Maturation, Activation, and Protection of Dendritic Cells Induced by Double-stranded RNA (original) (raw)
Abstract
The initiation of an immune response is critically dependent on the activation of dendritic cells (DCs). This process is triggered by surface receptors specific for inflammatory cytokines or for conserved patterns characteristic of infectious agents. Here we show that human DCs are activated by influenza virus infection and by double-stranded (ds)RNA. This activation results not only in increased antigen presentation and T cell stimulatory capacity, but also in resistance to the cytopathic effect of the virus, mediated by the production of type I interferon, and upregulation of MxA. Because dsRNA stimulates both maturation and resistance, DCs can serve as altruistic antigen-presenting cells capable of sustaining viral antigen production while acquiring the capacity to trigger naive T cells and drive polarized T helper cell type 1 responses.
Keywords: dendritic cell maturation and activation, influenza virus, double-stranded RNA, type I interferon, MxA
Dendritic cells (DCs)1 serve as the sentinels of the immune system (1, 2). In their so-called immature state, DCs reside in peripheral tissues, where they survey for incoming pathogens. Encounter with pathogens leads to DC activation and migration to secondary lymphoid organs where they trigger a specific T cell response. The capacity of DCs to recognize invading microbes and become activated therefore represents the first critical event in the initiation of the immune response. Because of the wide variety of pathogens, DCs should be equipped with receptors capable of recognizing all possible offending agents and should be able to react in such a way that they maximize the efficiency of antigen presentation.
Immature DCs, similar to those found in peripheral tissues, can be generated by culturing human monocytes with GM-CSF and IL-4 and have been used to identify the activation signals that induce DC maturation (3, 4). These cells, which have a high level of endocytic activity but low T cell stimulatory capacity, are activated to mature into immunostimulatory DCs by the inflammatory cytokines TNF-α and IL-1, by LPS and by CD40 ligand (CD40L). The maturation process results in upregulation of adhesion and costimulatory molecules and downregulation of the endocytic activity and provides an optimal window for loading exogenous antigens on MHC class II molecules (5, 6).
Influenza virus infection has been extensively used as a model to study MHC class I–restricted antigen presentation (7). Although the mechanisms and rules that direct loading of viral peptides on MHC class I molecules have been extensively clarified (8–10), it is less well understood how APCs can handle a cytopathic virus. DCs have been shown to be much more efficient than macrophages in generating CTL responses after infection with influenza virus (11–13) and to be able to present on class I molecules viral antigens taken up from infected apoptotic cells (14). In addition, it has been suggested that infection of DCs with influenza virus can overcome the requirements for T cell help in the stimulation of a CTL response (15), although the mechanism has not been clarified. A relevant question to ask is how immature DCs in peripheral sites can deal with infection by a cytopathic virus. If DCs have to sustain viral antigen presentation, they must be able to promptly recognize viral infection and compromise between production of viral antigens and resistance to the cytopathic effect of the virus.
We report here that human immature DCs are activated by influenza virus and by double-stranded (ds)RNA. In DCs, in contrast to other cells, this stimulus leads to increased protein synthesis and upregulation of MHC, adhesion, and costimulatory molecules. It also results in a rapid production of type I IFN leading to expression of MxA, a protein that protects the DCs from the lethal effect of the virus. By linking viral recognition to activation and resistance, DCs serve as altruistic APCs capable of sustaining production of viral antigens while acquiring the capacity to trigger a polarized Th1 response.
Materials and Methods
Generation and Stimulation of DCs.
To generate immature DCs, peripheral blood monocytes purified by centrifugal elutriation were cultured in RPMI-1640 supplemented with 10% FCS (Hyclone), 50 ng/ml GM-CSF, and 1,000 U/ml IL-4 for 6–7 d, as previously reported (3, 4). Immature DCs were challenged with LPS (1 μg/ml, from Salmonella abortus equi; Sigma Chemical Co.), recombinant TNF-α (50 ng/ml; R&D Systems), poly I:C (20 μg/ ml, Sigma Chemical Co.), and IFN-α (50–500 U/ml; Roferon-A, Roche), or infected with influenza virus strain PR8 (allantoic fluid containing 750 HAU/ml and 4 × 108 PFU/ml). Two neutralizing sheep antisera to human type I IFN were used: Iivari (450,000 neutralizing U/ml anti–IFN-α + 3,000 U/ml anti– IFN-β) and Kaaleppi (30,000 U/ml anti–IFN-α + 30,000 U/ml anti–IFN-β) (16).
Surface and Intracellular Staining.
DC maturation was evaluated by staining the cells with antibodies to HLA class I (W6/32; American Type Culture Collection [ATCC]), DR (L243; ATCC), CD86 (2331; PharMingen), CD83 (HB15a; Immunotech), CD115 (3-4A4; Santa Cruz Biotechnology), and CD38 (OKT10; ATCC), followed by FITC-labeled secondary antibodies. For intracellular staining, the cells were fixed for 30 min with 2% paraformaldehyde, and permeabilized for 30 min with PBS containing 0.5% saponin, 5% FCS, and 10 mM Hepes. The cells were stained with rabbit anti-MxA antibody (17) and biotinylated rat anti-PR8 HA mAb 1-10 (18), followed by appropriate secondary reagents (Southern Biotechnology Associates, Inc.). Intracellular staining for cytokine production was performed after stimulation of T cells for 6 h with 10−7 M PMA and 0.5 μg/ml ionomycin (Sigma Chemical Co.). 2 h after stimulation monensin was added at 2 μg/ml. Cells were fixed and permeabilized as above and stained with PE-labeled anti–IL-4 and FITC-labeled anti–IFN-γ antibodies (PharMingen).
Determination of Cell Viability.
