C‐type lectin domain group 14 proteins in vascular biology, cancer and inflammation (original) (raw)
Abstract
The C‐type lectin domain (CTLD) group 14 family of transmembrane glycoproteins consist of thrombomodulin, CD93, CLEC14A and CD248 (endosialin or tumour endothelial marker‐1). These cell surface proteins exhibit similar ectodomain architecture and yet mediate a diverse range of cellular functions, including but not restricted to angiogenesis, inflammation and cell adhesion. Thrombomodulin, CD93 and CLEC14A can be expressed by endothelial cells, whereas CD248 is expressed by vasculature associated pericytes, activated fibroblasts and tumour cells among other cell types. In this article, we review the current literature of these family members including their expression profiles, interacting partners, as well as established and speculated functions. We focus primarily on their roles in the vasculature and inflammation as well as their contributions to tumour immunology. The CTLD group 14 family shares several characteristic features including their ability to be proteolytically cleaved and engagement of some shared extracellular matrix ligands. Each family member has strong links to tumour development and in particular CD93, CLEC14A and CD248 have been proposed as attractive candidate targets for cancer therapy.
Keywords: cancer, CD248, CD93, CLEC14A, C‐type lectin, extracellular matrix, group XIV, immuno‐oncology, thrombomodulin, vascular targeting
The C‐type lectin domain‐containing group 14 family consists of thrombomodulin, CD248 (endosialin), CD93 and CLEC14A. These proteins exhibit a wide range of different functions in vascular biology, cancer and inflammation. In this review, we present and discuss what is known so far for each molecule and where current gaps in knowledge exist.
Abbreviations
ADAM10
a disintegrin and metalloproteinase‐10
ADC
antibody‐drug conjugate
CHO
Chinese hamster ovary
CTLD
C‐type lectin domain
ECD
extracellular domain
EGF
epidermal growth factor
EGFR1
epidermal growth factor receptor‐1
EMT
epithelial mesenchymal transition
EPCs
endothelial progenitor cells
ERK
extracellular‐signal regulated kinase
ERM
ezrin‐radixin‐moesin
GPR15
G protein‐coupled receptor‐15
GVHD
graft versus host disease
HCC
hepatocellular carcinoma
HMGB1
high‐mobility group protein B1
HUVEC
human umbilical vein endothelial cells
LLCs
Lewis lung carcinomas
LPS
lipopolysaccharide
MCAM
melanoma cell adhesion molecule
MMP9
matrix metalloproteinase‐9
MMRN2
multimerin‐2
NSCLC
nonsmall cell lung cancer
PDGF
platelet‐derived growth factor
PI3K
phosphoinositide 3‐kinase
RHBDL2
rhomboid like 2
TEM-1
tumour endothelial marker 1
TNFα
tumour necrosis factor‐α
VEGF
vascular endothelial growth factor
VEGFR2
vascular endothelial growth factor receptor 2
VSMCs
vascular smooth muscle cells
Introduction: C‐type lectin domain group 14 family
There are 17 families in the C‐type lectin domain (CTLD) containing superfamily described in humans. This superfamily comprises a range of remarkably diverse proteins that can be secreted or expressed on the cell surface. They mediate a wide range of functions including but not limited to inflammation, cell adhesion and carbohydrate recognition 1.
Thrombomodulin, CD248, CD93 and CLEC14A represent members of the CTLD group 14 family which share common domain architecture (Fig. 1). Each member is comprised of an N‐terminal signal peptide and a CTLD containing eight conserved cysteine residues. This is followed by a sushi‐like or complement control protein domain (also commonly referred to as a short consensus repeat), except for thrombomodulin which due to a lack of four conserved cysteine residues in this region does not satisfy the requirement for a sushi domain. Next are a number of EGF‐like domain repeats, thrombomodulin contains six, CD93 five, CD248 three and CLEC14A one. These are followed by a mucin‐like region of variable length which is proline, serine and threonine rich and encompasses many predicted O‐linked glycosylation sites. Finally, there is a single‐pass transmembrane region that connects to a cytoplasmic tail.
Figure 1.
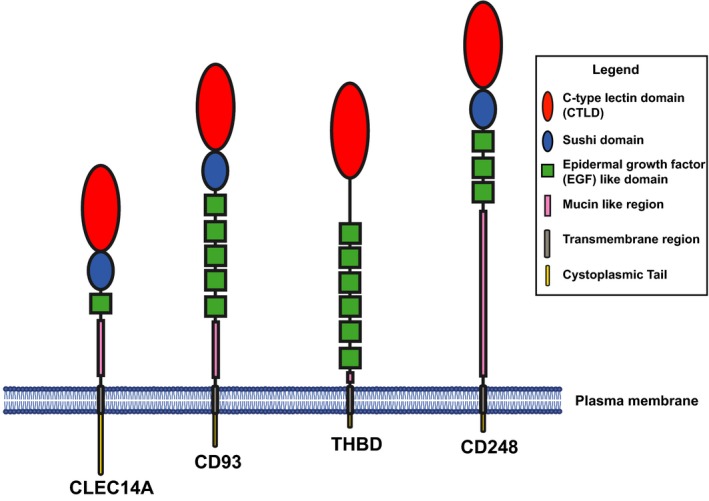
CTLD group 14 family proteins. Schematic diagrams of the CTLD group 14 family proteins. Each protein is drawn to relative scale based on primary amino acid sequence length. The CTLD is shown in red, the sushi in blue and the EGF repeats in green.
The CTLD was originally described as a calcium (Ca2+)‐dependent carbohydrate binding domain, although not all CTLDs require Ca2+ or demonstrate carbohydrate binding activity. The overall CTLD‐fold is characterised by a so called ‘loop in a loop’ structure stabilised by a conserved set of residues which contribute to a distinctive hydrophobic core 1. CTLD containing proteins have been widely described in many species and can even be found in the Bordetella bronchiseptica bacteriophage 2. Sushi domains exhibit extensive sequence variation but are generally characterised by four conserved cysteines (forming two disulfide linkages in a 1–4 and 2–3 pattern) and an invariant tryptophan, which contribute to preserving its tertiary structure 3. The sushi domain is an extracellular motif that can contribute to protein–protein interactions, best exemplified in interleukin‐15 receptor‐α (IL‐15Rα) recognition of IL‐15 4. EGF‐like domains are evolutionary conserved modules, which derive their name from the epidermal growth factor where they were originally described. EGF‐like domains are found in a wide range of proteins, chiefly of animal origin and are frequently observed in tandem repeats. Each EGF module typically consists of 30–40 amino acids and includes six conserved cysteines which form three intramolecular disulfide bonds 5. The highly glycosylated mucin region is commonly associated with adhesion proteins as described for CD164 6 and offers protection against protein degradation by preventing access to proteases. In addition, the presence of many O‐linked sugar moieties most likely allows proteins to adopt a more rigid and extended conformation 7. All of the CTLD group 14 family members have been detected at a much higher molecular weight than one would expect based on their primary amino acid sequences. These apparent disparities can be attributed to high degrees of post‐translational modifications, most likely glycosylation. Consistent with this hypothesis, when CD248 is treated with O‐glycanase and sialase, its molecular weight is reduced from 165 to 95 kDa when purified from human neuroblastoma cells 8. Similar findings have been reported when CD93 is treated with enzymes that remove O‐glycosylation 9. It is interesting to note that electron microscopy analysis of thrombomodulin revealed an elongated molecule with a large globular nodule at one end and a smaller nodule at the other 10. If we assume that the larger nodule is likely the CTLD, the smaller one is most likely comprised of the EGF repeats. Since the overall domain architecture of CTLD group 14 family members is relatively conserved, it is tempting to speculate that they all display a similar elongated structure with the membrane‐distal CTLD interacting with its cognate ligands. Additionally, the domain layout of CTLD, sushi and EGF modules are reminiscent of the CTLD group 4 selectin family of cell adhesion molecules, albeit in a different order 11. Similar to the group 4 family, there are numerous examples of the CTLD group 14 family mediating roles in adhesion (see below).
Based on whole protein sequence alignment, the family member with closest homology to CLEC14A is CD248 and CD93 is most closely related to thrombomodulin (Fig. 2). It has been suggested that CD93 could have arisen from thrombomodulin by gene duplication events as each is present on chromosome 20 in humans 12.
Figure 2.
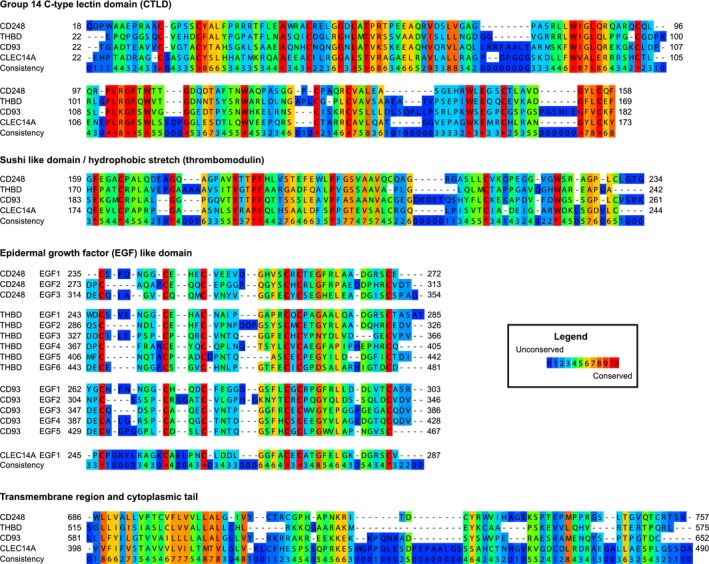
Alignments of CTLD group 14 family members based on amino acid sequence. Amino acid alignments of the whole primary sequence of each human family member using PRALINE 229. The following protein sequences were used thrombomodulin (http://www.uniprot.org/uniprot/P07204), CD93 (http://www.uniprot.org/uniprot/Q9NPY3), CLEC14A (http://www.uniprot.org/uniprot/Q86T13) and CD248 (http://www.uniprot.org/uniprot/Q9HCU0).
Thrombomodulin
Thrombomodulin (THBD or CD141) is the most extensively studied member of the CTLD group 14 family and is expressed by endothelium of all blood vessels and lymphatics 12, 13. It is also localised on a range of other cell types including but not restricted to monocytes, neutrophils and dendritic cells 14. Thrombomodulin is expressed early in development and mice lacking the gene show embryonic lethality 15. Interestingly, thrombomodulin‐deficient mouse embryos die at embryonic day 8.5 due to defects in nonendothelial tissue within the placenta, but reintroduction of thrombomodulin into the placenta allows normal development of embryos until day 12.5 16. This suggests two distinct roles for thrombomodulin during development, one in the placenta and the other in the embryo. Nevertheless, thrombomodulin is the only family member that following genetic deletion causes embryonic lethality, suggesting that it exhibits an indispensable role. This lethal phenotype is not dependent on the CTLD or the cytoplasmic tail, as mice that lack these modules remain viable 17, 18. Based on these considerations, such embryonic lethality is most likely due to disruptions in the thrombomodulin‐mediated coagulation cascade elicited by the EGF domain tandem repeats (see below).
Thrombomodulin and coagulation
One of the major roles for thrombomodulin is regulating the coagulation cascade by binding to the serine protease thrombin 19. The mode of recognition for this physiologically relevant co‐factor involves the EGF modules of thrombomodulin as determined by the crystal structure of the thrombomodulin–thrombin complex 20. This binding event inhibits procoagulant thrombin‐mediated hydrolysis of fibrinogen to fibrin, thereby inducing an anticoagulative effect 21. Thrombomodulin–thrombin binding also increases by approximately 1000‐fold the thrombin‐mediated cleavage and activation of the anticoagulant serine protease protein C 19. Activated protein C is involved in the inactivation of procoagulant factors FVa and FVIIIa 19, 22. In addition, thrombomodulin–thrombin complexes enhanced by approximately 1250‐fold the activation of the antifibrinolytic TAFI (thrombin activatable fibrinolysis inhibitor) 23. Therefore, by redirecting its cleavage activity towards the activation of anticoagulant and antifibrinolytic proteins, thrombomodulin can dampen the coagulation cascade by different mechanisms. Recently, the regulators of angiogenesis angiopoietin‐1 (Ang‐1) and angiopoietin‐2 (Ang‐2) have both been described as ligands for thrombomodulin in vitro 24. Ang‐2 binds with higher affinity than Ang‐1 but both, by competing with thrombin, can disrupt thrombomodulin–thrombin interactions leading to suppression of thrombin‐mediated anticoagulant functions. Thrombomodulin binding to heat shock protein 70 (HSP70‐1) on the endothelial cell surface can also inhibit thrombomodulin function in vitro by reducing protein C activation by as yet unknown mechanisms 25. The wide‐ranging roles of thrombomodulin in coagulation are well‐documented elsewhere 24, 26 and hence will not be discussed in any depth. We also direct the reader towards a recent extensive review exploring the ‘nontraditional roles’ of thrombomodulin 27.
Thrombomodulin and angiogenesis
Proangiogenic effects have been reported for a recombinant form of soluble thrombomodulin encompassing six contiguous EGF modules and the mucin‐like region (thrombomodulinEGF‐Mucin), resulting in increased endothelial proliferation, tube formation, migration and upregulation of matrix metalloprotease expression in vitro 28. This recombinant protein also elicited proangiogenic effects on endothelial progenitor cells (EPCs) through a phosphoinositide 3‐kinase (PI3K)‐dependent pathway 29. Furthermore, thrombomodulinEGF‐Mucin demonstrated endothelial protective roles chiefly by guarding against apoptosis again via the PI3K pathway 30. These roles are thought to be dependent on the EGF domains which can bind to and activate fibroblast growth factor receptor 1 (FGFR1) 31. The fifth EGF domain of thrombomodulin alone (thrombomodulinEGF5) has also demonstrated proangiogenic function as well as cytoprotective effects on endothelium 32. This cytoprotective phenomenon was suggested to be independent of thrombomodulin–thrombin interactions and instead due to upregulation of antiapoptotic protein myeloid‐cell leukaemia‐1 (MCL1) 33. A subsequent study revealed that this cytoprotective outcome was triggered by thrombomodulinEGF5 binding to G protein‐coupled receptor‐15 (GPR15) on endothelial cells, leading to the activation of endothelial nitric oxide synthase and extracellular signal‐regulated kinase (ERK) signalling, an effect that was abolished in GPR15‐deficient mice 34. Recently, the minimal fragment of thrombomodulinEGF5 necessary for binding to GPR15 and promoting proangiogenic function was identified as a 19‐amino acid peptide, that includes an intramolecular disulfide bond which adopts a loop structure similar to that observed for the prototypical EGF 31, 35. This peptide exhibited proangiogenic function and extended survival in mouse models of sinusoidal obstruction syndrome, a condition that is associated with injury of liver sinusoidal endothelium 35. However, whether thrombomodulin can bind to GPR15 while attached to the cell membrane, or if proteolytic processing is essential, is yet to be determined.
These proangiogenic signals mediated by the thrombomodulin EGF5 domain can be abolished when the soluble extracellular domain (ECD) contains the CTLD 36. The CTLD of thrombomodulin binds to Lewis Y antigen, which is a cell surface tetrasaccharide that is predominantly expressed during development and tumourigenesis 37. Relatedly, soluble CTLD alone can mediate aberrant effects in angiogenesis assays, presumably by virtue of its interactions with Lewis Y antigen localised on epidermal growth factor receptor‐1 (EGFR1), thereby inhibiting its activation 36. These findings suggest that the thrombomodulin CTLD exhibits roles distinct from the EGF domains and may be functionally dominant in its soluble form, due to its ability to negate EGF domain‐dependent effects. The CTLD of membrane‐bound thrombomodulin has been shown to bind to the extracellular matrix protein fibronectin, an interaction which activates focal adhesion kinase phosphorylation and upregulates matrix metalloproteinase‐9 (MMP9) 37, 38. The thrombomodulin–fibronectin interaction occurs on tumour blood vessels in murine melanoma suggesting that this interplay may serve as a putative target for antiangiogenic therapy, although an in‐depth understanding of this interaction in healthy tissues would first need to be considered. Thrombomodulin cell surface expression can be regulated by binding of the CTLD to Kringle 1–5, a proteolytically cleaved fragment of plasminogen 39. This binding event results in thrombomodulin internalisation and degradation, negating the proangiogenic roles of membrane‐bound thrombomodulin.
Thrombomodulin and cancer
Thrombomodulin expression has been described in multiple cancer types on the endothelium and tumour cells 40, 41. In genetically engineered mice expressing a mutant form of thrombomodulin with severely compromised thrombin binding, primary tumour growth was unaffected whereas lung metastasis was significantly enhanced 42. The authors suggested this observation was due to prolonged survival of tumour cells in the lung and demonstrated that this effect was attributed to the thrombin binding function and not the N‐terminal CTLD. A whole host of studies in different tumour settings (lung, colorectal, cervical, prostate and bladder) postulate a role for thrombomodulin overexpression in reversing epithelial mesenchymal transition (EMT) 43, 44, 45, 46, 47, 48. Upregulation of thrombomodulin may even enhance tumour sensitivity to chemotherapeutic agents, such as doxorubicin 49. Indeed, a more comprehensive review of the role of thrombomodulin in tumour biology has been documented 40. The overall findings seem to indicate that thrombomodulin expression correlates with a good prognosis and expression is abolished in more aggressive and highly metastatic tumour types.
More recently, soluble human thrombomodulin has been utilised as a potential cancer therapeutic agent and reductions in tumour growth were observed when administered to mice bearing pancreatic tumour xenografts 50. Furthermore, the soluble ECD of thrombomodulin has also been reported to reduce tumour growth in inflamed models of gastrointestinal tumours 51. Whether soluble thrombomodulin could have antitumour effects in patients has not been formally investigated; however, it has demonstrated clinical benefits in managing disseminated intravascular coagulation in cancer patients and has the potential for direct effects on tumour burden as well as in aberrant thrombosis 52.
Thrombomodulin and inflammation
Thrombomodulin has been described to have roles in inflammation some of which are linked to its anticoagulant function. This is best exemplified by protein C triggering an anti‐inflammatory signalling cascade by inhibiting tumour necrosis factor‐α (TNFα) production in response to lipopolysaccharide (LPS) 53. Independent of its roles in coagulation inhibition, thrombomodulin CTLD can bind to the proinflammatory molecule high‐mobility group protein B1 (HMGB1), leading to suppression of inflammation in vivo and protection against LPS‐induced lethality 54. A more recent study highlighted that the thrombomodulin–HMGB1 interaction allows the EGF domain bound thrombin to proteolytically cleave HMGB1 55. The inactivation of HMGB1 has potential implications on immunogenic cell death events following anticancer intervention invoked by chemotherapeutic agents or radiotherapy, whereby HMGB1 released by dying cells serves as damage‐associated molecular patterns activating antigen‐presenting cells and facilitating presentation of tumour antigens 56. The thrombomodulin CTLD has also been shown to reduce the adhesion of polymorphonuclear leucocytes to endothelium 57. The authors proposed that this process was dependent on thrombomodulin binding to Lewis Y antigen and therefore blocking its availability to bind leucocytes and aid subsequent transmigration. Conversely, human leucocytes have been suggested to directly recognise the mucin‐like region of thrombomodulin through leucocyte adhesion integrins lymphocyte function‐associated antigen‐1 (LFA‐1) and Mac‐1 in vitro 58. Thrombomodulin EGF domains were found to markedly suppress LPS‐induced inflammatory signalling pathways in macrophages by associating with the pattern recognition receptor CD14, and that this inhibitory effect was also dependent on the serine threonine‐rich domain 59.
