A1 Receptor and Adenosinergic Homeostatic Regulation of Sleep-Wakefulness: Effects of Antisense to the A1 Receptor in the Cholinergic Basal Forebrain (original) (raw)
Abstract
We hypothesized that adenosine, acting via the A1 receptor, is a key factor in the homeostatic control of sleep. The increase in extracellular levels of adenosine during prolonged wakefulness is thought to facilitate the transition to sleep by reducing the discharge activity of wakefulness-promoting neurons in the basal forebrain. Adenosine A1 receptor control of the homeostatic regulation of sleep was tested by microdialysis perfusion of antisense oligonucleotides against the mRNA of the A1 receptor in the magnocellular cholinergic region of the basal forebrain of freely behaving rats. After microdialysis perfusion of A1 receptor antisense in the basal forebrain, spontaneous levels of sleep-wakefulness showed a significant reduction in non-rapid eye movement (REM) sleep with an increase in wakefulness. After 6 hr of sleep deprivation, the antisense-treated animals spent a significantly reduced amount of time in non-REM sleep, with postdeprivation recovery sleep hours 2–5 showing a reduction of ∼50–60%. There was an even greater postdeprivation reduction in delta power (60–75%) and a concomitant increase in wakefulness. All behavioral state changes returned to control (baseline) values after the cessation of antisense administration. Control experiments with microdialysis perfusion of nonsense (randomized antisense) oligonucleotides and with artificial CSF showed no effect during postdeprivation recovery sleep or spontaneously occurring behavioral states. Antisense to the A1 receptor suppressed A1 receptor immunoreactivity but did not show any neurotoxicity as visualized by Fluoro-Jade staining. These data support our hypothesis that adenosine, acting via the A1 receptor, in the basal forebrain is a key component in the homeostatic regulation of sleep.
Keywords: non-REM sleep, adenosine, substantia innominata, cholinergic, basal forebrain, A1 receptor, antisense, microdialysis
Introduction
Prolonged wakefulness increases the propensity to sleep and decreases the degree of arousal. We suggested that adenosine, an inhibitory neuromodulator, is the key component in mediating the sleepiness associated with previous wakefulness (homeostatic regulation) (for review, see Strecker et al., 2000). Brain ATP metabolism is highest during wakefulness, and we hypothesize that, as a result of ATP breakdown, adenosine concentration increases, most markedly in the cholinergic basal forebrain (BF), a key anatomical substrate for the control of wakefulness and cortical activation (Szymusiak, 1995; Jones, 1998).
Several lines of evidence have supported this hypothesis. In vitro data indicated that BF neurons were under the tonic inhibitory control of adenosine (Rainnie et al., 1994). Microdialysis perfusion of adenosine in the BF reduced wakefulness in freely behaving cats (Portas et al., 1997). Extracellular concentrations of adenosine in the BF were higher during spontaneous wakefulness than during non-rapid eye movement (REM) sleep (Porkka-Heiskanen et al., 1997). In contrast to other brain regions, adenosine concentrations in the BF exhibited progressive increases during sustained, prolonged wakefulness, followed by a slow decline during recovery sleep (Porkka-Heiskanen et al., 2000). Microdialysis perfusion of an adenosine transport inhibitor _s_-(_p_-nitrobenzyl)-6-thioinosine not only doubled extracellular concentrations of adenosine in the BF but also mimicked the state changes seen with recovery sleep (Porkka-Heiskanen et al., 1997). Finally, adenosine inhibited the discharge activity of wakefulness-active neurons (Alam et al., 1999).
Data suggest that the A1 receptor mediates the adenosine effects in the BF. First, total sleep deprivation increased A1 receptor mRNA levels in BF, whereas A2A receptor mRNA–ligand binding remained undetectable (Basheer et al., 2001a). Second, in vivo, the A1 receptor agonist _N_6-cyclohexyladenosine inhibited the discharge activity of BF wakefulness-active neurons, but the A2A receptor agonist CGS-16284 (4-[2-[[6-amino-9-(_N_-ethyl-_b_-d-ribofuranuronamidosyl)-9_H_-purin-2-yl]amino]ethyl]benzenepropanoic acid hydrochloride) had no effect (Thakkar et al., 1999a). Finally, in vitro, an A1 antagonist increased the discharge rate of BF neurons (Rainnie et al., 1994).
However, additional and more specific evidence was needed to test whether the A1 receptor in the BF was a mediator of homeostatic changes in sleep. Pharmacological techniques were limited in specificity. Molecular techniques of constitutive receptor “knock-outs” are powerful but limited by developmental compensations and lack of specificity to brain areas of interest. Inducible knock-outs avoid some of the developmental constraints of constitutive knock-outs, but the technique is time consuming and difficult. Antisense technology is highly specific for receptor subtype, providing a localized, reversible receptor knock-down, and has been used successfully in many fields and recently by us in the study of behavioral state control (Thakkar et al., 1999b). Accordingly, we used antisense deoxyoligonucleotides (ODNs) directed against the mRNA for the adenosine A1 receptor. For better control of concentrations, local application of antisense ODNs in the BF was performed with microdialysis perfusion instead of microinjection. Experimental controls included the perfusion of nonsense ODNs and the immunohistochemical demonstration of antisense ODN-induced suppression of A1 receptor in the BF and the absence of Fluoro-Jade staining evidence for neurotoxicity.
Materials and Methods
Experimental design
Recovery sleep after sleep deprivation is an essential paradigm to study the homeostatic mechanisms that regulate sleep-wakefulness because it separates homeostatic from circadian influences. Therefore, our primary experiment (experiment 2) was to study the effects of A1 receptor antisense on recovery sleep after sleep deprivation. Another experiment (experiment 1) studied the effects of antisense ODNs on spontaneous sleep-wakefulness, whereas a third experiment evaluated the potential neurotoxicity caused by antisense administration and the loss of A1 receptor immunoreactivity after the microdialysis perfusion of A1 receptor antisense.
Animals and surgery
Adult male Sprague Dawley rats were housed under constant temperature with ad libitum access to food and water and a 12 hr light/dark cycle (light, 7:00 A.M. to 7:00 P.M.; dark, 7:00 P.M. to 7:00 A.M.). Under sterile conditions and according to a standard surgical protocol (for details, see Thakkar et al., 2001), the animals were implanted with electrodes for recording electroencephalograms (EEGs) and electromyograms (EMGs) for determination of behavioral state. Intracerebral guide cannulas, for later insertion of the microdialysis probes, were also implanted bilaterally at a 90° angle above the target site in the horizontal diagonal band/substantia innominata region of the BF. We chose the cholinergic horizontal diagonal band/substantia innominata as the target site on the basis of our previous adenosine studies (Porkka-Heiskanen et al., 1997; Portas et al., 1997; Thakkar et al., 1999a) and because a majority of neurons in this region have their highest discharge during wakefulness (wakefulness-active) (Szymusiak, 1995; Thakkar et al., 1999a). The bilateral target coordinates (Paxinos and Watson, 1997) for the tips of the 2 mm probes were as follows (in mm): anteroposterior, -0.35; mediolateral, ±2.0; and dorsoventral, -8.5 relative to bregma and skull surface at bregma. Every effort was made to minimize animal suffering and to reduce the number of animals used, and all animals were treated in accordance with the policy on care and use of laboratory animals by the American Association for Accreditation of Laboratory Animal Care.
