The matrix metalloproteinase stromelysin-1 acts as a natural mammary tumor promoter (original) (raw)
. Author manuscript; available in PMC: 2010 Sep 3.
Published in final edited form as: Oncogene. 2000 Feb 21;19(8):1102–1113. doi: 10.1038/sj.onc.1203347
Abstract
Extracellular matrix-degrading matrix metalloproteinases (MMPs) are invariably upregulated in epithelial cancers and are key agonists in angiogenesis, invasion and metastasis. Yet most MMPs are secreted not by the cancer cells themselves, but by stromal cells within and around the tumor mass. Because the stromal environment can influence tumor formation, and because MMPs can alter this environment, MMPs may also contribute to the initial stages of cancer development. Several recent studies in MMP-overexpressing and MMP-deficient mice support this possibility, but have required carcinogens or pre-existing oncogenic mutations to initiate tumorigenesis. Here we review the spontaneous development of premalignant and malignant lesions in the mammary glands of transgenic mice that express an autoactivating form of MMP-3/stromelysin-1 under the control of the whey acidic protein gene promoter. These changes were absent in nontransgenic littermates and were quenched by co-expression of a human tissue inhibitor of metalloproteinases-1 (TIMP-1) transgene. Thus by altering the cellular microenvironment, stromelysin-1 can act as a natural tumor promoter and enhance cancer susceptibility.
Keywords: matrix metalloproteinases, stromelysin, TIMP, mammary carcinogenesis, epithelial-to-mesenchymal conversion
Introduction
Matrix metalloproteinases (MMPs) are consistently upregulated in epithelial cancers, and considerable evidence indicates that they play an essential role in tumor angiogenesis, invasion and metastasis by virtue of their combined ability to degrade virtually all elements of the extracellular matrix (ECM) (Coussens and Werb, 1996). Indeed, without the help of ECM-degrading enzymes, cancer cells would probably be unable to cross the matrix barriers that otherwise contain their spread. This straightforward and conceptually appealing supposition forms the basis for current clinical trials of MMP inhibitors as anti-cancer agents. However, in addition to promoting cellular invasion by simply clearing away the surrounding matrix, MMPs can alter cellular signals (Lukashev and Werb, 1998; Werb, 1997) and may therefore influence initial tumor development. If so, then the inhibition of select MMPs during even the earliest stages of cancer progression may offer clinical benefit.
The MMP stromelysin-1 (MMP-3, Str1) exhibits a number of activities that would make it a particularly good tumor promoter. Like several other MMPs, Str1 was first cloned and later recloned as a cancer-specific gene (Matrisian et al., 1985; Muller et al., 1988; Ostrowski et al., 1988). In addition to degrading numerous ECM components, Str1 can activate gelatinase B and the collagenases, and can activate several serpin-type serine proteinase inhibitors (Sternlicht and Werb, 1999, for review). Moreover, it can release a number of cell surface molecules, including E-cadherin (Lochter et al., 1997a), a known contributor to cancer development (Christofori and Semb, 1999; Tlsty, 1998).
Str1 is expressed by stromal cells during normal mammary gland development, and is strongly upregulated during post-lactational mammary involution when considerable ECM remodeling and alveolar apoptosis occur (Lund et al., 1996; Thomasset et al., 1998; Witty et al., 1995). Interestingly, E-cadherin cleavage also occurs during involution and may induce apoptosis (Vallorosi et al., 1999). Alternatively, ECM degradation may induce the apoptosis that occurs during involution. Either way, Str1 could act as an apoptotic stimulus. Indeed, Str1 does induce apoptosis in differentiated mammary alveolar epithelial cells in culture and in vivo, however it also promotes the proliferation and branching of ductal epithelium (Alexander et al., 1996; Boudreau et al., 1995; Sympson et al., 1994; Thomasset et al., 1998; Witty et al., 1995). These seemingly contradictory effects can be reconciled by noting that ductal epithelial cells normally divide during branching morphogenesis and persist throughout involution, whereas alveolar epithelial cells do not. Thus the differentiation status of the target cell may determine its response to Str1. These effects were first observed in transgenic mice with an autoactivating rat Str1 transgene – (the autoactivating rat Str1 cDNA contained a Val92-to-Gly92 transition within its propeptide domain, thus destabilizing the ‘cysteine switch’ that otherwise maintains enzyme latency (Sanchez-Lopez et al., 1988)) – targeted to mammary epithelium by the whey acidic protein (WAP) gene promoter (Sympson et al., 1994) and mouse mammary tumor virus (MMTV) enhancer/promoter (Witty et al., 1995). In these mice, Str1 transgene expression resulted in increased ductal branching and precocious lobulo-alveolar development during puberty, basement membrane disruption and unscheduled involution during pregnancy, and alveolar collapse and low milk-protein production during lactation. Expression of the Str1 transgene during pregnancy and lactation also led to enhanced expression of endogenous Str1 by mammary fibroblasts, collagen accumulation (fibrosis), neovascularization, and tenascin-C expression (Thomasset et al., 1998). These changes are not only hallmarks of the reactive stroma seen during involution, but are also seen during cancer progression (Borsi et al., 1992; Rønnov-Jessen et al., 1996) and may even predispose toward neoplastic epithelial transformation (Jacobs et al., 1999; Jacoby et al., 1997; Kinzler and Vogelstein, 1998; Willenbucher et al., 1999). Furthermore, the proliferative effects of Str1 could support neoplastic transformation and its apoptotic effects could help select apoptosis-resistant clones. Thus Str1 triggers a number of changes (increased cell proliferation, apoptosis, angiogenesis, and an altered stromal environment) that could potentially promote mammary carcinogenesis.
The above effects, which might be viewed as a prelude to cancer, were observed in transgenic animals under 4 months of age. To further address the potential tumor promoting activity of Str1, mammary gland changes were monitored in WAP-Str1 transgenic mice from 6 – 24 months of age. We observed the development of spontaneous premalignant lesions and mammary cancers in these mice and the virtual absence of such changes in their nontransgenic littermates and in related bitransgenic mice that co-express a human tissue inhibitor of metalloproteinases (TIMP-1) transgene under the control of the same promoter (Sternlicht et al., 1999). These Str1-induced changes which occur in the absence of exogenous mutagens or endogenous oncogene or suppressor gene defects, offer strong evidence that Str1 can indeed act as a natural tumor promoter.
Str1 promotes mammary carcinogenesis
To evaluate the effects of prolonged Str1 expression in the mammary gland, WAP-Str1 transgenic mice from five independent CD-1 founder lines and nontransgenic controls were maintained under similar conditions for up to 2 years (Sternlicht et al., 1999). Only 12% of all WAP-Str1 transgenic mice had histologically normal mammary glands. Instead, about three-quarters had moderate-to-severe fibrosis, about half had epithelial hyperplasias, 20% had atypical hyperplasias (dysplasias) or ductal carcinoma in situ, and 7% developed malignant mammary carcinomas (Table 1). Lymphocytic infiltrates accompanied these lesions in about half of all transgenic mice. By comparison, 87% of the nontransgenic mice had entirely normal mammary glands, and the remaining 13% had only mild fibrosis, hyperplasia or lymphocytic infiltration, and none of the more severe lesions seen in the animals expressing the Str1 transgene. These genotype-specific differences were highly significant (P < 0.002 for carcinoma development and P< 0.0001 for all other pathologies).
Table 1.
Incidence of mammary gland pathologies in WAP-Str1 transgenic mice
| _Wap_-Str1 transgenic mice | Nontransgenic | ||||
|---|---|---|---|---|---|
| Virgin | Parous | All mice | All mice | P* | |
| N | 105 | 58 | 163 | 119 | |
| Median age(mos.) | 18 | 17 | 18 | 18 | |
| No pathology | 17 (16%) | 3 (5.2%) | 20 (12%) | 104 (87%) | <0.0001 |
| Fibrosis | 74 (70%) | 51 (88%) | 125 (77%) | 8 (6.7%) | <0.0001 |
| Lymphoid infiltrates | 51 (49%) | 36 (62%) | 87 (53%) | 10 (9.3%) | <0.0001 |
| Hyperplasia | 65 (62%) | 40 (69%) | 105 (64%) | 6 (5%) | <0.0001 |
| Dysplasia | 18 (17%) | 15 (26%) | 33 (20%) | 0 (0%) | <0.0001 |
| Carcinoma | 6 (5.7%) | 6 (10%) | 12 (7.4%) | 0 (0%) | 0.0016 |
Approximately one-third of the mice from each group were carried through pregnancy and lactation. Parity had no effect on the already low incidence of mammary changes seen in the nontransgenic mice, and slightly increased the incidence of each type of lesion in the transgenic mice (Table 1). The hyperplastic and fibrotic lesions also tended to be somewhat more severe in the parous subset of transgenic mice. The absence of more profound differences between parous and nulliparous mice, despite the use of a pregnancy-responsive promoter, probably reflects the low-level activity of the promoter during each estrus cycle which, in turn, would limit the increase in overall lifetime exposure to Str1 that would be gained through parity.
