A Genetic Screen for top3 Suppressors in Saccharomyces cerevisiae Identifies SHU1, SHU2, PSY3 and CSM2: Four Genes Involved in Error-Free DNA Repair (original) (raw)
Abstract
Helicases of the RecQ family and topoisomerase III are evolutionarily conserved proteins important for maintenance of genome stability. In Saccharomyces cerevisiae, loss of the TOP3 gene, encoding topoisomerase III, results in a phenotype of slow growth, DNA damage sensitivity, meiotic defects, and hyperrecombination. The sole RecQ helicase in budding yeast, Sgs1, interacts with Top3 both physically and genetically, and the two proteins are thought to act in concert in vivo. Much recent genetic and biochemical evidence points to the role of RecQ helicases and topoisomerase III in regulating homologous recombination (HR) during DNA replication. Previously, we found that mutations in HR genes partially suppress top3 slow growth. Here, we describe the analysis of four additional mutational suppressors of top3 defects: shu1, shu2, psy3, and csm2. These genes belong to one epistasis group and their protein products interact with each other, strongly suggesting that they function as a complex in vivo. Their mutant phenotype indicates that they are important for error-free repair of spontaneous and induced DNA lesions, protecting the genome from mutation. These mutants exhibit an epistatic relationship with rad52 and show altered dynamics of Rad52-YFP foci, suggesting a role for these proteins in recombinational repair.
ALL living cells possess mechanisms that ensure faithful reproduction of genetic material and protection of the genome from excessive mutation. Mutagenic DNA lesions can either arise spontaneously, for instance, during the process of DNA replication, or be induced by exogenous agents, such as chemicals or irradiation. One of the pathways used to repair both spontaneous and induced DNA double-strand breaks (DSBs) or single-strand gaps is homologous recombination (HR), a process of exchange of genetic material between two DNA molecules of nearly or perfectly identical sequence (Symington 2002). Since it uses a homologous sequence as template for DNA repair, normal HR is error free, thus playing an important role in protection of the genome from mutation. However, recombination has to be regulated, since excessive or inappropriate recombination can also contribute to genomic instability. A growing amount of evidence suggests that helicases of the RecQ family in conjunction with type III topoisomerases play crucial roles in regulating HR (Oakley and Hickson 2002; Khakhar et al. 2003).
Topoisomerase III was first identified in budding yeast as a mutation that causes a hyperrecombination phenotype at the _SUP4_-o locus (Wallis et al. 1989). In addition to causing increased recombination, mutation of TOP3 results in slow growth, hypersensitivity to genotoxic agents, such as hydroxyurea (HU) and methyl methanesulfonate (MMS), mitotic chromosome missegregation, and inability to undergo meiosis and sporulate (Wallis et al. 1989; Gangloff et al. 1999; Chakraverty et al. 2001). Mutation of SGS1, the gene encoding the sole budding yeast RecQ helicase, suppresses _top3_Δ slow growth and the two proteins physically interact with each other (Gangloff et al. 1994; Bennett et al. 2000; Fricke et al. 2001). Mutation of SGS1 in an otherwise wild-type background causes a phenotype reminiscent of that of the _top3_Δ strain, although milder in severity (Gangloff et al. 1994; Watt et al. 1995, 1996).
RecQ helicases and topoisomerase III are evolutionarily conserved proteins and play important roles in maintenance of genome stability in higher eukaryotes. For instance, mutations in three of five human RecQ homologs, BLM, WRN, and RECQL4, cause the cancer predisposition syndromes Bloom, Werner, and Rothmund-Thomson (reviewed in Hickson 2003). Werner syndrome and Rothmund-Thomson syndrome patients also exhibit symptoms of premature aging. At the cellular level, all of these syndromes are characterized by genomic instability that is, at least in part, caused by inappropriate recombination events. The functional interaction between RecQ-like helicases and topoisomerase III homologs also appears to be conserved in human cells. BLM physically interacts with an isoform of topoisomerase III, Top3α, and this interaction is necessary for BLM function (Wu et al. 2000). Mutation of topoisomerase III is not known to be associated with a human genetic disorder. However, deletion of _TOP3_α in the mouse results in embryonic lethality, while deletion of _TOP3_β produces mice with a shortened life span (Li and Wang 1998; Kwan and Wang 2001).
As described above, mutation of human or yeast RecQ helicases causes a genomic instability phenotype characterized by increased or inappropriate recombination events. Other evidence, both genetic and biochemical, indicates that RecQ family proteins have important roles in regulating HR. In Saccharomyces cerevisiae, _sgs1_Δ exhibits extreme synthetic sickness or lethality with mutation of the gene encoding another DNA helicase, Srs2 (Gangloff et al. 2000; McVey et al. 2001). Mutation of SGS1 also exhibits synthetic lethality with mutation of either of the two genes encoding the Mms4-Mus81 endonuclease (Kaliraman et al. 2001; Mullen et al. 2001). These synthetic defects are fully suppressed by deletion of HR genes RAD51, RAD52, RAD55, or RAD57 (Gangloff et al. 2000; Fabre et al. 2002). Additionally, mutation of HR genes partially suppresses the slow growth and DNA damage sensitivity of the _top3_Δ strain (Shor et al. 2002). Diploids lacking both copies of the TOP3 gene have a severe meiotic defect that can be bypassed by deleting SPO11, the gene responsible for initiating meiotic recombination (Gangloff et al. 1999). In fission yeast, several defects of the mutant lacking the RecQ homolog Rqh1 can be partially rescued by expression of bacterial Holliday junction (HJ) resolvase, RusA (Doe et al. 2000). These genetic data indicate that in the absence of the Sgs1-Top3 complex and other proteins that function in parallel pathways, HR plays a detrimental role, causing genomic instability and slow growth or lethality.
Biochemical analyses of purified proteins in vitro also suggest a role for RecQ helicases and associated factors in regulating HR. In addition to exhibiting “classic” helicase activity, that of unwinding dsDNA substrates, several RecQ homologs can unwind DNA structures that resemble HR intermediates, such as HJs and D-loops (Bennett et al. 1998, 1999; Constantinou et al. 2000; Karow et al. 2000; van Brabant et al. 2000). Topoisomerase III is a type I topoisomerase, acting on DNA that is negatively supercoiled and/or contains single-strand regions (Harmon et al. 1999; Champoux 2001). In several studies, the concerted action of RecQ helicase and topoisomerase III in vitro produces a novel activity not associated with either protein alone. For instance, Escherichia coli RecQ and topoisomerase III can catenate and decatenate covalently closed dsDNA circles (Harmon et al. 1999). Also, the BLM protein, in concert with human topoisomerase IIIα, can dissolve a double-HJ-containing DNA substrate to yield exclusively noncrossover products (Wu and Hickson 2003). As mentioned earlier, the SRS2 gene encodes another DNA helicase that exhibits a genetic interaction with sgs1 and is thus thought to act in a pathway parallel to Sgs1-Top3. Recently, two different laboratories showed that, in vitro, Srs2 can dislodge the yeast RecA homolog, Rad51, from ssDNA and prevent Rad51-mediated recombination reactions (Krejci et al. 2003; Veaute et al. 2003).
Regulation of HR is especially important during the process of DNA replication. Impediments to replication fork movement, such as DNA lesions, can result in fork pausing or arrest. Stalled forks contain regions of ssDNA, which can be used as substrate for Rad51 binding and initiation of HR (Sogo et al. 2002; Lucca et al. 2004). While HR is important for resumption of replication in certain contexts, for instance, when the fork collapses and forms a DSB, in the instance of replication arrest recombination can destabilize the fork and create untimely genomic rearrangement (Cox 2001; Michel et al. 2001; Lucca et al. 2004). In fact, analyses of yeast cells treated with HU, a chemical that inhibits replication progression, suggest that HR proteins are excluded from arrested replication forks (Lisby et al. 2004). Several lines of evidence suggest that RecQ helicases and associated factors provide a mechanism to regulate recombination during the process of DNA replication. In yeast and in human cells, RecQ homologs were shown to colocalize with sites of DNA synthesis and with other proteins involved in DNA replication (Brosh et al. 2000; Constantinou et al. 2000; Bischof et al. 2001). Also, both yeast and human cells carrying mutations in RecQ homologs exhibit various S-phase-specific defects. For instance, fission yeast rqh1 cells fail to recover normally from replication arrest and subsequently exhibit increased recombination and defects in chromosome segregation (Stewart et al. 1997). In S. cerevisiae, both Sgs1 protein levels and Top3 mRNA levels were shown to be cell cycle regulated, peaking during S phase (Frei and Gasser 2000; Chakraverty et al. 2001). Additionally, Sgs1 has a role in the intra-S checkpoint and is important for the stable association of DNA polymerases with replication forks during replication arrest (Frei and Gasser 2000; Cobb et al. 2003).
In a previous article, we described a genetic screen to isolate non-sgs1 suppressors of _top3_Δ slow growth (Shor et al. 2002). We reported that mutation of the HR genes RAD51, RAD52, RAD54, RAD55, or RAD57 partially suppresses _top3_Δ slow growth and DNA damage sensitivity. In addition to HR mutants, we isolated several other mutational suppressors of _top3_Δ defects. In this article, we describe our analyses of four such suppressors: shu1, shu2, psy3, and csm2. Through the efforts of several research groups performing mutant hunts using the S. cerevisiae deletion library, some of these mutants have been previously identified as causing a phenotype of MMS sensitivity, platinum sensitivity, increased forward mutation rates and gross chromosomal rearrangements, and meiotic chromosome segregation defects (Rabitsch et al. 2001; Hanway et al. 2002; Huang et al. 2003). In genome-wide two-hybrid screens, several interactions were observed among these proteins (Ito et al. 2001). In this article, we expand on some of these data and also analyze the genetic interactions of these genes with each other, with SGS1-TOP3, and with HR. We also analyze the impact of these mutations on recombination rates at several loci and on Rad52 focus dynamics. Our data show that these four proteins function as part of a single pathway, possibly a complex, in vivo. Genetic analyses place them downstream of RAD52 in MMS resistance and mutation avoidance. Analyses of Rad52 foci in the shu1 mutant indicate that recombinational repair is affected by mutation of genes in this group, especially after high doses of DNA damage. Overall, our data indicate that Shu1, Shu2, Psy3, and Csm2 promote efficient recombinational repair and function in the protection of the genome from spontaneous and induced DNA damage.
