Xeroderma pigmentosum group C protein interacts physically and functionally with thymine DNA glycosylase (original) (raw)
Abstract
The XPC–HR23B complex recognizes various helix-distorting lesions in DNA and initiates global genome nucleotide excision repair. Here we describe a novel functional interaction between XPC–HR23B and thymine DNA glycosylase (TDG), which initiates base excision repair (BER) of G/T mismatches generated by spontaneous deamination of 5-methylcytosine. XPC–HR23B stimulated TDG activity by promoting the release of TDG from abasic sites that result from the excision of mismatched T bases. In the presence of AP endonuclease (APE), XPC–HR23B had an additive effect on the enzymatic turnover of TDG without significantly inhibiting the subsequent action of APE. Our observations suggest that XPC–HR23B may participate in BER of G/T mismatches, thereby contributing to the suppression of spontaneous mutations that may be one of the contributory factors for the promotion of carcinogenesis in xeroderma pigmentosum genetic complementation group C patients.
Keywords: base excision repair/nucleotide excision repair/thymine DNA glycosylase/xeroderma pigmentosum/XPC–HR23B complex
Introduction
Organisms have multiple DNA repair pathways that remove various DNA lesions, thereby maintaining the integrity of the genome and cellular functions (Friedberg, 1995; Hoeijmakers, 2001). One such DNA repair pathway, base excision repair (BER), corrects a number of spontaneously and environmentally induced genotoxic or miscoding base lesions (McCullough et al., 1999; Krokan et al., 2000; Nilsen and Krokan, 2001). BER is initiated by a DNA glycosylase that releases the altered or inappropriate base from the phosphate–sugar backbone of DNA by hydrolyzing the _N_-glycosylic bond. In vertebrates, the resulting apurinic/apyrimidinic site (AP site) is then incised by AP endonuclease (APE; also called HAP1, APEX and Ref-1), leaving 5′-deoxyribose-5-phosphate and 3′-hydroxyl termini (Wilson and Barsky, 2001). Furthermore, a subset of the DNA glycosylase protein family possesses intrinsic AP lyase activity and thus can also make the 3′ incision to the AP site that it initially generated (McCullough et al., 1999). APE is probably involved further in processing the intermediate left behind as a result of this latter AP incision, given that it has additional enzymatic properties, including DNA 3′-phosphodiesterase and 3′-phosphatase activities. These latter activities can convert the 3′ end left behind by the AP lyase into a hydroxyl group, which can be then utilized as a primer for DNA repair synthesis catalyzed by DNA polymerases (Wilson and Barsky, 2001). Such DNA repair synthesis and strand rejoining involve at least two different mechanisms, namely short-patch repair involving DNA polymerase β or proliferating cell nuclear antigen (PCNA)-dependent long-patch repair (Nilsen and Krokan, 2001).
Hydrolytic deamination is one of the most frequently occurring spontaneous alterations of DNA bases. In vertebrate genomes, cytosines of CpG dinucleotide sites are often methylated at the C5 position, and deamination of the resulting 5-methylcytosines results in thymines being mispaired with guanines (Friedberg, 1995). Such a G/T mismatch, if not corrected, can lead to the alteration of the original G:C base pair into A:T after one cycle of DNA replication. CpG sites are thus highly susceptible to mutation. These mutations can be suppressed by correction of the G/T mismatches by the BER pathway initiated by thymine DNA glycosylase (TDG; for a review, see Hardeland et al., 2001). Mammalian TDG preferentially excises not only thymine in a G/T mismatch but also uracil opposite guanine: such G/U mismatch can result either from deamination of unmethylated cytosine, the frequency of which is four times less than that of 5-methylcytosine, or from misincorporation of dUMP during DNA replication. The crystal structures of the Escherichia coli mismatch-specific uracil DNA-glycosylase (MUG) pro tein, which belongs to the same DNA glycosylase subfamily as TDG, has been resolved in both the presence and absence of substrate DNAs (Barrett et al., 1998, 1999). The E.coli MUG is unique in that it binds to the widowed guanine residue while the mispaired uracil is flipped out of the DNA double helix. This feature is reminiscent of human TDG, as biochemical observations have indicated that it remains tightly bound to the AP site after excision of the mismatched T (or U) (Waters and Swann, 1998; Waters et al., 1999). There is increasing evidence that TDG may also play roles other than to initiate BER, as it is known to interact with several transcription factors. Thus, TDG may also function in transcriptional regulation (Um et al., 1998; Missero et al., 2001; Tini et al., 2002).
Nucleotide excision repair (NER) is another important DNA repair pathway. It can eliminate various structurally unrelated lesions that distort the double helix, including UV light-induced cyclobutane pyrimidine dimers (CPDs) and (6–4) photoproducts, as well as intrastrand cross-links and bulky adducts induced by numerous chemical compounds (de Laat et al., 1999). Impaired NER activity has been associated with several human genetic disorders, including xeroderma pigmentosum (XP). Patients suffering from XP are characterized clinically by cutaneous hypersensitivity to UV exposure and increased susceptibility to skin cancer. Thus far, seven NER-deficient genetic complementation XP groups (XP-A to G) have been identified, and for each of these the responsible gene has been cloned. Unlike most of the other XP groups, XP-C patients show defects only in one of the two NER subpathways, i.e. global genome NER that operates over the entire genome. The other subpathway, transcription-coupled NER, which specifically removes lesions on the transcribed strands of active genes, functions normally in XP-C patients. The gene responsible for the defect in XP-C patients encodes the XPC protein, which exists in vivo as a heterotrimeric complex with centrin 2 and HR23B, one of the two mammalian homologs of Saccharomyces cerevisiae Rad23p (Masutani et al., 1994; Shivji et al., 1994; Araki et al., 2001). The role of centrin 2 in the complex remains to be elucidated but it is known that the XPC–HR23B association is needed to reconstitute the cell-free NER reaction (Mu et al., 1995; Sugasawa et al., 1997; Araújo et al., 2000; Batty et al., 2000). Biochemical studies have revealed that XPC–HR23B specifically binds certain types of DNA lesions and initiates NER (Sugasawa et al., 1998; Batty et al., 2000; Kusumoto et al., 2001). Thus, XPC–HR23B appears to function as a damage recognition factor for global genome NER. Generally, XPC–HR23B functions by recognizing structural abnormalities introduced into double-stranded DNA by the lesions rather than recognizing any structural characteristics of the lesions themselves (Sugasawa et al., 2001, 2002). Furthermore, we have shown that specific DNA structures containing a junction of double- and single-stranded DNA appears to be recognized preferentially (Sugasawa et al., 2002). After XPC–HR23B binds DNA containing a helix distortion, TFIIH is recruited via protein–protein interactions, and unwinds the DNA helix by its DNA helicase activity probably together with XPG, XPA and RPA. Following the open complex formation, two structure-specific endonucleases, XPF-ERCC1 and XPG, incise the 5′ and 3′ sides of the lesion, respectively, releasing the oligonucleotide containing the damaged base(s). Finally, the gapped DNA region is resynthesized by PCNA-dependent DNA polymerase (δ or ε), and the resulting nick is rejoined by DNA ligase I (for a review, see de Laat et al., 1999).
Although significant advances have been made in understanding the biochemical properties of XPC– HR23B, little is known about the intracellular behavior of this complex. For instance, it is possible that XPC–HR23B may interact with unknown protein factors that assist its damage recognition or confer additional functions. In this study, we describe a novel functional interaction between XPC and TDG. This observation may help in understanding the mechanisms that promote mutagenesis and carcinogenesis in XP-C patients.
