Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility (original) (raw)
Abstract
Membrane-bounded organelles (MBOs) are delivered to different domains in neurons by fast axonal transport. The importance of kinesin for fast antero grade transport is well established, but mechanisms for regulating kinesin-based motility are largely unknown. In this report, we provide biochemical and in vivo evidence that kinesin light chains (KLCs) interact with and are in vivo substrates for glycogen synthase kinase 3 (GSK3). Active GSK3 inhibited anterograde, but not retrograde, transport in squid axoplasm and reduced the amount of kinesin bound to MBOs. Kinesin microtubule binding and microtubule-stimulated ATPase activities were unaffected by GSK3 phosphorylation of KLCs. Active GSK3 was also localized preferentially to regions known to be sites of membrane delivery. These data suggest that GSK3 can regulate fast anterograde axonal transport and targeting of cargos to specific subcellular domains in neurons.
Keywords: fast axonal transport/glycogen synthase kinase 3/GSK-3/kinesin/microtubule
Introduction
Owing to the lack of protein synthesis in axons, proteins needed in axons must be transported from cell bodies to sites of utilization. Membrane-bounded organelles (MBOs) are delivered to their destinations by fast axonal transport (Brady, 1993). Different membrane proteins are delivered to different subcompartments in axons, implying the existence of specific targeting mechanisms. For example, synaptic vesicles are delivered to presynaptic terminals, whereas vesicles with sodium channels are delivered to nodes of Ranvier. Little is known about how membrane proteins are enriched selectively in neuronal subdomains. Once an organelle reaches an appropriate destination, mechanisms must exist to prevent further translocation. Although the direction and rate of translocation for MBOs are determined by the activity of different microtubule (MT) motor proteins (i.e. kinesins and dyneins), molecular mechanisms that regulate trafficking of membrane proteins by these motors are poorly understood. This study looks at the role of kinesin phosphorylation in regulating its function.
The role of phosphorylation in regulating kinesin has been controversial. In vitro phosphorylation had either no or only modest effects on kinesin ATPase (Matthies et al., 1993). Other studies variously reported that phosphorylation was required for binding to (Lee and Hollenbeck, 1995) or detachment from (Sato-Yoshitake et al., 1992) MBOs. However, kinases analyzed for effects on kinesin function in vitro do not necessarily represent kinases relevant in vivo. For example, inhibition of protein kinase A (PKA), protein kinase C, Ca2+-dependent kinases or protein kinase G had no effect on fast axonal transport in either direction (Bloom et al., 1993). In contrast, inhibition of cdc2-like kinases inhibited fast anterograde transport (Ratner et al., 1998). Relevant kinases for in vivo phosphorylation of kinesin needed to be identified before studies on regulation of kinesin by phosphorylation were feasible.
The idea that phosphorylation regulates kinesin-mediated axonal transport was attractive because kinesin is a phosphoprotein in vivo (Hollenbeck, 1993), kinesin phosphorylation correlated with MBO binding in cultured cells (Lee and Hollenbeck, 1995) and local actions of kinases in isolated axoplasm affected transport of MBOs in vivo (McGuinness et al., 1989; Ratner et al., 1998). In rodent brain and cultured cells, kinesin is phosphorylated on serines with no detectable phosphothreonine or phosphotyrosine (Hollenbeck, 1993), but the responsible kinases had not been identified. To identify physiologically relevant kinases, in vivo phosphorylation sites on kinesin subunits were mapped. In vivo phosphorylation sites were found on both kinesin heavy chain (KHC) and kinesin light chain (KLC) subunits, implying that multiple kinases might differentially affect kinesin subunits and isoforms.
The importance of KLCs in kinesin binding to MBOs was established (Stenoien and Brady, 1997; Tsai et al., 2000) and KLCs were suggested to affect kinesin ATPase activity (Hackney et al., 1991; McIlvain et al., 1994; Verhey et al., 1998; Coy et al., 1999), so we focused on KLC phosphorylation. Several phosphorylated residues on KLCs matched consensus sites for glycogen synthase kinase 3 (GSK3), a widely expressed serine/threonine protein kinase. In vivo and in vitro experiments examined the ability of GSK3 to affect kinesin function. GSK3 phosphorylated and interacted with kinesin. In isolated axoplasm, GSK3 specifically and dramatically inhibited anterograde, but not retrograde, fast axonal transport. Inhibition required GSK3 kinase activity and correlated with reductions in kinesin bound to MBOs. Consistent with a role in regulating kinesin–MBO dissociation, active GSK3 levels were increased in regions of membrane delivery. We propose that phosphorylation of KLCs by GSK3 represents a regulatory pathway for kinesin-based motility that leads to delivery of its cargo to specific subcellular domains.
Results
To analyze phosphorylation of kinesin in axonal transport, proteins in optic nerve were pulse-labeled with either [32P]orthophosphate (for phosphoproteins) or [35S]methionine (for newly synthesized proteins). Retinal ganglion cells were labeled by injection of radiolabel into the vitreous of the eye and kinesin was immunoprecipitated from optic nerves 6 h after labeling (Figure 1A). At this time, kinesin associated with MBOs in fast axonal transport is enriched in optic nerve (Elluru et al., 1995). Immunoprecipitates (IPPs) of kinesin from optic nerve were visualized by fluorography or autoradiography and compared with Coomassie Blue-stained IPPs (lanes 2 and 5) and purified bovine brain kinesin (lanes 1 and 4). KHCs and KLCs were labeled by both 35S and 32P (Figure 1A). Lane 3 is a fluorograph of [35S]methionine-labeled kinesin from optic nerve showing newly synthesized kinesin transported in optic nerve. Lane 6 is an autoradiograph of 32P-labeled kinesin immunoprecipitated from optic nerve showing in situ phosphorylation of KHCs and KLCs in axons. Phosphoamino acid analysis detected only phosphoserine residues on kinesin (not shown), consistent with phosphorylated kinesin in cultured cells (Hollenbeck, 1993; Lee and Hollenbeck, 1995).
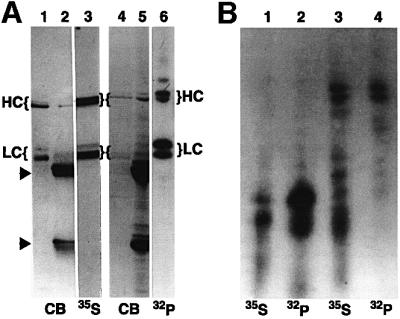
Fig. 1. Kinesin heavy and light chain isoforms show different phosphorylation patterns in vivo. (A) Autoradiographs of in vivo phosphorylation of axonal kinesin heavy (HC) and light chains (LC). Kinesin was immunoprecipitated from optic nerves labeled by fast axonal transport with 35S (lanes 2 and 3) or 32P (lanes 5 and 6). Proteins were visualized by Coomassie Blue (CB), fluorography (35S) or autoradiography (32P). Arrowheads indicate the position of IgG heavy and light chains. Purified bovine brain kinesin is included for comparison (lanes 1 and 4). (B) Peptide maps of 35S- and 32P-labeled KLC (lanes 1 and 2) and KHC (lanes 3 and 4) from optic nerve confirm the identification of kinesin phosphopeptides.
Peptide maps following limited proteolysis of 32P-labeled kinesin were compared with maps of [35S]methionine-labeled kinesin to verify identification of 32P-labeled peptides as KHC and KLC (Figure 1B). Multiple proteolytic fragments of [32P]phosphate-labeled kinesin co-migrate with [35S]methionine fragments. These peptide maps (Figure 1B) indicated that multiple in vivo phosphorylation sites exist on kinesin subunits and that isoforms of KHC and KLC are differentially phosphorylated (Figure 1A). Specifically, higher molecular weight variants of KHC and KLC2 were more heavily phosphorylated than lower molecular weight variants in vivo. Thus, kinesin moves down axons as a phosphoprotein.
Kinesin was tightly bound to purified vesicles but most kinesin was released if vesicles were incubated with ATP (Tsai et al., 2000) (Figure 2A). The ATP dependence of release suggested that a vesicle-associated kinase might modify KLCs. KLC released from vesicles showed an increase in apparent molecular weight (Figure 2A and B) and the shift was reversed by alkaline phosphatase treatment prior to electrophoresis (compare lanes 1 and 3, Figure 2B). KHC molecular weights did not shift consistently (compare lanes 1 and 2, Figure 2B). Thus, ATP-dependent release of kinesin from vesicles (Tsai et al., 2000) was accompanied by KLC phosphorylation.

Fig. 2. Kinesin light chains released from vesicles are phosphorylated. GSK3 is a kinase that specifically binds to kinesin and phosphorylates KLCs. (A) Washing vesicles with control buffers (–ATP) has little effect on the amount of kinesin (HC and LC, left) recovered in vesicle fractions (P), but incubation of vesicles with 1 mM ATP (+ATP) releases most kinesin (HC and LC, right) into the supernatant (S). Kinesin is released from vesicles in an ATP-dependent process, and released light chains (LC) migrate at a higher apparent molecular weight. (B) Increased apparent molecular weight of released KLC is due to phosphorylation. Immunoblots of kinesin vesicle fractions incubated without (lane 1) and with (lane 2) ATP. Lane 3 is with ATP but was treated with alkaline phosphatase prior to electrophoresis. Removal of phosphates eliminates the shift. (C) GSK3 specifically phosphorylates KLC subunits. SyproRed (SR) staining and western blotting (WB) with antibodies against KHCs (H2) and KLCs (63-90) indicate purity of rat brain kinesin. KLCs are phosphorylated by recombinant GSK3 (32P left lane). GSK3 is also autophosphorylated (32P right lane, no kinesin added). (D) Phosphorylation of the KLC2 C-terminal tail (KLC2t) requires pre-phosphorylation by CKII. Recombinant KLC2t was incubated with GSK3 and [γ-32P]ATP before (left lanes) or after (right lanes) incubation with CKII and unlabeled ATP. GSK3 phosphorylated KLC2t only after pre-phosphorylation with CKII. This confirms that KLC2t is a substrate for GSK3 and that kinesin purified from brain must be pre-phosphorylated at a priming site (see Table I). (E) GSK3 and kinesin co-immunoprecipitate. Brain lysate (lane 1), control immunoprecipitates with beads only, and beads/secondary antibody (lanes 2 and 3, respectively) and kinesin antibodies (lane 4) were immunoblotted with antibodies against KHC, GSK3 and PKA.