MATERIALS AND METHODS
Media and strains:
Yeast extract/peptone/dextrose medium (YPD) and synthetic complete (SC) media lacking specific amino acids or containing 5-fluoroorotic acid (5-FOA), geneticin, canavanine, or 3-aminotriazole (3-AT) were prepared as described (Rose et al. 1990) with twice the amount of leucine (60 μg/ml) in all SC-based media. 5-FOA was purchased from American Bioanalytical and the other chemicals from Sigma (St. Louis). Standard procedures were used for mating, sporulation, and dissection (Sherman et al. 1986). Yeast strains used in this study are listed in Table 1. In most experiments, multiple meiotic segregants of the same genotype were analyzed. When necessary, the figure legends state which diploid strains from Table 1 produced the haploid strains shown in the figure.
TABLE 1.
Strains
Replacements of the SHU1 ORF by the HIS3 gene and of the SHU2 ORF by the Kluyveromyces lactis URA3 gene were done using a PCR-based allele replacement method (Erdeniz et al. 1997). psy3::kanMX4, csm2::kanMX4, and rev3::kanMX4 constructs were amplified by PCR from the S. cerevisiae genome deletion library (Research Genetics, Huntsville, AL) and transformed into the W303 background (Thomas and Rothstein 1989). Transformants were selected on YPD medium containing 200 μg/ml geneticin and the gene deletions were verified by PCR.
Fusions of yellow fluorescent protein (YFP) at the Shu C termini were done using the PCR-based cloning-free chromosomal integration strategy as described (Lisby et al. 2001; Reid et al. 2002). The second YFP gene was subsequently integrated at the C terminus of Shu1-YFP using the same method. Shu1 and the two YFPs are each separated by a four-alanine linker.
Plasmids:
Construction of pWJ1323 expressing Nup49 fused to cyan fluorescent protein (Nup49-CFP) was described in M. Wagner, G. Price and R. Rothstein (unpublished results). Construction of the two-hybrid plasmids expressing Shu proteins fused to the Gal4 activation or binding domain was described as part of the genome-wide two-hybrid study (Ito et al. 2001).
Growth rate determination:
The OD600 of cultures in exponential growth phase in YPD was determined every hour for 5 hr. Three isolates were analyzed for every strain.
Cloning SHU1 and CSM2 by complementation of MMS sensitivity:
We transformed shu mutants with a genomic DNA library (Akada et al. 1997) and selected transformants on synthetic −Leu medium. The transformants were then replica plated onto −Leu medium containing 0.03% MMS. Library plasmids were isolated from colonies able to grow on MMS-containing medium. The plasmid inserts were sequenced using the primer CTTCTTTGCGTCCATCC. To confirm that the complementing insert indeed contains the SHU gene of interest, we deleted the corresponding ORF in the genome and showed that the resulting deletion has the same phenotype and fails to complement the shu mutant. We also sequenced the mutational alteration in each shu mutant.
Two-hybrid assays:
Strain PJ69-4A MATa was transformed with the Gal4 activation domain (GAD) plasmids and PJ69-4A _MAT_α with the Gal4-binding domain (GBD) plasmids containing the SHU ORFs (Ito et al. 2001). Each MATa transformant was crossed to each _MAT_α transformant and the diploids were selected on SC leucine- and tryptophan-dropout medium (−Leu −Trp; Bendixen et al. 1994). To use HIS3 as the two-hybrid reporter, each diploid was replica plated from −Leu −Trp onto −Leu −Trp −His and onto −Leu −Trp −His + 3 mm 3-AT. To use the lacZ gene as the reporter, the diploids were grown in liquid −Leu −Trp medium, the cells were permeabilized with chloroform and incubated with _o-_nitrophenyl-β-d-galactoside, and units of β-galactosidase activity were calculated as described (Ausubel et al. 1998).
Sensitivity to DNA-damaging agents:
To measure sensitivity to MMS, the appropriate numbers of cells were plated onto YPD medium containing various concentrations of MMS as described in Shor et al. (2002) and viable cell counts were calculated.
_SUP4-_o recombination assay:
The basis of the SUP4-o recombination assay has been described previously (Rothstein et al. 1987; Shor et al. 2002). Strains containing the SUP4-o::URA3 construct were grown overnight in YPD and plated onto SC arginine- and uracil-dropout plates containing 1 mg/ml 5-FOA, 60 μg/ml canavanine, and 10 μg/ml adenine. Three days later, red 5-FOA- and canavanine-resistant colonies were counted and SUP4 recombination rates were calculated on the basis of the method of the median (Lea and Coulson 1949). From 9 to 11 cultures were tested for each strain. SUP4 deletion class identification was performed for independently generated recombinants by the PCR-_Xho_I digestion approach as described (Shor et al. 2002).
Measuring the CAN1 forward mutation rate:
CAN1 strains were grown overnight in YPD and plated onto SC arginine-dropout medium containing 60 μg/ml canavanine. After 3 days, canavanine-resistant colonies were counted and mutation rates calculated on the basis of the method of the median (Lea and Coulson 1949). From five to nine cultures were tested for each strain. To measure the MMS-induced rate of mutagenesis, colonies were inoculated into YPD and pregrown for 8 hr. MMS was then added to a final concentration of 0.01% and the cultures were grown overnight. The rest of the assay was done as described above for the measurements of spontaneous mutation rates.
Microscopy:
Strains of the desired genotype carrying the RAD52-YFP fusion and the wild-type ADE2 allele were grown at 23° in SC medium supplemented with 50 μg/ml adenine and harvested for microscopy during midlog phase. The strain expressing Shu1-YFP-YFP and Nup49-CFP contained the ade2-1 allele but was otherwise treated the same as above. In the case of monitoring Rad52-YFP foci after DNA damage, MMS or HU were added to indicated concentrations and the cultures were further incubated at 23° for the indicated times prior to harvesting. Fluorescent images were captured with a Zeiss Axioplan equipped with a Hamamatsu Orca camera (Hamamatsu Photonics) and processed with Openlab 3.0.4 (Improvision) and Adobe Photoshop. Exposure times were 1000 msec for Rad52-YFP, 3000 msec for Shu-YFP-YFP, and 1000 msec for Nup49-CFP. For the time-lapse experiments, MMS was added to the culture to the final concentration of 0.01% immediately before mounting cells on the microscope slide. Images were captured every 5 min for the duration of 5 hr. For each field of cells, 11 fluorescent images were obtained at 0.4-μm intervals along the _z_-axis. Only cells that had exhibited growth and/or budding during the span of the time lapse were taken into account.
RESULTS
Identification of shu1, shu2, psy3, and csm2 as suppressors of _top3_Δ slow growth:
The screen for non-sgs1 suppressors of _top3_Δ slow growth was performed as previously described (Shor et al. 2002). All suppressor mutations not linked to SGS1 were backcrossed into a TOP3 background, tested for sensitivities to several genotoxic agents, and checked for complementation with other non-_sgs1 top3_Δ suppressors, such as HR mutants. A number of the new _top3_Δ suppressor mutations had a similar phenotype and fell into four complementation groups. In addition to suppressing the slow growth of the _top3_Δ mutant, these mutants partially suppress _top3_Δ sensitivity to HU and MMS (Figure 1B), the HU sensitivity of the _sgs1_Δ strain (Figure 1C), and the lethality/extreme sickness of the _sgs1_Δ _srs2_Δ and _sgs1_Δ _mus81_Δ double mutants (data not shown). The new mutants were named shu (_s_uppresses sgs1 HU sensitivity). In an otherwise wild-type background, the shu mutations resulted in moderate sensitivity to MMS (Figure 1D). This observation allowed us to clone two of the genes by complementation of the MMS sensitivity with a genomic DNA library (for details see materials and methods). In this manner, we identified yhl006c and csm2 as top3 slow-growth suppressors. YHL006C was named SHU1. CSM2 was previously identified and named as part of a genome-wide screen for mutations that affect _c_hromosome _s_egregation in _m_eiosis (csm; Rabitsch et al. 2001).
Figure 1.—

Genetic interactions among sgs1, top3, and the shu mutants. All the mutations shown are deletions of the respective genes. The haploid strains are meiotic segregants of diploids ESD50, ESD51, W4105, and W4173. For the DNA damage sensitivity assays in B–D, 10-fold serial dilutions of overnight cultures were spotted onto YPD medium containing the indicated drugs. (A) Deletion of SHU1, SHU2, PSY3, or CSM2 suppresses _top3_Δ slow growth. Doubling times in rich medium are shown. (B) Deletion of SHU1, SHU2, PSY3, or CSM2 suppresses _top3_Δ sensitivity to HU and MMS. (C) Deletion of SHU1, SHU2, PSY3, or CSM2 suppresses _sgs1_Δ HU sensitivity. (D) SHU1, SHU2, PSY3, and CSM2 are in one epistasis group for MMS resistance. The quadruple deletion mutant is no more MMS sensitive than any of the single mutants.
Genome-wide two-hybrid screens of S. cerevisiae gene products suggest that Shu1p and Csm2p are connected by a series of protein-protein interactions also involving the gene products of YDR078C and PSY3 (Ito et al. 2001). PSY3 was identified and named as part of a genome-wide search for mutants that confer _p_latinum s_ensitivit_y (psy; Wu et al. 2004). This network of two-hybrid interactions suggests that the proteins also interact functionally in vivo. Thus, we created chromosomal deletions of each of the four ORFs and tested their genetic interactions with _top3_Δ and _sgs1_Δ mutations. We find that each deletion mutant suppresses _top3_Δ slow growth, DNA damage sensitivity, and _sgs1_Δ HU sensitivity (Figure 1, A–C). Moreover, complementation analyses indicated and DNA sequencing confirmed that shu2 and shu3 mutants isolated in the screen were in fact alleles of YDR078C and PSY3. Thus, we gave YDR078C the name SHU2. Overall, we identified six alleles of shu1, one of shu2, one of psy3, and five of csm2 during the screen.