Abnormalities of varying severity were usually seen in all of the mammary glands examined in an individual transgenic mouse, and multiple abnormalities were often seen within individual mammary glands (Figures 1 and 2). Fibrotic changes included periductal, intralobular and diffuse accumulations of interstitial collagen and fibroblasts (Figure 1). In addition, fibrosis was often seen adjacent to or admixed with multi-loculated adipocytes (Figure 2), a feature that may reflect the dedifferentiation of adipocytes towards a matrix-producing fibroblastic phenotype. Hyperplastic lesions included discrete hyperplastic alveolar nodules (HANs), multifocal and diffuse alveolar hyperplasias, adenomatous hyperplasias, and papillary ductal hyperplasias (Figures 1 – 4). Alveolar-type hyperplasias were most common. These were packed with otherwise normal alveoli containing a single layer of luminal epithelial cells surrounded by a single layer of myoepithelial cells (Figure 3). Several alveolar hyperplasias displayed evidence of secretory activity with apical lipid vacuolization of the luminal cells, luminal eosinophilic concretions resembling residual (inspissated) milk, and enlarged (ectatic) ducts containing proteinaceous material and lipid droplets (Figures 2 and 3). Papillary lesions, on the other hand, contained multilayered mounds of cells within distended ducts (Figure 4). In addition, myoepithelial cells were not only present in their normal position between the luminal epithelial cells and basement membrane, but were also abnormally located within the ducts as a result of the inward growth and folding of the papillary projections (Figure 4c – e). Dysplastic lesions also showed multiple cell layering, but with attenuated myoepithelial cell staining in some areas.
Figure 1.
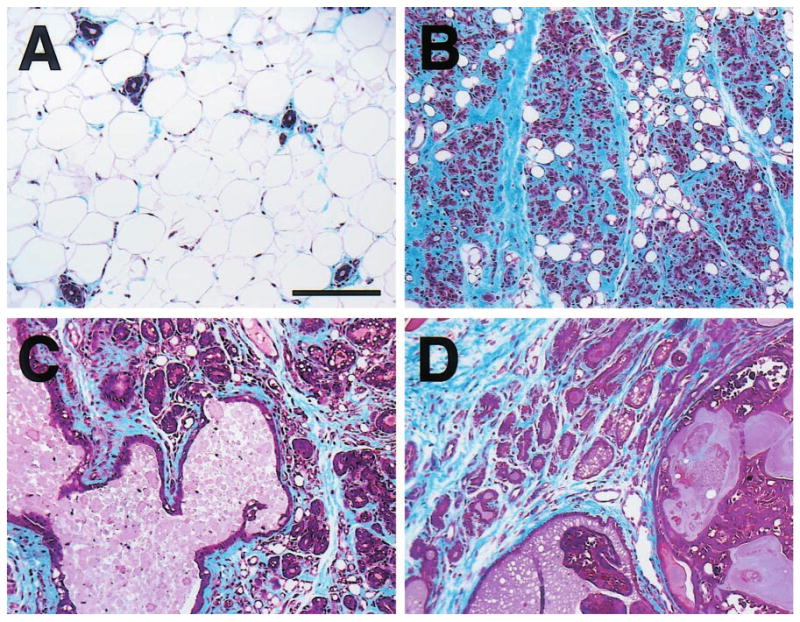
Masson's trichrome-stained mammary gland sections from (a) nontransgenic and (b – d) WAP-Str1 transgenic mice. The normal nontransgenic gland contains relatively few resting ducts surrounded by scant periductal collagen and embedded in an adipose stroma, whereas each transgenic gland exhibits extensive accumulation of blue-stained collagen (fibrosis) and few residual adipocytes. (b) This gland from a 7-month-old transgenic mouse contains numerous collapsed alveolar structures and extensive periglandular fibrosis. (c) A large dilated duct containing proteinaceous secretory material and hyperplastic alveolar epithelial cells with secretory vacuolization are apparent in this gland even though this 16-month-old transgenic mouse had never been pregnant. (d) This section from a 10-month-old transgenic mouse contains secretory hyperplastic epithelial cells and fibrosis adjacent to a secretory adenocarcinoma. Scale bar, 150 _μ_m
Figure 2.
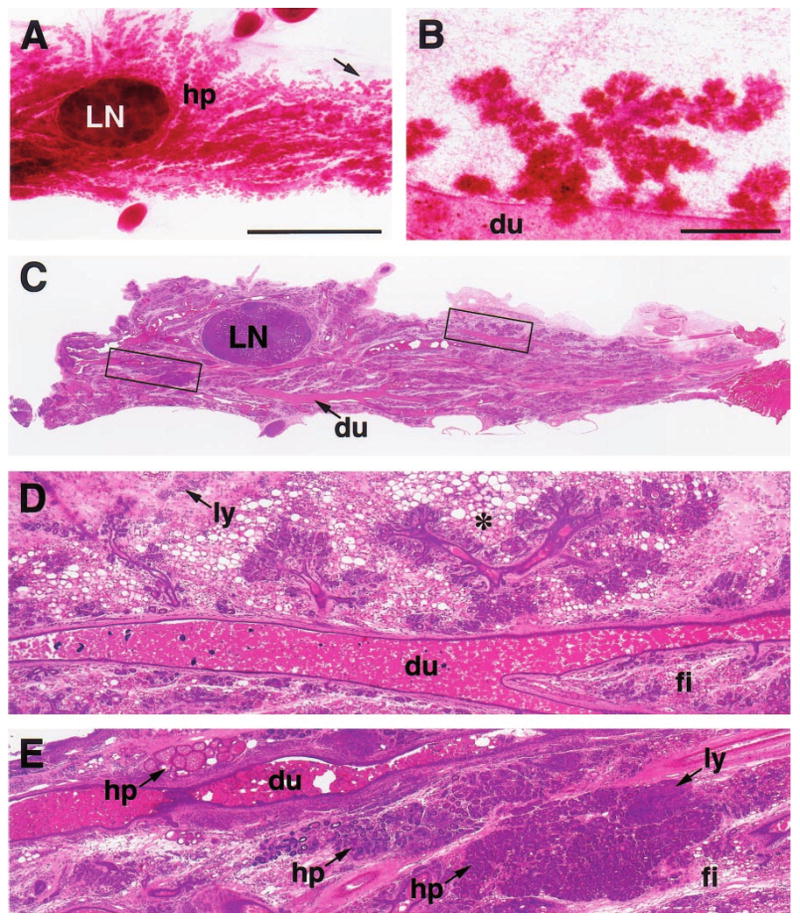
Carmine-stained wholemount (a,b) and H&E-stained paraffin section (c – e) of an abdominal (#4) mammary gland with diffuse hyperplasia (hp), fibrosis (fi) and lymphocytic infiltration (ly) from a 15-month-old parous WAP-Str1 mouse sacrificed 4 months after its pups were removed. The hyperplastic branches indicated by the arrow in (a) are outlined in (c) and are shown at higher magnification in (b and d). These sparse and disproportionately short secondary branches terminate in relatively well-developed lobuloalveolar structures and are surrounded by multilocular adipocytes (asterisk). The boxed area to the left of the central lymph node (LN) in c is enlarged in e and shows three hyperplastic areas, each with a distinct histologic appearance. Dilated (ectatic) primary ducts (du) containing considerable amounts of residual secretory material are also evident throughout the gland. Scale bars, 5 mm (a,c), 500 _μ_m (b,d,e)
Figure 4.
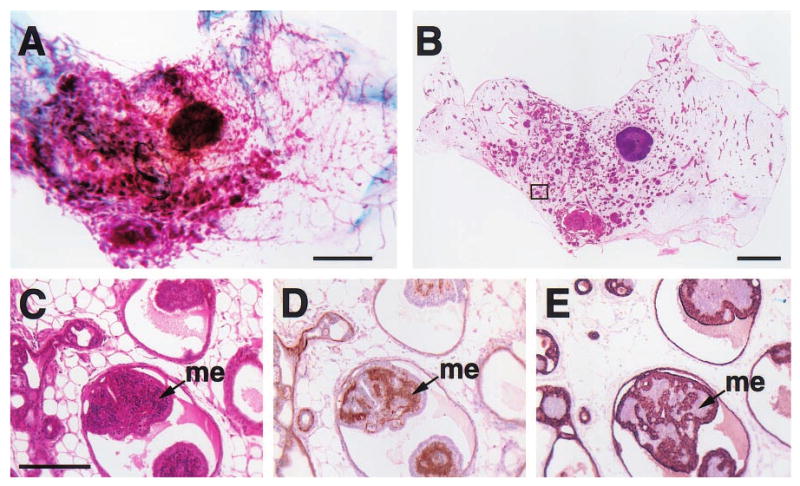
Histologic appearance of a florid papillary hyperplasia (intraductal papillomatosis) in the mammary gland of a 2-year-old virgin WAP-Str1 transgenic mouse as seen by wholemount (a), H&E (b,c), anti-smooth muscle actin (d) and anti-cytokeratin-8 (e) staining. The area outlined in (b) is shown at higher magnification in (c – e). The small, basophilic cells within the papillary projections (me) are smooth muscle actin-positive (d) and cytokeratin-8-negative (e), indicating the abnormal, internal presence of myoepithelial cells. Although small focal collections of lymphocytes were present (not shown), fibrosis was not observed and the far ends of the gland were essentially normal. Scale bars, 2 mm (a,b), 200 _μ_m (c – e)
Figure 3.