Mapping in vivo phosphorylation sites on kinesin can help identify physiologically relevant kinases. Phosphorylation sites on KHCs and KLCs were identified by mass spectrometry of tryptic peptides from immunopurified rat brain kinesin. Initial results identified multiple phosphorylation sites on KLCs, including several clusters of phosphorylated residues. One tryptic phosphopeptide identified was doubly phosphorylated and corresponded to a KLC2 C-terminal sequence (residues 606–616, Table IA). This domain contained two consensus phosphorylation sequences [X(S/T)XXX**Sp**] for GSK3, and this pattern was conserved in squid KLC (DDBJ/EMBL/GenBank accession No. P46825) (see Table IB), raising the possibility that KLCs could be phosphorylated by GSK3.
Table I. Kinesin light chain C-termini contain GSK3 consensus sites.
| (A) | |
|---|---|
| GTGLSDSR605/TL**SSSSMDLSR616/RSS**LVG | |
| (B) | |
| GTGLSDSRTL**SSSSMDLSRRSS**LVG | KLC2 (rat) |
| SIRSRSRTA**S** . . SDQL**S**SRPF | KLC2 (squid) |
| RPASVPPSPSLSRHSSPHQ**S**EDEE | Glycogen synthase |
| SYLDSGIHSGATTTAP**S** | β-catenin |
| STSTPAPSRTA**S** | ATP-citrate lyase |
The ability of GSK3 to phosphorylate kinesin was confirmed in vitro. Rat brain kinesin was purified under conditions to minimize post-lysis kinase/phosphatase modification (Hollenbeck, 1993). Purified rat brain kinesin was >95% pure by SyproRed staining (Figure 2C, SR) and the identity of SyproRed-stained bands was confirmed by immunoblot (Figure 2C, WB). No kinases appeared to co-purify with this kinesin as addition of [32P]ATP alone did not result in incorporation of label into either KHC or KLC. When purified rat brain kinesin was incubated with recombinant GSK3β, KLCs, but not KHCs, were specifically phosphorylated (Figure 2C, 32P panel). Incubation of GSK3 with [32P]ATP alone (Figure 2C, right lane 32P panel) showed autophosphorylation of GSK3 (Wang et al., 1994a). These results indicated that GSK3 selectively phosphorylates KLCs isolated from brain, suggesting that the priming phosphorylation required for GSK3 activity (Kennelly and Krebs, 1991) has already occurred. Consistent with this, GSK3 does not phosphorylate recombinant KLC2 tail domains comprising the last 95 residues of KLC2, unless KLC2 tail domains are pre-phosphorylated with casein kinase II (CKII; Figure 2D). Although the specific residues modified were not identified, these data indicate that KLC2 tail domains are substrates for GSK3 after a priming phosphorylation by CKII.
In vitro phosphorylation studies indicated that GSK3β could phosphorylate KLCs. To determine whether GSK3 interacts with kinesin in vivo, anti-kinesin antibodies cross-linked to protein A–agarose were used to probe rat brain lysates. Kinesin antibodies co-precipitated kinesin and GSK3β (Figure 2E, lane 4), whereas neither protein A–agarose alone nor normal mouse IgG– protein A–agarose precipitated kinesin or GSK3β (Figure 2E, lanes 2 and 3). IPPs of kinesin also phosphorylated CREB phosphopeptide, a specific substrate for GSK3 (not shown).
Adding radiolabeled ATP to kinesin IPPs resulted in phosphorylation of KLC2 (data not shown; De Vos et al., 2000). Recombinant KLC2 tail domain was also phosphorylated when added to control IPPs (G.Morfini and S.T.Brady, unpublished data). However, much of IPP-associated kinase activity was abolished by PKI, a specific PKA inhibitor, and enhanced by cAMP, suggesting that PKA might be present. This possibility was tested by immunoblot, and PKA was precipitated non-specifically (Figure 2E) under all conditions examined including various agarose-based affinity supports. In contrast, protein kinase B (PKB) immunoreactivity was detected only in lysates and failed to bind either kinesin or normal mouse IgG antibody beads (not shown). In light of non-specific binding of PKA to agarose affinity supports, the physiological significance of kinesin phosphorylation in IPPs is unclear unless controls for contaminating kinase activities are included. Both PKA and GSK3 phosphorylate kinesin, but only GSK3 co-precipitates specifically with kinesin.
Mechanisms for modulating axonal transport may involve proteins that interact with MTs and/or bind membranes. To analyze GSK3 on MBOs, three vesicle fractions and a soluble fraction were obtained by differential centrifugation of rat brain (Figure 3A). GSK3 exhibited a wide distribution by immunoblot analysis, but was enriched in membrane fractions containing synaptic vesicle proteins [synapsin (Syn); synaptophysin (p38)] and kinesin. In contrast, protein kinases such as PKA and PKB displayed very different distributions and were enriched in soluble fractions. The V1 fraction (100 000 g microsomal pellet) was enriched for both kinesin and GSK3, and was analyzed further on a continuous sucrose gradient. Kinesin and GSK3β distributions were assessed by immunoblot of pelleted vesicles from each fraction (Figure 3B) and found to overlap significantly in the gradient. A subset of GSK3-positive MBOs in V1 have identical buoyancy to kinesin-containing MBOs in this fraction and are likely to be present on the same MBOs.

Fig. 3. GSK3 and kinesin co-localize on a subpopulation of MBOs. (A) Immunoblots show kinesin, GSK3β, PKB, PKA and synaptic vesicle markers (Syn = synapsin and p38 = synaptophysin) in three vesicle fractions (V0, V1 and V2) and remaining supernatant (Cyt). GSK3 and kinesin are highly enriched in V1. (B) The upper panel shows immunoblots of V1 vesicles in continuous sucrose gradient fractions (0.32–2 M sucrose). Each fraction was diluted and pelleted to ensure that only vesicle-bound proteins were analyzed. Kinesin and GSK3β were present on a subset of V1 membranes of similar density. Scans show partial overlap of peaks. Arrows indicate peaks for kinesin (dark) and GSK3β (lighter). (C) KHC and GSK3β antibodies each labeled a single band in rat brain (R) and squid optic lobe (S). (D–F) Kinesin and GSK3 co-localize in double label immunofluorescence. Punctate immunostaining patterns for GSK3β (D) and kinesin (E) extensively overlap in the same cellular regions (F shows merge of D and E) in BHK21 cells. Scale bar: 25 µm. For higher resolution localization, GSK3β (G) and kinesin (H) distributions were determined in squid axoplasm. Both show punctate distributions characteristic of vesicles in squid axoplasm (Pfister et al., 1989; Stenoien and Brady, 1997). Most vesicles stained for both kinesin and GSK3β (I shows merge of G and H). However, some contained only kinesin (asterisk within inserts) or only GSK3β. Inserts in top right corners (G–I) are 4× digital magnifications of areas delineated by white squares.
Cell fractionation, co-immunoprecipitation and in vitro phosphorylation experiments indicated that GSK3β interacts with kinesin directly or indirectly, but the possibility remained that such associations resulted from redistribution of GSK3β or kinesin during homogenization (Tsai et al., 2000). To evaluate this possibility, kinesin and GSK3 distributions were examined by double immunofluorescence in cultured cells and isolated squid axoplasms. The antibodies used recognized single bands in both rat brain and squid extracts (Figure 3C). Double staining for endogenous GSK3β (Figure 3D) and kinesin (Figure 3E) showed similar punctate staining in BHK21 cells, with significant overlap in distribution (Figure 3F). Comparable results were obtained from immunofluorescence of GSK3 and kinesin in isolated squid axoplasm. In axoplasms (Figure 3G–I), both GSK3 (Figure 3G) and KHC (Figure 3H) antibodies labeled numerous punctate, vesicle-like structures (Figure 3I). Many, but not all, MBOs double-stained with both antibodies (see insets in Figure 3G–I). This punctate pattern of kinesin was shown previously to be Triton-soluble and consistent with MBOs (Pfister et al., 1989; Brady et al., 1990a). Omission of primary antibodies shows low levels of diffuse staining with no punctate structures. These results indicate that kinesin and GSK3 co-localized to a subpopulation of MBOs.
GSK3 interactions with cytoskeletal structures were also examined. GSK3β distributions in cultured hippocampal neurons (Figure 4A–C) and 3T3 fibroblasts (Figure 4D–F) were punctate (Figure 4A and D). GSK3β immunoreactivity was located preferentially in cellular regions containing MTs (Figure 4B and E), rather than actin-rich regions (Figure 4C and F). After mild detergent extractions to remove soluble GSK3β (Morfini et al., 2000), the punctate localization of GSK3β was even more pronounced (Figure 4G). Most GSK3β immunoreactivity aligned with MTs (Figure 4H and see inset), as previously seen with kinesin (Pfister et al., 1989; Morfini et al., 2000). Punctate staining for GSK3β could be due to direct binding of GSK3 to MTs, or to small MBOs that resist mild permeabilization, or both. Immunoblots of MT fractions from adult rat brain showed GSK3 immunoreactivity in MT pellets enriched in tubulin and MAP1A, an MT-associated protein (Figure 4I). This was consistent with observations of MT-associated GSK3 by immunofluorescence and indicated that GSK3 can interact directly or indirectly with MTs. PKB did not exhibit enrichment in MT pellets. Thus, GSK3β is present in cell domains appropriate to modify and regulate kinesin-based motility.