Shu1, Shu2, Psy3, and Csm2 are all relatively small proteins (under 250 aa). We attempted to glean information about their functions by searching for possible homologs in other organisms. However, BLAST searches identified no orthologs of any of the four proteins in bacteria, fission yeast, or higher eukaryotes. We did identify orthologs of all four proteins in sensu stricto and sensu lato Saccharomyces species, as well as those of Shu2 and Psy3 in several species of the genus Kluyveromyces. Since these genomes have not been completely sequenced, it is possible that they also contain orthologs of Shu1 and Csm2. A BLAST search against the fully sequenced Candida albicans genome revealed a possible ortholog of Csm2, with 21% identity and 35% similarity. Additionally, we have used the sequence analysis tools available at the Saccharomyces Genome Database (http://genome-www.stanford.edu/Saccharomyces) to identify possible motifs and domains within the Shu proteins. However, no significant matches were found.
Characterization of the physical and genetic interactions among Shu1, Shu2, Psy3, and Csm2:
To characterize the two-hybrid interactions among Shu1, Shu2, Psy3, and Csm2 in more detail, we obtained the relevant two-hybrid plasmids (Ito et al. 2001) and measured the two-hybrid interactions using HIS3 and β-galactosidase as reporters. The results of the two-hybrid assays are summarized in Table 2. We also sequenced the relevant ORFs on the plasmids and tested them for complementation of the MMS sensitivity of the corresponding deletion mutant. DNA sequencing revealed a −1 frameshift mutation at amino acid 7 (ATT to AT) of the Psy3 protein in the GBD-Psy3 construct. The resulting mutant Psy3 polypeptide is 22 aa long and fails to exhibit any two-hybrid interactions or to complement the MMS sensitivity of _psy3_Δ. The GAD-Psy3 construct, while failing to complement, is functional in the two-hybrid assay and does not contain errors at the sequence level. The rest of the constructs fully complement the MMS sensitivity of the respective _shu_Δ strains, do not contain any sequence errors, and interact with each other in the two-hybrid system (Table 2).
TABLE 2.
Two-hybrid Interactions
| GADa | GAD-Shu1 | GAD-Shu2 | GAD-Psy3 | GAD-Csm2 | |
|---|---|---|---|---|---|
| GBDa | — | — | — | — | — |
| GBD-Shu1 | — | — | 25 ± 7 | 10 ± 1 | ± |
| GBD-Shu2 | — | 28 ± 8 | — | 6 ± 1 | ± |
| GBD-Csm2 | — | — | ± | 19 ± 3 | — |
As described above, mutation of SHU1, SHU2, PSY3, or CSM2 leads to suppression of _top3_Δ slow growth and DNA damage sensitivity (Figure 1, A and B), suppression of _sgs1_Δ HU sensitivity (Figure 1C), and, in an otherwise wild-type background, a similar level of sensitivity to MMS (Figure 1D). Also, as indicated by the two-hybrid results, these proteins likely interact among themselves. These observations suggest that the four proteins function together in vivo, possibly as part of a single pathway or complex. To test this idea, we created double, triple, and quadruple deletion mutants and tested their MMS sensitivities. Indeed, we find that all of the mutant combinations exhibit the same degree of MMS sensitivity as any of the single mutants (Figure 1D; data not shown). Thus, shu1, shu2, psy3, and csm2 are in one epistasis group and likely function in the same pathway. For brevity, in the remainder of this article, we refer to them collectively as the Shu proteins or the SHU genes, even though two of the genes are named PSY3 and CSM2.
Mutation of SHU1 suppresses increased Rad52-YFP foci in top3 and sgs1 mutants:
HR proteins form spontaneous and DNA-damage-induced foci in both yeast and mammalian cells (Haaf et al. 1995; Lisby et al. 2001, 2004). These foci are sites of repair of multiple DNA lesions, such as DSBs (Lisby et al. 2003). In budding yeast, spontaneous Rad52-YFP foci are mostly observed in S and G2/M cells, suggesting that these foci are sites of replication fork repair (Lisby et al. 2001, 2003). Dynamics of focus formation can be affected by mutation of genes involved in DNA metabolism (Lisby et al. 2004; D. Alvaro and R. Rothstein, unpublished results; Figure 2). For instance, budding yeast _top3_Δ and _sgs1_Δ mutants, both of which exhibit hyperrecombination and increased genomic instability, also show increased incidence of spontaneous Rad52-YFP foci (M. Wagner, G. Price and R. Rothstein, unpublished results; Figure 2A). We tested whether deletion of SHU1 would influence this phenotype. The data, shown in Figure 2, are presented as the proportion of all G1 (i.e., unbudded) and S/G2/M (i.e., budded) cells that contain Rad52-YFP foci. The insets in B and C show the cell cycle distribution of all the strains tested (i.e., the fraction of the population in G1 or S/G2/M). We find that deletion of SHU1 does partially suppress the increased Rad52-YFP focus formation in the _top3_Δ mutant: 71% of all S/G2/M _top3_Δ cells contain Rad52-YFP foci, compared to 45% of all S/G2/M _top3_Δ _shu1_Δ cells (Figure 2B). This effect is not caused by a change in cell cycle distribution of the strain, as mutation of SHU1 does not rescue the abnormal cell cycle distribution of the _top3_Δ mutant (see inset in Figure 2B). We conclude that the SHU1 gene contributes to genomic instability and increased formation of Rad52-YFP foci in the absence of topoisomerase III.
Figure 2.—

Effects of _shu1_Δ on the formation of Rad52-YFP foci in the _top3_Δ and _sgs1_Δ mutants. The haploid strains for these experiments were generated from diploids ESD35 and ESD46. (A) The _top3_Δ strain exhibits an increase in Rad52-YFP foci compared to the wild-type strain. (B) _shu1_Δ partially suppresses increased Rad52 foci in the top3 mutant. The graph shows the percentage of cells in G1 (i.e., unbudded cells) or in S/G2/M (i.e., all budded cells) that contain Rad52-YFP foci in the indicated strains. The inset shows the cell cycle distribution of the same four strains. The graph represents data from at least 300 cells of each genotype. (C) _shu1_Δ partially suppresses increased Rad52-YFP foci in the _sgs1_Δ mutant after HU treatment. The graph shows the percentage of HU-treated and untreated cells in G1 or S/G2/M that contain Rad52-YFP foci. The two insets show the cell cycle distribution of untreated cells as well as of cells treated with 100 mm HU for 100 min. Between 150 and 300 cells were examined for every strain for either condition (HU or no HU) during this experiment.
In addition to cell cycle stage and mutation of DNA metabolism genes, Rad52 focus formation is affected by genotoxic agents, such as HU, which inhibit replication fork progression by depleting dNTP pools. There is a great decrease in Rad52-YFP foci in wild-type cells treated with HU, suggesting that active progression of replication forks is necessary for the formation of recombinational repair centers, or foci (Lisby et al. 2004). Replication forks that have arrested due to the action of HU are maintained and stabilized by processes that are checkpoint dependent. When cellular checkpoint mechanisms are perturbed, such as by mutation of the MEC1 gene, HU causes replication fork collapse (Lopes et al. 2001; Cha and Kleckner 2002). In mec1 cells, HU treatment results in a great increase in Rad52 foci, probably reflecting the formation of recombinational repair complexes at collapsed replication forks (Lisby et al. 2004). Mutation of SGS1 causes HU sensitivity, presumably because of the dual role of Sgs1 in the intra-S checkpoint activation and in replication fork restart (Frei and Gasser 2000; Cobb et al. 2003). As described above, deletion of any of the SHU genes suppresses _sgs1_Δ HU sensitivity (Figure 1C). Thus, we examined if deletion of SGS1 and/or SHU1 perturb the normal dynamics of Rad52-YFP foci after exposure to HU. The results of these experiments are shown in Figure 2C. We find that deletion of SHU1 does not significantly affect Rad52-YFP focus formation in either untreated or HU-treated cells, consistent with the observation that the shu mutants are not HU sensitive. Mutation of SGS1 causes an increased incidence of spontaneous Rad52-YFP foci, particularly in the S/G2/M cells (M. Wagner, G. Price and R. Rothstein, unpublished results). Deletion of SHU1 in the _sgs1_Δ strain slightly suppresses increased spontaneous Rad52-YFP foci (Figure 2C). In the wild-type strain, treatment with 100 mm HU reduces the focus-containing proportion of S/G2/M cells to 1% (Lisby et al. 2004; Figure 2C). This effect of HU on Rad52-YFP focus formation is perturbed by deletion of SGS1, with ∼22% of S/G2/M _sgs1_Δ cells containing Rad52-YFP foci after HU treatment. These are mostly large-budded cells and cells that appear arrested with the nucleus positioned between the mother and the bud (Figure 2C; data not shown). Deletion of SHU1 in the _sgs1_Δ strain reduces the proportion of S/G2/M cells that contain foci after HU treatment from 22% to 8%, reflecting the suppression of _sgs1_Δ HU sensitivity by _shu1_Δ.
Genetic analysis of Shu function:
We have shown that the Shu proteins function in the same pathway of MMS-induced DNA damage resistance (Figure 1D). Next, we asked how they interact genetically with other genes involved in MMS resistance. Because of the genetic interaction between the shu mutants and mutation of SGS1 on HU, we tested whether they have a similar relationship upon exposure to MMS. Thus, we analyzed the response of _sgs1_Δ _shu1_Δ and _sgs1_Δ _csm2_Δ double mutants to increasing concentrations of MMS compared to that of the single mutants (Figure 3A). Interestingly, we observed an additive effect of the mutations on the reduction in cell survival, indicating that, for repair of MMS-induced DNA damage, Sgs1 and the Shu proteins function in separate pathways.
