A Natural Killer T (NKT) Cell Developmental Pathway Involving a Thymus-dependent NK1.1−CD4+ CD1d-dependent Precursor Stage (original) (raw)
Abstract
The development of CD1d-dependent natural killer T (NKT) cells is poorly understood. We have used both CD1d/α-galactosylceramide (CD1d/αGC) tetramers and anti-NK1.1 to investigate NKT cell development in vitro and in vivo. Confirming the thymus-dependence of these cells, we show that CD1d/αGC tetramer-binding NKT cells, including NK1.1+ and NK1.1− subsets, develop in fetal thymus organ culture (FTOC) and are completely absent in nude mice. Ontogenically, CD1d/αGC tetramer-binding NKT cells first appear in the thymus, at day 5 after birth, as CD4+CD8−NK1.1−cells. NK1.1+ NKT cells, including CD4+ and CD4−CD8− subsets, appeared at days 7–8 but remained a minor subset until at least 3 wk of age. Using intrathymic transfer experiments, CD4+NK1.1− NKT cells gave rise to NK1.1+ NKT cells (including CD4+ and CD4− subsets), but not vice-versa. This maturation step was not required for NKT cells to migrate to other tissues, as NK1.1− NKT cells were detected in liver and spleen as early as day 8 after birth, and the majority of NKT cells among recent thymic emigrants (RTE) were NK1.1−. Further elucidation of this NKT cell developmental pathway should prove to be invaluable for studying the mechanisms that regulate the development of these cells.
Keywords: T lymphocyte, fetal thymus organ culture, cytokines, T cell development, natural killer T cell
Introduction
CD1d-dependent NKT cells are a distinct lineage of T cells with unique characteristics (for reviews, see references 1 and 2). These cells include CD4+ and CD4−CD8− double-negative (DN)* subsets and express a heavily biased TCR repertoire, with the majority expressing an invariant Vα14Jα281 TCR-α chain and either Vβ8.2, Vβ2, or Vβ7 TCR-β chains (3–7). NKT cells are potent cytokine producers and play a key role as immunoregulatory cells. Perhaps the best example is in the NOD mouse model for type-1 diabetes, in which a NKT cell deficiency is directly related to diabetes susceptibility (8–10). NKT cells have also been implicated in immunosuppression associated with anterior chamber–associated immune deviation (11–13), graft-versus-host disease (14), and allograft tolerance (15). They can also regulate antitumor responses, sometimes inhibiting (16) and sometimes promoting tumor rejection (17). Thus, abnormalities in NKT cell development that can alter the numbers of these cells may have a significant impact on immune responses in a range of diseases.
Although NKT cells are present in the thymus, the developmental origin of these cells is controversial. Some investigators have argued that NKT cells are present in thymus-deficient nude mice or in bone marrow–repopulated, thymectomized adult mice (18–21; for a review, see reference 1). However, several studies support a thymus-dependent origin for these cells, as they are significantly reduced in neonatally thymectomized mice (22), CD4+NK1.1+ cells are clearly absent from livers of nude mice (23), and canonical TCR Vα14Jα281 rearrangements are not detected in irradiated, thymectomized, and fetal liver–repopulated adult mice (24). Some of these conflicting results may be due to the fact that some NK1.1 expressing T cells exist that are quite distinct from “classical”, CD1d-dependent, NKT cells (2, 25, 26). These cells, sometimes referred to as type-II NKT cells, are CD1d independent, mostly CD4−, have diverse TCRs, may be thymus independent, and their functional significance is unknown. Given that conventional T cells can upregulate NK1.1 after activation (27, 28), CD1d-independent NK1.1+ T cells may be a highly diverse population including MHC-I– and MHC-II–restricted cells, and possibly represent an activation state rather than a unique lineage. In this paper, the term “NKT cells” refers only to the CD1d-dependent population. CD1d/α-GalCer tetrameric complexes, which bind stably and selectively to Vα14Jα281-expressing NKT cells, currently represent the best reagent for identifying CD1d-dependent NKT cells (29, 30). Although α-GalCer is a nonmammalian glycolipid, originally derived from marine sponges, NKT cells specifically interact with this molecule, in a strictly CD1d-dependent manner (31, 32). In this paper, CD1d/αGC tetramers have been used to identify NKT cells, thus avoiding complications due to promiscuous expression of, or lack of, NK1.1.
The intrathymic development of NKT cells depends on CD1d expression by CD4+CD8+ double positive (DP) thymocytes rather than thymic epithelial cells (23, 33–37). Although little information exists for a developmental pathway for NKT cells, two models have been proposed for their development (1). One model suggests that the generation of NKT cells results from programmed gene rearrangements that yield the invariant TCR-α chain; the other model suggests that this TCR is the result of random gene rearrangements and that NKT cells are selected from the mainstream T cell lineage at the DP stage of thymocyte development, as this is where TCR-α gene rearrangement, expression, and auditioning for selection occurs. In strong support of the latter model, the invariant TCR-α chain often carries N nucleotide additions that generate the canonical amino acid sequence; moreover, the homologous chromosome often carries random TCR-α gene rearrangements. (5, 38). Several studies support the concept that NKT cells are a developmentally distinct lineage. In contrast to conventional T cells, NKT cell development appears to require an interaction with membrane lymphotoxin expressing cells (39, 40). NKT cell development is also absolutely dependent on pre-Tα signaling (41) and at least partly dependent on GM-CSF signaling (42). Fyn-deficient mice show a selective defect in the development of CD1d-dependent NKT cells, but not of conventional T cells (43, 44). Analysis of NKT cells in common cytokine receptor γ chain–deficient mice revealed at least two stages in NKT cell development (45). Such mice generate thymocytes expressing normal amounts of Vα14Jα281 mRNA and develop IL-4–producing CD8− cells, suggesting the presence of NKT cells, yet these cells fail to express the NK receptors NK1.1 or Ly49 and are not exported to the periphery. Thymic stromal cell–derived cytokines IL-15 and IL-7 are required for development of normal numbers and IL-4–producing potential, respectively, of NKT cells (46, 47), and an intact thymic structure is also important (48).
In this study, we have examined NKT cell development in the thymus both in in vitro fetal thymus organ cultures (FTOC) and also during ontogeny in vivo. Using CD1d/αGC tetramers, NK1.1, and CD4 in combination, we have demonstrated the existence of a developmental pathway showing that the earliest CD1d-dependent NKT cells are CD4+CD8−NK1.1−. These cells are precursors of NK1.1+ NKT cells, including both CD4+ and DN subsets. This later maturation event is not required for thymic emigration, as immature NK1.1− cells are also present in spleen and liver of young mice, and these cells are abundant among recent thymic emigrants (RTE).
Materials and Methods
Mice.