Fig. 4. GSK3 was enriched in microtubule domains. Primary cultured hippocampal neurons (A–C) and 3T3 fibroblasts (D–F) were triple stained for GSK3β (A and D), tubulin (B and E) and actin (C and F). Perikarya and proximal neurites of a hippocampal neuron (A–C) and a 3T3 fibroblast (D–F) are shown. Scale bars in (C), (F) and (H) are 25 µm. GSK3β immunostaining is punctate and localized preferentially in MT-rich regions. (G–H) In detergent-permeabilized BHK21 cells, GSK3β staining aligned with MTs. Higher magnification (inserts of areas marked by arrows) revealed punctate staining closely matching MT distribution (asterisk). Scale bar for the insert is 3 µm. (I) GSK3β co-pellets with taxol-stabilized MTs. Total rat brain homogenate (lane 1) was centrifuged at 100 000 g. The supernatant (lane 2) was incubated with taxol and GTP then recentifuged to obtain an MT pellet (lane 3) and a final supernatant (lane 4). Immunoblots show GSK3α/β and MAP1A enriched in the MT pellet. PKB was absent from MT fractions.
To evaluate the effects of GSK3 on kinesin-based motility in vivo, fast axonal transport was analyzed in isolated axoplasm from squid. Extruded axoplasm is an excellent in vivo model to study fast axonal transport and was the basis for identification of kinesins (Brady, 1985; Vale et al., 1985). Bidirectional MBO movements continue in axoplasms with properties essentially unchanged from those in the intact axon (Brady et al., 1985). Typical anterograde rates in perfused axoplasms are 1.5–2.0 µm/s, while retrograde transport rates are 0.9–1.2 µm/s. These rates are maintained (±5–10%) for 1 h or more after perfusion with control buffers (Brady et al., 1985). The highly conserved GSK3 consensus sequences in the KLC tail (Table IB) and the presence of GSK3 on kinesin-containing organelles in squid axoplasm (Figure 3) suggested that these pathways would be conserved.
Perfusion of active GSK3 into axoplasm (10 nM; 250 U/ml; 1 U = 1 pM ATP/min), dramatically reduced anterograde transport rates, but not retrograde rates (Figure 5A and E). Average anterograde transport rates were reduced by 44% (P ≤0.0001; two-sample _t_-test). In addition, the amount of moving particles detectable was reduced, although particle numbers were not readily quantifiable due to resolution limits. This inhibition was not complete, as some particles continued to move anterogradely 60 min after perfusion, suggesting that not all MBOs were susceptible to GSK3 treatment. No changes in overall axoplasm structure or peripheral MTs were noted with GSK3 perfusion.

Fig. 5. Active GSK3β inhibited fast anterograde transport in isolated axoplasm. Each data point represents an average rate for particles moving in the specified direction in a field as described previously (see Brady et al., 1985, 1990a; McGuinness et al., 1989; Ratner et al., 1998). Anterograde (black arrows) and retrograde (gray arrows) transport were measured concurrently. Retrograde transport was unaffected by any of these treatments. (A) Perfusion of axoplasm with 10 nM GSK3β selectively inhibits anterograde axonal transport. The number of organelles that appear to move in anterograde transport was also reduced. (B) Perfusion with inactive, autophosphorylated GSK3 did not affect transport. (C) Co-perfusion of active GSK3 with millimolar CREB phosphopeptide (a specific GSK3β substrate that acts as a competitive inhibitor) abolished the inhibitory effects of GSK3β on transport. (D) Perfusion of axoplasm with PKA catalytic subunit had no effect on fast axonal transport in either direction. (E) Mean rates of particle movement for all measurements between 30 and 40 min (30m) with control buffers or PKA were not significantly different from preperfusion rates (0m) in either anterograde (gray bars on left) or retrograde (black bars on right) directions. Addition of GSK3β reduced anterograde transport at 30–40 min by 44% (#, significant at P ≤0.0001). Addition of GSK3 with 1 mM CREB phosphopeptide, a competitive inhibitor of GSK3, abolished the effect of GSK3 on anterograde transport. No other differences between experimental and matched control were statistically significant (P ≤0.05).
To determine whether GSK3 kinase activity was required to inhibit anterograde transport, GSK3 was incubated with ATP for 30 min before perfusion, resulting in autophosphorylation (Figure 2C) on GSK3β at Ser9 and inhibition of GSK3 kinase activity (Wang et al., 1994a). The effects of GSK3 on anterograde transport were abolished by autophosphorylation (Figure 5B). Similarly, perfusion with mutant GSK3β lacking kinase activity (kinase-dead; a gift from Dr H.Eldar-Finkelman) had no effect on transport (not shown). Co-perfusing a competitive inhibitor of GSK3, phosphoCREB peptide substrate (Bullock and Habener, 1998), also eliminated effects on anterograde transport (Figure 5C and E). Perfusion of millimolar phosphoCREB alone had no effect on axonal transport. If GSK3 kinase activity was inhibited (Figure 5B and C), average transport rates were not significantly different from control at P = 0.05 (Figure 5E).
Inhibition of anterograde axonal transport was not an effect of all protein kinases. Perfusion with micromolar cdk5 kinase and its activator p25 had no effect on axonal transport in either direction (not shown). Similarly, perfusion of axoplasm with PKA catalytic subunit (2 U/ml; 1 U = transfer of 1 µM phosphate/min) did not affect either direction of fast transport (Figure 5D and E), although PKA phosphorylates kinesin in vitro (Sato-Yoshitake et al., 1992). This result raises questions about the physiological significance of in vitro experiments on PKA phosphorylation of kinesin.
Active GSK3β inhibited fast anterograde transport in situ, but did not affect axonal structural elements, because perfusion of active GSK3 did not affect retrograde transport. One or more components essential for anterograde movement of many MBOs were affected by GSK3 kinase activity. The obvious candidate was the major anterograde motor kinesin, because KLCs are both in vivo and in vitro substrates for GSK3. This mechanism would require that phosphorylation of KLCs affects some aspect of kinesin function. Physiological functions of kinesin result from three activities: kinesin binding to MTs, MT-activated ATP hydrolysis and association with membrane surfaces. The effects of GSK3 on all three were measured using previously characterized in vitro assays (Stenoien and Brady, 1997).
First, the effect of GSK3 on kinesin binding to MTs was evaluated with ATP and AMP-PNP, a non-hydrolyzable ATP analog. Rat brain kinesin was labeled by GSK3β and [γ-32P]ATP, then 80 ng of kinesin was incubated with MTs in the presence of ATP or AMP-PNP (Figure 6A). Phosphorimaging showed that GSK3 phosphorylated KLCs bound to MTs in the presence of AMP-PNP, but little was bound to MTs in the presence of ATP, a signature property of kinesins. There were no detectable differences between labeled and unlabeled kinesin subunits in binding to or release from MTs whether visualized by SyproRed (not shown) or by phosphorimaging. The binding pattern of KHC to the MTs was identical to the KLCs shown here. Consistent with MT-binding studies, GSK3 phosphorylation of kinesin also failed to affect MT-activated ATPase activity (Figure 6B). Specific MT-activated ATPase activity for kinesin in these assays was comparable with previously published values (Wagner et al., 1991). Thus, inhibition of fast anterograde transport by GSK3β was not due to inhibition of kinesin MT binding or MT-activated ATPase activities.

Fig. 6. GSK3β phosphorylation of kinesin light chain releases kinesin from membranes without altering MT binding or ATPase activity. (A) Phosphorylation of kinesin by GSK3β did not affect kinesin interaction with MTs. GSK3β alone (lane 1) or GSK3β and kinesin (lanes 2–5) were labeled by incubation with [32P]ATP, then 80 ng aliquots were incubated with taxol-stabilized bovine brain MTs and either ATP (duplicate experiments shown in lanes 2 and 3) or AMP-PNP (duplicate experiments shown lanes 4 and 5), then pelleted. No effects on MT binding were seen between kinesin with KLCs phosphorylated by GSK3 and control kinesin fractions. (B) MT-activated ATPase activity of kinesin was also unaffected by GSK3 phosphorylation. Bars represent the percentage of ATP hydrolyzed by control, non-phosphorylated (white bars) and GSK3β-phosphorylated (gray bars) rat brain kinesin (n = 3). (C and D) Perfusion of active GSK3 in squid axoplasm dramatically reduced kinesin bound to membranes. (C) Membrane fractions from squid axoplasms perfused with buffer (Control), and with active GSK3 (GSK3) were immunoblotted for kinesin (KHC) and actin. (D) Quantitative immunoblots for kinesin and actin with [125I]protein A; values were quantitated by phosphoimager and kinesin levels normalized relative to actin (n = 4). GSK3 treatment reduced membrane-bound kinesin by 70%.
To evaluate whether phosphorylation of KLCs affected kinesin attachment to cargo, kinesin bound to MBOs was analyzed in axoplasms perfused with or without active GSK3 (Figure 6C). To ensure that pelleted kinesin was membrane associated, parallel reactions were carried out with 1% Triton X-100. No kinesin was detected in pellets with detergent. Kinesin bound to MBOs was reduced to one-third of controls in GSK3-perfused axoplasms (Figure 6D). This is an underestimate because kinesin release from MBOs is promoted by cytoplasmic factors during preparation of MBO fractions (Tsai et al., 2000). These results suggested that phosphorylation of KLCs by GSK3 increased release of kinesin from membranes.