Adenosine A1 receptor antisense
A 21 bp phosphorothioate oligonucleotide antisense designed against the mRNA of A1 receptor had been used previously to study ethanol-induced locomotor behaviors (Biggs and Myers, 1997; Phan et al., 1997, 1999) but had never been used to study its effect on sleep-wakefulness. The antisense sequence 5-GATGTAGGGC GGCATGGTGG G-3 was custom synthesized by Gene Link (Hawthorne, NY). The same sequence of bases was randomized and was used for the synthesis of the nonsense oligonucleotides (Gene Link). The antisense or nonsense ODNs were resuspended in Tris buffer, pH 8.0, per the instructions of the vendor (Gene Link) and divided into aliquots in sterile vials. The concentration of antisense perfused [20 μm in artifical CSF (ACSF)], the hours per day of perfusion, and the number of days of perfusion were determined on the basis of the results of initial dose-finding experiments. Previous in vitro recovery experiments done in our laboratory indicated that <1% of the antisense diffused through the dialysis membrane, thus delivering <0.2 μm immediately outside the probe (Thakkar et al., 1999b).
Antisense ODNs are short chains of deoxynucleic acids (∼20 nucleotides) targeted to a specific mRNA sequence (A1 receptor in the present report) of complementary bases. One possible mechanism of action is blocking the translation of the targeted mRNA by the nucleotide bases of the antisense ODNs that form hydrogen bonds with the complementary nucleotide bases of the targeted mRNA (Stein and Cohen, 1988; Walder and Walder, 1988). Another possible mechanism of action of ODNs is functioning as a substrate (ODN:mRNA complex) for enzymatic degradation by RNase H (Walder and Walder, 1988; Wahlestedt, 1994). Pinocytosis or receptor-mediated endocytosis is the primary means of ODN uptake (Loke et al., 1989; Yakubov et al., 1989; Stein et al., 1993), although other mechanisms are also involved (Krieg et al., 1993). The phosphorothioate analogs, wherein one sulfur molecule is substituted for one of the oxygen molecules, are known to be more resistant to degradation, have enhanced uptake and efficacy (Nicot and Pfaff, 1997), and have been infused continuously (both locally and intracerebroventricularly) without any toxic effects (Neckers and Whitesell, 1993; Thakkar et al., 1999b).
Methodological issues with antisense use
In most of the literature on antisense experiments, the preferred controls for specificity of the antisense sequence have included administration of “scrambled ODNs” (or nonsense, with the same bases as the antisense but a randomized sequence) and “sense ODNs” (sequence identical to the target mRNA). Lately, however, it has been suggested that sense controls do not provide any additional advantage over nonsense ODNs, especially because any potentially critical sequence motifs (recurring sequences of 3–8 bases in the mRNA) that may contribute to the structure or function of the targeted mRNA are lost (Stein, 1999). In addition, sense ODNs are known to produce nonspecific effects (Helene and Toulme, 1990; Nicot and Pfaff, 1997; Kalra and Kalra, 2000), including inhibition of the targeted protein (Georgieva et al., 1995), and therefore, several recent studies (in vivo and in vitro) have not used the sense control (Turchi and Sarter, 2001; Clayton et al., 2002; Frantseva et al., 2002; Grimpe et al., 2002; Torner et al., 2002). There also appears to be agreement that demonstration of antisense-induced suppression of the desired protein without any neurotoxicity and reversal of the behavioral changes after termination of antisense administration are essential to ascertain the efficacy and specificity of antisense (Nicot and Pfaff, 1997; Stein, 1999, 2001).
Use of microdialysis probe for local application of A1 receptor antisense
In some antisense application studies, including those of antisense to the A1 receptor (Biggs and Myers, 1997; Phan et al., 1997), multiple microinjections of antisense were performed; however, multiple microinjections, especially with high concentrations of antisense, may cause neurotoxicity (Chiasson et al., 1994). Microdialysis perfusion has several advantages over microinjection techniques (Quan and Blatteis, 1989; Portas et al., 1997; Thakkar et al., 1998, 1999b), including the ability to deliver very low and constant concentrations of antisense, thus likely reducing the probability of any neurotoxic damage.
Microdialysis perfusion and electrographic recordings of sleep-wakefulness Experiments were conducted in a sound-attenuated chamber that had the same temperature and light conditions as the animals' home cages, with food and water available ad libitum. After 5 d of postoperative recovery and habituation, the microdialysis probes were inserted through the guide cannula. After 12 hr of recovery from probe insertion, the experiment was begun. CMA/11 probes were used with a 2 mm length and 0.24 mm outer diameter dialyzing membrane (CMA Microdialysis, Acton, MA). Subjects were connected to a polygraph cable for behavioral state recording and then to the probe inlet and outlet tubing (1 m pieces of low dead volume polytetrafluorethylene tubing; CMA Microdialysis). ACSF (in mm: 147 NaCl, 3 KCl, 1.2 CaCl2, and 1.0 MgCl2, pH 7.2), antisense or nonsense ODNs, or both were perfused at a flow rate of 1.0 μl/min. The detailed protocol for each experiment is presented below.
Experiment 1: effect of microdialysis perfusion of antisense ODNs against A1 receptor on spontaneous sleep-wakefulness
The main aims of this experiment were to identify the effect of ODN perfusion on spontaneous sleep-wakefulness, the time of maximum effect, and the duration of the effect after antisense ODN perfusion. Two separate groups of animals were used for this experiment. Both of the groups received identical treatment (described in detail below), except that animals of group 1 (n = 6) were perfused with antisense ODNs, whereas the animals in group 2 (n = 6) were perfused with nonsense ODNs.
The experimental protocol consisted of 5 d of continuous electrographic recordings (24 hr each day, with each day scored from 7:00 A.M. to 7:00 A.M. the next day), during which there were 3 d of microdialysis perfusion. The detailed protocol is outlined below.
Control day. ACSF was continuously and bilaterally perfused for 8 hr (10:00 A.M. 6: 00 P.M.).
ODN days 1 and 2. The protocol was the same as the control day except that 20 μm of antisense (group I) or nonsense (group II) to the A1 receptor (ACSF vehicle) was perfused bilaterally for 8 hr (10:00 A.M. to 6:00 P.M.) instead of ACSF on each of the ODN days.
Post-ODN days 1 and 2. During post-ODN days 1 and 2, there was no microdialysis perfusion, but the behavior of the animal was recorded continuously.
Experiment 2: effect of antisense ODNs against A1 receptor on recovery sleep after 6 hr of sleep deprivation
The main aim of this experiment was to identify the effect of the A1 receptor on the homeostatic regulation of sleep-wakefulness. The animals were divided into three groups. All three groups received identical treatments (details are given below), except that group I animals (n = 6) were perfused with ACSF, group II animals (n = 4) were perfused with nonsense ODNs, and group III animals (n = 6) were perfused with antisense ODNs.
The experimental protocol consisted of 3 d of continuous electrographic recordings (24 hr each day) with 2 d of microdialysis perfusion (for scoring, an experimental day ran from 7:00 A.M. to 7:00 A.M. the next day). On the third day, total sleep deprivation was performed for 6 hr (1:00 P.M. to 7:00 P.M.), followed by 12 hr of undisturbed recovery sleep. Our preliminary studies on spontaneous sleep-wakefulness indicated that the effect of A1 receptor antisense on sleep-wakefulness peaked on the first day after perfusion of antisense ODNs to the A1 receptor. Therefore, the day after 2d of antisense perfusion was chosen to perform the sleep deprivation-recovery sleep experiment. The detailed protocol is outlined below.
Days 1 and 2. ACSF (group I) or nonsense (group II) or antisense (group III) ODNs were perfused continuously and bilaterally for 8 hr (10:00 A.M. to 6:00 P.M.) on each day.
Sleep-deprivation day. During the sleep-deprivation day, there was no microdialysis perfusion, but sleep deprivation was performed for 6 hr by means of gentle handling with sound and tactile stimulation, beginning at 1:00 P.M. and ending at 7:00 P.M., followed by an undisturbed recovery sleep period from 7:00 P.M. to 7:00 A.M. By performing sleep deprivation during the light (inactive) period, the normal sleeping period in rodents, sleep pressure was maximized, and by ending the deprivation just at the onset of the lights-off period (7:00 P.M., beginning the active period), any ceiling effects on sleep response to deprivation were minimized.