Viability of the cells after PR8 infection was evaluated by staining cells with FITC-labeled Annexin V (PharMingen) and propidium iodide. The samples were analyzed on a FACScalibur® using Cell Quest software (Becton Dickinson).
Immunoblotting.
Identical numbers of unstimulated DCs or DCs treated with different stimuli for 5 or 13 h were lysed in lysis buffer (100 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 5 mM EDTA, 1 mM PMSF, 10 μg/ml aprotinin, and 10 μg/ml leupeptin). Lysates were solved by 8% SDS-PAGE under reducing conditions, and transferred to nitrocellulose membranes (Amersham). After blocking with PBS/0.1% Tween/5% nonfat dry milk, the membranes were incubated with the rabbit anti-MxA antiserum (17), followed by horseradish peroxidase–conjugated goat anti-rabbit IgG (Southern Biotechnology Associates). Proteins were visualized by chemiluminescence using the ECL detection reagents (Amersham).
Cytokine Determination.
IL-12 p40 and p75 levels were measured by ELISA as previously described (19). Type I IFN was measured evaluating inhibition of Daudi cell proliferation (20) with reference to a standard IFN-α curve. The sensitivity of the assay was 0.2 U/ml.
Class I Synthetic Rate and Stability.
5 h after stimulation or infection, DCs were labeled with 35S[methionine]/cysteine (Amersham) for 1 h. Cells were chased for one additional hour or longer; identical number of cells were lysed and HLA class I molecules were immunoprecipitated using the W6/32 mAb (ATCC) followed by protein G–Sepharose (Pharmacia Biotech). The precipitates were resolved by SDS-PAGE and the radioactivity of specific bands was quantified by PhosphorImager (Molecular Dynamics). DCs do not divide during the assay. Total protein synthesis was estimated by counting the radioactivity incorporated by a fixed number of cells.
T Lymphocyte Stimulation.
Immature HLA-A2+ DCs either were pulsed for 1 h with 1 μg/ml Influenza Matrix 58-66 peptide or were infected with PR8. After 5 or 24 h, a graded number of cells were tested for their capacity to trigger proliferation of a specific T cell clone (21). In a different set of experiments, immature DCs were left untreated or were pretreated for 40 h with poly I:C, LPS, TNF-α, and IFN-α, or were infected with PR8. After irradiation (1,500 rad), a graded number of cells were cultured with allogeneic T cells and the proliferative response was measured on day 5 by [3H]thymidine incorporation (Amersham). Naive T cells were either derived from cord blood or from adult donors lymphocytes, negatively depleted of CD8, CD14, CD20, CD56, HLA class II, CD45 RO+ cells. The resulting population was 99% composed of CD4+CD45RA+ cells. Cells were stimulated with differently matured DCs, at a responder/stimulator ratio of 10:1, in the absence or presence of neutralizing anti–IL-12 antibodies (19).
Results
Susceptibility of DCs to Influenza Virus Infection Correlates with Expression of MxA.
Immature DCs can be generated by culturing peripheral blood monocytes with GM-CSF and IL-4 (3, 4). These cells are competent in antigen capture and mature when stimulated by LPS, TNF-α, or CD40L, losing their antigen-capturing capacity and acquiring T cell stimulatory capacity. Immature and mature DCs, obtained after LPS or TNF-α stimulation, were incubated with different doses of influenza virus strain PR8, and the proportion of infected cells was measured after 14 h by staining with a specific mAb. As shown in Fig. 1, there was a dose-dependent increase in the percentage of cells expressing viral proteins as well as in the level of viral proteins expressed by individual cells. Immature and TNF-α–matured DCs were highly susceptible to influenza virus infection: at high multiplicity of infection (MOI), all cells expressed high levels of viral protein, whereas at low multiplicity viral proteins were expressed in a bimodal distribution. In contrast, LPS-matured DCs were more, and in some experiments completely, resistant to infection; at high MOI they showed a bimodal protein expression, whereas at low multiplicity only a small proportion of cells was infected, expressing low levels of viral proteins. Finally, DCs pretreated for 40 h with IFN-α were almost completely resistant to infection. The different susceptibility of DCs to influenza infection could not be explained by their different levels of endocytic activity. TNF-α–matured DCs, which had completely lost endocytic activity, were equally, if not more, susceptible to infection than immature DCs. Conversely, IFN-α–treated DCs, which were as endocytic as immature DCs (data not shown), were completely resistant.
Figure 1.

Susceptibility to influenza virus infection of DC populations. (A and B) Percentage of DCs expressing viral proteins at high levels (A) or high plus low levels (B) 14 h after infection with different doses of PR8; immature DCs (▪), TNF-α–matured DCs (▵), LPS-matured DCs (⋄), IFN-α–treated DCs (○). The proportion of DCs expressing viral proteins and the level of expression were comparable when measured at 40 h (data not shown). (C and D) Level of expression of viral protein in DCs infected with PR8 at 5 HAU/ml (C) or 0.1 HAU/ml (D). The DCs were either untreated (1) or pretreated for 30 h with TNF-α (2), LPS (3), or IFN-α (4). Second antibody control (0).
In search for a mechanism that might modulate the susceptibility of DCs to influenza virus infection, we investigated the expression of MxA, a protein induced by type I IFN, that is known to mediate resistance to several viruses including influenza (22, 23). MxA was initially identified by two-dimensional gel analysis as a protein abundantly represented in LPS-matured but not immature DCs (Sakakibara, Y., unpublished data).