Inhibition of fast anterograde transport by GSK3 made it a candidate for regulation of kinesin-based motility. If so, GSK3 should be increased and/or preferentially activated in cellular domains where new membrane proteins are inserted, such as growth cones (Craig et al., 1995). To test this hypothesis, GSK3β and MT distributions were evaluated in nerve growth factor (NGF)-stimulated PC12 cells and primary cultured hippocampal neurons (Figure 7). GSK3 was high in cell bodies and detectable at moderate levels throughout neuritic processes, but showed increased levels in growing tips of neurites (Figure 7A–F). This concentration of GSK3 immunoreactivity did not match tubulin immunoreactivity in growth cones (see arrows in Figure 7C and F). At higher magnification, GSK3 immunoreactivity was concentrated in central regions of growth cones (Figure 7G), a region where MBOs accumulate distinct from both MTs and microfilament-enriched regions (Dailey and Bridgman, 1991) (Figure 7G–J).

Fig. 7. GSK3 was increased at sites of active membrane delivery. A hippocampal neuron (A–C) and a differentiating PC12 cell (D–F) were double immunostained for GSK3β (A and D) and tubulin (B and E). In both cell types, the level of GSK3 was high in cell bodies and lower amounts were distributed along neurites. Merged images (C and F) highlight relative enrichment of GSK3β relative to tubulin at neurite tips (arrows). GSK3 immunoreactivity appears concentrated distal to MT ends. (G–J) High magnification of a triple-stained PC12 growth cone. Anti-GSK3β labels vesicle-like structures in the growth cone core (G), with a pattern distinct from MTs (H) and actin (I). In the merged image (J), the relationship of GSK3β to cytoskeletal elements is emphasized. Asterisks serve as reference points. Immunolocalization with an antibody that recognizes GSK3β independently of phosphorylation (active and inactive forms) shows enrichment of immunoreactivity in growth cones (K and G). In contrast, an antibody that only recognizes GSK3β phosphoSer9 (inactive kinase) (Wang et al., 1994a) is not enriched in growth cones (L). PhosphoSer9 immunoreactivity is abolished by phosphatase treatment (M), while it has no effect on staining with the phosphorylation-independent GSK3β antibody (K). Immunoblotting on proteins isolated from growth cones (N) confirmed the results of immunostaining. Lane 1 contains homogenate from E18 rat brain, lane 2 a 3000 g supernatant from E18 brain and lane 3 purified growth cone particles. The antibodies used on each of the four blots are indicated on the right.
To determine whether growth cone GSK3 was activated, we compared immunoreactivity for total GSK3 (both active and inactive) with immunoreactivity of inactive GSK3 (phosphorylated on Ser9) (Wang et al., 1994a). Total GSK3 immunoreactivity was increased in growth cones (Figure 7G and K) and was not affected by phosphatase treatment (Figure 7K). In contrast, GSK3 phosphoSer9 immunoreactivity was not enriched in growth cones (Figure 7L) and phosphoSer9 immunoreactivity was comparable with levels in neighboring neurites. Both total GSK3 (Figure 7A and D) and phosphoSer9 GSK3 (and data not shown) immunoreactivity were higher in cell bodies. Staining for GSK3 phosphoSer9 in growth cones was abolished by phosphatase treatment (Figure 7M). These immunofluorescent localizations of active and inactive GSK3 were confirmed by immunoblot of purified growth cone particles (GCPs; Figure 7N). Total GSK3β, kinesin and GAP43 were all abundant in total brain homogenates (lanes 1), low speed supernatants (lanes 2) and GCP (lanes 3) fractions. In contrast, GSK3 Ser9 immunoreactivity was present in total homogenates and low speed supernatants of embryonic rat brain, but was almost undetectable in GCPs. This indicates that most GSK3β in growth cones is in an active state. These data are consistent with a model in which local concentrations and local regulation of GSK3 kinase activity at sites of active membrane insertion provide a molecular basis for regulating kinesin-based transport of MBOs.
Discussion
Fast anterograde axonal transport of many MBOs was inhibited by active GSK3. Mechanisms for inhibiting kinesin-based motility must exist to ensure MBO delivery to specific subcellular compartments. This is essential to generate and maintain polarized membrane protein distributions and ultimately for the ability of neurons to receive, process and transmit information. Regulation of kinesin-based motility has been poorly understood. Unlike most myosins and dyneins, no biochemical switch is yet known to turn off kinesin motor activity and MBO translocation at specific times and places in cells. The discovery that hsc70 could limit kinesin-based motility by removing kinesin from MBOs (Tsai et al., 2000) provided a plausible regulatory mechanism involving KLCs, but did not explain local control of kinesin activity.
Phosphorylation of kinesin in cells (Hollenbeck, 1993; Lee and Hollenbeck, 1995) implied that one or more kinases might regulate kinesin function, but did not identify physiologically relevant kinases. Phosphopeptide mapping suggested that GSK3 could modify KLCs in vivo. GSK3 is a ubiquitously expressed protein kinase that phosphorylates various substrates (Woodgett, 1994) including nuclear- (cJun, ATF transcription factors), cytoskeletal- (neurofilaments, tau, MAP1B), cytoplasmic- (glycogen synthase) and membrane-associated (kinesin) proteins. A distinctive characteristic of GSK3 consensus sites is a requirement for prior phosphorylation of a serine/threonine four residues carboxyl to the site, creating phosphorylation clusters like those identified on KLCs. KLCs but not KHCs purified from rat brain were phosphorylated by recombinant GSK3 in vitro without pre-phosphorylation by other kinases. This suggests that brain kinesin was primed for GSK3 phosphorylation. Interaction between kinesin and GSK3 was confirmed by co-immunoprecipitation of GSK3 from whole brain lysates with antibodies against kinesin.
Subcellular fractionation revealed that GSK3 was highly enriched in membrane fractions containing kinesin and synaptic vesicle markers. Such enrichment in membranes was not seen for other protein kinases such as PKA and PKB. Further fractionation suggested that a subpopulation of vesicles contained both GSK3 and kinesin. Immunolocalization in squid axoplasm showed extensive overlap in kinesin and GSK3 patterns. Lack of complete overlap between kinesin and GSK3 was not surprising, given that GSK3 can phosphorylate substrates in vivo at sites where kinesin is undetectable (i.e. nuclei or plasma membrane) (Leopold et al., 1992). Cytoplasmic GSK3 in cultured neuronal and non-neuronal cells was seen primarily in cellular regions rich in MTs rather than actin microfilaments (Figure 4). Following whole- cell permeabilization, GSK3 immunoreactivity clearly aligned with MTs. Similarly, GSK3 co-pelleted with taxol-stabilized MTs from brain. GSK3 can interact with both membranes and MTs either directly or indirectly; thus GSK3 is in places well suited to regulate MT-associated proteins and MT-based motility. These experiments document a subcellular distribution for GSK3 that includes interactions with MBOs, MTs and kinesin.
The squid axon has proven to be a powerful model for the study of neuronal functions ranging from the biophysics of the action potential to the discovery of kinesins. The squid axoplasm model has been a valuable, well-characterized in vivo model for study of kinesin-based motility, because the sequences for kinesin molecular motors are highly conserved from squid to human. Many domains in the KLCs have been particularly well conserved (Stenoien and Brady, 1997) and this extends to the C-terminal domain identified as a candidate for modification by GSK3 (see Table IB).
In vivo experiments with isolated axoplasm assessed effects of GSK3 on vesicle transport. Active GSK3 dramatically inhibited anterograde axonal transport. This effect required kinase activity, because neither Ser9-phosphorylated GSK3 (inactive) nor a kinase-dead GSK3 mutant affected transport. Similarly, co-perfusion of GSK3 and a competitive inhibitor, phosphoCREB, prevented GSK3 inhibition of anterograde transport. PhosphoCREB is a preferred substrate for GSK3 (Bullock and Habener, 1998) and has advantages over GSK3 inhibitors such as LiCl, because this peptide is not known to have other biological activities or to be a substrate for other kinases. In contrast, LiCl has low affinity for GSK3 (IC50 = 10 mM) (Woodgett, 1991). At effective concentrations in vivo, LiCl affects many things other than GSK3, including activation of PKB/Akt1 kinase (Chalecka-Franaszek and Chuang, 1999) and inhibition of Na+/H+ exchangers (Kobayashi et al., 2000).
The effects of GSK3 on anterograde transport were not shared by all kinases. Neither PKA (Figure 5D and E) nor cdk5 and its activator p25 had effects on either direction of transport in axoplasm. Phosphorylation of kinesin by PKA was proposed to regulate fast axonal transport (Okada et al., 1995) and kinesin binding to MBOs (Sato-Yoshitake et al., 1992). Although PKA phosphorylates kinesin in vitro (Sato-Yoshitake et al., 1992; Matthies et al., 1993), PKA had no effect on either anterograde or retrograde transport in axoplasm. Similarly, PC12 cells mutants lacking PKA and forskolin-stimulated wild-type PC12 cells exhibited no change in kinesin phosphorylation in vivo (Hollenbeck, 1993). Taken together, our data support a role for GSK3, but not PKA, in kinesin regulation in vivo.
Mechanisms by which GSK3 phosphorylation of KLCs inhibits kinesin function might include ATPase activity, MT binding or MBO binding. For example, KLCs inhibit ATPase activity in vitro when soluble kinesin is folded to allow tail domains to interact with its heads (Verhey et al., 1998; Coy et al., 1999; Seiler et al., 2000). Phosphorylation could affect folding or interaction of KLC sequences with motor domains. However, GSK3 phosphorylation of KLCs failed to alter kinesin MT-activated ATPase or binding to MTs under these conditions. KLCs play a role in binding to MBOs (Stenoien and Brady, 1997; Tsai et al., 2000). Comparing kinesin on MBOs in control and GSK3-perfused axoplasms indicated that GSK3 treatment dramatically decreases the amount of membrane-associated kinesin (Figure 6). This suggests that GSK3 phosphorylation of KLCs leads to detachment of kinesin from cargo and inhibition of fast anterograde transport.