Results
TDG interacts directly with XPC
To search for factors that interact with XPC, yeast two-hybrid screening of a mouse embryonic fibroblast cDNA library was performed using full-length human XPC as bait. One of the positive clones isolated from 7 × 106 transformants turned out to encode mouse TDG. We confirmed that human TDG could also interact with human XPC by performing the yeast two-hybrid assay with a human TDG cDNA.
To confirm that XPC and TDG interact physically, recombinant human TDG protein fused to the GST tag (GST–TDG) was expressed in E.coli and purified for use in ‘pull-down’ assays with the XPC–HR23B complex. After adding glutathione–Sepharose beads to assays containing XPC–HR23B and either GST or GST–TDG, the beads were centrifuged and washed extensively. Glutathione-bound proteins were then eluted with a buffer containing 10 mM glutathione and subjected to SDS–PAGE followed by immunoblotting with anti-XPC antibodies (Figure 1). Although only little binding to control GST was detected (lane 3), a significant amount of XPC–HR23B was co-precipitated with GST–TDG (lane 4). The addition of excess amounts of N-terminal His6-tagged TDG (His-TDG) abolished the binding in a dose-dependent manner (lanes 5 and 6). Together with the similar results obtained by using recombinant XPC protein instead of XPC–HR23B under our conditions (data not shown), the evidence for a direct interaction between XPC and TDG was verified. It should be noted that this direct protein–protein interaction was relatively weak: only ∼0.6% of input XPC–HR23B was pulled-down with GST–TDG (compare the band intensity in lane 4 with that in lane 1). Given that ∼30% of input GST–TDG was bound reproducibly to the glutathione–Sepharose beads under our experimental conditions (data not shown), the apparent association constant calculated was ∼1 × 106/M.
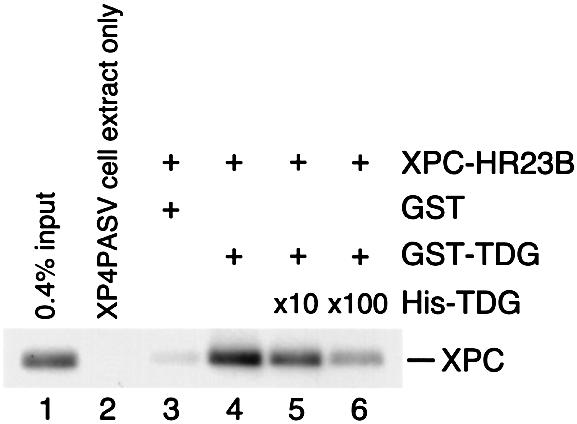
Fig. 1. Physical interaction of XPC–HR23B and TDG. Glutathione–Sepharose beads (20 µl) were incubated in 100 µl of the binding mixture with 16.6 nM of either GST (lane 3) or GST–TDG (lanes 4–6) in the presence of XPC–HR23B (6.7 nM). For lanes 5 and 6, 166 nM and 1.66 µM His-TDG, respectively, were also included as a competitor. After extensive washing, bound proteins were eluted with buffer containing 10 mM glutathione. A quarter of each eluate was mixed with whole-cell extract from XP4PASV cells, which do not express XPC, and subjected to 8% SDS–PAGE followed by immunoblotting with anti-XPC antibody. Lane 1, 0.4% of the input XPC; lane 2, XP4PASV cell extract only.
XPC–HR23B and TDG form a complex on DNA
Since TDG is involved in the initiation of BER that corrects G/T mismatches, it is of interest to know whether XPC–HR23B is involved in TDG-dependent BER as well as in NER. To examine this possibility, gel mobility shift analysis was carried out using a 32P-labeled double-stranded DNA fragment containing a site-specific G/T mismatch (Figure 2A). Although XPC–HR23B does recognize various structural abnormalities of double-stranded DNA (Sugasawa et al., 2001, 2002), very little specific XPC binding to the G/T-mismatched substrate could be observed under the conditions employed (Figure 3A, lanes 1–8). In marked contrast, TDG exhibited a band shift when incubated with the G/T-mismatched DNA but not with the control DNA containing a normal G/C pair at the corresponding position (Figure 3A, lanes 9–16; see also Neddermann and Jiricny, 1993). It should be noted that the binding reactions were carried out under conditions where TDG could exhibit its glycosylase activity. Therefore, at least a part of the observed TDG–DNA complex probably contained an AP site opposite guanine instead of a G/T mismatch.

Fig. 2. DNA substrates used in gel mobility shift analysis (A) and the nicking assays (B). X is A, C, G or T.

Fig. 3. XPC–HR23B is recruited to TDG-bound DNA and forms a ternary complex. (A) The indicated concentrations of XPC–HR23B or TDG were incubated at 30°C for 30 min with 0.35 nM of the 32P-labeled DNA fragment containing a single G/T mismatch or completely paired double-stranded DNA (G/C) as a control. The resulting DNA–protein complex was fixed with glutaraldehyde, and resolved by native PAGE. (B) The DNA substrate containing a single G/T mismatch (0.35 nM) was incubated at 30°C for 15 min in the presence or absence of His-TDG (16 nM). The indicated concentrations of XPC–HR23B were then added, incubated further at 30°C for 15 min and subjected to native PAGE after glutaraldehyde fixation. The asterisk shows the bands that newly appeared in the presence of both TDG and XPC–HR23B. (C) A supershift assay identifying the ternary complex containing TDG, XPC–HR23B and DNA. Sequential binding reactions were conducted as in (B) with 16 nM TDG, 6.7 nM XPC–HR23B and 0.35 nM DNA substrate containing a single G/T mismatch. Various amounts of anti-TDG or anti-XPC antibodies were then added and incubated further on ice for 15 min before cross-linking the DNA–protein complexes with glutaraldehyde.
Although the XPC–HR23B complex itself cannot bind to G/T-mismatched DNA, XPC–HR23B does bind to TDG-bound DNA. When XPC–HR23B was added after TDG was pre-incubated with the G/T-mismatched DNA, the amount of the TDG-bound DNA decreased in a dose-dependent manner and a new shifted band whose mobility was slower than the TDG-bound DNA appeared (shown by an asterisk in Figure 3B). To determine the protein components in this new band, a supershift assay was conducted with polyclonal antibodies raised against XPC and TDG (Figure 3C). As expected, the anti-XPC antibody did not supershift the TDG–DNA complex and the anti-TDG antibody did not supershift the XPC–HR23B–DNA complex (data not shown). However, addition of the anti-TDG antibody supershifted both the TDG–DNA complex and the new band (lanes 1–4). Moreover, the new band was also supershifted by the anti-XPC antibody (lanes 5–8). Although the direct interaction between TDG and XPC–HR23B was not so stable, the TDG–DNA complex appeared to be converted to the ternary complex quite efficiently (Figure 3B): the apparent association constant of XPC–HR23B and the TDG–DNA complex was ∼1 × 108/M, which is ∼100-fold higher than that obtained in the absence of DNA (Figure 1: it should be noted that we used the same protein concentrations and buffer conditions for both binding reactions). Therefore, DNA may stabilize the association between TDG and XPC–HR23B or, alternatively, TDG may induce certain conformational change in DNA, which may be then recognized by XPC–HR23B.