Evaluation of A1 receptor immunoreactivity and potential neurotoxicity at the site of A1 receptor antisense perfusion
The main aim of this experiment was to evaluate potential neurotoxicity and the loss of A1 receptor immunoreactivity after microdialysis perfusion of A1 receptor antisense ODNs. Our preliminary studies on spontaneous sleep-wakefulness indicated that the effect of A1 receptor antisense on sleep-wakefulness peaked on the first day after perfusion of antisense ODNs to the A1 receptor antisense for 2 d but returned to normal values on the second day after ODN perfusion stopped. This meant that, to assess the potential neurotoxicity and suppression of A1 receptor immunoreactivity, the animals would have to be killed on the day after ODN perfusion (the third day of the experiment). Because both experiment 1 and experiment 2 continued, at a minimum, until the beginning of day 4 (2 d after ODN perfusion), a separate group of animals was used for this experiment. These animals were implanted with bilateral probes and were perfused with antisense ODNs on one side and nonsense ODNs on the other side for 2 d and killed on day 3, as detailed below. No electrographic recordings were performed in these animals.
Days 1 and 2. Bilaterally implanted animals (n = 4) were perfused continuously with antisense ODNs on one side and nonsense ODNs on the other side for 8 hr (10:00 A.M. to 6:00 P.M.) on each day.
Day 3. All animals were killed on day 3 between 10:00 A.M. and 12:00 P.M. The brains were removed and processed for A1 receptor immunohistochemistry and Fluoro-Jade and cresyl violet staining. A1 receptor immunohistochemistry was performed to evaluate the loss of A1 receptor, whereas Fluoro-Jade and cresyl violet staining were performed to assess the potential neurotoxic effects of antisense administration.
Histology and identification of the perfusion site
After completion of the experiments, all of the animals were deeply anesthetized with sodium pentobarbital and then perfused with 500 ml of cold saline, followed by 2% formaldehyde in 0.1 m phosphate buffer. The BF was isolated, blocked, postfixed, and cryoprotected in 20% sucrose–0.1 m phosphate buffer. The blocked BF was cut in 40 μm coronal sections, and one in three sections was processed for choline acetyltransferase (ChAT) immunohistochemistry (Thakkar et al., 2001) with DAB as the chromogen (ChAT primary antibody from Chemicon, Temecula, CA) to localize the perfusion site (experiments 1 and 2).
The area of perfusion was processed to determine the loss of A1 receptor immunoreactivity after 2 d (16 hr) of antisense–nonsense ODN perfusion. A1 receptor immunohistochemistry (Thakkar et al., 2002a) (A1 receptor primary antibody from Chemicon) was performed in every third section with DAB as the chromogen.
Neurotoxicity assessment of antisense administration by Fluoro-Jade and cresyl violet staining
The exact mechanism of Fluoro-Jade staining of degenerating neurons is unclear, but it has been suggested that the acidic Fluoro-Jade dye binds to an undefined component of the degenerating neuron (Schmued et al., 1997; Eisch et al., 1998; Hopkins et al., 2000; Savaskan et al., 2000) and fluoresces. The details of the protocol are as follows. Sections adjacent (120 μm apart) to those processed for cresyl violet staining were processed for Fluoro-Jade staining, with a positive Fluoro-Jade label indicating neurodegeneration. First, the sections were mounted onto gelatin-coated slides and allowed to dry in the oven (35°C). Once the sections had dried, they were incubated in 100% ethanol for 3 min, followed by 1 min incubation in 70% ethanol and in distilled water. Sections were then incubated in 0.06% KMnO4 for 15 min with gentle shaking. This was followed by a 1 min rinse in distilled water, and then the sections were incubated in a solution composed of 0.001% Fluoro-Jade (Histo-Chem, Jefferson, AR) in 0.1% glacial acetic acid for 30 min. Sections were then washed (three successive 1 min rinses) in distilled water and air dried. Once the sections were dried, they were incubated in xylene and mounted under coverslips with DPX (a mixture of distyrene, tricresyl phosphate, and xylene) (Sigma-Aldrich, St. Louis, MO) neutral mounting medium. Cresyl violet staining was performed by a previously described protocol (Paxinos and Watson, 1997; Thakkar et al., 2002b).
Analysis of behavioral states
Behavioral state data were acquired and digitized with Harmonie software (Stellate Systems, Montreal, Canada). The electrographic data were scored separately (visually, 30 sec epochs) for the light period and the dark period (Thakkar et al., 2001). The behavior of the animals was divided into three states and scored as follows: (1) wakefulness (which included both active and quiet wakefulness), determined by the presence of low-amplitude, high-frequency desynchronized EEG with the concomitant presence of active muscle tone; (2) non-REM sleep, determined by the presence of low-frequency, high-amplitude, synchronized EEG with low EMG tone; or (3) REM sleep, determined by complete absence of muscle tone along with desynchronized EEG.
Spectral analysis
Increases in delta power (1–4 Hz) during recovery sleep are an important indicator of the homeostatic regulation of sleep (Franken et al., 1991). Therefore, in addition to behavioral state scoring, spectral analysis of the EEG (30 sec epochs; Harmonie software; Stellate Systems) was performed to investigate the effects of A1 receptor antisense perfusion on the total delta power during the first 5 hr (7:00 P.M. to 12:00 A.M.) of recovery sleep after sleep deprivation. Because of variability in recordings of delta activity across animals, the delta power values during recovery sleep were normalized and expressed as the percentage of delta power at the corresponding clock time during a nonrecovery day (previous day or ODN day 2) for each animal; the use of each animal as its own control in experiments affecting delta activity is standard in the literature (Tobler and Borbely, 1990). Our data on spontaneous sleep-wakefulness indicated that there was no effect of A1 receptor antisense on sleep-wakefulness on ODN day 2.
Statistics
Experiment 1. We used paired Student's t tests to compare the effect of antisense perfusion on spontaneous sleep-wakefulness. The percentage of time spent in wakefulness, non-REM sleep, and REM sleep during control day perfusion was compared with subsequent days of antisense and postantisense perfusion. Light and dark periods were analyzed separately. Similar analyses were performed to compare the effect of nonsense perfusion on spontaneous sleep-wakefulness.
Experiment 2. We used a Student's t test to compare the effect of antisense–nonsense perfusion on recovery sleep after 6 hr of sleep deprivation. The amount of time spent in wakefulness, non-REM sleep, and REM sleep was computed for each hour during the 12 hr of recovery sleep. Control groups (ACSF and nonsense) were compared with each other, and each group was also compared with the antisense group.
Results
Histology
All of the microdialysis probe tips were located in the cholinergic zone of the BF in or near the horizontal diagonal band/substantia innominata/magnocellular preoptic nucleus region (Fig. 1_A_). The presence of intact ChAT-immunoreactive neurons in and around the site of perfusion (Fig. 1_B_) suggests that there were no toxic effects of antisense or nonsense perfusion on the synthesis of this protein.
Figure 1.

A, Schematic representation of the anatomical location of the histologically identified probe tip sites in 28 animals. All sites were located between anteroposterior -0.2 and -0.8 mm and were mapped onto this one coronal brainstem section (-0.4 mm; adapted from Paxinos and Watson, 1997). Probes were placed bilaterally in each animal. The bilateral antisense perfusion sites (total on each side, n = 12; n = 6 each for experiments 1 and 2) are represented by solid red circles, the nonsense perfusion sites (total on each side, n = 10; n = 6 for experiment 1 and n = 4 for experiment 2) are represented by solid black circles, and the ACSF perfusion sites (n = 6 for experiment 2) are represented by solid blue circles. HDB, Nucleus of horizontal limb of the diagonal band; LPO, lateral preoptic nucleus; MCPO, magnocellular preoptic nucleus; SIB, substantia innominata basal; VP, ventral pallidum; VLPO, ventrolateral preoptic region. For identification of unlabeled structures, see Paxinos and Watson (1997). B, Representative photomicrograph of an antisense perfusion site in the cholinergic BF. The lesion (arrow) caused by the microdialysis probe is located in the midst of cholinergic neurons, the intact staining of which for ChAT indicates an absence of antisense-induced toxic effects on synthesis of this protein. Scale bar, 50 μm.