Figure 3.—
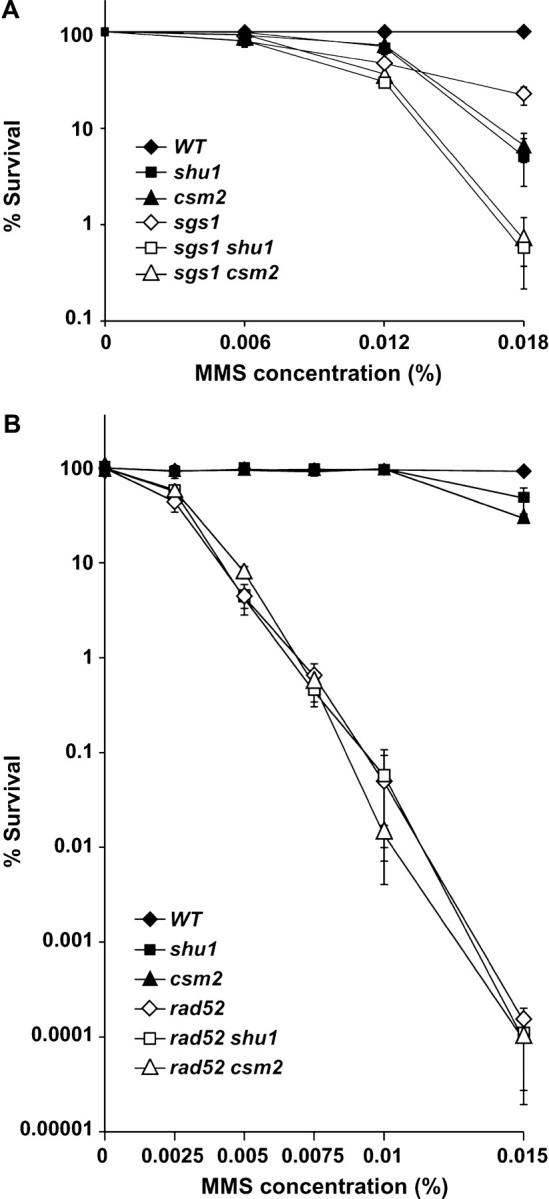
The SHU genes are in the RAD52 epistasis group for MMS resistance. All mutations shown are gene deletions. When comparing the data shown in A to those in B, note the difference in MMS concentrations. (A) Survival curves showing that SGS1 and the SHU genes are in different epistasis groups for MMS resistance. Deletion of SGS1 and either SHU1 or CSM2 results in additional MMS sensitivity compared to any of the single mutants. (B) Survival curves showing that _rad52_Δ _shu1_Δ and _rad52_Δ _csm2_Δ double mutants are no more MMS sensitive than the _rad52_Δ single mutant. At the highest MMS concentration used in these experiments (0.015%), deletion of SHU1 or CSM2 results in a two- to threefold decrease in survival compared to the wild-type strain.
Several lines of evidence suggest that the Shu proteins' function may be linked to HR. First, deletion of PSY3 was isolated in a screen for MMS-sensitive mutants and analyzed for genetic interactions with other mutants known to be involved in MMS resistance (Hanway et al. 2002). The only mutation found to be epistatic to _psy3_Δ was _rad52_Δ. Second, similarly to mutations in HR genes, shu mutants suppress top3 slow growth. Finally, recovery from HU-induced replication arrest in _sgs1_Δ mutants is thought to be a recombination-linked process. We have observed that this process is affected by shu mutations both on the level of cell survival and on the level of Rad52-YFP focus formation (Figures 1C and 2C). Thus, we examined the genetic interactions among _shu1_Δ, _csm2_Δ, and _rad52_Δ mutations by measuring the appropriate single- and double-mutant survival in various concentrations of MMS (Figure 3B). Indeed, we find that _rad52_Δ _shu1_Δ and _rad52_Δ _csm2_Δ double mutants are no more sensitive to MMS than _rad52_Δ single mutants, i.e., that _rad52_Δ is epistatic to mutation of SHU1 or CSM2 for MMS sensitivity. This observation provides further support for the notion that the Shu proteins' function is linked to HR.
The Shu proteins are predominantly nuclear:
Many proteins involved in recombinational DNA repair, when tagged with fluorescent markers, show diffuse nuclear localization but can relocalize to discrete foci either spontaneously or upon DNA damage (Lisby et al. 2001, 2003; Shor et al. 2002). To determine whether this pattern holds true for the Shu proteins, we C-terminally tagged each of them with YFP at the native chromosomal locus using a cloning-free allele replacement method and analyzed their localization using a fluorescent microscope (Reid et al. 2002). Shu2-YFP and Csm2-YFP did not yield a signal detectable above the background. However, Shu1-YFP and Psy3-YFP did produce a detectable, albeit very faint, fluorescent signal that colocalized with 4′,6-diamidino-2-phenylindole dye, indicating nuclear localization (data not shown). To enhance the signal of the Shu1-YFP fusion, we introduced another copy of YFP at the Shu1-YFP C terminus. The Shu1-YFP-YFP construct fully complements MMS sensitivity of the _shu1_Δ strain (data not shown). To precisely delineate the position of nuclei in Shu1-YFP-YFP-containing cells, we transformed them with a plasmid containing Nup49-CFP, a marker for the nuclear envelope. As shown in Figure 4, the Shu1-YFP-YFP signal is indeed predominantly nuclear. However, we did not observe any Shu1-YFP-YFP foci, either spontaneously or in response to MMS treatment.
Figure 4.—

Shu1-YFP-YFP exhibits predominantly nuclear localization. Differential interference contrast (DIC), YFP, and CFP images of a representative field of cells expressing Shu1-YFP-YFP and Nup49-CFP proteins are shown. Nup49 is a nuclear envelope protein. The Shu1-YFP-YFP fluorescent signal is strongest within the Nup49-CFP-defined nuclear boundaries.
Mutation of SHU1 or CSM2 affects recombination at the _SUP4-_o locus:
The epistatic relationship between rad52 and the shu mutants suggests a possible function of the Shu proteins in recombination. Thus, we measured recombination rates in the _shu1_Δ and _csm2_Δ mutants at several loci. Both spontaneous and MMS-induced marker loss at the rDNA locus and recombination between leu2 heteroalleles positioned on homologous chromosomes in the diploid were measured and neither was affected by deletion of either SHU1 or CSM2 (data not shown). Increased rDNA recombination has been linked to a shortened life span in yeast cells, e.g., in the _sgs1_Δ strain (Sinclair et al. 1997). Consistent with wild-type rDNA recombination rates, the shu mutants exhibit a normal life span (A. Falcon and J. Aris, unpublished results). We also measured recombination at the locus containing _SUP4_-o, the gene encoding a tyrosine tRNA ochre suppressor. This locus contains five δ-sequences (derived from long terminal repeats of the yeast Ty transposon) that are positioned as direct and inverted repeats (for details on the locus see Rothstein et al. 1987; Shor et al. 2002). Recombination between these δ-repeats by gene conversion (GC) between misaligned sister chromatids or by single-strand annealing (SSA) can result in deletion of the region containing SUP4-o. Insertion of the URA3 gene next to SUP4-o can facilitate detection and quantification of these deletion events as they result in the simultaneous loss of both genes (Rothstein et al. 1987; Shor et al. 2002). Both _top3_Δ and _sgs1_Δ mutants exhibit hyperrecombination at SUP4-o, with the degree of the defect greater in the _top3_Δ strain (Shor et al. 2002). We find that deletion of SHU1 partially suppresses _sgs1_Δ and _top3_Δ SUP4-o hyperrecombination, consistent with the observation that shu mutants also suppress other defects of _top3_Δ and _sgs1_Δ strains (Figure 5A). Interestingly, in an otherwise wild-type background, deletion of SHU1 or CSM2 results in a three- to sevenfold increase in the rate of SUP4-o recombination (Figure 5A). Overall, we conclude that the Shu proteins affect certain kinds of recombination events, such as those leading to deletion formation at the _SUP4_-o locus, both in an otherwise wild-type background and in the absence of Sgs1 or Top3.