XPC stimulates the activity of TDG
To test whether XPC–HR23B affects the activity of TDG, a ‘nicking assay’ was performed using a 60mer DNA fragment containing a single G/T mismatch, which was 32P-labeled at the 5′ end of the T-strand (Figure 2B). In this assay, the number of AP sites generated by TDG can be measured by treating the TDG-processed DNA with hot alkali and then separating the cleaved and non-cleaved T-strands by denaturing PAGE.
Purified XPC–HR23B by itself showed no detectable DNA glycosylase activity on the G/T mismatch in this assay (data not shown). However, when a limited amount of TDG was incubated with the substrate DNA, a significant number of the mismatched T residues were excised within the initial 15 min. After this initial burst of activity, the reaction slowed markedly (Figure 4A and B). This is probably due to the fact that once TDG is bound to a G/T mismatch and excises the T residue, it remains attached to the resulting AP site (Waters and Swann, 1998; Waters et al., 1999) and, consequently, after one cycle of TDG enzymatic action, most of the TDG molecules are sequestered by AP sites. However, when XPC–HR23B was added to the DNA simultaneously with TDG, the activity of TDG increased in a manner dependent on the dose of XPC–HR23B. Incubation with 4.4 nM XPC–HR23B resulted in a 3-fold stimulation of TDG activity after 120 min of incubation (Figure 4A and B). When >4.4 nM XPC–HR23B was added, the stimulatory effect was somewhat diminished (data not shown), probably because of non-specific binding of XPC–HR23B to the substrate DNA. To examine which subunit, XPC or HR23B, is responsible for the stimulation, the subunits were added individually to the reaction mix. As shown in Figure 4C, XPC alone (lanes 3–6) could achieve the same level of stimulation as that induced by XPC–HR23B (lane 2), while HR23B alone (lanes 7–10) had virtually no effect on TDG activity under similar conditions.

Fig. 4. XPC–HR23B stimulates the activity of TDG. (A) A nicking assay measuring the TDG activity in the presence of XPC–HR23B. The 60mer DNA substrate containing a single G/T mismatch, which was labeled at the 5′ end of the T-strand, was incubated at 30°C for the indicated time with 0.42 nM His-TDG in the presence or absence of various concentrations of XPC–HR23B. The DNA samples were purified and subjected to alkali treatment to cleave the resulting AP sites, after which denaturing PAGE was performed. (B) The percentage of the cleaved oligonucleotides in the 32P-labeled DNA substrate was calculated for each lane in (A) and plotted as a graph. The mean values and standard errors were calculated from at least two independent experiments. (C) A nicking assay using 0.42 nM His-TDG in the presence of various concentrations of XPC–HR23B (lane 2), XPC alone (lanes 3–6) or HR23B alone (lanes 7–10) as indicated. All reactions were incubated at 30°C for 120 min and the purified DNA samples were subjected to alkali treatment and denaturing PAGE.
To gain an insight into the mechanism by which XPC stimulates TDG activity, XPC–HR23B was added to the G/T-mismatched DNA at various time points. Pre-incubation of the G/T-mismatched DNA with XPC–HR23B before TDG was added did not alter the time course of the TDG reaction relative to when all the components were mixed simultaneously (data not shown). This is not surprising because XPC–HR23B is unable to bind the G/T mismatch (Figure 3B). Pre-incubation of TDG and XPC–HR23B prior to their addition to the substrate DNA also failed to enhance the initial reaction rate (data not shown), suggesting that pre-assembly of the TDG–XPC complex does not facilitate recognition of G/T mismatches. However, if XPC–HR23B was added after TDG had been allowed to react with the G/T-mismatched DNA, TDG activity was boosted dramatically (Figure 5). Given that once TDG excises the mismatched T residue it remains sequestered by the resulting AP site, our observations strongly suggest that XPC–HR23B promotes the dissociation of TDG from the AP site, thereby freeing TDG to react with remaining G/T mismatches.

Fig. 5. XPC–HR23B promotes the turnover of TDG. (A) The 32P-labeled DNA substrate containing a single G/T mismatch was incubated with 0.42 nM His-TDG at 30°C for various periods as indicated (lanes 1–5). After 120 min, the indicated concentrations of XPC–HR23B were added and incubated further at 30°C for 15, 30 or 60 min (total incubation time is shown above each lane). The purified DNA samples were subjected to alkali treatment and denaturing PAGE. (B) The percentage of the cleaved oligonucleotides in the 32P-labeled DNA substrate was calculated for each lane in (A). The mean values and standard errors were calculated from at least two independent experiments.
XPC–HR23B stimulates the activity of TDG in the presence of APE
It has been reported that APE, which acts in the BER pathway immediately after DNA glycosylase, also promotes the turnover of TDG by displacing it from AP sites (Waters et al., 1999). Therefore, we decided to test whether APE and XPC–HR23B interact functionally in promoting TDG turnover. APE (2.1 nM) was added with or without various concentrations of XPC–HR23B to the nicking assay reactions containing TDG. As described previously by Waters et al. (1999), TDG activity was significantly enhanced by the presence of APE, as 2-fold more AP sites were generated in the presence of APE after 120 min incubation under the conditions used (compare Figure 4A, lanes 2–5, with Figure 6A, lanes 2–5). If XPC–HR23B was then added to the reactions, it enhanced TDG activity still further in a manner that was dependent on the XPC–HR23B dose (Figure 6A and B). Thus, co-addition of 2.1 nM APE and 4.4 nM XPC–HR23B resulted in a 6-fold increase in the amount of AP sites generated after 120 min, indicating that the two factors have additive effects on TDG activity. However, it cannot be excluded that XPC–HR23B addition may influence the enzymatic action of APE, for instance, by binding to AP sites generated after the processing by TDG. In fact, the gel mobility shift assay using DNA substrates containing a 3-hydroxy-2-(hydroxymethyl)tetrahydrofuran (a synthetic AP site analog; Fujiwara et al., 1999) at a specific site revealed that XPC–HR23B can bind to AP sites. As shown in Figure 7A, binding affinity differs with the bases opposite the AP site in the order AP/C>AP/T>AP/G>AP/A. Although the binding was relatively weak when guanine was opposite the AP site, these findings suggest that XPC–HR23B may push out TDG from the AP site and promote its turnover. To examine whether the presence of XPC–HR23B interferes with the endonuclease activity of APE, the nicking assay was performed as done in Figure 6, except that the amounts of the labeled oligonucleotide cleaved by APE were measured by omitting the alkali treatment prior to denaturing PAGE. APE-mediated cleavage was actually increased by the addition of XPC–HR23B, although only some of the AP sites generated by TDG (<50%) were processed by APE (compare Figures 6B and 7C). Furthermore, when APE was omitted from the reactions, cleaved oligonucleotides could hardly be detected (data not shown), indicating that most of the detected cleavage was due to the activity of APE rather than spontaneous β-elimination. It is noteworthy that the low processivity of APE observed did not depend on the presence or absence of XPC–HR23B, which suggested that XPC–HR23B does not enhance or inhibit APE activity by binding to the AP site under our conditions. Therefore, these data indicated that XPC–HR23B stimulates the TDG-dependent BER reaction by promoting the activity of TDG without affecting that of APE.

Fig. 6. XPC–HR23B stimulates the activity of TDG in the presence of APE. (A) The 32P-labeled DNA substrate containing a single G/T mismatch was incubated with 0.42 nM His-TDG, 2.1 nM His-APE and the indicated concentrations of XPC–HR23B at 30°C for the specified time. The purified DNA samples were subjected to alkali treatment and denaturing PAGE. Due to the 3′→5′ exonuclease activity of APE, the bands of cleaved oligonucleotides appear as ladders. (B) The percentage of the cleaved oligonucleotides in the 32P-labeled DNA substrate was calculated for each lane in (A). The mean values and standard errors were calculated from at least two independent experiments.