Experiment 1: spontaneous sleep and wakefulness
Microdialysis perfusion of antisense ODNs against mRNA of the A1 receptor had significant effects on spontaneous duration of bouts of sleep-wakefulness during both the light and the dark period (Fig. 2). The maximum effect was seen on post-ODN day 1, whereas effects on ODN day 1 and 2 were comparable with those on the control day (except for REM sleep). In addition, most of the effects normalized on post-ODN day 2, which suggests the effects of antisense ODNs were reversible. Of note, the animals that received nonsense ODNs did not show any significant alterations in spontaneous bouts of sleep-wakefulness during either the light or the dark period.
Figure 2.

Effect of microdialys is perfusion of antisense oligonucleotides against the mRNA of the A1 receptor on spontaneous bouts of sleep-wakefulness in freely behaving animals during both the light period (top) and the dark period (bottom). During the control day (open bar), ACSF was perfused for 8 hr (from 10:00 A.M. to 6:00 P.M.). This was followed by microdialysis perfusion of antisense for 16 hr, delivered over 2d (8hr each day, from 10:00 A.M. to 6:00 P.M.). The top graph shows that, during the light period, there was a significant increase in wakefulness (p < 0.05; paired t test) and a significant decrease in non-REM sleep (p < 0.05; paired t test) on the first day after antisense perfusion (post-ODN day 1; solid bars) compared with the control (ACSF) day, whereas waking (W) and non-REM sleep were not different from control levels on post-ODN day 2 (gray bars). There was no effect on REM sleep. During the dark period (bottom), there was a significant (p < 0.05; paired t test) increase in wakefulness (W) but no change in non-REM sleep on post-ODN day 1 (solid bar) compared with control day (open bar), whereas both waking and non-REM sleep were not different from control levels on post-ODN day 2. There was a significant decrease in REM sleep (p < 0.05; paired t test) on both post-ODN day 1 and 2 (gray bar). For details, see Results. *p < 0.05.
During post-ODN day 1, the animals perfused with antisense ODN spent a significantly higher percentage of time in wakefulness during both the light (t = 3.01; df = 5; p < 0.05) and the dark (_t_ = 3.36; df = 5; _p_ < 0.05) period compared with the control (ACSF) day (Fig. 2). Although the mean values of wakefulness during post-ODN day 1 and post-ODN day 2 (dark cycle) were similar, the amount of time the animals spent in wakefulness during post-ODN day 2 compared with the control day did not reach statistical significance (_t_ = 1.98; df = 5; _p_ > 0.05) because of increased variability. Interestingly, the same group of animals spent a significantly reduced amount of time in non-REM sleep during the light period (t = 2.84; df = 5; p < 0.05), whereas during the dark period, non-REM sleep was unaffected.
REM sleep, on the other hand, did not show any significant change during the light period on any day. However, it was significantly reduced during the dark period on ODN day 2 (t = 2.5; df = 5; p < 0.05) and post-ODN day 1 (t = 2.73; df = 5; p < 0.05) compared with control day perfusion. In addition, although the effect on wakefulness and non-REM sleep returned to control values on post-ODN day 2 (during both the light and the dark period), the decrease in REM sleep seen during the dark period on ODN day 2 and post-ODN day 1 persisted during the dark period on post-ODN day 2 (t = 2.5; df = 5; p < 0.05) (Fig. 2). In two animals, we continued the electrographic recordings and observed that the decrease in REM sleep (during the dark period) returned to baseline levels on post-ODN day 3 (data not shown).
Experiment 2: controls
The nonsense ODN-treated (n = 4) and ACSF-treated (n = 6) animals did not show any significant difference in any of the behavioral states or delta power during any hour of the first 5 hr after deprivation (t test; all p values >0.05) (Fig. 3), when the experimental groups showed changes. Thus, for presentation of the results, the nonsense and ACSF control groups were pooled and compared with the antisense-treated group.
Figure 3.
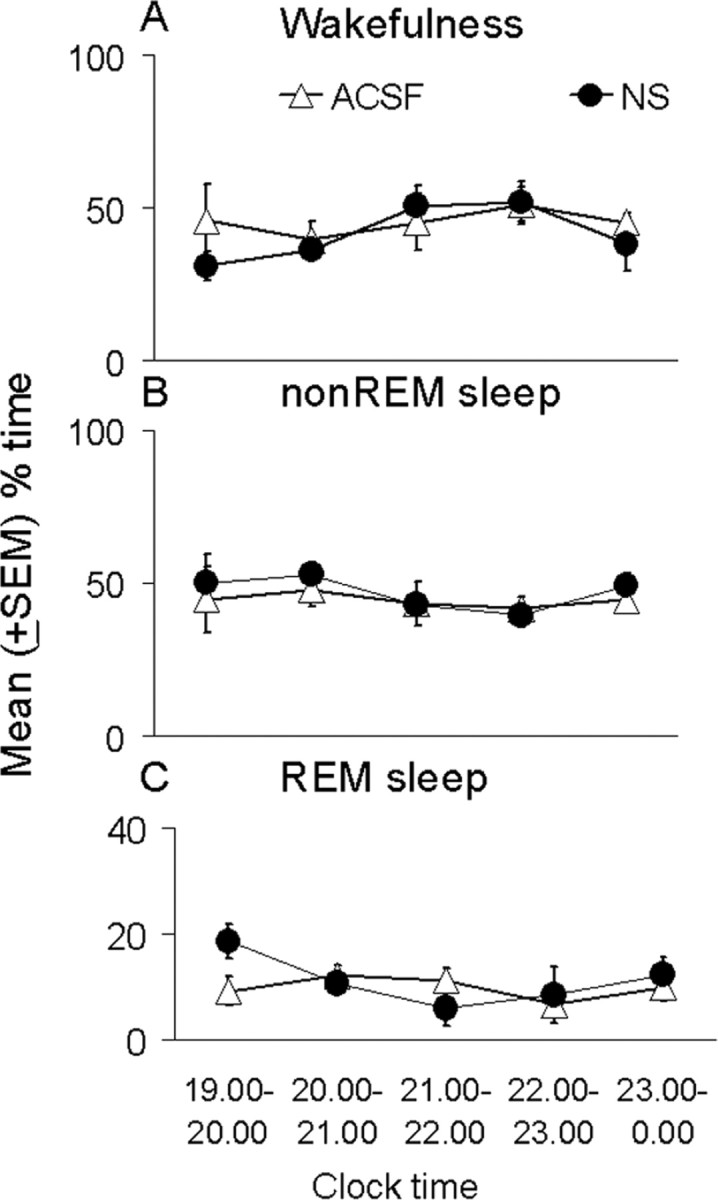
Absence of differences in the behavioral state effects of microdialysis perfusion of nonsense oligonucleotides against the mRNA of the adenosine A1 receptor (NS; solid circles) compared with ACSF perfusion (open triangles) during the first 5 hr of recovery sleep after 6 hr of sleep deprivation. Symbols indicate the mean ± SEM percentage of total time spent in wakefulness (A), non-REM sleep (B), and REM sleep (C). An hour-by-hour t test comparison for the two groups in each state showed no significant differences.
Postdeprivation recovery sleep
This experiment used postdeprivation recovery sleep as a measure of the homeostatic regulation of sleep.
