c-Abl is an effector of Src for growth factor-induced c-myc expression and DNA synthesis (original) (raw)
Abstract
The mechanism by which the ubiquitously expressed Src family kinases regulate mitogenesis is not well understood. Here we report that cytoplasmic tyrosine kinase c-Abl is an important effector of c-Src for PDGF- and serum-induced DNA synthesis. Inactivation of cytoplasmic c-Abl by the kinase- inactive Abl-PP-K– (AblP242E/P249E/K290M) or by microinjection of Abl neutralizing antibodies inhibited mitogenesis. The kinase-inactive SrcK295M induced a G1 block that was overcome by the constitutively active Abl-PP (AblP242E/P249E). Conversely, the inhibitory effect of Abl-PP-K– was not compensated by Src. c-Src-induced c-Abl activation involves phosphorylation of Y245 and Y412, two residues required for c-Abl mitogenic function. Finally, we found that p53 inactivation and c-myc expression, two cell cycle events regulated by Src during mitogenesis, also implied c-Abl: c-Abl function was dispensable in cells deficient in active p53 and inhibition of c-Abl reduced mitogen-induced c-myc expression. These data identify a novel function of cytoplasmic c-Abl in the signalling pathways regulating growth factor-induced c-myc expression and we propose the existence of a tyro sine kinase signalling cascade (PDGFR/c-Src/c-Abl) important for mitogenesis.
Keywords: c-Abl/c-Myc/c-Src/DNA synthesis/growth factors
Introduction
Growth factors transmit their mitogenic response by associating with receptors with intrinsic tyrosine kinase activity. Ligand binding triggers receptor dimerization and subsequent transphosphorylation allowing recruitment of signalling proteins for mitogenesis. These include proteins with catalytic activity such as the phospholipase Cγ, the phosphoinositide 3-kinase, the GTPase activating protein RasGAP, the tyrosine phosphatase Shp2, and adapters such as Nck, Shc and Grb2 (van der Geer et al., 1994). Most play important roles in the induction of DNA synthesis (van der Geer et al., 1994; Roche et al., 1996). We and others have found that in addition to these molecules, the Src family tyrosine kinases were also required for mitogenesis (Twamley-Stein et al., 1993; Roche et al., 1995; Broome and Hunter, 1996). However, the mechanism by which they transmit mitogenic signals is largely unknown. Recently, it has been reported that the expression of the SV40 large T-antigen can bypass the need for Src kinases in PDGF-induced mitogenesis (Broome and Courtneidge, 2000). This allowed the proposal of a functional link between the tumour suppressor p53 and c-Src. Therefore an important role of c-Src may be to inhibit a p53-dependent pathway in order for cells to enter S phase.
A key intermediate for mitogenesis is the expression of immediate early G1 genes, several of which are involved in G1 progression for S phase entry. Of particular interest are the proto-oncogenes encoding the transcription factors c-Fos and c-Myc. The mechanism by which growth factors induce c-fos expression is well established (Treisman, 1994). In contrast, the signalling proteins regulating c-myc expression are largely unknown. A Ras-dependent pathway has been reported for the stabilization and substantial accumulation of the protein in the cell (Sears et al., 1999). However, there is no evidence for a role of Ras in c-myc transcriptional expression. On the other hand, Src kinases have been reported to be critically involved in c-myc induction (Barone and Courtneidge, 1995; Blake et al., 2000; Chiariello et al., 2001). The role of Src during mitogenesis is thought to phosphorylate specific substrates. These include negative regulators such as the adapter c-Cbl and the protein kinase C δ, and positive regulators such as the adapter Shc and the transcription factor STAT3 (Abram and Courtneidge, 2000). So far only Shc and STAT3 have recently been linked to c-myc expression (Gotoh et al., 1997; Blake et al., 2000; Bowman et al., 2001).
The cytoplasmic tyrosine kinase c-Abl is another newly described Src substrate (Plattner et al., 1999). It belongs to the non-receptor tyrosine kinase family, distinct from the Src family, with a dual nuclear and cytoplasmic localization. Like c-Src, it comprises a myristoylation site, an SH2 and SH3 domain involved in protein–protein interaction and a catalytic domain (Van Etten, 1999). Additionally, it has a unique large C-terminus that contains polyproline-rich regions, nuclear localization and export signals, as well as F- and G-actin binding domains. c-Abl displays oncogenic activity when deregulated and human oncogenic forms have been identified with important functions in leukaemia (Sawyers, 1999). While its oncogenic function is well described (Van Etten, 1999), the role of c-Abl in regulation of normal cell responses remains elusive. A function has been ascribed in apoptosis, during DNA damage and cell growth, all of them referring to the nuclear pool of c-Abl (Van Etten, 1999). The role of c-Abl in cytoplasm is not clear. A role in cytoskeletal rearrangement has been ascribed to c-Abl (Van Etten, 1999), as well as in signal transduction initiated by cell adhesion (Lewis et al., 1996) and growth factors (Plattner et al., 1999). Plattner and colleagues showed the involvement of c-Abl in membrane ruffling induced by PDGF and that this kinase is a target of activated receptor tyrosine kinases and Src family kinases.
Oncogenic forms of Abl are excluded from the nucleus, indicating that c-Abl can relay positive signals for the promotion of DNA synthesis via its cytoplasmic function (Van Etten, 1999). This has recently been confirmed by entrapment of BCR-ABL to the nucleus, which results in induction of apoptosis instead of cell growth (Vigneri and Wang, 2001). Moreover, it has been suggested that like Src, activated forms of Abl can induce c-myc expression in fibroblasts, and this was required for cell transformation (Sawyers et al., 1992; Afar et al., 1994; Wong et al., 1995). Based on these data, we postulated that cytoplasmic c-Abl is an important effector of the Src pathway during growth factor-induced mitogenesis. Using various approaches that specifically affect cytoplasmic c-Abl activity, we report here that cytoplasmic c-Abl has a positive function during mitogenesis. We also show that it is an important c-Src effector for both inhibiting a p53-dependent function and inducing c-myc transcriptional expression. This highlights a new function of c-Abl in signal transduction and suggests the existence of a tyrosine kinase signalling cascade outside the Ras pathway that is also important for mitogenesis.
Results
Cytoplasmic c-Abl lies in the Src pathway during mitogenesis
We found that in NIH 3T3 fibroblasts, a double proline mutation in the regulatory SH2-CD linker (P242E/P249E, Abl-PP) conferring constitutive activity to c-Abl (Figure 1A) (Barila and Superti-Furga, 1998) is localized in the cytoplasm. NIH 3T3 fibroblasts were transiently transfected with constructs encoding Abl and Abl-PP, and were tested for ectopic protein localization by indirect immunostaining with specific antibody. An example of the images obtained is depicted in Figure 1B; in contrast to Abl, Abl-PP was strictly cytoplasmic. Similar results were obtained when protein localization was assessed by western blotting after subcellular fractionation (Figure 1C). A construct encoding the kinase-dead counterpart (AblP242E/P249E/K290M, Abl-PP-K–) was used to attempt to inhibit the c-Abl cytoplasmic function in vivo. Like Abl-PP, Abl-PP-K– displayed cytoplasmic localization (Figure 2A). The effect of Abl-PP-K– was tested on PDGF-induced mitogenic response. NIH 3T3 cells were transiently transfected with constructs expressing Abl- PP-K– or Abl-PP, synchronized in G0 by serum starvation and stimulated with PDGF for cell cycle re-entry. DNA synthesis was monitored by adding bromodeoxyuridine (BrdU), which would incorporate into DNA during S phase of the cell cycle. An example of such an experiment is shown in Figure 2A and the statistical analysis in Figure 2B. While Abl-PP did not affect BrdU incorporation induced by PDGF, Abl-PP-K– strongly inhibited this response. Similar results were obtained when constructs were microinjected into quiescent fibroblasts or transfected in mouse embryo fibroblasts (MEF) (Figure 3B and our unpublished data), indicating that this effect is specific neither for the cell line nor for the approach we used. In order to confirm the involvement of cytoplasmic c-Abl in this cellular response, we also tested the effect of Abl-PP and Abl-PP-K– mutants with inactivated nuclear localization signals (Abl-PP-NLS– and Abl-PP-K–NLS–) and we found that, like Abl-PP-K–, Abl-PP-K–NLS– inhibited PDGF mitogenic signalling (Figure 2B).