C57BL/6 mice, CD45.1 congenic C57BL/6, BALB/c, and BALB/c nu/nu mice (C57BL/6 nu/nu mice were not available in Australia) were obtained from either the Animal Resources Centre (Canning Vale, WA) or Monash University Central Animal House (Clayton, Victoria, Australia) and housed for 1 to 2 wk in micro-isolators in the Monash University Medical School Animal House (Prahran, Victoria, Australia). Embryonic thymuses for FTOC were derived from plug-timed pregnant mice where time of finding a plug was taken to be day 0. Perinatal mice were timed such that the day of birth was referred to as day 1. All mouse experiments were approved by the Monash University Animal Ethics Committee – Alfred Hospital branch.
FTOC.
Fetal thymus lobes were obtained at embryonic day 14 (E14) from plug-timed pregnant C57BL/6 mice as described previously (49). FTOC was performed in culture media consisting of RPMI-1640 (Life Technologies), 10% FCS (Commonwealth Serum Laboratories [CSL], Melbourne, Victoria, Australia), 2 mM GlutaMAX, 50 μM 2-ME, 100 IU/ml penicillin, and 100 μg/ml streptomycin, 15 mM HEPES buffer (Life Technologies), and 1 mM sodium pyruvate (Life Technologies). Lobes were cultured in groups of 4–6 per well of a 6-well plate for up to 18 d with a media change every 6 d of culture. At the end of culture, thymocytes were harvested from lobes, counted, and analyzed by flow cytometry.
Cell Suspensions.
Cell suspensions of thymus and spleen were prepared as described previously (26). Hepatic leukocytes were isolated by cutting individual livers into small pieces and gently pressing through a 200-gauge wire mesh. The cells were washed twice in ice-cold PBS with 2% FCS and 0.02% Azide and spun through 33.8% Percoll (Amersham Pharmacia Biotech) for 12 min at 693 g. Recovered leukocytes were washed and treated with red cell removal buffer (Sigma-Aldrich). Hepatic leukocytes from mice at days 5 and 8 were isolated by gently grinding the organ between frosted glass slides and staining without further enrichment, using a lymphocyte gate based on forward versus side light scatter for flow cytometry.
Flow Cytometry.
The following mAbs were used in multi-parameter flow cytometric analysis: anti-: αβTCR-allophycocyanin (APC) (clone H57–597), CD4-FITC or CD4-PerCP (clone RM4–5), CD8-biotin, CD8-PerCP or CD8-FITC (clone 53–6.7), NK1.1-PE or biotin (clone PK136, mouse IgG2a), CD45.2-FITC (clone 104; all purchased from BD PharMingen). Biotinylated mAb were detected with streptavidin-Alexa Fluor™ 488 (Molecular Probes), streptavidin-PerCP, or streptavidin APC (BD PharMingen). Fc-receptor block (anti-CD16/CD32, clone 2.4G2 culture supernatant) was always added to staining cocktails. PE-labeled, α-GalCer–loaded or –unloaded (control) mCD1d tetramers were produced in-house at La Jolla Institute for Allergy and Immunology (30). One fluorescence channel was often not used for specific staining but instead used for the exclusion of autofluorescent cells. Three and four-color analysis, as well as multi-color sorting, was performed using a FACSCalibur™ or FACStarPLUS™ (Becton Dickinson). CELLQuest™ software (Becton Dickinson) was used for analysis.
Isolation of NKT Cell Subsets for In Vitro Cytokine Assays.
Thymic NKT cells were enriched by depletion of CD24 (HSA)+ and CD8+ thymocytes with rat anti–mouse CD24 (clone J11D) and rat anti–mouse CD8 (clone 3.155), respectively, followed by rabbit complement (C-six Diagnostics Inc.). NK1.1+ or NK1.1− CD1d/αGC tetramer+ thymocytes were sorted to greater than 92% purity and stimulated by culturing in anti-CD3–coated microtitre plates (clone 145–2C11; BD PharMingen). Cells were cultured at a density of 105 cells in 100 μl tissue culture medium. Supernatants were harvested at 24 and 48 h, and IL-4 and IFN-γ levels were detected by sandwich ELISA as described previously (26). Generally the limit of detection for IL-4 was 2 U/ml, and IFN-γ 0.1 ng/ml. Quantitative differences between samples were compared with the Mann Whitney U (rank sum) test.
In Vivo Labeling of Thymocytes with FITC.
The technique for in vivo thymocyte labeling with FITC has been described previously in detail (50). Briefly, 6-wk-old mice were anesthetized and intrathymically injected with 10 μl of FITC (Sigma-Aldrich; 1 mg/ml in PBS) per lobe. The mice were subsequently injected subcutaneously with 0.03 mg of Buprenorphine analgesic (Reckitt and Coleman). Mice were killed 36 h later and their organs removed for flow cytometric analysis of RTE.
Intrathymic Injection of Cells.
NKT cell subsets derived from thymuses of 4-wk-old C57BL/6 mice were separated into NK1.1+ and NK1.1− fractions by FACS® sorting, and intrathymically injected into CD45.1 congenic C57BL/6 recipient mice. Intrathymic injections were performed in a similar fashion to that described for FITC injection, except cells were injected into one lobe of the thymus in a volume of 10 μl of PBS and mice were killed 7 or 14 d later. For these experiments, 2 × 105 NK1.1+CD4+HSA−/low cells, or 2 × 106 NK1.1−CD4+HSA−/low cells were injected. The latter population was injected in larger numbers, since upon restaining, it included more than just NK1.1−CD1d/αGC tetramer+ NKT cells, which were typically <10% of the injected cells. This approach was used in preference to sorting directly, using the tetramer, for CD4+CD1d/αGC tetramer+ NK1.1− cells, as the tetramer reagent would probably artificially stimulate the NKT cells through TCR cross-linking. To control for the possibility that other CD4+HSA−/low cells were giving rise to the NKT cells subsequently detected in the recipient mice, a separate group of mice were injected with CD4+HSA−/low cells that had been depleted of CD1d/αGC tetramer+ cells. Sorted cell purities were always >98% for the NK1.1− fraction and 94% for the NK1.1+ fraction.
Results
CD1d-dependent NKT Cell Development Is Thymus Dependent.