Although kinesin exists primarily as a membrane-associated protein in vivo (Pfister et al., 1989; Elluru et al., 1995; Tsai et al., 2000), tight association with MBOs is disrupted by hsc70 chaperones (Tsai et al., 2000) and by treatment with GSK3 (Figure 6). Hsc70 and GSK3 actions may be functionally connected in vivo (Figure 8). Hsc70 comprises a significant fraction of total brain protein (∼1%) and is enriched in growth cones and synapses. Therefore, hsc70-induced release of kinesin from MBOs must be regulated. GSK3-mediated phosphorylation of KLCs could be a regulatory switch. Consistent with this, KLC’s apparent molecular weight was increased with release from MBOs and this shift was abolished by alkaline phosphatase. Moreover, _N_-ethylmaleimide (NEM) treatment of vesicles blocked release of kinesin by hsc70 (Tsai et al., 2000), and NEM inhibits GSK3 kinase activity (data not shown).

Fig. 8. A simple model for the action of GSK3 on kinesin function. KLCs on many MBOs are pre-phosphorylated. When GSK3 is activated by action of one or more phosphatases, it phosphorylates KLCs further. Phosphorylation by GSK3 increases the accessibility of the J-domain motifs on KLCs to hsc70 (Tsai et al., 2000) and leads to removal of kinesin from a vesicle. Released kinesin appears to be degraded rapidly (Li et al., 1999) and vesicles become available for insertion into the plasma membrane. Given that membrane receptor pathways can regulate GSK3 activity locally, this would act as a targeting mechanism for membrane proteins to regions of local GSK3 activation. Local changes in kinase/phosphatase activity have been implicated in specific targeting of ion channels to nodes of Ranvier (de Waegh et al., 1992).
Some vesicles continue to move in isolated axoplasm after GSK3 treatment. Some vesicle populations may well use other motors for transport. A number of kinesin-related proteins were identified in vertebrate brain (Hirokawa, 1998), but the role of kinesin-related proteins in squid axoplasm is unknown. However, kinesin is far more abundant in the nervous system than any kinesin-related protein (Brady, 1991), and active GSK3 clearly reduces the amount of kinesin associated with vesicles (see Figure 6). Moreover, the evidence suggests that multiple pathways exist for regulating different kinesin isoforms on MBOs. Hsc70 and GSK3 affect kinesin association with MBOs, but are not likely to be the only pathway for regulating kinesin function in cells (Morfini et al., 2001).
We propose that localization and local activation of GSK3 are critical for regulating fast anterograde transport. Active GSK3 is increased in cellular domains, such as neuronal growth cones and synaptosomes (Yang et al., 1992), associated with delivery of membrane proteins (Figure 7). Inhibitory effects of GSK3 on kinesin-driven motility suggest that cellular compartments with active GSK3 will exhibit reduced amounts of kinesin-based motility. Therefore, much cellular GSK3 must be inactive, and increased GSK3 activity will alter MBO transport. Consistent with this, transgenic mice overexpressing GSK3 or expressing a constitutively active form of GSK3 exhibit increased mortality (Brownlees et al., 1997; Lucas et al., 2001), and sustained GSK3 activation correlates with neurite retraction and apoptosis (Hetman et al., 2000; Sanchez et al., 2001).
Multiple pathways for inactivating GSK3 are known, but relatively few are defined for its activation. Inhibitors of GSK3 include autophosphorylation and upstream kinases such as PKB/Akt (Cross et al., 1995) and PKCγ (Cook et al., 1996). These inactivate GSK3 by phosphorylation at Ser9 (GSK3β) or Ser21 (GSK3α) (Wang et al., 1994a). In addition, non-kinase effectors (i.e. axin, GBP and disheveled) (Ikeda et al., 1998; Yost et al., 1998; Krylova et al., 2000) exist. Activation of GSK3 is less well defined, but dephosphorylation of Ser9 on GSK3 is probably required for activating GSK3 (Wang et al., 1994a; Woodgett, 1994; Brady et al., 1998). Our findings suggest that fast anterograde transport depends upon a balance between kinases and phosphatases in specific cellular domains. Thus, various signaling pathways could alter this balance and lead to localized changes in kinesin-based motility (Figure 8). Local activation of GSK3 would promote delivery of newly synthesized membrane materials. Indeed, localized changes in the balance between kinases and phosphatases were noted at nodes of Ranvier and proposed to play a role in delivery of Na+ channels to the plasma membrane (de Waegh et al., 1992).
Disruption of axonal transport in neurons is implicated in pathogenesis for various neurodegenerative diseases, including diabetic neuropathy, Alzheimer’s disease, amyotrophic lateral sclerosis and Huntington’s disease. In diabetic neuropathy and Alzheimer’s disease, changes in kinase activities and phosphorylation patterns specifically implicate GSK3 (Mandelkow et al., 1992; Ishiguro et al., 1993; Cross et al., 1994, 1997; Baum et al., 1996; Eldar-Finkelman et al., 1999; Summers et al., 1999). Because GSK3 activity can be regulated through multiple signaling pathways, very different pathogenic mechanisms might converge to alter kinesin phosphorylation in axons, thereby disrupting kinesin-based motility. The unique reliance of neurons on protein transport to distant locations make them particularly vulnerable to decrements in fast axonal transport. Misregulation of fast anterograde transport in disease may suffice to produce neuropathies. Extracellular signaling molecules essential for development and oncogenesis also activate pathways that regulate GSK3 (Salinas, 1999; Polakis, 2000; Seidensticker and Behrens, 2000). Any pathway that alters GSK3 activity may affect kinesin-based motility. Specific alterations in kinesin-based MBO transport mediated by GSK3 may represent a key molecular component of cellular differentiation and pathology.
Materials and methods
Reagents
All reagents were from Sigma (St Louis, MO), except as noted. Protein concentration was measured by Coomassie Blue (Pierce). Commercial antibodies included monoclonal antibodies against GSK3β (Transduction Laboratories); phosphoSer9 GSK3 and PKB (Cell Signaling); PKAα and PKAβ (C-20) catalytic subunits (Santa Cruz); MAP1A (clone HM1A), tubulin and actin (Sigma); synaptophysin (Pierce); and synapsin (Serotec); and polyclonal antibodies against GSK3β (334-348) (Calbiochem). Antibodies against kinesin, H2 and 63-90, were characterized previously (Pfister et al., 1989; Stenoien and Brady, 1997). Recombinant GSK3β was from Sigma or New England Biolabs, and recombinant PKA catalytic subunit was from Boehringer Mannheim. The activity of recombinant kinases was verified by standard assays and substrates [phosphoCREB for GSK3 (Wang et al., 1994b); Kemptide (Slice and Taylor, 1989) (New England Biolabs) for PKA; and histone 1 for cdk5]. GAP-43 antibody was from Boehringer Mannheim. The GST–KLC2tail construct was kindly provided by Bruce Richards. Purified growth cone fractions were a generous gift from Keith Mikule and K.Pfenninger (University of Colorado Health Science Center).
In vitro and in vivo metabolic labeling of kinesin
[35S]Methionine (Trans 35S-label) or [32P]orthophosphate (ICN) was lyophilized and resuspended in water (0.25 or 0.5 mCi/µl, respectively). Four microliters of label were injected into the vitreous of Sprague–Dawley rat eyes as previously described (Elluru et al., 1995). Animals were anesthetized at specific times after injection and decapitated. Tissue samples were pooled from two rats per time point. Radiolabeled kinesin was immunoprecipitated from lysates and processed for SDS–PAGE and autoradiography (Elluru et al., 1995). Peptide mapping following limited proteolysis was by published methods (Cleveland et al., 1977). Recombinant rat GSK3 (Sigma) (200 nM) was incubated with or without 80 ng of rat brain kinesin in IME buffer plus 0.1 mM ATP and [γ-32P]ATP tracer. After 20 min at 34°C, reactions were used for ATPase and MT-binding assays or stopped by addition of 1 vol. of 2× Laemmli buffer for SDS–PAGE.
Kinesin purification from rat brain
Kinesin was purified from 20 rat brains (13 days old) as described previously (Wagner et al., 1991), with slight modifications. Homogenization buffer was supplemented with mammalian protease inhibitor cocktail, 0.1 mM ATP and a mix of phosphatase/kinase inhibitors (50 nM K252a, 50 nM staurosporine, 20 nM mycrocystin RR, 12 µM DRB, 200 nM roscovitine, all from Calbiochem). Kinesin was prepared by MT affinity with AMP-PNP, gel filtration and ion exchange chromatography. Kinesin samples eluted from ion exchange columns were dialyzed against IME (imidazole 15 mM, 2 mM MgCl2, EGTA 1 mM pH 7.0) and concentrated with Ultrafree Centrifuge Filters (Millipore). Sucrose was added to a final concentration of 10%, then aliquots were flash-frozen in liquid N2 and stored at –80°C.
Recombinant KLC tail phosphorylation
The C-terminal tail comprising the last 95 amino acids of rat KLC2 was subcloned into the PGEX vector (Pharmacia), expressed in Escherichia coli and purified using glutathione–Sepharose affinity resin (Sigma). A 6 mg aliquot of GST–KLC2tail bound to glutathione–Sepharose beads was first incubated with or without 500 U of recombinant CKII (New England Biolabs) in 20 ml of K buffer [20 mM Tris–HCl pH 7.5, 10 mM MgCl2, 5 mM dithiothreitol (DTT)] plus 100 mM ATP for 40 min at 30°C. Experiments performed with radiolabeled ATP indicated strong phosphate incorporation in KLC2tail (data not shown). The beads were then washed three times with 1 ml of K buffer, and finally resuspended in 20 µl of K buffer plus 200 nM recombinant rat GSK3 (Sigma). Reactions were started by addition of 100 µM radiolabeled ATP (200 mCi/mmol). After 60 min at 30°C, reactions were stopped by adding 50 µl of 2× Laemmli buffer. Reactions were separated by SDS–PAGE, the gels dried and exposed to a phosphoimager screen. Control experiments with GST showed no incorporation by either kinase.