Fig. 2 shows the level of MxA expression in relation to the level of production of viral HA (A–G) and to the viability of the infected cells (H–P). Immature DCs did not express MxA, but became MxA+ after infection with PR8, explaining their capacity to resist the cytopathic effect of the virus. At low MOI (1:1), immature DCs could be efficiently infected (61%) with no increase in cell death over the background, whereas at higher MOI they were infected more efficiently and produced higher protein levels, but showed progressively lower viability. In contrast, LPS-matured DCs, which already expressed MxA, required ∼10-fold higher doses of virus to produce comparable amounts of proteins, but were far more resistant to the cytopathic effect even at an MOI of 100:1. Taken together, these results suggest that susceptibility to influenza virus infection is modulated in DCs by the expression of MxA, which is induced by LPS stimulation as well as by infection with influenza virus.
Figure 2.

LPS-matured DCs express MxA protein and are more resistant than immature DCs to the cytopathic effect of influenza virus at high MOI. Immature DCs were left untreated (A and H) or were infected with PR8 at 1 HAU/ml (B and I), 10 HAU/ml (C and L) or 30 HAU/ ml (D and M). Mature DCs (40 h after LPS) were left untreated (E and N) or were infected with PR8 at 10 HAU/ml (F and O) or 100 HAU/ml (G and P). After 24 h the cells were tested for the expression of MxA and influenza HA proteins (A–G). Viable, early and late apoptotic cells were evaluated in the same samples by staining with FITC-labeled Annexin V and propidium iodide (H–P).
MxA Is Induced by Autocrine Production of Type I IFN.
The time course of MxA protein expression was analyzed in DCs challenged with different stimuli, either by immunofluorescence on fixed and permeabilized cells or by immunoblotting (Fig. 3, A and B). MxA was induced with comparable fast kinetics by IFN-α, LPS, and PR8 infection, as well as by poly I:C, a synthetic source of dsRNA. In contrast, TNF-α failed to induce MxA expression, and CD40L induced it only to a low extent and with a slower kinetics.
Figure 3.

MxA expression is rapidly induced in DCs by LPS, poly I:C and viral infection. Time course of MxA upregulation as detected by intracellular staining (A) or immunoblotting (B). (C) Time course of type I IFN production in culture supernatant. DCs were stimulated with the following: 50 U/ml IFN-α (○, panel A only), LPS (▵), 20 μg/ml poly I:C (▪), 1 HAU PR8 (•), TNF-α (▿), and CD40L (⋄). (D) MxA induction after 5 h of stimulation in the absence (black bar) or in the presence of two neutralizing sheep antisera to human type I IFN: Iivari, hatched bars, and Kaaleppi, empty bars.
The capacity of different stimuli to induce MxA correlated with their capacity to induce production of type I IFN by DCs (Fig. 3 C). Both poly I:C and LPS led to a rapid production of type I IFN that reached a plateau at 4 h, whereas PR8 infection induced an equally rapid, but more sustained and consistently higher production. In contrast, TNF-α and CD40L stimulation did not induce production of detectable levels of IFN.
These results demonstrate that DCs can produce type I IFN in response to specific stimuli, which in turn leads to upregulation of MxA gene expression via an autocrine loop. To address this possibility, we evaluated the capacity of neutralizing antibodies to type I IFNs to inhibit MxA expression (Fig. 3 D). The antisera we used completely abrogated MxA upregulation at 5 h induced by recombinant IFN-α, PR8 infection, and CD40L. However, they also inhibited significantly, although not completely (at least 50%), MxA induction after LPS or poly I:C stimulation. Although these results do not rule out the possibility of an IFN-α–independent MxA upregulation, they suggest that autocrine production of type I IFN by DCs represents a major mechanism to ensure the rapid build-up of a resistance state to the infecting virus.
dsRNA and PR8 Upregulate Protein Synthesis in Immature and IFN-α–treated DCs.
dsRNA has been shown to trigger PKR, an IFN-α–induced kinase that inactivates eIF2, thereby reducing protein synthesis (24). This mechanism may be important to ensure reduction of viral replication and elimination of infected cells. However, PKR has also been shown to activate NF-κB (25) and transcription. PKR was expressed constitutively in DCs and its level was not further increased upon IFN-α treatment (data not shown). We thus compared the effect of poly I:C and PR8 on protein synthesis in DCs and Hela cells before and after treatment with IFN-α (Fig. 4). As expected, PR8 infection and poly I:C induced a marked decrease in the rate of total and MHC class I protein synthesis in IFN-α–pretreated, but not in untreated, Hela cells. In contrast, in DCs the same treatments resulted in a strong boost of total, and particularly MHC class I, protein synthesis, irrespective of whether or not the cells had been pretreated with IFN-α. These results demonstrate that, as compared with other cell types, DCs have a unique capacity to respond to dsRNA, a property that may be important to allow efficient presentation of viral antigens.
Figure 4.

dsRNA and PR8 infection induce upregulation of total protein and HLA class I synthesis in untreated as well as in IFN-α–pretreated DCs. Total protein synthesis (hatched bars) and HLA class I synthesis (black bars) were measured in DCs (A and C) or Hela cells (B and D) 5 h after stimulation with 20 μg/ml poly I:C, 5 HAU PR8, or medium alone. The cells were either untreated (A and B) or pretreated for 24 h with 500 U/ml IFN-α (C and D).
Influenza Virus Infection Promotes and Sustains Generation of Peptide–MHC Class I Complexes.
We next investigated whether stimulation of DCs by infectious virus might optimize loading of class I molecules with antigenic peptides. In PR8-infected DCs there was a strong upregulation of HLA class I synthesis (5–10-fold in four experiments) (Fig. 5 A). Furthermore, the class I molecules synthesized during the initial stages of infection displayed a two- to threefold longer half-life than those synthesized in immature or in LPS-treated DCs (Fig. 5 B). This increased stability may be related to the abundant supply of high affinity peptides derived from degradation of viral proteins, since it was not noticed in DCs stimulated with LPS.