The thymus dependence of NKT cells is an ongoing area of debate. Although NK1.1 expressing T cells have been identified in FTOC (4), we sought to revisit this technique using CD1d/αGC tetramer, which would permit identification of NKT cells without relying on NK1.1 expression, thus including NK1.1− CD1d-dependent NKT cells (2, 29, 30). As shown in Fig. 1 , and in support of the earlier study by Bendelac et al. (4), CD1d/αGC tetramer-binding NKT cells develop in FTOC, although they did not appear until between 11 and 13 d of culture, whereas conventional αβTCRhigh cells were present as early as 6 d of FTOC (not shown). The majority of these NKT cells were CD8−, and unexpectedly, NK1.1− (Fig. 1). In contrast, most NKT cells in adult thymus were NK1.1+, with a minor subset of CD4+ NK1.1− cells (Fig. 1), as has been reported previously (29, 30). CD1d/αGC tetramer-reactive NKT cells accumulated with time in these cultures from day 12 to day 18 (Fig. 1 B). Parallel analysis of NK1.1+αβTCR+ cells in these cultures gave significantly different results. These cells appeared at a similar rate and time, however, the majority did not stain with CD1d/αGC tetramer (Fig. 1 B) and although some NK1.1+ CD1d/αGC tetramer− cells were CD4+CD8− and CD4−CD8−, between 10–30% were CD4−CD8+ (data not shown), a phenotype that was never seen for CD1d/αGC tetramer+ cells. Taken together, these findings support and significantly expand on results from the previous study (4), showing that both NK1.1**+** and NK1.1− NKT cells can develop in FTOC, and also reveal that NK1.1 in isolation is not an ideal marker for studying NKT cell development in FTOC. Nonetheless, these FTOC experiments confirmed that NKT cells can develop in the thymus in the absence of extrathymic factors, but did not exclude an alternative, extrathymic pathway for the development of these cells.
Figure 1.

CD1d-dependent NKT cell development is thymus dependent. (A) Thymocytes were harvested from FTOC after 11, 13, 15, and 18 d of culture and stained for multi-parameter flow cytometric analysis. An adult thymus sample is included as a labeling control. The first column shows CD1d/αGC tetramer+αβTCR+ cells with the percentages indicated. The second column shows CD4 versus NK1.1 expression on NKT cells, gated as shown in the first column. The third column shows CD4 versus CD8 expression on NKT cells, gated as shown in the first column. (B) The data from all FTOC experiments were graphed and means ± standard error plotted as shown. In some cases where the error was very low, no bars are visible. Results are from 1–5 different experiments with at least four separate cultures per experiment. Tetramer, CD1d/αGC tetramer.
Several studies have investigated whether NKT cells exist in athymic nude mice, with highly ambiguous results (for a review, see reference 1). Therefore, we examined spleen and liver lymphocytes of nude mice for the presence of CD1d/αGC tetramer-binding NKT cells (Fig. 2) . These experiments, performed using young (13-d-old), and aged (11-wk-old) mice, revealed a complete absence of NKT cells in the absence of a thymus. Taken together with the FTOC data, this clearly demonstrates that the thymus is both necessary and sufficient for the development of CD1d-dependent NKT cells.
Figure 2.

NKT cells are absent from spleen and liver of nude mice. Spleens and livers were harvested from BALB/c nude (nu/nu) mice at day 13 after birth and at 11 wk of age. Cells were counted and labeled for flow cytometric analysis. Day 13 heterozygous (+/nu) littermates and 11-wk-old wild-type (+/+) BALB/c spleen and liver cells were examined in parallel as a control to show that labeling for CD1d/αGC tetramer+ αβTCR+ cells was as expected. Dotplots are representative of five day 13 and three 11-wk-old BALB/c nude (nu/nu) mice. Three nude mice were also screened at 7 wk of age with similar results (not shown).
Developmentally Regulated Appearance of NKT Cell Subsets.
Given that many NKT cells exhibited an NK1.1− phenotype in FTOC, it was important to investigate whether this represented an immature stage in the development of these cells. Thus, thymus, spleen, and liver lymphocytes were examined for CD1d/αGC tetramer-binding NKT cells from a range of developmental stages between day 3 after birth to adult (Figs. 3 and 4 ; day of birth = day 1). NKT cells were first detected in very low percentages and numbers in the thymus, but not other tissues, at day 5 and increased steadily from then on (Fig. 3). These cells were detected in spleen and liver at day 8, and also accumulated at a similar rate to that seen in the thymus. Multi-parameter flow cytometric analysis of thymic NKT cells at day 5 revealed that nearly all of these cells were CD4+CD8−NK1.1− (Fig. 4 A). NK1.1+ cells gradually emerged in the thymus over the next few days, but remained in the minority until at least day 28. This supports the concept that NK1.1− NKT cells represent a precursor stage in the development of NKT cells. Similar populations of NKT cells appeared in spleen and liver from day 8 onwards (Fig. 4, B and C), including a majority of NK1.1− NKT cells. This suggested that less mature NK1.1− NKT cells are able to leave the thymus before acquisition of NK1.1 expression, and may subsequently acquire a more mature phenotype in the peripheral organs. Regardless of the stage of development, a very minor subset of cells within the NKT cell gate in the thymus appeared to express both CD4 and CD8 (Fig. 4 A). Similar cells have been previously observed (30) and the possibility has been raised that they represent an early precursor stage in the NKT cell lineage (51). However, we did not detect a stage during NKT cell ontogeny at which these cells appeared to be significantly more abundant, and careful analysis of these cells showed that they appeared to be larger than other NKT cells based on forward light scatter (data not shown), suggesting that they might be doublets. Furthermore, when these cells were sorted from adult thymus and vigorously resuspended before rerunning through the flow cytometer, nearly all of these appeared to be derived from doublets that had formed between DP thymocytes and CD4+CD8− or DN NKT cells (data not shown).
Figure 3.

Ontogeny of NKT cells. Thymuses, spleens, and livers were removed from C57BL/6 mice at various ages and harvested cells counted and labeled for flow cytometric analysis. The first column shows mean percentages of CD1d/αGC tetramer+ αβTCR+ NKT cells, and the second column shows mean numbers of these cells. The error bars represent standard error of the mean. In some cases where the error was very low, no bars are visible. Results are from 4–12 mice per time point.
Figure 4.
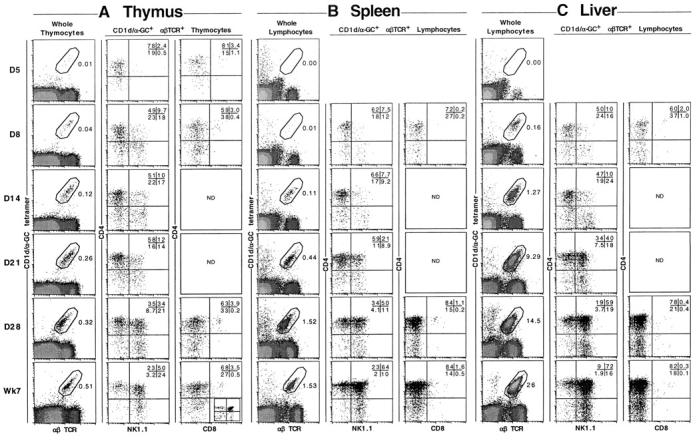
Phenotype of NKT cells during development. C57BL/6 mice were killed at various ages and (A) thymus, (B) spleen, and (C) liver harvested, cells counted, and labeled for flow cytometric analysis. The first column shows CD1d/αGC tetramer+αβTCR+ cells with the percentages indicated. The second column shows CD4 versus NK1.1 expression on NKT cells, gated as shown in the first column. The third column shows CD4 versus CD8 expression on NKT cells, gated as shown in the first column. As a staining control, CD4 versus CD8 labeling on total thymocytes is shown in the inset dotplot in the bottom row (Adult) of the third column. Dotplots are representative of 4–12 mice per time point. ND, not determined.
