Sequence Determinants in Hypoxia-inducible Factor-1α for Hydroxylation by the Prolyl Hydroxylases PHD1, PHD2, and PHD3 (original) (raw)
. Author manuscript; available in PMC: 2007 Mar 15.
Published in final edited form as: J Biol Chem. 2002 Aug 13;277(42):39792–39800. doi: 10.1074/jbc.M206955200
Abstract
Hypoxia-inducible factor (HIF) is a heterodimeric transcription factor induced by hypoxia. Under normoxic conditions, site-specific proline hydroxylation of the α subunits of HIF allows recognition by the von Hippel-Lindau tumor suppressor protein (VHL), a component of an E3 ubiquitin ligase complex that targets these subunits for degradation by the ubiquitin-proteasome pathway. Under hypoxic conditions, this hydroxylation is inhibited, allowing the α subunits of HIF to escape VHL-mediated degradation. Three enzymes, prolyl hydroxylase domain-containing proteins 1, 2, and 3 (PHD1, -2, and -3; also known as HIF prolyl hydroxylase 3, 2, and 1, respectively), have recently been identified that catalyze proline hydroxylation of HIF α subunits. These enzymes hydroxylate specific prolines in HIF α subunits in the context of a strongly conserved L_XX_LA_P_ sequence motif (where X indicates any amino acid and P indicates the hydroxylacceptor proline). We report here that PHD2 has the highest specific activity toward the primary hydroxylation site of HIF-1α. Furthermore, and unexpectedly, mutations can be tolerated at the −5, −2, and −1 positions (relative to proline) of the L_XX_LAP motif. Thus, these results provide evidence that the only obligatory residue for proline hydroxylation in HIF-1α is the hydroxylacceptor proline itself.
A critical means by which cells respond to stress is by the modulation of gene expression. The cellular response to hypoxia provides a cardinal example of this (1–3). Low oxygen tension (1% O2) induces a battery of genes that allow cells to increase their uptake of glucose (through up-regulation of specific isoforms of the glucose transporter) and shift utilization of this energy source from aerobic to glycolytic metabolism (through up-regulation of genes such as phosphofructokinase, aldolase, and glyceraldehyde-3-phosphate dehydrogenase, among others) (4). Low oxygen tension also induces the up-regulation of genes involved in local and systemic responses to hypoxia. An example of the former is vascular endothelial growth factor, a potent angiogenic agent, whereas an example of the latter is erythropoietin, the central regulator of red blood cell maturation.
Hypoxia-inducible factor (HIF)1 is a transcription factor that is the master regulator of all of the above genes (1, 2). HIF is heterodimeric and comprises an α and a β subunit, both of which in turn are members of the PAS family of transcription factors. The β subunit is a member of the aryl hydrocarbon nuclear translocator family of proteins. Three α subunit isoforms have been identified, HIF-1α, HIF-2α, and HIF-3α (5–9). The central mechanism by which HIF is activated by hypoxia is by regulated stabilization of the α subunit. In the case of HIF-1α, the most intensively studied isoform, two proline residues, Pro-402 and Pro-564, are constitutively hydroxylated under normoxic conditions (10–13). This hydroxylation allows binding by the von Hippel-Lindau tumor suppressor protein (VHL) (14), the substrate recognition component of an E3 ubiquitin ligase complex (15). This, in turn, is followed by the ubiquitination (16–19) and subsequent degradation of HIF-1α by the proteasome (20–22). Structural studies have implicated the formation of specific hydrogen bonds between the 4-hydroxyl group of hydroxylproline and residues in the β domain of VHL as being critical to recognition of this post-translational modification (23, 24). Hydroxylation of an asparagine residue in the C-terminal transactivation domain of HIF-1α provides a distinct mechanism by which transcriptional activity of HIF-1α is regulated (25–29).
The site-specific hydroxylation of the proline residues in HIF is catalyzed by a recently described family of enzymes, PHD1/ HPH3, PHD2/HPH2, and PHD3/HPH1 (hereafter referred to in the PHD nomenclature) (30, 31). These enzymes bear homology to the collagen hydroxylases that catalyze the hydroxylation of proline residues in collagen and to a larger family of iron-containing dioxygenases that catalyze a variety of hydroxylation reactions using both protein and nonprotein substrates (32). These enzymes employ molecular oxygen and 2-oxoglutarate as substrates in addition to the hydroxylacceptor proline residue (33). In this reaction, 2-oxoglutarate undergoes stoichiometric decarboxylation that is coupled to hydroxylation of the proline substrate. Ascorbic acid serves as a distinct cosubstrate, providing an alternative oxygen acceptor in uncoupled decarboxylation cycles (33). This enzymatic reaction provides a means by which oxygen tension can be coupled to HIF stabilization. Under normoxic conditions, proline hydroxylation occurs constitutively, thereby promoting the VHL-mediated degradation of HIF. Under hypoxic conditions, proline hydroxylation of HIF does not proceed, allowing HIF to escape degradation (30, 31).
Mutagenesis studies have provided evidence that PHDs require only a short stretch of HIF amino acids for selective recognition (10–12). HIF-1α peptides as short as 20 residues in length, in fact, can support both site-specific proline hydroxylation and subsequent binding to VHL. The sites of proline hydroxylation reside within an L_XX_LAP sequence motif that is strongly conserved between the two hydroxylation target sites of HIF-1α and HIF-2α and between HIF isoforms from different species (10–13,31). These considerations raise the possibility that PHDs recognize with absolute specificity the L_XX_LAP motif, implying that the alanine and two leucines in this motif may be strictly required for recognition by PHDs. Using assays for site-specific hydroxylation of HIF-1α, we report here that mutations can unexpectedly be tolerated at each of these three positions. Indeed, proline appears to be the only residue in this motif that is absolutely required. We furthermore show that, among the PHD isoforms, PHD2 has the highest activity toward the HIF-1α peptide.
EXPERIMENTAL PROCEDURES
Cell Cultures and Reagents
COS-1 cells were cultured as described previously (34). Iron chloride (FeCl2), cobalt chloride (CoCl2), 2-oxoglutarate, and ascorbic acid were obtained from Sigma.
Plasmids
pcDNA3-FLAG-PHD1 was constructed by first subcloning the 1.1-kb _Sma_I fragment of Gene Connection Discovery Clone C10485 (Stratagene) into the _Xho_I (blunt) site of pcDNA3-FLAG, a vector derived from pcDNA3 containing the coding sequence for the FLAG epitope. Sequencing revealed the presence of a codon encoding for serine at residue 30 in this particular clone. Inspection of the published sequences for PHD1 (30, 31), as well as multiple expressed sequence tags, reveals that they contain a codon encoding for glycine at this position. Therefore, we changed the codon for residue 30 to one encoding for glycine using a QuikChange mutagenesis kit (Stratagene). The final product was designated pcDNA3-FLAG-PHD1. pcDNA3-FLAG-PHD2 was constructed in several steps. First, the _Nco_I (blunt)/_Not_I fragment of IMAGE clone 3916773 was subcloned into the _Xho_I (blunt)/_Not_I site of pcDNA3-FLAG. An oligonucleotide duplex comprised of the following sequences: 5′-GGCCGCGTCGCCCTGCAGAGCGGCCGGAGACGT-3′ and 5′-CTAGACGTCTCCGGCCGCTCTGCAGGGCGACGC-3′ was then subcloned into the _Not_I/_Xba_I site of the product from the previous step to yield pcDNA3-FLAG-PHD2-N. Next, the 0.4-kb _Nco_I/_Xba_I fragment of IMAGE clone 2094232 was subcloned into the _Nco_I/_Xba_I site of IMAGE clone 2448834. The 0.9-kb _Not_I/_Xba_I fragment of the product from this step was then subcloned into the _Not_I/_Xba_I site of pcDNA3 to yield pcDNA3-PHD2-C. Finally, the 1.3-kb _Bgl_II/_Bsm_BI fragment of pcDNA3-FLAG-PHD2-N was subcloned into the _Bgl_II/_Not_I site of pcDNA3-PHD2-C to yield pcDNA3-FLAG-PHD2. pcDNA3-FLAG-PHD3 was constructed by subcloning the 0.8-kb _Bam_HI (partial)/_Eco_RI fragment of Gene Connection Discovery Clone C20430 (Stratagene) into the _Bam_HI/_Eco_RI site of pcDNA3-FLAG. The H297A, R383A, and H135A mutants of PHD1, PHD2, and PHD3, respectively, were prepared using a QuikChange mutagenesis kit (Stratagene).
