol. 2010;518:1879–1891. doi: 10.1002/cne.22357. [DOI] [PubMed] [Google Scholar]
- Benvegnù S, Roncaglia P, Agostini F, Casalone C, Corona C, Gustincich S, Legname G. Developmental influence of the cellular prion protein on the gene expression profile in mouse hippocampus. Physiol Genomics. 2011;43:711–725. doi: 10.1152/physiolgenomics.00205.2010. [DOI] [PubMed] [Google Scholar]
- Büeler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C. Normal development and behavior of mice lacking the neuronal cell-surface Prp protein. Nature. 1992;356:577–582. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- Caiati MD, Sivakumaran S, Cherubini E. In the developing rat hippocampus, endogenous activation of presynaptic kainate receptors reduces GABA release from mossy fiber terminals. J Neurosci. 2010;30:1750–1759. doi: 10.1523/JNEUROSCI.4566-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carleton A, Tremblay P, Vincent JD, Lledo PM. Dose-dependent, prion protein (PrP)-mediated facilitation of excitatory synaptic transmission in the mouse hippocampus. Pflugers Archiv. 2001;442:223–229. doi: 10.1007/s004240100523. [DOI] [PubMed] [Google Scholar]
- Cherubini E, Conti F. Generating diversity at GABAergic synapses. Trends Neurosci. 2001;24:155–162. doi: 10.1016/s0166-2236(00)01724-0. [DOI] [PubMed] [Google Scholar]
- Coitinho AS, Freitas ARO, Lopes MH, Hajj GN, Roesler R, Walz R, Rossato JI, Cammarota M, Izquierdo I, Martins VR, Brentani RR. The interaction between prion protein and laminin modulates memory consolidation. Eur J Neurosci. 2006;24:3255–3264. doi: 10.1111/j.1460-9568.2006.05156.x. [DOI] [PubMed] [Google Scholar]
- Colby DW, Prusiner SB. Prions. Cold Spring Harb Perspect Biol. 2011;3:a006833. doi: 10.1101/cshperspect.a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colling SB, Collinge J, Jefferys JG. Hippocampal slices from prion protein null mice: disrupted Ca(2+)-activated K+ currents. Neurosci Lett. 1996;209:49–52. doi: 10.1016/0304-3940(96)12596-9. [DOI] [PubMed] [Google Scholar]
- Collinge J, Whittington MA, Sidle KC, Smith CJ, Palmer MS, Clarke AR, Jefferys JG. Prion protein is necessary for normal synaptic function. Nature. 1994;370:295–297. doi: 10.1038/370295a0. [DOI] [PubMed] [Google Scholar]
- Duffy SN, Nguyen PV. Postsynaptic application of a peptide inhibitor of cAMP-dependent protein kinase blocks expression of long-lasting synaptic potentiation in hippocampal neurons. J Neurosci. 2003;23:1142–1150. doi: 10.1523/JNEUROSCI.23-04-01142.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann M, Bittner T, Mitteregger G, Haider N, Moosmang S, Kretzschmar H, Herms J. Loss of the cellular prion protein affects the Ca2+ homeostasis in hippocampal CA1 neurons. J Neurochem. 2006;98:1876–1885. doi: 10.1111/j.1471-4159.2006.04011.x. [DOI] [PubMed] [Google Scholar]
- Gutiérrez R, Romo-Parra H, Maqueda J, Vivar C, Ramìrez M, Morales MA, Lamas M. Plasticity of the GABAergic phenotype of the “glutamatergic” granule cells of the rat dentate gyrus. J Neurosci. 2003;23:5594–5598. doi: 10.1523/JNEUROSCI.23-13-05594.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas P, Major G, Sakmann B. Quantal components of unitary EPSCs at the mossy fiber synapse on CA3 pyramidal cells of rat hippocampus. J Physiol. 1993;472:615–663. doi: 10.1113/jphysiol.1993.sp019965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaani J, Prusiner SB, Diacovo J, Baekkeskov S, Legname G. Recombinant prion protein induces rapid polarization and development of synapses in embryonic rat hippocampal neurons in vitro. J Neurochem. 2005;95:1373–1386. doi: 10.1111/j.1471-4159.2005.03469.x. [DOI] [PubMed] [Google Scholar]