Most NKT Cells Leave the Thymus as NK1.1− Cells.
Recent thymic emigrants were identified by intrathymic injection of FITC dye, and RTE were examined 36 h later for CD1d/αGC tetramer and NK1.1 staining (Fig. 5) . NKT cells were clearly identifiable as CD1d/αGC tetramer+ cells within the RTE population of both spleen and liver. Furthermore, the majority of these cells (FITC+ NKT cells) were NK1.1−, despite the fact that resident (FITC−) NKT cells in these tissues were mostly NK1.1+. Similar results were obtained when NKT RTE from blood were examined (data not shown).
Figure 5.

Preferential thymic emigration of NK1.1− NKT cells. RTE were tracked following intrathymic injection of FITC dye. 36 h after injection, lymphocytes were harvested from thymus, spleen, and liver, counted, and stained for flow cytometric analysis. FITC+ cells were gated above the line shown in the first column. The second column shows that FITC+ (FL-1+) events in the spleen and liver do not occur when FITC has not been injected intrathymically. The third column shows CD1d/αGC tetramer+ αβTCR+ NKT cells within the RTE population. The third and fourth columns show NK1.1 expression on NKT cells within the FITC+ RTE and FITC− resident cell populations in each tissue. Dotplots are representative of four separate mice from one experiment.
Thymic NK1.1− NKT Cells Are Progenitors of NK1.1+ NKT Cells.
Analysis of NKT cells through ontogeny (Fig. 4) strongly suggested that CD4+NK1.1− NKT cells represent an intermediate stage in their development. To directly test this, we separated CD4+ NKT cells into NK1.1+ and NK1.1− subsets and adoptively transferred these into CD45.1 congenic recipient mice by intrathymic injection (Fig. 6) . It was important to avoid using the CD1d/αGC tetramer to sort these cells, which might lead to TCR stimulation and artificially lead to their activation. Therefore, donor cells were sorted as CD4+HSA−/low NK1.1− or NK1.1+ fractions. Donor-derived NKT cells were clearly identifiable in thymuses screened at 1 wk after transfer (Fig. 6). Whereas nearly all NK1.1+CD4+ NKT cells remained NK1.1+CD4+, the majority of the NK1.1− NKT cell fraction yielded NK1.1+ NKT cells within 1 wk, including CD4+ and CD4− subsets. Very similar results were observed two weeks after transfer (data not shown). Importantly, when CD4+HSA−/low cells were depleted of CD1d/αGC tetramer-binding cells before transfer, NKT cells were not detected in recipient mice, indicating that NK1.1− NKT cells, and not other CD4+HSA−/low cells, were responsible for the emergence of donor-derived NKT cell populations in these experiments. These results strongly suggest that thymic NK1.1− NKT cells represent a precursor stage in the development of this lineage.
Figure 6.

NK1.1− NKT cells are precursors of NK1.1**+** NKT cells. NK1.1−CD4+HSAlow and NK1.1+CD4+HSAlow populations from 4-wk-old C57BL/6 (CD45.2+) thymuses were purified by FACS® sorting and intrathymically injected into CD45.1 congenic C57BL/6 recipient mice. 1 wk later, recipient thymuses were harvested and donor-derived cells identified based on CD45.2 expression. A gate was placed around CD1d/αGC tetramer+ CD45.2+ NKT cells which were examined for CD4 versus NK1.1 expression. The percentage of acquisition-gated CD45.2+ cells that were CD1d/αGC tetramer+ is shown. These results are representative of three experiments with 1–3 recipient mice per group per experiment. Controls included transfer of CD4+HSA−/low cells that had previously been depleted of CD1d/αGC tetramer+ cells (third row) and PBS-injected thymuses (last row). Tetramer, CD1d/αGC tetramer.
NK1.1− NKT Cells Produce High Levels of IL-4.
NK1.1− and NK1.1+ subsets of NKT cells were separated by FACS® sorting and examined for cytokine production in the presence of plate-bound anti-CD3 antibodies (Table I). NK1.1+ NKT cells made high levels of both IL-4 and IFN-γ, as expected from previous studies (26). Remarkably, at both 24 and 48 h, NK1.1− NKT cells produced higher levels of IL-4, and lower levels of IFN-γ, than their NK1.1+ counterparts. Thus, although NK1.1− NKT cells seem to be developmentally less mature, the capacity to produce high levels of cytokines, particularly IL-4, appears to be an early event in the development of this lineage.
Table I.
Cytokine Production by NK1.1+ and NK1.1− CD1d/α-GC Tetramer+ Thymocytes
| IL-4 | IFN-γ | |||
|---|---|---|---|---|
| NK cellsubset | Day 1 | Day 2 | Day 1 | Day 2 |
| U/ml/105 cellsa | ng/ml/105 cells | |||
| NK1.1− | 4,400 ± 500b | 13,000 ± 1,000 | 2.2 ± 0.3 | 15 ± 3 |
| NK1.1+ | 1,160 ± 80c | 2,900 ± 200c | 16 ± 2c | 150 ± 20c |
Discussion
Despite the identification of NKT cells over 14 years ago, the developmental origin and pathway has remained an unresolved and controversial area (for reviews, see references 1 and 2). Whereas several studies have provided evidence in support of an extrathymic origin for NKT cells (for example, references 18–20 and 52–54), others provided evidence to the contrary (4, 22–24, 55–57). Some of this controversy may be explained by the absence of reliable and specific reagents to identify NKT cells. Even NK1.1, which has traditionally been used for the identification of NKT cells in C57BL/6 mice, is not ideal for two reasons: not all CD1d-dependent NKT cells express NK1.1 (even in C57BL/6 mice [29, 30, 58]), and not all NK1.1-expressing αβTCR+ cells exhibit the characteristics normally associated with NKT cells (25, 26, 29, 30; for a review, see reference 2). CD1d/αGC tetramers represent the first reagent that clearly and specifically defines CD1d-dependent NKT cells (29, 30). A potential limitation with this reagent may lie in the existence of CD1d-dependent NKT cells that do not recognize α-GalCer, although it appears to encompass the majority of invariant NKT cells (29) and is therefore a great improvement over previous approaches. There is currently no reagent available that can reliably detect all CD1d-dependent NKT cells, regardless of their specificity for α-GalCer. Using CD1d/αGC tetramers, we have verified, and expanded upon, earlier studies (4, 59), showing that CD1d/α-GalCer-dependent NKT cells (including both NK1.1**+** and NK1.1− subsets) can develop in the thymus in isolation (FTOC), and are absent in the spleen and liver of nude mice. This data firmly supports the contention that the thymus is both necessary and sufficient for the development of these cells.