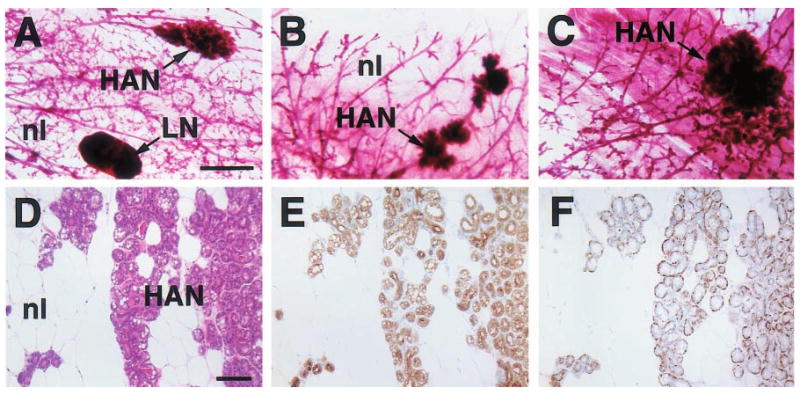
Histologic appearance of hyperplastic alveolar nodules (HANs) from 23- (a), 16- (b), 24- (c) and 12-month-old (d – f) virgin WAP-Str1 transgenic mice as seen by wholemount (a – c), H&E (d), anti-cytokeratin-8 immunoperoxidase (e) and anti-smooth muscle actin immunoperoxidase (f) staining. The multiple alveolar structures are composed of an internal layer of cytokeratin-8-positive luminal epithelial cells with lipid vacuolization (e) surrounded by a single smooth muscle actin-positive myoepithelial cell layer (f). Adjacent normal areas (nl) contain mostly adipocytes and sparse ducts with the same bilayered luminal and myoepithelial cell staining, but without secretory vacuoles. LN, lymph node. Scale bars, 2 mm (a – c), 100 _μ_m (d – f)
Twelve mammary carcinomas developed in the transgenic mice with only two arising before 1 year of age and an average tumor latency of 18.7 months. Hyperplastic or dysplastic lesions and a fibrotic (scirrhous or desmoplastic) stroma were consistently found adjacent to the malignant tumors (Figures 1 and 5). For the most part, however, the tumors were histologically and cytologically diverse. One large adenocarcinoma with adjacent papillary lesions contained unusual, internally located myoepithelial cell islands (Figure 5). Otherwise, myoepithelial cells were uniformly absent in the tumors. The mesenchymal intermediate filament marker vimentin was absent in the nine tumors that were well- or moderately well-differentiated, except, of course, in their surrounding and intervening stroma (Figure 5h). The three remaining undifferentiated tumors, however, each exhibited vimentin immunoreactivity in addition to epithelial cytokeratin staining. One of these tumors gave rise to multiple lung and kidney metastases and stained positive for both vimentin and cytokeratins (Figure 6a – c). It also gave rise to a tumor cell line (TCL-1) (Lochter et al., 1997b) that continued to express both intermediate filament markers in culture (Figure 6d – f) and formed highly metastatic spindle-cell tumors that remained cytokeratin- and vimentin-positive in nude mice (Figure 6g – i). The other undifferentiated tumors were carcinosarcomas (carcinomas with sarcomatous metaplasia) that contained distinct epithelial-like (carcinomatous) and mesenchymal-like (sarcomatous) cell populations (Figure 6j – l). The fibroblast-like sarcomatous cells had malignant cytologic features, composed the majority of some parts of the tumor, and contained similar genomic changes to those seen in the carcinomatous cells (Sternlicht et al., 1999), thus indicating that they did not merely represent a stromal response to the malignant epithelial-like cells. Furthermore, both cell populations persisted after serial transplantation to immunocompromised mice. Thus, even though carcinosarcomas are extremely rare in humans and in mice, one-sixth of the tumors in WAP-Str1 mice were of this type, and one-quarter of all tumors exhibited some degree of epithelial-to-mesenchmal phenotypic conversion. This incidence is intriguing in light of recent data indicating that phenotypically normal mammary epithelial cells undergo epithelial-to-mesenchymal conversion in response to Str1 in culture and in vivo (Lochter et al., 1997a; Sternlicht et al., 1999). This phenomenon has been associated with more aggressive malignant behavior (Birchmeier et al., 1996; Gilles and Thompson, 1996), and careful examination reveals that a large percentage of human tumors, and perhaps all poorly differentiated tumors, exhibit some degree of epithelial-to-mesenchymal conversion (Oft et al., 1998). Because most MMPs are stromal (mesenchymal) cell products, cancer cells begin to secrete their own MMPs only when they undergo such an epithelial-to-mesenchymal phenotypic transition (Ahmad et al., 1998; Martorana et al., 1998; Wright et al., 1994). Thus, Str1 may represent both a cause and a consequence of epithelial-to-mesenchymal conversion.
Figure 5.
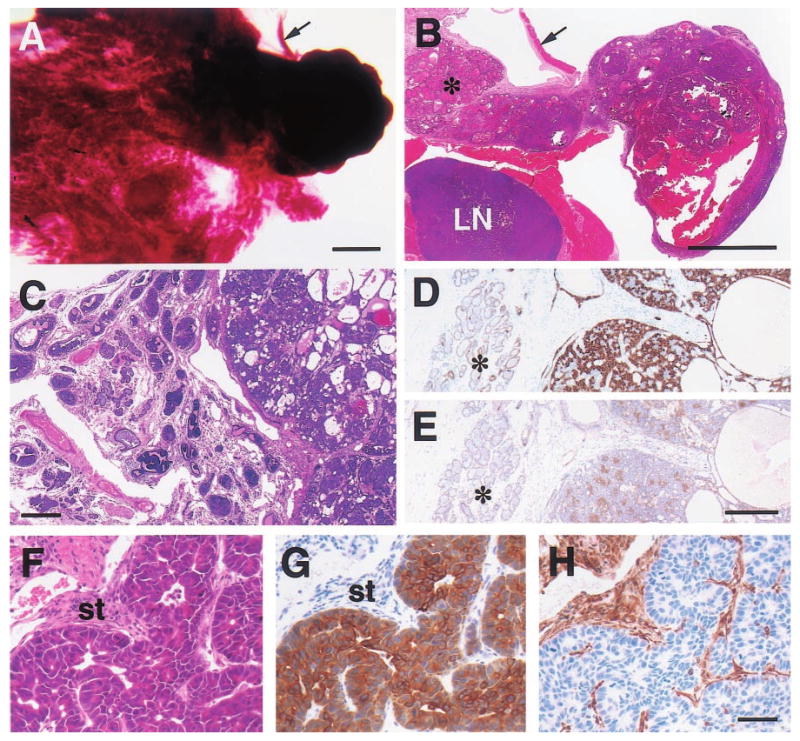
Histologic appearance of moderately well-differentiated mammary adenocarcinomas from 15- (a,b), 19- (c – e) and 23-month-old (f – h) WAP-Str1 transgenic mice as seen after wholemount (a), H&E (b,c,f), anti-cytokeratin-8 (d,g), anti-smooth muscle actin (e) and anti-vimentin (h) staining. (a,b) The tumor at right contains cystic spaces and necrotic debris, and sits adjacent to a diffuse lactational-like hyperplasia (asterisk) and a lymph node (LN) that is surrounded by muscle. The strip of connective tissue and skeletal muscle (arrow) is indicated for orientation. (c – e) This complex tumor (at right in each panel) contains numerous cystic spaces and tumor cell nests composed of mixed small and large cell populations. The small cells are smooth muscle actin-positive (e) and have small nuclei with a dense chromatin structure. The larger cells are cytokeratin-8 positive (d), have larger nuclei with a more open chromatin structure, and exhibit distinct intercellular bridges. The tumor is surrounded by a fibrotic stroma, atypical papillary lesions (c) and areas of secretory hyperplasia (asterisk). (f – h) These serial sections show a papillary adenocarcinoma with large cytokeratin-positive tumor cells, numerous mitotic figures, and an abundant vimentin-positive stroma (st). Scale bars, 2 mm (a,b), 300 _μ_m (c), 200 _μ_m (d,e), 50 _μ_m (f – h)
Figure 6.