Fig. 1. Subcellular localization of Abl and Abl-PP. (A) Schematic representation of Abl and Abl-PP. Abl-PP contains a point mutation in the linker region between the SH2 domain and the catalytic sequence, replacing two prolines with two glutamates (P242E/P249E), and resulting in constitutive activity of Abl. The myristoylation site (\/\/), SH2, SH3, linker, catalytic and C-terminal sequences are indicated. (B) Immunostaining of Abl and Abl-PP expressed in fibroblasts. NIH 3T3 cells were transiently transfected with Abl and Abl-PP, and fixed and stained for Abl expression by indirect immunofluorescence. Shown is a representative example of images that were obtained after digital restoration (Huygens software) and visualized using shadow projection (Imaris software) as described in Materials and methods. (C) Abl and Abl-PP levels in various subcellular fractions. Lysates of NIH 3T3 cells overexpressing Abl and Abl-PP were fractionated into cytoplasmic/membrane (membrane+cyto) and nuclear (Nuc) fractions as described in Materials and methods. Equal quantities of proteins were subjected to SDS–PAGE and the Abl level was assessed by western blotting using 24-21 monoclonal antibody. The position of Abl and the fraction used are indicated.

Fig. 2. Abl-PP-K–, but not Abl-PP, inhibits PDGF-induced DNA synthesis. NIH 3T3 cells plated onto coverslips were transiently transfected with the indicated Abl-PP or Abl-PP-NLS– mutants for 24 h. After serum starvation, cells were stimulated or not with PDGF in the presence of BrdU. Eighteen hours later, cells were fixed and stained for Abl expression and BrdU incorporation by double immunostaining, and analysed by microscopy. (A) An example of double immunostaining analysis. The white arrowheads mark the position of cells expressing the indicated ectopic protein. (B) Statistical analysis. Shown is the percentage of BrdU-positive cells present in expressing and non-expressing cells under the specified conditions. For each coverslip, the percentage of BrdU incorporation was calculated according to the following formula: % of BrdU-positive cells = (number of BrdU-positive cells/number of cells) × 100. For each coverlip, approximately 150–200 cells were counted. The data from four independent experiments have been averaged, and the mean and standard deviation are shown.



Fig. 3. Like SrcK–, Abl-PP-K– G1 block is overcome when inhibiting p53 function. (A) Inactivation of p53 rescues the G1 block induced by kinase-inactive Src (SrcK–) in MEF cells. Cells were plated onto coverslips and transiently co-transfected with SrcK– and the indicated construct. After serum starvation, cells were stimulated or not with PDGF (top panel) or serum (bottom panel) in the presence of BrdU for 18 h. (B) Inactivation of p53 rescues the G1 block induced by kinase-inactive Abl (Abl-PP-K–) in MEF cells. Cells were plated onto coverslips and transiently co-transfected with Abl-PP-K– and the indicated construct, and treated as in (A). (C) Inactivation of p53 rescues the PDGF mitogenic response inhibited by SrcK– or Abl-PP-K– in NIH 3T3 cells. Cells were plated onto coverslips and transiently transfected either with SrcK– or Abl-PP-K– in the presence or absence of E1B, then treated as in (A). Cells were fixed, stained for kinase expression and BrdU incorporation by double immunostaining, and analysed by microscopy. Shown is the percentage of BrdU-positive cells present in expressing and non-expressing cells under the specified conditions, as calculated in the legend to Figure 2. The data from three to five independent experiments have been averaged, and the mean and standard deviation are shown.
We next investigated whether Abl-PP-K– could affect an Src-dependent pathway. We first confirmed that c-Src regulates a p53-dependent event in fibroblasts (Broome and Courtneidge, 2000). To this end, we used the adenovirus early 1B (E1B) 55K protein that inhibits p53 transactivation (Yew and Berk, 1992). As shown in Figure 3A (top panel), PDGF induced a 30% BrdU incorporation in MEF cells that was abrogated when overexpressing the kinase-inactive form of Src (K295M) (SrcK–). Co-expression of E1B circumvents the G1 arrest induced by SrcK–. This was attributed to the capacity of the viral protein to target p53, since the mutant E1BR309 that no longer inhibits p53 function had no effect. The functional link between c-Src and p53 was reinforced by similar rescue effects obtained with the transcriptionally inactive form of p53, p53H273. Collectively, this data confirms that PDGF-induced DNA synthesis requires the inhibition of a p53-dependent pathway that is controlled by c-Src. Interestingly, similar results were obtained with serum response (Figure 3A, bottom panel): serum induced a 50% BrdU incorporation that was abrogated by SrcK–. Like PDGF, the G1 block was overcome by E1B and p53H273, but not by E1BR309. Therefore, inactivation of a p53-dependent pathway may be a general function of Src during mitogenesis. We next analysed whether Abl-PP-K– also affects a similar pathway. As shown in Figure 3B, Abl-PP-K– indeed inhibited PDGF- and serum-induced mitogenic response, and both E1B and p53H273 co-expression counteracted this inhibition. The effect of E1B was due to inactivation of endogenous p53, as the E1BR309 mutant could not rescue the G1 block. The rescue effects of E1B and p53H273 still required the presence of growth factor, indicating that they did not induce DNA synthesis by themselves. Since we used an early passage of primary culture of MEF cells, we tested whether this pathway also exists in spontaneously immortalized NIH 3T3 cells. As shown in Figure 3C, neither SrcK– nor Abl-PP-K– inhibited PDGF-induced DNA synthesis in the presence of E1B, confirming the functional interaction between SrcK–, Abl-PP-K– and p53.
One interpretation of these observations is that Abl-PP-K– associates with important c-Src substrates for mitogenic signalling. If this is true, then c-Src co-expression may bypass the inhibitory effect induced by Abl-PP-K–. However, neither wild-type Src nor the activated form SrcY527F restored PDGF signalling (Figure 4A). In contrast, Abl-PP largely rescued the G1 block induced by SrcK– (Figure 4B); this was specific for c-Abl, since co-expression of Tec, a cytoplasmic tyrosine kinase distinct from the Abl family, had no effect. Therefore, c-Abl may phosphorylate specific substrates for mitogenic signalling. We also found that the compensatory effect of Abl-PP was specific for the Src pathway, as it did not restore the signalling blocked by the dominant-negative form of Ras, RasN17. The domains of c-Abl involved in this response were analysed next (Figure 4B). While still active in vivo (unpublished data), mutants with an inactive SH2 domain (Abl-PP-SH2*) or a C-terminal deletion (Abl-PP-ΔC) did not significantly restore PDGF signalling. From this data we concluded that c-Abl lies downstream of c-Src and that its function requires both the SH2 and C-terminal sequences.

