The domain of the Bacillus subtilis DEAD-box helicase YxiN that is responsible for specific binding of 23S rRNA has an RNA recognition motif fold (original) (raw)
Abstract
The YxiN protein of Bacillus subtilis is a member of the DbpA subfamily of prokaryotic DEAD-box RNA helicases. Like DbpA, it binds with high affinity and specificity to segments of 23S ribosomal RNA as short as 32 nucleotides (nt) that include hairpin 92. Several experiments have shown that the 76-residue carboxy-terminal domain of YxiN is responsible for the high-affinity RNA binding. The domain has been crystallized and its structure has been solved to 1.7 Å resolution. The structure reveals an RNA recognition motif (RRM) fold that is found in many eukaryotic RNA binding proteins; the RRM fold was not apparent from the amino acid sequence. The domain has two solvent exposed aromatic residues at sites that correspond to the aromatic residues of the ribonucleoprotein (RNP) motifs RNP1 and RNP2 that are essential for RNA binding in many RRMs. However, mutagenesis of these residues (Tyr404 and Tyr447) to alanine has little effect on RNA affinity, suggesting that the YxiN domain binds target RNAs in a manner that differs from the binding mode commonly found in many eukaryotic RRMs.
Keywords: DEAD-box RNA helicase, RNA recognition motif, RNA binding, X-ray crystallography, mutagenesis
INTRODUCTION
Proteins of the DEx(D/H)-box RNA helicase family participate in a broad spectrum of activities involving rearrangement of RNA structure and modulation of protein–RNA interactions (Rocak and Linder 2004). Their activities range from relatively nonspecific with respect to the sequence and structure of the RNA substrate, such as the “melting out” of secondary structure in mRNAs by eukaryotic initiation factor 4A (eIF4A) (Pause and Sonenberg 1992; Rogers et al. 1999), to highly selective, as illustrated by participation of several DEx(D/H)-box proteins in the sequential multistep recognition and rearrangements of RNA structures around the exon junction in the yeast spliceosome (Staley and Guthrie 1998). Consistent with the diversity of activities, DEx(D/H)-box helicase proteins are variable with respect to amino acid sequence and size, ranging from fewer than 400 to more than 1200 residues. Additionally, accessory proteins cooperate with some DEx(D/H)-box helicases in their activity (Silverman et al. 2003); for example, initiation factor 4B (eIF4B) enhances the helicase activity of eIF4A (Jaramillo et al. 1991). Nonetheless, proteins of the family share a highly conserved fragment of ∼400 residues that embraces seven short sequence motifs that were originally used to identify them as belonging to the broader family of RNA and DNA helicases (Gorbalenya and Koonin 1993), including the Walker ATP binding motifs (Walker et al. 1982).
Within the DEx(D/H)-box helicase family, DbpA (DEAD-box protein A), has been shown to have a high (∼10 nM) affinity for 23S ribosomal RNA (rRNA) (Fuller-Pace et al. 1993; Nicol and Fuller-Pace 1995; Tsu et al. 2001), and appears to facilitate ribosome maturation in a manner that is not yet well understood. The high-affinity RNA binding can be accomplished by short oligonucleotides that include hairpin 92 of 23S rRNA with a short single-strand extension (Tsu et al. 2001). The primary structure of proteins in the DbpA subfamily consists of the minimal helicase fragment connected to a small, 70- to 80-residue domain at the carboxy terminus by a peptide linker of variable length. It was recently shown that the 76-residue carboxy-terminal fragment (residues 404–479) of YxiN, the Bacillus subtilis ortholog of DbpA (Kossen and Uhlenbeck 1999), binds oligonucleotides derived from 23S rRNA with essentially the same affinity and sequence specificity as the full-length protein (Karginov et al. 2005). It was also shown that the ATPase activity of the minimal helicase fragment of YxiN (residues 1–368) is similar to that of eIF4A, a helicase of minimal length that is unrelated to YxiN in biological function. Additionally, it has been shown that when the carboxy-terminal fragment of the Escherichia coli SrmB protein, a DEAD-box helicase with no observed sequence specificity in RNA binding, is replaced with the carboxy-terminal RNA binding domain (RBD) of YxiN, the resulting chimera has the RNA binding affinity and specificity of YxiN but retains the RNA-dependent ATPase activity of SrmB (Kossen et al. 2002). These data support a model in which this class of proteins is modular with the peripheral domains targeting the catalytic helicase domains to the desired site on rRNA.
Structures have been reported for yeast eIF4A (Caruthers et al. 2000), a putative DEAD-box helicase from Methanococcus jannaschi (Story et al. 2001), and fragments of the human UAP56 protein (Shi et al. 2004) and yeast Dhh1p protein (Cheng et al. 2005), each of which represents a minimal helicase fragment. Consistent with the general scheme of modular helicase architecture (Caruthers and McKay 2002), these structures have two domains, each of which has a folding topology related to that of the RecA protein, with linkers of various lengths between the domains. However, structures that include peripheral domains have not emerged. Here, we report the structure of the carboxy-terminal RBD of the B. subtilis YxiN protein. The tertiary fold is similar to that of RNA recognition motifs (RRMs) that are prevalent in eukaryotes, such as the spliceosomal U1A protein (Nagai et al. 1990). However, RNA binding assays of mutant YxiN RBD protein fragments suggest that the mode in which this domain binds RNA differs substantially from that of the eukaryotic RRMs.