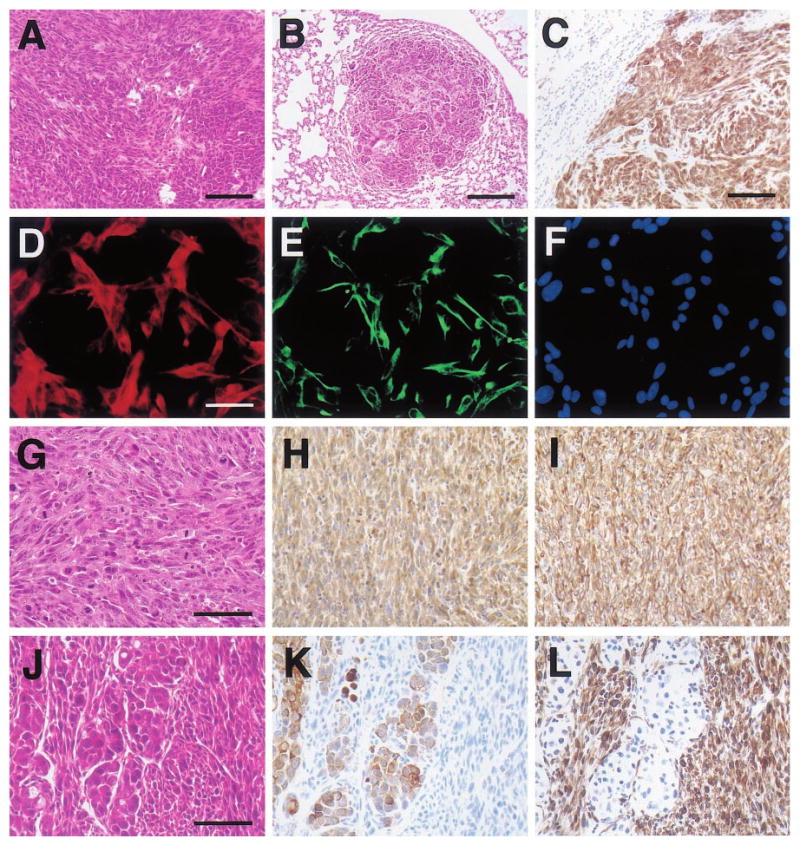
Histologic appearance of poorly-differentiated mammary tumors and a tumor-derived cell line from WAP-Str1 transgenic mice. (a) This H&E-stained area of primary mammary cancer from a 16-month-old virgin mouse contains spindle-shaped cells (left) and polygonal, epithelial-like tumor cells at lower right. (b,c) These two lung metastases from the tumor in (a) also contain spindled and polygonal cancer cells and stain positive for both cytokeratins (c) and vimentin (not shown). (d – f) The tumor cell line (TCL-1) established from the tumor in (a) exhibits a spindle-cell morphology and cytokeratin (d) and vimentin (e) immunoreactivity by dual immunocytochemistry and DAPI counterstaining (f). (g – i) TCL-1-derived tumors in immunocompromised mice show a pure spindle-cell morphology and numerous mitotic figures by H&E (g) and continue to stain positive for both cytokeratins (h) and vimentin (i). (j – l) Serial sections of this carcinosarcoma from a 17-month-old virgin transgenic mouse reveal a mixed cellular morphology by H&E (j) with polygonal carcinomatous cells that are cytokeratin-positive (k) and spindle-shaped sarcomatous cells that are vimentin-positive (l). Scale bars 100 _μ_m (a,c), 200 _μ_m (b), 50 _μ_m (d – i), 75 _μ_m (j – l)
TIMP-1 inhibits mammary neoplasia in Str1 transgenic mice
If the proteolytic activity of Str1 is responsible for the development of premalignant and malignant neoplasms in Str1 transgenic mice, then these changes should be quenched by overexpression of its natural inhibitor, TIMP-1. We previously showed that mating the Str1 transgenic mice with mice that overexpress a human TIMP1 transgene driven by the constitutive _β_-actin gene promoter abolishes the ECM degradation and unscheduled apoptosis otherwise seen in young pregnant Str1 transgenic mice (Alexander et al., 1996). To test the ability of TIMP-1 to counter the long-term effects of Str1 in the mammary gland, WAP-Str1 transgenic mice were crossed with mice that expressed the human TIMP1 transgene under the control of the same WAP promoter. Using mammary hyperplasia as a surrogate end-point, 73% of 10 – 16-month-old offspring carrying the Str1 transgene alone, but only 19% of age-matched bitransgenic mice carrying both the Str1 and TIMP1 transgenes developed hyperplasia (P<0.02, two-tailed Fisher's exact test). The double-transgenic mice also had a significantly lower incidence of hyperplasia than the whole cohort of WAP-Str1 mice included in Table 1 (P<0.0006). The few hyperplasias that did develop in the bitransgenic mice were considerably milder than those observed in the single-transgenic littermates with WAP-Str1 alone. Parity did not significantly alter the incidence or severity of mammary hyperplasia in the single or double-transgenic mice. Mammary hyperplasias were also uniformly absent in those littermates with only the TIMP1 transgene and in nontransgenic littermates. Thus it is the enzymatic activity of Str1 that is required to promote mammary neoplasia.
Other MMPs can promote carcinogenesis
Several recent observations support a role for MMPs early in cancer development. For example, mice that express a human collagenase-1 (MMP-1) transgene in squamous epithelium develop hyperproliferative skin lesions, and although they fail to form tumors spontaneously, they are more sensitive to chemical carcinogens than their non-transgenic littermates (D'Armiento et al., 1995). Conversely, mice that lack stromelysin-3 (MMP-11) form fewer and smaller DMBA-induced tumors than wild-type mice (Masson et al., 1998). Moreover, wild-type fibroblasts foster the tumorigenicity of human MCF-7 breast cancer cells in nude mice, whereas fibroblasts without MMP-11 do not (Masson et al., 1998). Because ECM-associated growth factors are also required for MCF-7 tumorigenicity, the authors propose that MMP-11 may promote tumor formation by causing the release or activation of sequestered growth factors. The lack of matrilysin (MMP-7) in mice carrying the Apcmin mutation hinders the development of benign intestinal adenomas (Wilson et al., 1997), and its overexpression in mammary tissue accelerates mammary tumor formation in mice carrying an MMTV promoter-driven ErbB-2/neu transgene (Rudolph-Owen et al., 1998). In addition, MMTV-MMP-7 transgenic mice develop premalignant hyperplastic alveolar nodules (HANs) even in the absence of MMTV-neu, whereas their non-transgenic littermates do not (Rudolph-Owen et al., 1998). The lack of either Str1 or gelatinase B (MMP-9) inhibits the development of human papilloma virus-16-induced squamous cell carcinomas in transgenic mice (LM Coussens, D Hanahan and Z Werb, unpublished observations). Furthermore, those tumors that develop despite the lack of MMP-9 tend to be more aggressive than usual, suggesting that MMP-9-deficiency provides pressure for the selection of less differentiated cancers that are better able to overcome the absence of MMP-9 (unpublished observations). Other MMPs that are highly expressed in malignant disease, such as MMP-19 (Grant et al., 1999), may also influence cancer progression, but remain essentially unexplored. (Because the GenBank sequences submitted as human MMP-18 and 19 are identical but substantially different from the Xenopus MMP-18 sequence, they are designated as MMP-19).
Although the above studies support a role for MMPs in early tumor progression and indicate that MMPs may increase neoplastic risk, they still required pre-existing oncogene or suppressor gene mutations or the administration of chemical carcinogens to achieve tumorigenesis. Here, however, we have described the spontaneous development of Str1 transgene-induced lesions in the absence of such mutations or mutagens. These changes, which failed to occur in non-transgenic controls, were also quenched by coexpression of a TIMP-1 transgene. Thus their spontaneous development lends even greater support to the likelihood that MMPs profoundly influence early tumor initiation and development.