ThrombomodulinEGF5 also has the capacity to engage T‐cells bearing GPR15 60. This results in immunosuppression of T‐cell responses and facilitates the differentiation of regulatory T‐cells (Tregs). In addition, recombinant thrombomodulinEGF5 was shown to inhibit dendritic cell activation. Taken together, this provides a possible rationale for recombinant thrombomodulin‐mediated alleviation of graft versus host disease (GVHD) in mouse models of haematopoietic stem cell transplantation 32. More importantly, it reconciles with clinical observations that increased expression of thrombomodulin can reduce GVHD in patients 32, 61. This immunosuppressive role of thrombomodulin could also have relevance in tumour immunology as thrombomodulin expressed in the tumour microenvironment has the potential to expand anti‐inflammatory and protumour Tregs, a cell type that contributes to tumour immune evasion mechanisms 62. However, these findings are contrary to observations reporting thrombomodulin as a good prognostic factor, and its role in immunosuppression may be outweighed by its function in EMT and aggressiveness. Thrombomodulin also exhibits anti‐inflammatory properties by regulating the alternative pathway of complement activation. Complement activation is a critical process in inflammation of the innate immune system and involves the cleavage of various proteins resulting in proteolytic release of chemotactic factors and activation of further cleavage events ultimately leading to the assembly of pathogen destruction complexes 63. Thrombomodulin–thrombin‐mediated activation of TAFI cleaves complement proteins C3a and C5a leading to their inactivation 64 which has been demonstrated both in vitro and in vivo 65, 66. Thrombomodulin binds to complement protein C3b and enhances its factor H‐ and factor I‐mediated degradation into its inactive form (iC3b), another mechanism of negative regulation of the alternative complement pathway 67.
Thrombomodulin shedding
There are many transmembrane proteins that are specifically shed from the cell membrane to activate or deactivate distinct protein functions in angiogenesis and other physiological processes. Examples include the membrane‐bound EGF precursor proteins, which are cleaved by metalloproteinases such as ADAM10 (a disintegrin and metalloproteinase‐10) and ADAM17 resulting in growth factor activation 68. Conversely, the EGF receptor itself can be subjected to proteolytic cleavage thereby suppressing downstream signalling functions 69.
Thrombomodulin can be cleaved by the serine protease rhomboid‐like 2 (RHBDL2) at a site proximal to the transmembrane domain, resulting in release of the entire ECD 70. This RHBDL2 cleaved form of thrombomodulin can increase migration and wound healing in keratinocytes in vitro 71. Also, the full‐length thrombomodulin‐ECD has also been shown to be cleaved from the endothelial cell surface after incubation with the neutrophil proteases elastase, cathepsin G and proteinase 3 72. Soluble thrombomodulin has been detected in human blood, urine and synovial fluid 73, 74, 75. Indeed, monitoring serum levels of soluble thrombomodulin may be important as it can positively correlate with disease status such as systemic lupus erythematosus 76.
The thrombomodulin CTLD can also be cleaved leaving the remainder of the molecule intact upon the cell surface, an event that is most likely facilitated by matrix metalloproteinases (MMPs) 77. Although, these cleavage events are yet to be explicitly shown in vivo, it is noteworthy that two forms of thrombomodulin have been isolated from human urine 74. Characterisation of these fragments by N‐terminal sequencing revealed that one form encompasses the EGF repeats and the mucin‐rich region and retained the ability to bind thrombin. In contrast, the second fragment corresponded to the equivalent molecular weight for the N‐terminal CTLD and failed to bind thrombin. Furthermore, four different versions of thrombomodulin were detected in human plasma suggesting even more potential cleavage sites 78. Indeed, the physiological relevance of these different fragments requires further investigation. Although not resulting in shedding, neutrophil proteases have been shown to inactivate human thrombomodulin by oxidation of a key methionine between EGF4 and EGF5 79.
In summary, these findings of differential shedding along with the distinct biological function of each thrombomodulin subdomain offers a scenario where one molecule can be proteolytically processed in different ways to elicit opposing effects. The shedding of thrombomodulin is likely a tightly regulated process in which specific domains are released depending on the requirement for pro or antiangiogenic signals, as well as pro or anti‐inflammatory outcomes.
Additional roles for thrombomodulin
Along with its vital roles in regulating blood coagulation and inflammation, thrombomodulin also reportedly contributes to cell–cell adhesion in vitro, an event which is dependent upon the CTLD 80. This proadhesion role was Ca2+ dependent and could be abolished with CTLD‐specific antibodies or addition of mannose, chondroitin sulfate A or C. This suggests that the thrombomodulin CTLD serves as a conventional Ca2+‐dependent carbohydrate‐binding lectin.
The cytoplasmic tail of thrombomodulin has been proposed as a ligand for the intracellular adaptor protein ezrin 81, a member of the ezrin‐radixin‐moesin (ERM) family of proteins that link transmembrane proteins to the actin cytoskeleton 82. This reinforces the likelihood of thrombomodulin‐mediating cell adhesion events. Consistent with this, knockdown of thrombomodulin can compromise the integrity of E‐cadherin‐mediated cell–cell contacts, potentially implicating thrombomodulin downregulation in the induction of EMT in cancer 43. A summary of thrombomodulin protein interactions is displayed in Fig. 3.
Figure 3.
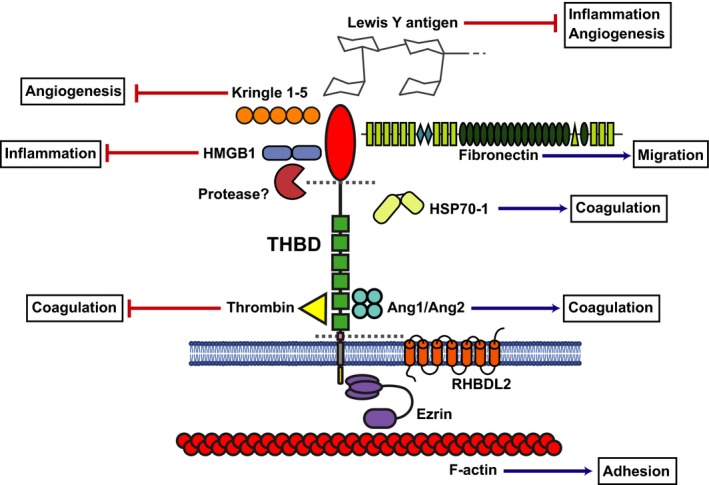
Schematic of thrombomodulin protein structure with ligand binding partners. Thrombomodulin CTLD has been shown to interact with fibronectin, HMGB1, Kringle 1–5, Lewis Y antigen and HSP70‐1. The CTLD may be proteolytically cleaved by an as yet unidentified MMP. Thrombin binds to the 5th and 6th EGF domains, this binding is in competition with Ang1 and/or Ang2. RHBDL2 can cleave the whole ECD of thrombomodulin as can neutrophil elastase, cathepsin G and proteinase 3. The cytoplasmic tail binds to ezrin which in turn links thrombomodulin to the actin cytoskeleton.
CD248
CD248 also known as endosialin or tumour endothelial marker‐1 (TEM‐1) is the prototypical member of the CTLD group 14 family. It was first discovered as an antigen detected by the antibody FB5, which stained human tumour sections with patterns resembling blood vessels, but not healthy tissues 8. This led the authors to describe CD248 as a marker for tumour endothelium, although it could not be detected in cultured human umbilical vein endothelial cells (HUVEC). The study did however demonstrate that it was a highly glycosylated cell surface glycoprotein leading to its proposed name at the time; endosialin. CD248 was later identified as a marker of tumour endothelium in studies involving serial analysis of gene expression of vessels purified from human colorectal cancers in comparison to healthy colon vessels, hence its alternative name TEM‐1 83. Despite this, it is now widely accepted that CD248 is expressed by perivascular cells, stromal fibroblasts (especially in cancer and inflammation), mesenchymal stem cells and some tumour cells but not adult endothelium 84, 85, 86. The expression of CD248 on perivascular cells but not on endothelium in vivo was unequivocally demonstrated using multiple fluorescent labelling of human glioma sections 87. The study by St. Croix and colleagues which originally identified CD248 as TEM‐1 utilised CD146 or melanoma cell adhesion molecule (MCAM) antibodies to enrich the endothelium. Since MCAM also serves as a marker for pericytes, these samples likely contained perivascular cells as well as endothelium explaining the enrichment of CD248 84, 88. The proposed expression of CD248 on EPCs may have also added to this confusion 89.
CD248 expression
CD248 is expressed during development and is first detected in mice at embryonic day 9.5 90. CD248 expression is mostly diminished in postnatal organs except for the kidney glomeruli and the uterus. Mice deficient in CD248 are viable and display no obvious defects, suggesting compensatory mechanisms may be employed during development 91. However, a marked decrease in tumour growth, metastasis and invasion was observed when CD248 deficient mice were challenged with human colorectal cancer xenografts. This defect in tumour growth and metastasis was only evident with abdominally implanted tumour cells, whereas subcutaneous implants displayed no difference relative to control animals. Further studies revealed that expression of CD248 exhibited negligible effects on primary tumour growth but increased metastasis formation in mouse models of breast cancer 92. Such prometastatic effects were attributed to CD248 expressing pericytes enhancing tumour cell intravasation. Elevated‐CD248 expression also correlated with greater metastasis and poorer survival in human breast cancer patients.
CD248 expression has been reported to be induced by hypoxia, predominantly involving the transcription factor hypoxia inducible factor‐2α (HIF‐2α) 93. This could explain the high levels of CD248 observed in the tumour microenvironment which is often poorly perfused and contains areas of hypoxia 94. Upregulation of CD248 can also arise in response to the growth factors FGF‐2, EGF and platelet‐derived growth factor‐BB (PDGF‐BB), which is further enhanced under hypoxic conditions 95.
CD248 expression has been described on naïve human CD8+ T‐cells, where it can negatively regulate proliferation 96. CD248 is expressed on stromal cells in secondary lymphoid organs and is required for correct secondary lymph node expansion in models of vaccination 97. However, CD248 was not essential for correct spatial organisation of T and B cells in this model.
CD248 interaction partners and biology
CD248 has been reported to interact with the ECM proteins fibronectin and collagens I and IV 98. The interaction of CD248 with fibronectin increased cell adhesion of Chinese hamster ovary (CHO) cells overexpressing CD248 and was dependent upon the N terminus of fibronectin and the CTLD of CD248. Consistent with these data, the CTLD‐specific monoclonal antibody MORAb‐004 (ontuxizumab) could block CD248 binding to fibronectin and collagen I. Also, siRNA‐mediated knockdown of CD248 resulted in reduced migration and proliferation of fibroblasts, reinforcing a putative role in adhesion 99. Interestingly, a characteristic feature identified in CHO cells overexpressing CD248 is the upregulation of MMP9, thereby implicating CD248 in ECM degradation, a key step in sprouting angiogenesis as well as tumour metastasis and invasion 98. Further evidence in support of CD248 associating with the ECM stemmed from immunofluorescent staining with CD248 ECD fused to an Fc tag (CD248‐ECD‐Fc), this staining was only observed in the ECM from endothelial cells (HUVEC) and could partially co‐localise with fibronectin 100. More recently, we have shown direct interaction of CD248 with the endothelial ECM protein multimerin‐2 (MMRN2) 101. This interaction was dependent upon the CTLD of CD248 and CD248‐ECD‐Fc staining could partially co‐localise with MMRN2 on HUVEC; this may clarify previous findings involving the CD248‐ECD binding to the endothelial ECM 100.
Another ligand identified for CD248 was the secreted galectin‐3 (Mac‐2) binding protein Mac‐2BP and this interaction proved to be carbohydrate and Ca2+ independent 102. The CD248 interaction was mapped to two C‐terminal domains of Mac‐2BP and these have been previously implicated in binding galectin‐3, collagens V and VI, and nidogen, suggesting overlapping binding sites 103. This interaction invokes repulsion of human fibroblasts and HeLa cells expressing CD248 and Mac‐2BP, respectively. Moreover, this phenomenon was reduced following siRNA induced gene‐silencing of either molecule. Mac‐2BP is upregulated in the tumour cells of many different types of cancer and has been associated with increased metastasis and decreased survival in lung cancer patients 104. These findings strengthen the likelihood of CD248‐Mac‐2BP interactions occurring during tumorigenesis. It is currently unknown whether the therapeutic antibody ontuxizumab can block CD248 binding Mac‐2BP or MMRN2, a question that will likely impact novel future clinical interventions that target CD248.
There is evidence to suggest that the cytoplasmic tail of CD248 is involved in tumour development, as mice lacking this domain display reduced tumour growth in T241 fibrosarcomas and Lewis lung carcinomas (LLCs) 105. The cytoplasmic tail has also been predicted to contain a PDZ binding site and three potential phosphorylation sites, although to date identification of CD248 intracellular domain interactors have proved elusive 85, 106. A summary of CD248 protein interactions is summarised in Fig. 4.
Figure 4.
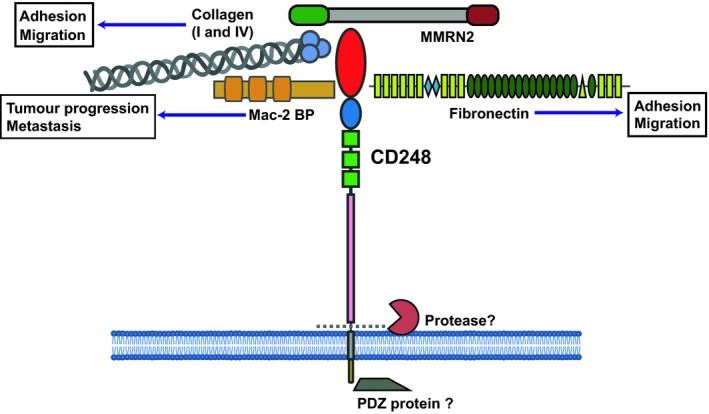
Schematic of CD248 structure with ligand binding partners. CD248 CTLD binds to fibronectin, Mac‐2 BP, Collagens I and IV and MMRN2. There are currently no known direct intracellular interaction partners for CD248.
CD248 implications in angiogenesis
The role of CD248 in angiogenesis is complex and there is evidence to suggest it can both promote and inhibit angiogenesis depending on the circumstances. When CD248 is overexpressed in HeLa cells (normally lacking expression of the protein), multiple pro and antiangiogenic molecules are produced 95. CD248‐deficient mice displayed no gross defects in developmental angiogenesis or wound healing, but abnormalities were clearly apparent in tumour models of both the full gene deletion and the cytoplasmic deletion, resulting in smaller tumours exhibiting increased vessel density, although larger vessels were reduced 91, 105. Curiously, defects in tumour growth were not observed in all tumour models and the underlying mechanism remains unclear. Increased vascularity is also found in CD248‐deficient mouse models of glioblastoma multiforme, but there are no differences in tumour growth compared to wild‐type animals 107. These observations of increased vessel density may be rationalised by findings connecting CD248 with regulation of vascular patterning 87. This function of CD248 was uncovered when HUVEC treated with plate bound CD248 (to mimic pericyte expression), exhibited higher levels of apoptosis. This study highlighted the prospect of CD248 mediating a key role in vessel regression and pruning and emphasised for the first time that pericytes could be linked to such functions. Therefore, such defects in vascular pruning can result in an increase in microvessels that may have aberrant function. Moreover, these observations underline the possible therapeutic potential of the CD248‐ECD for inducing vessel regression and vascular normalisation, which might conceivably increase the delivery of chemotherapeutic agents into tumour tissue 108. Likewise, this vessel normalisation effect has been shown to allow more efficient infiltration of effector immune cells into tumours 109.
CD248 has also been implicated in the platelet‐derived growth factor (PDGF) signalling cascade 110. For example, following CD248 knockdown in pericytes, PDGF‐mediated proliferation is reduced in vitro. Furthermore, CD248 knockout mice displayed defects in sprouting angiogenesis but not splitting (intussusceptive) angiogenesis in skeletal muscle 111. Such defects could be recapitulated in mice treated with PDGFRβ inhibitors reinforcing a role for CD248 in PDGF signalling.
CD248 in cancer
Elevated CD248 expression levels on tumour‐associated stroma have been reported in various primary tumour types including glioma, colorectal, melanoma as well as brain metastases 87, 112, 113, 114. CD248 expression has also been associated with worse outcome in patients with breast or colorectal cancer and could serve as a prognostic marker 115, 116. CD248 is expressed in numerous tumour cell lines and clinical samples of sarcomas and neuroblastomas, but is absent in cancer cells of epithelial origin 86. Indeed, highly malignant ‘side population’ sarcoma cells with some characteristics of cancer stem cells express CD248 117. These highly invasive side populations are also CD248+ in osteosarcoma 118. For these reasons, there has been a substantial drive into developing innovative strategies of targeting CD248 for tumour therapy.
Targeting CD248 has been attempted mainly by antibody‐based therapeutic approaches. One of the first preclinical attempts utilised single‐chain variable fragment (scFv) antibody‐like molecules generated against CD248 to successfully direct cytotoxic agents to neuroblastoma cells in vitro 119. Internalising antibodies against CD248 coupled with anti‐human IgG toxin‐conjugated antibodies revealed cell cytotoxic effects on CD248 expressing cancer cell lines in vitro 86. Such antibodies were developed as full antibody‐drug conjugate (ADC) molecules utilising conjugation to tubulin‐inhibiting drugs 120. Administration of these ADCs retarded tumour growth in multiple xenograft models. Another ADC against CD248 has been developed conjugated to a DNA‐binding duocarmycin derivative which has shown therapeutic efficacy in a human osteosarcoma xenograft model 121. The previously described CTLD‐specific CD248 antibody ontuxizumab has been utilised as a possible diagnostic imaging tool through use of 125Iodine conjugation and positron‐emission tomography 122. This technique resulted in rapid tumour uptake and real‐time visualisation of tumour burden and CD248 localisation in mice. Some more recent developments have involved the generation of human CD248 knock‐in mice to study the in vivo effects of ontuxizumab 123. Indeed, upon administration of ontuxizumab into B16 melanoma‐bearing mice, tumour growth was significantly reduced by up to 70%. This was presumably due to increase in microvessel density and the presence of nonfunctioning tumour blood vessels; phenocopying previous findings in CD248 knockout animals. This study also showed downregulation of surface expression of CD248 on pericytes by internalisation after ontuxizumab treatment in vitro and in vivo. Despite some preclinical efficacies, the ontuxizumab‐humanised CD248 antibody has recently completed two phase II clinical trials which failed to show improvements from standard of care or placebo in metastatic colorectal cancer and neglected to meet a progression‐free survival (PFS) goal of 35% in metastatic melanoma 124, 125 No clinical benefit was also observed in phase II trials involving metastatic soft tissue sarcoma treated with a combination with ontuxizumab and chemotherapy (gemcitabine and docetaxel) 126.
DNA vaccine approaches against CD248 have also been attempted preclinically, with antitumour effects being reported in both the prophylactic and therapeutic vaccine setting 127. The DNA construct consisted of murine CD248 fused to a fragment of tetanus toxoid, which circumvents tolerance to the self‐protein allowing triggering of an adaptive immune response. The authors described CD4+ and CD8+ T‐cell clones that were specific for CD248 epitopes as well as tumour specific antigens. The vaccination did not detrimentally affect wound healing or reproduction.
The targeting of CD248 may even be detrimental in some tumour types, as CD248 expression upregulated in hepatocellular carcinoma (HCC) patients in hepatic stellate cells (specialised pericytes found in the liver vasculature) was found to be protective correlating with better outcomes 128. Furthermore, inducible models of HCC in CD248 knockout mice displayed enhanced liver tumour progression relative to wild‐type controls.
CD248 in inflammation and fibrosis
CD248 is expressed in mesenchymal stromal cells of developing mouse lymphoid tissues such as the spleen, thymus and lymph nodes 129, 130 expression in the adult mouse spleen is low, but is enhanced in stromal cells upon Salmonella enterica infection coinciding with spleen remodelling and repair 129. CD248 expression in mice is required for postnatal development of the thymus as well as regeneration of the thymus after S. enterica infection, in CD248 deficient animals this postinfection regeneration is impaired 130.
CD248 is expressed in vascular smooth muscle cells (VSMCs) undergoing proliferation and remodelling in apolipoprotein‐E (ApoE) KO mouse models of atherosclerosis as well as atherosclerotic lesions from patients 131. CD248 was shown to promote atherosclerosis as ApoE and CD248 double KO mice displayed less atherosclerosis when fed a Western style diet. These CD248‐deficient animals also exhibited marked reductions in macrophage infiltration into atherosclerotic plaques, due to reduced chemokine expression in VSMCs. Reduced macrophage recruitment in CD248 KO mice was also shown in other models of inflammation not involving atherosclerosis.
CD248 is expressed in the fibroblasts and pericytes of synovial tissue from patients with rheumatoid arthritis and psoriatic arthritis 132. CD248 expression has also been identified in the sublining layer of a distinct subset of synovial fibroblasts 133. CD248 knockout mice and mice lacking the cytoplasmic domain of CD248 both showed reductions in experimental arthritis compared to wild‐type animals, and displayed a marked reduction in synovial inflammation 132. CD248 also has roles in bone formation, it is expressed by bone forming osteoblasts but not bone removing osteoclasts in both mice and humans 134. CD248 knockout mice display denser bones most likely due to the hyperactivation of osteoblasts and increased mineral formation that would normally be inhibited by PDGF signalling, which is disrupted in CD248‐deficient cells. These findings suggest that targeting CD248 therapeutically may not only reduce inflammation in arthritis but also reduce bone loss associated with arthritis.