Fig. 4. Abl-PP largely restores the PDGF mitogenic response inhibited by SrcK–, while Src does not overcome the Abl-PP-K– inhibition. (A) Src does not rescue the G1 block induced by Abl-PP-K–. NIH 3T3 cells were plated onto coverslips and transiently co-transfected with Abl-PP-K– and the indicated Src construct. After serum starvation, cells were stimulated or not with PDGF in the presence of BrdU for 18 h. (B) Abl-PP largely rescues the G1 block induced by SrcK– and this compensatory effect requires both the SH2 and C-terminal domains of Abl. NIH 3T3 cells were plated onto coverslips and transiently co-transfected either with SrcK– or RasN17 and the indicated construct. After serum starvation, cells were stimulated or not with PDGF in the presence of BrdU for 18 h. Cells were then fixed and stained for Abl (A), Src (B) or Ras (B) expression and BrdU incorporation, as described in Materials and methods. The data are presented as the percentage of BrdU-positive cells present in expressing and non-expressing cells under the specified conditions and was calculated as in Figure 2. The data from three to five independent experiments have been averaged, and the mean and standard deviation are shown.
We next confirmed the role of cytoplasmic c-Abl with an antibody microinjection approach. Three antibodies were used in this study: two specific for the SH2 and SH3 domains, respectively, and 19-110 specific for the catalytic sequence. All of these recognized the native protein when translated in vitro and formed immunocomplexes with endogenous c-Abl expressed in MEF cells (Figure 5A). While anti-SH2 and anti-SH3 antibodies recognized the relative kinase Arg, we found that Abl is the major kinase of this class expressed in murine fibroblasts (data not shown). The effect of these antibodies was next analysed in vivo by injection into the cytoplasm of quiescent MEF cells that were then stimulated with PDGF in the presence of BrdU. Anti-AblSH2 and anti-AblSH3 antibodies are expected to inhibit the association of respective AblSH2 and AblSH3 ligands, while 19-110 antibody may reduce catalytic activity in vivo. This notion was supported by the fact that 19-110 antibody reduced 50% of Abl catalytic activity in vitro, while anti-AblSH2 and anti-AblSH3 antibodies had no effect (Figure 5A). The cst.1 antibody that inhibits Src kinases (Roche et al., 1995) was used as a positive control and, as expected, antibody injection abrogated PDGF-induced DNA synthesis. Both anti-AblSH2 and 19-110 antibodies gave 70 and 65% inhibition, respectively (Figure 5B). In contrast, anti-AblSH3 antibody did not affect signalling, showing specificity of antibody inhibition. We next tested their effect in MEF cells that do not express p53 (p53–/– MEF). As shown in Figure 5C, the cst.1 antibody failed to inhibit PDGF-induced DNA synthesis as described previously (Broome and Courtneidge, 2000), and no significant inhibition was observed with the anti-AblSH2 antibody. From these experiments, we concluded that cytoplasmic c-Abl is required for PDGF mitogenic response and that one of its important functions is to inhibit a p53-dependent pathway.

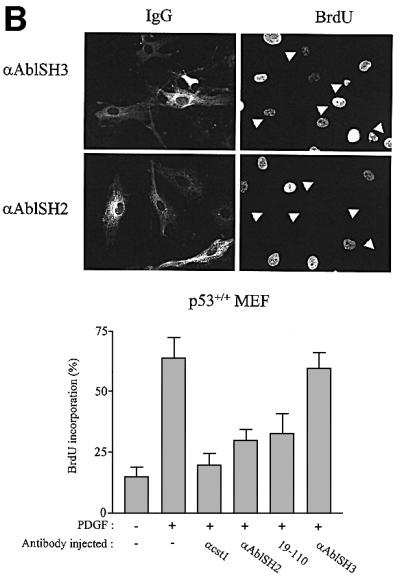
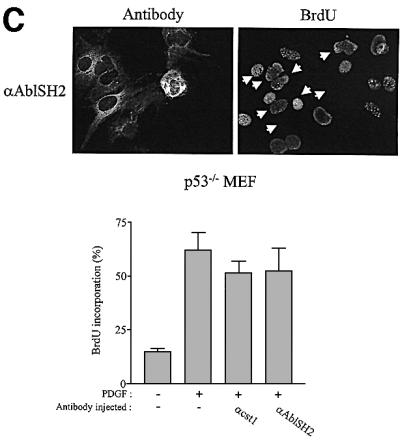
Fig. 5. Cytoplasmic microinjection of antibodies specific for Abl inhibited PDGF mitogenic response in p53+/+, but not in p53–/– MEF cells. (A) Characterization of Abl antibodies. c-Abl that was in vitro translated in reticulocytes lysates in the presence of [35S]methionine was immunoprecipitated (ip) with the indicated antibody, followed by SDS–PAGE and autoradiography (top panel). Endogenous c-Abl was immunoprecipitated (ip) from a MEF cell lysate with the indicated antibody and revealed by western blotting (wb) (middle panel) using the Abl monoclonal 24-21 antibody. The position of Abl is shown. Purified Abl was incubated or not with the indicated antibody and subjected to an in vitro kinase assay using GST–Crk as a substrate (bottom panel). Indicated is the position of radiolabelled GST–Crk as well as the kinase activity obtained relative to the control (c-Abl activity in the absence of any antibody). This data represents one of two independent experiments. (B) Abl antibodies inhibit PDGF mitogenic response in p53+/+ MEF cells. p53+/+ MEF cells plated onto coverslips and made quiescent by serum starvation were microinjected into the cytoplasm with the indicated antibody and stimulated with PDGF in the presence of BrdU for 18 h. Cells were then fixed and stained for BrdU incorporation. The injected antibodies were visualized with a FITC-conjugated antibody. An example of a representative antibody microinjection experiment is shown in the top panel. White arrowheads mark the position of injected cells. The bottom panel represents the percentage of BrdU-positive cells present in injecting and non-injecting cells under the specified conditions, calculated as in Figure 2. The data were obtained from four or five independent experiments (with >80 injected cells for each experiment) that had been averaged, and the mean and standard deviation are shown. (C) Inhibition of endogenous cytoplasmic c-Abl fails to inhibit PDGF mitogenesis in MEF deficient in p53. p53–/– MEF cells plated onto coverslips and made quiescent by serum starvation were microinjected into the cytoplasm with the indicated antibody and stimulated with PDGF in the presence of BrdU for 18 h. Cells were fixed and stained for BrdU incorporation. The injected antibodies were visualized with a FITC-conjugated antibody. An example of a representative antibody microinjection experiment is shown on top panel. White arrows mark the position of injected cells. The bottom panel represents the percentage of BrdU-positive cells present in injecting and non-injecting cells under the specified conditions, as calculated in Figure 2. The data were obtained from four or five independent experiments (with >80 injected cells for each experiment) that had been averaged, and the mean and standard deviation are shown.