Figure 5.—
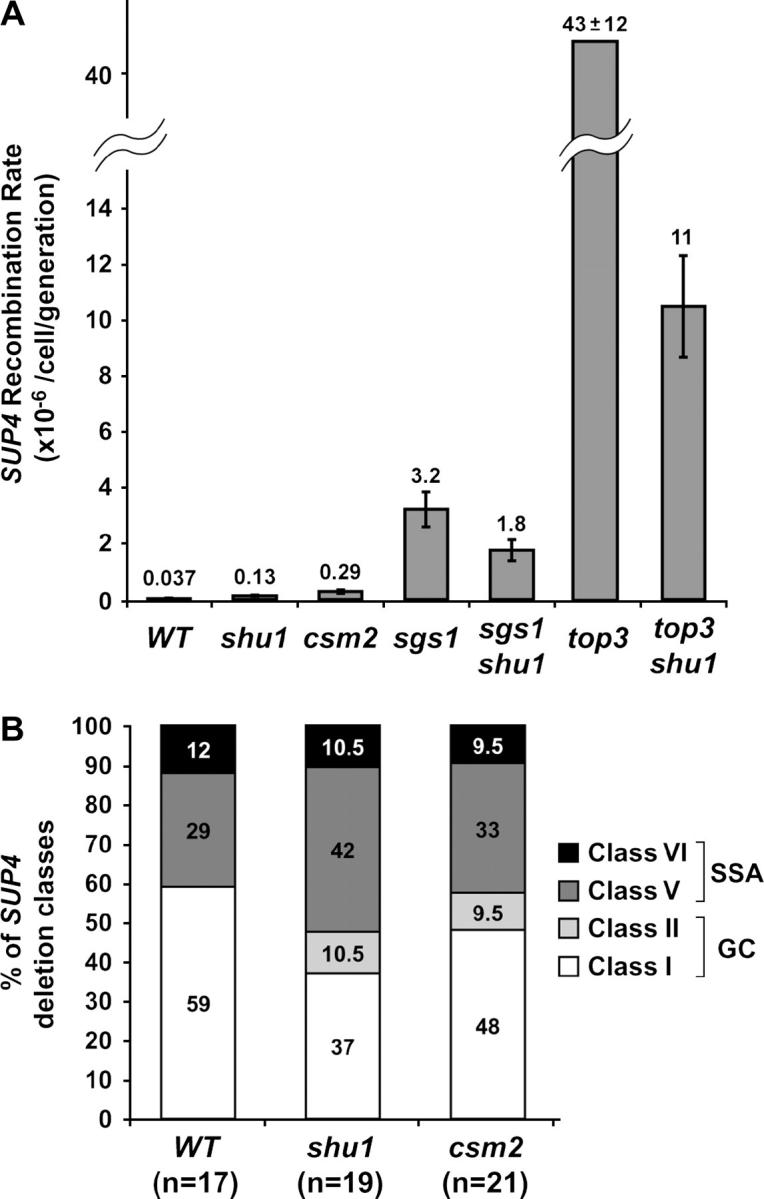
Effect of deletion of SHU1 or CSM2 on recombination at the _SUP4_-o locus in an otherwise wild-type background, as well as in _sgs1_Δ and _top3_Δ mutants. Diploids ESD45, ESD47, and ESD48 produced the haploid strains shown. (A) _shu1_Δ and _csm2_Δ mutants show a three- to sevenfold increase in the _SUP4_-o recombination rate. Deletion of SHU1 partially suppresses the hyperrecombination phenotype of _sgs1_Δ and _top3_Δ mutants. (B) _shu1_Δ and _csm2_Δ mutants do not affect SUP4 deletion class distribution. n, number of independently generated SUP4 recombinants tested for each strain.
In addition to measuring the rate of recombination at the SUP4-o locus, it is possible to analyze the structure of the locus in the recombinants by PCR and restriction enzyme digestion. In this manner, seven SUP4-o deletion classes can be distinguished (Rothstein et al. 1987; Shor et al. 2002). On the basis of the sequence and relative positioning of the δ-sequences in the recombinants, it was concluded that five of these classes arise via GC and two via direct repeat recombination by SSA (Rothstein et al. 1987). In the wild-type strain, GC is the predominant mechanism of deletion formation. HR mutants _rad51_Δ, _rad52_Δ, _rad54_Δ, _rad55_Δ, and _rad57_Δ are severely impaired in performing GC, so virtually all SUP4-o deletions generated in these strains belong to the SSA classes (Shor et al. 2002). On the other hand, the relative proportions of GC and SSA classes are not significantly affected by deletion of either SGS1 or TOP3 (Shor et al. 2002). We analyzed the SUP4-o locus structure in a number of independently generated recombinants in the _shu1_Δ and _csm2_Δ strains. We find that the spectrum of SUP4-o deletion classes is not significantly affected by mutation of SHU1 or CSM2 (Figure 5B).
Genetic characterization of the shu mutator phenotype:
A study of new genes involved in protecting the genome from mutagenesis demonstrated that _shu1_Δ, _shu2_Δ, _psy3_Δ, and _csm2_Δ strains exhibit increased spontaneous forward mutation rates at the CAN1 locus (Huang et al. 2003). The so-called “mutator” phenotype is characteristic of mutations that impair any one of several pathways involved in error-free repair of spontaneous DNA damage, such as base excision repair (BER), the error-free branch of postreplicative repair (PRR), or HR (Swanson et al. 1999; Broomfield and Xiao 2002). Elimination of the HR pathway by deletion of RAD52 increases the CAN1 forward mutation rate approximately sevenfold, which is similar to the rate observed in the _shu_Δ strains (Swanson et al. 1999; Huang et al. 2003; Figure 6A). We have shown that mutation of RAD52 is epistatic to _shu1_Δ and _csm2_Δ for MMS sensitivity, suggesting that the Shu proteins' function is connected to HR. To address whether the mutator phenotype of the shu mutants is due to a HR defect, we measured the CAN1 forward mutation rate in _rad52_Δ, _shu1_Δ, _csm2_Δ, _rad52_Δ _shu1_Δ, and _rad52_Δ _csm2_Δ mutants. The mutation rates that we obtained for the single mutants are similar in magnitude to the ones published by other groups (Swanson et al. 1999; Huang et al. 2003; Figure 6A). Interestingly, the mutation rates of the _rad52_Δ _shu1_Δ and _rad52_Δ _csm2_Δ double mutants are similar to those of the single mutants, indicating an epistatic relationship between rad52 and the shu mutations (Figure 6A). Thus, we conclude that the mutator phenotype of the _shu_Δ strains is due to a defect in a _RAD52_-dependent process, such as HR.
Figure 6.—

Deletion of SHU1 or CSM2 results in a mutator phenotype. The forward mutation rates of the CAN1 gene are shown. The strains shown are meiotic segregants of diploids ESD41, ESD44, and ESD49. (A) Spontaneous CAN1 mutation rates. rad52 is epistatic to shu1 and csm2: CAN1 mutation rate in the _rad52_Δ _shu1_Δ and _rad52_Δ _csm2_Δ double mutants is similar to that in the single mutants. This graph also shows that the mutator phenotype of _shu1_Δ and _csm2_Δ mutants is dependent on the REV3 gene. (B) MMS-induced CAN1 mutation rates. Treatment with 0.01% MMS stimulates the CAN1 mutation rate ∼35-fold in the wild-type and the mutant strains.
In HR mutants, the majority of mutations introduced into the CAN1 gene are produced by translesion synthesis (TLS), an error-prone DNA repair process carried out by the low-fidelity polymerase ζ (Pol ζ) (Morey et al. 2003). Pol ζ, a complex of Rev3p and Rev7p, can insert nucleotides across damaged bases or fill in gaps left by the replicative polymerase unable to replicate across a DNA lesion (Murakumo 2002). To test whether TLS is responsible for the increased mutation rate observed in the shu mutants, we asked whether the mutator phenotype of the _shu1_Δ strain is REV3 dependent. We combined _rev3_Δ with _shu1_Δ and measured the CAN1 mutation rate in the double mutant. Indeed, we found that deletion of REV3 virtually abrogates the mutator phenotype of the _shu1_Δ mutant, indicating that most of the mutations introduced into the CAN1 gene in the _shu1_Δ strain are produced by TLS (Figure 6A).
Certain mutants of the PRR pathway exhibit a spontaneous mutator phenotype but are defective for damage-induced mutagenesis (Cassier-Chauvat and Fabre 1991; Giot et al. 1997). Thus, we measured the rate of MMS-induced CAN1 forward mutation in the _shu1_Δ and _csm2_Δ mutants, as well as in the wild-type strain (Figure 6B). We found that treatment with 0.01% MMS increases the rate of mutagenesis ∼35-fold in all three strains tested, indicating that mutation of SHU1 or CSM2 does not impair the cellular ability to induce mutagenesis in response to DNA damage by MMS.
Rad52 foci dynamics after DNA damage are affected by mutation of SHU1:
Genetic analyses of MMS sensitivity and the mutator phenotype of the shu mutants place them in the rad52 epistasis group. This genetic relationship indicates that mutation of the SHU genes affects a _RAD52_-dependent process, presumably HR. As mentioned above, Rad52-YFP focus dynamics are altered by mutations that affect HR and genome stability, such as _top3_Δ and _sgs1_Δ, and by exposure to DNA-damaging agents. We find that mutation of SHU1 does not significantly affect Rad52-YFP focus formation in untreated cells (Figure 2B). However, since shu mutants are MMS sensitive, we considered the possibility that their effect on Rad52-YFP focus dynamics could become evident upon MMS treatment. Thus, we treated wild-type and shu1Δ cells expressing Rad52-YFP with increasing concentrations of MMS and determined the percentage of cells containing Rad52-YFP foci. In agreement with published data, we find that MMS treatment causes an increase in the number of Rad52-YFP foci in wild-type cells (Lisby et al. 2003; Figure 7B). Interestingly, mutation of SHU1 exacerbates this effect: MMS-treated _shu1_Δ cells form more Rad52-YFP foci than do wild-type cells, especially at higher concentrations of the drug (Figure 7, A and B).
Figure 7.—

Deletion of SHU1 alters Rad52-YFP focus dynamics after DNA damage by MMS. (A) Deletion of SHU1 results in an increase in Rad52-YFP foci upon MMS treatment. Representative samples of wild-type and _shu1_Δ cells treated with 0.05% MMS are shown. (B) The graph shows the response of the wild-type strain and the _shu1_Δ mutant to increasing concentrations of MMS. The percentages of S/G2/M (i.e., all budded) cells containing Rad52-YFP foci are plotted. Fluorescent images were captured after 100 min of growth in the presence of MMS. At least 150 cells were analyzed for each strain under each condition. (C) Time-lapse analyses of Rad52-YFP foci in cells exposed to 0.01% MMS for 100 min. Duration of individual foci is plotted on the _x_-axis vs. the number of foci that lasted for that amount of time. While images of cells were captured every 5 min, the data are plotted in 10-min intervals to reduce complexity. Thus, in the graph, the bars over the 10-min mark correspond to the number of foci that lasted <10 min, the bars over the 20-min mark correspond to the number of foci that lasted between 10 and 20 min, etc. The median duration of Rad52-YFP foci is increased from 15 min in the wild-type strain to 25 min in the _shu1_Δ mutant. The inset shows the data from untreated cells, where the median duration of Rad52-YFP foci is unchanged by _shu1_Δ mutation.