Fig. 7. XPC–HR23B does not inhibit the enzymatic action of APE. (A) The 32P-labeled DNA substrate containing a synthetic AP site analog opposite four different bases or a normal G/C pair at the corresponding position (0.35 nM each) were incubated at 30°C for 30 min with the indicated concentrations of XPC–HR23B. The resulting DNA–protein complex was fixed with glutaraldehyde, and resolved by native PAGE. (B) The 32P-labeled DNA substrate containing a single G/T mismatch was incubated with 0.42 nM His-TDG, 2.1 nM His-APE and the indicated concentrations of XPC–HR23B at 30°C for the specified periods. The reactions were stopped by addition of EDTA and subjected to denaturing PAGE without alkali treatment. (C) The percentage of the cleaved oligonucleotides in the 32P-labeled DNA substrate was calculated for each lane in (B). The mean values and standard errors were calculated from at least two independent experiments.
Discussion
NER factors such as XPC–HR23B are involved in other DNA repair pathways
The XPC–HR23B protein complex plays a key role in the initiation of NER by recognizing various types of DNA lesions (Sugasawa et al., 1998, 2001, 2002; Batty et al., 2000; Kusumoto et al., 2001). On the other hand, only limited information is available for factors that interact with XPC, except for TFIIH, another essential NER protein complex (Li et al., 1998; Yokoi et al., 2000; Araújo et al., 2001). In this report, we show a novel interaction between XPC and TDG, a molecule that initiates BER. This interaction was also observed in the presence of the G/T-mismatched DNA substrates: XPC–HR23B was recruited to the TDG-bound DNA and formed a ternary complex. Furthermore, XPC–HR23B stimulated the activity of TDG by promoting its enzymatic turnover. APE exhibited an additive effect on the TDG turnover, whereas XPC–HR23B did not influence the activity of APE initiated by TDG. Our data suggest the possibility that XPC may play an additional role outside NER.
Evidence has accumulated suggesting that some components of the NER pathway participate in the BER pathway. For example, XPG protein, a structure-specific NER endonuclease that makes an incision on the 3′ side of a lesion, stimulates in vitro the activity of human Nth1 (hNth1) protein, a bifunctional DNA glycosylase–AP lyase that initiates BER of oxidized pyrimidines, such as thymine glycol (Bessho, 1999; Klungland et al., 1999). In this reaction, it was shown that the stimulatory activity of XPG did not reside in its ability to incise the damaged DNA, but in its ability to promote hNth1 binding to the oxidative DNA damage site. Moreover, together with TFIIH and the Cockayne syndrome group B (CSB) protein, XPG appears to be involved in transcription-coupled BER of oxidative DNA lesions such as thymine glycol and 8-oxoguanine (Cooper et al., 1997; Klungland et al., 1999; Le Page et al., 2000). Another example of NER proteins functioning in BER are the two human Rad23p homologs, HR23A and HR23B, which have been shown to interact with and stimulate the activity of the BER protein 3-methyladenine-DNA glycosylase (MPG protein; Miao et al., 2000). However, XPC seems to act in a different manner in BER compared with XPG and the HR23 proteins. XPG and the HR23 proteins appear specifically to aid a certain type of DNA glycosylase to recognize the DNA damage. In contrast, our data reveal that XPC–HR23B stimulates TDG activity by promoting its enzymatic turnover, similar to the enhancement of TDG activity by APE (Waters et al., 1999).
Functional protein–protein interactions involving TDG
TDG has been shown to interact with several transcription factors, such as the retinoid receptors (RAR/RXR), the thyroid transcription factor-1 (TTF-1) and the CREB-binding protein (CBP/p300), suggesting that TDG is somehow involved in transcriptional regulation (Um et al., 1998; Missero et al., 2001; Tini et al., 2002). In marked contrast to these transcription factors, XPC–HR23B significantly stimulates the activity of TDG. Several structural studies, as well as biochemical analyses, have suggested that TDG tightly binds the AP site generated by its own enzymatic action, and thus a specific factor may be required to facilitate its release from one substrate so that it can bind to another site (Barrett et al., 1998, 1999; Waters and Swann, 1998; Waters et al., 1999). One obvious candidate for such a releasing factor is APE, which functions in BER after TDG (Waters et al., 1999). APE was shown to stimulate the TDG activity in vitro and, in a gel mobility shift analysis, addition of APE efficiently converted the TDG–DNA complex to the APE–DNA complex. However, an intermediate ternary complex involving TDG, APE and DNA could not be detected, and thus no interaction between TDG and APE on the DNA substrate has been demonstrated. This raised the argument that the stimulation may be due to competition of TDG and APE at the AP site rather than an active displacement of TDG by APE (Hardeland et al., 2001). We also failed to detect a physical interaction between TDG and APE under the same binding conditions as used in Figure 1 (data not shown), indicating that the interaction between APE and TDG is much weaker, if present at all, than that between XPC and TDG. Because we show that XPC–HR23B can bind specifically to AP sites (Figure 7A), it is not excluded that the stimulation of TDG turnover by the complex may also be due to competition at AP sites. Unlike the case of APE, however, XPC–HR23B can form a detectable amount of the ternary complex with TDG and DNA. We have shown recently that XPC–HR23B recognizes structural abnormalities of double-stranded DNA, but not any structural characteristics of lesions themselves (Sugasawa et al., 2001). The presence of DNA bases that do not form normal Watson–Crick pairs is a crucial factor for specific binding of XPC–HR23B, and specific DNA secondary structure containing a junction of double- and single-stranded regions appears to be recognized preferably (Sugasawa et al., 2002). Based on such binding properties, one can reasonably assume that, when bound to the AP site, XPC–HR23B interacts with the widowed base rather than the AP site itself. This is consistent with the observed different affinity toward AP sites with four kinds of opposite bases (Figure 7A), although the physiological relevance of this base preference remains to be elucidated. Crystal structures of E.coli MUG complexed with DNA substrates strongly suggest that the stable binding of TDG to the AP site may be due, at least partly, to its interaction with the widowed guanine residue. Therefore, the competitive interaction of XPC–HR23B with the guanine may attenuate the binding of TDG to the AP site (Figure 8). Since the observed protein–protein interaction was somewhat weak, it remains unclear whether the direct interaction could be important for recruitment of XPC–HR23B by TDG bound to the AP site. It is also possible that structural changes in DNA induced by TDG may be more crucial for recruiting XPC–HR23B, and the protein–protein interaction may play some role in displacement of TDG from the AP site. Determination of the binding domain in each protein together with mutagenesis studies would be helpful to answer these questions.