- Kasyanov AM, Safiulina VF, Voronin LL, Cherubini E. GABA-mediated giant depolarizing potentials as coincidence detectors for enhancing synaptic efficacy in the developing hippocampus. Proc Natl Acad Sci U S A. 2004;101:3967–3972. doi: 10.1073/pnas.0305974101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauri SE, Segerstråle M, Vesikansa A, Maingret F, Mulle C, Collingridge GL, Isaac JT, Taira T. Endogenous activation of kainate receptors regulates glutamate release and network activity in the developing hippocampus. J Neurosci. 2005;25:4473–4484. doi: 10.1523/JNEUROSCI.4050-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Magueresse C, Safiulina V, Changeux JP, Cherubini E. Nicotinic modulation of network and synaptic transmission in the immature hippocampus investigated with genetically modified mice. J Physiol. 2006;576:533–546. doi: 10.1113/jphysiol.2006.117572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerma J. Kainate receptor physiology. Curr Opin Pharmacol. 2006;6:89–97. doi: 10.1016/j.coph.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Maggi L, Sola E, Minneci F, Le Magueresse C, Changeux JP, Cherubini E. Persistent decrease in synaptic efficacy induced by nicotine at Schaffer collateral-CA1 synapses in the immature rat hippocampus. J Physiol. 2004;559:863–874. doi: 10.1113/jphysiol.2004.067041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. Long-term potentiation—a decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- Mallucci GR, Ratté S, Asante EA, Linehan J, Gowland I, Jefferys JG, Collinge J. Post-natal knockout of prion protein alters hippocampal CA1 properties, but does not result in neurodegeneration. EMBO J. 2002;21:202–210. doi: 10.1093/emboj/21.3.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manson JC, Hope J, Clarke AR, Johnston A, Black C, MacLeod N. PrP gene dosage and long term potentiation. Neurodegeneration. 1995;4:113–114. doi: 10.1006/neur.1995.0014. [DOI] [PubMed] [Google Scholar]
- Manson J, West JD, Thomson V, McBride P, Kaufman MH, Hope J. The prion protein gene: a role in mouse embryogenesis? Development. 1992;115:117–122. doi: 10.1242/dev.115.1.117. [DOI] [PubMed] [Google Scholar]
- McKinley MP, DeArmond SJ, Torchia M, Mobley WC, Prusiner SB. Acceleration of scrapie in neonatal Syrian hamsters. Neurology. 1989;39:1319–1324. doi: 10.1212/wnl.39.10.1319. [DOI] [PubMed] [Google Scholar]
- Mohajerani MH, Sivakumaran S, Zacchi P, Aguilera P, Cherubini E. Correlated network activity enhances synaptic efficacy via BDNF and the ERK pathway at immature CA3-CA1 connections in the hippocampus. Proc Natl Acad Sci U S A. 2007;104:13176–13181. doi: 10.1073/pnas.0704533104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obrietan K, van den Pol AN. GABA activity mediating cytosolic Ca2+ rises in developing neurons is modulated by cAMP-dependent signal transduction. J Neurosci. 1997;17:4785–4799. doi: 10.1523/JNEUROSCI.17-12-04785.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prestori F, Rossi P, Bearzatto B, Lainé J, Necchi D, Diwakar S, Schiffmann SN, Axelrad H, D'Angelo E. Altered neuron excitability and synaptic plasticity in the cerebellar granular layer of juvenile prion protein knock-out mice with impaired motor control. J Neurosci. 2008;28:7091–7103. doi: 10.1523/JNEUROSCI.0409-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Moreno A, Sihra TS. Metabotropic actions of kainate receptors in the CNS. J Neurochem. 2007;103:2121–2135. doi: 10.1111/j.1471-4159.2007.04924.x. [DOI] [PubMed] [Google Scholar]