Wakefulness
During the 12 hr of recording after 6 hr of deprivation, the antisense-treated animals spent a significantly greater amount of time in wakefulness than the pooled (control and nonsense) groups (p < 0.01), with the significant differences occurring during the first 5 hr of recovery sleep (Fig. 4_A_). Although the pooled controls and antisense groups did not differ in the first hour (_t_ = 0.08; df = 14; _p_ > 0.05), there was significantly greater wakefulness in the antisense group during the second (t = 3.37; df = 14; p < 0.01), third (t = 3.58; df = 14; p < 0.01), fourth (t = 3.12; df = 14; p < 0.01), and fifth (t = 3.36; df = 14; p < 0.01) hours, with the maximal difference of 77% more wakefulness in the antisense group occurring during the second hour. The antisense-treated animals spent ∼50% more time in wakefulness during the third, fourth, and fifth hours than the pooled controls. The wakefulness values in the antisense-treated animals during the subsequent 7 hr of recovery sleep were comparable with those of pooled controls.
Figure 4.
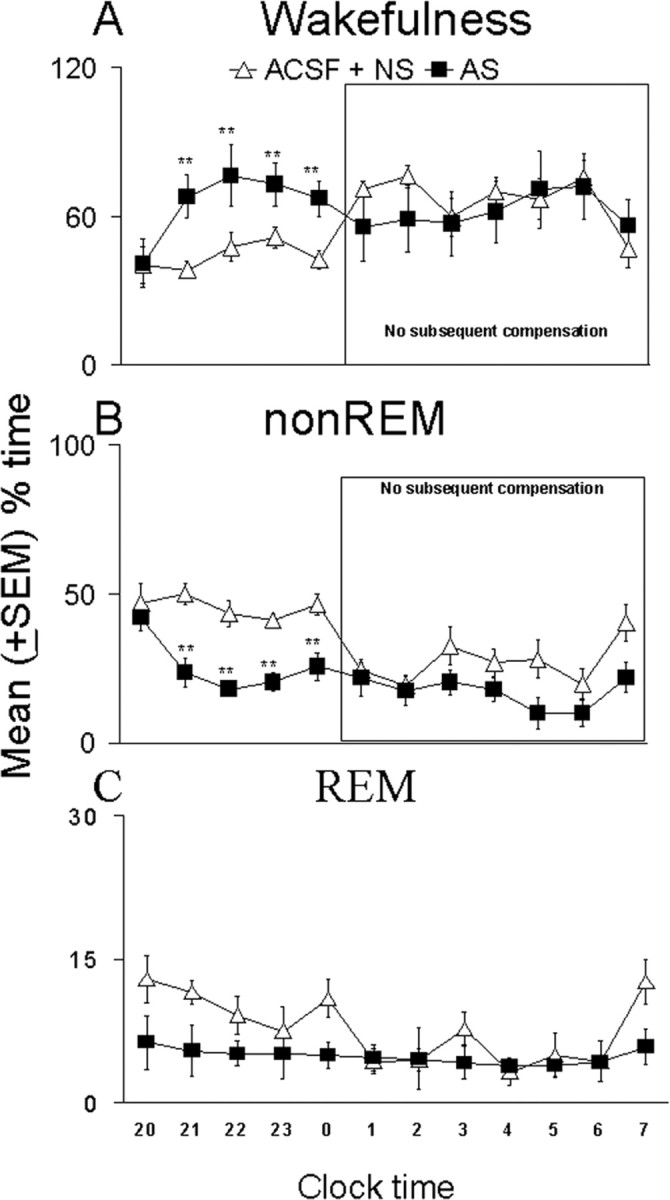
Increased wakefulness (A) and decreased non-REM sleep (B) effects of antisense oligonucleotides against the mRNA of the adenosine A1 receptor during the first 5 hr of the recovery sleep period (left part of each graph) after 6 hr of sleep deprivation compared with control animals (pooled ACSF and nonsense treatment groups, ACSF + NS). There was a significant increase in wakefulness and a decrease in non-REM sleep during the second, third, fourth, and fifth hours. REM sleep (C) did not show significant differences. The right part of the graphs (within box) shows that, for the subsequent 7 hr, there was no compensation for the antisense-induced changes in wakefulness and non-REM. Ordinate is mean ± SEM percentage of time spent in each behavioral state, and abscissa is time of day, with lights off occurring at 7:00 P.M. and lights on occurring at 7:00 A.M. **p < 0.01.
Non-REM sleep
The amount of non-REM sleep in the antisense group was significantly reduced compared with the pooled control groups during the 12 hr of recovery sleep after the 6 hr of sleep deprivation. The time course of differences in the first 5 hr paralleled and were inverse to those of wakefulness (Fig. 4_B_). During the first hour, the groups did not differ in time spent in non-REM sleep (t = 0.5; df = 14; p > 0.05), but there were significantly lower amounts of non-REM sleep in the antisense group during the second (t = 4.06; df = 14; p < 0.01), third (t = 4.08; df = 14; p < 0.01), fourth (t = 4.85; df = 14; p < 0.01), and fifth (t = 3.3; df = 14; p < 0.01) hours. The maximal difference of ∼60% less non-REM sleep in the antisense group was observed during the third hour, whereas 40–50% differences in non-REM sleep were seen during hours 2, 4, and 5. During the last 7 hr of recovery sleep, the amount of time spent in non-REM sleep by the antisense-treated animals was comparable with the values of the pooled controls.
REM sleep
Although the antisense group showed mean values of REM sleep that were smaller than those of the control group, particularly in the first 5 hr after deprivation (Fig. 4_C_), these were not statistically significant. This was true for both the hour-by-hour comparisons and the 12 hr time period after deprivation.
Spectral analysis
Because of technical difficulties, data were available in only five of the six ACSF-treated animals. Delta power was expressed as the percentage of delta power at the corresponding clock time (7:00 P.M. to 12:00 A.M.) during ODN day 2 (the previous day). There were significant decreases in the percentage of delta power values in antisense-treated animals during all 5 hr of recovery sleep compared with the pooled controls (Fig. 5). During the first hour, the antisense animals showed almost a 60% decrease in total delta power (t = 3.61; df = 13; p < 0.01) compared with pooled controls. The maximum decrease in the delta power of ∼75% was observed during the third hour, whereas hours 2, 4, and 5 showed between 60 and 70% reduction in delta power in antisense-treated animals compared with pooled controls (Fig. 5).
Figure 5.

EEG delta power (1–4 Hz, mean ± SEM) after sleep deprivation, expressed as percentage of each animal's delta power during the same time period on a nondeprivation day (previous day). Open triangles indicate control animals (ACSF and nonsense), and solid squares indicate antisense-treated animals. There was a significant decrease in delta activity in antisense-treated animals during all 5 hr of recovery sleep compared with the pooled controls (**p < 0.01). For details, see Results.
Immunohistochemistry and potential neurotoxicity
Immunohistochemical analyses of the effects of ODN perfusion revealed extensive suppression of A1 receptor only in response to antisense perfusion (Fig. 6); nonsense perfusion did not show any effect. There was a substantial loss of A1 receptor immunoreactivity near the site of perfusion, extending ∼1–2 mm around the perfusion site. Fluoro-Jade staining and cresyl violet (Nissl) staining did not indicate neurotoxic damage in the area surrounding the probe tip after the perfusion of antisense (Fig. 7), because Fluoro-Jade fluorescence was present only in a restricted region immediately proximate to the tip.
Figure 6.