pBXG-HIF-1α-(531–652)-P564A was constructed by subcloning the 0.36-kb _Eco_RI/_Spe_I fragment of pcDNA3-HA-HIF-1α P564A (12) into the _Eco_RI/_Spe_I site of pBXG1. pcDNA3-GAL4-HIF-1α-(531–652)-P564A was constructed by subcloning the 0.9-kb _Hin_dIII/_Bcl_I fragment of pBXG-HIF-1α-(531–652)-P564A into the _Hin_dIII/_Bam_HI site of pcDNA3. pcDNA3-GAL4-HIF-1α-(531–652)-P564A ΔMfeΔ_Bst_XI (which lack vector _Mfe_I and _Bst_XI sites) was constructed by partial digestion with _Mfe_I, blunting with the Klenow fragment of Escherichia coli DNA polymerase I, and self ligation; followed by digestion with _Xba_I, blunting with T4 DNA polymerase, and self ligation. pcDNA3-GAL4-HIF-1α-(531–652) was constructed by subcloning the 0.6-kb _Hin_dIII/ _Mfe_I fragment of pBXG-HIF-1α-(531–575) (12) into the _Hin_dIII/_Mfe_I site of pcDNA3-GAL4-HIF-1α-(531–652)-P564A. The L562A, A563G, and Y565F mutants of pcDNA3-GAL4-HIF-1α-(531–652) were prepared in an analogous manner using corresponding mutants of pBXG-HIF-1α-(531–575) (12). The L559V, L562V, A563S, D558A/E560Q, D569N/D570N/D571N, D569A/D570A/D571A, and D558A/E560Q/ D569N/D570N/D571N mutants were prepared by subcloning synthetic oligonucleotide duplexes encoding the desired mutations into the _Xba_I/ _Mfe_I site of pcDNA3-GAL4-HIF-1α-(531–652).
pcDNA3-HA-VHL, which contains the coding sequence for a hemagglutinin tag fused to the N terminus of VHL, was prepared by standard recombinant DNA methods. Sequences of recombinant plasmids were confirmed using a Big Dye Terminator kit and an ABI automated sequencer. The sources of pSX/g7F, pcDNA3-HA-HIF-1α, pGEX-HIF-1α-(531–575), pcDNA3-FLAG-VHL, and (eHRE)3-Luc have been described previously (12, 34).
Recombinant Proteins
The coding sequences of PHD1, PHD2, and PHD2 (both FLAG-tagged and untagged versions) were subcloned into pFastBacHT (Invitrogen). Preparation of recombinant baculovirus and purification of recombinant proteins using nickel-nitrilotriacetic acid-agarose were performed according the manufacturer’s instructions.
VBC was expressed as a polyhistidine- and FLAG-tagged protein using the baculovirus system. pFastBacHT-VHL was constructed in the following manner. First, the 0.7-kb _Bsm_I (blunt)/_Sal_I fragment of pUC18-VHL (a gift of Dr. Richard Klausner, NCI, National Institutes of Health) was subcloned into the _Eco_RI (blunt)/_Sal_I site of pGEX-5X-1 to yield pGEX-VHL. Next, the 0.2-kb _Bam_HI/_Not_I fragment of pGEX-VHL was subcloned into the _Bam_HI/_Not_I site of pFastBacHTc. Finally, the 0.5-kb _Not_I/_Bam_HI (blunt) fragment of pSX/g7F was subcloned into the _Not_I/_Xba_I (blunt) site of the product from the previous step to yield pFastBacHT-FLAG-VHL.
pFastBacDUAL-Elongin B-Elongin C was constructed by sequentially subcloning the 0.35-kb _Xba_I/_Hin_dIII and 0.43-kb _Bam_HI fragments of pST37-HisTrxVBC (35) (a gift of Dr. Song Tan, Pennsylvania State University) that encode Elongin C (residues 17–112) and Elongin B, respectively, into the _Xba_I/_Hin_dIII and _Bbs_I sites of pFastBacDUAL, respectively. In the resulting construct, the expression of Elongin B and that of Elongin C are driven by separate polyhedrin and p10 promoters, respectively. His6-FLAG-VBC was purified using nickel-nitrilotriacetic acid-agarose from Sf9 cells coinfected with recombinant baculoviruses expressing His6-FLAG-VHL and Elongin B/Elongin C. GST-HIF-1α-(531–575) was purified from E. coli DH5α transformed with pGEX-HIF-1α-(531–575) (12) by affinity chromatography on GSH-agarose (36).
In Vitro Translation
For most experiments, 35S-labeled, in vitro translated wild type or mutant GAL4-HIF-1α-(531–652) or HA-HIF-1α was prepared using a wheat germ TnT kit (Promega) and the appropriate pcDNA3-GAL4-HIF-1α-(531–652) vector or pcDNA3-HA-HIF-1α, respectively. In the experiment shown in Fig. 1B, it was prepared using a reticulocyte lysate TnT Quick kit (Promega). 35S-labeled, in vitro translated VHL or PHD was prepared using a reticulocyte lysate TnT Quick kit and either pcDNA3-HA-VHL or the appropriate pcDNA3-FLAG-PHD vector, respectively.
Fig. 1. PHD-induced mobility shift of HIF-1α-(531–652).
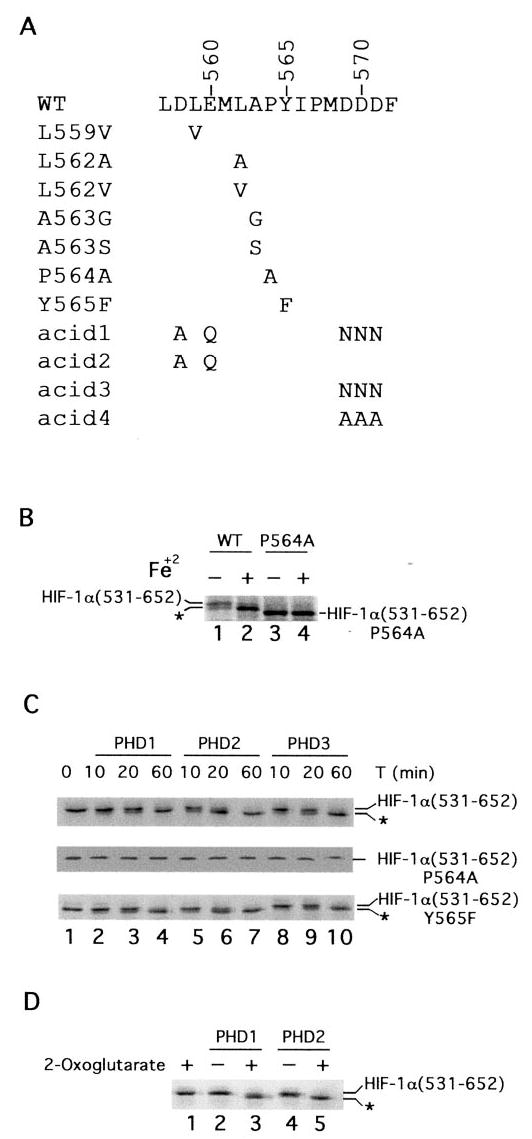
A, sequence of HIF-1α from residue 557–572. Wild type sequence is shown at the top, and mutants are indicated below. B, reticulocyte lysate-induced mobility shift of HIF-1α-(531–652). 35S-labeled GAL4-HIF-1α-(531–652) or GAL4-HIF-1α-(531–652)-P564A was prepared by in vitro transcription and translation with reticulocyte lysates in the absence or presence of 100 μM FeCl2, as shown. Products were subjected to SDS-PAGE and autoradiography. C, PHD-induced mobility shift of HIF-1α-(531–652). In vitro translated, 35S-labeled wild type (top panel), P564A (middle panel), or Y565F (bottom panel) GAL4-HIF-1α-(531–652) was prepared using wheat germ extracts and incubated with 0.5 μg of His6-FLAG-PHD1, His6-FLAG-PHD2, or His6-FLAG-PHD3 for the indicated times at 30 °C. The reaction products were subjected to SDS-PAGE and autoradiography. One of four representative results is shown. D, 2-oxoglutarate dependence of PHD-induced mobility shift. His6-FLAG-PHD1 or His6-FLAG-PHD2 was incubated with 35S-labeled, in vitro translated GAL4-HIF-1α-(531–652) for 1 h at 30 °C in the absence or presence of 1 mM 2-oxoglutarate. Reaction products were subjected to SDS-PAGE and autoradiography. B–D, the positions of the in vitro translated proteins are indicated, and asterisks indicate more rapidly migrating species (see text).