CD248 is expressed in healthy human and mouse kidney mainly in the mesangial cells of the glomerulus, but also in pericytes and fibroblasts. CD248 is upregulated in chronic kidney disease and inflamed kidney on a population of myofibroblasts and may be a useful predictor of renal failure 135. CD248 was later shown to have a potential role in the development of kidney fibrosis in mice 136, 137. CD248‐deficient mice undergoing renal damage were protected against fibrosis, this was not as a result of reduced inflammation but possibly due to CD248 KO fibroblasts producing less collagen and CD248 KO pericytes displaying impaired differentiation into myofibroblasts (a major cell type involved in fibrosis). Similar to fibrosis in kidney, CD248 deficiency also protected mice against liver fibrosis following liver injury, and these KO mice displayed reductions in collagen but no change in inflammation 138. CD248 expression was also detected in human samples of liver fibrosis on myofibroblasts and perivascular cells and was elevated in human liver fibrosis samples compared to healthy controls as well as correlating with levels of collagen deposition. Idiopathic pulmonary fibrosis patients also display high expression of CD248 in fibroblasts and it may serve as a disease severity marker 139.
CD248 has also been described as being highly expressed in skin samples of patients with systemic sclerosis in comparison to healthy controls 140. When CD248 expression was silenced by means of siRNA in mesenchymal stem cells from systemic sclerosis patients, TGF‐β and PDGF profibrotic signalling was reduced.
These described roles of CD248 promoting aberrant inflammation and fibrosis suggest it may be a suitable therapeutic target in certain diseases. However, it is possible that such CD248 inhibition may also have detrimental effects on lymphoid tissue remodelling and repair following infection.
CD248 shedding
There are numerous reports describing soluble variants of CD248, suggesting its ECD may be shed from the cell surface as highlighted for other CTLD group 14 family proteins. CD248 has been suggested as a possible biomarker after it was purified from ascites fluid of patients with stage IV ovarian cancer 141, and pancreatic cancer 142. CD248 can be immuno‐precipitated from human serum in a fully glycosylated form of around 150–120 kDa, likely corresponding to the full ECD 143. In this same study, a highly sensitive and specific assay was developed using two different CD248 monoclonal antibodies to evaluate CD248 levels in patient blood. However, there was no significant difference in serum levels of soluble CD248 from colorectal cancer patients compared with healthy controls, which may limit its utility as a predictive biomarker particularly in this tumour setting. A protease capable of cleaving CD248 from the cell surface has yet to be identified.
CD93
CD93 was first described as a receptor for the complement component C1q, hence its alternative name C1q receptor‐1 (C1qR1 or C1qRp) 144, 145. A subsequent study revealed that CD93 failed to engage C1q, but was instead implicated in cellular adhesion events 146. CD93 is expressed predominantly by endothelial cells and has been reported to be expressed by neurons in a rat model of inflammation and various cells of the haematopoietic system, including monocytes, neutrophils, B cells, natural killer (NK) cells, naïve T‐cells, platelets and haematopoietic stem cells 147, 148, 149, 150, 151, 152. It is also highly expressed on the tumour‐associated vasculature. In a recent example, elevated levels of CD93 expression were detected on human colorectal carcinoma sections 153. Interestingly, this study also examined soluble levels of CD93 within patient plasma and found a 30% reduction in colorectal carcinoma patients compared with healthy controls. CD93 has been described as a key gene in a proposed ‘tumour angiogenesis signature’ determined by meta‐analysis of 959 breast cancers, 170 renal cancers and 121 head and neck cancers 154. Moreover, CD93 has been identified as a member of a group of genes that are vastly upregulated in high‐grade glioblastoma tumour vasculature 155. This high expression profile was later confirmed at the protein level and correlated with poorer survival 156. Upregulated vascular expression of CD93 has also been described in nasopharyngeal carcinoma, as well as tumours of the eye including retinoblastoma and choroidal melanoma 157, 158, and correlates with a worse survival outcome. More recently, the CD93 CTLD has been derived from Escherichia coli expression systems that allow disulfide bond formation and has been purified to homogeneity allowing preliminary structural analyses using nuclear magnetic resonance approaches 159. This study revealed the CD93 CTLD does not bind Ca2+ and ongoing experiments will undoubtedly resolve the three‐dimensional structure and provide further molecular and functional insights into this family member.
CD93 expression
During mouse development CD93 is expressed at embryonic day 9 and is detected in the vasculature including the inter‐segmental vessels 160. CD93‐deficient mice were viable and displayed no obvious abnormalities, but macrophages from deficient mice exhibited reductions in phagocytosis and reduced clearance of apoptotic cells in both in vivo and in vitro apoptotic clearance assays 161. A defect in antibody secretion in plasma cells was also a characteristic feature of CD93 knockout mice 152. Intriguingly, only CD93‐deficient female mice display aberrations in tumour growth and perfusion in orthotopic glioblastoma and fibrosarcoma models 156.
CD93 interaction partners and biology
Silencing of CD93 by RNA interference in HUVEC‐impaired proliferation, migration, adhesion and sprout formation 162. Subsequent studies validated these effects with disruptions observed in adhesion, migration and tube formation 156, 157. A monoclonal antibody raised against human CD93 (clone 4E1) which binds between the CTLD and sushi domains demonstrated antiangiogenic activity in Matrigel assays both in vitro and in vivo, reiterating its roles in endothelial biology 162. CD93 has been identified as a gene that is downregulated upon VEGF blockade by using bevacizumab in patented studies performed by Genentech 163. Similarly, another report highlighted that CD93 protein expression was diminished upon pharmacological inhibition of VEGFR2 and fibroblast growth factor‐1 with brivanib alaninate 164. While VEGF could be having effects on CD93 independent of angiogenesis, the loss of function experiments involving CD93 seem to steer towards a proangiogenic function. Ligand binding studies of CD93 with a variety of ECM proteins revealed a lack of binding to all proteins tested in vitro including; collagen I and IV, gelatin, laminin, vitronectin and fibronectin 165. The only known extracellular interacting partner for CD93 was recently identified as the endothelial specific ECM protein MMRN2 101. This interaction is dependent on the CTLD of CD93 and by combining structural modelling with site‐directed mutagenesis a predicted long‐loop region of this structure was proposed to be critical for binding to MMRN2. This offers a platform for developing innovative therapeutics that specifically target CD93 to interrupt this interaction. The CD93‐MMRN2 interaction was later independently validated and surface plasmon resonance was used to characterise the interaction and determine binding affinities 166. A key residue within the coiled‐coil domain of MMRN2 (F238) was proposed as being integral for CD93 binding. Interestingly, this study also provided an explanation for the previously described antiangiogenic effects of the CD93 antibody 4E1, as it could interrupt the CD93‐MMRN2 interaction.
The CD93‐MMRN2 interaction was also shown to be involved in the proper deposition and organisation of fibronectin a process termed fibrogenesis 167. In CD93‐deficient mice the fibronectin matrix was disrupted in vessels of postnatal retinas and vessels in orthotopic models of glioblastoma 167. In the same study, the use of specific antibodies that detect activated α5β1 integrins revealed disruption of this activated integrin in CD93 knockout mice. During postnatal retinal angiogenesis, CD93 is expressed on filopodia while MMRN2 expression is absent from these protrusions but present in the surrounding ECM. Finally the authors showed that MMRN2 and fibronectin expressions are upregulated in high grade human glioma 167. Co‐localisation of CD93 and MMRN2 expressions has been demonstrated in vessels of a range of different solid human tumours including melanoma, Ewing's sarcoma, ovarian carcinoma and glioma amongst others 166, 167. There are likely other partner proteins for CD93 as a study showed that the recombinant form of the CD93 ECD can engage the cell surface of THP‐1 cells indicating the expression of a currently unknown CD93 ligand in this monocyte cell line 168.
The cytoplasmic domain of CD93 encompasses a positively charged juxtamembrane region that binds to the adaptor protein moesin 169. Moesin is a member of the ERM family of proteins, which like ezrin and radixin, anchor proteins to the actin cytoskeleton 82. In knockdown studies involving CD93, adherens junctions were disrupted 156. Strikingly, reintroduction of wild‐type CD93 but not CD93 lacking the moesin binding motif, restored adhesion junctions and highlighted the importance of CD93‐moesin interactions in maintaining the integrity of endothelial cell adhesion in vitro. Relatedly, CD93‐deficient mice display increased permeability in blood vessels possibly due to disruptions in tight junctions 170. Another intracellular binding partner for CD93 has been defined as GIPC, (Gα interacting protein (GAIP)‐interacting protein C‐terminus), 171 an adapter protein that contributes to arterial maturation and mural cell coverage 172. The binding of GIPC was dependent on the positively charged juxtamembrane as well as the final C‐terminal 11 amino acids of the cytoplasmic tail. CD93 was originally predicted to bind to the E3 ubiquitin ligase Cbl, due to the CD93 cytoplasmic domain containing a binding motif that is also found in the Cbl binding protein APS (adapter with pleckstrin homology and Src homology‐2 domains) 173. CD93 binding to Cbl was proved experimentally by co‐immunoprecipitation in HUVEC, and this interaction was abolished upon knockdown of the ECM adhesion molecule β‐dystroglycan 165. This study proposed that the cross‐talk between the laminin‐binding protein β‐dystroglycan and CD93 led to endothelial cell adhesion and migration. The authors suggested that upon laminin binding to β‐dystroglycan src kinase phosphorylates specific tyrosine residues in the cytoplasmic tail of CD93, which in turn facilitates binding to Cbl. In this setting, Cbl may serve as an adapter protein rather than a ubiquitin ligase. A summary of the protein interaction partners of CD93 are summarised in Fig. 5.
Figure 5.
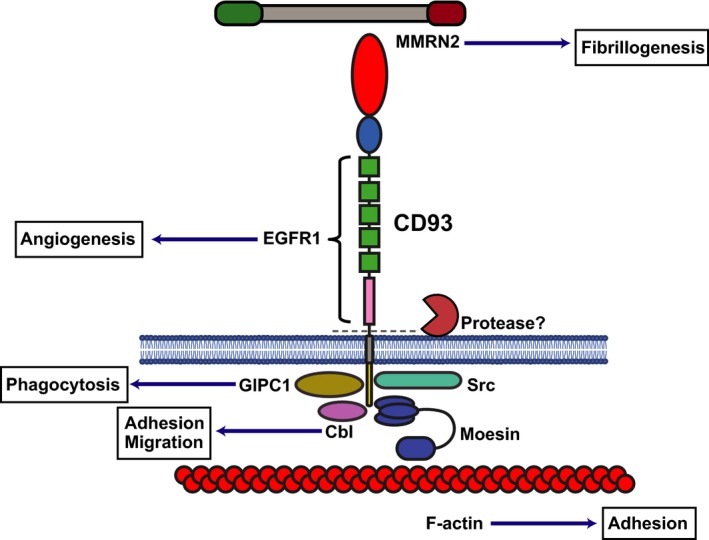
Schematic of CD93 structure with ligand binding partners. CD93 CTLD binds to MMRN2. The whole ECD has been shown to be cleaved by an as yet unidentified metalloproteinase. The intracellular cytoplasmic domain binds to moesin which in turn links CD93 to the actin cytoskeleton. The cytoplasmic domain also binds to Cbl and GIPC1 and src.
CD93 and inflammation
Mice deficient in CD93 when subjected to experimental peritonitis displayed increased leucocyte infiltration, and this effect was not restricted to a particular cell type 170. CD93 also has been suggested to have neuroprotective roles and as it is upregulated in murine models of stroke 174. This upregulation effect was also observed at the protein level in several cell types including endothelial cells, microglia and macrophages. Moreover, cerebral ischaemia in CD93 knockout mice resulted in enhanced neuro‐inflammation compared to wild‐type animals. CD93 knockout mice also displayed increased brain and spinal cord inflammation when compared to wild‐type mice in two different models of encephalomyelitis 175. Based on the CD93 expression profile within the tumour vasculature and its potential anti‐inflammatory roles, it is plausible to contemplate that CD93 may serve as an immunosuppressive molecule in the tumour microenvironment, limiting immune cell infiltration and facilitating tumour immune evasion mechanisms.
CD93 shedding
Soluble CD93 has been detected in human plasma, described to be a protein released from HUVEC and also a component of their ECM 171, 176, 177. Several studies have highlighted that levels of soluble CD93 directly correlate with disease status; in plasma it has been proposed as a potential biomarker for coronary artery disease and is elevated in synovial fluid of rheumatoid arthritis patients 168, 178. In another study, soluble CD93 was proposed as a marker for inflammation as it is reportedly shed from the cell surface of monocytes and neutrophils, which is likely to be dependent on metalloproteinases, although the major sheddase ADAM17 is not involved 179. This cleavage event most likely liberates the entire ECD of CD93 and can be stimulated by TNFα or LPS. This inflammation induced shedding of CD93 was subsequently confirmed by in vivo experiments, and macrophages were suggested as the main source of soluble CD93 180. Conversely, elevated levels of soluble CD93 in peritonitis fluid were shown to be dependent on nonhaematopoietic cells, likely from endothelium 170. Soluble CD93 has been suggested to induce differentiation of monocytes by as yet undefined mechanisms 168. A role for soluble CD93 in the process of efferocytosis has been proposed, whereby cleaved CD93 binds to apoptotic cells via the CTLD in a Ca2+ independent manner 181. Soluble CD93 then acts as an opsonin‐coating apoptotic cells and is in turn bound by αxβ2 integrin on macrophages via the CD93 EGF repeats aiding phagocytosis.
Soluble recombinant CD93 encompassing solely the five tandem EGF repeats and mucin domain of CD93 mediate proangiogenic effects on endothelial cells, increasing proliferation and migration of HUVEC and promoting angiogenesis in vivo 182. Although these proangiogenic signals were also induced with the full‐length CD93‐ECD, constructs lacking the CTLD elicited more potent effects by enhancing the EGFR1 mediated PI3K signalling pathway; similar results were observed with recombinant soluble forms of thrombomodulin encompassing the EGF and mucin domains 36.
The O‐glycosylation modifications within the mucin‐like domain of CD93 contribute to stabilising its cell surface expression 183. Intriguingly, the lack of O‐linked glycosylation enhanced proteolytic cleavage of CD93 from the cell membrane and increased levels in culture medium. This provides a possible role for the mucin‐like region within all the CTLD group 14 family members in preventing proteolytic cleavage, and also offers a potential mechanism of modulating surface cleavage events. The cell‐surface expression of CD93 is regulated by protein kinase C isoenzymes 184, and shedding could be enhanced by phorbol 12‐myristate 13‐acetate (PMA), a potent activator of protein kinase C 185.
Upon knockdown of MMRN2, together with inhibition of new protein synthesis by cycloheximide treatment, cell surface CD93 levels were shown to be diminished whereas soluble CD93 levels increased 167. This suggests that the interaction with MMRN2 may render CD93 less susceptible to proteolytic cleavage and hence this recognition event is important for regulating stable cell surface expression of CD93.
Additional roles for CD93
A study examining CD93 expression in neurons and microglia revealed that upon response to LPS mediated inflammation, the cytoplasmic tail of CD93 could be detected in the cytoplasm and nucleus 148. This is the first instance that a possible gene expression modulating role has been inferred for the CD93 cytoplasmic region. As CD93 ECD cleavage is enhanced by LPS, and the cytoplasmic domain can be detected even after cleavage 171, it is possible that it translocates to the nucleus after ECD shedding, similar to that described for notch ECD 186. The authors did not confirm whether nuclear localisation followed CD93 ECD cleavage and further work is warranted in order to define the precise molecular mechanisms underlying this effect. Notably, similarities have been proposed between CD44 and CD93 171 as the CD44 ECD can be cleaved by ADAM10 and its intracellular domain by γ‐secretase, similar to that described for notch receptors 187.
CLEC14A
C‐type lectin family 14 member A (CLEC14A) is a type‐I single‐pass transmembrane glycoprotein and considered to be endothelial specific. It was described as a novel endothelial‐specific gene identified by microarray analysis and data mining, and referred to as an unidentified expression sequence tag (EST; accession number http://www.ncbi.nlm.nih.gov/protein/AW770514) 188. CLEC14A was initially classified as a tumour endothelial marker based on immunohistochemical staining of multiple distinct tumour types, with strong staining on tumour‐associated vessels in contrast to a near absence of staining in healthy tissues 189. Upregulation of CLEC14A at the mRNA level was also described in nonsmall cell lung cancer (NSCLC) tissues compared to healthy lung 190. Interestingly, high expression of CLEC14A in this cancer type correlated with improved clinical outcomes. A further study indicated that the methylation status of CLEC14A strongly correlated with its expression levels in NSCLC, and CLEC14A protein levels were reduced in tumour tissues compared to healthy adjacent tissue 191. Similar to CD93, CLEC14A was described as a key gene in a proposed ‘tumour angiogenesis signature’ determined by meta‐analysis of over 1000 tumour samples including breast, renal and head and neck cancers 154. It was subsequently found to be upregulated at the protein level and increased with tumour progression in two different spontaneous mouse tumour models, namely cervical and pancreatic 177. More recently, CLEC14A overexpression on the vasculature in ovarian cancer has been reported but did not correlate with survival in this tumour type 192. The authors also demonstrated that CLEC14A expression was undetectable along with reductions in microvessel density in patients receiving neoadjuvant chemotherapy prior to surgery.
CLEC14A expression
CLEC14A (or C1qrl in zebrafish) is thought to be located downstream of the master endothelial and haematopoietic regulatory transcription factor etsrp in zebrafish (ETV2 in humans) 193. The etsrp transcription factor has recently been implicated in tumour angiogenesis in xenograft models of melanoma and sarcoma in zebrafish embryos 194. During zebrafish development, clec14a is expressed at 24 h postfertilisation and morpholino knockdown of gene expression can have detrimental effects on vasculature formation 189. Interestingly, following reintroduction of human CLEC14A mRNA into these knockdown zebrafish embryos, the vasculature reverted back to a normal phenotype showing the correct zebrafish homologue was targeted and highlighting the conserved nature of these genes. Zebrafish homozygous for clec14a null alleles develop normally into viable adults 195, these zebrafish as embryos do however display delays in vasculogenesis and angiogenesis, and these defects can be heightened with knockdown of C1qr/CD93.
Mouse embryos display expression of CLEC14A in inter‐segmental vessels and vessels in the developing brain, at embryonic day 10.5 196. Expression was also detected in the vessels of mouse retinas at postnatal day 12, which are constantly undergoing development after birth. CLEC14A has also been described as being upregulated when EPCs differentiate into outgrowth endothelial cells 197. Despite early expression in embryogenesis, CLEC14A is not completely critical to angiogenesis as CLEC14A deficient animals develop normally with no gross defects in vessel formation.
CLEC14A has been described to be upregulated by low shear stress 189. Indeed, upon application of 2 pascal (Pa) of flow induced laminar shear stress to HUVEC in culture, this leads to a significant reduction (> 90%) of CLEC14A expression when compared to static culture (low shear stress). This may explain the expression of CLEC14A observed within the ill‐formed vessels of tumours that experience irregular blood flow and low shear stress 198. Upstream regions of the CLEC14A gene in humans contain predicted Sp1 transcription factor binding sites. Interestingly, Sp1 is phosphorylated in response to shear stress and can inhibit the expression of membrane type‐I matrix metalloproteinase (MT1‐MMP) in endothelium 199. Microarray analysis of atherosclerosis patient samples revealed upregulation of CLEC14A in vessels that display high levels of stenosis 200. This is consistent with previous findings, as shear stress is lower in blood vessels containing atherosclerotic plaques when compared with healthy controls 201. CLEC14A expression has also been linked with hypoxia in HUVEC, and could explain its greater expression in the tumour vasculature 202.
CLEC14A expression has been demonstrated in two different human lung cancer cell lines in vitro and when CLEC14A was further overexpressed in these cell lines, this led to reductions in proliferation, migration and invasion as well as reductions of in vivo tumour formation as xenografts in nude mice 191. However, the physiological relevance of CLEC14A expression in tumour cells themselves remains to be seen as such expression has not been reported in clinical specimens or in tumour cells by any other group.