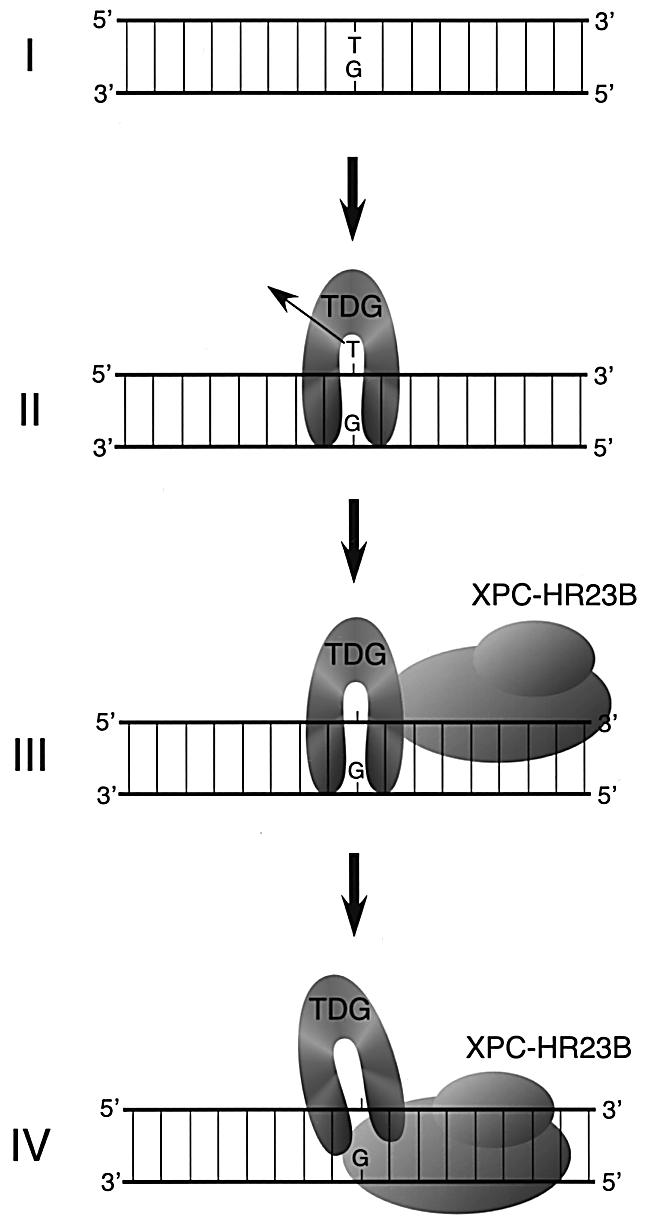
Fig. 8. A schematic model depicting a possible mechanism for TDG stimulation by XPC–HR23B. A G/T mismatch can be generated by spontaneous deamination of 5-methylcytosine (I). After the removal of the mismatched T (II), TDG remains attached to the resulting AP site through interaction with the opposite guanine residue. XPC–HR23B is then recruited via protein–protein interaction and/or structural changes of DNA induced by TDG (III). It has been shown that XPC–HR23B preferentially recognizes DNA secondary structure containing a junction with a single-stranded arm, branching away from duplex DNA in the 3′ direction (Sugasawa et al., 2002). Therefore, XPC–HR23B probably interacts with the unpaired guanine and binds to the 3′ side of the AP site. XPC–HR23B may thus compete with TDG for the guanine residue and push it out from the AP site (IV).
It is also of interest whether XPC–HR23B plays any role in the following BER reaction after release of TDG. In particular, APE could incise only some of the AP sites generated by TDG, regardless of the presence or absence of XPC–HR23B (Figure 7), suggesting the requirement for additional factors for efficient engagement of APE. XPC–HR23B left behind on the AP site may also interact with such factors, thereby contributing to the recruitment of APE. Not only a search for novel protein–protein interactions, but also involvement of other known BER factors (e.g. DNA polymerase β, XRCC1, etc.) should be considered in future studies.
Implications of the XPC–TDG interaction for carcinogenesis in XP patients and mice
XP is characterized by cutaneous and cellular hypersensitivity to UV, and the risk of skin cancer in XP patients is elevated several thousand-fold compared with normal individuals (Friedberg, 1995). The tumors that develop in XP patients have been analyzed for mutations in the p53 tumor suppressor gene, which is often mutated in human cancers. The majority of the p53 mutations identified were C to T transitions at dipyrimidinic sites, including CC to TT dinucleotide transitions, which are characteristic of UV-induced mutations (Giglia et al., 1998). Moreover, many of these mutations were found at CpG sites, where the cytosine often is methylated (Giglia et al., 1998). This may be related to the observation that CPDs are induced preferentially at sites containing 5-methylcytosine (Tommasi et al., 1997).
An extensive study on the spectrum of p53 gene mutations in UVB-induced skin cancers has been conducted in mice bearing various combinations of p53 and Xpc genotypes (Reis et al., 2000). In addition to the typical C (CC) to T (TT) transitions at dipyrimidinic sites, C to T transitions at a specific non-dipyrimidinic CpG site in the p53 gene (in codon 122Thr) were found. This type of mutation was never found in tumors from Xpc+/+ mice and, in the p53+/+ background, it was only found in _Xpc_–/–, but not in Xpc+/– tumors. When one of the wild-type p53 alleles was deleted, some tumors from Xpc+/– mice contained this mutation and, in p53+/– _XPC_–/– tumors, the majority of p53 mutations were located at this site. Analysis of the p53 gene mutations in UVB-induced skin tumors from _Xpa_–/– mice failed to detect the same type of mutation (Takeuchi et al., 1998). Although we do not exclude any other possibilities, one of the explanations for the C to T transitions at non-dipyrimidinic CpG sites could be the deamination of the methylated cytosine and inefficient correction of the resulting G/T mismatch, presumably because of the absence of XPC. Based on the hypothesis that XPC participates in both the NER and BER pathways, it is possible that upon UV irradiation, most of the XPC molecules are engaged in NER and, consequently, few are available for BER. Moreover, it has been shown recently in vitro that p53 can directly stimulate BER (Zhou et al., 2001), providing a possible explanation for the observed enhancement of the mutation in codon 122Thr of the p53 gene in the p53+/– background. Thus, the increased predisposition to UVB-induced skin cancer in the Xpc+/– and _Xpc_–/– background could be due not only to reduced NER activity but also to some impairment of BER.
Since spontaneous deamination is known to occur frequently, reduced TDG activity in XPC-deficient patients and mice may lead to an increase in mutations without UV irradiation. In fact, Wijnhoven et al. (2000) have reported that _Xpc_–/– mice underwent age-dependent spontaneous mutations in the Hprt gene, although the mutations identified were not restricted to C to T transitions. As plasmid reactivation assays suggest that the repair of oxidative DNA damage may be impaired in several XP-C cell lines (Rünger et al., 1995), it is also possible that XPC may be involved in other BER pathways. This could account for the p53 gene mutations identified in internal tumors from XP-C patients, which are not the C to T transitions typical of the UV-induced mutations (Giglia et al., 1998). So far, the functional interaction between XPC–HR23B and TDG has been observed only with purified proteins. Probably because the interaction appears to be rather unstable in the absence of DNA, detection of complex formation in soluble cell extracts has been unsuccessful and thus in vivo evidence for the relevance of this interaction remains to be obtained. It would be of great interest to measure the activity of G/T mismatch correction in normal as well as XP-C cells, and also systematically to re-investigate which DNA glycosylases are stimulated by XPC and to which DNA-damaging agents XP-C cells are sensitive.
Materials and methods
Yeast two-hybrid screening
To identify proteins interacting with XPC, yeast two-hybrid screening was performed with the Matchmaker LexA two-hybrid system (Clontech). The cDNA encoding the full-length human XPC was fused in-frame to the GAL4 DNA-binding domain in the bait vector, pLexA. The yeast strain EGY48 was co-transformed with the resulting plasmid, pLexA-XPC, and the reporter plasmid p8op-lacZ, and transformants were selected on SD medium lacking His and Ura.