Finally, the mitogenic function of cytoplasmic Abl was also confirmed in spontaneously immortalized 3T3 cells deficient in abl and arg genes. While no apparent cell growth defect was previously reported when incubated in the presence of 15% serum (Koleske et al., 1998), we noticed that Abl/Arg–/– 3T3 cells grow more slowly in the presence of a slightly reduced concentration of mitogens (data not shown). This biological defect was confirmed when looking at the mitogenic response induced by PDGF or 10% serum. As shown in Figure 6A, a 4–6 h delay into S phase entry was observed in Abl/Arg–/– 3T3 cells relative to parental cells. We next generated Abl/Arg–/– 3T3 fibroblasts stably expressing Abl-NLS– and observed that, in agreement with a function for Abl in morphological change (Plattner et al., 1999), the introduction of Abl-NLS– in Abl/Arg–/– 3T3 cells at a similar level to cytoplasmic Abl expressed in Abl/Arg+/+ cells restored the dorsal/circular ruffling induced by PDGF (Figure 6B). Most importantly, the number of cells that incorporated BrdU after 16 h of PDGF or serum stimulation was significantly higher (Figure 6C), confirming that cytoplasmic Abl relay the mitogenic signal for S phase entry.


Fig. 6. Quiescent Abl/Arg–/–3T3 fibroblasts exhibit a 4–6 h delay in S phase entry when induced by PDGF or serum. (A) Cells deficient in Abl and Arg exhibit a 4–6 h delay in S phase entry induced by mitogens. Abl/Arg+/+ 3T3 (grey bars) and Abl/Arg–/– 3T3 (black bars) cells were grown on coverslips and made quiescent by serum starvation. Cells were stimulated or not with PDGF (top panel) or serum (bottom panel), as indicated in the presence of BrdU for indicated times, fixed, stained for BrdU incorporation by immunostaining and analysed by microscopy. Shown is the percentage of BrdU-positive cells under the specified conditions, calculated as in Figure 2. These data are representative of two independent experiments. (B) Abl-NLS– restores the actin reorganization induced by PDGF in Abl/Arg–/– 3T3 cells. Top panel: Abl/Arg–/– 3T3 and Abl/Arg–/– 3T3 expressing the indicated constructs grown onto coverslips were made quiescent by serum starvation, stimulated with PDGF for 5 min and fixed. Actin was visualized by staining with rhodamin–phalloidin. Pictures are representative of two independent experiments. Bottom panel: level of immunoprecipitated Abl from the cytoplasmic fraction of the indicated cells. (C) Abl-NLS– restores mitogenesis in Abl/Arg–/– 3T3 cells. Cells were stimulated or not with PDGF (20 ng/ml) or serum (10%), as indicated, in the presence of BrdU for 16 h, then fixed and stained for BrdU incorporation and analysed by microscopy. Shown is the percentage of BrdU-positive cells under the specified conditions, calculated as described in Figure 2. The data from three independent experiments have been averaged, and the mean and standard deviation are shown.
Src-induced Abl activation involves phosphorylation of two residues requiredfor c-Abl function
The mechanism by which c-Src induced c-Abl activation during mitogenesis was next investigated. Brasher and Van Etten (2000) suggested the involvement of Y412 and Y245 in the regulation of intrinsic activity. We thus tested whether these residues are involved in c-Src-induced c-Abl activation. This study required the use of a regulated cytoplasmic allele of c-Abl, therefore excluding Abl-PP due to its constitutive kinase activity (Barila and Superti-Furga, 1998). Instead, a regulated form of c-Abl with inactivated nuclear localization signals (Abl-NLS–) was used. Abl-NLS– constructs were transiently expressed in human embryonic kidney (HEK)-293 cells in the presence or absence of activated Src, and Abl mutants were tested for their tyrosine phosphorylation content and in vitro activity. As shown in Figure 7A, Src induced Abl-NLS– tyrosine phosphorylation concomitant with an 8-fold increase in activity. The replacement of Y412 with a Phe (AblY412F-NLS–) strongly reduced Abl-NLS– tyrosine phosphorylation and Src-induced activity. Moreover, Abl-NLS– with a double Y412/Y245 mutation was neither tyrosine phosphorylated nor activated by Src. We concluded that Y412 and Y245 are two residues involved in c-Src-mediated c-Abl phosphorylation and catalytic activation. We also found that a similar mechanism participates in PDGF-induced c-Abl activation in fibroblasts. Since endogeneous c-Abl tyrosine phosphorylation is downregulated by the PEST-type tyrosine phosphatase (Cong et al., 2000), cells were pretreated with the potent inhibitor of tyrosine phosphatases, pervanadate. Such a treatment leads to a significant increase in c-Abl tyrosine phosphorylation and catalytic activity (Cong et al., 2000; Dorey et al., 2001). Nevertheless, PDGF further increased tyrosine phosphorylation and catalytic activity in Abl/Arg–/– 3T3 cells expressing Abl-NLS–, but not in cells expressing AblY245F/Y412F-NLS– (Figure 7B). These biochemical events were largely reduced by a low dose of the Src inhibitor PP1, implicating Src in this cellular process. Note that at 1 µM this drug reduced Src activity but did not affect Abl or PDGF receptor activity in vivo (Chiariello et al., 2001; and data not shown). Finally, the role of these residues was investigated in vivo (Figure 7C). Abl-NLS– did not affect PDGF mitogenic response in MEF cells, confirming the positive function of cytoplasmic c-Abl. In contrast, AblY412F-NLS– partially inhibited PDGF signalling (∼40%) and AblY245F/Y412F-NLS– acted as a dominant negative. Moreover, AblY245F/Y412F-NLS– was unable to overcome the defect of dorsal/circular ruffling and S phase entry in Abl/Arg–/– 3T3 cells (Figure 6B and C). We concluded that both Y245 and Y412 are required for c-Abl function.


Fig. 7. Src-induced Abl activation involves phosphorylation of two residues required for c-Abl mitogenic function. (A and B) Src activates cytoplasmic Abl via phosphorylation of both Y412 and Y245. (A) HEK-293 cells were transiently transfected with the indicated Abl-NLS– construct together with SrcY527F or empty vector. (B) Quiescent Abl/Arg–/– 3T3 cells expressing Abl-NLS– or Abl-Y245F/Y412F-NLS–, as indicated, were pre-treated for 15 min with vanadate (100 µM) in the presence or absence of PP1 (1 µM), then stimulated or not with PDGF (25 ng/ml) for 10 min. Abl mutants were immunoprecipitated and analysed for Abl protein level and Abl tyrosine phosphorylation content by western blotting using 24-21 and 4G10 antibodies respectively, and for their in vitro kinase activity using GST–Crk as a substrate. The position of Abl, tyrosine phosphorylated Abl and radiolabelled GST–Crk are shown. (A, bottom panel) Src-induced Abl mutant activation obtained from three independent experiments that were averaged; the mean and standard deviation are shown. Kinase activation (fold control) was calculated as described in Materials and methods. Abl activation relative to the control (c-Abl activity from unstimulated cells) obtained in (B) is indicated, and is representative of two independent experiments. (C) Abl mitogenic function requires both Y412 and Y245. MEF cells plated onto coverslips were transiently transfected with the indicated Abl-NLS– construct. After serum starvation, cells were stimulated or not with PDGF in the presence of BrdU for 18 h. Cells were fixed and stained for Abl expression and BrdU incorporation by double immunostaining, and analysed by microscopy. Shown is the percentage of BrdU-positive cells present in expressing and non-expressing cells under the specified conditions, calculated as in Figure 2. The data from four independent experiments have been averaged, and the mean and standard deviation are shown.