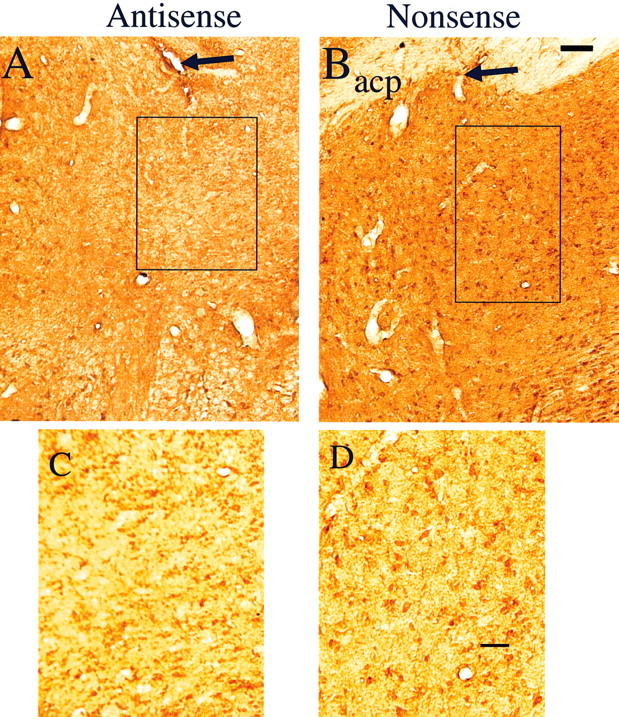
Effect of A1 receptor antisense ODN perfusion (A, C) compared with nonsense ODN perfusion (B, D) on A1 receptor immunoreactivity in the BF. Note the substantial suppression of A1 receptor immunoreactivity in and around the probe lesion site (black arrow) on the antisense-perfused side compared with the nonsense-perfused side (B; arrow, probe lesion site). The rectangular area in A and B is magnified and displayed in C and D. The cell bodies with A1 receptor immunoreactivity are clearly visible (D) in the side that received nonsense ODNs, whereas the side with antisense treatment (C) showed marked suppression of cell body A1 immunoreactivity, leaving fuzzy-appearing background activity. acp, Anterior commissure posterior part (Paxinos and Watson, 1997). Scale bars: A, B, 100 μm; C, D, 50 μm
Figure 7.

Effects of A1 receptor antisense perfusion on the integrity of neurons. A, Fluoro-Jade labeling is constrained to the immediate vicinity of the microdialysis probe track. This restricted location in and just around the track was present regardless of whether ACSF or antisense was perfused. B, Cresyl violet staining of a section 120 μm from the Fluoro-Jade section revealed the presence of intact neurons (arrows) outside the probe track. Scale bar, 20 μm.
Discussion
In this study, we examined the role of adenosine, acting at the A1 receptor, as a homeostatic regulator of sleep-wakefulness in the cholinergic BF. The data revealed the following. (1) Microdialysis perfusion of antisense ODNs against the adenosine A1 receptor in the cholinergic zone of the BF after 6 hr of sleep deprivation reduced both time in recovery non-REM sleep and delta power, enhanced wakefulness, and, during spontaneous bouts of sleep-wakefulness, reduced time in sleep while enhancing wakefulness. (2) In contrast, microdialysis perfusion of nonsense ODNs in the same region had no effect on recovery sleep after 6 hr of sleep deprivation or on spontaneous bouts of sleep-wakefulness. (3) The effects of antisense were not confounded by neurotoxicity, as measured by Fluoro-Jade staining and the continued presence of ChAT staining. These data provide important new evidence implicating adenosine, acting at the A1 receptor, as a key component in the regulation of behavioral states and point to the BF as a key anatomical site.
Use of antisense to suppress A1 receptor expression
These experiments are, to the best of our knowledge, the first to use adenosine A1 receptor antisense in elucidating the role of adenosine in behavioral state control and using antisense ODNs to create a reversible knock-out or knock-down of the A1 receptor. Antisense technology appears to provide a significant advantage over various other techniques, especially in understanding the role of specific receptor subtypes, and, when accompanied by adequate controls, it provides a highly specific and reversible means for inhibition of particular gene expression.
Effect of A1 receptor antisense on A1 receptor immunoreactivity and absence of evidence for neurotoxic effects
Evidence of the predicted action on the targeted protein was evinced by A1 receptor immunohistochemistry showing a marked loss of A1 receptor immunoreactivity in and around the site of antisense perfusion compared with that of nonsense perfusion (Fig. 6). Variability in immunohistochemical staining was minimized by performance of both the nonsense and the antisense perfusions in the same animal on homotopic contralateral BF sites. The higher-magnification photomicrographs show clearly visible A1 receptor-labeled cell bodies on the nonsense side (Fig. 6_D_), whereas mainly background staining is visible on the antisense side (Fig. 6_C_).
Fluoro-Jade staining for neuron degeneration provided no evidence of any antisense-induced neurotoxic damage (Fig. 7), because the only damage observed was immediately adjacent to the probe, was as prominent with application of antisense or nonsense, and was not present in the region in which immunohistochemistry staining indicated loss of the A1 receptor (Figs. 6, 7). Moreover, the continued presence of intact ChAT-labeled neurons in and around the antisense perfusion site also suggested that antisense perfusion did not cause any neurotoxic damage.
Effect of A1 receptor antisense on spontaneous sleep-wakefulness
The primary effect of A1 receptor antisense perfusion in the BF during spontaneous sleep-wakefulness was to increase wakefulness (∼20%) and decrease spontaneous non-REM sleep (∼20%). However, the effect on spontaneous wakefulness–non-REM sleep was not as strong as that seen during postdeprivation sleep, which indicates a primary role of the A1 receptor in homeostatic mediation.
The timing of the maximum effect of adenosine A1 receptor antisense in the present study (post-ODN day 1, which indicates a 24 hr turnover time for the A1 receptor) showed a similar time course range as in the other A1 receptor antisense study (Phan et al., 1999). As an indicator of reversibility, most of the alteration in the behavioral states seen on post-ODN day 1 had returned to baseline values on post-ODN day 2, and all returned to baseline in animals recorded beyond this time point.
Effect of A1 receptor antisense in the BF on recovery sleep after sleep deprivation
We considered recovery sleep after sleep deprivation to be an ideal experimental paradigm to study the homeostatic regulation of sleep-wakefulness because it is a direct measure of sleep homeostasis, and therefore it constituted our primary experiment. Sleep deprivation was performed on the day (post-ODN day 1) when we had found the A1 receptor antisense showed the maximum effect on spontaneous sleep-wakefulness. To build up maximum sleep pressure and allow its optimal measurement, we timed the sleep deprivation to occur from 1:00 P.M. to 7:00 P.M., during the light period when the animals normally slept, and to end coincident with lights off, the time when the animals were normally awake, and thus allow a measure of postdeprivation sleep without the “ceiling effect” that might have occurred had we used sleep deprivation at the start of the normal sleep period (7:00 A.M. to 1:00 P.M.).
Microdialysis perfusion of A1 receptor antisense caused a statistically significant and large (∼50%) reduction in the amount of time the animals spent in non-REM sleep during the second through the fifth hour after deprivation, with a concomitant increase in wakefulness. Importantly, spectral analysis revealed that the antisense-treated animals showed a significant, even larger (60–75%) decrease in percentage of delta power during all 5 hr of recovery sleep.
The increase in delta power during recovery sleep after sleep deprivation is considered to be an indicator of the homeostatic regulation of sleep (Franken et al., 1991). Thus, the decrease in delta power in antisense-treated animals further strongly supports our hypothesis that adenosine and the A1 receptor are the key components in the homeostatic regulation of sleep-wakefulness. Our expression of delta power values during recovery sleep as the percentage of delta power at the corresponding clock time during a nonrecovery day (previous day or ODN day 2) is standard in the literature as a way of overcoming interanimal variation in strength of the recorded delta (Tobler and Borbely, 1990). The decrease in delta power during the first hour of recovery sleep in the antisense-treated animals without a reduction in the time spent in non-REM sleep suggests that delta power is a more sensitive indicator of the homeostatic regulation of sleep than is duration of non-REM sleep. That non-REM duration did not change in the first hour might be attributable to transient adenosine accumulation at other brain sites, especially cortex, which might have exerted a short-term homeostatic effect (Porkka-Heiskanen et al., 2000). The magnitude of the observed change in sleep and delta power provides a conservative estimate of adenosinergic influence on homeostatic control of sleep, because it is unlikely that the microdialysis application of antisense reached all of the cholinergic BF. One of the more noteworthy findings is that, despite the large differences in sleep and wakefulness in the 5 hr after deprivation between the antisense-treated and control animals, the antisense-treated animals did not show any compensatory changes during the subsequent 7 hr after deprivation. The antisense effect thus did not represent a postponement of changes but rather an absence of marked alterations, as if part of the coupling mechanism for signaling homeostatic changes had been disconnected. This, of course, is consistent with our postulated homeostatic signaling role for BF adenosine acting at the A1 receptor.