Proline Hydroxylase Assays
Mobility Shift Assay
Recombinant PHD was incubated with 35S-labeled, in vitro translated wild type or mutant GAL4-HIF-1α-(531–652) or HA-HIF-1α in 10 μl of buffer A (20 mM Hepes, pH 7.9, 100 mM KCl, 10% glycerol, 1.4 mM β-mercaptoethanol) supplemented with 100 μM FeCl2, 1 mM 2-oxoglutarate, and 5 mM ascorbate for 1 h at 30 °C. Reaction products were subjected to SDS-PAGE and autoradiography.
VBC Pull-down Assay
In vitro translated, 35S-labeled wild type or mutant GAL4-HIF-1α-(531–652) or HA-HIF-1α was incubated with recombinant PHD as just described. Hydroxylated reaction product was then isolated by first incubating with 1 μg of His6-FLAG-VBC in 500 μl of buffer A for 1 h at 4 °C with rocking, and then immunoprecipitating the His6-FLAG-VBC complex with 10 μl of anti-FLAG-agarose (M2-agarose, Sigma) for 1 h at 4 °C with rocking. The resins were washed three times with buffer A, eluted with 2× SDS-PAGE loading buffer, and the eluates then subjected to SDS-PAGE and autoradiography.
Endogenous PHD Assay
COS-1 cells were seeded in six-well plates at 80% confluency, and some cells were then treated with 100 μM CoCl2 for 2 h. Cells were then washed once with phosphate-buffered saline (PBS) and then lysed in 1 ml of buffer B (20 mM Tris, pH 7.6, 150 mM NaCl, 25 mM β-glycerophosphate, 2 mM sodium pyrophosphate, 10% glycerol, 1% Triton X-100, 1.4 mM β-mercaptoethanol) supplemented with 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, and 10 μg/ml leupeptin. Following centrifugation at 13,000 × g for 10 min at 4 °C, the whole cell extracts were incubated with GST-HIF-1α-(531–575) (20 μg) prebound to GSH-agarose for 1 h at 4 °C with rocking. The resins were washed two times with buffer B and then two times with buffer A. The resins were then incubated in a total volume of 20 μl of buffer A supplemented with 100 μM FeCl2, 1 mM 2-oxoglutarate, and 5 mM ascorbate for 1 h at 30 °C. Resins were then washed once with buffer A and incubated with 5 μl of 35S-labeled, in vitro translated VHL in 500 μl of buffer A supplemented with 0.2% Nonidet P-40 and 0.5 mg/ml bovine serum albumin (BSA) for 1 h at 4 °C with rocking. Resins were washed three times with buffer A supplemented with 0.2% Non-idet P-40, eluted with 2× SDS-PAGE loading buffer, and the eluates then subjected to SDS-PAGE and autoradiography.
Protein Interaction Assays
PHD Pull-down Assay
Five microliters of 35S-labeled, in vitro translated wild type or mutant GAL4-HIF-1α-(531–652) was incubated with 1 μg of His6-FLAG-PHD1, His6-FLAG-PHD2, or His6-FLAG-PHD3 in 500 μl of buffer A supplemented with 100 μM FeCl2, 0.2% Nonidet P-40, and 0.5 mg/ml BSA for 1 h at 4 °C with rocking. The PHD was then immunoprecipitated by the addition of 10 μl of M2-agarose, followed by incubation for an additional 1 h at 4 °C. The resins were washed three times with buffer A, eluted with 2× SDS-PAGE loading buffer, and the eluates were subjected to SDS-PAGE and autoradiography.
GST Pull-down Assay
Twenty micrograms of GST or GST-HIF-1α-(531–575) prebound to 10 μl of GSH-agarose was incubated with rocking for 1 h at 4 °C with 5 μl of in vitro translated, 35S-labeled PHD in 500 μl of buffer A supplemented with 0.2% Nonidet P-40 and 0.5 mg/ml BSA. The resins were washed three times with buffer A, eluted with 2× SDS loading buffer, and the eluates were subjected to SDS-PAGE and autoradiography.
Luciferase Assay
The day prior to transfection, COS-1 cells were seeded in 24-well plates so as to achieve 80% confluency. The cells were then cotransfected using Fugene6 (Roche Molecular Biochemicals). Cells were harvested typically after 24 h. Luciferase activities were measured using a Dual-Luciferase Reporter Assay System (Promega) and a Wallac LB9507 luminometer. Aliquots of cell extracts were also analyzed by immunoblotting using anti-GAL4 (RK5C1, Santa Cruz Biotechnology), anti-HIF-1α (Transduction Laboratories), or anti-FLAG (M2, Sigma) monoclonal antibodies as described previously (34).
Immunofluorescence Staining
COS-1 cells were plated on glass culture slides (Falcon) at 80% confluency and then cotransfected using Fugene6 with various combinations of pcDNA3-FLAG-PHD1, pcDNA3-FLAG-PHD2, pcDNA3-FLAG-PHD3, pcDNA3-HA-HIF-1α, pcDNA3-HA-VHL, or pcDNA3. Twenty hours post-transfection, the cells were fixed (4% paraformaldehyde for 20 min), permeabilized (0.2% Triton X-100 for 5 min), and blocked (2% BSA/PBS for 2 h) (37). Cells were then incubated with anti-FLAG monoclonal antibodies (M2, Sigma) and/or anti-HA rabbit polyclonal antibodies (Y-11, Santa Cruz Biotechnology) for 1 h. After three washes with PBS, the cells were incubated with fluorescein iso-thiocyanate-labeled anti-rabbit IgG and/or Texas Red-labeled anti-mouse IgG. Preparations were mounted in Vectashield mounting medium (Vector Laboratories). Confocal images of a single color or an overlay of two colors were taken using Bio-Rad 1024-ES and Nikon Eclipse E-600 confocal microscope system (Biomedical Imaging Core Laboratory, University of Pennsylvania School of Medicine).
RESULTS
The hydroxylation of HIF-1α was first identified at Pro-564 (10–12), which is present in the context of a L_XX_LAP sequence motif that is also present at the secondary site of hydroxylation (Pro-402) (13). We employed the sequence surrounding Pro-564 as the basis of our studies, and focused on the −1, −2, and −5 positions (relative to Pro-564) containing alanine, leucine, and leucine, respectively. Toward this end, we prepared the following point mutants in the context of a GAL4-HIF-1α-(531–652) fusion protein: L559V, L562A, L562V, A563G, A563S, and P564A (Fig. 1_A_). In addition, a Y565F mutant was examined to determine the stringency for Tyr at the +1 position. Finally, we prepared the following four mutants to examine potential roles for acidic residues, which are characteristically seen in the vicinity of the sites of proline hydroxylation in HIF: D558A/ E560Q/D569N/D570N/D571N (hereafter referred to as acid1), D558A/E560Q (acid2), D569N/D570N/D571N (acid3), and D569A/D570A/D571A (acid4).