Figure 5.

Increase in MHC class I biosynthesis and stability induced by PR8 infection allows efficient presentation of viral antigen to cytotoxic T cells. (A) Synthetic rate and stability of HLA class I molecules in immature DCs and in DCs that were stimulated for 5 h with 5 HAU/ml PR8 or LPS. Labeled class I molecules were precipitated after 1 h of chase (time 0) or after 12 or 24 h. (B) Half-life of labeled HLA class I molecules quantitated by PhosphorImager in immature DCs (○), LPS-treated DC (•), or PR8-infected DC (▴). (C) Proliferative response of an HLA-A2– restricted M58-66–specific T cell clone cultured with graded numbers of HLA-A2+ DCs. DCs were either pulsed with 1 μM M58-66 peptide (circles) or infected with 5 HAU/ml PR8 with (triangles) or without (squares) IFN-α (500 U/ml) pretreatment. DCs were tested 5 h (open symbols) or 24 h after pulsing (filled symbols).
PR8-infected immature DCs rapidly acquired a high capacity to trigger an HLA-A2–restricted T cell clone specific for the influenza matrix peptide M58-66. Furthermore, the stimulatory capacity increased with time after infection (Fig. 5 C). In contrast, although immediately stimulatory, immature DCs pulsed with a high dose of M58-66 peptide lost the capacity to stimulate specific T cells by 20 h of culture, a finding consistent with the short half-life of MHC class I–peptide complexes. In addition, IFN-α–pretreated DCs, which were highly resistant to viral infection, did not stimulate the virus-specific T cell clone even at late time points.
Taken together, these results indicate that in PR8- infected DCs the increased synthesis and loading of MHC class I molecules results in a rapid and continuous production of large numbers of complexes containing viral peptides. These events allow presentation to T cells to be sustained over a long period of time.
Influenza Virus Infection and dsRNA Trigger DC Maturation and Prime for a Th1 Response.
We compared dsRNA and influenza virus infection to known stimuli for their capacity to induce DC maturation. As a readout we analyzed the expression of costimulatory molecules and the production of Th1 polarizing cytokines (i.e., IL-12 [26]), and evaluated the capacity of DCs to stimulate and polarize allogeneic naive T cells. As shown in Table I, poly I:C and PR8 induced a marked upregulation of MHC class I and class II, CD80, CD86, CD83, and CD38, and downregulation of CD115, a phenotype characteristic of mature DCs (19, 27). In addition, poly I:C and PR8 induced the production of low but significant levels of IL-12 p75 (1–2 ng/ml), although the amount detected was always lower (at least 10– 50-fold) than that induced via CD40L (19). Although TNF-α stimulation never led to production of bioactive IL-12, low levels of IL-12 p75 (0.1–0.2 ng/ml) were detectable after LPS stimulation. On the other hand, IFN-α tested over a broad range of concentrations (10–500 U/ml) did not induce DC maturation but only upregulation of HLA class I and a modest shift in the expression of CD38 and CD86 with no significant increase in CD83, HLA class II, or other costimulatory and adhesion molecules.
Table I.
DC Maturation and IL-12 p75 Production Are Induced by dsRNA and Influenza Virus Infection
| Stimuli* | Class I | HLA-DR | CD86 | CD83 | CD38 | CD115 | IL-12 p75§ |
|---|---|---|---|---|---|---|---|
| Untreated | 370 | 575 | 11 | 3 | 17 | 71 | 10 |
| PR8‡ | 1,576 | 1,441 | 261 | 133 | 134 | 11 | 527 |
| Poly I:C | 729 | 866 | 264 | 112 | 108 | 5 | 1,218 |
| LPS | 973 | 1,087 | 200 | 157 | 121 | 8 | 185 |
| TNF-α | 634 | 1,012 | 67 | 69 | 15 | 4 | 11 |
| IFN-α | 525 | 519 | 32 | 4 | 37 | 34 | 16 |
Consistent with the effect on DC maturation and cytokine production, poly I:C and PR8 increased ∼30-fold the capacity of DCs to stimulate a proliferative response by allogeneic CD45RA+CD4+ T cells, whereas IFN-α treatment did not show a significant effect. Poly I:C-matured DCs were as efficient as DCs induced to mature by TNF-α or LPS (Fig. 6). Although the ability to trigger T cell proliferation was comparable, the capacity to polarize T cells was remarkably different, depending on the nature of the maturative stimulus applied. Alloreactive T cells expanded by TNF-α–matured DCs were poorly polarized (only 32% of cells producing either IFN-γ or IL-4) and consisted of both Th1 and Th2 (Fig. 7 A). In contrast, T cells expanded by poly I:C-matured DCs were highly polarized (70–90%, in different experiments) with most of the cells producing high amounts of IFN-γ, consistent with a dominant Th1 phenotype (Fig. 7 B). Addition of neutralizing antibodies to IL-12 prevented Th1 polarization, skewing the cultures towards a Th2 phenotype (Fig. 7 C). In some experiments, the inhibition of Th1 development required the simultaneous neutralization of type I IFN and IL-12 (data not shown). Taken together, these results indicate that dsRNA and influenza virus infection can induce DC maturation, and confer the capacity to prime a Th1 response through the production of IL-12 and type I IFN.
Figure 6.

PR8 infection and poly I:C increase the T cell stimulatory capacity of DCs. Proliferative response of cord blood naive T cells stimulated with graded numbers of irradiated allogeneic DCs. DCs were untreated (•) or pretreated for 24 h with 50 U/ml IFN-α (×), LPS (⋄), TNF-α (▵), or 20 μg/ml poly I:C (□), or infected with 3 HAU/ml PR8 (○).