How can this observation be explained in light of the epistatic relationship between shu and rad52 mutants? Mutation of the SHU genes presumably impairs some aspect of _RAD52_-dependent recombinational repair. Impaired function of the recombinational repair factories, or foci, can result in a delay in focus disassembly. Thus, an increase in the observed number of Rad52-YFP foci could conceivably be due to the fact that individual foci last longer in MMS-treated _shu1_Δ cells than in wild-type cells. To test the idea of longer-lasting foci directly, we performed time-lapse experiments measuring the duration of Rad52-YFP foci in untreated and MMS-treated wild-type and _shu1_Δ cells. MMS was added to exponentially growing cells to a concentration of 0.01%. The cells were immediately mounted onto slides and placed under the fluorescent microscope, with images taken every 5 min for 5 hr. The acquired time-lapse images were analyzed, noting the time of appearance and disappearance of a particular Rad52-YFP focus. Only cells that were budding and/or growing were taken into account (for every condition used, at least 75% of cells present at the beginning of the time lapse satisfied this requirement). The results of these experiments are graphed in Figure 7C. In agreement with previously published data, we find that the median duration of Rad52-YFP foci in untreated wild-type cells is 10 min (Lisby et al. 2003). Deletion of SHU1 does not affect this Rad52-YFP property. Treatment with MMS increases the median duration of the foci in wild-type cells to 15 min. Interestingly, we find that the foci do persist longer in MMS-treated _shu1_Δ cells (the median is 25 min) than in wild-type cells. This observation supports the hypothesis that an increase in the overall number of Rad52-YFP foci in MMS-treated _shu1_Δ cells is due to a longer persistence of the foci, reflecting the importance of Shu1 in the timely completion of recombinational repair.
DISCUSSION
We have isolated four genes, SHU1, SHU2, PSY3, and CSM2, as mutational suppressors of _top3_Δ slow growth. Mutation of any of these genes also partially rescues other defects associated with loss of TOP3 or SGS1, such as sensitivity to genotoxic agents, increased formation of Rad52-YFP foci, and hyperrecombination at the SUP4-o locus (Figures 1, 2, and 5). Mutation of these genes in an otherwise wild-type background causes sensitivity to MMS and an increased rate of forward mutation in the CAN1 gene (Hanway et al. 2002; Huang et al. 2003; Figures 1 and 6). CSM2 was originally identified as a mutation affecting meiotic chromosome segregation (Rabitsch et al. 2001) and PSY3 as a gene important for platinum resistance (Wu et al. 2004). These observations indicate that the four proteins function in DNA metabolism and are presumably involved in error-free repair of spontaneous and induced DNA lesions. Moreover, several lines of evidence suggest that the four proteins function together in vivo: (1) the mutants have a similar phenotype (Figure 1); (2) all four genes are in the same epistasis group (Figure 1); and (3) the proteins interact among themselves in the two-hybrid assay (Ito et al. 2001; Table 2). We also show that rad52 is epistatic to the shu mutants for both MMS sensitivity and mutator phenotype, indicating that mutation of the Shu proteins impairs a _RAD52_-dependent process, presumably HR (Figure 3). Finally, we find that the dynamics of Rad52-YFP foci are affected by mutation of SHU1, particularly after MMS treatment (Figure 7). We observe an increased percentage of cells with Rad52-YFP foci after MMS treatment of the _shu1_Δ strain, and time-lapse experiments suggest that this phenotype is due to the increased duration of individual foci. Taken together, these data strongly suggest that the Shu proteins' function is important for error-free repair of DNA lesions via HR.
The MMS sensitivity and mutator phenotype of the shu mutants suggest an important role for the Shu proteins in protecting the genome from spontaneous and induced DNA damage and in promoting error-free DNA repair. MMS-induced DNA lesions can be repaired by error-free mechanisms, such as PRR or HR (which may be interlinked) or by error-prone TLS involving polymerase ζ (Barbour and Xiao 2003). Since several repair systems can work on the same lesion, mutation of one system increases the load on the others. This principle is illustrated by the observations that mutation of BER or TLS leads to an increase in recombination, while mutation of BER or HR causes a mutator phenotype (Swanson et al. 1999). Consistent with this pattern, mutation of the SHU genes causes a mutator phenotype that is dependent on Pol ζ, showing that when the Shu pathway is abolished, the load on TLS is increased. Importantly, we find that mutation of RAD52 is epistatic to mutation of the SHU genes for both MMS sensitivity and the mutator phenotype. This epistatic relationship suggests that the Shu proteins are involved in error-free repair via HR.
In a previous article, we showed that HR is “toxic” in the _top3_Δ mutants (Shor et al. 2002). We now show that toxicity of HR genes in the _top3_Δ mutant can be alleviated by mutation of the SHU genes. Previously, we hypothesized that, in the absence of Top3, Sgs1 creates a recombinogenic intermediate that is acted upon by HR proteins, causing hyperrecombination, genome instability, and slow growth. This model is complicated by the notion that HR is also thought to act upstream of Sgs1 (Gangloff et al. 2000; Wu et al. 2001). Thus, taking into account the genetic data showing that SHU mutations impair some aspect of HR, several nonmutually exclusive reasons for suppression of _top3_Δ by the shu mutants exist. For example, in the wild-type strain, the Shu proteins may be involved in generating the substrate for Sgs1. Thus, in the shu mutants, the requirement for a functional Sgs1-Top3 pathway may be at least partially bypassed. Alternatively, the Shu proteins may be involved in the processing of the Sgs1-generated intermediate in the _top3_Δ mutant with detrimental consequences. In the shu mutants, this intermediate may be channeled into other pathways for resolution, accounting for the suppression of _top3_Δ defects.
The observation that mutation of the SHU genes rescues _sgs1_Δ HU sensitivity indicates that Shu protein function is detrimental in the absence of Sgs1 when replication forks pause en masse. The reasons for _sgs1_Δ HU sensitivity are probably manifold since several roles have been proposed for Sgs1 during DNA replication. Sgs1 contributes to both the intra-S checkpoint and the stabilization of DNA polymerases at stalled forks (Frei and Gasser 2000; Cobb et al. 2003). Also, Sgs1 is thought to function in maturation and/or resolution of recombination intermediates at replication forks, preventing excessive recombination that can be initiated at stalled forks and cause genome rearrangement (Kaliraman et al. 2001; Fabre et al. 2002; Fricke and Brill 2003). We imagine that the reasons for the shu suppression of sgs1 HU sensitivity largely mirror the ones proposed above with respect to suppression of top3 slow growth. In sgs1 shu mutants, recombination intermediates that are normally resolved by Sgs1 may not form as readily or may be channeled into another, less detrimental, pathway for resolution. This hypothesis is supported by the partial rescue of increased Rad52-YFP foci in HU-treated _sgs1_Δ cells by mutation of SHU1 (Figure 2C).
Although the epistatic relationship between rad52 and the shu mutations indicates that the Shu proteins are important for some aspect of HR, they clearly do not play a direct or essential role in this process. This conclusion is illustrated by several significant differences between the shu phenotype and the phenotype of bona fide recombination mutants, like _rad51_Δ or _rad52_Δ. One important difference concerns the mutants' sensitivity to DNA-damaging agents: unlike HR mutants, the shu mutants are not sensitive to HU and exhibit only mild gamma-ray sensitivity to high doses of radiation (>1000 Gy; data not shown). These observations indicate that the Shu proteins are not essential for repair of DSBs or of stalled replication forks. Also, unlike proteins directly involved in HR, the Shu proteins (or, at least, Shu1-YFP and Psy3-YFP, the two Shu-YFPs that produced a fluorescent signal in our hands) do not form foci either spontaneously or in response to gamma-ray or MMS treatment. There are several possible explanations for this observation. Judging by the low intensity of fluorescence produced by the Shu-YFP constructs, the Shu proteins are not abundant. Even if there is some aggregation of these proteins at sites of damage, it may not produce enough localized fluorescence to qualify as a “focus.” Alternatively, the Shu proteins may not need to aggregate to function, unlike HR protein Rad51, which forms a nucleo-protein filament on DNA, and Rad52, which forms 7- to 11-mer ring structures (Radding 1993; Stasiak et al. 2000; Kagawa et al. 2002). Finally, it is conceivable that the Shu-YFPs do form foci upon DNA damage, but these foci are short lived (i.e., under 5 min) and disappear by the time the cells are visualized under the microscope.
Proteins that have established roles in HR have been assigned these roles both by analyses of their biochemical activities on DNA substrates in vitro and by measuring recombination rates at various chromosomal loci in the mutants. Mutation of RAD52 affects several kinds of HR, such as heteroallelic recombination between homologous chromosomes in the diploid, GC, SSA, and break-induced replication (BIR; reviewed in Symington 2002). At the _SUP4_-o locus, recombination is virtually eliminated in the _rad52_Δ mutant, and the few recombinants that arise belong to the SSA classes (Rothstein et al. 1987; Shor et al. 2002). On the other hand, mutation of RAD51 causes a drastic reduction in GC, but SSA and BIR can still take place. Interestingly, the overall rate of SUP4 recombination increases severalfold in the _rad51_Δ mutant, but all the recombinants belong to the SSA classes (Shor et al. 2002). This observation is explained by the idea that, in the wild-type strain, _SUP4_-o recombination occurs exclusively via GC using the sister chromatid as substrate, virtually always resulting in accurate repair and generating very few deletions. Elimination of GC by _rad51_Δ channels all DNA lesions into the SSA pathway, accounting for the observed increase in deletion formation (McDonald and Rothstein 1994). Similarly, deletion formation at the SUP4-o locus is increased three- to sevenfold in the shu mutants compared to the wild-type strain. However, unlike the situation in the _rad51_Δ mutant, distribution of recombinant classes is not significantly altered in the shu mutants, indicating that both GC and SSA can still occur. We conclude that, in the absence of Shu proteins, HR substrates and/or intermediates are processed in a way that is less accurate and more rearrangement prone compared to the wild-type strain. Moreover, time-lapse analyses of Rad52-YFP foci in the _shu1_Δ mutant suggest that shu mutations impair not only the accuracy, but also the efficiency of recombinational repair.