Reticulocyte lysates, in comparison to wheat germ extracts, induce a more rapidly migrating species upon SDS-PAGE when employed to prepare in vitro translated, 35S-labeled GAL4-HIF-1α-(531–652) (10). This more rapidly migrating species, in contrast to the more slowly migrating species, binds VHL (10), consistent with this species representing the proline-hydroxylated species generated by one or more PHDs resident in the reticulocyte lysate. This notion is supported by the following observations. First, we show that inclusion of FeCl2 to the in vitro translation reaction converts the more slowly migrating species to the more rapidly migrating species (indicated by asterisk in Fig. 1_B_, compare lanes 1 and 2); previous studies have shown that the PHD(s) resident in the reticulocyte lysate are deficient in iron and that their activity can be augmented by inclusion of iron (11). Interestingly, we have not been able to detect a mobility shift when the substrate employed was GAL4 fused to shorter fragments of HIF-1α-(531–575) or -(549–575) (data not shown). Second, this iron-inducible mobility shift is abolished by the P564A mutation (Fig. 1_B_, lanes 3 and 4). Third, we show that this mobility shift can be reproduced in a time-dependent manner by recombinant PHD1, PHD2, and PHD3 isolated from baculovirus-infected Sf9 cells (Fig. 1_C_, top panel, lanes 2–10). Consistent with previous results, the more rapidly migrating species produced in this manner binds to VHL (data not shown). Fourth, the mobility shift induced by recombinant PHD is abolished when 2-oxoglutarate is omitted (Fig. 1_D_, compare lanes 2 with 3, and lanes 4 with 5).
The experiment of Fig. 1_C_ also suggests that PHD2 has the highest specific activity toward this HIF-1α peptide. Thus, after 20 min of incubation, most of the GAL4-HIF-1α-(531–652) substrate has been converted to the more rapidly migrating species by PHD2 (top panel, lane 6). In the case of PHD1 and PHD3, substantially less of the substrate has been converted at the same time point (top panel, lanes 3 and 9, respectively). When a Y565F mutant of HIF-1α is examined, a similar mobility shift is seen (bottom panel). This mobility shift appears with kinetics comparable to that observed with the wild type substrate, strongly suggesting that this particular mutation does not adversely affect hydroxylation by the PHDs. As expected the P564A mutant does not display any mobility shift with any of the PHD isoforms (middle panel).
We attempted to employ this mobility shift-based assay to examine other HIF-1α mutants and indeed, we were able to detect subtle PHD-induced mobility shifts (to more rapidly migrating species) with the L559V, L562A, L562V, and A563S mutants (data not shown). However, the mobility shifts were substantially smaller in degree than that seen with the wild type substrate; moreover, it was not possible to reliably determine if the more rapidly migrating species, as opposed to the more slowly migrating species, had the capacity to bind VHL (data not shown).
We therefore employed a pull-down assay that relies on the ability of the VHL to selectively recognize proline-hydroxylated HIF-1α (10, 11). For purposes of these experiments, it should be noted that this recognition occurs irrespective of mutations at the −2, −1, and +1 positions (10, 31). Indeed, the structures of hydroxylproline HIF-1α peptide bound to the VHL·Elongin B·Elongin C (VBC) complex reveals that the salient feature of this interaction consists of multiple, specific contacts with the hydroxylproline moiety, with two particularly noteworthy hydrogen bonds linking the hydroxyl group to highly conserved VHL residues (Ser-111 and His-115) (23, 24). The residues either N- or C-terminal to the hydroxylproline lay in an extended conformation, and these residues make more limited contacts with VHL.
Accordingly, we incubated the 35S-labeled HIF-1α substrates with PHD, and subsequently isolated the hydroxylated substrate by coimmunoprecipitation with the VBC complex. The hydroxylated substrate was then analyzed by SDS-PAGE. As shown in Fig. 2_A_, all three PHDs can be assayed in a time-dependent manner using this assay. In the case of PHD2, for example, hydroxylation-dependent binding to VHL can be detected at 20 min and more strongly at 60 min (lanes 6 and 7, top panel). Importantly, this experiment confirms that PHD2 has the highest specific activity of the PHDs toward this HIF-1α peptide (top panel, compare lanes 3, 7, and 11). This experiment also indicates that the Y565F mutant behaves in a manner almost indistinguishable from the wild type substrate (fourth panel from bottom), as might be predicted from the results from the mobility shift assay (Fig. 1_C_). As expected, the P564A mutant fails to serve as a substrate in this assay (fifth panel from bottom). Consistent with previous findings that the A563G mutation abolishes the regulated in vivo interaction with VHL (12) and hydroxylation by in vitro translated PHD2 and PHD3 (31), we find here that it also fails to serve as a substrate for all three PHDs in this assay (fifth panel from top). It has been previously shown that a hydroxylproline-containing HIF-1α peptide harboring this mutation can still bind VHL (31), thereby ruling out the possibility that the finding is accounted for by this mutation simply abolishing VHL binding.
Fig. 2. PHD-induced hydroxylation of wild type and mutant HIF-1α-(531–652).
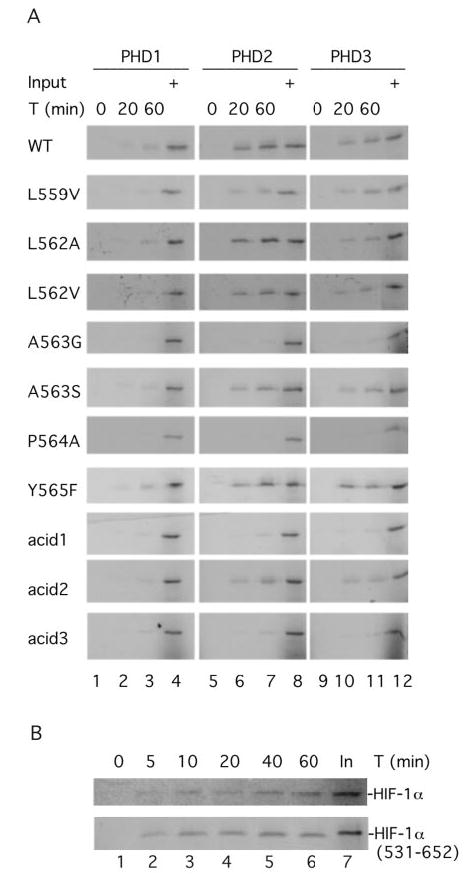
A, in vitro translated, 35S-labeled wild type or mutant GAL4-HIF-1α-(531–652) was incubated with 0.5 μg of His6-PHD1, His6-PHD2, or His6-PHD3 for the indicated times. Hydroxylated reaction products were then isolated by first incubating with His6-FLAG-VBC and then by immunoprecipitating with anti-FLAG antibodies coupled to agarose. The immunoprecipitated reaction products were subjected to SDS-PAGE and autoradiography. B, same as in A except that His6-PHD2 and either HA-HIF-1α or GAL4-HIF-1α-(531–652) were employed. Input (In) designates 20% of the substrate analyzed at a given time point. One of three representative results is shown.