In addition to MMPs, closely related metalloproteinases, such as the membrane-anchored ADAM (a disintegrin and metalloproteinase domain) and the secreted ADAMTS (thrombospondin domain-containing) proteins, are likely to influence cancer progression (Vazquez et al., 1999; Werb and Yan, 1998). For example, tumor necrosis factor-α converting enzyme (TACE, ADAM-17) can clearly influence cancer progression. Recent data also suggest that an unidentified metalloproteinase causes Fas ligand to be shed from cells, thus enabling them to avoid Fas-mediated apoptosis (Mitsiades et al., 1999). In addition, a unique metalloproteinase that is inhibited by TIMP-1 but not TIMP-2 causes cleavage and shedding of the extracellular domain of the ErbB2/neu growth factor receptor (Codony-Servat et al., 1999). Such shedding, which is often observed in breast cancer patients, may have oncogenic consequences and may limit the efficacy of anti-_ErbB2_-directed therapy. Other ADAM and ADAMTS domains may also influence cancer progression. For example, the cysteine-rich domain of meltrin-α (ADAM-12) can support tumor cell adhesion (Iba et al., 1999). Some members of these multi-gene families may even play conflicting roles in cancer due to the presence of domains with distinct biologic activities. For example, the potent anti-angiogenic activity of some ADAMTS (metallospondin) family members (Vazquez et al., 1999) may exert tumor suppressive effects, while other domains may promote tumor progression.
TIMPs may promote and suppress carcinogenesis by distinct mechanisms
If, in fact, MMPs promote carcinogenesis, then their endogenous inhibitors, the TIMPs, should defy cancer development. However, whereas some studies do suggest that TIMPs suppress tumor development, others do not. In support of a tumor suppressive role, antisense depletion of TIMP-1 renders murine 3T3 cells tumorigenic in vivo (Khokha et al., 1989). In addition, the transformation-promoting activity of the prototypic tumor promoter 12-_O_-tetradecanoylphorbol-13-acetate (TPA) is inhibited by TIMPs 1 and 2 in culture (Shoji et al., 1997). Thus, the well-known ability of TPA to promote tumors in vivo may be partly due to its ability to upregulate MMP gene expression (Gack et al., 1994; Reichardt et al., 1998). On the other hand, TPA also upregulates TIMP-1 gene expression (Logan et al., 1996; Lu et al., 1991). In double transgenic mice, TIMP-1 overexpression inhibits simian virus 40 T antigen-induced hepatocellular carcinogenesis by inhibiting hepatocyte proliferation and angiogenesis (Martin et al., 1996, 1999). TIMP-1 overexpression also inhibits the tumorigenicity of melanoma and lymphoma cells (Khokha, 1994; Krüger et al., 1997). However, in an experimental metastasis assay, certain tumor cell lines were better able to grow in the presence of tumor-associated TIMP-1, suggesting that it may protect ECM or cell surface molecules that are critical for cell viability (Soloway et al., 1996). TIMP-1 overexpression also appears to promote intestinal adenoma formation in Min mice, yet a synthetic MMP inhibitor decreases tumor multiplicity in this same model (Heppner Goss et al., 1998). This discrepancy may reflect the growth-promoting activity of TIMP-1, a function that point-mutation studies indicate is independent of its MMP-inhibitory activity (Chesler et al., 1995). Indeed, TIMP-1 was initially cloned as ‘EPA’ by virtue of its erythroid-potentiating activity (Docherty et al., 1985) and has been shown to act as a mitogen for other cell types (Bertaux et al., 1991). Thus it is not entirely counterintuitive that TIMP-1 is often upregulated in human cancers (Kossakowska et al., 1996; Lindsay et al., 1997; Yoshiji et al., 1996) and that such upregulation is predictive for metastatic progression and a poor prognosis (Jung et al., 1997; McCarthy et al., 1999; Mimori et al., 1997; Ree et al., 1997; Zeng et al., 1995). Although TIMP-1 upregulation may simply be a consequence of the increased matrix remodeling that occurs during invasion, and would certainly hinder the pro-oncogenic and pro-invasive effects of MMPs, emerging evidence indicates that TIMP upregulation could also benefit tumors. In addition to its growth-stimulatory activity, recent studies indicate that TIMP-1 can upregulate vascular endothelial growth factor expression (Yoshiji et al., 1998), that it can exert anti-apoptotic activity (Guedez et al., 1998a,b), and that it may even be internalized by cells and translocated to the nucleus (Ritter et al., 1999).
Like TIMP-1, TIMP-2 promotes cell growth in culture (Hayakawa et al., 1994; Nemeth et al., 1996; Stetler-Stevenson et al., 1992) and appears to inhibit tumor growth in vivo (Imren et al., 1996). Although some studies indicate that TIMP-2 expression tends to be similar in tumors and matched normal tissues (Stetler-Stevenson et al., 1990), others have found a significant correlation between TIMP-2 expression and the development of distant metastases (Ree et al., 1997). Unlike TIMP-1, TIMP-2 expression is down-regulated by TGF-_β_1 and is unaffected by serum and phorbol esters, each of which increase TIMP-1 expression (Leco et al., 1992).
The role of TIMP-3 in cancer is also unclear. Some studies indicate that TIMP-3 is upregulated in human tumors (Uría et al., 1994) and may provide an early marker for malignant disease (SP Hawkes, personal communication). Others, however, indicate that the TIMP-3 gene promoter is epigenetically downregulated during cancer development (Bachman et al., 1999). Like TIMP-1, TIMP-3 is induced during cell transformation in culture (Lu et al., 1991; Staskus et al., 1991). TIMP-3 is also transiently induced by hepatocyte growth factor (HGF) (Castagnino et al., 1998), which, in turn, has been implicated in mesenchymal-to-epithelial cellular conversion (Tsarfaty et al., 1994). Interestingly, ectopic overexpression of TIMP-3 can also induce mesenchymal-to-epithelial conversion and loss of malignant characteristics in cultured sarcoma cells, and its antisense depletion has the opposite effect, suggesting that it may be a mediator of HGF activity (Castagnino et al., 1998). In addition, TIMP-3 is the only known endogenous inhibitor of TACE (Amour et al., 1998). Thus, it may also influence cancer development by inhibiting TACE and other relevant ADAM and ADAMTS family members. Alternatively, some ADAM and ADAMTS proteins may be inhibited by other TIMPs. For example, aggrecanase-1 (ADAMTS-4) is inhibited by TIMP-1 (Tortorella et al., 1999). Finally, the most recently discovered TIMP, TIMP-4, has been shown to inhibit mammary tumor growth and may be downregulated in human breast cancers (Wang et al., 1997). Thus it appears that the issue of whether TIMPs defy or exacerbate the effects of cancer-causing agents and mutations is confounded by their multiple and independent functions. Ultimately, the TIMPs may both defy carcinogenicity through their metalloproteinase-inhibitory activity and promote it through their capacity to affect cellular behavior in a metalloproteinase-independent manner.
How MMPs might promote tumor development
Although MMPs are not oncogenic or mutagenic per se, there are several routes whereby they can alter signaling and thus affect the process of neoplastic transformation. By degrading extracellular matrices, MMPs alter cell-matrix interactions and cause the release of bioactive ECM fragments (Lukashev and Werb, 1998). MMPs can also cleave a growing list of cell surface molecules, including the tumor suppressor E-cadherin (Sternlicht and Werb, 1999). They can release active growth factors, angiogenic factors and angiogenic inhibitors from the cell surface and ECM (Patterson and Sang, 1997; Suzuki et al., 1997). They can cleave growth factor binding proteins (Fowlkes et al., 1994) and cell surface growth factor receptors (Levi et al., 1996). They can generate an α1-antitrypsin cleavage product that assists tumor growth and invasion, possibly by modulating NK cell cytotoxicity (Kataoka et al., 1999). They can foster the recruitment of various host cells by altering the stromal environment (Thomasset et al., 1998; Werb, 1997), and they may alter cell cycle checkpoint controls and promote genomic instability by affecting cell adhesion (Tlsty, 1998). MMPs can also induce programmed cell death in anchorage-dependent cells (Alexander et al., 1996; Thomasset et al., 1998), which could either defy tumor progression or exert pressure for the selection of anchorage-independent and apoptosis-resistant subpopulations, and thus promote progression. Therefore, MMPs may contribute in multiple ways to all stages of cancer progression, including initiation.
The evolution of epithelial cancers is also profoundly reliant on the stromal cells that help make up the tumor mass (Rønnov-Jessen et al., 1996). In addition, an altered stromal environment may actually promote neoplastic transformation (Jacobs et al., 1999; Jacoby et al., 1997; Kinzler and Vogelstein, 1998; Willenbucher et al., 1999). Indeed, stromal changes appeared to presage malignant epithelial changes in the WAP-Str1 transgenic mice (Thomasset et al., 1998). Thus, because Str1 can alter the extracellular environment and is itself a stromal product, it may be partly responsible for the tumorigenic effects of an altered stroma.