CLEC14A roles in vascular biology and cancer
The involvement of CLEC14A in angiogenesis is reinforced by in vivo experiments performed in homozygous CLEC14A knockout mice 203. These mice remained viable and displayed no gross developmental defects. Nevertheless, when challenged with subcutaneous LLC, tumour growth and tumour angiogenesis were reduced relative to wild‐type controls. Similarly, in subcutaneous sponge implants FGF‐2‐induced angiogenesis was also impaired. However, another report has suggested that CLEC14A may not serve as a viable antivascular target; this study demonstrated that although implanted tumour growth of LLC and B16F10 melanoma was markedly impaired in CLEC14A knockout mice in comparison to wild‐type littermates, tumour bearing CLEC14A knockout mice died earlier 204. These deleterious effects were attributed to reduced pericyte coverage and CLEC14A‐deficient vessels displaying increased permeability. Furthermore, this study revealed that CLEC14A deficiency led to increased lung metastasis burden when B16F10 cells were injected intravenously or into the foot pad.
There are conflicting results regarding vessel sprouting from aortic ring assays from CLEC14A KO mice. Noy et al. described reduced sprouting in these CLEC14A‐deficient mice, but Lee et al. described increased sprouting in aortic ring assays in comparison to control 203, 204. The reasons for these described conflicting roles for CLEC14A in angiogenesis have not been elucidated. It is possible that discrepancies are due to differences in methodology of these assays or dissimilarities in background strains of these CLEC14A KO mice.
The requirement for CLEC14A in various in vitro angiogenesis assays were reported by two independent groups utilising siRNA‐mediated knockdown of CLEC14A 189, 196. Based on these knockdown experiments, the ability of HUVEC to form tubes and close wounded monolayers in scratch assays was compromised. In addition, involvement of CLEC14A in sprouting angiogenesis was demonstrated by siRNA knockdown of CLEC14A in HUVEC which led to marked reduction in sprout formation based on spheroid assays, CLEC14A deficient cells were also less likely to be found as tip cells in these sprouts 203. Ectopic expression of CLEC14A in cells that do not normally express it, results in the formation of filopodia‐like protrusions 189. Altogether these findings implicate CLEC14A in filopodia formation, a vital step in sprouting angiogenesis.
The CLEC14A CTLD has been implicated in cell–cell adhesion interactions, since CLEC14A overexpressing HEK293F cells have the ability to initiate preliminary cell–cell aggregates, which can be abolished following incubation with CTLD‐specific CLEC14A antibodies 205. These antibodies were reactive against both human and mouse CLEC14A forms and could downregulate CLEC14A levels on the surface of HUVEC, posing a potential for internalisation of antibodies, and possible utilisation as ADCs carrying a cytotoxic payload. Finally, these antibodies could reduce HUVEC cell migration and tube formation based on in vitro assays. Further studies optimised the solubility and stability of the CLEC14A CTLD‐targeting antibodies and showed that they could block angiogenesis in mouse models utilising Matrigel plugs injected with recombinant VEGF or human tumour cells 206. Collectively, these results suggest that the CTLD of CLEC14A has functional roles in angiogenesis.
CLEC14A interaction partners
CLEC14A has been described as a component of HUVEC ECM which binds to the ECM glycoprotein MMRN2 177. Like CLEC14A, MMRN2 protein was upregulated with tumour progression of two different spontaneous mouse cancer models, highlighting importance of this interaction and potential as therapeutic tumour vascular targets 177. The CLEC14A–MMRN2 interaction could be blocked by a monoclonal antibody specific for CLEC14A, and when administered intraperitoneally retarded growth of subcutaneously implanted LLC in mice 203. This interaction was dependent upon a predicted long‐loop region encompassing residues 97–108 within the CLEC14A CTLD 101. The CLEC14A‐MMRN2 interaction could also be targeted using a minimal peptide fragment derived from MMRN2. This peptide reduced endothelial tube formation and also decreased tumour growth when expressed by LLC cells in vivo 101.
The CLEC14A CTLD also has the capacity to bind other ligands including the heat shock protein 70 kDa 1A (HSP70‐1A) which increased HUVEC adhesion, aggregation and ERK phosphorylation 207. This finding may rationalise the cell aggregation effects observed in HEK293F cells overexpressing CLEC14A, with HSP70‐1A forming oligomeric complexes and creating a bridge between CLEC14A expressed on different cells. This binding phenomenon was dependent on amino acids 43–69 of the CLEC14A CTLD 207, which based on its predicted structure encompasses an alpha helical region that is distal to the MMRN2 binding site. However, at present, it is unclear whether HSP70‐1A and MMRN2 are mutually exclusive binding events or if they compete with each other 101. The same group previously discovered that HSP70‐1A could serve as a potent proangiogenic factor 208. The active HSP70‐1A binding region of CLEC14A fused to an Fc tag was used to create a novel peptibody which could inhibit HSP70‐1A–stimulated tubule formation of HUVEC in vitro 207. In the same study, stimulation of HUVEC with HSP70‐1A increased ERK phosphorylation, and this effect was reduced when incubating with CLEC14A CTLD‐Fc fusion proteins. This suggests that CLEC14A may have signalling roles, although the authors did not probe whether HSP70‐1A–mediated ERK phosphorylation was blocked with knockdown of CLEC14A.
The intracellular domain of CLEC14A reportedly interacts with vascular endothelial growth factor receptor‐3 (VEGFR‐3) 204. There are currently no other known interactors for the CLEC14A cytoplasmic domain, although global phosphoproteomic analysis of HUVEC has revealed the presence of five serine residues that can be phosphorylated, namely S437, S445, S483 S487 and S488 209, 210. The phosphorylated S483 was also found in other proteomic analyses and was described as being close to a predicted PDZ binding domain in the CLEC14A cytoplasmic domain 177. Since these residues are not conserved in mouse CLEC14A the relevance of these post‐translational modifications will need to be determined experimentally. A summary of the protein interactions of CLEC14A are shown in Fig. 6.
Figure 6.
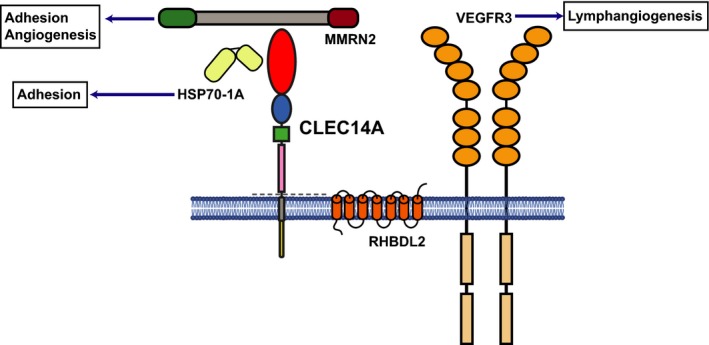
Schematic of CLEC14A protein with ligand binding partners. CLEC14A CTLD binds to MMRN2 and to HSP70‐1A. The whole ECD can be cleaved by RHBDL2. There are currently no known direct intracellular partners for CLEC14A.
CLEC14A shedding
CLEC14A can be shed from the endothelial cell membrane by the intramembrane serine protease RHBDL2 211. RHBDL2 cleaves at a site close to the transmembrane domain, liberating the intact ECD of CLEC14A to regulate sprouting angiogenesis. The CLEC14A‐ECD can mediate antiangiogenic effects in vitro and in vivo when utilised as an Fc‐tagged recombinant protein. Intriguingly, when used as a staining reagent the CLEC14A‐ECD‐Fc fusion bound to sprouting endothelial cells, predominantly tip cells. Therefore, one could propose a scenario in which shedding of CLEC14A may aid in regulation of sprouting angiogenesis. Such cleaved CLEC14A predominantly by stalk cells in an angiogenic sprout would then be able to bind to tip cells.
CTLD group 14 family summary
The CTLD group 14 family members all mediate effects upon the vasculature and some share remarkable similarities with respect to binding partners localised within the extracellular matrix. Further similarities are also observed with respect to expression patterns and regulation by predicted transcription factors. A summary of similarities and differences are displayed in Table 1.
Table 1.
Biological comparisons of CTLD group 14 family members.
| Biology | Thrombomodulin | CD248 | CD93 | CLEC14A | References |
|---|---|---|---|---|---|
| Knockout mouse | Embryonic lethal | Viable | Viable | Viable | 15, 91, 161, 203 |
| Knockout mouse tumour development | N/A | Reduced growth | Reduced growth | Reduced growth | 91, 156, 203 |
| Extracellular binding partners | Thrombin, Protein C, Lewis Y antigen, FGFR1, Fibronectin, GPR15 (EGF5), Ang‐1, Ang‐2, CD14, HSP70‐1 | Mac‐2BP, Fibronectin, Collagens I & IV, MMRN2 | EGFR1 (EGF domains), MMRN2, αxβ2 | MMRN2, HSP70‐1A | 19, 24, 25, 31, 34, 37, 38, 59, 60, 98, 101, 102, 177, 181, 182, 207 |
| Intracellular binding partners | Ezrin | None reported | Moesin, GIPC, Cbl, src | VEGFR3 | 81, 148, 165, 169, 171, 204 |
| Expression | Endothelial, Haematopoietic | Pericytes, Fibroblasts, CD8+ T cells | Endothelial, Haematopoietic, Neural | Endothelial | 12, 14, 27, 84, 95, 96, 148, 156, 189, 196 |
| Shear‐induced expression | Downregulated with shear | Not reported | Not reported | Downregulated with shear | 57, 189 |
| Cleavage | Whole ECD, Possibly CTLD | Not reported | Whole ECD | Whole ECD | 70, 77, 179, 211 |
| Cleavage enzyme | RHBDL2, MMPs neutrophil elastase, cathepsin G, proteinase 3 | Not reported | Metalloproteinases | RHBDL2 | 70, 72, 179, 211 |
| Location of soluble form | Culture medium, Blood, Urine, Synovial fluid | Blood, Ascites | Culture medium, Blood, Synovial fluid | Culture medium, Urine | 30, 55, 73, 74, 75, 76, 141, 143, 153, 158, 178, 179, 180, 211, 228 |
| Solved structures | EGF domains in complex with thrombin | Not reported | Not reported | Not reported | 20 |
Expression localisation of CTLD group 14 family members in vivo
To gain an in‐depth understanding of gene expression of all four CTLD group 14 family members in vivo we used the recently described Tabula Muris database, which consists of single‐cell transcriptomic analyses of over 100 000 cells derived from 20 different healthy adult mouse organs and tissues from C57BL/6 strain mice 212. This allowed graphical representation of gene expression by use of t‐SNE plots and revealed that thrombomodulin is mainly expressed in endothelium, epithelium and mesenchymal cell types, as well as some myeloid, pro‐B cell and bladder cells (Fig. 7). CD248 is restricted mainly to mesenchymal cells, fibroblasts, pericytes and bladder cells, and importantly there was a lack of expression of CD248 in endothelial cells from multiple organs. CD93 showed mainly endothelial, as well as myeloid, pro‐B cell and haematopoietic progenitor cell expression, but a lack of expression in neurons. Finally, CLEC14A exhibited the most endothelial specific expression of the four family members but also localised in bladder cells and leucocytes from the thymus. Bladder cells described here include mesenchymal cell types. The endothelial expression of CTLD group 14 family members was then investigated further, t‐SNE plots of all endothelial cells from different organs as well as pericytes from brain were created, showing that thrombomodulin is expressed in mostly all endothelial cell types, CD248 is not expressed in endothelial cells (but is expressed in pericytes) and CD93 and CLEC14A are expressed to a varied degree in most endothelial cells (Fig. 8A). As CD93 and CLEC14A share the ligand MMRN2, they are both expressed by endothelium, share similar endothelial phenotypes and have been suggested previously to compensate for lack of the other, we investigated whether endothelial cells in certain organs displayed differential expression of each gene. This revealed that CD93 is expressed higher than CLEC14A in a majority of organs except kidney (no significant difference) and liver, lung and pancreas endothelium, where CLEC14A is expressed significantly higher (Fig. 8B). Interestingly, there appeared to be a subset of endothelial cells within the lung that do not express CD93 but do express CLEC14A, t‐SNE plots solely of lung endothelium showed that there was a clustering of these cells suggesting an unknown endothelial subtype that does not express CD93 in mouse lung (Fig. 8C). The Tabula Muris database provides novel interesting insights into expression patterns, at least at the gene expression level, in an adult healthy mouse, although this is not an exhaustive list of all mouse cell types that express these genes as only 20 major organs and tissues were analysed. Similar studies analysing single‐cell gene expression of mice in different disease states such as cancer or inflammation would be an extremely valuable resource.
Figure 7.
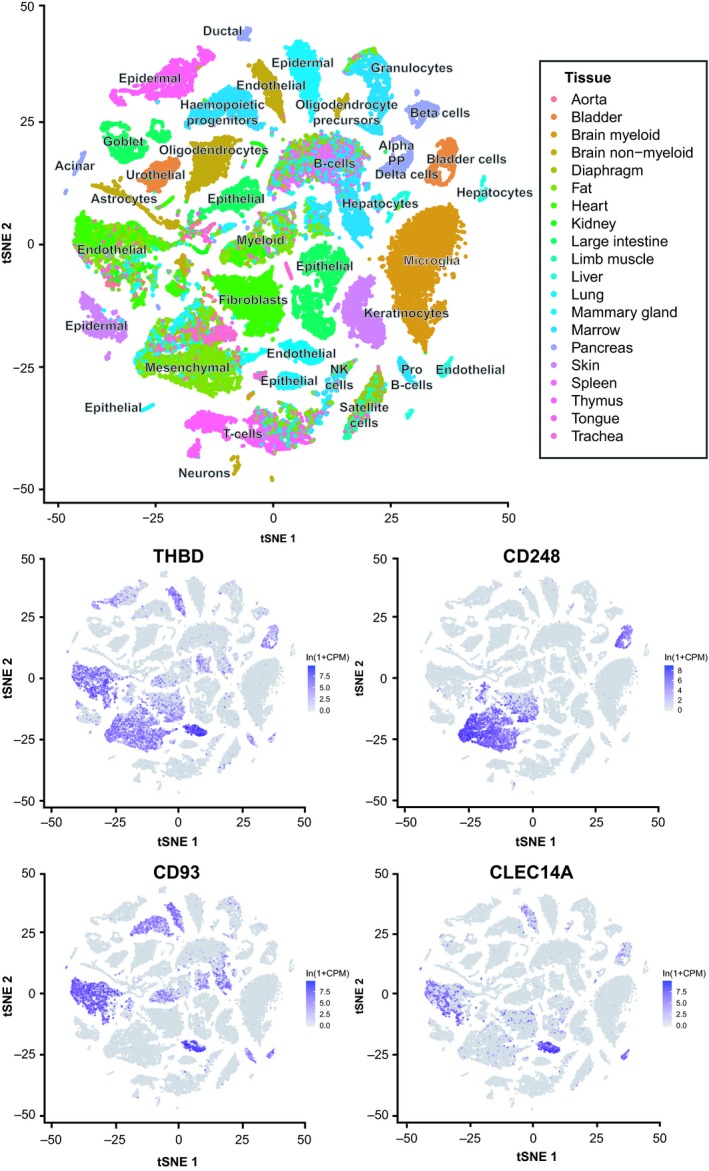
Expression of CTLD group 14 family members in mouse tissues. The Tabula Muris database was used to determine which mouse cell types expressed each CTLD group 14 family gene from data acquired through fluorescence activated cell sorting and single‐cell gene expression analysis. The t‐SNE plot at the top displays annotations of each cell type and shows a legend of colours corresponding to which organ or tissue type that cell was from. The lower t‐SNE plots display in which cell types each family member was expressed (purple), ln(1 + CPM) is the natural logarithm of counts per million + 1.
Figure 8.
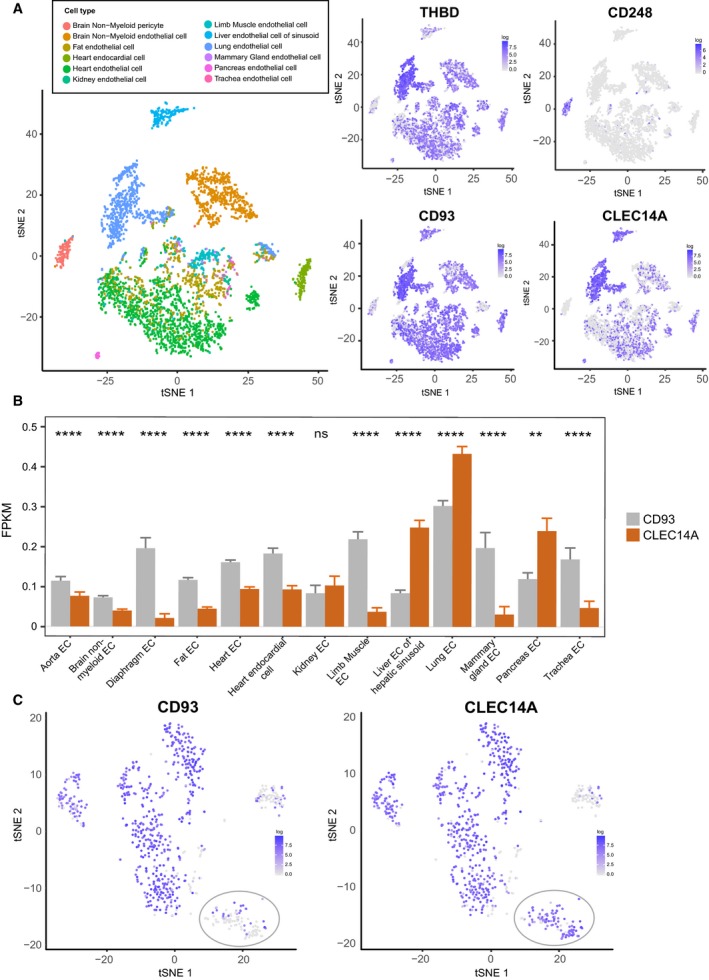
Endothelial expression of CTLD group 14 family members in mouse tissues. (A) The Tabula Muris database was used to create t‐SNE plots of all endothelial cells from different organs as well as brain pericytes. The t‐SNE plot at the top left displays a legend of colours corresponding to which organ or tissue type that cell was from. Expression of each CTLD group 14 family member within these cell types are displayed as t‐SNE plots. (B) Single‐cell sequencing data analysed as fragments per kilobase million was used to compare CD93 and CLEC14A expression in different endothelial cells from different organs. Wilcoxon statistical test was used to compare ****P ≤ 0.0001 Aorta ECs n = 262, Brain nonmyeloid EC n = 1250, Diaphragm EC n = 154, Fat EC n = 1180, Heart EC n = 2274, Heart endocardial cell n = 350, Kidney EC n = 80, Limb Muscle EC n = 258, Liver EC n = 392, Lung EC n = 1476, Mammary gland EC n = 98, Pancreas EC n = 98, Trachea EC n = 66. (C) t‐SNE plots of lung endothelium alone were created which revealed the presence of a cluster of cells expressing low levels of CD93 when compared with all other lung endothelial cells but similar levels of CLEC14A (grey ellipse).
Considerations and perspectives
Similarities between CTLD group 14 family members
All CTLD group 14 family members comprise six canonical cysteines in the CTLD that are predicted to form disulfide bonds and support the CTLD scaffold. They also encompass two noncanonical cysteines located within the predicted long‐loop regions which due to their close proximity may also form disulfide links. Interestingly, such noncanonical cysteines within the long‐loop region are found only in three other CTLD families; group 8 containing layilin and chondrolectin, groups 11 and 12. Disulfide bond formation within this long loop appears to be essential for the interaction of CLEC14A and CD93 with MMRN2. Upon point mutation of these long‐loop cysteines, the CLEC14A CTLD folds correctly as it is recognised by conformation‐specific monoclonal antibodies, but completely diminishes its binding capability with MMRN2 101. It is possible that the corresponding cysteines in CD248 and thrombomodulin are similarly important for CTLD‐mediated recognition events and constructs containing point mutations of these residues could represent invaluable tools in determining functional relevance of the CTLD within this family.