Screening was then performed by further transforming the transformed EGY48 yeast with a mouse embryonic fibroblast LexA matchmaker library (Clontech), in which expression of cDNAs fused to the GAL4 activation domain is regulated by the GAL1 promoter. About 7 × 106 transformants were selected on the medium containing galactose and raffinose but lacking His, Leu, Trp and Ura. Colonies that appeared after 2 weeks incubation at 30°C were selected further for the presence of β-galactosidase activity. Positive clones were grown in SD medium lacking Trp, and the prey plasmids were rescued and electroporated into E.coli strain KC8. The DNA recovered from bacteria was sequenced to identify the candidate proteins that interact with XPC.
Purification of recombinant proteins
Purification of recombinant human XPC and His-tagged HR23B (HR23B-His) proteins was carried out as described previously (Sugasawa et al., 1996; Masutani et al., 1997). The XPC–HR23B-His heterodimer was reconstituted in vitro and purified as described previously (Sugasawa et al., 2001). His-TDG was expressed in the E.coli strain BL21 (DE3) using the pET28a vector (Novagen) and was purified as described elsewhere (Hardeland et al., 2000), except that Talon resin (Clontech) was used instead of nickel-NTA–agarose. GST–TDG and GST were expressed in the E.coli strain JM109 (DE3) using the pGEX4T-3 vector (Amersham Biosciences). Protein expression was induced with 0.2 mM IPTG at 16°C for 19 h (for GST–TDG), or with 1 mM IPTG at 37°C for 3 h (for GST). The bacterial cell pellets were sonicated in buffer A [25 mM Tris–HCl pH 7.5, 1 mM EDTA, 25 mM NaCl, 0.01% NP-40, 1 mM DTT, 0.25 mM PMSF and a protease inhibitor cocktail (1× Complete; Roche Diagnostics)] and centrifuged for 30 min at 100 000 g. The supernatant was applied to a GSTrap column (Amersham Biosciences) equilibrated with buffer B (25 mM Tris–HCl pH 7.5, 1 mM EDTA, 0.01% Triton X-100, 10% glycerol, 1 mM DTT, 0.25 mM PMSF) containing 0.1 M NaCl. After the column was washed successively with buffer B containing 0.5 and 1 M NaCl, bound proteins were eluted with buffer B containing 0.1 M NaCl and 10 mM glutathione. Fractions containing GST–TDG were loaded further onto a Mono S PC 1.6/5 column (Amersham Biosciences) equilibrated with buffer C (25 mM sodium phosphate pH 7.0, 1 mM EDTA, 10% glycerol, 0.01% Triton X-100, 1 mM DTT, 0.25 mM PMSF) containing 0.1 M NaCl, and bound proteins were eluted with a linear gradient of 0.1–0.8 M NaCl in buffer C. GST–TDG appeared in fractions eluted around 0.4 M NaCl. Finally, both GST–TDG and GST were subjected to gel filtration chromatography using a Superdex 75 PC 3.2/30 column (Amersham Biosciences) equilibrated with buffer B containing 0.2 M NaCl. His-APE was expressed in the E.coli strain BL21 (DE3) using the pET28a vector, and a bacterial cell lysate was prepared as described previously (Chou and Cheng, 2002). The sample was applied to a phosphocellulose (Whatman P11) column equilibrated with buffer D (20 mM sodium phosphate pH 7.8, 10% glycerol, 0.01% Triton X-100 and 0.25 mM PMSF) containing 0.1 M NaCl, and bound proteins were eluted stepwise with buffer D containing 0.2, 0.6 and 1 M NaCl. Proteins eluted with 1 M NaCl were loaded onto a column packed with Talon resin (Clontech) that had been equilibrated with buffer D containing 0.1 M NaCl and 5 mM imidazole. After washing extensively with the same buffer, bound proteins were eluted by increasing the imidazole concentration to 20, 100 and 250 mM. The 100 mM imidazole fraction containing His-APE subsequently was loaded onto a Mono S PC 1.6/5 column equilibrated with buffer E (50 mM HEPES–NaOH pH 7.6, 1 mM EDTA, 0.01% Triton X-100, 10% glycerol, 1 mM DTT, 0.25 mM PMSF) containing 0.1 M NaCl. The column was developed with a linear gradient of 0.1–0.7 M NaCl in buffer E and His-APE was eluted at ∼0.25 M NaCl.
GST pull-down assay
Glutathione–Sepharose 4 fast flow (Amersham Biosciences: 20 µl) was mixed with recombinant XPC–HR23B (6.7 nM) and either GST or GST–TDG (each 16.6 nM) in 100 µl of the binding buffer containing 20 mM sodium phosphate pH 7.4, 1 mM EDTA, 150 mM NaCl, 1 mM DTT, 5% glycerol, 0.01% Triton X-100 and 100 µg/ml of BSA. Where indicated, His-TDG was also included as a competitor. After incubation on ice for 45 min followed by 30°C for 15 min with occasional agitation, the beads were washed seven times with 500 µl of the binding buffer and the proteins retained on the beads were eluted using 20 µl of the binding buffer containing 10 mM glutathione. Since purified XPC has a tendency to aggregate when boiled in the presence of SDS, each eluate was mixed before denaturation with a whole-cell extract (0.4 µg protein) from XP4PASV cells that do not express endogenous XPC. This procedure prevents aggregation of XPC and enables more quantitative estimation of the protein. An aliquot of each eluate mixed with XP4PASV cell extract was subjected to 8% SDS–PAGE and analyzed by immunoblotting using polyclonal anti-XPC antibody.
Gel mobility shift assay
The covalently closed circular DNAs shown in Figure 2A and 32P-labeled, blunt-ended DNA fragments were prepared exactly as described previously (Sugasawa et al., 2001). For gel mobility shift analysis, binding reactions (10 µl) were carried out also as described previously (Sugasawa et al., 2001), except that MgCl2 was omitted and the amount of covalently closed circular plasmid DNA as competitor DNA was increased to 8 ng. For supershift assays, affinity-purified polyclonal antibodies were added after the standard binding reactions and incubated on ice for 15 min before glutaraldehyde fixation.
Nicking assay
The enzymatic activities of the recombinant His-TDG and His-APE proteins were monitored by a ‘nicking assay’, as described previously (Neddermann and Jiricny, 1993), with some modifications.
To prepare the substrate DNA that contains a single G/T mismatch, the T-strand oligonucleotide (the bottom strand shown in Figure 2B) was 5′ end-labeled by treatment with T4 polynucleotide kinase and [γ-32P]ATP. After heat inactivation of the enzyme at 65°C for 1 h, an equimolar amount of the upper strand oligonucleotide was added and annealed by heating at 65°C for 1 h and then gradually cooling to room temperature. The annealed 60 bp DNA was purified by native PAGE. The nicking reactions were performed in mixtures (25 µl) containing 25 mM HEPES–KOH pH 7.8, 0.5 mM EDTA, 50 mM NaCl, 2 mM MgCl2, 0.5 mM DTT, BSA (9 µg) and the substrate DNA (1.6 nM). After pre-incubation at 30°C for 1 h, purified His-TDG was added and incubated further at 30°C. XPC–HR23B-His and/or His-APE were also added at various time points, as indicated. Reactions were stopped by adding an equal volume of stop buffer (50 mM Tris–HCl pH 7.5, 25 mM EDTA, 0.3 mg/ml yeast tRNA, 2% SDS, 0.8 mg/ml proteinase K) and incubated further at 30°C for >1 h. To convert the resulting AP sites into single strand breaks, NaOH was added to a final concentration of 90 mM and heated at 100°C for 10 min. The DNA was purified by phenol/chloroform extraction and ethanol precipitation and subjected to 16% denaturing PAGE followed by autoradiography. To measure the cleavage by APE, the reactions were performed as described above but stopped by adding 1 µl of 0.5 M EDTA. Subsequently, the reactions were mixed with an equal volume of solution containing 90% formamide, 0.04% bromophenol blue and 0.04% xylene cyanol FF, heated at 65°C for 3 min, and analyzed by 16% denaturing PAGE followed by autoradiography. For both assays, radioactivity in the cleaved and non-cleaved oligonucleotides was quantified using the BAS2500 bioimaging analyzer (Fujifilm).