c-Abl controls growth factor-induced c-myc expression
Finally, we analysed whether c-Abl lies in the c-Src signalling cascade inducing c-myc expression during mitogenesis. First, we found that c-Myc expression can bypass the inhibitory effect induced by Abl-PP-K– during PDGF stimulation, revealing a functional interaction between c-Abl and c-Myc (Figure 8A). This was dependent on growth factor stimulation, since c-Myc per se did not induce DNA synthesis. Secondly, we investigated whether c-Abl is involved in PDGF-induced c-myc expression. To this end, the c-myc mRNA level was determined by northern blotting from quiescent Abl/ Arg+/+ 3T3 cells stimulated with PDGF and compared with the response obtained from Abl/Arg–/– 3T3 cells and Abl/Arg–/– 3T3 cells expressing Abl-NLS–. As shown in Figure 8B, PDGF induced a 10-fold increase in mRNA level, a response that was largely impaired in cells that do not express Abl kinases (∼80% reduction). This inhibition was largely attributed to the lack of cytoplasmic Abl, as a 7-fold increase in c-myc expression was obtained in Abl/Arg–/– 3T3 cells expressing Abl-NLS–. Similarly, a strong decline in c-myc expression was also observed in Abl/Arg–/– 3T3 cells when stimulated with serum. In order to confirm this data, we analysed the effect of the inhibitor of Abl activity, STI 571, on this cellular response (Druker et al., 1996). This drug was not used in the case of PDGF response since it also affects PDGF receptor kinase activity (Buchdunger et al., 2000). Nevertheless, this approach was still valuable for investigating the serum response. As shown in Figure 8C, STI 571 largely reduced serum-induced DNA synthesis in NIH 3T3 cells. Inhibition was specific for Abl as cell treatment with the PDGF receptor inhibitor AG 1296 had no effect, precluding an involvement of PDGF in serum-induced mitogenesis. The effect of STI 571 on c-myc expression was then analysed (Figure 8D): serum induced a 9-fold increase in c-myc mRNA level within 1 h of stimulation, and STI 571 treatment reduced this response by 30–45%. Collectively, this data provides strong evidence for a role of c-Abl on c-myc transcriptional expression during mitogenesis.
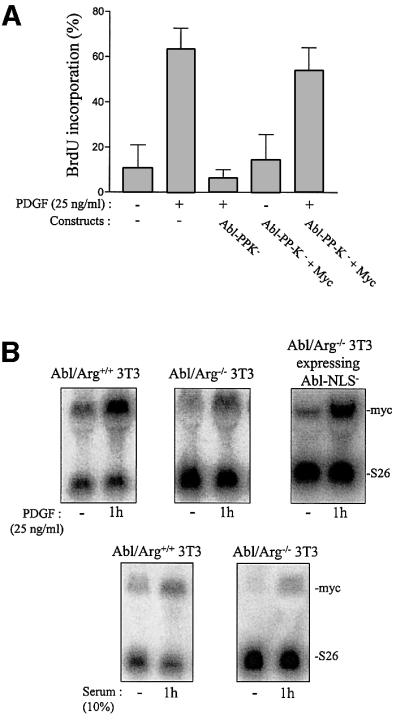

Fig. 8. c-Abl regulates PDGF- and serum-induced c-myc expression. (A) Functional interaction between Abl and Myc during PDGF mitogenesis. NIH 3T3 cells were plated onto coverslips and transiently co-transfected with Abl-PP-K– and Myc. After serum starvation, cells were stimulated or not with PDGF in the presence of BrdU for 18 h. Cells were fixed and stained for Abl expression and BrdU incorporation by indirect immunostaining, and analysed by microscopy. Shown is the percentage of BrdU-positive cells present in expressing and non-expressing cells under the specified conditions, calculated as in Figure 2. The data from four independent experiments have been averaged, and the mean and standard deviation are shown. (B) c-myc mRNA expression during mitogenesis is reduced in 3T3 cells deficient in both Abl and Arg. After serum starvation, Abl/Arg+/+, Abl/Arg–/– or Abl/Arg–/– 3T3 cells expressing Abl-NLS– were stimulated or not with PDGF (top) or serum (bottom) for 1 h. Total RNA was isolated from all cell types and was blotted and probed with a probe specific for c-myc, or S26 as a control for RNA level. Shown is a nothern blot analysis representative of three independent experiments. The positions of c-myc and S26 mRNA are shown. (C and D) The small molecule inhibitor of Abl, STI 571, reduces serum-induced BrdU incorporation (C) and c-myc mRNA expression (D). NIH 3T3 cells were serum starved and treated with vehicle (DMSO), STI 571 (1–10 µM) or the small PDGF receptor inhibitor AG 1296 (10 µM) 2 h before serum stimulation. Cells were stimulated for 18 h in the presence of BrdU for DNA synthesis analysis (C) or for the indicated times for c-myc expression (D). (C) Shown is the percentage of BrdU incorporation of cells treated with the indicated inhibitor and analysed as described in Materials and methods. The data from four independent experiments have been averaged, and the mean and standard deviation are shown. (D) Top panel represents an example of a northern blot analysis from cells treated with the indicated drug and stimulated or not with 10% serum for 1 h. Bottom panel represents the statistical analysis obtained from cells treated with DMSO (open bars) or 10 µM STI 571 (grey bars). c-myc mRNA level was calculated as described in Materials and methods. The data from three independent experiments have been averaged, and the mean and standard deviation are shown.
Discussion
Growth factor receptors initiate several signalling cascades important for DNA synthesis, including those regulated by the small GTPase Ras and the Src family tyrosine kinases (Abram and Courtneidge, 2000). While the Ras pathway has been documented extensively (van der Geer et al., 1994), the mechanism by which c-Src regulates mitogenesis remains elusive. However, it is clear that both pathways are largely independent, at least when initiated by PDGF (Abram and Courtneidge, 2000). Here we have identified a new element of the latter, with the use of the cytoplasmic tyrosine kinase c-Abl. This led us to postulate the existence of a tyrosine kinases cascade (PDGF receptor/c-Src/c-Abl) that is also important for mitogenesis. Our conclusion is based on the following: inhibition of cytoplasmic c-Abl by microinjection of specific antibodies, dominant-negative approaches or the use of a small-molecule inhibitor all affect mitogenesis. Furthermore, fibroblasts deficient in Abl and Arg proteins exhibit a 4–6 h delay in S phase entry when stimulated by mitogens, a defect that is overcome by expressing Abl-NLS–. Our evidence for c-Abl as an effector of c-Src is based on the fact that PDGF-induced c-Abl activation and phosphorylation is largely dependent on c-Src activity and that this kinase does not rescue the G1 block induced by Abl-PP-K–. This is also supported by the Abl-PP rescue effect on the SrcK– G1 block and by the fact that both kinases lie in the same pathway.