Although effects of antisense to the A1 receptor on the REM phase of sleep were not statistically significant on an hour-by-hour basis, for the first 5 hr after deprivation, the antisense-perfused animals had consistently less REM sleep than the control animals, and larger numbers might have produced a significant effect. The slightly decreased REM sleep could be a simple reflection of more wakefulness with less opportunity for REM, a result of BF-to-brainstem projections thought to be activated during wakefulness and to be REM suppressive (Strecker et al., 2000), or both.
Finally, recent data indicate that adenosine, acting via the A1 receptor, may have long-term effects, including activation of the nuclear transcription factor-κB and the upregulation of A1 adenosine receptor mRNA (Basheer et al., 2001b), effects that may be mediated via IP3 receptor-regulated calcium release from intracellular stores (Basheer et al., 2002), a release that occurred only in cholinergic neurons.
Implications for the homeostatic regulation of sleep
Sleep occurs as a result of the time of day (circadian influences), being awake for a long time (homeostatic influences), or both. Numerous studies done in our laboratory and by others have suggested that adenosine is a sleep factor (Radulovacki et al., 1984; Thakkar and Mallick, 1996; Strecker et al., 2000). We suggested that, with gradual increases in metabolic demands during wakefulness, there is an increase in ATP breakdown, which causes an increase in the intracellular levels of adenosine. This adenosine is transported out and acts on the A1 receptor to inhibit the discharge activity of the target neurons, especially the wakefulness-maintaining neurons of the BF, and thereby leads to sleep.
What would happen if A1 receptors were removed or inactivated? Our hypothesis suggests that, although there would be an extracellular buildup of adenosine during prolonged wakefulness, adenosine would not be able to inhibit the activity of wakefulness-promoting neurons and thus would not suppress wakefulness and promote non-REM sleep. This is, of course, what was observed after microdialysis of A1 receptor antisense in the cholinergic BF. We conclude that A1 receptors in the BF are necessary to mediate the homeostatic influences of adenosine in controlling sleep-wakefulness.
Footnotes
This work was supported by the Department of Veterans Affairs Medical Research Service (R.W.M.), the National Institute of Mental Health Grants R37MH39683 (R.W.M.) and KO1MH01798 (M.M.T.), and the Sleep Medicine Education and Research Foundation (M.M.T.).We thank Dr. Robert Strecker for his helpful intellectual input, John Franco of the Veterans Affairs Medical Center Animal Research Facility for providing care to the animals, and Kristen Winston for helping with data analysis.
Correspondence should be addressed to either Dr. Mahesh M. Thakkar or Dr. Robert W. McCarley, Laboratory of Neuroscience, Department of Psychiatry, Harvard Medical School, Veterans Affairs Medical Center, 940 Belmont Street, Brockton, MA 02301. E-mail: mthakkar@hms.harvard.edu or robert_mccarley@hms.harvard.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/234278-10$15.00/0
References
- Alam MN, Szymusiak R, Gong H, King J, McGinty D ( 1999) Adenosinergic modulation of rat basal forebrain neurons during sleep and waking: neuronal recording with microdialysis. J Physiol (Lond) 521: 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basheer R, Rainnie DG, Porkka-Heiskanen T, Ramesh V, McCarley RW ( 2001a) Adenosine, prolonged wakefulness, and A1-activated NF-kB DNA binding in the basal forebrain of the rat. Neuroscience 104: 731–739. [DOI] [PubMed] [Google Scholar]
- Basheer R, Halldner L, Alanko L, McCarley RW, Fredholm BB, Porkka-Heiskanen T ( 2001b) Opposite changes in adenosine A1 and A2A receptor mRNA in the rat following sleep deprivation. NeuroReport 12: 1577–1580. [DOI] [PubMed] [Google Scholar]
- Basheer R, Arrigoni E, Thatte HS, Greene RW, Ambudkar IS, McCarley RW ( 2002) Adenosine induces IP3 receptor-mediated mobilization of intracellular calcium stores. J Neurosci 22: 7680–7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggs TA, Myers RD ( 1997) Adenosine A1 receptor antisense infused in striatum of rats: actions on alcohol-induced locomotor impairment, blood alcohol, and body temperature. Alcohol 14: 617–621. [DOI] [PubMed] [Google Scholar]
- Chiasson BJ, Armstrong JN, Hooper ML, Murphy PR, Robertson HA ( 1994) The application of antisense oligonucleotide technology to the brain: some pitfalls. Cell Mol Neurobiol 14: 507–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton DA, Mesches MH, Alvarez E, Bickford PC, Browning MD ( 2002) A hippocampal NR2B deficit can mimic age-related changes in long-term potentiation and spatial learning in the Fischer 344 rat. J Neurosci 22: 3628–3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisch AJ, Schmued LC, Marshall JF ( 1998) Characterizing cortical neuron injury with Fluoro-Jade labeling after a neurotoxic regimen of methamphetamine. Synapse 30: 329–333. [DOI] [PubMed] [Google Scholar]
- Franken P, Dijk DJ, Tobler I, Borbely AA ( 1991) Sleep deprivation in rats: effects on EEG power spectra, vigilance states, and cortical temperature. Am J Physiol 261: R198–R208. [DOI] [PubMed] [Google Scholar]
- Frantseva MV, Kokarovtseva L, Naus CG, Carlen PL, MacFabe D, Perez Velazquez JL ( 2002) Specific gap junctions enhance the neuronal vulnerability to brain traumatic injury. J Neurosci 22: 644–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgieva J, Heilig M, Nylander I, Herrera-Marschitz M, Terenius L ( 1995) In vivo antisense inhibition of prodynorphin expression in rat striatum: dose-dependence and sequence specificity. Neurosci Lett 192: 69–71. [Google Scholar]
- Grimpe B, Dong S, Doller C, Temple K, Malouf AT, Silver J ( 2002) The critical role of basement membrane-independent laminin gamma 1 chain during axon regeneration in the CNS. J Neurosci 22: 3144–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helene C, Toulme JJ ( 1990) Specific regulation of gene expression by antisense, sense and antigene nucleic acids. Biochim Biophys Acta 1049: 99–125. [DOI] [PubMed] [Google Scholar]
- Hopkins KJ, Wang G, Schmued LC ( 2000) Temporal progression of kainic acid induced neuronal and myelin degeneration in the rat forebrain. Brain Res 864: 69–80. [DOI] [PubMed] [Google Scholar]
- Jones BE ( 1998) The neural basis of consciousness across the sleep-waking cycle. In: Consciousness: at the frontiers of neuroscience, advances in neurology (Jasper HH, Descarries L, Castellucci VF, Rossignol S, eds), pp 75–94. Philadelphia: Lippincott-Raven. [PubMed]
- Kalra PS, Kalra SP ( 2000) Use of antisense oligodeoxynucleotides to study the physiological functions of neuropeptide Y. Methods 22: 249–254. [DOI] [PubMed] [Google Scholar]