Thrombomodulin and CD93 are both anchored to the actin cytoskeleton by associating with ERM adapter proteins; thrombomodulin to ezrin and CD93 to moesin. The thrombomodulin–ezrin interaction was initially described in epithelial cells and this is not surprising given that ezrin in this cell type is the highest expressed ERM protein. In contrast in endothelial cells, moesin is the most abundantly expressed ERM protein 213. Due to the high sequence homology between ezrin and moesin (~ 75% sequence identity) 214, it is tempting to speculate that thrombomodulin also interacts with moesin in the endothelium. However, the ability of thrombomodulin to bind to ezrin or moesin within endothelial cells is yet to be assessed. Likewise, CD93 may be able to bind to multiple ERM adapter proteins as is the case for CD44 binding to ezrin, radixin and moesin 215. Both thrombomodulin and CD93 interactions with ERM adapter proteins are dependent upon positively charged residues within the cytoplasmic tail, which are absent in the corresponding regions of CD248 and CLEC14A. This motif comprises of RKK in thrombomodulin and RKR in CD93. Strikingly, the RKE motif in CLEC14A could potentially abolish binding to ERM proteins due to repulsion effects attributable to the negatively charged glutamic acid side chain. Nevertheless, there is a distinct possibility that the intracellular domain of CLEC14A makes direct or indirect contacts with the cytoskeleton, due to its proposed roles in filopodia formation and cell migration. Also, the corresponding region in the CD248 intracellular domain consists of the NKR motif, and it is unclear whether the noncharged asparagine residue can compensate for binding to ERM adapter proteins. Finally, a related point to consider is that in both CD248 and CLEC14A, the three‐amino acid motif is preceded by a proline residue which may cause rigidity and/or conformational alterations that could affect interactions with ERM adapter proteins.
Evidence for CLEC14A along with thrombomodulin acting as potential cell adhesion molecules is observed following overexpression of each protein and leads to induced cell aggregation. Such effects are dependent upon the CTLD of each protein 80, 196, 205. HUVEC plated on immobilised fragments of MMRN2 are sufficient to allow adherence of HUVEC in cell binding assays; however, at present, it is unclear whether CLEC14A, CD93 or both mediate this adhesive function 101. Similarly, when CD248 is overexpressed in CHO cells, this enables them to bind to fibronectin and Matrigel in cell adhesion assays 98.
CLEC14A and CD93 both bind MMRN2 as does CD248, this to our knowledge is the first example of an endothelial protein binding to an extracellular matrix protein which in turn interacts with a pericyte‐expressed protein of the same family. This raises an interesting question of how MMRN2 has evolved to bind two distinct CTLD group 14 family members in nonoverlapping regions of the same molecule. This binding event may have roles in already proposed CD248‐dependent vascular regression caused by pericytes 87. With regard to CLEC14A expression and MMRN2 interaction, this may flag areas of the newly formed vasculature that is experiencing low blood flow and low shear stress. Upon binding to MMRN2 through CD248, pericytes could then selectively cause vascular regression through unknown mechanisms. Interestingly, pericyte coverage of endothelium is reduced in the brain, retina and melanoma tumour vasculature of CLEC14A knockout mice 204. However, no defects were reported in pericyte coverage of vessels in models of gliomas between CD93 knockout and wild‐type mice 156. This suggests that CLEC14A may have more predominant roles in pericyte attachment, or there could be differences in the requirement of CLEC14A or CD93 in pericyte attachment in different tissues.
MMRN2 has been shown to be a substrate for MMP9, although it is unclear whether the subsequent cleaved fragments can still bind to members of the CTLD group 14 family and clearly warrants further investigation 216. Intriguingly, as mentioned previously, CD248 overexpression results in upregulation of MMP9 posing a scenario in which MMRN2 could be processed by MMP9 potentially regulating CD248–MMRN2 binding. Alternatively, CD248‐mediated upregulation of MMP9 may allow cleavage of MMRN2 and detachment of the endothelial–pericyte interaction.
The EGF repeats and mucin‐like regions of both CD93 and thrombomodulin have been reported to have proangiogenic effects. In the case of thrombomodulin, this mitogenic ability was abolished if the CTLD was present on the soluble protein (i.e. including the CTLD, sushi and EGF repeats), although it is unclear whether this also applies for CD93. Nevertheless, one could speculate a scenario where differential proteolytic cleavage of such proteins results in diverse outcomes upon the endothelium and other cell types, allowing fine tuning of cellular events. As discussed previously, there is evidence that the CTLD of thrombomodulin can be shed from the full‐length molecule or from the cleaved ECD. Additionally, there is likely a second cleavage event in the CLEC14A ECD generating a fragment smaller than the full‐length ECD which encompasses the CTLD 211. Multiple proteolytic cleavage events may be true for other CTLD group 14 family members.
Potential roles in immunosuppression
Angiogenesis and immunosuppression are two tightly regulated processes that often occur in unison. They have been described as parallel processes especially in the context of tumour angiogenesis and tumour immunosuppression 217. Many proangiogenic proteins also mediate immunosuppressive effects upon the vasculature as well as immune cells directly 218. Here we describe some examples of CTLD group 14 family members that elicit immunosuppressive roles. For example, expression of CD248 on naïve T‐cells correlated with decreased cell proliferation. In this setting, CD248 binding to its ligands that are upregulated in tumour angiogenesis (i.e. MMRN2 or fibronectin etc.) may inhibit T‐cell proliferation. Similarly, thrombomodulin expression on the vasculature or perhaps in soluble form can mediate immunosuppressive functions upon binding GPR15 on T‐cells as well as a whole host of other anti‐inflammatory roles as described above. Although high expression of thrombomodulin has been reported by multiple groups in diverse cancer indications, whether thrombomodulin can actually elicit an immunosuppressive function in the context of cancer remains to be elucidated and the finding that low thrombomodulin leads to improved prognosis seems to contradict this theory. CD93 has also been described to trigger anti‐inflammatory events, such as limiting leucocyte migration in peritonitis 170. Other members of the CTLD group 14 family may invoke broader effects upon distinct components of the immune system, and potentially contribute to immunosuppression especially in the context of tumours evading the immune system.
Potential use as therapeutic targets
So far clinical trials targeting CD248 have proved to be very disappointing, and such agents may only be effective in certain tumour types or may need to be combined with other therapeutics for optimal clinical benefit. However, CD248 targeting antibodies still have promise as therapies for inflammatory or fibrotic diseases where CD248 has a pathological role.
CLEC14A as a therapeutic target of the tumour vasculature has been investigated by many different preclinical strategies using antibodies as well as fragments of its known ligands and even chimeric antigen receptor T‐cells 219, 220. Since it is well established that CLEC14A is expressed on vessels that experience low shear stress and aberrant blood flow, it is conceivable that only nonfunctional tumour vessels will be targeted by such agents. This could prove beneficial as vascular normalisation effects would likely take place, lowering hypoxia, which could lead to better accumulation and delivery of other drugs used in combination such as chemotherapy. Additionally, such CLEC14A targeting could be combined with immunotherapies which rely on infiltration of effector immune cells into the tumour mass, where functional and more ‘normal’ vasculature would likely be advantageous 109.
In studies investigating the use of CLEC14A CTLD specific antibodies, Kim et al. tested a human colorectal cancer cell line HCT116 as well as a bevacizumab‐resistant version of this line. Both cell lines showed significant reductions in in vivo angiogenesis following treatment with CLEC14A antibodies when these cells were embedded in Matrigel and injected subcutaneously 206. This suggests a possible use for targeting of CLEC14A in patients that have acquired resistance to VEGF blockade. More importantly these findings suggest that although targeting of CLEC14A can reduce VEGF‐dependent angiogenesis in various models, it may also ablate angiogenesis induced by VEGF‐independent pathways. However, the authors did not assess the efficacy of these antibodies in targeting this resistant cell line in tumour xenograft studies; therefore, the tangible benefit of CLEC14A targeting in tumour types resistant to VEGF blockade is yet to be fully established.
Dual targeting of CLEC14A and CD93 was achieved by use of a MMRN2 fragment that contained the CTLD‐binding region (amino acid residues 495–674 in human and 495–678 in mouse) fused to an Fc tag 101. This resulted in a decrease in syngeneic tumour growth in vivo and disruptions in angiogenesis in vitro. Blocking CLEC14A and CD93 in this manner will likely inhibit endothelial cells binding to endogenous MMRN2 and may even interrupt the fibrogenesis of fibronectin, as has been described with genetic ablation of CD93 167. Furthermore, such targeting strategies may destabilise the binding of CD248 expressing pericytes to the tumour vasculature, although whether this affects pericyte coverage remains to be investigated. There is scepticism in the field in terms of whether such pericyte targeting approaches provide meaningful clinical benefit 221. The use of such dual‐targeting approaches negates the ability of one protein compensating the loss of the other. However, it is important to note that this dual‐targeting MMRN2 fragment Fc fusion protein was expressed directly in the tumour microenvironment by genetically engineered tumour cells. With less restricted expression of the targeting fragment, we cannot rule out the possibility that this agent could display off target effects by binding to other CD93 expressing cell types such as monocytes or B‐cells.
The likelihood of CTLD group 14 family members serving as viable targets in cancer therapy will ultimately depend on the expression profile of these proteins, which if not tumour or tumour vasculature specific could result in toxicity‐related issues in patients. In this regard, a seminal paper investigating targeting the tumour endothelial marker and immunomodulatory molecule CD276 (also referred to as B7‐H3) described that the most important determining factor for avoiding toxicity is in fact level of expression 222, 223 . Indeed, even though CD276 displays a widespread expression pattern in mouse and human tissues, the fact that it is so highly expressed by tumour cells and the associated tumour vasculature, resulted in ADCs against CD276 only having substantial effects upon the tumour microenvironment. In light of this data, experiments that determine levels of target protein expression will likely become paramount. Relatedly, low‐affinity, high avidity therapeutic agents could be used against targets that are highly expressed on tumour‐associated tissue but still expressed lowly on normal tissue; in this way, agents would preferentially bind to highly expressing cell types. This approach was demonstrated with low‐affinity, high avidity HER‐2/CD3‐binding bispecific agents that redirect T‐cells towards breast cancer cells 224. These high‐avidity bispecific antibodies induced negligible effects on in vivo tumour models expressing low levels of HER‐2 but successfully eradicated high‐expressing tumour lines, suggesting that normal tissues expressing HER‐2 at low levels may be avoided and toxicity minimised.
As mentioned previously, the cleavage of the CTLD group 14 family members may negatively impact antibody targeting therapies, as the soluble forms may sequester the antibodies in the blood rendering them incapable of binding to the cell surface receptors. However, this issue will likely be addressed in preclinical models and presumably be overcome in phase I dose escalation studies of CTLD group 14 family targeted agents.
High priority areas of future research
There are many unanswered questions relating to the physiological and pathological functions of this family of molecules, especially for CD248, CD93 and CLEC14A. In particular, information on intracellular signalling events is currently lacking.
It is currently unknown whether CLEC14A and CD93 share other extracellular binding partners as is the case for MMRN2. Likewise, other CTLD group 14 members may share additional binding proteins as has been shown for HSP70‐1 binding both CLEC14A and thrombomodulin 25, 207. The region of human CLEC14A CTLD that engages HSP70‐1A exhibits 25.9% sequence identity to thrombomodulin, 29.6% identity to CD93 and 33.3% to CD248; however, the domain important for the thrombomodulin‐HSP70‐1 interaction has not yet been defined. Clearly further binding experiments to extensively characterise interactions between other CTLD group 14 family members and newly described ligands will need to be conducted.
It is uncertain whether CLEC14A and CD93 compete for binding with MMRN2 or whether they have independent or similar roles. Also, the signalling outcomes following MMRN2 binding to CLEC14A, CD93 or CD248 are not fully established. CLEC14A and CD93 have been postulated to have redundant roles in zebrafish angiogenesis but not vasculogenesis 225. Simultaneous knockout of both CLEC14A and CD93 led to more severe defects in intersegmental vessel formation compared with single‐gene knockouts, now reported by two different groups 195. VE‐cadherin expression was absent in vessels that lacked CLEC14A and CD93, suggesting abnormalities in endothelial cell–cell adhesion, when VE‐cadherin was replaced this rescued the detrimental phenotype. Knockdown of CD93 has also been shown to reduce VE‐cadherin levels in HUVEC 156, although phenotypic outcomes following double knockdown of both CLEC14A and CD93 have not yet been reported in mammalian cell types. Currently there are no data on whether CLEC14A and CD93 display redundancy in mammals, and a double KO mouse would begin to address this important question.
Finally, as CLEC14A and CD93 have been described as proteins highly expressed in the tumour vasculature, with roles in angiogenesis, it remains to be investigated whether these molecules are also expressed and have roles in other routes in which a tumour can acquire a blood supply. Two such mechanisms involve (a) vessel co‐option, whereby tumour cells hijack the existing vasculature in highly vascularised organs such as the lung, liver and brain 226, or (b) vascular mimicry, where tumour cells can exhibit endothelial‐like properties and form functional vessel like structures 227.
Concluding remarks
The CTLD group 14 is a family of molecules with diverse roles in the vasculature, inflammation as well as tumour progression. The increasing interest in these molecules including elucidation of their normal biology as well as their potential as therapeutic targets in cancer will likely continue to be explored in the future.
Conflict of interest
KAK and RB are inventors on patents WO/2016/116760 entitled ‘Inhibitors of the interaction between CLEC14A and Multimerin‐2 for inhibition of angiogenesis’ and the related filed patents GB1612860.5, GB1612534.6 and GB1702926.5.
Author contributions
KAK designed and wrote the manuscript as well as directed analysis of the Tabula Muris data. JLM analysed the Tabula Muris data and edited and reviewed the manuscript. FM and RB aided in writing, editing and review of the manuscript.
Acknowledgements
We thank Dr Spyros Darmanis, Professor Tony Wyss‐Coray and Professor Stephen R Quake for their kind permission to use the Tabula Muris database and Professor Kurt Drickamer for compiling a useful database on human and mouse CTLD containing proteins (https://www.imperial.ac.uk/research/animallectins/ctld/mammals/mammals.html). KAK was funded by a Medical Research Council PhD Studentship and is currently funded by a Canadian Institute of Health Research Banting Postdoctoral Fellowship. JLM is funded by a CRUK PhD studentship. FM is funded by Wellcome Trust (grant reference 099266/Z/12/Z).
Contributor Information
Kabir A. Khan, Email: kkhan@sri.utoronto.ca.
Roy Bicknell, Email: r.bicknell@bham.ac.uk.
References
- 1.Zelensky AN & Gready JE (2005) The C‐type lectin‐like domain superfamily. FEBS J 272, 6179–6217. [DOI] [PubMed] [Google Scholar]
- 2.McMahon SA, Miller JL, Lawton JA, Kerkow DE, Hodes A, Marti‐Renom MA, Doulatov S, Narayanan E, Sali A, Miller JF_et al_ (2005) The C‐type lectin fold as an evolutionary solution for massive sequence variation. Nat Struct Mol Biol 12, 886–892. [DOI] [PubMed] [Google Scholar]
- 3.Norman DG, Barlow PN, Baron M, Day AJ, Sim RB & Campbell ID (1991) Three‐dimensional structure of a complement control protein module in solution. J Mol Biol 219, 717–725. [DOI] [PubMed] [Google Scholar]
- 4.Wei X‐Q, Orchardson M, Gracie JA, Leung BP, Gao BM, Guan H, Niedbala W, Paterson GK, McInnes IB & Liew FY (2001) The sushi domain of soluble IL‐15 receptor is essential for binding IL‐15 and inhibiting inflammatory and allogenic responses in vitro and in vivo. J Immunol 167, 277–282. [DOI] [PubMed] [Google Scholar]
- 5.Wouters MA, Rigoutsos I, Chu CK, Feng LL, Sparrow DB & Dunwoodie SL (2005) Evolution of distinct EGF domains with specific functions. Protein Sci 14, 1091–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doyonnas R, Yi‐Hsin Chan J, Butler LH, Rappold I, Lee‐Prudhoe JE, Zannettino ACW, Simmons PJ, Buhring HJ, Levesque JP & Watt SM (2000) CD164 monoclonal antibodies that block hemopoietic progenitor cell adhesion and proliferation interact with the first mucin domain of the CD164 receptor. J Immunol 165, 840–851. [DOI] [PubMed] [Google Scholar]
- 7.Jentoft N (1990) Why are proteins O‐glycosylated? Trends Biochem Sci 15, 291–294. [DOI] [PubMed] [Google Scholar]
- 8.Rettig WJ, Garin‐Chesa P, Healey JH, Su SL, Jaffe EA & Old LJ (1992) Identification of endosialin, a cell surface glycoprotein of vascular endothelial cells in human cancer. Proc Natl Acad Sci USA 89, 10832–10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nepomuceno RR, Ruiz S, Park M & Tenner AJ (1999) C1qRP is a heavily O‐glycosylated cell surface protein involved in the regulation of phagocytic activity. J Immunol 162, 3583–3589. [PubMed] [Google Scholar]
- 10.Weisel JW, Nagaswami C, Young TA & Light DR (1996) The shape of thrombomodulin and interactions with thrombin as determined by electron microscopy. J Biol Chem 271, 31485–31490. [DOI] [PubMed] [Google Scholar]
- 11.Etzioni A (1996) Adhesion molecules‐their role in health and disease. Pediatr Res 39, 191–198. [DOI] [PubMed] [Google Scholar]
- 12.Conway EM (2012) The type XIV family of C‐type lectin‐like domain (CTLD) containing proteins. Curr Drug Targets 13, 409–410. [DOI] [PubMed] [Google Scholar]