This study has also revealed that NKT cells are clearly identifiable before their acquisition of the NK1.1 marker, and strongly suggests that CD1d-dependent NK1.1− NKT cells in the thymus represent a precursor stage in the development of this lineage. Whereas the thymus appears to be important for NKT cells to reach the intermediate NK1.1− stage, further maturation appears to be able to occur outside of the thymus, and indeed, the majority of NKT cells leaving the thymus are NK1.1−. Our data indicate that thymic CD4+NK1.1− NKT cells give rise to CD4+NK1.1+ and CD4−NK1.1+ subsets. These may each develop directly from CD4+NK1.1− population, as most CD4+NK1.1+ NKT cells retained that phenotype following transfer. This pathway contrasts an earlier pathway proposed by MacDonald (51), based on NK1.1+ NKT cell ontogeny, suggesting that DN NKT cells develop before CD4+ NKT cells. Indeed, our data in Fig. 4 of this paper would support the argument that NK1.1+ DN are more abundant than NK1.1+CD4+ NKT cells early in thymic ontogeny. However, these data need to be reinterpreted in light of our results; although DN NKT cells appear to predominate among the more mature NK1.1+ fraction (suggesting that they mature faster than the CD4+NK1.1+ fraction), the earliest detectable NKT cells are CD4+ and NK1.1−. In support of our data demonstrating the existence of an NK1.1− NKT cell precursor stage, an earlier study by Lantz et al. (45) demonstrated that NKT cells appeared to partly develop in common γ-chain–deficient mouse thymus, based on normal frequency of Vα14Jα281 expressing, IL-4–producing cells, yet these cells failed to acquire NK1.1 and Ly49 expression. However, these mice also lacked NKT cells in the periphery, leading the authors to suggest that full maturation to the NK1.1+ stage was required for emigration of NKT cells. Although our data differs on this point, an alternate view that could satisfy both studies is that the common γ-chain is required for the emigration of less mature NK1.1− NKT cells, as well as for their maturation within the thymus. The ability of the earlier NK1.1− NKT cells to produce very high levels of IL-4 and moderate IFN-γ might suggest that these cells are functionally competent and may participate in immune responses, as these cells also exist in the periphery (29, 30, 60), and peripheral NK1.1− NKT cells have also been shown to have a similar phenotype (58, 61). Alternatively, this characteristic may be related to the development of this lineage, possibly related to their exposure to intrathymic IL-7, a cytokine which potentiates IL-4 production by NKT cells (46, 58). A trivial explanation for the difference in IL-4 and IFN-γ production by these two subsets of NKT cells might be related to cross-linking of NK1.1 during the purification of the NK1.1+ fraction, which may enhance IFN-γ (62) and in turn suppress IL-4 synthesis. Although our ontogeny and transfer data indicate that NK1.1− NKT cells give rise to NK1.1**+** NKT cells, it remains possible that some NK1.1− NKT cells are not precursors, and may be mature NKT cells, particularly in the periphery. In line with this possibility, NK1.1+ NKT cells can at least transiently downregulate NK1.1 expression after TCR-mediated activation (63), although it remains to be determined whether this happens when NKT cells are activated in vivo.
While this paper was under review, another study was published providing evidence that early CD1d-dependent NKT cells were CD4lowCD8low (64). This clearly contrasts with our data, and we are unable to easily explain the basis for this difference. An unusual approach in the other study was that CD1d/αGC tetramer-binding thymocytes were preenriched using anti-PE conjugated magnetic beads, whereas our study involved labeling of whole, unenriched thymocytes. Another difference is that Gapin et al. did not appear to include colabeling with anti-αβTCR to exclude CD1d/αGC tetramer-binding non-T cells, as have been previously reported (29, 30, 60) and as were observed in our results in this paper (Fig. 4). Finally, we should add that we first detected tetramer+ NKT cells at day 5 after birth, when nearly all were NK1.1−CD4+CD8−, whereas the CD4lowCD8low cells reported in the other study (at days 6 and 9) did not exceed 30%. At no time did we observe a distinct population of cells with this phenotype. Taken together, we believe that our data provides a more reliable phenotype for early CD1d/αGC tetramer-binding NKT cells.
The precise factors that regulate the developmental steps in the NKT cell lineage remain to be determined. Expression of CD1d by DP thymocytes (23, 34) is clearly required to reach the first NK1.1− stage, as CD1d tetramer-binding cells were not detected in the thymus of β2m or CD1d-deficient mice (30). Factors that might be important in the progression to NK1.1 expression are unknown, but may involve signaling through the common γ-chain (45). Given that NKT cells seed to the periphery at the immature NK1.1− stage, these maturation signals may be thymus independent. Until recently, the stage at which NKT cells branch away from the mainstream αβ T cell lineage was unknown. It was hypothesized to occur at the DP stage, as this is where TCR-α gene rearrangement begins and thymocytes first start to express surface TCR-αβ and audition for intrathymic selection. As NKT cells appear to arise through randomly generated TCR-α gene rearrangements, the most likely model is that their uncommitted precursors receive an instructive signal via CD1d at the DP precursor stage that diverts developing thymocytes from the mainstream αβ T cell lineage as they express cell surface TCR. The study by Gapin et al. (64), provides direct evidence for such a pathway, showing that intrathymic adoptive transfer of DP thymocytes leads to the generation of NKT cells. Although, as mentioned above, we were unable to detect a clear population of NKT cells with a DP phenotype at any stage of ontogeny, it is possible that CD8 down-regulation occurs before TCR upregulation to levels that are sufficiently high to be detectable by CD1d/αGC tetramer staining. Apart from the clear necessity for CD1d expression by DP thymocytes (23, 33–37), the signals that control this branch-point are poorly defined. Whether CD4 or CD8 molecules are actually involved in this step is an important question. NKT cells develop in both CD4−/− and CD8−/− mice, although NKT cells in CD8−/− mice showed an altered TCR Vβ usage (4), whereas forced (transgenic) expression of CD8 prevents the development of NKT cells. This suggests that although CD8 might play a minor role in NKT cell selection, its down-regulation after selection is vital for their survival.