- Krieg AM, Tonkinson J, Matson S, Zhao Q, Saxon M, Zhang LM, Bhanja U, Yakubov L, Stein CA ( 1993) Modification of antisense phosphodiester oligodeoxynucleotides by a 5' cholesteryl moiety increases cellular association and improves efficacy. Proc Natl Acad Sci USA 90: 1048–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loke SL, Stein CA, Zhang XH, Mori K, Nakanishi M, Subasinghe C, Cohen JS, Neckers LM ( 1989) Characterization of oligonucleotide transport into living cells. Proc Natl Acad Sci USA 86: 3474–3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neckers L, Whitesell L ( 1993) Antisense technology: biological utility and practical considerations. Am J Physiol 265: L1–L12. [DOI] [PubMed] [Google Scholar]
- Nicot A, Pfaff DW ( 1997) Antisense oligodeoxynucleotides as specific tools for studying neuroendocrine and behavioral functions: some prospects and problems. J Neurosci Methods 71: 45–53. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C ( 1997) The rat brain in stereotaxic coordinates. Boston: Academic. [DOI] [PubMed]
- Phan TA, Gray AM, Nyce JW ( 1997) Intrastriatal adenosine A1 receptor antisense oligodeoxynucleotide blocks ethanol-induced motor incoordination. Eur J Pharmacol 323: R5–R7. [DOI] [PubMed] [Google Scholar]
- Phan TA, Gray AM, Nyce JW, McGinty JF ( 1999) Autoradiographic evidence that intrastriatal administration of adenosine A1 receptor antisense oligodeoxynucleotide decreases adenosine A1 receptors in the rat striatum and cortex. Brain Res Mol Brain Res 72: 226–230. [DOI] [PubMed] [Google Scholar]
- Porkka-Heiskanen T, Strecker RE, Thakkar M, Bjorkum AA, Greene RW, McCarley RW ( 1997) Adenosine: a mediator of the sleep-inducing effects of prolonged wakefulness. Science 276: 1265–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porkka-Heiskanen T, Strecker RE, McCarley RW ( 2000) Brain site-specificity of extracellular adenosine concentration changes during sleep deprivation and spontaneous sleep: an in vivo microdialysis study. Neuroscience 99: 507–517. [DOI] [PubMed] [Google Scholar]
- Portas CM, Thakkar M, Rainnie DG, Greene RW, McCarley RW ( 1997) Role of adenosine in behavioral state modulation: a microdialysis study in the freely moving cat. Neuroscience 79: 225–235. [DOI] [PubMed] [Google Scholar]
- Quan N, Blatteis CM ( 1989) Microdialysis: a system for localized drug delivery into the brain. Brain Res Bull 22: 621–625. [DOI] [PubMed] [Google Scholar]
- Radulovacki M, Virus RM, Djuricic-Nedelson M, Green RD ( 1984) Adenosine analogs and sleep in rats. J Pharmacol Exp Ther 228: 268–274. [PubMed] [Google Scholar]
- Rainnie DG, Grunze HC, McCarley RW, Greene RW ( 1994) Adenosine inhibition of mesopontine cholinergic neurons: implications for EEG arousal. Science 263: 689–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savaskan NE, Eyupoglu IY, Brauer AU, Plaschke M, Ninnemann O, Nitsch R, Skutella T ( 2000) Entorhinal cortex lesion studied with the novel dye fluoro-jade. Brain Res 864: 44–51. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Albertson C, Slikker Jr W ( 1997) Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neurnal degeneration. Brain Res 751: 37–46. [DOI] [PubMed] [Google Scholar]
- Stein CA ( 1999) Two problems in antisense biotechnology: in vitro delivery and the design of antisense experiments. Biochim Biophys Acta 1489: 45–52. [DOI] [PubMed] [Google Scholar]
- Stein CA ( 2001) The experimental use of antisense oligonucleotides: a guide for the perplexed. J Clin Invest 108: 641–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein CA, Cohen JS ( 1988) Oligodeoxynucleotides as inhibitors of gene expression: a review. Cancer Res 48: 2659–2668. [PubMed] [Google Scholar]
- Stein CA, Tonkinson JL, Zhang LM, Yakubov L, Gervasoni J, Taub R, Rotenberg SA ( 1993) Dynamics of the internalization of phosphodiester oligodeoxynucleotides in HL60 cells. Biochemistry 32: 4855–4861. [DOI] [PubMed] [Google Scholar]
- Strecker RE, Morairty S, Thakkar MM, Porkka-Heiskanen T, Basheer R, Dauphin LJ, Rainnie DG, Portas CM, Greene RW, McCarley RW ( 2000) Adenosinergic modulation of basal forebrain and preoptic/anterior hypothalamic neuronal activity in the control of behavioral state. Behav Brain Res 115: 183–204. [DOI] [PubMed] [Google Scholar]
- Szymusiak R ( 1995) Magnocellular nuclei of the basal forebrain: substrates of sleep and arousal regulation. Sleep 18: 478–500. [DOI] [PubMed] [Google Scholar]
- Thakkar M, Mallick BN ( 1996) Effect of rapid eye movement sleep deprivation on 5' nucleotidase activity in the rat brain. Neurosci Lett 206: 177–180. [DOI] [PubMed] [Google Scholar]
- Thakkar MM, Strecker RE, McCarley RW ( 1998) Behavioral state control through differential serotonergic inhibition in the mesopontine cholinergic nuclei: a simultaneous unit recording and microdialysis study. J Neurosci 18: 5490–5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakkar MM, Delgiacco RA, Strecker RE, McCarley RW ( 1999a) Adenosinergic A1 inhibition of basal forebrain wake-active neurons: a combined unit recording and microdialysis study in freely behaving cats. Sleep 22: 9. [DOI] [PubMed] [Google Scholar]
- Thakkar MM, Ramesh V, Cape EG, Winston S, Strecker RE, McCarley RW ( 1999b) REM sleep enhancement and behavioral cataplexy following orexin (hypocretin)-II receptor antisense perfusion in the pontine reticular formation. Sleep Res Online 2: 112–120. [PubMed] [Google Scholar]
- Thakkar MM, Ramesh V, Strecker RE, McCarley RW ( 2001) Microdialysis perfusion of orexin-A in the basal forebrain increases wakefulness in freely behaving rats. Arch Ital Biol 139: 313–328. [PubMed] [Google Scholar]
- Thakkar MM, Winston S, McCarley RW ( 2002a) Orexin neurons of the hypothalamus express adenosine A1 receptors. Brain Res 944: 190–194. [DOI] [PubMed] [Google Scholar]
- Thakkar MM, Strecker RE, McCarley RW ( 2002b) Phasic but not tonic REM-selective discharge of periaqueductal gray neurons in freely behaving animals: relevance to postulates of GABAergic inhibition of monoaminergic neurons. Brain Res 945: 276–280. [DOI] [PubMed] [Google Scholar]
- Tobler I, Borbely AA ( 1990) The effect of 3-h and 6-h sleep deprivation on sleep and EEG spectra of the rat. Behav Brain Res 36: 73–78. [DOI] [PubMed] [Google Scholar]
- Torner L, Toschi N, Nava G, Clapp C, Neumann ID ( 2002) Increased hypothalamic expression of prolactin in lactation: involvement in behavioural and neuroendocrine stress responses. Eur J Neurosci 15: 1381–1389. [DOI] [PubMed] [Google Scholar]
- Turchi J, Sarter M ( 2001) Antisense oligodeoxynucleotide-induced sup pression of basal forebrain NMDA-NR1 subunits selectively impairs visual attentional performance in rats. Eur J Neurosci 14: 103–117. [DOI] [PubMed] [Google Scholar]
- Wahlestedt C ( 1994) Antisense oligonucleotide strategies in neuropharmacology. Trends Pharmacol Sci 15: 42–46. [DOI] [PubMed] [Google Scholar]
- Walder RY, Walder JA ( 1988) Role of RNase H in hybrid-arrested translation by antisense oligonucleotides. Proc Natl Acad Sci USA 85: 5011–5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakubov LA, Deeva EA, Zarytova VF, Ivanova EM, Ryte AS, Yurchenko LV, Vlassov VV ( 1989) Mechanism of oligonucleotide uptake by cells: involvement of specific receptors? Proc Natl Acad Sci USA 86: 6454–6458. [DOI] [PMC free article] [PubMed] [Google Scholar]