Figure 7.
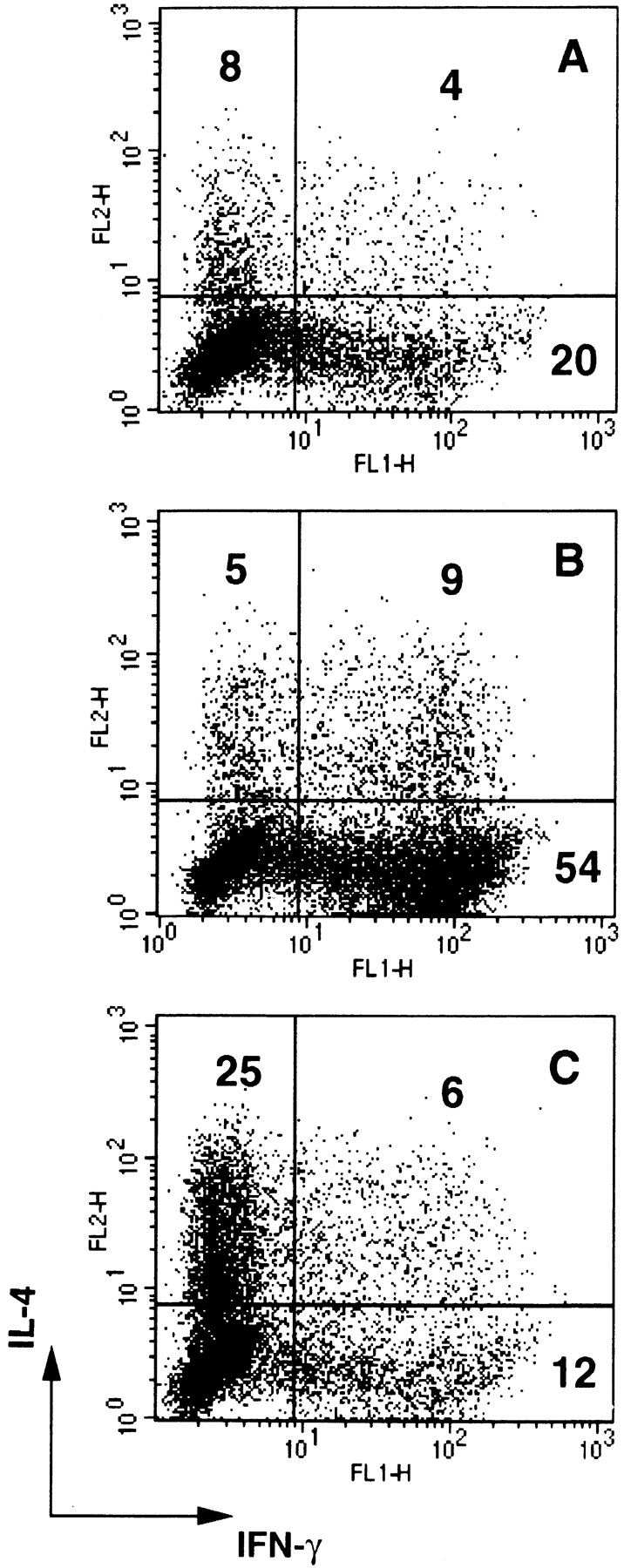
Poly I:C primes for a Th1 response. IFN-γ and IL-4 production by polyclonal T cell lines generated by stimulating purified CD45RA+CD4+ T cells with allogeneic DCs. A, TNF-α–matured DCs; B, poly I:C-matured DCs; C, poly I:C-matured DCs in the presence of neutralizing antibodies to IL-12. A strong Th1 response was also obtained with LPS-matured DCs (data not shown). In some experiments both anti–IL-12 and anti–IFN-α antibodies were required to inhibit Th1 development. Anti–IL-12 antibodies did not affect polarization induced by TNF-α–matured or immature DCs.
Discussion
Protection and Maturation of DCs Induced by the Viral Pattern dsRNA.
We have shown that dsRNA induces two functionally distinct responses in DC: (i) protection from the viral cytopathic effect, and (ii) maturation, leading to increased capacity to prime and polarize T cells. In the course of viral infection, the simultaneous induction of protection and maturation optimizes loading of viral antigens on MHC class I molecules.
dsRNA is a classical inducer of type I IFN (28–30), which plays a critical role in antiviral responses (31, 32). DCs represent a strategic source of type I IFN and we have shown that it is produced not only in response to dsRNA or virus infection (33), but also in response to LPS stimulation. However, IFN is not produced in response to other stimuli such as TNF-α, and only in minute amounts in response to CD40L. Type I IFN produced by DCs results in rapid induction of MxA, a cytoplasmic protein that has been shown to protect cells from the cytopathic effect of some viruses (22, 23, 34). Neutralizing antibodies to type I IFN completely inhibited MxA induction by recombinant exogenous IFN-α, influenza virus infection, and CD40L, but only partially inhibited MxA upregulation by poly I:C or LPS, suggesting a possible direct activation of MxA expression independent from type I IFN (35).
dsRNA and viral infection also induced a rapid maturation of DCs with upregulation of MHC, adhesion, and costimulatory molecules. These phenotypic changes were to a large extent comparable to those induced by other maturation stimuli such as LPS, TNF-α, or CD40L, although a different pattern in cytokine production was observed. These results may well account for the increased capacity of influenza virus–infected DCs to stimulate CTL responses (15). Although a minor shift in CD38 and CD86 was reproducibly observed, IFN-α per se was unable to support a complete DC maturation. The maturation effect of type I IFN reported in other studies was observed in a different culture system and was dependent on the simultaneous presence of TNF-α, which by itself is a DC maturation factor (36).