In conclusion, we have identified mutations in SHU1, SHU2, PSY3, and CSM2 as suppressors of several defects associated with mutation of Top3 or Sgs1, such as hyperrecombination at the SUP4-o locus, increased incidence of Rad52-YFP foci, _top3_Δ slow growth and HU and MMS sensitivity, and _sgs1_Δ HU sensitivity. Our results suggest that the Shu proteins function together in vivo in protecting the genome from spontaneous and induced DNA damage. Genetic analyses place the SHU genes in the RAD52 epistasis group, implicating them in HR. However, they do not play a direct or essential role in HR, as evidenced by several notable differences between the shu phenotype and that of bona fide HR mutants. The Shu proteins, on the other hand, do play an important role in error-free DNA repair, since in the absence of Shu proteins DNA lesions are channeled into TLS and rearrangement-prone recombinational repair routes. We have not been able to identify Shu1, Shu2, Psy3, and Csm2 homologs in organisms other than closely related yeast species. This lack of homologs could be due either to other proteins taking on Shu functions in higher eukaryotes or to a high degree of divergence between S. cerevisiae Shu proteins and their counterparts in other organisms. Since the Shu proteins do not contain easily identifiable motifs that yield information about their molecular roles, biochemical analyses are necessary to understand how the Shu proteins function at the molecular level. Also, crystallization and determination of 3-D protein structure can help find structural, if not sequence, homologs of the Shu proteins.
Acknowledgments
We gratefully acknowledge the gifts of two-hybrid plasmids from the Ito laboratory, of a genomic DNA library from the Yamashita laboratory, and of plasmid pWJ1323 from Michael Lisby. We thank Marian Carlson, Michael Cox, Catherine Fox, James Keck, Michael Lisby, Robert Reid, Lorraine Symington, and Marisa Wagner for useful discussions. We also thank John Aris and Alaric Falcon for communication of unpublished results. E.S. is indebted to the Craig, Fox, Kimble, and, especially, Cox laboratories at the University of Wisconsin for providing laboratory materials and support during 2002 and 2003. This work was supported by a National Science Foundation Graduate Fellowship to E.S. and by National Institutes of Health grant GM50237 to R.R.
References
- Akada, R., J. Yamamoto and I. Yamashita, 1997. Screening and identification of yeast sequences that cause growth inhibition when overexpressed. Mol. Gen. Genet. 254**:** 267–274. [DOI] [PubMed] [Google Scholar]
- Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman et al., 1998 Current Protocols in Molecular Biology. John Wiley & Sons, New York.
- Barbour, L., and W. Xiao, 2003. Regulation of alternative replication bypass pathways at stalled replication forks and its effects on genome stability: a yeast model. Mutat. Res. 532**:** 137–155. [DOI] [PubMed] [Google Scholar]
- Bendixen, C., S. Gangloff and R. Rothstein, 1994. A yeast mating-selection scheme for detection of protein-protein interactions. Nucleic Acids Res. 22**:** 1778–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, R. J., J. A. Sharp and J. C. Wang, 1998. Purification and characterization of the Sgs1 DNA helicase activity of Saccharomyces cerevisiae. J. Biol. Chem. 273**:** 9644–9650. [DOI] [PubMed] [Google Scholar]
- Bennett, R. J., J. L. Keck and J. C. Wang, 1999. Binding specificity determines polarity of DNA unwinding by the Sgs1 protein of S. cerevisiae. J. Mol. Biol. 289**:** 235–248. [DOI] [PubMed] [Google Scholar]
- Bennett, R. J., M. F. Noirot-Gros and J. C. Wang, 2000. Interaction between yeast Sgs1 helicase and DNA topoisomerase III. J. Biol. Chem. 275**:** 26898–26905. [DOI] [PubMed] [Google Scholar]
- Bischof, O., S. H. Kim, J. Irving, S. Beresten, N. A. Ellis et al., 2001. Regulation and localization of the Bloom syndrome protein in response to DNA damage. J. Cell Biol. 153**:** 367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broomfield, S., and W. Xiao, 2002. Suppression of genetic defects within the RAD6 pathway by srs2 is specific for error-free post-replication repair but not for damage-induced mutagenesis. Nucleic Acids Res. 30**:** 732–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosh, R. M., Jr., J. L. Li, M. K. Kenny, J. K. Karow, M. P. Cooper et al., 2000. Replication protein A physically interacts with the Bloom's syndrome protein and stimulates its helicase activity. J. Biol. Chem. 275**:** 23500–23508. [DOI] [PubMed] [Google Scholar]
- Cassier-Chauvat, C., and F. Fabre, 1991. A similar defect in UV-induced mutagenesis conferred by the rad6 and rad18 mutations of Saccharomyces cerevisiae. Mutat. Res. 254**:** 247–253. [DOI] [PubMed] [Google Scholar]
- Cha, R. S., and N. Kleckner, 2002. ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science 297**:** 602–606. [DOI] [PubMed] [Google Scholar]
- Chakraverty, R. K., J. M. Kearsey, T. J. Oakley, M. Grenon, M. A. de La Torre Ruiz et al., 2001. Topoisomerase III acts upstream of Rad53p in the S-phase DNA damage checkpoint. Mol. Cell. Biol. 21**:** 7150–7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champoux, J. J., 2001. DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem. 70**:** 369–413. [DOI] [PubMed] [Google Scholar]
- Cobb, J. A., L. Bjergbaek, K. Shimada, C. Frei and S. M. Gasser, 2003. DNA polymerase stabilization at stalled replication forks requires Mec1 and the RecQ helicase Sgs1. EMBO J. 22**:** 4325–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinou, A., M. Tarsounas, J. K. Karow, R. M. Brosh, V. A. Bohr et al., 2000. Werner's syndrome protein (WRN) migrates Holliday junctions and co-localizes with RPA upon replication arrest. EMBO Rep. 1**:** 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox, M. M., 2001. Historical overview: searching for replication help in all of the rec places. Proc. Natl. Acad. Sci. USA 98**:** 8173–8180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doe, C. L., J. Dixon, F. Osman and M. C. Whitby, 2000. Partial suppression of the fission yeast rqh1(-) phenotype by expression of a bacterial Holliday junction resolvase. EMBO J. 19**:** 2751–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdeniz, N., U. H. Mortensen and R. Rothstein, 1997. Cloning-free PCR-based allele replacement methods. Genome Res. 7**:** 1174–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre, F., A. Chan, W. D. Heyer and S. Gangloff, 2002. Alternate pathways involving Sgs1/Top3, Mus81/Mms4, and Srs2 prevent formation of toxic recombination intermediates from single-stranded gaps created by DNA replication. Proc. Natl. Acad. Sci. USA 99**:** 16887–16892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frei, C., and S. M. Gasser, 2000. The yeast Sgs1p helicase acts upstream of Rad53p in the DNA replication checkpoint and colocalizes with Rad53p in S-phase-specific foci. Genes Dev. 14**:** 81–96. [PMC free article] [PubMed] [Google Scholar]
- Fricke, W. M., and S. J. Brill, 2003. Slx1-Slx4 is a second structure-specific endonuclease functionally redundant with Sgs1-Top3. Genes Dev. 17**:** 1768–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke, W. M., V. Kaliraman and S. J. Brill, 2001. Mapping the DNA topoisomerase III binding domain of the Sgs1 DNA helicase. J. Biol. Chem. 276**:** 8848–8855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff, S., J. P. McDonald, C. Bendixen, L. Arthur and R. Rothstein, 1994. The yeast type I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: a potential eukaryotic reverse gyrase. Mol. Cell. Biol. 14**:** 8391–8398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff, S., B. de Massy, L. Arthur, R. Rothstein and F. Fabre, 1999. The essential role of yeast topoisomerase III in meiosis depends on recombination. EMBO J. 18**:** 1701–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff, S., C. Soustelle and F. Fabre, 2000. Homologous recombination is responsible for cell death in the absence of the Sgs1 and Srs2 helicases. Nat. Genet. 25**:** 192–194. [DOI] [PubMed] [Google Scholar]
- Giot, L., R. Chanet, M. Simon, C. Facca and G. Faye, 1997. Involvement of the yeast DNA polymerase delta in DNA repair in vivo. Genetics 146**:** 1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haaf, T., E. I. Golub, G. Reddy, C. M. Radding and D. C. Ward, 1995. Nuclear foci of mammalian Rad51 recombination protein in somatic cells after DNA damage and its localization in synaptonemal complexes. Proc. Natl. Acad. Sci. USA 92**:** 2298–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanway, D., J. K. Chin, G. Xia, G. Oshiro, E. A. Winzeler et al., 2002. Previously uncharacterized genes in the UV- and MMS-induced DNA damage response in yeast. Proc. Natl. Acad. Sci. USA 99**:** 10605–10610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmon, F. G., R. J. DiGate and S. C. Kowalczykowski, 1999. RecQ helicase and topoisomerase III comprise a novel DNA strand passage function: a conserved mechanism for control of DNA recombination. Mol. Cell 3**:** 611–620. [DOI] [PubMed] [Google Scholar]
- Hickson, I. D., 2003. RecQ helicases: caretakers of the genome. Nat. Rev. Cancer 3**:** 169–178. [DOI] [PubMed] [Google Scholar]