Immunoprecipitation and immunoblots
Adult rat brains were homogenized in lysis buffer (100 mM NaCl, 50 mM Tris–HCl pH 7.4, 0.5% Triton X-100, 0.5% sodium deoxycholate, 1 mM orthovanadate, 50 mM NaF, 5 mM EDTA, 40 mM β-glycerophosphate, 0.01% SDS, 0.02% saponin) and 0.5% (v/v) mammalian protease inhibitor cocktail. Lysates were spun at 40 000 _g_max for 1 h and pellets discarded. Aliquots (15 mg of total protein) were pre-cleared with protein A–agarose (20 µl bed volume). H2 antibody or normal mouse IgG were covalently cross-linked to protein A–agarose beads using dimethilpimelimidate (Harlow and Lane, 1988) at 2 mg IgG/ml bed volume. Brain extracts were incubated at 4°C with 30 µg of cross-linked antibody. Controls include lysate incubated with protein A–agarose beads without antibody. Beads were centrifuged as above and washed twice with lysis buffer, twice with lysis buffer plus 200 mM NaCl, and once with lysis buffer to remove salt, then resuspended in Laemmli buffer. For immunoblots, samples were transferred to Zetaprobe nylon membranes or nitrocellulose as described previously (Stenoien and Brady, 1997) and visualized by chemiluminescence (ECL, Amersham).
Motility studies in isolated axoplasm
Axoplasm was extruded from squid giant axons (Loligo pealii; Marine Biological Laboratory) as described previously (Brady et al., 1985). Axons (400–600 µm in diameter) provided ∼5 µl of axoplasm. Recombinant enzymes, peptides or inhibitors were diluted into X/2 buffer (175 mM potassium aspartate, 65 mM taurine, 35 mM betaine, 25 mM glycine, 10 mM HEPES, 6.5 mM MgCl2, 5 mM EGTA, 1.5 mM CaCl2, 0.5 mM glucose pH 7.2) supplemented with 2–5 mM ATP and 20 µl perfused into chambers (Brady et al., 1990a). Motility was analyzed on a Zeiss Axiomat with a ×100, 1.3 n.a. objective, and DIC optics. A Hamamatsu Argus 20 system and Model 2400 CCD camera were used for image processing and analysis. Organelle velocities were measured with a Photonics Microscopy C2117 video manipulator (Hamamatsu) by matching calibrated cursor movements to the speed of vesicles moving in the axoplasm. Limits to resolution in the light microscope preclude following individual organelles in axoplasm, but this method has proved highly reproducible. Rates obtained in this fashion are comparable with rates obtained with individual organelles moving along isolated microtubules (Ratner et al., 1998). Resolution limits also prevent accurate counts of the numbers of vesicle moving in axoplasm, but qualitative comparisons are easily accomplished and are reproducible (Brady et al., 1990b, 1993). Because the velocity measurements represent a sampling of vesicle movements in and out of the plane of focus, the average rate correlates with the number of vesicles moving in a given treatment, i.e. high rates also mean high numbers of vesicles, but low rates reflect reduced numbers as well as slower mean velocities (Brady et al., 1990b, 1993; Ratner et al., 1998).
Subcellular fractionation procedures
Subcellular fractions were as described previously (Morfini et al., 2000). Adult rat brains were homogenized in 10 ml of homogenization buffer (HB; 300 mM sucrose, 10 mM HEPES pH 7.4, 5 mM EDTA and 2% mammalian protease inhibitor cocktail). Homogenates were centrifuged at 12 500 _g_max to eliminate cell debris, nuclei and most mitochondria. Supernatants were centrifuged for 40 min at 39 800 _g_max, for V0 pellets. S0 was recentrifuged for 40 min at 120 000 _g_max, for V1 pellets, and S1 was centrifuged for 2 h at 260 000 _g_max for V2 pellets. The final supernatant was termed cytosol. All vesicle pellets were resuspended in homogenization buffer. Equal amounts of protein from V0, V1, V2 and cytosol fractions were processed for immunoblots. MT fractions were prepared from rat brains by standard methods (Vallee, 1982).
For sucrose gradient centrifugation, V1 vesicle pellets were resuspended in HB by three passages through a 25 g hypodermic needle to disperse vesicles. Approximately 1 mg of V1 vesicles was resuspended in 500 µl of HB and loaded on an 11 ml sucrose linear gradient (0.32–2 M sucrose in HB). Samples were spun at 286 000 _g_max for 4 h at 4°C. Eighteen fractions of 660 µl were collected from the bottom to the top with a peristaltic pump and diluted with 2 ml of HB. Diluted fractions were pelleted at 260 000 _g_max for 30 min at 4°C. Resulting membrane pellets were resuspended in Laemmli buffer and analyzed by immunoblot.
Four axoplasms per condition were incubated with or without 10 nM recombinant GSK3 in 150 µl of X/2 buffer plus 5mM ATP. After 40 min, 20 µl of EDTA (0.5 M) was added to each reaction. Axoplasms were homogenized with a micropestle in low protein binding Eppendorf tubes (USA Scientific). Homogenates were loaded on a 40 µl cushion of 0.32 M sucrose in X/2 buffer, and centrifuged at 120 000 _g_max for 40 min at 4°C. Pellets were resuspended in Laemmli buffer and processed for quantitative immunoblots with [125I]protein A. Relative amounts of kinesin in each condition were normalized to actin immunoreactivity.
Purification of embryonic growth cones
Fetal rat brains (E18) were fractionated according to published methods (Pfenninger et al., 1983; Quiroga et al., 1995) to obtain GCPs. Briefly, the low speed supernatant (L) of fetal brain homogenate (H) was loaded on a discontinuous sucrose gradient in which the 0.75 and 1 M sucrose layers were replaced with a single 0.83 M sucrose step. This facilitated collection of the interface and increased GCP yield without decreasing purity (Lohse et al., 1996). The 0.32 M–0.83 M interface (A fraction) was collected, diluted with 0.32 M sucrose and pelleted to give the GCP fraction. This was resuspended in 0.32 M sucrose for experimentation. Similar GCP preparations have been characterized extensively by electron microscopy and biochemical methods (Pfenninger et al., 1983). These studies have revealed that GCPs contain significant amounts of c-src (Helmke and Pfenninger, 1996), tau, GAP-43 (Lohse et al., 1996) and insulin-like growth factor-1 receptor subunits (Quiroga et al., 1995), but lack significant amounts of high molecular weight MAP2, glial fibrillary acidic protein and vimentin (Lohse et al., 1996). Equal amounts of total brain homogenate (30 mg) were separated by SDS–PAGE and analyzed by western immunoblot.
ATPase and microtubule-binding studies
ATPase assays were as described previously (Tsai et al., 2000). Purified rat brain kinesin (0.02 mg/ml) either phosphorylated or not by GSK3 was incubated with taxol-stabilized MAP-free MTs (1 mg/ml) and 1 mM ATP ([γ-32P]ATP, ICN Biochemicals, diluted to 5000 c.p.m./nmol with cold ATP) in a 5 µl volume of IME buffer for 20 min at 37°C. Reactions were stopped by adding 2 µl of 10% SDS. A 2 µl aliquot was spotted on PEI-cellulose plates and developed in 0.5 M LiCl/1 M formic acid. Chromatogram spots corresponding to [32P]phosphate and [γ-32P]ATP were excised and counted. ATP hydrolysis was expressed as the percentage total radioactivity recovered as free phosphate. To determine the effects of GSK3 phosphorylation on kinesin ATPase activity, kinesin was pre-phosphorylated by GSK3 as described above. To control for contributions from residual GSK3 and ATPase activities in the MT fractions, samples containing ATP, GSK3 and MTs or ATP and MTs, but no kinesin, were assayed in parallel and counts subtracted from assays.
MT-binding assays were performed by incubating 80 ng of GSK3-phosphorylated rat brain kinesin (32P-labeled) with taxol-stabilized MT (1 mg/ml) and 1 mM AMP-PNP in PEM buffer for 20 min at 37°C. Samples were centrifuged for 30 min at 4°C at 108 920 _g_max over a cushion of 20% sucrose in PEM. Pellets were collected by resuspending in sample buffer and phosphorylated kinesin detected in each fraction by phosphoimager.
Cell culture, immunocytochemistry and cell imaging
BHK21 and 3T3 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Gibco-BRL) supplemented with 5% fetal bovine serum (FBS; Hyclone) and antibiotics (Cellgro). PC12 cells were cultured in 50% DMEM/50% F12 plus1% FBS and induced to differentiate by 50 ng/ml NGF. Hippocampal cells were cultured as described previously (Goslin et al., 1998). Cells were cultured for 3 days on dishes coated with 0.5 mg/ml poly-d-lysine, then fixed in 2% paraformaldehyde/0.01% glutaraldehyde at 37°C for 10 min. Some cultures were extracted with 0.05% Triton X-100 prior to fixation (Morfini et al., 2000). After fixation, cells were extracted with 0.2% Triton X-100 for 10 min, then blocked in 2.5% bovine serum albumin/2.5% gelatin for 1 h. Primary antibodies were diluted in block solution: anti-kinesin (H2, 20 µg/ml), polyclonal rabbit anti-GSK3β (5 µg/ml) or anti-tubulin (DM1A, 1:400). Secondary antibodies were anti-mouse or anti-rabbit IgG-labeled with OG488 or Texas Red (Molecular Probes) used at 1:400 dilution in block solution. Images were acquired with an Orca CCD camera (Hamamatsu) controlled by Openlab software (Improvision) and processed for presentation in Adobe Photoshop.