Importantly, the data now also provide evidence that the L559V, L562A, L562V, and A563S mutants can also serve as substrates for the PHDs. For example, with PHD2, the L562A, L562V, and A563S mutants appear to be utilized only marginally less efficiently than the wild type substrate (lane 7, third, fourth, and sixth panels from top, respectively); the L559V mutant is utilized somewhat less effectively (lane 7, second panel from top). With PHD1 and PHD3, the trends are similar. Thus, with PHD3, the L559V, L562A, L562V, and A563S mutants can all serve as substrates (lane 11, second, third, fourth, and sixth panels from top, respectively). With PHD1, which has a lower activity toward HIF-1α than PHD2, the L562A, L562V, and A563S mutants demonstrate the ability to serve as substrates in this assay (lane 3, third, fourth, and sixth panels from top, respectively).
The unexpected capacity of the HIF-1α substrate to tolerate mutations at these positions prompted us to continue to search for residues that might be important for substrate recognition. In this regard, the sites of proline hydroxylation in HIF are typically seen in the context of acidic residues. Although these residues are generally not as strongly conserved in position as Leu at the −5 and −2 positions, and Ala at the −1 position, they are frequently seen in the vicinity of the hydroxylacceptor proline. We therefore first prepared a mutant, acid1, that abolishes all five acidic residues in the region encompassing residues 558–571 (Fig. 1_A_). As shown in Fig. 2_A_, this particular mutant is, at best, a weak substrate in this assay (third panel from bottom). We then prepared additional mutants targeting the acidic residues N-terminal (Asp-558, Glu-560) or C-terminal (Asp-569, Asp-570, Asp-571) to the hydroxylacceptor proline. As shown in Fig. 2_A_, the first of these (acid2) can now be demonstrated to serve as a substrate for PHD (second from bottom panel), albeit less efficiently than wild type, whereas the second (acid3) of these fails to demonstrate any significant evidence in this capacity (bottom panel). The latter result appears to be unlikely to be accounted for by this mutation simply interfering with VBC binding, because a hydroxylproline-containing HIF-1α-(558–568)-peptide lacking all three of the C-terminal acidic residues can still bind VBC (23).
We also compared the hydroxylation of GAL4-HIF-1α-(531–652) with that of full-length wild type HIF-1α. As shown in Fig. 2_B_, both are hydroxylated by PHD2 with comparable kinetics. The rate of in vitro hydroxylation with both, it should be noted, appears to be significantly lower than that which likely occurs in vivo (on the order of seconds to minutes) (34, 38). This raises the possibility that there may be additional factors within the cell that facilitate the hydroxylation reaction.
Enzyme-substrate interactions can sometimes be sufficiently stable to allow their detection in protein-protein interaction assays. Indeed, these interactions provide a coarse gauge of the Michaelis constant that is a major factor in determining the catalytic efficiency of an enzyme toward a particular substrate. We sought to determine whether these interactions could be employed to corroborate the assays employed above. Thus, we incubated the wild type or mutant, in vitro translated, 35S-labeled GAL4-HIF-1-(531–652) with recombinant FLAG-tagged PHD1, PHD2, or PHD3, immunoprecipitated the latter with anti-FLAG antibodies, and then examined the immunoprecipitates for bound HIF-1α. As shown in Fig. 3_A_, wild type GAL4-HIF-1α-(531–652) coimmunoprecipitates with all three PHDs (top panel). The strongest coimmunoprecipitation seen with PHD2 (lane 3) is consistent with this particular isoform displaying the highest activity toward HIF-1α-(531–652). These interactions with the PHDs are essentially abolished by the P564A mutation (seventh panel from top). In addition, and in a converse approach, 35S-labeled, in vitro translated PHD1, PHD2, or PHD3 was incubated with either GST or GST-HIF-1α-(531–575) immobilized on GSH-agarose, washed, and then eluted. As shown in Fig. 3_B_, all three PHD isoforms were found to specifically interact with GST-HIF-1α-(531–575) (lanes 3, 6, and 9, respectively).
Fig. 3. Interaction between PHDs and HIF-1α.
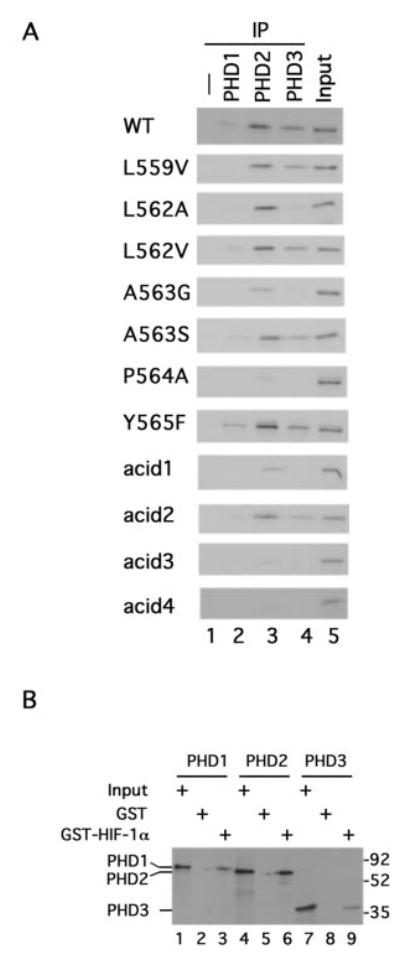
A, PHD pull-down analysis. In vitro translated, 35S-labeled wild type or mutant GAL4-HIF-1α-(531–652) prepared with wheat germ extracts were incubated with 1 μg of His6-FLAG-PHD1, His6-FLAG-PHD2, or His6-FLAG-PHD3. The PHDs were then immunoprecipitated with anti-FLAG antibodies coupled to agarose. Coimmunoprecipitating wild type or mutant GAL4-HIF-1α-(531–652) was then subjected to SDS-PAGE and autoradiography. B, in vitro translated, 35S-labeled FLAG-PHD1, FLAG-PHD2, or FLAG-PHD3 was incubated with GST or GST-HIF-1α-(531–575) prebound to GSH-agarose. After washing, bound PHD was subjected to SDS-PAGE and autoradiography. The positions of the PHDs are shown to the left, and those of molecular weight markers are to the right. In A and B, Input designates 10% of input.
We then examined the mutants described above for their capacity to coimmunoprecipitate with the PHDs. As shown in Fig. 3_A_, the capacity to serve as a substrate in the VBC pull-down assay is closely correlated with their binding affinity in the PHD coimmunoprecipitation assays. Thus, the L559V, L562A, L562V, A563S, Y565F, and acid2 mutants interact with the PHDs in a manner similar to or only marginally weaker than wild type (for example, compare lane 3 of the relevant panels), whereas the A563G mutant displays either negligible or weak (in the case of PHD2) interaction. Moreover, the various HIF substrates coimmunoprecipitate with PHD2 more effectively than with either PHD1 or PHD3, consistent with their behavior in the kinetic assays. The acid1 and acid3 mutants both displayed either negligible or weak (in the case of the acid 1 mutant and PHD2) interaction, consistent with their failure to demonstrate activity in the kinetic assays. The failure of the acid3 mutant to display binding to the PHDs is not simply due to the particular substitutions made (Asp → Asn); this failure is also seen with an additional mutant in which Asp → Ala substitutions are made at identical sites (acid4, Figs. 1_A_ and 3_A_, bottom panel).
The capacity of these HIF-1α peptides to form complexes with the PHDs prompted us to examine whether they might be able to serve as dominant negative inhibitors in the HIF degradation pathway. In particular, overexpression of these peptides might be predicted to inhibit endogenous PHD activity, resulting in hypohydroxylation of endogenous HIF and consequent activation of a HIF reporter gene. Indeed, it has previously been shown that overexpression of wild type HIF peptides can efficiently activate HIF reporter genes (34, 39). We therefore cotransfected COS-1 cells with a hypoxia response element (HRE) reporter gene containing three tandem HIF binding sites and constructs for wild type or mutant GAL4-HIF-1α-(531–562) and subsequently measured reporter gene (luciferase) activity.