The signal transduction pathway triggered by dsRNA has been studied in detail. It has been shown that dsRNA activates PKR, a serine-threonine kinase that phosphorylates and inactivates eIF2, thereby shutting off protein synthesis (24). PKR can also activate NF-κB (25) and consequently may induce transcription of genes involved in DC maturation. The fact that PKR-deficient mice can still respond to dsRNA suggests that there may be additional pathways of signal transduction that may be operative in DCs (37). The relative role and function of the pathways activated by dsRNA may vary in different cells. Our experiments show that the response to dsRNA in DCs and Hela cells is considerably different. In Hela cells dsRNA induces a downregulation of protein synthesis, whereas in immature DCs dsRNA actually upregulates total protein synthesis, while fully inducing the maturation process.
It has been suggested that recognition of conserved molecular patterns characteristic of pathogens is a property of the innate immune system, which is instrumental to initiating and regulating the adaptive immune response (38). Our results clearly indicates that dsRNA behaves as one of such molecular patterns. dsRNA can directly signal to the APCs the presence of an infectious agents and, by activating the DCs, can lead to activation of lymphocytes bearing clonally specific antigen receptors, thus triggering an adaptive immune response. The relevance of this recognition system is also underlined by the fact that many viruses specifically target the dsRNA-binding protein PKR to escape immune recognition (39–41).
Sustained Synthesis of Viral Antigens and Class I Molecules Maximizes Antigen Presentation.
We have shown that efficient presentation influenza virus is the result of a delicate compromise. On one hand, immature DCs are highly susceptible to infection and thus produce large amounts of viral proteins. On the other hand, they can rapidly build up resistance to the virus, thus limiting its cytopathic effect. The importance of synchronizing these two functions is illustrated by the fact that IFN-α–pretreated DCs, which are resistant to influenza infection, are extremely inefficient at presenting viral antigens.
The dramatic upregulation of class I synthesis (up to 10-fold) induced by viral infection is an important mechanism that ensures effective presentation of viral antigens. MHC class I synthesis is sustained after induction of maturation, allowing continuous accumulation of peptide–MHC complexes and thus compensating for the relatively short half-life of class I molecules (10–20 h). The importance of the sustained antigen loading is exemplified by the fact that although the capacity to stimulate class I–restricted, virus-specific T cells is rapidly lost in peptide-pulsed DCs, it actually increases with time in virus-infected DCs. This fact should be taken into account when considering the use of DCs as vaccines to induce class I–restricted responses.
We have previously shown that in maturing DCs the synthesis of class II molecules is transiently upregulated and subsequently stopped, while stable peptide class II complexes are retained with extremely long half-lives (>100 h) (5). In contrast, in DCs MHC class I molecules have a short half-life that is not significantly affected by the maturation process. The different regulation of class I and class II biosynthesis and stability in DCs makes good sense. Class II molecules present antigens that are transiently encountered in the surrounding environment, so it is important for a DC to be able to load antigenic peptides over a short period of time and retain the antigen as a stable complex. Instead, the short half-life of class I molecules is instrumental to allow a continuous monitoring of early and late viral antigens, as long as the cell remains infected.
Flexibility in Cytokine Production Determines the T Cell Polarizing Capacity of DCs.
Although DCs induced to mature by different stimuli share several common features, such as the increase in MHC, costimulatory molecules, and T cell stimulatory capacity (3, 19), consistent differences are observed in the pattern of cytokines produced in response to the different stimuli. These in turn determine both the extent and the type of T cell polarization, suggesting that mature DCs can exist in different functional states. TNF-α–matured DCs, which produce neither IL-12 nor type I IFN, were highly stimulatory, but induced only a low percentage of fully polarized cells. In contrast, dsRNA- or LPS-matured DCs, which produced both type I IFN and IL-12 (although in different proportions) elicited strong Th1 polarization, with 70–90% of T cells producing high levels of IFN-γ. The Th1 polarizing effect was inhibited in most cases by neutralization of IL-12, although in some cases neutralization of type I IFN was also required, as previously reported for monocytes (30). These results indicate a flexibility of DCs in the use of Th1 polarizing cytokines and suggest that a multiplicity of responses can be generated by distinct environmental signals. In addition, it is possible that the relative production of IL-12 and type I IFN may play a more subtle role in modulating T cell polarization. Although both IL-12 (26) and type I IFN in humans (42) are known to drive Th1 polarization, it is possible that they may play distinct roles by differentially regulating IL-4 and IL-13 production by T cells (43).
Our results also imply that the nature of the signal inducing DC maturation may determine the capacity of the DCs to generate polarized immune responses. On one hand, stimuli from viral or bacterial patterns (dsRNA or LPS) or T cell help, through CD40–CD40L interaction, generate DCs capable of priming strong Th1 responses. On the other hand, an endogenous inflammatory stimulus, such as TNF-α, generates DCs capable of inducing a more balanced response, comprising both Th1 and Th2 cells. The fact that the same DCs can mature to different functional states capable of stimulating polarized Th1 or Th2 responses makes good sense, since it allows the APCs to initiate a response that is appropriate for the signal received and has practical implications for the therapeutic use of DCs.
Acknowledgments
We thank K. Karjalainen and M. Colonna for critical reading and comments; M. Dessing and A. Pickert for expert assistance in FACS® analysis; and Drs. A. Vitiello (R.W. Johnson Pharmaceutical Research Institute, San Diego, CA), R. Maccario (University of Pavia, IRCCS Policcinico, San Matteo, Italy), W. Gerhard (Wistar Institute, Philadelphia, PA), and M. Gately (Hoffmann-La Roche Inc., Nutley, NJ) for kindly sharing reagents.