- Safiulina VF, Cherubini E. At immature mossy fibers-CA3 connections, activation of presynaptic GABA(B) receptors by endogenously released GABA contributes to synapses silencing. Front Cell Neurosci. 2009;3:1. doi: 10.3389/neuro.03.001.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safiulina VF, Kasyanov AM, Sokolova E, Cherubini E, Giniatullin R. ATP contributes to the generation of network-driven giant depolarizing potentials in the neonatal rat hippocampus. J Physiol. 2005;565:981–992. doi: 10.1113/jphysiol.2005.085621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safiulina VF, Fattorini G, Conti F, Cherubini E. GABAergic signaling at mossy fiber synapses in neonatal rat hippocampus. J Neurosci. 2006;26:597–608. doi: 10.1523/JNEUROSCI.4493-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salès N, Hässig R, Rodolfo K, Di Giamberardino L, Traiffort E, Ruat M, Frétier P, Moya KL. Developmental expression of the cellular prion protein in elongating axons. Eur J Neurosci. 2002;15:1163–1177. doi: 10.1046/j.1460-9568.2002.01953.x. [DOI] [PubMed] [Google Scholar]
- Scanziani M, Salin PA, Vogt KE, Malenka RC, Nicoll RA. Use-dependent increases in glutamate concentration activate presynaptic metabotropic glutamate receptors. Nature. 1997;385:630–634. doi: 10.1038/385630a0. [DOI] [PubMed] [Google Scholar]
- Sciancalepore M, Cherubini E. PKA-dependent increase in frequency of miniature GABAergic currents in rat CA3 hippocampal neurons. Neurosci Lett. 1995;187:91–94. doi: 10.1016/0304-3940(95)11348-7. [DOI] [PubMed] [Google Scholar]
- Sivakumaran S, Mohajerani MH, Cherubini E. At immature mossy-fiber–CA3 synapses, correlated presynaptic and postsynaptic activity persistently enhances GABA release and network excitability via BDNF and cAMP-dependent PKA. J Neurosci. 2009;29:2637–2647. doi: 10.1523/JNEUROSCI.5019-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele AD, Emsley JG, Ozdinler PH, Lindquist S, Macklis JD. Prion protein (PrPc) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proc Natl Acad Sci U S A. 2006;103:3416–3421. doi: 10.1073/pnas.0511290103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele AD, Lindquist S, Aguzzi A. The prion protein knockout mouse: a phenotype under challenge. Prion. 2007;1:83–93. doi: 10.4161/pri.1.2.4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voronin LL, Cherubini E. 'Deaf, mute and whispering' silent synapses: their role in synaptic plasticity. J Physiol. 2004;557:3–12. doi: 10.1113/jphysiol.2003.058966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker MC, Ruiz A, Kullmann DM. Monosynaptic GABAergic signaling from dentate to CA3 with a pharmacological and physiological profile typical of mossy fiber synapses. Neuron. 2001;29:703–715. doi: 10.1016/s0896-6273(01)00245-8. [DOI] [PubMed] [Google Scholar]
- Yasuda H, Barth AL, Stellwagen D, Malenka RC. A developmental switch in the signaling cascades for LTP induction. Nat Neurosci. 2003;6:15–16. doi: 10.1038/nn985. [DOI] [PubMed] [Google Scholar]
- Zhang JM, Wang HK, Ye CQ, Ge W, Chen Y, Jiang ZL, Wu CP, Poo MM, Duan S. ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron. 2003;40:971–982. doi: 10.1016/s0896-6273(03)00717-7. [DOI] [PubMed] [Google Scholar]
- Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]