- 13.Maruyama I, Salem HH, Ishii H & Majerus PW (1985) Human thrombomodulin is not an efficient inhibitor of the procoagulant activity of thrombin. J Clin Invest 75, 987–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Conway EM (2012) Thrombomodulin and its role in inflammation. Semin Immunopathol 34, 107–125. [DOI] [PubMed] [Google Scholar]
- 15.Healy AM, Rayburn HB, Rosenberg RD & Weiler H (1995) Absence of the blood‐clotting regulator thrombomodulin causes embryonic lethality in mice before development of a functional cardiovascular system. Proc Natl Acad Sci USA 92, 850–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Isermann B, Hendrickson SB, Hutley K, Wing M & Weiler H (2001) Tissue‐restricted expression of thrombomodulin in the placenta rescues thrombomodulin‐deficient mice from early lethality and reveals a secondary developmental block. Development 128, 827–838. [DOI] [PubMed] [Google Scholar]
- 17.Conway EM, Pollefeyt S, Cornelissen J, DeBaere I, Steiner‐Mosonyi M, Weitz JI, Weiler‐Guettler H, Carmeliet P & Collen D (1999) Structure‐function analyses of thrombomodulin by gene‐targeting in mice: the cytoplasmic domain is not required for normal fetal development. Blood 93, 3442–3450. [PubMed] [Google Scholar]
- 18.Conway EM, Van de Wouwer M, Pollefeyt S, Jurk K, Van Aken H, De Vriese A, Weitz JI, Weiler H, Hellings PW, Schaeffer P_et al_ (2002) The lectin‐like domain of thrombomodulin confers protection from neutrophil‐mediated tissue damage by suppressing adhesion molecule expression via nuclear factor kappaB and mitogen‐activated protein kinase pathways. J Exp Med 196, 565–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Esmon NL, Owen WG & Esmon CT (1982) Isolation of a membrane‐bound cofactor for thrombin‐catalyzed activation of protein C. J Biol Chem 257, 859–864. [PubMed] [Google Scholar]
- 20.Fuentes‐Prior P, Iwanaga Y, Huber R, Pagila R, Rumennik G, Seto M, Morser J, Light DR & Bode W (2000) Structural basis for the anticoagulant activity of the thrombin-thrombomodulin complex. Nature 404, 518–525. [DOI] [PubMed] [Google Scholar]
- 21.Esmon CT, Esmon NL & Harris KW (1982) Complex formation between thrombin and thrombomodulin inhibits both thrombin‐catalyzed fibrin formation and factor V activation. J Biol Chem 257, 7944–7947. [PubMed] [Google Scholar]
- 22.Marlar RA, Kleiss AJ & Griffin JH (1982) Mechanism of action of human activated protein C, a thrombin‐dependent anticoagulant enzyme. Blood 59, 1067–1072. [PubMed] [Google Scholar]
- 23.Bajzar L, Morser J & Nesheim M (1996) TAFI, or plasma procarboxypeptidase B, couples the coagulation and fibrinolytic cascades through the thrombin‐thrombomodulin complex. J Biol Chem 271, 16603–16608. [DOI] [PubMed] [Google Scholar]
- 24.Daly C, Qian X, Castanaro C, Pasnikowski E, Jiang X, Thomson BR, Quaggin SE, Papadopoulos N, Wei Y, Rudge JS_et al_ (2018) Angiopoietins bind thrombomodulin and inhibit its function as a thrombin cofactor. Sci Rep 8, 505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Araujo TLS, Venturini G, Moretti AIS, Tanaka LY, Pereira AC & Laurindo FRM (2019) Cell‐surface HSP70 associates with thrombomodulin in endothelial cells. Cell Stress Chaperones 24, 273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anastasiou G, Gialeraki A, Merkouri E, Politou M & Travlou A (2012) Thrombomodulin as a regulator of the anticoagulant pathway: implication in the development of thrombosis. Blood Coagul Fibrinolysis 23, 1–10. [DOI] [PubMed] [Google Scholar]
- 27.Loghmani H & Conway EM (2018) Exploring traditional and nontraditional roles for thrombomodulin. Blood 132, 148–158. [DOI] [PubMed] [Google Scholar]
- 28.Shi C‐S, Shi G‐Y, Chang Y‐S, Han H‐S, Kuo C‐H, Liu C, Huang H‐C, Chang Y‐J, Chen P‐S & Wu H‐L (2005) Evidence of human thrombomodulin domain as a novel angiogenic factor. Circulation 111, 1627–1636. [DOI] [PubMed] [Google Scholar]
- 29.Li J‐Y, Su C‐H, Wu Y‐J, Tien T‐Y, Hsieh C‐L, Chen C‐H, Tseng Y‐M, Shi G‐Y, Wu H‐L, Tsai C‐H_et al_ (2011) Therapeutic angiogenesis of human early endothelial progenitor cells is enhanced by thrombomodulin. Arterioscler Thromb Vasc Biol 31, 2518–2525. [DOI] [PubMed] [Google Scholar]
- 30.Chao T‐H, Tsai W‐C, Chen J‐Y, Liu P‐Y, Chung H‐C, Tseng S‐Y, Kuo C‐H, Shi G‐Y, Wu H‐L & Li Y‐H (2014) Soluble thrombomodulin is a paracrine anti‐apoptotic factor for vascular endothelial protection. Int J Cardiol 172, 340–349. [DOI] [PubMed] [Google Scholar]
- 31.Kuo C‐H, Sung M‐C, Chen P‐K, Chang B‐I, Lee F‐T, Cho C‐F, Hsieh T‐T, Huang Y‐C, Li Y‐H, Shi G‐Y_et al_ (2015) FGFR1 mediates recombinant thrombomodulin domain‐induced angiogenesis. Cardiovasc Res 105, 107–117. [DOI] [PubMed] [Google Scholar]
- 32.Ikezoe T, Yang J, Nishioka C & Yokoyama A (2015) Thrombomodulin alleviates murine GVHD in association with an increase in the proportion of regulatory T cells in the spleen. Bone Marrow Transplant 50, 113–120. [DOI] [PubMed] [Google Scholar]
- 33.Ikezoe T, Yang J, Nishioka C, Honda G, Furihata M & Yokoyama A (2012) Thrombomodulin protects endothelial cells from a calcineurin inhibitor‐induced cytotoxicity by upregulation of extracellular signal‐regulated kinase/myeloid leukemia cell‐1 signaling. Arterioscler Thromb Vasc Biol 32, 2259–2270. [DOI] [PubMed] [Google Scholar]
- 34.Pan B, Wang X, Nishioka C, Honda G, Yokoyama A, Zeng L, Xu K & Ikezoe T (2017) G‐protein coupled receptor 15 mediates angiogenesis and cytoprotective function of thrombomodulin. Sci Rep 7, 692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang X, Pan B, Honda G, Wang X, Hashimoto Y, Ohkawara H, Xu K, Zeng L & Ikezoe T (2018) Cytoprotective and pro‐angiogenic functions of thrombomodulin are preserved in the C loop of the fifth epidermal growth factor‐like domain. Haematologica 103, 1730–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuo C‐H, Chen P‐K, Chang B‐I, Sung M‐C, Shi C‐S, Lee J‐S, Chang C‐F, Shi G‐Y & Wu H‐L (2012) The recombinant lectin‐like domain of thrombomodulin inhibits angiogenesis through interaction with Lewis Y antigen. Blood 119, 1302–1313. [DOI] [PubMed] [Google Scholar]
- 37.Shi C‐S, Shi G‐Y, Hsiao H‐M, Kao Y‐C, Kuo K‐L, Ma C‐Y, Kuo C‐H, Chang B‐I, Chang C‐F, Lin C‐H_et al_ (2008) Lectin‐like domain of thrombomodulin binds to its specific ligand Lewis Y antigen and neutralizes lipopolysaccharide‐induced inflammatory response. Blood 112, 3661–3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsu Y‐Y, Shi G‐Y, Wang K‐C, Ma C‐Y, Cheng T‐L & Wu H‐L (2016) Thrombomodulin promotes focal adhesion kinase activation and contributes to angiogenesis by binding to fibronectin. Oncotarget 7, 68122–68139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cho C‐F, Chen P‐K, Chang P‐C, Wu H‐L & Shi G‐Y (2013) Human plasminogen kringle 1‐5 inhibits angiogenesis and induces thrombomodulin degradation in a protein kinase A‐dependent manner. J Mol Cell Cardiol 63, 79–88. [DOI] [PubMed] [Google Scholar]
- 40.Hanly A & Winter D (2007) The role of thrombomodulin in malignancy. Semin Thromb Hemost 33, 673–679. [DOI] [PubMed] [Google Scholar]
- 41.Iqbal S (2000) Role of thrombomodulin in cancer biology. Breast 9, 264–266. [DOI] [PubMed] [Google Scholar]
- 42.Horowitz NA, Blevins EA, Miller WM, Perry AR, Talmage KE, Mullins ES, Flick MJ, Queiroz KCS, Shi K, Spek CA_et al_ (2011) Thrombomodulin is a determinant of metastasis through a mechanism linked to the thrombin binding domain but not the lectin‐like domain. Blood 118, 2889–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kao Y‐C, Wu L‐W, Shi C‐S, Chu C‐H, Huang C‐W, Kuo C‐P, Sheu H‐M, Shi G‐Y & Wu H‐L (2010) Downregulation of thrombomodulin, a novel target of Snail, induces tumorigenesis through epithelial‐mesenchymal transition. Mol Cell Biol 30, 4767–4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zheng N, Huo Z, Zhang B, Meng M, Cao Z, Wang Z & Zhou Q (2016) Thrombomodulin reduces tumorigenic and metastatic potential of lung cancer cells by up‐regulation of E‐cadherin and down‐regulation of N‐cadherin expression. Biochem Biophys Res Commun 476, 252–259. [DOI] [PubMed] [Google Scholar]
- 45.Wu C‐T, Chang Y‐J, Chen M‐F, Liu J‐J, Wei P‐L, Wang W & Liu H‐H (2014) Thrombomodulin mediates the migratory ability of hormone‐independent prostate cancer cells through the regulation of epithelial‐to‐mesenchymal transition biomarkers. Tumour Biol 35, 6047–6054. [DOI] [PubMed] [Google Scholar]
- 46.Wu C‐T, Chang Y‐H, Lin P‐Y, Chen W‐C & Chen M‐F (2014) Thrombomodulin expression regulates tumorigenesis in bladder cancer. BMC Cancer 14, 375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tai C‐J, Cheng C‐W, Su H‐Y, Chen W‐Y, Wu C‐T, Lin F‐Y, Wang C‐K, Tai C‐J & Wei P‐L (2013) Thrombomodulin mediates the migration of cervical cancer cells through the regulation of epithelial–mesenchymal transition biomarkers. Tumor Biol 35, 47–54. [DOI] [PubMed] [Google Scholar]
- 48.Chang Y‐J, Cheng Y‐W, Lin R‐K, Huang C‐C, Chen WT‐L, Ke T‐W & Wei P‐L (2016) Thrombomodulin influences the survival of patients with non‐metastatic colorectal cancer through epithelial‐to‐mesenchymal transition (EMT). PLoS One 11, e0160550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang Y, Cheng B‐J & Lu S (2017) Thrombomodulin regulates doxorubicin sensitivity through epithelial‐mesenchymal transition in non‐small cell lung cancer. Eur Rev Med Pharmacol Sci 21, 95–101. [PubMed] [Google Scholar]
- 50.Shirai Y, Uwagawa T, Shiba H, Shimada Y, Horiuchi T, Saito N, Furukawa K, Ohashi T & Yanaga K (2017) Recombinant thrombomodulin suppresses tumor growth of pancreatic cancer by blocking thrombin‐induced PAR1 and NF‐κB activation. Surgery 161, 1675–1682. [DOI] [PubMed] [Google Scholar]
- 51.Amada E, Fukuda K, Kumagai K, Suda K, Takeuchi H & Kitagawa Y (2017) Abstract 1681: The antitumor effect of a soluble recombinant human thrombomodulin as growth suppression against gastrointestinal tumor in murine peritonitis model. Can Res 77, 1681. [Google Scholar]
- 52.Kashiwagi S, Asano Y, Takahashi K, Shibutani M, Amano R, Tomita S, Hirakawa K & Ohira M (2019) Clinical outcomes of recombinant human‐soluble thrombomodulin treatment for disseminated intravascular coagulation in solid tumors. Anticancer Res 39, 2259–2264. [DOI] [PubMed] [Google Scholar]
- 53.Yuksel M, Okajima K, Uchiba M, Horiuchi S & Okabe H (2002) Activated protein C inhibits lipopolysaccharide‐induced tumor necrosis factor‐alpha production by inhibiting activation of both nuclear factor‐kappa B and activator protein‐1 in human monocytes. Thromb Haemost 88, 267–273. [PubMed] [Google Scholar]
- 54.Abeyama K, Stern DM, Ito Y, Kawahara K‐I, Yoshimoto Y, Tanaka M, Uchimura T, Ida N, Yamazaki Y, Yamada S_et al_ (2005) The N‐terminal domain of thrombomodulin sequesters high‐mobility group‐B1 protein, a novel antiinflammatory mechanism. J Clin Invest 115, 1267–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hayashi Y, Tsujita R, Tsubota M, Saeki H, Sekiguchi F, Honda G & Kawabata A (2018) Human soluble thrombomodulin‐induced blockade of peripheral HMGB1‐dependent allodynia in mice requires both the lectin‐like and EGF‐like domains. Biochem Biophys Res Commun 495, 634–638. [DOI] [PubMed] [Google Scholar]
- 56.Kono K, Mimura K & Kiessling R (2013) Immunogenic tumor cell death induced by chemoradiotherapy: molecular mechanisms and a clinical translation. Cell Death Dis 4, e688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin W‐L, Chang C‐F, Shi C‐S, Shi G‐Y & Wu H‐L (2013) Recombinant lectin‐like domain of thrombomodulin suppresses vascular inflammation by reducing leukocyte recruitment via interacting with Lewis Y on endothelial cells. Arterioscler Thromb Vasc Biol 33, 2366–2373. [DOI] [PubMed] [Google Scholar]
- 58.Kawamoto E, Okamoto T, Takagi Y, Honda G, Suzuki K, Imai H & Shimaoka M (2016) LFA‐1 and Mac‐1 integrins bind to the serine/threonine‐rich domain of thrombomodulin. Biochem Biophys Res Commun 473, 1005–1012. [DOI] [PubMed] [Google Scholar]
- 59.Ma C‐Y, Chang W‐E, Shi G‐Y, Chang B‐Y, Cheng S‐E, Shih Y‐T & Wu H‐L (2015) Recombinant thrombomodulin inhibits lipopolysaccharide‐induced inflammatory response by blocking the functions of CD14. J Immunol 194, 1905–1915. [DOI] [PubMed] [Google Scholar]
- 60.Pan B, Wang X, Kojima S, Nishioka C, Yokoyama A, Honda G, Xu K & Ikezoe T (2017) The fifth epidermal growth factor‐like region of thrombomodulin alleviates murine graft‐versus‐host disease in a G‐protein coupled receptor 15 dependent manner. Biol Blood Marrow Transplant 23, 746–756. [DOI] [PubMed] [Google Scholar]
- 61.Nomoto H, Takami A, Espinoza JL, Matsuo K, Mizuno S, Onizuka M, Kashiwase K, Morishima Y, Fukuda T, Kodera Y_et al_ (2015) A donor thrombomodulin gene variation predicts graft‐versus‐host disease development and mortality after bone marrow transplantation. Int J Hematol 102, 460–470. [DOI] [PubMed] [Google Scholar]
- 62.Chaudhary B & Elkord E (2016) Regulatory T cells in the tumor microenvironment and cancer progression: role and therapeutic targeting. Vaccines (Basel) 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Noris M & Remuzzi G (2013) Overview of complement activation and regulation. Semin Nephrol 33, 479–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Campbell WD, Lazoura E, Okada N & Okada H (2002) Inactivation of C3a and C5a octapeptides by carboxypeptidase R and carboxypeptidase N. Microbiol Immunol 46, 131–134. [DOI] [PubMed] [Google Scholar]
- 65.Myles T, Nishimura T, Yun TH, Nagashima M, Morser J, Patterson AJ, Pearl RG & Leung LLK (2003) Thrombin activatable fibrinolysis inhibitor, a potential regulator of vascular inflammation. J Biol Chem 278, 51059–51067. [DOI] [PubMed] [Google Scholar]
- 66.Nishimura T, Myles T, Piliponsky AM, Kao PN, Berry GJ & Leung LLK (2007) Thrombin‐activatable procarboxypeptidase B regulates activated complement C5a in vivo. Blood 109, 1992–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Delvaeye M, Noris M, De Vriese A, Esmon CT, Esmon NL, Ferrell G, Del‐Favero J, Plaisance S, Claes B, Lambrechts D_et al_ (2009) Thrombomodulin mutations in atypical hemolytic‐uremic syndrome. N Engl J Med 361, 345–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sahin U, Weskamp G, Kelly K, Zhou H‐M, Higashiyama S, Peschon J, Hartmann D, Saftig P & Blobel CP (2004) Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol 164, 769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cassel D & Glaser L (1982) Proteolytic cleavage of epidermal growth factor receptor. A Ca2+‐dependent, sulfhydryl‐sensitive proteolytic system in A431 cells. J Biol Chem 257, 9845–9848. [PubMed] [Google Scholar]
- 70.Lohi O, Urban S & Freeman M (2004) Diverse substrate recognition mechanisms for rhomboids; thrombomodulin is cleaved by Mammalian rhomboids. Curr Biol 14, 236–241. [DOI] [PubMed] [Google Scholar]
- 71.Cheng T‐L, Wu Y‐T, Lin H‐Y, Hsu F‐C, Liu S‐K, Chang B‐I, Chen W‐S, Lai C‐H, Shi G‐Y & Wu H‐L (2011) Functions of rhomboid family protease RHBDL2 and thrombomodulin in wound healing. J Invest Dermatol 131, 2486–2494. [DOI] [PubMed] [Google Scholar]
- 72.Boehme MWJ, Galle P & Stremmel W (2002) Kinetics of thrombomodulin release and endothelial cell injury by neutrophil‐derived proteases and oxygen radicals. Immunology 107, 340–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Oida K, Takai H, Maeda H, Takahashi S, Tamai T, Nakai T, Miyabo S & Ishii H (1990) Plasma thrombomodulin concentration in diabetes mellitus. Diabetes Res Clin Pract 10, 193–196. [DOI] [PubMed] [Google Scholar]
- 74.Jackson DE, Tetaz TJ, Salem HH & Mitchell CA (1994) Purification and characterization of two forms of soluble thrombomodulin from human urine. Eur J Biochem 221, 1079–1087. [DOI] [PubMed] [Google Scholar]
- 75.Conway EM & Nowakowski B (1993) Biologically active thrombomodulin is synthesized by adherent synovial fluid cells and is elevated in synovial fluid of patients with rheumatoid arthritis. Blood 81, 726–733. [PubMed] [Google Scholar]
- 76.Boehme MW, Raeth U, Galle PR, Stremmel W & Scherbaum WA (2000) Serum thrombomodulin‐a reliable marker of disease activity in systemic lupus erythematosus (SLE): advantage over established serological parameters to indicate disease activity. Clin Exp Immunol 119, 189–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wu H‐L, Lin C‐I, Huang Y‐L, Chen P‐S, Kuo C‐H, Chen M‐S, Wu GC‐C, Shi G‐Y, Yang H‐Y & Lee H (2008) Lysophosphatidic acid stimulates thrombomodulin lectin‐like domain shedding in human endothelial cells. Biochem Biophys Res Commun 367, 162–168. [DOI] [PubMed] [Google Scholar]
- 78.Takano S, Kimura S, Ohdama S & Aoki N (1990) Plasma thrombomodulin in health and diseases. Blood 76, 2024–2029. [PubMed] [Google Scholar]
- 79.Glaser CB, Morser J, Clarke JH, Blasko E, McLean K, Kuhn I, Chang RJ, Lin JH, Vilander L, Andrews WH_et al_ (1992) Oxidation of a specific methionine in thrombomodulin by activated neutrophil products blocks cofactor activity. A potential rapid mechanism for modulation of coagulation. J Clin Invest 90, 2565–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Huang H‐C, Shi G‐Y, Jiang S‐J, Shi C‐S, Wu C‐M, Yang H‐Y & Wu H‐L (2003) Thrombomodulin‐mediated cell adhesion: involvement of its lectin‐like domain. J Biol Chem 278, 46750–46759. [DOI] [PubMed] [Google Scholar]
- 81.Hsu Y‐Y, Shi G‐Y, Kuo C‐H, Liu S‐L, Wu C‐M, Ma C‐Y, Lin F‐Y, Yang H‐Y & Wu H‐L (2012) Thrombomodulin is an ezrin‐interacting protein that controls epithelial morphology and promotes collective cell migration. FASEB J 26, 3440–3452. [DOI] [PubMed] [Google Scholar]
- 82.Clucas J & Valderrama F (2014) ERM proteins in cancer progression. J Cell Sci 127, 267–275. [DOI] [PubMed] [Google Scholar]
- 83.St Croix B, Rago C, Velculescu V, Traverso G, Romans KE, Montgomery E, Lal A, Riggins GJ, Lengauer C, Vogelstein B_et al_ (2000) Genes expressed in human tumor endothelium. Science 289, 1197–1202. [DOI] [PubMed] [Google Scholar]
- 84.MacFadyen JR, Haworth O, Roberston D, Hardie D, Webster M‐T, Morris HR, Panico M, Sutton‐Smith M, Dell A, van der Geer P_et al_ (2005) Endosialin (TEM1, CD248) is a marker of stromal fibroblasts and is not selectively expressed on tumour endothelium. FEBS Lett 579, 2569–2575. [DOI] [PubMed] [Google Scholar]
- 85.Valdez Y, Maia M & Conway EM (2012) CD248: reviewing its role in health and disease. Curr Drug Targets 13, 432–439. [DOI] [PubMed] [Google Scholar]
- 86.Rouleau C, Curiel M, Weber W, Smale R, Kurtzberg L, Mascarello J, Berger C, Wallar G, Bagley R, Honma N_et al_ (2008) Endosialin protein expression and therapeutic target potential in human solid tumors: sarcoma versus carcinoma. Clin Cancer Res 14, 7223–7236. [DOI] [PubMed] [Google Scholar]
- 87.Simonavicius N, Robertson D, Bax DA, Jones C, Huijbers IJ & Isacke CM (2008) Endosialin (CD248) is a marker of tumor‐associated pericytes in high‐grade glioma. Mod Pathol 21, 308–315. [DOI] [PubMed] [Google Scholar]
- 88.Li Q, Yu Y, Bischoff J, Mulliken JB & Olsen BR (2003) Differential expression of CD146 in tissues and endothelial cells derived from infantile haemangioma and normal human skin. J Pathol 201, 296–302. [DOI] [PubMed] [Google Scholar]
- 89.Bagley RG, Rouleau C, St Martin T, Boutin P, Weber W, Ruzek M, Honma N, Nacht M, Shankara S, Kataoka S_et al_ (2008) Human endothelial precursor cells express tumor endothelial marker 1/endosialin/CD248. Mol Cancer Ther 7, 2536–2546. [DOI] [PubMed] [Google Scholar]
- 90.Huang H‐P, Hong C‐L, Kao C‐Y, Lin S‐W, Lin S‐R, Wu H‐L, Shi G‐Y, You L‐R, Wu C‐L & Yu I‐S (2011) Gene targeting and expression analysis of mouse Tem1/endosialin using a lacZ reporter. Gene Expr Patterns 11, 316–326. [DOI] [PubMed] [Google Scholar]