Antibodies
The polyclonal antibody against XPC was raised as described previously (Sugasawa et al., 1996). The anti-TDG antibody was raised against recombinant TDG fused to the N-terminal FLAG tag (FLAG-TDG) and expressed in E.coli. The antiserum from an immunized rabbit was subjected to ammonium sulfate precipitation (50% saturation) and subsequent affinity purification with GST–TDG covalently coupled to NHS-activated Sepharose 4 fast flow (Amersham Biosciences).
Acknowledgments
Acknowledgements
We are grateful to all the members of Cellular Physiology Laboratory for helpful discussions and encouragement. We also thank Y.Ichikawa and R.Nakazawa (Bioarchitect Research Group) for DNA sequencing, and the Laboratory Animal Research Center, RIKEN, for preparing the anti-TDG antibody. This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and from the Core Research for Evolutional Science and Technology (CREST) from the Japan Science and Technology Corporation. The work was also supported by a grant from the Bioarchitect Research Project of RIKEN. Y.S. was supported by the fellowship of Junior Research Associate (JRA) from RIKEN.
References
- Araki M., Masutani,C., Takemura,M., Uchida,A., Sugasawa,K., Kondoh,J., Ohkuma,Y. and Hanaoka,F. (2001) Centrosome protein centrin 2/caltractin 1 is part of the xeroderma pigmentosum group C complex that initiates global genome nucleotide excision repair. J. Biol. Chem., 276, 18665–18672. [DOI] [PubMed] [Google Scholar]
- Araújo S.J., Tirode,F., Coin,F., Pospiech,H., Syväoja,J.E., Stucki,M., Hübscher,U., Egly,J.-M. and Wood,R.D. (2000) Nucleotide excision repair of DNA with recombinant human proteins: definition of the minimal set of factors, active forms of TFIIH and modulation by CAK. Genes Dev., 14, 349–359. [PMC free article] [PubMed] [Google Scholar]
- Araújo S.J., Nigg,E.A. and Wood,R.D. (2001) Strong functional interactions of TFIIH with XPC and XPG in human DNA nucleotide excision repair, without a preassembled repairosome. Mol. Cell. Biol., 21, 2281–2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett T.E., Savva,R., Panayotou,G., Barlow,T., Brown,T., Jiricny,J. and Pearl,L.H. (1998) Crystal structure of a G:T/U mismatch-specific DNA glycosylase: mismatch recognition by complementary-strand interactions. Cell, 92, 117–129. [DOI] [PubMed] [Google Scholar]
- Barrett T.E., Schärer,O.D., Savva,R., Brown,T., Jiricny,J., Verdine,G.L. and Pearl,L.H. (1999) Crystal structure of a thwarted mismatch glycosylase DNA repair complex. EMBO J., 18, 6599–6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batty D., Rapic’-Otrin,V., Levine,A.S. and Wood,R.D. (2000) Stable binding of human XPC complex to irradiated DNA confers strong discrimination for damaged sites. J. Mol. Biol., 300, 275–290. [DOI] [PubMed] [Google Scholar]
- Bessho T. (1999) Nucleotide excision repair 3′ endonuclease XPG stimulates the activity of base excision repair enzyme thymine glycol DNA glycosylase. Nucleic Acids Res., 27, 979–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou K.M. and Cheng,Y.C. (2002) An exonucleotic activity of human apurinic/apyrimidinic endonuclease on 3′ mispaired DNA. Nature, 415, 655–659. [DOI] [PubMed] [Google Scholar]
- Cooper P.K., Nouspikel,T., Clarkson,S.G. and Leadon,S.A. (1997) Defective transcription-coupled repair of oxidative base damage in Cockayne syndrome patients from XP group G. Science, 275, 990–993. [DOI] [PubMed] [Google Scholar]
- de Laat W.L., Jaspers,N.G.J. and Hoeijmakers,J.H.J. (1999) Molecular mechanism of nucleotide excision repair. Genes Dev., 13, 768–785. [DOI] [PubMed] [Google Scholar]
- Friedberg E.C., Walker,G.C. and Siede,W. (1995) DNA Repair and Mutagenesis. ASM Press, Washington, DC.
- Fujiwara Y., Masutani,C., Mizukoshi,T., Kondo,J., Hanaoka,F. and Iwai,S. (1999) Characterization of DNA recognition by the human UV-damaged DNA-binding protein. J. Biol. Chem., 274, 20027–20033. [DOI] [PubMed] [Google Scholar]
- Giglia G., Dumaz,N., Drougard,C., Avril,M.F., Daya-Grosjean,L. and Sarasin,A. (1998) p53 mutations in skin and internal tumors of xeroderma pigmentosum patients belonging to the complementation group C. Cancer Res., 58, 4402–4409. [PubMed] [Google Scholar]
- Hardeland U., Bentele,M., Jiricny,J. and Schär,P. (2000) Separating substrate recognition from base hydrolysis in human thymine DNA glycosylase by mutational analysis. J. Biol. Chem., 275, 33449–33456. [DOI] [PubMed] [Google Scholar]
- Hardeland U., Bentele,M., Lettieri,T., Steinacher,R., Jiricny,J. and Schär,P. (2001) Thymine DNA glycosylase. Prog. Nucleic Acid Res. Mol. Biol., 68, 235–253. [DOI] [PubMed] [Google Scholar]
- Hoeijmakers J.H.J. (2001) Genome maintenance mechanisms for preventing cancer. Nature, 411, 366–374. [DOI] [PubMed] [Google Scholar]
- Klungland A., Höss,M., Gunz,D., Constantinou,A., Clarkson,S.G., Doetsch,P.W., Bolton,P.H., Wood,R.D. and Lindahl,T. (1999) Base excision repair of oxidative DNA damage activated by XPG protein. Mol. Cell, 3, 33–42. [DOI] [PubMed] [Google Scholar]
- Krokan H.E., Nilsen,H., Skorpen,F., Otterlei,M. and Slupphaug,G. (2000) Base excision repair of DNA in mammalian cells. FEBS Lett., 476, 73–77. [DOI] [PubMed] [Google Scholar]
- Kusumoto R., Masutani,C., Sugasawa,K., Iwai,S., Araki,M., Uchida,A., Mizukoshi,T. and Hanaoka,F. (2001) Diversity of the damage recognition step in the global genomic nucleotide excision repair in vitro. Mutat. Res., 485, 219–227. [DOI] [PubMed] [Google Scholar]
- Le Page F., Kwoh,E.E., Avrutskaya,A., Gentil,A., Leadon,S.A., Sarasin,A. and Cooper,P.K. (2000) Transcription-coupled repair of 8-oxoguanine: requirement for XPG, TFIIH and CSB and implications for Cockayne syndrome. Cell, 101, 159–171. [DOI] [PubMed] [Google Scholar]
- Li R.Y., Calsou,P., Jones,C.J. and Salles,B. (1998) Interactions of the transcription/DNA repair factor TFIIH and XP repair proteins with DNA lesions in a cell-free repair assay. J. Mol. Biol., 281, 211–218. [DOI] [PubMed] [Google Scholar]