In summary, we have identified the CD4+NK1.1− CD1d/αGC tetramer-binding population as a thymus-dependent precursor stage in NKT cell development. These cells are able to seed to the periphery before acquisition of an NK1.1+ phenotype, suggesting that subsequent maturation to the mature NK1.1+ subsets may occur both within and outside of the thymus. This proposed pathway of NKT cell development should prove to be invaluable framework for understanding the mechanisms that control the development of this important lineage of cells.
Acknowledgments
The authors are very grateful to Prof. Mitchell Kronenberg and Dr. Olga Naidenko for providing us with CD1d/αGC reagents, Dr. Elise Randle-Barrett for assistance with flow cytometry, and Ms. Samantha Fennell, Ms. Karen Sterling, and Mr. Dale Morris for animal husbandry assistance.
This work was supported by the National Health and Medical Research Council (NHMRC), ADCORP-Diabetes Australia, and donations from Rothschild Australia. A.P. Uldrich is a recipient of an Australian Post-Graduate Award. A.G. Baxter, M.J. Smyth, and D.I. Godfrey are recipients of NHMRC research fellowships and D.I. Godfrey was a recipient of an ADCORP-Diabetes Australia Research Fellowship.
Footnotes
*
Abbreviations used in this paper: DN, double negative; DP, double positive; FTOC, fetal thymus organ culture(s); RTE, recent thymic emigrant(s).
References
- 1.Bendelac, A., M.N. Rivera, S.H. Park, and J.H. Roark. 1997. Mouse CD1-specific NK1 T cells - development, specificity, and function. Annu. Rev. Immunol. 15:535–562. [DOI] [PubMed] [Google Scholar]
- 2.Godfrey, D.I., K.J.L. Hammond, L.D. Poulton, M.J. Smyth, and A.G. Baxter. 2000. NKT cells: facts, functions and fallacies. Immunol. Today. 21:573–583. [DOI] [PubMed] [Google Scholar]
- 3.Arase, H., N. Arase, K. Ogasawara, R.A. Good, and K. Onoe. 1992. An NK1.1+ CD4+8− single-positive thymocyte subpopulation that expresses a highly skewed T-cell antigen receptor V-beta family. Proc. Natl. Acad. Sci. USA. 89:6506–6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bendelac, A., N. Killeen, D.R. Littman, and R.H. Schwartz. 1994. A subset of CD4+ thymocytes selected by MHC class I molecules. Science. 263:1774–1778. [DOI] [PubMed] [Google Scholar]
- 5.Lantz, O., and A. Bendalac. 1994. An invariant T cell receptor α chain is used by a unique subset of MHC class I specific CD4+ and CD4−8− T cells in mice and humans. J. Exp. Med. 180:1097–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Makino, Y., R. Kanno, T. Ito, K. Higashino, and M. Taniguchi. 1995. Predominant expression of invariant Vα14+ TCR alpha chain in NK1.1+ T cell populations. Int. Immunol. 7:1157–1161. [DOI] [PubMed] [Google Scholar]
- 7.Ohteki, T., and H.R. MacDonald. 1996. Stringent Vβ requirement for the development of NK1.1+ T cell receptor-alpha/beta+ cells in mouse liver. J. Exp. Med. 183:1277–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hammond, K.J.L., L.D. Poulton, L.J. Palmisano, P.A. Silveira, D.I. Godfrey, and A.G. Baxter. 1998. Alpha/beta-T cell receptor (TCR)+ CD4−CD8− (NKT) thymocytes prevent insulin-dependent diabetes mellitus in nonobese diabetic (NOD/Lt) mice by the influence of interleukin (IL)-4 and/or IL-10. J. Exp. Med. 187:1047–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lehuen, A., O. Lantz, L. Beaun, V. Laloux, C. Carnaud, A. Bendelac, J.F. Bach, and R.C. Monteiro. 1998. Overexpression of natural killer T cells protects Vα14-Jα281 transgenic nonobese diabetic mice against diabetes. J. Exp. Med. 188:1831–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang, B., Y.B. Geng, and C.R. Wang. 2001. CD1-restricted NKT cells protect nonobese diabetic mice from developing diabetes. J. Exp. Med. 194:313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang, Y., I. Goldschneider, D. Foss, D.Y. Wu, J. O'Rourke, and R.E. Cone. 1997. Direct thymic involvement in anterior chamber-associated immune deviation: evidence for a nondeletional mechanism of centrally induced tolerance to extrathymic antigens in adult mice. J. Immunol. 158:2150–2155. [PubMed] [Google Scholar]
- 12.Sonoda, K.H., M. Exley, S. Snapper, S.P. Balk, and J. Stein-Streilein. 1999. CD1-reactive natural killer T cells are required for development of systemic tolerance through an immune-privileged site. J. Exp. Med. 190:1215–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sonoda, K.H., D.E. Faunce, M. Taniguchi, M. Exley, S. Balk, and J. Stein-Streilein. 2001. NK T cell-derived IL-10 is essential for the differentiation of antigen- specific T regulatory cells in systemic tolerance. J. Immunol. 166:42–50. [DOI] [PubMed] [Google Scholar]
- 14.Zeng, D.F., D. Lewis, S. Dejbakhsh-Jones, F.S. Lan, M. Garcia-Ojeda, R. Sibley, and S. Strober. 1999. Bone marrow NK1.1− and NK1.1+ T cells reciprocally regulate acute graft versus host disease. J. Exp. Med. 189:1073–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seino, K.-I., K. Fukao, K. Muramoto, K. Yanagisawa, Y. Takada, S. Kakuta, Y. Iwakura, L. van Kaer, K. Takeda, T. Nakayama, et al. 2001. Requirement for natural killer T (NKT) cells in the induction of allograft tolerance. Proc. Natl. Acad. Sci. USA. 98:2577–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Terabe, M., S. Matsui, N. Noben-Trauth, H. Chen, C. Watson, D.D. Donaldson, D.P. Carbone, W.E. Paul, and J.A. Berzofsky. 2000. NKT cell-mediated repression of tumor immunosurveillance by IL-13 and the IL-4R-STAT6 pathway. Nat. Immunol. 1:515–520. [DOI] [PubMed] [Google Scholar]