Our experiments have also revealed a new function of cytoplasmic c-Abl in signal transduction. Thus, not only does c-Abl regulate growth factor-induced membrane ruffling (Plattner et al., 1999), but also the promotion of DNA synthesis. Of note, a negative function for nuclear c-Abl during G1/S progression has been proposed previously (Sawyers et al., 1994; Wen et al., 1996) based on the G1 block induced by overexpressing Abl in the nucleus. This is particularly the case for oncogenic forms that, when overexpressed, may exhibit sufficient kinase activity in the nucleus for cell growth inhibition (Wen et al., 1996). The positive function we observed for c-Abl in the cytoplasm is supported by the fact that cytoplasmic Abl alleles (Abl-PP-NLS– and Abl-NLS–) did not inhibit mitogenesis. This was also true for the oncogenic Abl-PP mutant, which, when expressed at a moderate level, displayed cytoplasmic localization and did not affect mitogenesis. This notion is reinforced by the effect of antibody injection, leading to specific inhibition of endogenous cytoplasmic c-Abl. While endogenous c-Abl may have distinct functions depending on its subcellular localization, it has a globally positive role as illustrated by the inhibitory effect of STI 571 that does not discriminate between cytoplasmic and nuclear pools of c-Abl, and by the delay in S phase entry observed in Abl/Arg-deficient cells.
Our report also sheds new light on the mechanism by which c-Src may activate cytoplasmic c-Abl. It appears to involve a transphosphorylation mechanism, as previously suggested, with the phosphorylation of Y412 present in the activation loop (Plattner et al., 1999). Nevertheless, our results also show that the Y245 present in the SH2-CD linker may also be required for full activation. This largely supports the model proposed by Brasher and van Etten (2000), with the involvement of both residues for maximal activity, but in addition our data strongly suggest that these residues are phosphorylated by c-Src during PDGF stimulation and are required for Abl mitogenic function. It should be noted that while phosphorylation of these sites may be required for Abl activation, we cannot exclude that they also have a function outside the enzymatic activity, such as the association with an SH2-containing signalling protein. Similarly, neither can the phosphorylation of additional tyrosines be excluded.
Finally, we have identified two targets of c-Abl in this response: the tumour suppressor p53 and the transcription factor c-Myc. The functional link between p53 and the Src pathway was originally established as during PDGF mitogenic response (Broome and Courtneidge, 2000). Our results obtained with the adenoviral E1B protein and a dominant-negative p53 mutant largely confirm these observations. This reveals a novel function of p53 that might be related to the change in p53 level reported in murine fibroblasts during mitogenesis (Reich and Levine, 1984). Whether Src kinases regulate p53 expression and/or activity during G1 is entirely unknown. Our observations show that, like Src, cytoplasmic c-Abl regulates a p53-dependent event. While an association has been identified in the nucleus during DNA damage (Yuan et al., 1996), we believe that the interaction between c-Abl and p53 is not direct, as it concerns the cytoplasmic pool of c-Abl. Therefore, c-Abl may phosphorylate a cytoplasmic substrate involved in the inhibition of p53 activity. We have also identified cytoplasmic c-Abl as a new signalling protein, regulating growth factor-induced c-myc transcriptional expression. This allowed us to confirm that c-Abl lies in the Src pathway, probably by phosphorylating specific substrates. Our mutagenesis analyses together with the microinjection experiments implicate the SH2 and C-terminal domains for substrate association and/or phosphorylation. However, the nature of these substrates is unknown. Few signalling proteins have recently been involved in c-myc expression, including the adaptors Shc (Gotoh et al., 1997; Blake et al., 2000), STAM (Takeshita et al., 1997), STAM2 (Endo et al., 2000; Pandey et al., 2000), the small GTPases of the Rho family (Chiariello et al., 2001) and the transcription factor STAT3 (Bowman et al., 2001). One attractive hypothesis is that Abl may phosphorylate and/or activate some of these signalling proteins; this is currently under investigation.
Materials and methods
DNA constructs and antibodies
Constructs expressing c-Src, SrcY527F, SrcK– (K295M), RasN17, c-Myc and c-Fos have been described previously (Barone and Courtneidge, 1995; Manes et al., 2000), as have those expressing Abl-PP (P242E/249E), Abl-PP-SH2*, Abl-PP-ΔC, c-Abl, Abl-NLS– and Abl-PP-K– (Barila et al., 2000). AblY412F-NLS–, AblY412F/Y245F-NLS–, Abl-PP-NLS– and Abl-PP-K–NLS– were generated by PCR using the Quick Change site-directed mutagenesis system (Stratagene, La Jolla, CA) and pcDNA3-Abl-NLS– (Barila et al., 2000) as a template. The pcDNA3 constructs expressing p53 and p53H273 were described in Lassus et al. (1999), the E1B and E1BR309 constructs were described in Yew and Berk (1992), pGEX-AblSH2 was from B.Mayer (Harvard Medical School) and pGEX-AblSH3 was from A.Musacchio (European Institute of Oncology). Cst.1 and Ras Y13 259 antibodies were described in Roche et al. (1995), as was Abl monoclonal antibody 24-21 in Barila et al. (2000), 19-110 and anti-p53 FL-293 antibodies were purchased from Oncogene Science (Cambridge, MA) and Santa Cruz (Santa-Cruz, CA), respectively, Arg antibody was from G.D.Kruh (University of Pennsylvania), the Tec construct and antibody were from Dr Pucéat (CNRS), and 4G10 antibody was from Dr Mangeat (University of Montpellier II). Anti-AblSH2 and anti-AblSH3 polyclonal antibodies were raised against respective glutathione _S_-transferase (GST) fusion proteins. GST fusion proteins and antibodies were generated, purified and concentrated as described previously (Roche et al., 1996). Antibodies coupled to fluorescein isothiocyanate (FITC) or Texas Red isothiocyanate and AG-1296 were purchased from Sigma (St Louis, MO), and anti-BrdU monoclonal antibody was from PharMingen (La Jolla, CA). STI 571 was from B.Willi (Novartis Pharma AG, Basel, Switzerland).
Cell culture, transfection, microinjection and DNA synthesis analysis
Primary mouse embryo fibroblasts (MEFs) were prepared from 13-day-old embryos as described (Zindy et al., 1997). All the experiments using MEFs were performed at passage 3 or 4. p53–/– MEF cells were from P.Roux (CNRS), Abl/Arg+/+ 3T3 and Abl/Arg–/– 3T3 from T.Koleske (Yale University). Abl/Arg–/– 3T3 cells expressing Abl-NLS– and AblY412F/Y245F-NLS– were generated by pooling parental cells that were stably transfected with the corresponding construct. Cell culture and transfection methods were described previously (Manes et al., 2000). For PDGF-induced Abl tyrosine phosphorylation experiments, quiescent cells were pre-treated with 100 µM vanadate for 15 min before stimulation in order to inhibit tyrosine phosphatase activity in vivo. For DNA synthesis experiments, cells were transfected 24 h before serum starvation, and in the case of biochemical analysis, 40 h before lysis of the cells. Cells microinjection studies, actin staining and DNA synthesis analysis were described previously (Manes et al., 2000). For drug treatment, cells were treated with the indicated inhibitor or vehicle (DMSO) 2 h before stimulation. Cells were fixed and analysed by immunofluorescence as in Roche et al. (1995). Actin was visualized by staining with rhodamin–phalloidin (Molecular Probe, Eugene, OR) as in Manes et al. (2000). Fixed cells were observed with a DMR A oil immersion microscope PL APO 63× (Leica Microsystems, Deerfield, IL). Images were captured with Micromax camera (ROPER Scientific Inc., Trenton, NJ) driven by MetaMorph (V. 4.7; Universal Imaging Corp, Downingtown, PA). The percentage of injected (or transfected) cells that incorporated BrdU for each coverslip was calculated using the following formula: % BrdU-positive cells = [number of BrdU-positive injected (or transfected) cells]/[number of injected (or transfected) cells] × 100. For each coverlip, ∼150–200 cells were counted. In the case of Figure 1B (Abl and Abl-PP), xyz stacks of images were restored with Huygens 1 System 2.2.1-64 (Scientific Volume Imaging b.v, Hilversum, The Netherlands) using the MLE (Maximal Likelihood Estimation based restoration of images) algorithm. Restored stacks were processed with Imaris 3.0 (Bitplane, Zurich, Switzerland) for rendering and analysis.