RESULTS
We have solved the crystallographic structure of the YxiN RBD to 1.7 Å resolution. The majority of the polypeptide is unambiguous in the experimental electron density map, as are several well-ordered solvent molecules (Fig. 1A). The register of the sequence placement was further confirmed by the sites of three selenium atoms in selenomethionine (SeMet)-labeled protein. Despite this, two segments of polypeptide remained recalcitrant to model building and refinement, presumably due to multiple conformations. The first segment includes residues 410–423, spanning a glycine, lysine-rich loop leading to an α-helix (α1, Fig. 1B). Gly423, in the middle of the helix, is a break point between somewhat poorly defined electron density leading up to it and well-defined electron density following it. Ultimately, the first part of this helix was modeled as two conformations of equal occupancy; this is probably an approximate description of multiple conformations that are significantly more complex. The second segment spans residues 468–470, for which it was not possible to model conformations with acceptable geometries. As a consequence, these two regions of the molecule remain ambiguous.
FIGURE 1.

Structure of the YxiN RBD. (A) Experimental solvent-flipped electron density map contoured at 1.8σ. Molecular model shown for reference. (B) Ribbon drawing of the RBD structure. Dotted lines indicate segments of polypeptide that could not be traced. The side chains of Tyr407 and Tyr447 are also shown. (C) Superposition of the U1A RNA binding domain (PDB ID 1URN) and YxiN RBD, represented as a coil passing through Cα positions. Segments of polypeptide for which the distance between equivalent Cα’s of the two structures is <1.5 Å are shown in yellow; segments for which the distance is >1.5 Å, in magenta (YxiN) and cyan (U1A). Side chains of YxiN residues Tyr407 and Tyr447 are shown in red; side chains of the equivalent U1A residues Tyr13 and Phe56, in green. (D) Manner in which RNA nucleotide bases stack on aromatic side chains of U1A. RNA nucleotides C10 and A11, magenta; U1A Tyr13 and Phe56 side chains, green. A, C, D, and Figure 3 were made with Pymol (http://pymol.sourceforge.net/index.php); B was made with Molscript (Kraulis 1991) and rendered with Raster3D (Merritt and Bacon 1997).
The YxiN RNA binding domain has the architecture of an RRM, also called the ribonucleoprotein (RNP) motif, which was originally described for the eukaryotic RNA binding protein U1A (Nagai et al. 1990) and which has subsequently been found in a large number of proteins involved in sequence-specific RNA binding (Fig. 1). As with other RRMs, the YxiN RBD is an α–β sandwich with four antiparallel β-strands on one face and two helices on the other. The order of secondary structure elements in the polypeptide sequence is β–α–β–β–α–β; topology of strands in the β-sheet is 4–1–3–2. These elements of the YxiN RBD secondary structure superimpose well on those of other RRMs. For example, the U1A domain superimposes on the YxiN domain with a root mean square difference (RMSD) in α-carbon position of 1.4 Å for 51 closest proximity residues out of a total of 76 (Fig. 1C). Loops connecting the α-helices and β-strands differ substantially between YxiN and the eukaryotic RRMs. For example, in most RRMs the connection from β-strand 2 to strand 3 is an extended loop that forms part of a “cradle” for RNA binding, while in the YxiN RBD, the two strands are connected by a short β-turn.
In structures of complexes between RRMs and their cognate RNAs, two surface-exposed aromatic residues that are structurally equivalent to Tyr407 and Tyr447 of YxiN are usually involved in ssRNA binding through stacking interactions with RNA bases (Maris et al. 2005; Fig. 1C–D). Within the DbpA/YxiN subfamily of DEAD-box RNA helicases, tyrosine or phenylalanine is always found at the position equivalent to Tyr447 of YxiN (Karginov et al. 2005). The amino acid at position 407 is more variable; tyrosine and cysteine are the most common amino acids at this position, but serine, arginine, glutamine, and glutamic acid are also found. In this context, we prepared the Y407C, Y407A, and Y447A mutations of the YxiN RBD. Since cysteine is found at position 407 in E. coli DbpA, the Y407C mutation should not significantly impair RNA binding, but the latter two would be expected to if these residues participate in RNA binding through stacking of nucleotide bases on the aromatic side chains.