Abbreviations used in this paper
CD40L
CD40 ligand
DC
dendritic cell
ds
double-stranded
MOI
multiplicity of infection
Footnotes
M. Salio holds an EC fellowship (EUNIDI). The Basel Institute for Immunology was founded and is supported by Hoffmann-La Roche, Basel, Switzerland.
References
- 1.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 2.Steinman RM. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol. 1991;9:271–296. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- 3.Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor α. J Exp Med. 1994;179:1109–1118. doi: 10.1084/jem.179.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sallusto F, Cella M, Danieli C, Lanzavecchia A. Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartment: downregulation by cytokines and bacterial products. J Exp Med. 1995;182:389–400. doi: 10.1084/jem.182.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cella M, Engering A, Pinet V, Pieters J, Lanzavecchia A. Inflammatory stimuli induce accumulation of MHC class II complexes on dendritic cells. Nature. 1997;388:782–787. doi: 10.1038/42030. [DOI] [PubMed] [Google Scholar]
- 6.Pierre P, Turley SJ, Gatti E, Hull M, Meltzer J, Mirza A, Inaba K, Steinman RM, Mellman I. Developmental regulation of MHC class II transport in mouse dendritic cells. Nature. 1997;388:787–792. doi: 10.1038/42039. [DOI] [PubMed] [Google Scholar]
- 7.Braciale TJ, Yap KL. Role of viral infectivity in the induction of influenza virus-specific cytotoxic T cells. J Exp Med. 1978;147:1236–1252. doi: 10.1084/jem.147.4.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yewdell JW, Bennink JR, Hosaka Y. Cells process exogenous proteins for recognition by cytotoxic T lymphocytes. Science. 1988;239:637–640. doi: 10.1126/science.3257585. [DOI] [PubMed] [Google Scholar]
- 9.Townsend AR, Rothbard J, Gotch FM, Bahadur G, Wraith D, McMichael AJ. The epitopes of influenza nucleoprotein recognized by cytotoxic T lymphocytes can be defined with short synthetic peptides. Cell. 1986;44:959–968. doi: 10.1016/0092-8674(86)90019-x. [DOI] [PubMed] [Google Scholar]
- 10.Gotch F, Rothbard J, Howland K, Townsend A, McMichael A. Cytotoxic T lymphocytes recognize a fragment of influenza virus matrix protein in association with HLA-A2. Nature. 1987;326:881–882. doi: 10.1038/326881a0. [DOI] [PubMed] [Google Scholar]
- 11.Bhardwaj N, Bender A, Gonzalez N, Bui LK, Garrett MC, Steinman RM. Influenza virus-infected dendritic cells stimulate strong proliferative and cytolytic responses from human CD8+T cells. J Clin Invest. 1994;94:797–807. doi: 10.1172/JCI117399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bender A, Albert M, Reddy A, Feldman M, Sauter B, Kaplan G, Hellman W, Bhardwaj N. The distinctive features of influenza virus infection of dendritic cells. Immunobiology. 1998;198:552–567. doi: 10.1016/S0171-2985(98)80078-8. [DOI] [PubMed] [Google Scholar]
- 13.Bender A, Bui LK, Feldman MA, Larsson M, Bhardwaj N. Inactivated influenza virus, when presented on dendritic cells, elicits human CD8+cytolytic T cell responses. J Exp Med. 1995;182:1663–1671. doi: 10.1084/jem.182.6.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Albert ML, Sauter B, Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature. 1998;392:86–89. doi: 10.1038/32183. [DOI] [PubMed] [Google Scholar]
- 15.Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4 T helper and a T killer cell. Nature. 1998;393:474–478. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- 16.Mogensen KE, Pyhala L, Cantell K. Raising antibodies to human leukocyte interferon. Acta Pathol Microbiol Scand Suppl. 1995;83:443–448. doi: 10.1111/j.1699-0463.1975.tb00123.x. [DOI] [PubMed] [Google Scholar]
- 17.Ronni T, Melen K, Malygin A, Julkunen I. Control of IFN-inducible MxA gene expression in human cells. J Immunol. 1993;150:1715–1726. [PubMed] [Google Scholar]
- 18.Caton AJ, Swartzentruber JR, Kuhl AL, Carding SR, Stark SE. Activation and negative selection of functionally distinct subsets of antibody-secreting cells by influenza hemagglutinin as a viral and a neo-self-antigen. J Exp Med. 1996;183:13–26. doi: 10.1084/jem.183.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cella M, Scheidegger D, Palmer K, Lehmann, Lane P, Lanzavecchia A, Alber G. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin 12 and enhances T cell stimulatory capacity: T-T help via APC activation. J Exp Med. 1996;184:747–752. doi: 10.1084/jem.184.2.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nederman T, Karlstrom E, Sjodin L. An in vitro bioassay for quantitation of human interferons by measurements of antiproliferative activity on a continuous human lymphoma cell line. Biologicals. 1990;18:29–34. doi: 10.1016/1045-1056(90)90066-9. [DOI] [PubMed] [Google Scholar]