Biochemistry
Cell lysates were prepared as described in Roche et al. (1996). In vitro translation of Abl was performed with the Transcription and Transduction Coupled Reticulocyte Lysates System (TNT; Promega, Madison, WI) according to manufacturer’s instructions and labelled with [35S]methionine (ICN, Irvine, CA). Abl was immunoprecipitated either from reticulocyte lysates expressing labelled Abl or from mammalian cell lysates using the indicated antibody. Immunocomplexes were processed for SDS–PAGE and subjected to autoradiography in the case of labelled Abl, or to western blotting with the indicated antibody (Manes et al., 2000). In vitro kinase assays were performed as described previously (Dorey et al., 2001). Radiolabelled GST–Crk was quantified using a PhosphorImager (Molecular Dynamics, Inc., Sunnyvale, CA). Abl activation was calculated as follows: (relative Abl activity in the presence of SrcY527F)/(relative Abl activity in the absence of SrcY527F). Subcellular fractionation was performed as described in Wang et al. (1994).
Northern blot analysis
Northern blot analysis was performed as described in Vincent et al. (1993). An _Eco_RV–_Hin_dIII fragment of pSP65Myc54 (II+III) was used as a probe. Autoradiographic images were acquired and quantified using a PhosphorImager. As a control for RNA level, the blots were hybridized with a cDNA encoding the 26S ribosomal protein (using the _Hin_dIII–_Bam_HI fragment of pT7T3318U-S26 as a probe) (Vincent et al., 1993). The c-myc mRNA level was calculated as follows: [c-myc mRNA level (arbitrary units)]/[S26 mRNA level (arbitrary units)].
Acknowledgments
Acknowledgements
We thank P.Roux for the gift of p53–/– MEF cells, U.Hibner for p53 constructs, A.J.Berk for E1B constructs, M.Pucéat for Tec constructs and antibody, G.D.Kruh for Arg constructs and antibody, B.Meyer and A.Musacchio for pGEX-AblSH2 and -AblSH3 constructs, B.Willi for the STI 571 inhibitor, P.Mangeat for 4G10 antibody, and A.J.Koleske for Abl/Arg+/+ and Abl/Arg–/– 3T3 cells. We also thank P.Travo, Head of IFR 24 Integrated Imaging Facility, for his constant interest and support. We thank P.Bello, A.Davy and C.Bénistant for critically reading the manuscript. This work was supported by the CNRS and the Association pour la Recherche contre le Cancer, ARC. S.R. was supported by INSERM and ARC, O.F. was supported by MRES and ARC, and D.B. was supported by the Antonio Castelnuovo Foundation and EMBL.
References
- Abram C.L. and Courtneidge,S.A. (2000) Src family tyrosine kinases and growth factor signaling. Exp. Cell Res., 254, 1–13. [DOI] [PubMed] [Google Scholar]
- Afar D.E., Goga,A., McLaughlin,J., Witte,O.N. and Sawyers,C.L. (1994) Differential complementation of Bcr-Abl point mutants with c-Myc. Science, 264, 424–426. [DOI] [PubMed] [Google Scholar]
- Barila D. and Superti-Furga,G. (1998) An intramolecular SH3-domain interaction regulates c-Abl activity. Nature Genet., 18, 280–282. [DOI] [PubMed] [Google Scholar]
- Barila D., Mangano,R., Gonfloni,S., Kretzschmar,J., Moro,M., Bohmann,D. and Superti-Furga,G. (2000) A nuclear tyrosine phosphorylation circuit: c-Jun as an activator and substrate of c-Abl and JNK. EMBO J., 19, 273–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone M.V. and Courtneidge,S.A. (1995) Myc but not Fos rescue of PDGF signalling block caused by kinase-inactive Src. Nature, 378, 509–512. [DOI] [PubMed] [Google Scholar]
- Blake R.A., Broome,M.A., Liu,X., Wu,J., Gishizky,M., Sun,L. and Courtneidge,S.A. (2000) SU6656, a selective Src family kinase inhibitor, used to probe growth factor signaling Mol. Cell. Biol., 20, 9018–9027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman T., Broome,M.A., Sinibaldi,D., Wharton,W., Pledger,W.J., Sedivy,J.M., Irby,R., Yeatman,T., Courtneidge,S.A. et al. (2001) Stat3-mediated Myc expression is required for Src transformation and PDGF-induced mitogenesis. Proc. Natl Acad. Sci. USA, 98, 7319–7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasher B.B. and Van Etten,R.A. (2000) c-Abl has high intrinsic tyrosine kinase activity that is stimulated by mutation of the Src homology 3 domain and by autophosphorylation at two distinct regulatory tyrosines. J. Biol. Chem., 275, 35631–35637. [DOI] [PubMed] [Google Scholar]
- Broome M.A. and Courtneidge,S.A. (2000) No requirement for Src family kinases for PDGF signaling in fibroblasts expressing SV40 large T antigen. Oncogene, 19, 2867–2869. [DOI] [PubMed] [Google Scholar]
- Broome M.A. and Hunter,T. (1996) Requirement for c-Src catalytic activity and the SH3 domain in platelet-derived growth factor BB and epidermal growth factor mitogenic signaling. J. Biol. Chem., 271, 16798–16806. [DOI] [PubMed] [Google Scholar]
- Buchdunger E., Cioffi,C.L., Law,N., Stover,D., Ohno-Jones,S., Druker,B.J. and Lydon,N.B. (2000) Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J. Pharmacol. Exp. Ther., 295, 139–145. [PubMed] [Google Scholar]
- Chiariello M., Marinissen,M.J. and Gutkind,J.S. (2001) Regulation of c-myc expression by PDGF through Rho GTPases. Nature Cell Biol., 3, 580–586. [DOI] [PubMed] [Google Scholar]
- Cong F., Spencer,S., Cote,J.F., Wu,Y., Tremblay,M.L., Lasky,L.A. and Goff,S.P. (2000) Cytoskeletal protein PSTPIP1 directs the PEST-type protein tyrosine phosphatase to the c-Abl kinase to mediate Abl dephosphorylation. Mol. Cell, 6, 1413–1423. [DOI] [PubMed] [Google Scholar]
- Dorey K., Engen,J., Kretzschmar,J., Wilm,M., Neubard,G., Schindler,T. and Superti-Furga,G. (2001) phosphorylation and structure-based functional studies reveal a positive and a negative role for the activation loop of the c-Abl tyrosine kinase. Oncogene, 20, 8075–8084. [DOI] [PubMed] [Google Scholar]
- Druker B.J., Tamura,S., Buchdunger,E., Ohno,S., Segal,G.M., Fanning,S., Zimmermann,J. and Lydon,N.B. (1996) Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nature Med., 2, 561–566. [DOI] [PubMed] [Google Scholar]
- Endo K. et al. (2000) STAM2, a new member of the STAM family, binding to the Janus kinases. FEBS Lett., 477, 55–61. [DOI] [PubMed] [Google Scholar]