- 91.Nanda A, Karim B, Peng Z, Liu G, Qiu W, Gan C, Vogelstein B, St Croix B, Kinzler KW & Huso DL (2006) Tumor endothelial marker 1 (Tem1) functions in the growth and progression of abdominal tumors. Proc Natl Acad Sci USA 103, 3351–3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Viski C, König C, Kijewska M, Mogler C, Isacke CM & Augustin HG (2016) Endosialin‐expressing pericytes promote metastatic dissemination. Cancer Res 76, 5313–5325. [DOI] [PubMed] [Google Scholar]
- 93.Ohradanova A, Gradin K, Barathova M, Zatovicova M, Holotnakova T, Kopacek J, Parkkila S, Poellinger L, Pastorekova S & Pastorek J (2008) Hypoxia upregulates expression of human endosialin gene via hypoxia‐inducible factor 2. Br J Cancer 99, 1348–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hockel M & Vaupel P (2001) Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst 93, 266–276. [DOI] [PubMed] [Google Scholar]
- 95.Kontsekova S, Polcicova K, Takacova M & Pastorekova S (2016) Endosialin: molecular and functional links to tumor angiogenesis. Neoplasma 63, 183–192. [DOI] [PubMed] [Google Scholar]
- 96.Hardie DL, Baldwin MJ, Naylor A, Haworth OJ, Hou TZ, Lax S, John Curnow S, Willcox N, MacFadyen J, Isacke CM_et al_ (2011) The stromal cell antigen CD248 (endosialin) is expressed on naive CD8 human T cells and regulates proliferation. Immunology 133, 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lax S, Hardie DL, Wilson A, Douglas MR, Anderson G, Huso D, Isacke CM & Buckley CD (2010) The pericyte and stromal cell marker CD248 (endosialin) is required for efficient lymph node expansion. Eur J Immunol 40, 1884–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tomkowicz B, Rybinski K, Foley B, Ebel W, Kline B, Routhier E, Sass P, Nicolaides NC, Grasso L & Zhou Y (2007) Interaction of endosialin/TEM1 with extracellular matrix proteins mediates cell adhesion and migration. Proc Natl Acad Sci USA 104, 17965–17970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Christian S, Winkler R, Helfrich I, Boos AM, Besemfelder E, Schadendorf D & Augustin HG (2008) Endosialin (Tem1) is a marker of tumor‐associated myofibroblasts and tumor vessel‐associated mural cells. Am J Pathol 172, 486–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Simonavicius N, Ashenden M, van Weverwijk A, Lax S, Huso DL, Buckley CD, Huijbers IJ, Yarwood H & Isacke CM (2012) Pericytes promote selective vessel regression to regulate vascular patterning. Blood 120, 1516–1527. [DOI] [PubMed] [Google Scholar]
- 101.Khan KA, Naylor AJ, Khan A, Noy PJ, Mambretti M, Lodhia P, Athwal J, Korzystka A, Buckley CD, Willcox BE_et al_ (2017) Multimerin‐2 is a ligand for group 14 family C‐type lectins CLEC14A, CD93 and CD248 spanning the endothelial pericyte interface. Oncogene 36, 6097–6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Becker R, Lenter MC, Vollkommer T, Boos AM, Pfaff D, Augustin HG & Christian S (2008) Tumor stroma marker endosialin (Tem1) is a binding partner of metastasis‐related protein Mac‐2 BP/90K. FASEB J 22, 3059–3067. [DOI] [PubMed] [Google Scholar]
- 103.Hellstern S, Sasaki T, Fauser C, Lustig A, Timpl R & Engel J (2002) Functional studies on recombinant domains of Mac‐2‐binding protein. J Biol Chem 277, 15690–15696. [DOI] [PubMed] [Google Scholar]
- 104.Marchetti A, Tinari N, Buttitta F, Chella A, Angeletti CA, Sacco R, Mucilli F, Ullrich A & Iacobelli S (2002) Expression of 90K (Mac‐2 BP) correlates with distant metastasis and predicts survival in stage I non‐small cell lung cancer patients. Cancer Res 62, 2535–2539. [PubMed] [Google Scholar]
- 105.Maia M, DeVriese A, Janssens T, Moons M, Lories RJ, Tavernier J & Conway EM (2011) CD248 facilitates tumor growth via its cytoplasmic domain. BMC Cancer 11, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Opavsky R, Haviernik P, Jurkovicova D, Garin MT, Copeland NG, Gilbert DJ, Jenkins NA, Bies J, Garfield S, Pastorekova S_et al_ (2001) Molecular characterization of the mouse Tem1/endosialin gene regulated by cell density in vitro and expressed in normal tissues in vivo. J Biol Chem 276, 38795–38807. [DOI] [PubMed] [Google Scholar]
- 107.Carson‐Walter EB, Winans BN, Whiteman MC, Liu Y, Jarvela S, Haapasalo H, Tyler BM, Huso DL, Johnson MD & Walter KA (2009) Characterization of TEM1/endosialin in human and murine brain tumors. BMC Cancer 9, 417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D & Jain RK (2011) Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev 91, 1071–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Huang Y, Goel S, Duda DG, Fukumura D & Jain RK (2013) Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res 73, 2943–2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tomkowicz B, Rybinski K, Sebeck D, Sass P, Nicolaides NC, Grasso L & Zhou Y (2010) Endosialin/TEM‐1/CD248 regulates pericyte proliferation through PDGF receptor signaling. Cancer Biol Ther 9, 908–915. [DOI] [PubMed] [Google Scholar]
- 111.Naylor AJ, McGettrick HM, Maynard WD, May P, Barone F, Croft AP, Egginton S & Buckley CD (2014) A differential role for CD248 (Endosialin) in PDGF‐mediated skeletal muscle angiogenesis. PLoS One 9, e107146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rmali KA, Puntis MCA & Jiang WG (2005) Prognostic values of tumor endothelial markers in patients with colorectal cancer. World J Gastroenterol 11, 1283–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Huber MA, Kraut N, Schweifer N, Dolznig H, Peter RU, Schubert RD, Scharffetter‐Kochanek K, Pehamberger H & Garin‐Chesa P (2006) Expression of stromal cell markers in distinct compartments of human skin cancers. J Cutan Pathol 33, 145–155. [DOI] [PubMed] [Google Scholar]
- 114.Brady J, Neal J, Sadakar N & Gasque P (2004) Human endosialin (tumor endothelial marker 1) is abundantly expressed in highly malignant and invasive brain tumors. J Neuropathol Exp Neurol 63, 1274–1283. [DOI] [PubMed] [Google Scholar]
- 115.Davies G, Cunnick GH, Mansel RE, Mason MD & Jiang WG (2004) Levels of expression of endothelial markers specific to tumour‐associated endothelial cells and their correlation with prognosis in patients with breast cancer. Clin Exp Metastasis 21, 31–37. [DOI] [PubMed] [Google Scholar]
- 116.O'Shannessy DJ, Somers EB, Chandrasekaran LK, Nicolaides NC, Bordeaux J & Gustavson MD (2014) Influence of tumor microenvironment on prognosis in colorectal cancer: tissue architecture‐dependent signature of endosialin (TEM‐1) and associated proteins. Oncotarget 5, 3983–3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rouleau C, Sancho J, Campos‐Rivera J & Teicher BA (2012) Endosialin expression in side populations in human sarcoma cell lines. Oncol Lett 3, 325–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sun D‐X, Liao G‐J, Liu K‐G & Jian H (2015) Endosialin‐expressing bone sarcoma stem‐like cells are highly tumor‐initiating and invasive. Mol Med Rep 12, 5665–5670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Marty C, Langer‐Machova Z, Sigrist S, Schott H, Schwendener RA & Ballmer‐Hofer K (2006) Isolation and characterization of a scFv antibody specific for tumor endothelial marker 1 (TEM1), a new reagent for targeted tumor therapy. Cancer Lett 235, 298–308. [DOI] [PubMed] [Google Scholar]
- 120.Rouleau C, Gianolio DA, Smale R, Roth SD, Krumbholz R, Harper J, Munroe KJ, Green TL, Horten BC, Schmid SM_et al_ (2015) Anti‐endosialin antibody‐drug conjugate: potential in sarcoma and other malignancies. Mol Cancer Ther 14, 2081–2089. [DOI] [PubMed] [Google Scholar]
- 121.Capone E, Piccolo E, Fichera I, Ciufici P, Barcaroli D, Sala A, De Laurenzi V, Iacobelli V, Iacobelli S & Sala G (2017) Generation of a novel antibody‐drug conjugate targeting endosialin: potent and durable antitumor response in sarcoma. Oncotarget 8, 60368–60377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chacko A‐M, Li C, Nayak M, Mikitsh JL, Hu J, Hou C, Grasso L, Nicolaides NC, Muzykantov VR, Divgi CR_et al_ (2014) Development of 124I immuno‐PET targeting tumor vascular TEM1/endosialin. J Nucl Med 55, 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rybinski K, Imtiyaz HZ, Mittica B, Drozdowski B, Fulmer J, Furuuchi K, Fernando S, Henry M, Chao Q, Kline B_et al_ (2015) Targeting endosialin/CD248 through antibody‐mediated internalization results in impaired pericyte maturation and dysfunctional tumor microvasculature. Oncotarget 6, 25429–25440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Grothey A, Strosberg JR, Renfro LA, Hurwitz HI, Marshall JL, Safran H, Guarino MJ, Kim GP, Hecht JR, Weil SC_et al_ (2018) A randomized, double‐blind, placebo‐controlled phase II study of the efficacy and safety of monotherapy ontuxizumab (MORAb‐004) plus best supportive care in patients with chemorefractory metastatic colorectal cancer. Clin Cancer Res 24, 316–325. [DOI] [PubMed] [Google Scholar]
- 125.D'Angelo SP, Hamid OA, Tarhini A, Schadendorf D, Chmielowski B, Collichio FA, Pavlick AC, Lewis KD, Weil SC, Heyburn J_et al_ (2018) A phase 2 study of ontuxizumab, a monoclonal antibody targeting endosialin, in metastatic melanoma. Invest New Drugs 36, 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jones RL, Chawla SP, Attia S, Schöffski P, Gelderblom H, Chmielowski B, Le Cesne A, Van Tine BA, Trent JC, Patel S_et al_ (2019) A phase 1 and randomized controlled phase 2 trial of the safety and efficacy of the combination of gemcitabine and docetaxel with ontuxizumab (MORAb‐004) in metastatic soft‐tissue sarcomas. Cancer 125, 2445–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Facciponte JG, Ugel S, De Sanctis F, Li C, Wang L, Nair G, Sehgal S, Raj A, Matthaiou E, Coukos G_et al_ (2014) Tumor endothelial marker 1‐specific DNA vaccination targets tumor vasculature. J Clin Invest 124, 1497–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mogler C, König C, Wieland M, Runge A, Besemfelder E, Komljenovic D, Longerich T, Schirmacher P & Augustin HG (2017) Hepatic stellate cells limit hepatocellular carcinoma progression through the orphan receptor endosialin. EMBO Mol Med 9, 741–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lax S, Hou TZ, Jenkinson E, Salmon M, MacFadyen JR, Isacke CM, Anderson G, Cunningham AF & Buckley CD (2007) CD248/endosialin is dynamically expressed on a subset of stromal cells during lymphoid tissue development, splenic remodeling and repair. FEBS Lett 581, 3550–3556. [DOI] [PubMed] [Google Scholar]
- 130.Lax S, Ross EA, White A, Marshall JL, Jenkinson WE, Isacke CM, Huso DL, Cunningham AF, Anderson G & Buckley CD (2012) CD248 expression on mesenchymal stromal cells is required for post‐natal and infection‐dependent thymus remodelling and regeneration. FEBS Open Bio 2, 187–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hasanov Z, Ruckdeschel T, König C, Mogler C, Kapel SS, Korn C, Spegg C, Eichwald V, Wieland M, Appak S_et al_ (2017) Endosialin promotes atherosclerosis through phenotypic remodeling of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 37, 495–505. [DOI] [PubMed] [Google Scholar]
- 132.Maia M, de Vriese A, Janssens T, Moons M, van Landuyt K, Tavernier J, Lories RJ & Conway EM (2010) CD248 and its cytoplasmic domain: a therapeutic target for arthritis. Arthritis Rheum 62, 3595–3606. [DOI] [PubMed] [Google Scholar]
- 133.Croft AP, Naylor AJ, Marshall JL, Hardie DL, Zimmermann B, Turner J, Desanti G, Adams H, Yemm AI, Müller‐Ladner U_et al_ (2016) Rheumatoid synovial fibroblasts differentiate into distinct subsets in the presence of cytokines and cartilage. Arthritis Res Ther 18, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Naylor AJ, Azzam E, Smith S, Croft A, Poyser C, Duffield JS, Huso DL, Gay S, Ospelt C, Cooper MS_et al_ (2012) The mesenchymal stem cell marker CD248 (endosialin) is a negative regulator of bone formation in mice. Arthritis Rheum 64, 3334–3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Smith SW, Eardley KS, Croft AP, Nwosu J, Howie AJ, Cockwell P, Isacke CM, Buckley CD & Savage COS (2011) CD248+ stromal cells are associated with progressive chronic kidney disease. Kidney Int 80, 199–207. [DOI] [PubMed] [Google Scholar]
- 136.Smith SW, Croft AP, Morris HL, Naylor AJ, Huso DL, Isacke CM, Savage COS & Buckley CD (2015) Genetic deletion of the stromal cell marker CD248 (endosialin) protects against the development of renal fibrosis. Nephron 131, 265–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chang‐Panesso M & Humphreys BD (2015) CD248/endosialin: a novel pericyte target in renal fibrosis. Nephron 131, 262–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wilhelm A, Aldridge V, Haldar D, Naylor AJ, Weston CJ, Hedegaard D, Garg A, Fear J, Reynolds GM, Croft AP_et al_ (2016) CD248/endosialin critically regulates hepatic stellate cell proliferation during chronic liver injury via a PDGF‐regulated mechanism. Gut 65, 1175–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bartis D, Crowley LE, D'Souza VK, Borthwick L, Fisher AJ, Croft AP, Pongrácz JE, Thompson R, Langman G, Buckley CD_et al_ (2016) Role of CD248 as a potential severity marker in idiopathic pulmonary fibrosis. BMC Pulm Med 16, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Di Benedetto P, Liakouli V, Ruscitti P, Berardicurti O, Carubbi F, Panzera N, Di Bartolomeo S, Guggino G, Ciccia F, Triolo G_et al_ (2018) Blocking CD248 molecules in perivascular stromal cells of patients with systemic sclerosis strongly inhibits their differentiation toward myofibroblasts and proliferation: a new potential target for antifibrotic therapy. Arthritis Res Ther 20, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kuk C, Kulasingam V, Gunawardana CG, Smith CR, Batruch I & Diamandis EP (2009) Mining the ovarian cancer ascites proteome for potential ovarian cancer biomarkers. Mol Cell Proteomics 8, 661–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kosanam H, Makawita S, Judd B, Newman A & Diamandis EP (2011) Mining the malignant ascites proteome for pancreatic cancer biomarkers. Proteomics 11, 4551–4558. [DOI] [PubMed] [Google Scholar]
- 143.O'Shannessy DJ, Smith MF, Somers EB, Jackson SM, Albone E, Tomkowicz B, Cheng X, Park Y, Fernando D, Milinichik A_et al_ (2016) Novel antibody probes for the characterization of endosialin/TEM‐1. Oncotarget 7, 69420–69435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Nepomuceno RR, Henschen‐Edman AH, Burgess WH & Tenner AJ (1997) cDNA cloning and primary structure analysis of C1qR(P), the human C1q/MBL/SPA receptor that mediates enhanced phagocytosis in vitro. Immunity 6, 119–129. [DOI] [PubMed] [Google Scholar]
- 145.Steinberger P, Szekeres A, Wille S, Stöckl J, Selenko N, Prager E, Staffler G, Madic O, Stockinger H & Knapp W (2002) Identification of human CD93 as the phagocytic C1q receptor (C1qRp) by expression cloning. J Leukoc Biol 71, 133–140. [PubMed] [Google Scholar]
- 146.McGreal EP, Ikewaki N, Akatsu H, Morgan BP & Gasque P (2002) Human C1qRp is identical with CD93 and the mNI‐11 antigen but does not bind C1q. J Immunol 168, 5222–5232. [DOI] [PubMed] [Google Scholar]
- 147.Nepomuceno RR & Tenner AJ (1998) C1qRP, the C1q receptor that enhances phagocytosis, is detected specifically in human cells of myeloid lineage, endothelial cells, and platelets. J Immunol 160, 1929–1935. [PubMed] [Google Scholar]
- 148.Liu C, Cui Z, Wang S & Zhang D (2014) CD93 and GIPC expression and localization during central nervous system inflammation. Neural Regen Res 9, 1995–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Løvik G, Larsen Sand K, Iversen JG & Rolstad B (2001) C1qRp elicits a Ca++ response in rat NK cells but does not influence NK‐mediated cytotoxicity. Scand J Immunol 53, 410–415. [DOI] [PubMed] [Google Scholar]
- 150.Danet GH, Luongo JL, Butler G, Lu MM, Tenner AJ, Simon MC & Bonnet DA (2002) C1qRp defines a new human stem cell population with hematopoietic and hepatic potential. Proc Natl Acad Sci USA 99, 10441–10445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ikewaki N, Yamao H, Kulski JK & Inoko H (2010) Flow cytometric identification of CD93 expression on naive T lymphocytes (CD4(+)CD45RA (+) cells) in human neonatal umbilical cord blood. J Clin Immunol 30, 723–733. [DOI] [PubMed] [Google Scholar]
- 152.Chevrier S, Genton C, Kallies A, Karnowski A, Otten LA, Malissen B, Malissen M, Botto M, Corcoran LM, Nutt SL_et al_ (2009) CD93 is required for maintenance of antibody secretion and persistence of plasma cells in the bone marrow niche. Proc Natl Acad Sci USA 106, 3895–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Olsen RS, Lindh M, Vorkapic E, Andersson RE, Zar N, Löfgren S, Dimberg J, Matussek A & Wågsäter D (2015) CD93 gene polymorphism is associated with disseminated colorectal cancer. Int J Colorectal Dis 30, 883–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Masiero M, Simões FC, Han HD, Snell C, Peterkin T, Bridges E, Mangala LS, Wu SY‐Y, Pradeep S, Li D_et al_ (2013) A core human primary tumor angiogenesis signature identifies the endothelial orphan receptor ELTD1 as a key regulator of angiogenesis. Cancer Cell 24, 229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Dieterich LC, Mellberg S, Langenkamp E, Zhang L, Zieba A, Salomäki H, Teichert M, Huang H, Edqvist P‐H, Kraus T_et al_ (2012) Transcriptional profiling of human glioblastoma vessels indicates a key role of VEGF‐A and TGFβ2 in vascular abnormalization. J Pathol 228, 378–390. [DOI] [PubMed] [Google Scholar]
- 156.Langenkamp E, Zhang L, Lugano R, Huang H, Elhassan TEA, Georganaki M, Bazzar W, Lööf J, Trendelenburg G, Essand M_et al_ (2015) Elevated expression of the C‐type lectin CD93 in the glioblastoma vasculature regulates cytoskeletal rearrangements that enhance vessel function and reduce host survival. Cancer Res 75, 4504–4516. [DOI] [PubMed] [Google Scholar]
- 157.Bao L, Tang M, Zhang Q, You B, Shan Y, Shi S, Li L, Hu S & You Y (2016) Elevated expression of CD93 promotes angiogenesis and tumor growth in nasopharyngeal carcinoma. Biochem Biophys Res Commun 476, 467–474. [DOI] [PubMed] [Google Scholar]
- 158.Tosi GM, Caldi E, Parolini B, Toti P, Neri G, Nardi F, Traversi C, Cevenini G, Marigliani D, Nuti E_et al_ (2017) CD93 as a potential target in neovascular age‐related macular degeneration. J Cell Physiol 232, 1767–1773. [DOI] [PubMed] [Google Scholar]
- 159.Nativel B, Figuester A, Andries J, Planesse C, Couprie J, Gasque P, Viranaicken W & Iwema T (2016) Soluble expression of disulfide‐bonded C‐type lectin like domain of human CD93 in the cytoplasm of Escherichia coli. J Immunol Methods 439, 67–73. [DOI] [PubMed] [Google Scholar]
- 160.Petrenko O, Beavis A, Klaine M, Kittappa R, Godin I & Lemischka IR (1999) The molecular characterization of the fetal stem cell marker AA4. Immunity 10, 691–700. [DOI] [PubMed] [Google Scholar]
- 161.Norsworthy PJ, Fossati‐Jimack L, Cortes‐Hernandez J, Taylor PR, Bygrave AE, Thompson RD, Nourshargh S, Walport MJ & Botto M (2004) Murine CD93 (C1qRp) contributes to the removal of apoptotic cells in vivo but is not required for C1q‐mediated enhancement of phagocytosis. J Immunol 172, 3406–3414. [DOI] [PubMed] [Google Scholar]
- 162.Orlandini M, Galvagni F, Bardelli M, Rocchigiani M, Lentucci C, Anselmi F, Zippo A, Bini L & Oliviero S (2014) The characterization of a novel monoclonal antibody against CD93 unveils a new antiangiogenic target. Oncotarget 5, 2750–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Bais C, Singh M, Kaminker J & Brauer M (2011) Biological markers for monitoring patient response to vegf antagonists. World Patent.