- 17.Smyth, M.J., K.Y.T. Thia, S.E.A. Street, E. Cretney, J.A. Trapani, M. Taniguchi, T. Kawano, S.B. Pelikan, N.Y. Crowe, and D.I. Godfrey. 2000. Differential tumor surveillance by natural killer (NK) and NKT cells. J. Exp. Med. 191:661–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kikly, K., and G. Dennert. 1992. Evidence for extrathymic development of TNK cells. NK1+ CD3+ cells responsible for acute marrow graft rejection are present in thymus-deficient mice. J. Immunol. 149:403–412. [PubMed] [Google Scholar]
- 19.Makino, Y., N. Yamagata, T. Sasho, Y. Adachi, R. Kanno, H. Koseki, M. Kanno, and M. Taniguchi. 1993. Extrathymic development of Vα14-positive T cells. J. Exp. Med. 177:1399–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Makino, Y., R. Kanno, H. Koseki, and M. Taniguchi. 1996. Development of Vα14+ NK T cells in the early stages of embryogenesis. Proc. Natl. Acad. Sci. USA. 93:6516–6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sato, K., K. Ohtsuka, K. Hasegawa, S. Yamagiwa, H. Watanabe, H. Asakura, and T. Abo. 1995. Evidence for extrathymic generation of intermediate T cell receptor cells in the liver revealed in thymectomized, irradiated mice subjected to bone marrow transplantation. J. Exp. Med. 182:759–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hammond, K.J.L., W.E. Cain, I.R. Van Driel, and D.I. Godfrey. 1998. Three day neonatal thymectomy selectively depletes NK1.1+ T cells. Int. Immunol. 10:1491–1499. [DOI] [PubMed] [Google Scholar]
- 23.Coles, M.C., and D.H. Raulet. 2000. NK1.1+ T cells in the liver arise in the thymus and are selected by interactions with class I molecules on CD4+CD8+ cells. J. Immunol. 164:2412–2418. [DOI] [PubMed] [Google Scholar]
- 24.Tilloy, F., J.P. Di Santo, A. Bendelac, and O. Lantz. 1999. Thymic dependence of invariant Vα14+ natural killer-T cell development. Eur. J. Immunol. 29:3313–3318. [DOI] [PubMed] [Google Scholar]
- 25.Eberl, G., R. Lees, S.T. Smiley, M. Taniguchi, M.J. Grusby, and H.R. MacDonald. 1999. Tissue-specific segregation of CD1d-dependent and CD1d-independent NK T cells. J. Immunol. 162:6410–6419. [PubMed] [Google Scholar]
- 26.Hammond, K.J.L., S.B. Pelikan, N.Y. Crowe, E. Randle-Barrett, T. Nakayama, M. Taniguchi, M.J. Smyth, I.R. van Driel, R. Scollay, A.G. Baxter, and D.I. Godfrey. 1999. NKT cells are phenotypically and functionally diverse. Eur. J. Immunol. 29:3768–3781. [DOI] [PubMed] [Google Scholar]
- 27.Assarsson, E., T. Kambayashi, J.K. Sandberg, S. Hong, M. Taniguchi, L. Van Kaer, H.G. Ljunggren, and B.J. Chambers. 2000. CD8+ T cells rapidly acquire NK1.1 and NK cell-associated molecules upon stimulation in vitro and in vivo. J. Immunol. 165:3673–3679. [DOI] [PubMed] [Google Scholar]
- 28.Slifka, M.K., R.R. Pagarigan, and J.L. Whitton. 2000. NK markers are expressed on a high percentage of virus-specific CD8+ and CD4+ T cells. J. Immunol. 164:2009–2015. [DOI] [PubMed] [Google Scholar]
- 29.Benlagha, K., A. Weiss, A. Beavis, L. Teyton, and A. Bendelac. 2000. In vivo identification of glycolipid antigen-specific T cells using fluorescent CD1d tetramers. J. Exp. Med. 191:1895–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsuda, J.L., O.V. Naidenko, L. Gapin, T. Nakayama, M. Taniguchi, C.R. Wang, Y. Koezuka, and M. Kronenberg. 2000. Tracking the response of natural killer T cells to a glycolipid antigen using CD1d tetramers. J. Exp. Med. 192:741–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kawano, T., J.Q. Cui, Y. Koezuka, I. Toura, Y. Kaneko, K. Motoki, H. Ueno, R. Nakagawa, H. Sato, E. Kondo, et al. 1997. CD1d-restricted and TCR-mediated activation of Vα14 NKT cells by glycosylceramides. Science. 278:1626–1629. [DOI] [PubMed] [Google Scholar]
- 32.Burdin, N., L. Brossay, Y. Koezuka, S.T. Smiley, M.J. Grusby, M. Gui, M. Taniguchi, K. Hayakawa, and M. Kronenberg. 1998. Selective ability of mouse CD1 to present glycolipids - α-galactosylceramide specifically stimulates Vα14+ NKT lymphocytes. J. Immunol. 161:3271–3281. [PubMed] [Google Scholar]
- 33.Adachi, Y., H. Koseki, M. Zijlstra, and M. Taniguchi. 1995. Positive selection of invariant Vα14+ T cells by non-major histocompatibility complex-encoded class I-like molecules expressed on bone marrow derived cells. Proc. Natl. Acad. Sci. USA. 92:1200–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bendelac, A. 1995. Positive selection of mouse NK1+ T cells by CD1-expressing cortical thymocytes. J. Exp. Med. 182:2091–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bix, M., M. Coles, and D. Raulet. 1993. Positive selection of Vβ8+CD4-8- thymocytes by class I molecules expressed by hematopoietic cells. J. Exp. Med. 178:901–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coles, M.C., and D.H. Raulet. 1994. Class I dependence of the development of CD4+ CD8− NK1.1+ thymocytes. J. Exp. Med. 180:395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohteki, T., and H.R. MacDonald. 1994. Major histocompatibility complex class I related molecules control the development of CD4+8− and CD4−8− subsets of NK1.1+ T cell receptor αβ cells in the liver of mice. J. Exp. Med. 180:699–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shimamura, M., T. Ohteki, U. Beutner, and H.R. MacDonald. 1997. Lack of directed Vα14Jα281 rearrangements in NK1+ T cells. Eur. J. Immunol. 27:1576–1579. [DOI] [PubMed] [Google Scholar]
- 39.Iizuka, K., D.D. Chaplin, Y. Wang, Q. Wu, L.E. Pegg, W.M. Yokoyama, and Y.X. Fu. 1999. Requirement for membrane lymphotoxin in natural killer cell development. Proc. Natl. Acad. Sci. USA. 96:6336–6340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Elewaut, D., L. Brossay, S.M. Santee, O.V. Naidenko, N. Burdin, H. De Winter, J. Matsuda, C.F. Ware, H. Cheroutre, and M. Kronenberg. 2000. Membrane lymphotoxin is required for the development of different subpopulations of NK T cells. J. Immunol. 165:671–679. [DOI] [PubMed] [Google Scholar]