- Masutani C. et al. (1994) Purification and cloning of a nucleotide excision repair complex involving the xeroderma pigmentosum group C protein and a human homologue of yeast RAD23. EMBO J., 13, 1831–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masutani C. et al. (1997) Identification and characterization of XPC-binding domain of hHR23B. Mol. Cell. Biol., 17, 6915–6923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough A.K., Dodson,M.L. and Lloyd,R.S. (1999) Initiation of base excision repair: glycosylase mechanisms and structures. Annu. Rev. Biochem., 68, 255–285. [DOI] [PubMed] [Google Scholar]
- Miao F., Bouziane,M., Dammann,R., Masutani,C., Hanaoka,F., Pfeifer,G. and O’Connor,T.R. (2000) 3-Methyladenine-DNA glycosylase (MPG protein) interacts with human RAD23 proteins. J. Biol. Chem., 275, 28433–28438. [DOI] [PubMed] [Google Scholar]
- Missero C., Pirro,M.T., Simeone,S., Pischetola,M. and Di Lauro,R. (2001) The DNA glycosylase T:G mismatch-specific thymine DNA glycosylase represses thyroid transcription factor-1-activated transcription. J. Biol. Chem., 276, 33569–33575. [DOI] [PubMed] [Google Scholar]
- Mu D., Park,C.H., Matsunaga,T., Hsu,D.S., Reardon,J.T. and Sancar,A. (1995) Reconstitution of human DNA repair excision nuclease in a highly defined system. J. Biol. Chem., 270, 2415–2418. [DOI] [PubMed] [Google Scholar]
- Neddermann P. and Jiricny,J. (1993) The purification of a mismatch-specific thymine-DNA glycosylase from HeLa cells. J. Biol. Chem., 268, 21218–21224. [PubMed] [Google Scholar]
- Nilsen H. and Krokan,H.E. (2001) Base excision repair in a network of defence and tolerance. Carcinogenesis, 22, 987–998. [DOI] [PubMed] [Google Scholar]
- Reis A.M., Cheo,D.L., Meira,L.B., Greenblatt,M.S., Bond,J.P., Nahari,D. and Friedberg,E.C. (2000) Genotype-specific Trp53 mutational analysis in ultraviolet B radiation-induced skin cancers in Xpc and Xpc Trp53 mutant mice. Cancer Res., 60, 1571–1579. [PubMed] [Google Scholar]
- Rünger T.M., Epe,B. and Möller,K. (1995) Repair of ultraviolet B and singlet oxygen-induced DNA damage in xeroderma pigmentosum cells. J. Invest. Dermatol., 104, 68–73. [DOI] [PubMed] [Google Scholar]
- Shivji M.K.K., Eker,A.P.M. and Wood,R.D. (1994) DNA repair defect in xeroderma pigmentosum group C and complementing factor from HeLa cells. J. Biol. Chem., 269, 22749–22757. [PubMed] [Google Scholar]
- Sugasawa K., Masutani,C., Uchida,A., Maekawa,T., van der Spek,P.J., Bootsma,D., Hoeijmakers,J.H.J. and Hanaoka,F. (1996) HHR23B, a human Rad23 homolog, stimulates XPC protein in nucleotide excision repair in vitro. Mol. Cell. Biol., 16, 4852–4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugasawa K. et al. (1997) Two human homologs of Rad23 are functionally interchangeable in complex formation and stimulation of XPC repair activity. Mol. Cell. Biol., 17, 6924–6931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugasawa K., Ng,J.M.Y., Masutani,C., Iwai,S., van der Spek,P.J., Eker,A.P.M., Hanaoka,F., Bootsma,D. and Hoeijmakers,J.H.J. (1998) Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol. Cell, 2, 223–232. [DOI] [PubMed] [Google Scholar]
- Sugasawa K., Okamoto,T., Shimizu,Y., Masutani,C., Iwai,S. and Hanaoka,F. (2001) A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes Dev., 15, 507–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugasawa K., Shimizu,Y., Iwai,S. and Hanaoka,F. (2002) A molecular mechanism for DNA damage recognition by the xeroderma pigmentosum group C protein complex. DNA Repair, 1, 95–107. [DOI] [PubMed] [Google Scholar]
- Takeuchi S., Nakatsu,Y., Nakane,H., Murai,H., Hirota,S., Kitamura,Y., Okuyama,A. and Tanaka,K. (1998) Strand specificity and absence of hot spots for p53 mutations in ultraviolet B-induced skin tumors of XPA-deficient mice. Cancer Res., 58, 641–646. [PubMed] [Google Scholar]
- Tini M., Benecke,A., Um,S., Torchia,J., Evans,R.M. and Chambon,P. (2002) Association of CBP/p300 acetylase and thymine DNA glycosylase links DNA repair and transcription. Mol. Cell, 9, 265–277. [DOI] [PubMed] [Google Scholar]
- Tommasi S., Denissenko,M.F. and Pfeifer,G.P. (1997) Sunlight induces pyrimidine dimers preferentially at 5-methylcytosine bases. Cancer Res., 57, 4727–4730. [PubMed] [Google Scholar]
- Um S., Harbers,M., Benecke,A., Pierrat,B., Losson,R. and Chambon,P. (1998) Retinoic acid receptors interact physically and functionally with the T:G mismatch-specific thymine-DNA glycosylase. J. Biol. Chem., 273, 20728–20736. [DOI] [PubMed] [Google Scholar]
- Waters T.R. and Swann,P.F. (1998) Kinetics of the action of thymine DNA glycosylase. J. Biol. Chem., 273, 20007–20014. [DOI] [PubMed] [Google Scholar]
- Waters T.R., Gallinari,P., Jiricny,J. and Swann,P.F. (1999) Human thymine DNA glycosylase binds to apurinic sites in DNA but is displaced by human apurinic endonuclease 1. J. Biol. Chem., 274, 67–74. [DOI] [PubMed] [Google Scholar]
- Wijnhoven S.W.P., Kool,H.J.M., Mullenders,L.H.F., van Zeeland,A.A., Friedberg,E.C., van der Horst,G.T.J., van Steeg,H. and Vrieling,H. (2000) Age-dependent spontaneous mutagenesis in Xpc mice defective in nucleotide excision repair. Oncogene, 19, 5034–5037. [DOI] [PubMed] [Google Scholar]
- Wilson D.M. III and Barsky,D. (2001) The major human abasic endonuclease: formation, consequences and repair of abasic lesions in DNA. Mutat. Res., 485, 283–307. [DOI] [PubMed] [Google Scholar]
- Yokoi M., Masutani,C., Maekawa,T., Sugasawa,K., Ohkuma,Y. and Hanaoka,F. (2000) The xeroderma pigmentosum group C protein complex XPC–HR23B plays an important role in the recruitment of transcription factor IIH to damaged DNA. J. Biol. Chem., 275, 9870–9875. [DOI] [PubMed] [Google Scholar]
- Zhou J., Ahn,J., Wilson,S.H. and Prives,C. (2001) A role for p53 in base excision repair. EMBO J., 20, 914–923. [DOI] [PMC free article] [PubMed] [Google Scholar]