As shown in Fig. 4_A_, the wild type HIF peptide induced robust activation of the HRE reporter gene, as expected, whereas the P564A mutant, which does not interact with the PHDs (Fig. 3_A_), failed to. The significant finding here is the strong correlation between PHD binding affinity (Fig. 3_A_) and the capacity to induce HRE reporter gene activity. Thus, the L562V and Y565F mutants induce strong activity, comparable to wild type. The L559V, L562A, A563S, and acid2 mutants also induce substantial activity (53–78% of wild type). Other mutants, including the A563G, acid1, acid3, and acid4 mutants induce either no or only marginally detectable activity. Hence, the affinity of a particular peptide for the PHDs is a strong predictor of its capacity to serve as a dominant negative inhibitor of the HIF degradation pathway in this reporter gene assay.
Fig. 4.
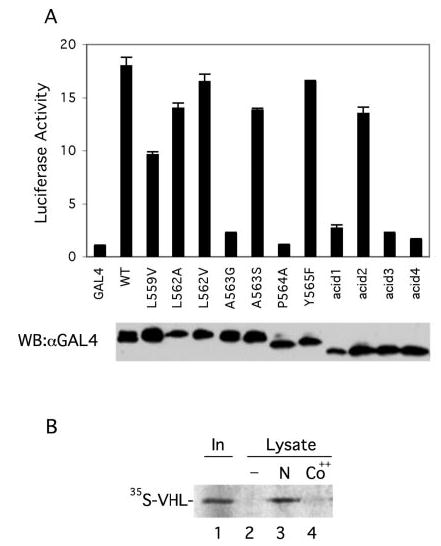
A, dominant negative behavior of wild type and mutant HIF-1α-(531–652). COS-1 cells were cotransfected with 0.5 μg of (eHRE)3-Luc, 0.05 μg of pRL-TK (Promega), and 0.5 μg of wild type or mutant pcDNA3-GAL4-HIF-1α-(531–652), or pcDNA3-GAL4. Cellular extracts were assayed for luciferase activity, normalizing to that of the Renilla luciferase internal transfection control. One of three representative results, performed in triplicate, is shown. Aliquots of the extracts were analyzed by Western blotting with anti-GAL4 antibodies (bottom). B, stimulus-sensitive endogenous PHD activity. COS-1 cells were mock (N) or cobalt (Co++)-treated for 2 h. Cellular lysates were prepared, and endogenous PHD from these lysates was then isolated by incubation with GST-HIF-1α-(531–575) immobilized on GSH-agarose. Hydroxylation reactions were performed on the resins, and the extent of HIF-1α-(531–575) hydroxylation was then assayed by subsequent incubation with 35S-labeled, in vitro translated VHL. Bound VHL was subjected to SDS-PAGE and autoradiography. In a control reaction, lysates were omitted. The position of VHL is shown, whereas “In ” designates 10% of the input VHL. One of two representative results is shown.
The discovery of proline hydroxylation as the key regulatory event governing HIF-1α activity, and the subsequent identification of the enzymes that catalyze this unusual modification not only provides an explanation for the mechanism by which hypoxia might regulate HIF-1α but also an explanation for the mechanism by which treatment of cells with cobalt might regulate HIF-1α, i.e. through the inhibition of PHD activity. We employed the substantial affinity of PHD for the HIF-1α peptide (Fig. 3_B_) to test this experimentally. Cells were first either mock- or cobalt-treated for 2 h. Whole cell lysates were prepared and incubated with GST-HIF-1α-(531–575) immobilized on agarose to capture endogenous PHD. The resins were then washed, incubated in the presence of iron, 2-oxoglutarate, and ascorbic acid to allow the proline hydroxylation assay to proceed, followed by incubation with in vitro translated, 35S-labeled VHL. Eluates from the resins were subjected to SDS-PAGE to determine the degree of VHL binding. As shown in Fig. 4_B_, extracts from mock-treated cells display HIF-1α proline hydroxylase activity that is readily detected by the subsequent binding of VHL (lane 3). In contrast, extracts from cobalt-treated cells are defective in their capacity to promote this hydroxylation (lane 4). This therefore provides evidence that the PHDs are indeed the target of cobalt in the HIF activation pathway. Conversely, it also provides evidence that the PHD activity is stimulus-sensitive. The data are consistent with the finding that cobalt inhibits the activity of recombinant Caenorhabditis elegans PHD or in vitro translated human PHD (30).
All three PHDs have the capacity to hydroxylate HIF in vitro. In vivo, this hydroxylation then targets HIF for degradation by the ubiquitin-proteasome pathway. To examine this experimentally, we transfected COS-1 cells with an expression vector for HA-tagged full-length HIF-1α with or without ones for FLAG-tagged versions of PHD1, PHD2, or PHD3. As shown in Fig. 5, overexpressed PHD1 is predominantly nuclear (Fig. 5, D and G), whereas overexpressed PHD2 and PHD3 appears predominantly cytoplasmic (J and M, and P and S, respectively). Further analysis will be required to determine if either PHD2 or PHD3 displays mitochondrial localization, as is the case for rat Sm-20, a homologue of human PHD3 (40, 41). Overexpressed HIF-1α is predominantly nuclear (B) and, upon expression with PHD2, HIF-1α levels are dramatically reduced (K). Coexpression with PHD1 or PHD3 also reduces HIF-1α levels (E and Q, respectively) but more modestly. The specificity of this reduction in HIF-1α levels is supported by the observation that none of the PHDs significantly affects the levels of coexpressed VHL (H, N, and T), which is predominantly cytoplasmic (C).
Fig. 5. Cellular localization of PHDs, HIF-1α, and VHL in transfected COS-1 cells.
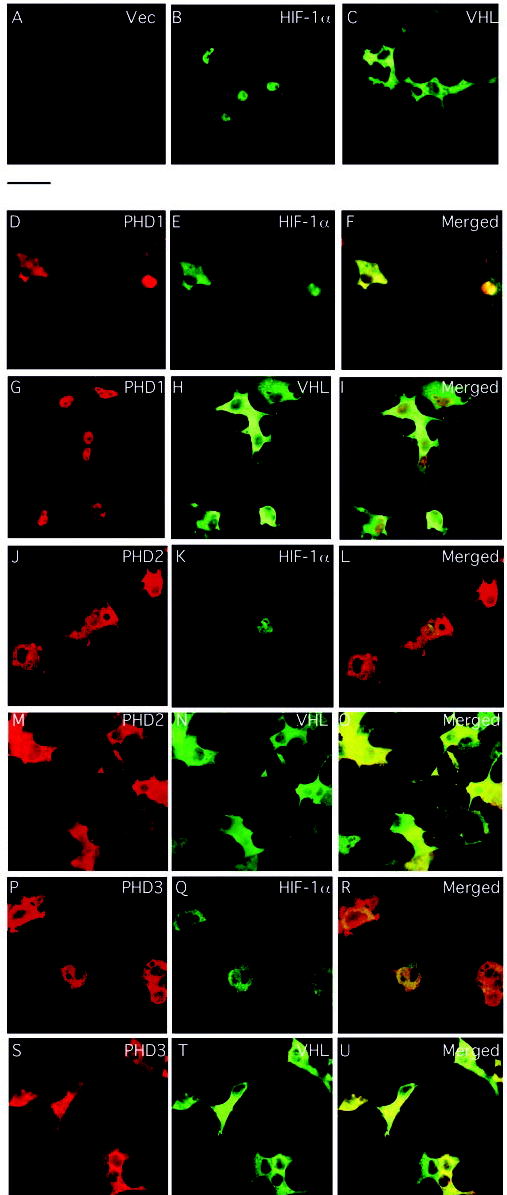
COS-1 cells were cotransfected with constructs encoding HA-HIF-1α (B, D–F, J–L, and P–R) or HA-VHL (C, G–I, M–O, and S–U) with or without ones for FLAG-tagged PHD1 (D–I), PHD2 (J–O), or PHD3 (P–U), or pcDNA3 alone (A). At 20-h post-transfection, the cells were fixed, permeabilized, and stained with anti-FLAG or anti-HA antibodies, or both. Confocal images of single color and an overlay of two colors (Merged) are shown. The bar between panels A and D indicates 50 μM.