- 164.Ayers M, Fargnoli J, Lewin A, Wu Q & Platero JS (2007) Discovery and validation of biomarkers that respond to treatment with brivanib alaninate, a small‐molecule VEGFR‐2/FGFR‐1 antagonist. Cancer Res 67, 6899–6906. [DOI] [PubMed] [Google Scholar]
- 165.Galvagni F, Nardi F, Maida M, Bernardini G, Vannuccini S, Petraglia F, Santucci A & Orlandini M (2016) CD93 and dystroglycan cooperation in human endothelial cell adhesion and migration adhesion and migration. Oncotarget 7, 10090–10103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Galvagni F, Nardi F, Spiga O, Trezza A, Tarticchio G, Pellicani R, Andreuzzi E, Caldi E, Toti P, Tosi GM_et al_ (2017) Dissecting the CD93‐multimerin 2 interaction involved in cell adhesion and migration of the activated endothelium. Matrix Biol 64, 112–127. [DOI] [PubMed] [Google Scholar]
- 167.Lugano R, Vemuri K, Yu D, Bergqvist M, Smits A, Essand M, Johansson S, Dejana E & Dimberg A (2018) CD93 promotes β1 integrin activation and fibronectin fibrillogenesis during tumor angiogenesis. J Clin Invest 128, 3280–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Jeon J‐W, Jung J‐G, Shin E‐C, Choi HI, Kim HY, Cho M‐L, Kim S‐W, Jang Y‐S, Sohn M‐H, Moon J‐H_et al_ (2010) Soluble CD93 induces differentiation of monocytes and enhances TLR responses. J Immunol 185, 4921–4927. [DOI] [PubMed] [Google Scholar]
- 169.Zhang M, Bohlson SS, Dy M & Tenner AJ (2005) Modulated interaction of the ERM protein, moesin, with CD93. Immunology 115, 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Greenlee‐Wacker MC, Briseño C, Galvan M, Moriel G, Velázquez P & Bohlson SS (2011) Membrane‐associated CD93 regulates leukocyte migration and C1q‐hemolytic activity during murine peritonitis. J Immunol 187, 3353–3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Bohlson SS, Zhang M, Ortiz CE & Tenner AJ (2005) CD93 interacts with the PDZ domain‐containing adaptor protein GIPC: implications in the modulation of phagocytosis. J Leukoc Biol 77, 80–89. [DOI] [PubMed] [Google Scholar]
- 172.Paye JMD, Phng L‐K, Lanahan AA, Gerhard H & Simons M (2009) Synectin‐dependent regulation of arterial maturation. Dev Dyn 238, 604–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hu J & Hubbard SR (2005) Structural characterization of a novel Cbl phosphotyrosine recognition motif in the APS family of adapter proteins. J Biol Chem 280, 18943–18949. [DOI] [PubMed] [Google Scholar]
- 174.Harhausen D, Prinz V, Ziegler G, Gertz K, Endres M, Lehrach H, Gasque P, Botto M, Stahel PF, Dirnagl U_et al_ (2010) CD93/AA4.1: a novel regulator of inflammation in murine focal cerebral ischemia. J Immunol 184, 6407–6417. [DOI] [PubMed] [Google Scholar]
- 175.Griffiths MR, Botto M, Morgan BP, Neal JW & Gasque P (2018) CD93 regulates central nervous system inflammation in two mouse models of autoimmune encephalomyelitis. Immunology 155, 346–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Tunica DG, Yin X, Sidibe A, Stegemann C, Nissum M, Zeng L, Brunet M & Mayr M (2009) Proteomic analysis of the secretome of human umbilical vein endothelial cells using a combination of free‐flow electrophoresis and nanoflow LC‐MS/MS. Proteomics 9, 4991–4996. [DOI] [PubMed] [Google Scholar]
- 177.Zanivan S, Maione F, Hein MY, Hernández‐Fernaud JR, Ostasiewicz P, Giraudo E & Mann M (2013) SILAC‐based proteomics of human primary endothelial cell morphogenesis unveils tumor angiogenic markers. Mol Cell Proteomics 12, 3599–3611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Mälarstig A, Silveira A, Wågsäter D, Öhrvik J, Bäcklund A, Samnegård A, Khademi M, Hellenius M‐L, Leander K, Olsson T_et al_ (2011) Plasma CD93 concentration is a potential novel biomarker for coronary artery disease. J Intern Med 270, 229–236. [DOI] [PubMed] [Google Scholar]
- 179.Bohlson SS, Silva R, Fonseca MI & Tenner AJ (2005) CD93 is rapidly shed from the surface of human myeloid cells and the soluble form is detected in human plasma. J Immunol 175, 1239–1247. [DOI] [PubMed] [Google Scholar]
- 180.Greenlee MC, Sullivan SA & Bohlson SS (2009) Detection and characterization of soluble CD93 released during inflammation. Inflamm Res 58, 909–919. [DOI] [PubMed] [Google Scholar]
- 181.Blackburn JWD, Lau DHC, Liu EY, Ellins J, Vrieze AM, Pawlak EN, Dikeakos JD & Heit B (2019) Soluble CD93 is an apoptotic cell opsonin recognized by αxβ2. Eur J Immunol 49, 600–610. [DOI] [PubMed] [Google Scholar]
- 182.Kao Y‐C, Jiang S‐J, Pan W‐A, Wang K‐C, Chen P‐K, Wei H‐J, Chen W‐S, Chang B‐I, Shi G‐Y & Wu H‐L (2012) The epidermal growth factor‐like domain of CD93 is a potent angiogenic factor. PLoS One 7, e51647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Park M & Tenner AJ (2003) Cell surface expression of C1qRP/CD93 is stabilized by O‐glycosylation. J Cell Physiol 196, 512–522. [DOI] [PubMed] [Google Scholar]
- 184.Ikewaki N, Kulski JK & Inoko H (2006) Regulation of CD93 cell surface expression by protein kinase C isoenzymes. Microbiol Immunol 50, 93–103. [DOI] [PubMed] [Google Scholar]
- 185.Ikewaki N, Sonoda T & Inoko H (2013) Unique properties of cluster of differentiation 93 in the umbilical cord blood of neonates. Microbiol Immunol 57, 822–832. [DOI] [PubMed] [Google Scholar]
- 186.Andersson ER, Sandberg R & Lendahl U (2011) Notch signaling: simplicity in design, versatility in function. Development 138, 3593–3612. [DOI] [PubMed] [Google Scholar]
- 187.Zöller M (2011) CD44: can a cancer‐initiating cell profit from an abundantly expressed molecule? Nat Rev Cancer 11, 254–267. [DOI] [PubMed] [Google Scholar]
- 188.Ho M, Yang E, Matcuk G, Deng D, Sampas N, Tsalenko A, Tabibiazar R, Zhang Y, Chen M, Talbi S_et al_ (2003) Identification of endothelial cell genes by combined database mining and microarray analysis. Physiol Genomics 13, 249–262. [DOI] [PubMed] [Google Scholar]
- 189.Mura M, Swain RK, Zhuang X, Vorschmitt H, Reynolds G, Durant S, Beesley JFJ, Herbert JMJ, Sheldon H, Andre M_et al_ (2012) Identification and angiogenic role of the novel tumor endothelial marker CLEC14A. Oncogene 31, 293–305. [DOI] [PubMed] [Google Scholar]
- 190.Pircher A, Fiegl M, Untergasser G, Heidegger I, Medinger M, Kern J & Hilbe W (2013) Favorable prognosis of operable non‐small cell lung cancer (NSCLC) patients harboring an increased expression of tumor endothelial markers (TEMs). Lung Cancer 81, 252–258. [DOI] [PubMed] [Google Scholar]
- 191.Su C, Shi K, Cheng X, Han Y, Li Y, Yu D & Liu Z (2018) Methylation of CLEC14A is associated with its expression and lung adenocarcinoma progression. J Cell Physiol 234, 2954–2962. [DOI] [PubMed] [Google Scholar]
- 192.Krishna Priya S, Kumar K, Hiran KR, Bindhu MR, Nagare RP, Vijaykumar DK & Ganesan TS (2017) Expression of a novel endothelial marker, C‐type lectin 14A, in epithelial ovarian cancer and its prognostic significance. Int J Clin Oncol 22, 107–117. [DOI] [PubMed] [Google Scholar]
- 193.Wong KS, Proulx K, Rost MS & Sumanas S (2009) Identification of vasculature‐specific genes by microarray analysis of Etsrp/Etv2 overexpressing zebrafish embryos. Dev Dyn 238, 1836–1850. [DOI] [PubMed] [Google Scholar]
- 194.Baltrunaite K, Craig MP, Palencia Desai S, Chaturvedi P, Pandey RN, Hegde RS & Sumanas S (2017) ETS transcription factors Etv2 and Fli1b are required for tumor angiogenesis. Angiogenesis 20, 307–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Pociute K, Schumacher JA & Sumanas S (2019) Clec14a genetically interacts with Etv2 and Vegf signaling during vasculogenesis and angiogenesis in zebrafish. BMC Dev Biol 19, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Rho S‐S, Choi H‐J, Min J‐K, Lee H‐W, Park H, Park H, Kim Y‐M & Kwon Y‐G (2011) Clec14a is specifically expressed in endothelial cells and mediates cell to cell adhesion. Biochem Biophys Res Commun 404, 103–108. [DOI] [PubMed] [Google Scholar]
- 197.Maeng Y‐S, Choi H‐J, Kwon J‐Y, Park Y‐W, Choi K‐S, Min J‐K, Kim Y‐H, Suh P‐G, Kang K‐S, Won M‐H_et al_ (2009) Endothelial progenitor cell homing: prominent role of the IGF2‐IGF2R‐PLCbeta2 axis. Blood 113, 233–243. [DOI] [PubMed] [Google Scholar]
- 198.Wragg JW, Durant S, McGettrick HM, Sample KM, Egginton S & Bicknell R (2014) Shear stress regulated gene expression and angiogenesis in vascular endothelium. Microcirculation 21, 290–300. [DOI] [PubMed] [Google Scholar]
- 199.Yun S, Dardik A, Haga M, Yamashita A, Yamaguchi S, Koh Y, Madri JA & Sumpio BE (2002) Transcription factor Sp1 phosphorylation induced by shear stress inhibits membrane type 1‐matrix metalloproteinase expression in endothelium. J Biol Chem 277, 34808–34814. [DOI] [PubMed] [Google Scholar]
- 200.Hägg S, Skogsberg J, Lundström J, Noori P, Nilsson R, Zhong H, Maleki S, Shang M‐M, Brinne B, Bradshaw M_et al_ (2009) Multi‐organ expression profiling uncovers a gene module in coronary artery disease involving transendothelial migration of leukocytes and LIM domain binding 2: the Stockholm Atherosclerosis Gene Expression (STAGE) study. PLoS Genet 5, e1000754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Gnasso A, Irace C, Carallo C, De Franceschi MS, Motti C, Mattioli PL & Pujia A (1997) In vivo association between low wall shear stress and plaque in subjects with asymmetrical carotid atherosclerosis. Stroke 28, 993–998. [DOI] [PubMed] [Google Scholar]
- 202.Delcourt N, Quevedo C, Nonne C, Fons P, O'Brien D, Loyaux D, Diez M, Autelitano F, Guillemot J‐C, Ferrara P_et al_ (2015) Targeted identification of sialoglycoproteins in hypoxic endothelial cells and validation in zebrafish reveal roles for proteins in angiogenesis. J Biol Chem 290, 3405–3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Noy PJ, Lodhia P, Khan K, Zhuang X, Ward DG, Verissimo AR, Bacon A & Bicknell R (2015) Blocking CLEC14A‐MMRN2 binding inhibits sprouting angiogenesis and tumour growth. Oncogene 34, 5821–5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Lee S, Rho S‐S, Park H, Park JA, Kim J, Lee I‐K, Koh GY, Mochizuki N, Kim Y‐M & Kwon Y‐G (2017) Carbohydrate‐binding protein CLEC14A regulates VEGFR‐2‐ and VEGFR‐3‐dependent signals during angiogenesis and lymphangiogenesis. J Clin Invest 127, 457–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Ki MK, Jeoung MH, Choi JR, Rho S‐S, Kwon Y‐G, Shim H, Chung J, Hong HJ, Song BD & Lee S (2013) Human antibodies targeting the C‐type lectin‐like domain of the tumor endothelial cell marker clec14a regulate angiogenic properties in vitro. Oncogene 32, 5449–5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kim T‐K, Park CS, Jang J, Kim MR, Na H‐J, Lee K, Kim HJ, Heo K, Yoo BC, Kim Y‐M_et al_ (2018) Inhibition of VEGF‐dependent angiogenesis and tumor angiogenesis by an optimized antibody targeting CLEC14a. Mol Oncol 12, 356–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Jang J, Kim MR, Kim T‐K, Lee WR, Kim JH, Heo K & Lee S (2017) CLEC14a‐HSP70‐1A interaction regulates HSP70‐1A‐induced angiogenesis. Sci Rep 7, 10666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Kim T‐K, Na HJ, Lee WR, Jeoung MH & Lee S (2016) Heat shock protein 70‐1A is a novel angiogenic regulator. Biochem Biophys Res Commun 469, 222–228. [DOI] [PubMed] [Google Scholar]
- 209.Meijer LAT, Zhou H, Chan OYA, Altelaar AFM, Hennrich ML, Mohammed S, Bos JL & Heck AJR (2013) Quantitative global phosphoproteomics of human umbilical vein endothelial cells after activation of the Rap signaling pathway. Mol BioSyst 9, 732–749. [DOI] [PubMed] [Google Scholar]
- 210.van den Biggelaar M, Hernández‐Fernaud JR, van den Eshof BL, Neilson LJ, Meijer AB, Mertens K & Zanivan S (2014) Quantitative phosphoproteomics unveils temporal dynamics of thrombin signaling in human endothelial cells. Blood 123, e22–e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Noy PJ, Swain RK, Khan K, Lodhia P & Bicknell R (2016) Sprouting angiogenesis is regulated by shedding of the C‐type lectin family 14, member A (CLEC14A) ectodomain, catalyzed by rhomboid‐like 2 protein (RHBDL2). FASEB J 30, 2311–2323. [DOI] [PubMed] [Google Scholar]
- 212.Tabula Muris Consortium , Overall Coordination , Logistical Coordination , Organ Collection and Processing , Library Preparation and Sequencing , Computational Data Analysis , Cell Type Annotation , Writing Group , Supplemental Text Writing Group & Principal Investigators (2018) Single‐cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Amieva MR & Furthmayr H (1995) Subcellular localization of moesin in dynamic filopodia, retraction fibers, and other structures involved in substrate exploration, attachment, and cell‐cell contacts. Exp Cell Res 219, 180–196. [DOI] [PubMed] [Google Scholar]
- 214.Pearson MA, Reczek D, Bretscher A & Karplus PA (2000) Structure of the ERM protein moesin reveals the FERM domain fold masked by an extended actin binding tail domain. Cell 101, 259–270. [DOI] [PubMed] [Google Scholar]
- 215.Tsukita S, Oishi K, Sato N, Sagara J, Kawai A & Tsukita S (1994) ERM family members as molecular linkers between the cell surface glycoprotein CD44 and actin‐based cytoskeletons. J Cell Biol 126, 391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Andreuzzi E, Colladel R, Pellicani R, Tarticchio G, Cannizzaro R, Spessotto P, Bussolati B, Brossa A, De Paoli P, Canzonieri V_et al_ (2017) The angiostatic molecule multimerin 2 is processed by MMP‐9 to allow sprouting angiogenesis. Matrix Biol 64, 40–53. [DOI] [PubMed] [Google Scholar]
- 217.Motz GT & Coukos G (2011) The parallel lives of angiogenesis and immunosuppression: cancer and other tales. Nat Rev Immunol 11, 702–711. [DOI] [PubMed] [Google Scholar]
- 218.Khan KA & Kerbel RS (2018) Improving immunotherapy outcomes with anti‐angiogenic treatments and vice versa. Nat Rev Clin Oncol 15, 310–324. [DOI] [PubMed] [Google Scholar]
- 219.Knoblich K, Cruz Migoni S, Siew SM, Jinks E, Kaul B, Jeffery HC, Baker AT, Suliman M, Vrzalikova K, Mehenna H_et al_ (2018) The human lymph node microenvironment unilaterally regulates T‐cell activation and differentiation. PLoS Biol 16, e2005046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Zhuang X, Kaul B, Bentley M, Nagy Z, Giraudo E, Bendle G, Gilham D, Bicknell R & Lee SP (2014) Abstract LB‐256: Immunotherapy using genetically modified T lymphocytes to target CLEC14A on the tumor vasculature. Cancer Res 74, LB–256. [Google Scholar]
- 221.Meng M‐B, Zaorsky NG, Deng L, Wang H‐H, Chao J, Zhao L‐J, Yuan Z‐Y & Ping W (2015) Pericytes: a double‐edged sword in cancer therapy. Future Oncol 11, 169–179. [DOI] [PubMed] [Google Scholar]
- 222.Seaman S, Zhu Z, Saha S, Zhang XM, Yang MY, Hilton MB, Morris K, Szot C, Morris H, Swing DA_et al_ (2017) Eradication of tumors through simultaneous ablation of CD276/B7‐H3‐positive tumor cells and tumor vasculature. Cancer Cell 31, 501–515.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Khan KA & Kerbel RS (2017) A CD276 antibody guided missile with one warhead and two targets: the tumor and its vasculature. Cancer Cell 31, 469–471. [DOI] [PubMed] [Google Scholar]
- 224.Slaga D, Ellerman D, Lombana TN, Vij R, Li J, Hristopoulos M, Clark R, Johnston J, Shelton A, Mai E_et al_ (2018) Avidity‐based binding to HER2 results in selective killing of HER2‐overexpressing cells by anti‐HER2/CD3. Sci Transl Med 10. [DOI] [PubMed] [Google Scholar]
- 225.Du J, Yang Q, Luo L & Yang D (2017) C1qr and C1qrl redundantly regulate angiogenesis in zebrafish through controlling endothelial Cdh5. Biochem Biophys Res Commun 483, 482–487. [DOI] [PubMed] [Google Scholar]
- 226.Kuczynski EA, Vermeulen PB, Pezzella F, Kerbel RS & Reynolds AR (2019) Vessel co‐option in cancer. Nat Rev Clin Oncol. [DOI] [PubMed] [Google Scholar]
- 227.Angara K, Borin TF & Arbab AS (2017) Vascular mimicry: a novel neovascularization mechanism driving anti‐angiogenic therapy (AAT) resistance in glioblastoma. Transl Oncol 10, 650–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Ambrose S, Gordon N, Goldsmith J, Wei W, Zeegers M, James N, Knowles M, Bryan R & Ward D (2015) Use of Aleuria alantia lectin affinity chromatography to enrich candidate biomarkers from the urine of patients with bladder cancer. Proteomes 3, 266–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Bawono P & Heringa J. (2014) PRALINE: a versatile multiple sequence alignment toolkit. Methods Mol. Biol 1079, 245–262. [DOI] [PubMed] [Google Scholar]