One of the more appealing prospective mechanisms that might be responsible for the tumor promoting capacity of Str1 is that it may alter E-cadherin/_β_-catenin signaling (Figure 7). According to this putative scenario, cleavage of E-cadherin by Str1 or another MMP may increase the cytosolic levels of its intracellular partner, _β_-catenin. Cytosolic _β_-catenin, in turn, can be phosphorylated and degraded, or translocated into the nucleus where it then partners with TCF/LEF transcription factors in order to regulate the transcription of genes that contain functional LEF recognition sequences within their promoters (Tlsty, 1998). In support of this mechanism, Str1-induced epithelial-to-mesenchymal conversion is accompanied by E-cadherin cleavage and a rapid redistribution of _β_-catenin away from cell-cell contacts towards a more cytoplasmic and perinuclear location (Lochter et al., 1997a). Furthermore, Str1 induces an early and sustained upregulation of cyclin-D1 (ME Lukashev and Z Werb, unpublished results). This is consistent with the above mechanism, because cyclin-D1 is regulated by _β_-catenin (Tetsu and McCormick, 1999) and can exert oncogenic effects in the mammary gland (Wang et al., 1994). The c-myc proto-oncogene is also regulated by _β_-catenin/LEF transactivation in colon cancer cells (He et al., 1998), however significant changes in c-myc expression were not observed by us during Str1-induced phenotypic conversion in mammary epithelial cells (unpublished results). Finally, matrilysin (MMP-7) gene expression is also regulated by _β_-catenin/LEF transactivation (Crawford et al., 1999), and this same pathway may also account for our observation that a number of other MMPs are upregulated during Str1-induced epithelial-to-mesenchymal conversion (Lochter et al., 1997a).
Figure 7.
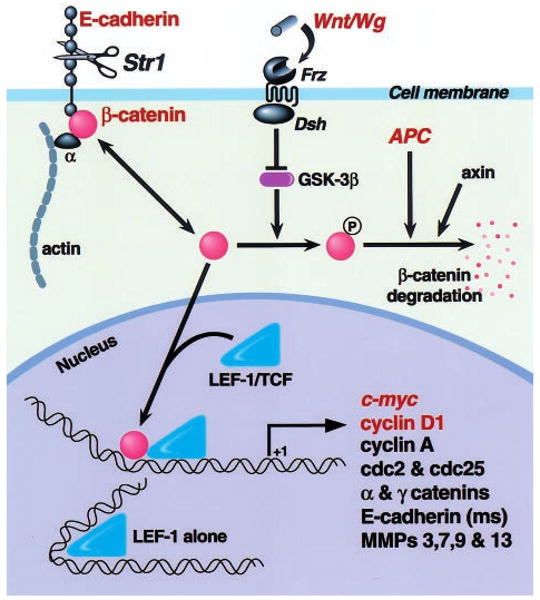
A hypothetical model of how Str1 may affect cellular behavior via the β_-catenin/LEF signal transduction pathway. Following E-cadherin cleavage by Str1 or another metalloproteinase, free cytosolic β_-catenin pools are increased. In the absence of Wnt signaling, glycogen synthase kinase 3_β (GST-3_β) phosphorylates _β_-catenin, thus targeting it for association with the adenomatous polyposis coli (APC) gene product and axin, ubiquitination, and proteosomal degradation. Alternatively, unphosphorylated _β_-catenin enters the nucleus where it interacts with LEF/TCF transcription factors, thus regulating the transcription of genes containing functional LEF recognition sites. A number of potential target genes are shown, although only c-myc, cyclin-D1 and matrilysin (MMP-7) have been so far shown to respond to _β_-catenin/LEF transactivation. Molecules indicated in red have been shown to play a causal role in cancer development. Frz, Frizzled family Wnt/Wg receptor; Dsh, Disheveled family or other GSK inhibitor; Wg, Wingless; ms, mouse
The ability of MMPs to release growth factors from the cell surface and ECM is also likely to play a critical role in cancer development. Some of these growth factors may influence tumor cells directly, while others may influence neighboring cells, such as endothelial cells, that are required to support tumor growth. Indeed, there is a growing awareness that MMPs promote tumor angiogenesis. In a transgenic model of pancreatic islet cell carcinogenesis, broad-range MMP inhibition suppresses the ‘angiogenic switch’ that occurs during premalignant cancer progression and slows tumor growth during later stages of progression (Bergers et al., 1999). Gelatinase B is probably an important target of such inhibition, in light of its association with premalignant angiogenesis (Coussens et al., 1999) and its critical role in angiogenesis during bone development (Vu et al., 1998). On the other hand, some MMPs, such as metalloelastase (MMP-12), matrilysin and gelatinase B, can cleave plasminogen to generate the angiogenesis inhibitor angiostatin (Patterson and Sang, 1997).
MMPs could also conceivably promote genomic instability by affecting adhesion-dependent cell cycle checkpoint controls (Tlsty, 1998). Interestingly, statistically nonrandom genomic changes were observed by comparative genomic hybridization in both premalignant and malignant mammary gland lesions in WAP-Str1 transgenic mice (Sternlicht et al., 1999). The most prevalent change was a deletion in the mid-distal region of mouse chromosome 4 that was present in 70% of the examined WAP-Str1 mammary lesions. This is consistent with the high incidence of chromosome 4 losses seen in two other models of mouse mammary cancer (Donehower et al., 1995; Ritland et al., 1997). In addition, a more recent study indicates that, in one of these models, the highest incidence of loss of heterozygosity occurs in the same mid-distal region of chromosome 4, thus further implicating this region as a putative tumor suppressor locus (Cool and Jolicoeur, 1999). Our own data are also consistent with the possibility that Str1 promotes the accumulation of genetic mutations or the selection and clonal expansion of mutant cells.
MMPs clearly do more than just degrade extracellular matrices, and such matrices are not just passive structures. MMPs can influence cell-matrix, cell-cell and paracrine signals that, in turn, control such basic processes as cellular growth, differentiation, morphogenesis, migration and death. Thus, the importance of MMPs in normal physiologic processes and in pathologic processes other than cancer may also partly stem from their ability to alter cellular signals. Moreover, the role of MMPs in normal physiologic processes and the potential for untoward effects must be considered when designing and undertaking clinical interventions that target the MMPs. A better understanding of the molecular mechanisms responsible for their expanding role in cancer can only benefit the development of more effective therapeutics and therapeutic stratagies.
Acknowledgments
We thank R Boudreau, J Xie, DR Williams, and Drs CJ Sympson, A McMillan and RD Cardiff for their assistance. This work was supported by grants from the National Cancer Institute (CA57621 and CA72006), the US Army Medical Research and Materiel Command (DAMD17-97-1-7246), and the US Department of Energy, Office of Health and Environmental Research (DE-AC03-76-SF00098).