Acknowledgments
Acknowledgements
This paper is dedicated to Irene Iscla. The authors want to express gratitude to Robin Wray, Milena Gould and Hannah Brown for invaluable technical assistance, Ross Payne for expert imaging advice, Bruce Richards for sharing unpublished data on rat KLC primary sequence, Clive Slaughter (UT Southwestern) for mass spectrometry analysis, and Dr Joseph Albanesi for critically reading the manuscript. Rat hippocampal neuron cultures were a gift from Dr Anne Marie Craig (University of Illinois at Urbana). GSK3β and GSK3β kinase-dead constructs were a generous gift from Dr H.Eldar-Finkelman (Harvard Medical School). Research was supported in part by grants to S.T.B. from the National Institutes of Health (NS23868, NS23320, NS41170 and AG12646), NASA (NAG2-962), Juvenile Diabetes and Welch Foundations. G.M. was supported in part by a Pew Fellowship from the Latin American Fellowship Program. Support for N.R. at the MBL was generously provided by the University of Cincinnati.
References
- Baum L., Hansen,L., Masliah,E. and Saitoh,T. (1996) Glycogen synthase kinase 3 alteration in Alzheimer disease is related to neurofibrillary tangle formation. Mol. Chem. Neuropathol., 29, 253–261. [DOI] [PubMed] [Google Scholar]
- Bloom G.S., Richards,B.W., Leopold,P.L., Ritchey,D.M. and Brady,S.T. (1993) GTPγS inhibits organelle transport along axonal microtubules. J. Cell Biol., 120, 467–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady M.J., Bourbonais,F.J. and Saltiel,A.R. (1998) The activation of glycogen synthase by insulin switches from kinase inhibition to phosphatase activation during adipogenesis in 3T3-L1 cells. J. Biol. Chem., 273, 14063–14066. [DOI] [PubMed] [Google Scholar]
- Brady S.T. (1985) A novel brain ATPase with properties expected for the fast axonal transport motor. Nature, 317, 73–75. [DOI] [PubMed] [Google Scholar]
- Brady S.T. (1991) Molecular motors in the nervous system. Neuron, 7, 521–533. [DOI] [PubMed] [Google Scholar]
- Brady S.T. (1993) Axonal dynamics and regeneration. In Gorio,A. (ed.), Neuroregeneration. Raven Press, New York, NY, pp. 7–36.
- Brady S.T., Lasek,R.J. and Allen,R.D. (1985) Video microscopy of fast axonal transport in isolated axoplasm: a new model for study of molecular mechanisms. Cell Motil., 5, 81–101. [DOI] [PubMed] [Google Scholar]
- Brady S.T., Pfister,K.K. and Bloom,G.S. (1990a) A monoclonal antibody against kinesin inhibits both anterograde and retrograde fast axonal transport in squid axoplasm. Proc. Natl Acad. Sci. USA, 87, 1061–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady S.T., Pfister,K.K., Leopold,P.L. and Bloom,G.S. (1990b) Fast axonal transport in isolated axoplasm. Cell Motil. Cytoskeleton, 17**(Video Suppl. 2)**, 22. [DOI] [PubMed] [Google Scholar]
- Brady S.T., Richards,B.W. and Leopold,P.L. (1993) Assay of vesicle motility in squid axoplasm. Methods Cell Biol., 39, 191–202. [DOI] [PubMed] [Google Scholar]
- Brownlees J., Irving,N.G., Brion,J.P., Gibb,B.J., Wagner,U., Woodgett,J. and Miller,C.C. (1997) Tau phosphorylation in transgenic mice expressing glycogen synthase kinase-3β transgenes. Neuroreport, 8, 3251–3255. [DOI] [PubMed] [Google Scholar]
- Bullock B.P. and Habener,J.F. (1998) Phosphorylation of the cAMP response element binding protein CREB by cAMP-dependent protein kinase A and glycogen synthase kinase-3 alters DNA-binding affinity, conformation and increases net charge. Biochemistry, 37, 3795–3809. [DOI] [PubMed] [Google Scholar]
- Chalecka-Franaszek E. and Chuang,D.M. (1999) Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proc. Natl Acad. Sci. USA, 96, 8745–8750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland D.W., Fischer,S., Kirschner,M. and Laemmli,R. (1977) Peptide mapping by limited proteolysis in sodium dodecyl sulfate and analysis by electrophoresis. J. Biol. Chem., 254, 12610–12678. [PubMed] [Google Scholar]
- Cook D., Fry,M.J., Hughes,K., Sumathipala,R., Woodgett,J.R. and Dale,T.C. (1996) Wingless inactivates glycogen synthase kinase-3 via an intracellular signalling pathway which involves a protein kinase C. EMBO J., 15, 4526–4536. [PMC free article] [PubMed] [Google Scholar]
- Coy D.L., Hancock,W.O., Wagenbach,M. and Howard,J. (1999) Kinesin’s tail domain is an inhibitory regulator of the motor domain. Nature Cell Biol., 1, 288–292. [DOI] [PubMed] [Google Scholar]
- Craig A.M., Wyborski,R.J. and Banker,G. (1995) Preferential addition of newly synthesized membrane protein at axonal growth cones. Nature, 375, 592–594. [DOI] [PubMed] [Google Scholar]
- Cross D.A., Alessi,D.R., Vandenheede,J.R., McDowell,H.E., Hundal,H.S. and Cohen,P. (1994) The inhibition of glycogen synthase kinase-3 by insulin or insulin-like growth factor 1 in the rat skeletal muscle cell line L6 is blocked by wortmannin, but not by rapamycin: evidence that wortmannin blocks activation of the mitogen-activated protein kinase pathway in L6 cells between Ras and Raf. Biochem. J., 303, 21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross D.A., Alessi,D.R., Cohen,P., Andjelkovich,M. and Hemmings, B.A. (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature, 378, 785–789. [DOI] [PubMed] [Google Scholar]
- Cross D.A., Watt,P.W., Shaw,M., van der Kaay,J., Downes,C.P., Holder,J.C. and Cohen,P. (1997) Insulin activates protein kinase B, inhibits glycogen synthase kinase-3 and activates glycogen synthase by rapamycin-insensitive pathways in skeletal muscle and adipose tissue. FEBS Lett., 406, 211–215. [DOI] [PubMed] [Google Scholar]
- Dailey M.E. and Bridgman,P.C. (1991) Structure and organization of membrane organelles along distal microtubule segments in growth cones. J. Neurosci. Res., 30, 242–258. [DOI] [PubMed] [Google Scholar]
- De Vos K., Severin,F., Van Herreweghe,F., Vancompernolle,K., Goossens,V., Hyman,A. and Grooten,J. (2000) Tumor necrosis factor induces hyperphosphorylation of kinesin light chain and inhibits kinesin-mediated transport of mitochondria. J. Cell Biol., 149, 1207–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Waegh S.M., Lee,V.M.-Y. and Brady,S.T. (1992) Local modulation of neurofilament phosphorylation, axonal caliber and slow axonal transport by myelinating Schwann cells. Cell, 68, 451–463. [DOI] [PubMed] [Google Scholar]
- Eldar-Finkelman H., Schreyer,S.A., Shinohara,M.M., LeBoeuf,R.C. and Krebs,E.G. (1999) Increased glycogen synthase kinase-3 activity in diabetes- and obesity-prone C57BL/6J mice. Diabetes, 48, 1662–1666. [DOI] [PubMed] [Google Scholar]
- Elluru R., Bloom,G.S. and Brady,S.T. (1995) Fast axonal transport of kinesin in the rat visual system: functionality of the kinesin heavy chain isoforms. Mol. Biol. Cell, 6, 21–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goslin K., Asmussen,H. and Banker,G. (1998) Rat hippocampal neurons in low density culture. In Goslin,K. and Banker,G. (eds), Culturing Nerve Cells. MIT Press, Cambridge, MA, pp. 339–370.