- Huang, M. E., A. G. Rio, A. Nicolas and R. D. Kolodner, 2003. A genomewide screen in Saccharomyces cerevisiae for genes that suppress the accumulation of mutations. Proc. Natl. Acad. Sci. USA 100**:** 11529–11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito, T., T. Chiba, R. Ozawa, M. Yoshida, M. Hattori et al., 2001. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl. Acad. Sci. USA 98**:** 4569–4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James, P., J. Halladay and E. A. Craig, 1996. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics 144**:** 1425–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagawa, W., H. Kurumizaka, R. Ishitani, S. Fukai, O. Nureki et al., 2002. Crystal structure of the homologous-pairing domain from the human Rad52 recombinase in the undecameric form. Mol. Cell 10**:** 359–371. [DOI] [PubMed] [Google Scholar]
- Kaliraman, V., J. R. Mullen, W. M. Fricke, S. A. Bastin-Shanower and S. J. Brill, 2001. Functional overlap between Sgs1-Top3 and the Mms4-Mus81 endonuclease. Genes Dev. 15**:** 2730–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karow, J. K., A. Constantinou, J. L. Li, S. C. West and I. D. Hickson, 2000. The Bloom's syndrome gene product promotes branch migration of Holliday junctions. Proc. Natl. Acad. Sci. USA 97**:** 6504–6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakhar, R. R., J. A. Cobb, L. Bjergbaek, I. D. Hickson and S. M. Gasser, 2003. RecQ helicases: multiple roles in genome maintenance. Trends Cell Biol. 13**:** 493–501. [DOI] [PubMed] [Google Scholar]
- Krejci, L., S. Van Komen, Y. Li, J. Villemain, M. S. Reddy et al., 2003. DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature 423**:** 305–309. [DOI] [PubMed] [Google Scholar]
- Kwan, K. Y., and J. C. Wang, 2001. Mice lacking DNA topoisomerase IIIbeta develop to maturity but show a reduced mean lifespan. Proc. Natl. Acad. Sci. USA 98**:** 5717–5721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lea, D. E., and C. A. Coulson, 1949. The distribution in the numbers of mutants in bacterial populations. J. Genet. 49**:** 264–285. [DOI] [PubMed] [Google Scholar]
- Li, W., and J. C. Wang, 1998. Mammalian DNA topoisomerase IIIalpha is essential in early embryogenesis. Proc. Natl. Acad. Sci. USA 95**:** 1010–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby, M., R. Rothstein and U. H. Mortensen, 2001. Rad52 forms DNA repair and recombination centers during S phase. Proc. Natl. Acad. Sci. USA 98**:** 8276–8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby, M., U. H. Mortensen and R. Rothstein, 2003. Colocalization of multiple DNA double-strand breaks at a single Rad52 repair centre. Nat. Cell Biol. 5**:** 572–577. [DOI] [PubMed] [Google Scholar]
- Lisby, M., J. H. Barlow, R. C. Burgess and R. Rothstein, 2004. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell 118**:** 699–713. [DOI] [PubMed] [Google Scholar]
- Lopes, M., C. Cotta-Ramusino, A. Pellicioli, G. Liberi, P. Plevani et al., 2001. The DNA replication checkpoint response stabilizes stalled replication forks. Nature 412**:** 557–561. [DOI] [PubMed] [Google Scholar]
- Lucca, C., F. Vanoli, C. Cotta-Ramusino, A. Pellicioli, G. Liberi et al., 2004. Checkpoint-mediated control of replisome-fork association and signalling in response to replication pausing. Oncogene 23**:** 1206–1213. [DOI] [PubMed] [Google Scholar]
- McDonald, J. P., and R. Rothstein, 1994. Unrepaired heteroduplex DNA in Saccharomyces cerevisiae is decreased in _RAD1 RAD52_-independent recombination. Genetics 137**:** 393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVey, M., M. Kaeberlein, H. A. Tissenbaum and L. Guarente, 2001. The short life span of Saccharomyces cerevisiae sgs1 and srs2 mutants is a composite of normal aging processes and mitotic arrest due to defective recombination. Genetics 157**:** 1531–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel, B., M. J. Flores, E. Viguera, G. Grompone, M. Seigneur et al., 2001. Rescue of arrested replication forks by homologous recombination. Proc. Natl. Acad. Sci. USA 98**:** 8181–8188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morey, N. J., P. W. Doetsch and S. Jinks-Robertson, 2003. Delineating the requirements for spontaneous DNA damage resistance pathways in genome maintenance and viability in Saccharomyces cerevisiae. Genetics 164**:** 443–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen, J. R., V. Kaliraman, S. S. Ibrahim and S. J. Brill, 2001. Requirement for three novel protein complexes in the absence of the Sgs1 DNA helicase in Saccharomyces cerevisiae. Genetics 157**:** 103–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakumo, Y., 2002. The property of DNA polymerase zeta: REV7 is a putative protein involved in translesion DNA synthesis and cell cycle control. Mutat. Res. 510**:** 37–44. [DOI] [PubMed] [Google Scholar]
- Oakley, T. J., and I. D. Hickson, 2002. Defending genome integrity during S-phase: putative roles for RecQ helicases and topoisomerase III. DNA Repair 1**:** 175–207. [DOI] [PubMed] [Google Scholar]
- Rabitsch, K. P., A. Toth, M. Galova, A. Schleiffer, G. Schaffner et al., 2001. A screen for genes required for meiosis and spore formation based on whole-genome expression. Curr. Biol. 11**:** 1001–1009. [DOI] [PubMed] [Google Scholar]
- Radding, C. M., 1993. A universal recombination filament. Curr. Biol. 3**:** 358–360. [DOI] [PubMed] [Google Scholar]
- Reid, R. J., M. Lisby and R. Rothstein, 2002. Cloning-free genome alterations in Saccharomyces cerevisiae using adaptamer-mediated PCR. Methods Enzymol. 350**:** 258–277. [DOI] [PubMed] [Google Scholar]
- Rose, M. D., F. Winston and P. Heiter, 1990 Methods in Yeast Genetics: A Laboratory Course Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Rothstein, R., C. Helms and N. Rosenberg, 1987. Concerted deletions and inversions are caused by mitotic recombination between delta sequences in Saccharomyces cerevisiae. Mol. Cell. Biol. 7**:** 1198–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman, F., G. R. Fink and J. B. Hicks, 1986 Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Shor, E., S. Gangloff, M. Wagner, J. Weinstein, G. Price et al., 2002. Mutations in homologous recombination genes rescue top3 slow growth in Saccharomyces cerevisiae. Genetics 162**:** 647–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair, D. A., K. Mills and L. Guarente, 1997. Accelerated aging and nucleolar fragmentation in yeast sgs1 mutants. Science 277**:** 1313–1316. [DOI] [PubMed] [Google Scholar]
- Sogo, J. M., M. Lopes and M. Foiani, 2002. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 297**:** 599–602. [DOI] [PubMed] [Google Scholar]
- Stasiak, A. Z., E. Larquet, A. Stasiak, S. Muller, A. Engel et al., 2000. The human Rad52 protein exists as a heptameric ring. Curr. Biol. 10**:** 337–340. [DOI] [PubMed] [Google Scholar]
- Stewart, E., C. R. Chapman, F. Al-Khodairy, A. M. Carr and T. Enoch, 1997. rqh1+, a fission yeast gene related to the Bloom's and Werner's syndrome genes, is required for reversible S phase arrest. EMBO J. 16**:** 2682–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson, R. L., N. J. Morey, P. W. Doetsch and S. Jinks-Robertson, 1999. Overlapping specificities of base excision repair, nucleotide excision repair, recombination, and translesion synthesis pathways for DNA base damage in Saccharomyces cerevisiae. Mol. Cell. Biol. 19**:** 2929–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington, L. S., 2002. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol. Mol. Biol. Rev. 66**:** 630–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, B. J., and R. Rothstein, 1989. Elevated recombination rates in transcriptionally active DNA. Cell 56**:** 619–630. [DOI] [PubMed] [Google Scholar]
- van Brabant, A. J., T. Ye, M. Sanz, I. J. German, N. A. Ellis et al., 2000. Binding and melting of D-loops by the Bloom syndrome helicase. Biochemistry 39**:** 14617–14625. [DOI] [PubMed] [Google Scholar]
- Veaute, X., J. Jeusset, C. Soustelle, S. C. Kowalczykowski, E. Le Cam et al., 2003. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature 423**:** 309–312. [DOI] [PubMed] [Google Scholar]
- Wallis, J. W., G. Chrebet, G. Brodsky, M. Rolfe and R. Rothstein, 1989. A hyper-recombination mutation in S. cerevisiae identifies a novel eukaryotic topoisomerase. Cell 58**:** 409–419. [DOI] [PubMed] [Google Scholar]
- Watt, P. M., E. J. Louis, R. H. Borts and I. D. Hickson, 1995. Sgs1: a eukaryotic homolog of E. coli RecQ that interacts with topoisomerase II in vivo and is required for faithful chromosome segregation. Cell 81**:** 253–260. [DOI] [PubMed] [Google Scholar]
- Watt, P. M., I. D. Hickson, R. H. Borts and E. J. Louis, 1996. SGS1, a homologue of the Bloom's and Werner's syndrome genes, is required for maintenance of genome stability in Saccharomyces cerevisiae. Genetics 144**:** 935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, H. I., J. A. Brown, M. J. Dorie, L. Lazzeroni and J. M. Brown, 2004. Genome-wide identification of genes conferring resistance to the anticancer agents cisplatin, oxaliplatin, and mitomycin C. Cancer Res. 64**:** 3940–3948. [DOI] [PubMed] [Google Scholar]
- Wu, L., and I. D. Hickson, 2003. The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature 426**:** 870–874. [DOI] [PubMed] [Google Scholar]
- Wu, L., S. L. Davies, P. S. North, H. Goulaouic, J. F. Riou et al., 2000. The Bloom's syndrome gene product interacts with topoisomerase III. J. Biol. Chem. 275**:** 9636–9644. [DOI] [PubMed] [Google Scholar]
- Wu, L., S. L. Davies, N. C. Levitt and I. D. Hickson, 2001. Potential role for the BLM helicase in recombinational repair via a conserved interaction with RAD51. J. Biol. Chem. 276**:** 19375–19381. [DOI] [PubMed] [Google Scholar]