References
- Ahmad A, Hanby A, Dublin E, Poulsom R, Smith P, Barnes D, Rubens R, Anglard P, Hart I. Am J Pathol. 1998;152:721–728. [PMC free article] [PubMed] [Google Scholar]
- Alexander CM, Howard EW, Bissell MJ, Werb Z. J Cell Biol. 1996;135:1669–1677. doi: 10.1083/jcb.135.6.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amour A, Slocombe PM, Webster A, Butler M, Knight CG, Smith BJ, Stephens PE, Shelley C, Hutton M, Knauper V, Docherty AJ, Murphy G. FEBS Lett. 1998;435:39–44. doi: 10.1016/s0014-5793(98)01031-x. [DOI] [PubMed] [Google Scholar]
- Bachman KE, Herman JG, Corn PG, Merlo A, Costello JF, Cavenee WK, Baylin SB, Graff JR. Cancer Res. 1999;59:798–802. [PubMed] [Google Scholar]
- Bergers G, Javaherian K, Lo KM, Folkman J, Hanahan D. Science. 1999;284:808–812. doi: 10.1126/science.284.5415.808. [DOI] [PubMed] [Google Scholar]
- Bertaux B, Hornebeck W, Eisen AZ, Dubertret L. J Invest Dermatol. 1991;97:679–685. doi: 10.1111/1523-1747.ep12483956. [DOI] [PubMed] [Google Scholar]
- Birchmeier C, Birchmeier W, Brand-Saberi B. Acta Anat (Basel) 1996;156:217–226. doi: 10.1159/000147848. [DOI] [PubMed] [Google Scholar]
- Borsi L, Carnemolla B, Nicolò G, Spina B, Tanara G, Zardi L. Int J Cancer. 1992;52:688–692. doi: 10.1002/ijc.2910520504. [DOI] [PubMed] [Google Scholar]
- Boudreau N, Sympson CJ, Werb Z, Bissell MJ. Science. 1995;267:891–893. doi: 10.1126/science.7531366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castagnino P, Soriano JV, Montesano R, Bottaro DP. Oncogene. 1998;17:481–492. doi: 10.1038/sj.onc.1201957. [DOI] [PubMed] [Google Scholar]
- Chesler L, Golde DW, Bersch N, Johnson MD. Blood. 1995;86:4506–4515. [PubMed] [Google Scholar]
- Christofori G, Semb H. Trends Biochem Sci. 1999;24:73–76. doi: 10.1016/s0968-0004(98)01343-7. [DOI] [PubMed] [Google Scholar]
- Codony-Servat J, Albanell J, Lopez-Talavera JC, Arribas J, Baselga J. Cancer Res. 1999;59:1196–1201. [PubMed] [Google Scholar]
- Cool M, Jolicoeur P. Cancer Res. 1999;59:2438–2444. [PubMed] [Google Scholar]
- Coussens LM, Raymond WW, Bergers G, Laig-Webster M, Behrendtsen O, Werb Z, Caughey GH, Hanahan D. Genes Dev. 1999;13:1382–1397. doi: 10.1101/gad.13.11.1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens LM, Werb Z. Chem Biol. 1996;3:895–904. doi: 10.1016/s1074-5521(96)90178-7. [DOI] [PubMed] [Google Scholar]
- Crawford HC, Fingleton BM, Rudolph-Owen LA, Goss KJH, Rubinfeld B, Polakis P, Matrisian LM. Oncogene. 1999;18:2883–2891. doi: 10.1038/sj.onc.1202627. [DOI] [PubMed] [Google Scholar]
- D'Armiento J, DiColandrea T, Dalal SS, Okada Y, Huang MT, Conney AH, Chada K. Mol Cell Biol. 1995;15:5732–5739. doi: 10.1128/mcb.15.10.5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docherty AJP, Lyons A, Smith BJ, Wright EM, Stephens PE, Harris TJR, Murphy G, Reynolds JJ. Nature. 1985;318:66–69. doi: 10.1038/318066a0. [DOI] [PubMed] [Google Scholar]
- Donehower LA, Godley LA, Aldaz CM, Pyle R, Shi YP, Pinkel D, Gray J, Bradley A, Medina D, Varmus HE. Genes Dev. 1995;9:882–895. doi: 10.1101/gad.9.7.882. [DOI] [PubMed] [Google Scholar]
- Fowlkes JL, Enghild JJ, Suzuki K, Nagase H. J Biol Chem. 1994;269:25742–25746. [PubMed] [Google Scholar]
- Gack S, Vallon R, Schaper J, Ruther U, Angel P. J Biol Chem. 1994;269:10363–10369. [PubMed] [Google Scholar]
- Gilles C, Thompson EW. Breast J. 1996;2:83–96. [Google Scholar]
- Grant GM, Giambernardi TA, Grant AM, Klebe RJ. Matrix Biol. 1999;18:145–148. doi: 10.1016/s0945-053x(99)00003-7. [DOI] [PubMed] [Google Scholar]
- Guedez L, Courtemanch L, Stetler-Stevenson M. Blood. 1998a;92:1342–1349. [PubMed] [Google Scholar]
- Guedez L, Stetler-Stevenson WG, Wolff L, Wang J, Fukushima P, Mansoor A, Stetler-Stevenson M. J Clin Invest. 1998b;102:2002–2010. doi: 10.1172/JCI2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa T, Yamashita K, Ohuchi E, Shinagawa A. J Cell Science. 1994;107:2373–2379. doi: 10.1242/jcs.107.9.2373. [DOI] [PubMed] [Google Scholar]
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- Heppner Goss KJ, Brown D, Matrisian LM. Int J Cancer. 1998;78:629–635. doi: 10.1002/(sici)1097-0215(19981123)78:5<629::aid-ijc17>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Iba K, Albrechtsen R, Gilpin BJ, Loechel F, Wewer UM. Am J Pathol. 1999;154:1489–1501. doi: 10.1016/s0002-9440(10)65403-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imren S, Kohn DB, Shimada H, Blavier L, DeClerck YA. Cancer Res. 1996;56:2891–2895. [PubMed] [Google Scholar]
- Jacobs TW, Byrne C, Colditz G, Connolly JL, Schnitt SJ. N Engl J Med. 1999;340:430–436. doi: 10.1056/NEJM199902113400604. [DOI] [PubMed] [Google Scholar]
- Jacoby RF, Schlack S, Cole CE, Skarbek M, Harris C, Meisner LF. Gastroenterology. 1997;112:1398–1403. doi: 10.1016/s0016-5085(97)70156-2. [DOI] [PubMed] [Google Scholar]
- Jung K, Nowak L, Lein M, Priem F, Schnorr D, Loening SA. Int J Cancer. 1997;74:220–223. doi: 10.1002/(sici)1097-0215(19970422)74:2<220::aid-ijc14>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Kataoka H, Uchino H, Iwamura T, Seiki M, Nabeshima K, Koono M. Am J Pathol. 1999;154:457–468. doi: 10.1016/s0002-9440(10)65292-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khokha R. J Natl Cancer Inst. 1994;86:299–304. doi: 10.1093/jnci/86.4.299. [DOI] [PubMed] [Google Scholar]
- Khokha R, Waterhouse P, Yagel S, Lala PK, Overall CM, Norton G, Denhardt DT. Science. 1989;243:947–950. doi: 10.1126/science.2465572. [DOI] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B. Science. 1998;280:1036–1037. doi: 10.1126/science.280.5366.1036. [DOI] [PubMed] [Google Scholar]
- Kossakowska AE, Huchcroft SA, Urbanski SJ, Edwards DR. Br J Cancer. 1996;73:1401–1408. doi: 10.1038/bjc.1996.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger A, Fata JE, Khokha R. Blood. 1997;90:1993–2000. [PubMed] [Google Scholar]
- Leco KJ, Hayden LJ, Sharma RR, Rocheleau H, Greenberg AH, Edwards DR. Gene. 1992;117:209–217. doi: 10.1016/0378-1119(92)90731-4. [DOI] [PubMed] [Google Scholar]
- Levi E, Fridman R, Miao HQ, Ma YS, Yayon A, Vlodavsky I. Proc Natl Acad Sci USA. 1996;93:7069–7074. doi: 10.1073/pnas.93.14.7069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay CK, Thorgeirsson UP, Tsuda H, Hirohashi S. Human Pathol. 1997;28:359–366. doi: 10.1016/s0046-8177(97)90136-2. [DOI] [PubMed] [Google Scholar]
- Lochter A, Galosy S, Muschler J, Freedman N, Werb Z, Bissell MJ. J Cell Biol. 1997a;139:1861–1872. doi: 10.1083/jcb.139.7.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochter A, Srebrow A, Sympson CJ, Terracio N, Werb Z, Bissell MJ. J Biol Chem. 1997b;272:5007–5015. doi: 10.1074/jbc.272.8.5007. [DOI] [PubMed] [Google Scholar]
- Logan SK, Garabedian MJ, Campbell CE, Werb Z. J Biol Chem. 1996;271:774–782. doi: 10.1074/jbc.271.2.774. [DOI] [PubMed] [Google Scholar]
- Lu XQ, Levy M, Weinstein IB, Santella RM. Cancer Res. 1991;51:6231–6235. [PubMed] [Google Scholar]
- Lukashev ME, Werb Z. Trends Cell Biol. 1998;8:437–441. doi: 10.1016/s0962-8924(98)01362-2. [DOI] [PubMed] [Google Scholar]
- Lund LR, Rømer J, Thomasset N, Solberg H, Pyke C, Bissell MJ, Danø K, Werb Z. Development. 1996;122:181–193. doi: 10.1242/dev.122.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DC, Rüther U, Sanchez-Sweatman OH, Orr FW, Khokha R. Oncogene. 1996;13:569–576. [PubMed] [Google Scholar]
- Martin DC, Sanchez-Sweatman OH, Ho AT, Inderdeo DS, Tsao MS, Khokha R. Lab Invest. 1999;79:225–234. [PubMed] [Google Scholar]