- 41.Eberl, G., H.J. Fehling, H. von Boehmer, and H.R. MacDonald. 1999. Absolute requirement for the pre-T cell receptor alpha chain during NK1.1+ TCR alpha beta cell development. Eur. J. Immunol. 29:1966–1971. [DOI] [PubMed] [Google Scholar]
- 42.Sato, H., T. Nakayama, Y. Tanaka, Y. Yamashita, Y. Saito, and M. Taniguchi. 1999. Induction of differentiation of pre-NKT cells to mature Vα14 NKT cells by granulocyte macrophage colony-stimulating factor. Proc. Natl. Acad. Sci. USA. 96:7439–7444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eberl, G., B. Lowin-Kropf, and H.R. MacDonald. 1999. Cutting edge: NKT cell development is selectively impaired in Fyn-deficient mice. J. Immunol. 163:4091–4094. [PubMed] [Google Scholar]
- 44.Gadue, P., N. Morton, and P.L. Stein. 1999. The Src family tyrosine kinase Fyn regulates natural killer T cell development. J. Exp. Med. 190:1189–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lantz, O., L.I. Sharara, F. Tilloy, A. Andersson, and J.P. DiSanto. 1997. Lineage relationships and differentiation of natural killer (NK) T cells - intrathymic selection and interleukin (IL)-4 production in the absence of NKR-P1 and Ly49 molecules. J. Exp. Med. 185:1395–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vicari, A.P., A. Herbelin, and R.D. Leite de Moraes. 1996. NK1.1+ T cells from IL-7-deficient mice have a normal distribution and selection but exhibit impaired cytokine production. Int. Immunol. 8:1759–1766. [DOI] [PubMed] [Google Scholar]
- 47.Ohteki, T., S. Ho, H. Suzuki, T.W. Mak, and P.S. Ohashi. 1997. Role for IL-15/IL-15 receptor beta-chain in NK1.1+ T cell receptor-alpha-beta+ cell development. J. Immunol. 159:5931–5935. [PubMed] [Google Scholar]
- 48.Nakagawa, K., K. Iwabuchi, K. Ogasawara, M. Ato, M. Kajiwara, H. Nishihori, C. Iwabuchi, H. Ishikura, R.A. Good, and K. Onoe. 1997. Generation of NK1.1+ T cell antigen receptor alpha/beta+ thymocytes associated with intact thymic structure. Proc. Natl. Acad. Sci. USA. 94:2472–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Godfrey, D.I., J. Kennedy, M.K. Gately, J. Hakimi, B.R. Hubbard, and A. Zlotnik. 1994. IL-12 influences intrathymic T cell development. J. Immunol. 152:2729–2735. [PubMed] [Google Scholar]
- 50.Scollay, R.G., E.C. Butcher, and I.L. Weissman. 1980. Thymus cell migration. Quantitative aspects of cellular traffic from the thymus to the periphery in mice. Eur. J. Immunol. 10:210–218. [DOI] [PubMed] [Google Scholar]
- 51.MacDonald, H.R. 2000. CD1d-glycolipid tetramers: A new tool to monitor natural killer T cells in health and disease. J. Exp. Med. 192:F15–F20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koseki, H., H. Asano, T. Inaba, N. Miyashita, K. Moriwaki, K.F. Lindahl, Y. Mizutani, K. Imai, and M. Taniguchi. 1991. Dominant expression of a distinctive V14+ T-cell antigen receptor alpha chain in mice. Proc. Natl. Acad. Sci. USA. 88:7518–7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shimamura, M., T. Ohteki, P. Launois, A.M. Garcia, and H.R. Macdonald. 1997. Thymus-independent generation of NK1+ T cells in vitro from fetal liver precursors. J. Immunol. 158:3682–3689. [PubMed] [Google Scholar]
- 54.Shimamura, M., Y.Y. Huang, Y. Suda, S. Kusumoto, K. Sato, M.J. Grusby, H. Sato, T. Nakayama, and M. Taniguchi. 1999. Positive selection of NKT cells by CD1(+), CD11c(+) non-lymphoid cells residing in the extrathymic organs. Eur. J. Immunol. 29:3962–3970. [DOI] [PubMed] [Google Scholar]
- 55.Emoto, M., Y. Emoto, and S.H.E. Kaufmann. 1995. Il-4 producing cd4(+) tcr-alpha-beta(int) liver lymphocytes - influence of thymus, beta(2)-microglobulin and nk1.1 expression. Int. Immunol. 7:1729–1739. [DOI] [PubMed] [Google Scholar]
- 56.Levitsky, H.I., P.T. Golumbek, and D.M. Pardoll. 1991. The fate of CD4−8− T cell receptor-ab+ thymocytes. J. Immunol. 146:1113–1117. [PubMed] [Google Scholar]
- 57.Sykes, M. 1990. Unusual T cell populations in adult murine bone marrow. Prevalence of CD3+CD4−CD8− and abTCR+ NK1.1+ cells. J. Immunol. 145:3209–3215. [PubMed] [Google Scholar]
- 58.Hameg, A., C. Gouarin, J.M. Gombert, S.M. Hong, L. van Kaer, J.F. Bach, and A. Herbelin. 1999. IL-7 up-regulates IL-4 production by splenic NK1.1+ and NK1.1− MHC class I-like/CD1-dependent CD4+ T cells. J. Immunol. 162:7067–7074. [PubMed] [Google Scholar]
- 59.Ceredig, R., F. Lynch, and P. Newman. 1987. Phenotypic properties, interleukin 2 production, and developmental origin of a “mature” subpopulation of Lyt-2- L3T4- mouse thymocytes. Proc. Natl. Acad. Sci. USA. 84:8578–8582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hammond, K.J.L., D.G. Pellicci, L.D. Poulton, O.V. Naidenko, A.A. Scalzo, A.G. Baxter, and D.I. Godfrey. 2001. CD1d-restricted NKT cells: an interstrain comparison. J. Immunol. 167:1164–1173. [DOI] [PubMed] [Google Scholar]
- 61.Hameg, A., I. Apostolou, M. Leite de Moraes, J.M. Gombert, C. Garcia, Y. Koezuka, J.F. Bach, and A. Herbelin. 2000. A subset of NKT cells that lacks the NK1.1 marker, expresses CD1d molecules, and autopresents the α-galactosylceramide antigen. J. Immunol. 165:4917–4926. [DOI] [PubMed] [Google Scholar]
- 62.Arase, H., N. Arase, and T. Saito. 1996. Interferon gamma production by natural killer (NK) cells and NK1.1+ T cells upon NKR-P1 cross-linking. J. Exp. Med. 183:2391–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen, H.J., H. Huang, and W.E. Paul. 1997. NK1.1+CD4+ T cells lose NK1.1 expression upon in vitro activation. J. Immunol. 158:5112–5119. [PubMed] [Google Scholar]
- 64.Gapin, L., J.L. Matsuda, C.D. Surh, and M. Kronenberg. 2001. NKT cells derive from double-positive thymocytes that are positively selected by CD1d. Nat. Immunol. 2:971–978. [DOI] [PubMed] [Google Scholar]