- Hackney D.D., Levitt,J.D. and Wagner,D.D. (1991) Characterization of α2β2 and α2 forms of kinesin. Biochem. Biophys. Res. Commun., 174, 810–815. [DOI] [PubMed] [Google Scholar]
- Harlow E. and Lane,D. (1988) Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Helmke S. and Pfenninger,K. (1996) Growth cone enrichment and cytoskeletal association of non-receptor tyrosine kinases. Cell Motil. Cytoskeleton, 30, 194–207. [DOI] [PubMed] [Google Scholar]
- Hetman M., Cavanaugh,J.E., Kimelman,D. and Xia,Z. (2000) Role of glycogen synthase kinase-3β in neuronal apoptosis induced by trophic withdrawal. J. Neurosci., 20, 2567–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N. (1998) Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science, 279, 519–526. [DOI] [PubMed] [Google Scholar]
- Hollenbeck P.J. (1993) Phosphorylation of neuronal kinesin heavy and light chains in vivo. J. Neurochem., 60, 2265–2275. [DOI] [PubMed] [Google Scholar]
- Ikeda S., Kishida,S., Yamamoto,H., Murai,H., Koyama,S. and Kikuchi,A. (1998) Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3β and β-catenin and promotes GSK-3β-dependent phosphorylation of β-catenin. EMBO J., 17, 1371–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro K., Shiratsuchi,A., Sato,S., Omori,A., Arioka,M., Kobayashi,S., Uchida,T. and Imahori,K. (1993) Glycogen synthase kinase 3β is identical to tau protein kinase I generating several epitopes of paired helical filaments. FEBS Lett., 325, 167–172. [DOI] [PubMed] [Google Scholar]
- Kennelly P.J. and Krebs,E.G. (1991) Consensus sequences as substrate specificity determinants for protein kinases and protein phosphatases. J. Biol. Chem., 266, 15555–15558. [PubMed] [Google Scholar]
- Kobayashi Y., Pang,T., Iwamoto,T., Wakabayashi,S. and Shigekawa,M. (2000) Lithium activates mammalian Na+/H+ exchangers: isoform specificity and inhibition by genistein. Pflugers Arch., 439, 455–462. [DOI] [PubMed] [Google Scholar]
- Kreegipuu A., Blom,N. and Brunak,S. (1999) PhosphoBase, a database of phosphorylation sites: release 2.0. Nucleic Acids Res., 27, 237–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krylova O., Messenger,M.J. and Salinas,P.C. (2000) Dishevelled-1 regulates microtubule stability: a new function mediated by glycogen synthase kinase-3β. J. Cell Biol., 151, 83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.-D. and Hollenbeck,P.J. (1995) Phosphorylation of kinesin in vivo correlates with organelle association and neurite outgrowth. J. Biol. Chem., 270, 5600–5605. [DOI] [PubMed] [Google Scholar]
- Leopold P.L., McDowall,A.W., Pfister,K.K., Bloom,G.S. and Brady,S.T. (1992) Association of kinesin with characterized membrane-bounded organelles. Cell Motil. Cytoskeleton, 23, 19–33. [DOI] [PubMed] [Google Scholar]
- Li J.-Y., Pfister,K.K., Brady,S.T. and Dahlström,A. (1999) Axonal transport and distribution of immunologically distinct kinesin heavy chains in rat neurons. J. Neurosci. Res., 58, 226–241. [PubMed] [Google Scholar]
- Lohse K., Helmke,S.M., Wood,M.R., Quiroga,S., de la Houssaye,B.A., Miller,V.E., Negre-Aminou,P. and Pfenninger,K.H. (1996) Axonal origin and purity of growth cones isolated from fetal rat brain. Brain Res. Dev. Brain Res., 96, 83–96. [DOI] [PubMed] [Google Scholar]
- Lucas J.J., Hernandez,F., Gomez-Ramos,P., Moran,M.A., Hen,R. and Avila,J. (2001) Decreased nuclear β-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3β conditional transgenic mice. EMBO J., 20, 27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow E.M., Drewes,G., Biernat,J., Gustke,N., Van Lint,J., Van den heede,J.R. and Mandelkow,E. (1992) Glycogen synthase kinase-3 and the Alzheimer-like state of microtubule-associated protein tau. FEBS Lett., 314, 315–321. [DOI] [PubMed] [Google Scholar]
- Matthies H.J., Miller,R.J. and Palfrey,H.C. (1993) Calmodulin binding to and cAMP-dependent phosphorylation of kinesin light chains modulate kinesin ATPase activity. J. Biol. Chem., 268, 11176–11187. [PubMed] [Google Scholar]
- McGuinness T.L., Brady,S.T., Gruner,J., Sugimori,M., Llinas,R. and Greengard,P. (1989) Phosphorylation-dependent inhibition by synapsin I of organelle movement in squid axoplasm. J. Neurosci., 9, 4138–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlvain J.M., Burkhardt,J.K., Hamm-Alvarez,S., Argon,Y. and Sheetz,M.P. (1994) Regulation of kinesin activity by phosphorylation of kinesin associated proteins. J. Biol. Chem., 269, 19176–19182. [PubMed] [Google Scholar]
- Morfini G., Tsai,M., Szebenyi,G. and Brady,S.T. (2000) Approaches to study interactions between kinesin motors and membranes. In Vernos,I. (ed.), Kinesin Protocols. Vol. 164. Humana Press, Totowa, NJ, pp. 147–162. [DOI] [PubMed]
- Morfini G., Szebenyi,G., Richards,B. and Brady,S.T. (2001) Regulation of kinesin: implications for neuronal development. Dev. Neurosci., 23, 364–376. [DOI] [PubMed] [Google Scholar]
- Okada Y., Sato-Yoshitake,R. and Hirokawa,N. (1995) The activation of protein kinase A pathway selectively inhibits anterograde axonal transport of vesicles but not mitochondria transport or retrograde transport in vivo. J. Neurosci., 15, 3053–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfenninger K., Ellis,L., Johnson,M., Friedman,L. and Somlo,S. (1983) Nerve growth cones isolated from fetal rat brain. I. Subcellular fractionation and characterization. Cell, 35, 573–584. [DOI] [PubMed] [Google Scholar]
- Pfister K.K., Wagner,M.C., Stenoien,D., Bloom,G.S. and Brady,S.T. (1989) Monoclonal antibodies to kinesin heavy and light chains stain vesicle-like structures, but not microtubules, in cultured cells. J. Cell Biol., 108, 1453–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plyte S.E., Hughes,K., Nikolakaki,E., Pulverer,B.J. and Woodgett,J.R. (1992) Glycogen synthase kinase-3: functions in oncogenesis and development. Biochim. Biophys. Acta, 1114, 147–162. [DOI] [PubMed] [Google Scholar]
- Polakis P. (2000) Wnt signaling and cancer. Genes Dev., 14, 1837–1851. [PubMed] [Google Scholar]
- Quiroga S., Garofalo,R.S. and Pfenninger,K.H. (1995) Insulin-like growth factor I receptors of fetal brain are enriched in nerve growth cones and contain a β-subunit variant. Proc. Natl Acad. Sci. USA, 92, 4309–4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratner N., Bloom,G.S. and Brady,S.T. (1998) A role for Cdk5 kinase in fast anterograde axonal transport: novel effects of olomoucine and the APC tumor suppressor protein. J. Neurosci., 18, 7717–7726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas P.C. (1999) Wnt factors in axonal remodelling and synaptogenesis. Biochem. Soc. Symp., 65, 101–109. [PubMed] [Google Scholar]
- Sanchez S., Sayas,C.L., Lim,F., Diaz-Nido,J., Avila,J. and Wandosell,F. (2001) The inhibition of phosphatidylinositol-3-kinase induces neurite retraction and activates GSK3. J. Neurochem., 78, 468–481. [DOI] [PubMed] [Google Scholar]
- Sato-Yoshitake R., Yorifuji,H., Inagaki,M. and Hirokawa,N. (1992) The phosphorylation of kinesin regulates its binding to synaptic vesicles. J. Biol. Chem., 267, 23930–23936. [PubMed] [Google Scholar]
- Seidensticker M.J. and Behrens,J. (2000) Biochemical interactions in the wnt pathway. Biochim. Biophys. Acta, 1495, 168–182. [DOI] [PubMed] [Google Scholar]
- Seiler S., Kirchner,J., Horn,C., Kallipolitou,A., Woehlke,G. and Schliwa,M. (2000) Cargo binding and regulatory sites in the tail of fungal conventional kinesin. Nature Cell Biol., 2, 333–338. [DOI] [PubMed] [Google Scholar]
- Slice L.W. and Taylor,S.S. (1989) Expression of the catalytic subunit of cAMP-dependent protein kinase in Escherichia coli. J. Biol. Chem., 264, 20940–20946. [PubMed] [Google Scholar]
- Stenoien D.S. and Brady,S.T. (1997) Immunochemical analysis of kinesin light chain function. Mol. Biol. Cell, 8, 675–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers S.A., Kao,A.W., Kohn,A.D., Backus,G.S., Roth,R.A., Pessin,J.E. and Birnbaum,M.J. (1999) The role of glycogen synthase kinase 3β in insulin-stimulated glucose metabolism. J. Biol. Chem., 274, 17934–17940. [DOI] [PubMed] [Google Scholar]
- Tsai M.-Y., Morfini,G., Szebenyi,G. and Brady,S.T. (2000) Modulation of kinesin–vesicle interactions by Hsc70: implications for regulation of fast axonal transport. Mol. Biol. Cell, 11, 2161–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale R.D., Reese,T.S. and Sheetz,M.P. (1985) Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell, 42, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallee R.B. (1982) A taxol-dependent procedure for the isolation of microtubules and microtubule-associated proteins. J. Cell Biol., 92, 435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhey K.J., Lizotte,D.L., Abramson,T., Barenboim,L., Schnapp,B.J. and Rapoport,T.A. (1998) Light chain-dependent regulation of kinesin’s interaction with microtubules. J. Cell Biol., 143, 1053–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner M.C., Pfister,K.K., Brady,S.T. and Bloom,G.S. (1991) Purification of kinesin from bovine brain and assay of microtubule-stimulated ATPase activity. Methods Enzymol., 196, 157–175.1851939 [Google Scholar]
- Wang Q.M., Fiol,C.J., DePaoli-Roach,A.A. and Roach,P.J. (1994a) Glycogen synthase kinase-3β is a dual specificity kinase differentially regulated by tyrosine and serine/threonine phosphorylation. J. Biol. Chem., 269, 14566–14574. [PubMed] [Google Scholar]
- Wang Q.M., Roach,P.J. and Fiol,C.J. (1994b) Use of a synthetic peptide as a selective substrate for glycogen synthase kinase 3. Anal. Biochem., 220, 397–402. [DOI] [PubMed] [Google Scholar]
- Woodgett J.R. (1991) cDNA cloning and properties of glycogen synthase kinase-3. Methods Enzymol., 200, 564–577. [DOI] [PubMed] [Google Scholar]
- Woodgett J.R. (1994) Regulation and functions of the glycogen synthase kinase-3 subfamily. Semin. Cancer Biol., 5, 269–275. [PubMed] [Google Scholar]
- Yang S.D., Yu,J.S., Fong,Y.L. and Liu,J.S. (1992) The mechanism of activation of protein kinase FA (the activator of type-1 protein phosphatase) in brain synaptosomes. Biochem. Biophys. Res. Commun., 182, 129–136. [DOI] [PubMed] [Google Scholar]
- Yost C., Farr,G.H.,III, Pierce,S.B., Ferkey,D.M., Chen,M.M. and Kimelman,D. (1998) GBP, an inhibitor of GSK-3, is implicated in Xenopus development and oncogenesis. Cell, 93, 1031–1041. [DOI] [PubMed] [Google Scholar]