- Martorana AM, Zheng G, Crowe TC, O'Grady RL, Lyons JG. Cancer Res. 1998;58:4970–4979. [PubMed] [Google Scholar]
- Masson R, Lefebvre O, Noël A, Fahime ME, Chenard MP, Wendling C, Kebers F, LeMeur M, Dierich A, Foidart JM, Basset P, Rio MC. J Cell Biol. 1998;140:1535–1541. doi: 10.1083/jcb.140.6.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matrisian LM, Glaichenhaus N, Gesnel MC, Breathnach R. EMBO J. 1985;4:1435–1440. doi: 10.1002/j.1460-2075.1985.tb03799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy K, Maguire T, McGreal G, McDermott E, O'Higgins N, Duffy MJ. Int J Cancer. 1999;84:44–48. doi: 10.1002/(sici)1097-0215(19990219)84:1<44::aid-ijc9>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Mimori K, Mori M, Shiraishi T, Fujie T, Baba K, Haraguchi M, Abe R, Ueo H, Akiyoshi T. Br J Cancer. 1997;76:531–536. doi: 10.1038/bjc.1997.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsiades N, Poulaki V, Leone A, Tsokos M. Proc Am Assoc Cancer Res. 1999;40:722. [Google Scholar]
- Muller D, Quantin B, Gesnel MC, Millon-Collard R, Abecassis J, Breathnach R. Biochem J. 1988;253:187–192. doi: 10.1042/bj2530187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeth JA, Rafe A, Steiner M, Goolsby CL. Exp Cell Res. 1996;224:110–115. doi: 10.1006/excr.1996.0117. [DOI] [PubMed] [Google Scholar]
- Oft M, Heider KH, Beug H. Current Biol. 1998;8:1243–1252. doi: 10.1016/s0960-9822(07)00533-7. [DOI] [PubMed] [Google Scholar]
- Ostrowski LE, Finch J, Krieg P, Matrisian L, Patskan G, O'Connell JF, Phillips J, Slaga TJ, Breathnach R, Bowden GT. Mol Carcinog. 1988;1:13–19. doi: 10.1002/mc.2940010106. [DOI] [PubMed] [Google Scholar]
- Patterson BC, Sang QXA. J Biol Chem. 1997;272:28823–28825. doi: 10.1074/jbc.272.46.28823. [DOI] [PubMed] [Google Scholar]
- Ree AH, Florenes VA, Berg JP, Maelandsmo GM, Nesland JM, Fodstad O. Clin Cancer Res. 1997;3:1623–1628. [PubMed] [Google Scholar]
- Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, Wessely O, Bock R, Gass P, Schmid W, Herrlich P, Angel P, Schutz G. Cell. 1998;93:531–541. doi: 10.1016/s0092-8674(00)81183-6. [DOI] [PubMed] [Google Scholar]
- Ritland SR, Rowse GJ, Chang Y, Gendler SJ. Cancer Res. 1997;57:3520–3525. [PubMed] [Google Scholar]
- Ritter LM, Garfield SH, Thorgeirsson UP. Biochem Biophys Res Comm. 1999;257:494–499. doi: 10.1006/bbrc.1999.0408. [DOI] [PubMed] [Google Scholar]
- Rønnov-Jessen L, Petersen OW, Bissell MJ. Physiol Rev. 1996;76:69–125. doi: 10.1152/physrev.1996.76.1.69. [DOI] [PubMed] [Google Scholar]
- Rudolph-Owen LA, Chan R, Muller WJ, Matrisian LM. Cancer Res. 1998;58:5500–5506. [PubMed] [Google Scholar]
- Sanchez-Lopez R, Nicholson R, Gesnel MC, Matrisian LM, Breathnach R. J Biol Chem. 1988;263:11892–11899. [PubMed] [Google Scholar]
- Shoji A, Sakamoto Y, Tsuchiya T, Moriyama K, Kaneko T, Okubo T, Umeda M, Miyazaki K. Carcinogenesis. 1997;18:2093–2100. doi: 10.1093/carcin/18.11.2093. [DOI] [PubMed] [Google Scholar]
- Soloway PD, Alexander CM, Werb Z, Jaenisch R. Oncogene. 1996;13:2307–2314. [PubMed] [Google Scholar]
- Staskus PW, Masiarz FR, Pallanck LJ, Hawkes SP. J Biol Chem. 1991;266:449–454. [PubMed] [Google Scholar]
- Sternlicht MD, Lochter A, Sympson CJ, Huey B, Rougier JP, Gray JW, Pinkel D, Bissell MJ, Werb Z. Cell. 1999;98:137–146. doi: 10.1016/s0092-8674(00)81009-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternlicht MD, Werb Z. In: Guidebook to the Extracellular Matrix and Adhesion Proteins. Kreis T, Vale R, editors. Oxford University Press; Oxford: 1999. pp. 503–562. [Google Scholar]
- Stetler-Stevenson WG, Bersch N, Golde DW. FEBS Lett. 1992;296:231–234. doi: 10.1016/0014-5793(92)80386-u. [DOI] [PubMed] [Google Scholar]
- Stetler-Stevenson WG, Brown PD, Onisto M, Levy AT, Liotta LA. J Biol Chem. 1990;265:13933–13938. [PubMed] [Google Scholar]
- Suzuki M, Raab G, Moses MA, Fernandez CA, Klagsburn M. J Biol Chem. 1997;272:31730–31737. doi: 10.1074/jbc.272.50.31730. [DOI] [PubMed] [Google Scholar]
- Sympson CJ, Talhouk RS, Alexander CM, Chin JR, Clift SM, Bissell MJ, Werb Z. J Cell Biol. 1994;125:681–693. doi: 10.1083/jcb.125.3.681. erratum in J. Cell Biol., 132, 752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetsu O, McCormick F. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- Thomasset N, Lochter A, Sympson CJ, Lund LR, Williams DR, Behrendtsen O, Werb Z, Bissell MJ. Am J Pathol. 1998;153:457–467. doi: 10.1016/S0002-9440(10)65589-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tlsty TD. Curr Opin Cell Biol. 1998;10:647–653. doi: 10.1016/s0955-0674(98)80041-0. [DOI] [PubMed] [Google Scholar]
- Tortorella MD, Burn TC, Pratta MA, Abbaszade I, Hollis JM, Liu R, Rosenfeld SA, Copeland RA, Decicco CP, Wynn R, Rockwell A, Yang F, Duke JL, Solomon K, George H, Bruckner R, Nagase H, Itoh Y, Ellis DM, Ross H, Wiswall BH, Murphy K, Hillman MC, Hollis GF, Newton RC, Magolda RL, Trzaskos JM, Arner EC. Science. 1999;284:1664–1666. doi: 10.1126/science.284.5420.1664. [DOI] [PubMed] [Google Scholar]
- Tsarfaty I, Rong S, Resau JH, Rulong S, da Silva PP, Vande Woude GF. Science. 1994;263:98–101. doi: 10.1126/science.7505952. [DOI] [PubMed] [Google Scholar]
- Uría JA, Ferrando AA, Velasco G, Freije JM, López-Otín C. Cancer Res. 1994;54:2091–2094. [PubMed] [Google Scholar]
- Vallorosi CJ, Day KC, Zhao X, Day ML. Proc Am Assoc Cancer Res. 1999;40:721. [Google Scholar]
- Vazquez F, Hastings G, Ortega MA, Lane TF, Oikemus S, Lombardo M, Iruela-Arispa ML. J Biol Chem. 1999;274:23349–23357. doi: 10.1074/jbc.274.33.23349. [DOI] [PubMed] [Google Scholar]
- Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, Shapiro SD, Senior RM, Werb Z. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Liu YE, Greene J, Sheng S, Fuchs A, Rosen EM, Shi YE. Oncogene. 1997;14:2767–2774. doi: 10.1038/sj.onc.1201245. [DOI] [PubMed] [Google Scholar]
- Wang TC, Cardiff RD, Zuckerberg L, Lees E, Arnold A, Schmidt EV. Nature. 1994;369:669–671. doi: 10.1038/369669a0. [DOI] [PubMed] [Google Scholar]
- Werb Z. Cell. 1997;91:439–442. doi: 10.1016/s0092-8674(00)80429-8. [DOI] [PubMed] [Google Scholar]
- Werb Z, Yan Y. Science. 1998;282:1279–1280. doi: 10.1126/science.282.5392.1279. [DOI] [PubMed] [Google Scholar]
- Willenbucher RF, Aust DE, Chang CG, Zelman SJ, Ferrell LD, Moore DH, Waldman FM. Am J Pathol. 1999;154:1825–1830. doi: 10.1016/S0002-9440(10)65438-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CL, Heppner KJ, Labosky PA, Hogan BL, Matrisian LM. Proc Natl Acad Sci (USA) 1997;94:1402–1407. doi: 10.1073/pnas.94.4.1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witty JP, Wright JH, Matrisian LM. Mol Biol Cell. 1995;6:1287–1303. doi: 10.1091/mbc.6.10.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright JH, McDonnell S, Portella G, Bowden GT, Balmain A, Matrisian LM. Mol Carcinog. 1994;10:207–215. doi: 10.1002/mc.2940100405. [DOI] [PubMed] [Google Scholar]
- Yoshiji H, Gomez DE, Thorgeirsson UP. Int J Cancer. 1996;69:131–134. doi: 10.1002/(SICI)1097-0215(19960422)69:2<131::AID-IJC11>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Yoshiji H, Harris SR, Raso E, Gomez DE, Lindsay CK, Shibuya M, Sinha CC, Thorgeirsson UP. Int J Cancer. 1998;75:81–87. doi: 10.1002/(sici)1097-0215(19980105)75:1<81::aid-ijc13>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Zeng ZS, Cohen AM, Zhang ZF, Stetler-Stevenson W, Guillem JG. Clin Cancer Res. 1995;1:899–906. [PubMed] [Google Scholar]