To pursue this further, COS-1 cells were cotransfected with an HRE reporter gene, and expression vectors for full-length HIF-1α with or without ones for PHD1, PHD2, or PHD3. As shown in Fig. 6, overexpression of full-length HIF-1α activates the HRE reporter gene, as expected. In contrast, coexpression of HIF-1α with PHD1, PHD2, or PHD3 dramatically reduces activity from the reporter gene as well as levels of the coexpressed HIF-1α (lanes 3, 5, and 7, respectively). Mutation of residues that are predicted to chelate the active site iron (His-297 in PHD1 or His-135 in PHD3) (30, 31) or bind the 2-oxoglutarate cosubstrate (Arg-383 in PHD2) (42) substantially reverse PHD-induced inhibition of HRE reporter gene activity. Thus, overexpressed PHDs reduce the protein levels (Fig. 5) and, in turn, the transcriptional activity (Fig. 6) derived from coexpressed HIF-1α, and this effect depends on the catalytic activity of the PHDs.
Fig. 6. Inhibition of HIF-1α transcriptional activity by PHD.

COS-1 cells were cotransfected with 0.25 μg of (eHRE)3-Luc, 0.025 μg of pRL-TK (Promega), 0.5 μg of pcDNA3-HA-HIF-1α, and 0.1 μg of pcDNA3-FLAG-PHD1, pcDNA3-FLAG-PHD1 (H297A), pcDNA3-FLAG-PHD2, pcDNA3-FLAG-PHD2 (R383A), pcDNA3-FLAG-PHD3, or pcDNA3-FLAG-PHD3 (H135A). The total DNA dose was adjusted to 0.875 μg with pcDNA3. Cellular extracts were assayed for luciferase activity, normalizing to that of the Renilla luciferase internal transfection control. One of three representative results, performed in triplicate, is shown. Aliquots of extracts were also subjected to Western blotting with anti-HIF-1α or anti-FLAG antibodies. The positions of the PHDs and a nonspecific (NS) band are shown to the right, and those of molecular weight markers are to the left. WT and M indicate the wild type and mutant forms of PHD described above, respectively.
DISCUSSION
The recent identification of a family of enzymes, the PHDs, that catalyze the site-specific proline hydroxylation of HIF (30, 31), and the central importance of this pathway in the cellular response to hypoxia, makes it imperative to understand the molecular determinants that govern this enzyme-substrate interaction. In HIF, the sites of proline hydroxylation reside in a conserved L_XX_LAP sequence motif, but the unexpected result of these studies is that mutations can actually be tolerated at each of these positions except for the hydroxylacceptor proline. The present findings support and extend our previous studies examining the behavior of these mutants in VHL coimmunoprecipitation assays in vivo (12). In those studies, we found that mutations at both Leu-559 and Leu-562 appear to weaken but do not abolish the stimulus-sensitive coimmunoprecipitation with VHL, implying that the mutations neither abolish hydroxylation by PHD nor abolish subsequent binding by VHL. Those studies also indicated that D558A, E560D, and Y565F mutations could be tolerated, again consistent with the present results. Interestingly, a more substantial mutation of Tyr-565 (to alanine) abolishes hydroxylation by PHD (11, 31).
The current data provides evidence that recognition of HIF by these PHDs results from multiple interactions, none of which is absolutely critical, except for the obligatory hydroxylacceptor proline. Indeed, the results suggest that the interaction of PHD with HIF is probably analogous to the interaction of hydroxylated HIF with VHL (23, 24), in which the principal core interaction with the hydroxylproline is surrounded by multiple additional interactions; hence, mutations can be tolerated at positions surrounding this central interaction. Given that the PHDs likely possess the three-dimensional jelly roll topology seen with other members of the 2-oxoglutarate dioxygenase family (30, 42), it is possible that HIF-1α will bind to the PHDs in an extended conformation, just as it binds in an extended conformation to VHL. However, it is also possible that PHD recognition of HIF-1α may require distinct features, and thus, the detailed interactions between HIF-1α and VHL on the one hand and PHD on the other may not necessarily be identical. As one example of this, our studies raise the possibility that acidic residues C-terminal to the hydroxylacceptor proline may be important for recognition by PHD. In the case of VHL, although up to two of these residues (Asp-571 with or without Asp-569) make direct contact with VHL (23, 24), these contacts are not essential for the HIF-1α-VHL interaction (23).
Thus, the PHDs might recognize their substrates with a stringency that might be somewhat analogous to proline-directed kinases such as the MAPKs. With these kinases, the minimal recognition motif at the phosphoacceptor site is actually quite small, Ser/Thr-Pro (underlining indicates the phosphoacceptor residue), and substrate specificity is in large part governed by additional substrate residues (43, 44). In the case of the PHDs, the stringency in this respect is even lower, with proline being the minimal recognition motif. There are differences, however, as well. In the case of MAPKs, the additional residues that govern substrate specificity are typically distant in primary structure from the phosphoacceptor residue (43, 44). In the case of the PHDs and HIF-1α, these residues are in the vicinity of the hydroxylacceptor residue, because HIF-1α peptides encompassing the hydroxylacceptor residue that are as short as 20 residues are sufficient to serve as PHD substrates (10, 11). It also must be emphasized that, even though proline is the minimal recognition motif for PHDs, this is clearly not sufficient. As just one example, in HIF-1α there is a proline (Pro-567) that is just three residues away from the hydroxylacceptor proline targeted by the PHDs, but this proline is not hydroxylated by the PHDs (10, 11).
Our studies reveal an absence of a rigid consensus sequence for hydroxylation by PHDs, raising the possibility that there might be novel substrates for the PHDs. Such substrates, if they were to exist, could potentially broaden the use of this unusual modification in the cellular response to hypoxia. Indeed, in the amoeba Dictyostelium, the Skp1 protein is not only proline-hydroxylated, but this hydroxylproline is further glycosylated (45). Although the specific hydroxylacceptor proline is not conserved in vertebrates, it is intriguing to note that Skp1 is a component of the Skp1·Cul1·F-box protein (SCF) E3 ubiquitin ligase complex that is functionally analogous to the VHL-containing E3 complex that targets hydroxylated HIF for degradation (46).
Acknowledgments
We are grateful to Drs. Richard Klausner and Song Tan for gifts of plasmids. We thank Irina Chernysh for assistance with the confocal microscopy and Zheng Tu for a critical reading of the manuscript.