- Gotoh N., Toyoda,M. and Shibuya,M. (1997) Tyrosine phosphorylation sites at amino acids 239 and 240 of Shc are involved in epidermal growth factor-induced mitogenic signaling that is distinct from Ras/mitogen-activated protein kinase activation. Mol. Cell. Biol., 17, 1824–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koleske A.J., Gifford,A.M., Scott,M.L., Nee,M., Bronson,R.T., Miczek,K.A. and Baltimore,D. (1998) Essential roles for the Abl and Arg tyrosine kinases in neurulation. Neuron, 21, 1259–1272. [DOI] [PubMed] [Google Scholar]
- Lassus P., Bertrand,C., Zugasti,O., Chambon,J.P., Soussi,T., Mathieu-Mahul,D. and Hibner,U. (1999) Anti-apoptotic activity of p53 maps to the COOH-terminal domain and is retained in a highly oncogenic natural mutant. Oncogene, 18, 4699–4709. [DOI] [PubMed] [Google Scholar]
- Lewis J.M., Baskaran,R., Taagepera,S., Schwartz,M.A. and Wang,J.Y. (1996) Integrin regulation of c-Abl tyrosine kinase activity and cytoplasmic-nuclear transport. Proc. Natl Acad. Sci. USA, 93, 15174–15179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manes G., Bello,P. and Roche,S. (2000) Slap inhibits the Src-mitogenic function but does not affect Src-induced cell transformation. Mol. Cell. Biol., 20, 3396–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey A., Fernandez,M., Steen,H., Blagoev,B., Nielsen,M., Roche,S., Mann,M. and Lodish,H.F. (2000) Identification of a novel immunoreceptor tyrosine-based activation motif (ITAM)-containing molecule, STAM2, in growth factor and cytokine receptor signaling pathways by mass spectrometry. J. Biol. Chem., 275, 38633–38639. [DOI] [PubMed] [Google Scholar]
- Plattner R., Kadlec,L., DeMali,K.A., Kazlauskas,A. and Pendergast,A.M. (1999) c-Abl is activated by growth factors and Src family kinases and has a role in the cellular response to PDGF. Genes Dev., 13, 2400–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich N.C. and Levine,A.J. (1984) Growth regulation of a cellular tumour antigen, p53, in nontransformed cells. Nature, 308, 199–201. [DOI] [PubMed] [Google Scholar]
- Roche S., Koegl,M., Barone,M.V., Roussel,M.F. and Courtneidge,S.A. (1995) DNA synthesis induced by some but not all growth factors requires Src family protein tyrosine kinases. Mol. Cell. Biol., 15, 1102–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche S., McGlade,J., Jones,M., Gish,G.D., Pawson,T. and Courtneidge,S.A. (1996) Requirement of phospholipase C γ, the tyrosine phosphatase Syp and the adaptor proteins Shc and Nck for PDGF-induced DNA synthesis: evidence for the existence of Ras-dependent and Ras-independent pathways. EMBO J., 15, 4940–4948. [PMC free article] [PubMed] [Google Scholar]
- Sawyers C.L. (1999) Chronic myeloid leukemia. N. Engl. J. Med., 340, 1330–1340. [DOI] [PubMed] [Google Scholar]
- Sawyers C.L., Callahan,W. and Witte,O.N. (1992) Dominant negative MYC blocks transformation by ABL oncogenes. Cell, 70, 901–910. [DOI] [PubMed] [Google Scholar]
- Sawyers C.L., McLaughlin,J., Goga,A., Havlik,M. and Witte,O. (1994) The nuclear tyrosine kinase c-Abl negatively regulates cell growth. Cell, 77, 121–131. [DOI] [PubMed] [Google Scholar]
- Sears R., Leone,G., DeGregori,J. and Nevins,J.R. (1999) Ras enhances Myc protein stability. Mol. Cell, 3, 169–179. [DOI] [PubMed] [Google Scholar]
- Takeshita T., Arita,T., Higuchi,M., Asao,H., Endo,K., Kuroda,H., Tanaka,N., Murata,K., Ishii,N. and Sugamura,K. (1997) STAM, signal transducing adaptor molecule, is associated with Janus kinases and involved in signaling for cell growth and c-myc induction. Immunity, 6, 449–457. [DOI] [PubMed] [Google Scholar]
- Treisman R. (1994) Ternary complex factors: growth factor regulated transcriptional activators. Curr. Opin. Genet. Dev., 4, 96–101. [DOI] [PubMed] [Google Scholar]
- Twamley-Stein G.M., Pepperkok,R., Ansorge,W. and Courtneidge,S.A. (1993) The Src family tyrosine kinases are required for platelet-derived growth factor-mediated signal transduction in NIH 3T3 cells. Proc. Natl Acad. Sci. USA, 90, 7696–7700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Geer P., Hunter,T. and Lindberg,R.A. (1994) Receptor protein-tyrosine kinases and their signal transduction pathways. Annu. Rev. Cell Biol., 10, 251–337. [DOI] [PubMed] [Google Scholar]
- Van Etten R.A. (1999) Cycling, stressed-out and nervous: cellular functions of c-Abl. Trends Cell Biol., 9, 179–186. [DOI] [PubMed] [Google Scholar]
- Vigneri P. and Wang,J.Y. (2001) Induction of apoptosis in chronic myelogenous leukemia cells through nuclear entrapment of BCR-ABL tyrosine kinase. Nature Med., 7, 228–234. [DOI] [PubMed] [Google Scholar]
- Vincent S., Marty,L. and Fort,P. (1993) S26 ribosomal protein RNA: an invariant control for gene regulation experiments in eucaryotic cells and tissues. Nucleic Acids Res., 21, 1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Sato,R., Brown,M.S., Hua,X. and Goldstein,J.L. (1994) SREBP-1, a membrane-bound transcription factor released by sterol-regulated proteolysis. Cell, 77, 53–62. [DOI] [PubMed] [Google Scholar]
- Wen S.T., Jackson,P.K. and Van Etten,R.A. (1996) The cytostatic function of c-Abl is controlled by multiple nuclear localization signals and requires the p53 and Rb tumor suppressor gene products. EMBO J., 15, 1583–1595. [PMC free article] [PubMed] [Google Scholar]
- Wong K.K., Zou,X., Merrell,K.T., Patel,A.J., Marcu,K.B., Chellappan,S. and Calame,K. (1995) v-Abl activates c-myc transcription through the E2F site. Mol. Cell. Biol., 15, 6535–6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yew P.R. and Berk,A.J. (1992) Inhibition of p53 transactivation required for transformation by adenovirus early 1B protein. Nature, 357, 82–85. [DOI] [PubMed] [Google Scholar]
- Yuan Z.M., Huang,Y., Whang,Y., Sawyers,C., Weichselbaum,R., Kharbanda,S. and Kufe,D. (1996) Role for c-Abl tyrosine kinase in growth arrest response to DNA damage. Nature, 382, 272–274. [DOI] [PubMed] [Google Scholar]
- Zindy F., Quelle,D.E., Roussel,M.F. and Sherr,C.J. (1997) Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene, 15, 203–211. [DOI] [PubMed] [Google Scholar]