- 21.Valitutti S, Mueller S, Dessing M, Lanzavecchia A. Different responses are elicited in cytotoxic T lymphocytes by different levels of T cell receptor occupancy. J Exp Med. 1996;183:1917–1921. doi: 10.1084/jem.183.4.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pavlovic J, Arzet HA, Hefti HP, Frese M, Rost D, Ernst B, Kolb E, Staeheli P, Haller O. Enhanced virus resistance of transgenic mice expressing the human MxA protein. J Virol. 1995;69:4506–4510. doi: 10.1128/jvi.69.7.4506-4510.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pavlovic J, Haller O, Staeheli P. Human and mouse Mx proteins inhibit different steps of the influenza virus multiplication cycle. J Virol. 1992;66:2564–2569. doi: 10.1128/jvi.66.4.2564-2569.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meurs E, Chong K, Galabru J, Thomas NS, Kerr IM, Williams BR, Hovanessian AG. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell. 1990;62:379–390. doi: 10.1016/0092-8674(90)90374-n. [DOI] [PubMed] [Google Scholar]
- 25.Kumar A, Haque J, Lacoste J, Hiscott J, Williams BR. Double-stranded RNA-dependent protein kinase activates transcription factor NF-kappa B by phosphorylating I kappa B. Proc Natl Acad Sci USA. 1994;91:6288–6292. doi: 10.1073/pnas.91.14.6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O'Garra A. Cytokines induce the development of functionally heterogeneous T helper cell subsets. Immunity. 1998;8:275–283. doi: 10.1016/s1074-7613(00)80533-6. [DOI] [PubMed] [Google Scholar]
- 27.Bender A, Sapp M, Schuler G, Steinman RM, Bhardwaj N. Improved methods for the generation of dendritic cells from nonproliferating progenitors in human blood. J Immunol Methods. 1996;196:121–135. doi: 10.1016/0022-1759(96)00079-8. [DOI] [PubMed] [Google Scholar]
- 28.Field AK, Tytell AA, Lampson GP, Hilleman MR. Inducers of interferon and host resistance. II. Multistranded synthetic polynucleotide complexes. Proc Natl Acad Sci USA. 1967;58:1004–1010. doi: 10.1073/pnas.58.3.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pestka S, Langer JA, Zoon KC, Samuel CE. Interferons and their actions. Annu Rev Biochem. 1987;56:727–777. doi: 10.1146/annurev.bi.56.070187.003455. [DOI] [PubMed] [Google Scholar]
- 30.Manetti R, Annunziato F, Tomasevic L, Gianno V, Parronchi P, Romagnani S, Maggi E. Polyinosinic acid: polycytidylic acid promotes T helper type 1-specific immune responses by stimulating macrophage production of interferon-alpha and interleukin-12. Eur J Immunol. 1995;25:2656–2660. doi: 10.1002/eji.1830250938. [DOI] [PubMed] [Google Scholar]
- 31.van den Broek MF, Muller U, Huang S, Aguet M, Zinkernagel RM. Antiviral defense in mice lacking both alpha/beta and gamma interferon receptors. J Virol. 1995;69:4792–4796. doi: 10.1128/jvi.69.8.4792-4796.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 33.Perussia B, Fanning V, Trinchieri G. A leukocyte subset bearing HLA-DR antigens is responsible for in vitro alpha interferon production in response to viruses. Nat Immun Cell Growth Regul. 1985;4:120–137. [PubMed] [Google Scholar]
- 34.Pavlovic J, Zurcher T, Haller O, Staeheli P. Resistance to influenza virus and vesicular stomatitis virus conferred by expression of human MxA protein. J Virol. 1990;64:3370–3375. doi: 10.1128/jvi.64.7.3370-3375.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ronni T, Sareneva T, Pirhonen J, Julkunen I. Activation of IFN-alpha, IFN-gamma, MxA, and IFN regulatory factor 1 genes in influenza A virus-infected human peripheral blood mononuclear cells. J Immunol. 1995;154:2764–2774. [PubMed] [Google Scholar]
- 36.Luft T, Pang KC, Thomas E, Hertzog P, Hart DN, Trapani J, Cebon J. Type I IFNs enhance the terminal differentiation of dendritic cells. J Immunol. 1998;161:1947–1953. [PubMed] [Google Scholar]
- 37.Yang YL, Reis LF, Pavlovic J, Aguzzi A, Schafer R, Kumar A, Williams BR, Aguet M, Weissmann C. Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. EMBO (Eur Mol Biol Organ) J. 1995;14:6095–6106. doi: 10.1002/j.1460-2075.1995.tb00300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Medzhitov R, Janeway CA., Jr Innate immunity: the virtues of a nonclonal system of recognition. Cell. 1997;91:295–298. doi: 10.1016/s0092-8674(00)80412-2. [DOI] [PubMed] [Google Scholar]
- 39.Kitajewski J, Schneider RJ, Safer B, Munemitsu SM, Samuel CE, Thimmappaya B, Shenk T. Adenovirus VAI RNA antagonizes the antiviral action of interferon by preventing activation of the interferon-induced eIF-2 alpha kinase. Cell. 1986;45:195–200. doi: 10.1016/0092-8674(86)90383-1. [DOI] [PubMed] [Google Scholar]
- 40.Katze MG. Regulation of the interferon-induced PKR: can viruses cope? . Trends Microbiol. 1995;3:75–78. doi: 10.1016/s0966-842x(00)88880-0. [DOI] [PubMed] [Google Scholar]
- 41.Jacobs BL, Langland JO. When two strands are better than one: the mediators and modulators of the cellular responses to double-stranded RNA. Virology. 1996;219:339–349. doi: 10.1006/viro.1996.0259. [DOI] [PubMed] [Google Scholar]
- 42.Rogge L, Barberis-Maino L, Biffi M, Passini N, Presky DH, Gubler U, Sinigaglia F. Selective expression of an interleukin-12 receptor component by human T helper 1 cells. J Exp Med. 1997;185:825–831. doi: 10.1084/jem.185.5.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaser A, Molnar C, Tilg H. Differential regulation of interleukin 4 and interleukin 13 production by interferon alpha. Cytokine. 1998;10:75–81. doi: 10.1006/cyto.1997.0270. [DOI] [PubMed] [Google Scholar]