The abbreviations used are
HIF
hypoxia-inducible factor
BSA
bovine serum albumin
GSH
glutathione
GST
glutathione _S_-transferase
HPH
HIF prolyl hydroxylase
PBS
phosphate-buffered saline
PHD
prolyl hydroxylase domain-containing protein
Hyp
4-hydroxylproline
VBC
von Hippel-Lindau tumor suppressor protein·Elongin B·Elongin C complex
VHL
von Hippel-Lindau tumor suppressor protein
WT
wild type
E3
ubiquitin-protein isopeptide ligase
HA
hemagglutinin
HRE
hypoxia response element
MAPK
mitogen-activated protein kinase
References
- 1.Semenza GL. Curr Opin Cell Biol. 2001;13:167–171. doi: 10.1016/s0955-0674(00)00194-0. [DOI] [PubMed] [Google Scholar]
- 2.Maxwell PH, Pugh CW, Ratcliffe PJ. Adv Exp Med Biol. 2001;502:365–376. [PubMed] [Google Scholar]
- 3.Kaelin WG., Jr Genes Dev. 2002;16:1441–1445. doi: 10.1101/gad.1003602. [DOI] [PubMed] [Google Scholar]
- 4.Semenza GL. Annu Rev Cell Dev Biol. 1999;15:551–578. doi: 10.1146/annurev.cellbio.15.1.551. [DOI] [PubMed] [Google Scholar]
- 5.Wang GL, Jiang BH, Rue EA, Semenza GL. Proc Natl Acad Sci U S A. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ema M, Taya S, Yokotani N, Sogawa K, Matsuda Y, Fujii-Kuriyama Y. Proc Natl Acad Sci U S A. 1997;94:4273–4278. doi: 10.1073/pnas.94.9.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tian H, McKnight SL, Russell DW. Genes Dev. 1997;11:72–82. doi: 10.1101/gad.11.1.72. [DOI] [PubMed] [Google Scholar]
- 8.Hogenesch JB, Chan WK, Jackiw VH, Brown RC, Gu YZ, Pray-Grant M, Perdew GH, Bradfield CA. J Biol Chem. 1997;272:8581–8593. doi: 10.1074/jbc.272.13.8581. [DOI] [PubMed] [Google Scholar]
- 9.Gu YZ, Moran SM, Hogenesch JB, Wartman L, Bradfield CA. Gene Expr. 1998;7:205–213. [PMC free article] [PubMed] [Google Scholar]
- 10.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 11.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 12.Yu F, White SB, Zhao Q, Lee FS. Proc Natl Acad Sci U S A. 2001;98:9630–9635. doi: 10.1073/pnas.181341498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Masson N, Willam C, Maxwell PH, Pugh CW, Ratcliffe PJ. EMBO J. 2001;20:5197–5206. doi: 10.1093/emboj/20.18.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 15.Ivan M, Kaelin WG. Curr Opin Genet Dev. 2001;11:27–34. doi: 10.1016/s0959-437x(00)00152-0. [DOI] [PubMed] [Google Scholar]
- 16.Cockman ME, Masson N, Mole DR, Jaakkola P, Chang GW, Clifford SC, Maher ER, Pugh CW, Ratcliffe PJ, Maxwell PH. J Biol Chem. 2000;275:25733–25741. doi: 10.1074/jbc.M002740200. [DOI] [PubMed] [Google Scholar]
- 17.Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V, Kaelin WG. Nat Cell Biol. 2000;2:423–427. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]
- 18.Tanimoto K, Makino Y, Pereira T, Poellinger L. EMBO J. 2000;19:4298–4309. doi: 10.1093/emboj/19.16.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamura T, Sato S, Iwai K, Czyzyk-Krzeska M, Conaway RC, Conaway JW. Proc Natl Acad Sci U S A. 2000;97:10430–10435. doi: 10.1073/pnas.190332597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salceda S, Caro J. J Biol Chem. 1997;272:22642–22647. doi: 10.1074/jbc.272.36.22642. [DOI] [PubMed] [Google Scholar]
- 21.Huang LE, Gu J, Schau M, Bunn HF. Proc Natl Acad Sci U S A. 1998;95:7987–7992. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kallio PJ, Wilson WJ, O’Brien S, Makino Y, Poellinger L. J Biol Chem. 1999;274:6519–6525. doi: 10.1074/jbc.274.10.6519. [DOI] [PubMed] [Google Scholar]
- 23.Min JH, Yang H, Ivan M, Gertler F, Kaelin WG, Jr, Pavletich NP. Science. 2002;296:1886–1889. doi: 10.1126/science.1073440. [DOI] [PubMed] [Google Scholar]
- 24.Hon WC, Wilson MI, Harlos K, Claridge TD, Schofield CJ, Pugh CW, Maxwell PH, Ratcliffe PJ, Stuart DI, Jones EY. Nature. 2002;417:975–978. doi: 10.1038/nature00767. [DOI] [PubMed] [Google Scholar]
- 25.Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Science. 2002;295:858–861. doi: 10.1126/science.1068592. [DOI] [PubMed] [Google Scholar]
- 26.Mahon PC, Hirota K, Semenza GL. Genes Dev. 2001;15:2675–2686. doi: 10.1101/gad.924501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sang N, Fang J, Srinivas V, Leshchinsky I, Caro J. Mol Cell Biol. 2002;22:2984–2992. doi: 10.1128/MCB.22.9.2984-2992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. Genes Dev. 2002;16:1466–1471. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hewitson KS, McNeill LA, Riordan MV, Tian YM, Bullock AN, Welford RW, Elkins JM, Oldham NJ, Bhattacharya S, Gleadle JM, Ratcliffe PJ, Pugh CW, Schofield CJ. J Biol Chem. 2002;277:26351–26355. doi: 10.1074/jbc.C200273200. [DOI] [PubMed] [Google Scholar]
- 30.Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 31.Bruick RK, McKnight SL. Science. 2001;294:1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 32.Schofield CJ, Zhang Z. Curr Opin Struct Biol. 1999;9:722–731. doi: 10.1016/s0959-440x(99)00036-6. [DOI] [PubMed] [Google Scholar]
- 33.Kivirikko KI, Pihlajaniemi T. Adv Enzymol Relat Areas Mol Biol. 1998;72:325–398. doi: 10.1002/9780470123188.ch9. [DOI] [PubMed] [Google Scholar]
- 34.Yu F, White SB, Zhao Q, Lee FS. Cancer Res. 2001;61:4136–4142. [PubMed] [Google Scholar]
- 35.Tan S. Protein Expr Purif. 2001;21:224–234. doi: 10.1006/prep.2000.1363. [DOI] [PubMed] [Google Scholar]
- 36.Smith DB, Johnson KS. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- 37.Marchetti S, Gimond C, Iljin K, Bourcier C, Alitalo K, Pouyssegur J, Pages G. J Cell Sci. 2002;115:2075–2085. doi: 10.1242/jcs.115.10.2075. [DOI] [PubMed] [Google Scholar]
- 38.Jewell UR, Kvietikova I, Scheid A, Bauer C, Wenger RH, Gassmann M. FASEB J. 2001;15:1312–1314. [PubMed] [Google Scholar]
- 39.Srinivas V, Zhang LP, Zhu XH, Caro J. Biochem Biophys Res Commun. 1999;260:557–561. doi: 10.1006/bbrc.1999.0878. [DOI] [PubMed] [Google Scholar]
- 40.Lipscomb EA, Sarmiere PD, Freeman RS. J Biol Chem. 2001;276:5085–5092. doi: 10.1074/jbc.M008407200. [DOI] [PubMed] [Google Scholar]
- 41.Taylor MS. Gene. 2001;275:125–132. doi: 10.1016/s0378-1119(01)00633-3. [DOI] [PubMed] [Google Scholar]
- 42.McNeill LA, Hewitson KS, Gleadle JM, Horsfall LE, Oldham NJ, Maxwell PH, Pugh CW, Ratcliffe PJ, Schofield CJ. Bioorg Med Chem Lett. 2002;12:1547–1550. doi: 10.1016/s0960-894x(02)00219-6. [DOI] [PubMed] [Google Scholar]
- 43.Sharrocks AD, Yang SH, Galanis A. Trends Biochem Sci. 2000;25:448–453. doi: 10.1016/s0968-0004(00)01627-3. [DOI] [PubMed] [Google Scholar]
- 44.Enslen H, Davis RJ. Biol Cell. 2001;93:5–14. doi: 10.1016/s0248-4900(01)01156-x. [DOI] [PubMed] [Google Scholar]
- 45.West CM, van Der Wel H, Gaucher EA. Glycobiology. 2002;12:17R–27R. doi: 10.1093/glycob/12.2.17r. [DOI] [PubMed] [Google Scholar]
- 46.Tyers M, Rottapel R. Proc Natl Acad Sci U S A. 1999;96:12230–12232. doi: 10.1073/pnas.96.22.12230. [DOI] [PMC free article] [PubMed] [Google Scholar]