A gel mobility shift assay was used to measure affinities of the wild-type and mutant RBD proteins for RNA, in the same manner as done previously with the wild-type protein (Karginov et al. 2005). RNAs used in the binding experiments were a wild-type 32-mer fragment of 23S RNA that has been shown previously to bind with high affinity to DbpA and YxiN (sequences A and B, Fig. 2A), and mutant oligonucleotides derived from this sequence (sequences C and D, Fig. 2A; Tsu et al. 2001). Gel shifts of oligonucleotide mobility of a protein concentration range of 0–500 nM were measured (representative data, Fig. 2B). The wild-type RBD binds the wild-type 32-mer RNA (oligonucleotide A) with essentially the same affinity as reported previously in Karginov et al. (2005). Somewhat surprisingly, all three of the mutant RBDs bind RNA A with affinities that do not differ significantly from that of the wild-type protein (Table 1; representative data, Fig. 2C). The binding of the three mutant domains to RNA A was also measured in 200 mM KCl to attenuate sequence-nonspecific binding; while the higher salt reduced the affinities modestly, the values of the dissociation constant were not substantially different from the wild-type domain under lower ionic strength conditions.
FIGURE 2.

Binding of the YxiN RBD to RNA. (A) RNA 32-mer oligonucleotides used for binding measurements. RNA A is the wild-type sequence derived from 23S RNA, which includes hairpin H92. Mutations in oligonucleotides B–D are shown with large outline characters. (B) Representative gel shift assay data showing the binding of wild-type and Y447A mutant RBD proteins to RNA B. (C) Representative binding curves, plotting the binding of YxiN RBD wild-type (filled symbols) and Y447A mutant (open symbols) protein to RNA B (circles) and RNA D (triangles).
TABLE 1.
Measured values of dissociation constants for binding of RNA oligonucleotides to wild-type and mutant YxiN RBDs

The pattern of binding of mutant RNA oligonucleotides by the mutant proteins also paralleled that of the wild-type protein. Mutations in the single-strand extension (RNA B) do not impair affinity, while mutations in the loop or base of the stem of the hairpin (RNAs C and D) abolish high affinity binding for the mutant proteins as well as for the wild-type protein. Thus the three mutant domains have affinity and specificity for RNA that is indistinguishable from that of the wild-type domain.
DISCUSSION
The RRM domain occurs frequently in eukaryotic genomes with ∼100 examples in the Caenorhabditis elegans genome (Washington University Genome Sequencing Center 1998) and nearly 200 in the Arabidopsis genome (Lorković and Barta 2002). The Pfam family for RRMs (Pfam 00,076) (Bateman et al. 2000) currently lists >7000 sequences. These sequences are predominantly eukaryotic in origin, but they include at least 135 from bacteria (of which approximately half are identified in cyanobacteria) and three from viruses. These RRMs were originally defined by two short, conserved “ribonucleoprotein (RNP)” peptide sequences, of length eight and six amino acid residues, called RNP1 and RNP2, respectively (for review, see Maris et al. 2005). The YxiN/DbpA RNA binding domain (Pfam 03,880) does not readily fit the consensus RRM sequence profile, with only a very weak similarity in a region around RNP1 (Karginov et al. 2005). Structures of eukaryotic RRM domains of the Pfam 00,076 family have four antiparallel β-strands with two α-helices on one face in a βαββαβ topology, with RNP1 and RNP2 embedded in β-strands 3 and 1, respectively (Nagai et al. 1990). The structure of the YxiN RBD presented here reveals that, despite the lack of apparent sequence similarity, it has the same tertiary fold as RRMs of the Pfam 00,076 family.
Structures of several eukaryotic RRM proteins or protein fragments complexed with their RNA targets have been reported. These include the spliceosomal U1A protein (Oubridge et al. 1994), and also its homolog U2B′′ in conjunction with U2A′ (Price et al. 1998), a poly(A) binding protein (Deo et al. 1999), the Drosophila sex-lethal protein (Handa et al. 1999), and the HuD protein (Wang and Tanaka Hall 2001). In all these examples, a segment of single-strand RNA binds across the face of the β-sheet. A shared feature in these structures is the specific interaction of a tandem pair of nucleotides with a subset of conserved amino acids localized to positions 1, 3, and 5 of the eight-residue RNP1 and position 2 of the six-residue RNP2 (Maris et al. 2005). Interactions between the amino acids of RNP1 and the 3′ nucleotide of the tandem pair typically include arginine or lysine at position 1 making a salt bridge to the phosphate, a hydrophobic residue at position 3 interacting with the ribose, and the side chain of tyrosine or phenylalanine at position 5 forming a stacking interaction with the nucleotide base. The base of the 5′ nucleotide of the pair stacks with the side chain of tyrosine or phenylalanine at RNP2 position 2. All of the structures of eukaryotic RRMs complexed with RNA manifest some, and in several cases all, of these four interactions. Additional interactions with the RNA or DNA typically employ residues of the polypeptide backbone of β-strand 4 (described in more detail in Wang and Tanaka Hall 2001). A similar binding scheme for single-strand DNA is seen in the structure of hnRNP A1 protein complexed with single-stranded telomeric DNA (Ding et al. 1999). These data illustrate a common mode of binding of single strand segments of RNA or DNA by RRMs.
Superposition of the YxiN RBD domain on structures of other RRMs assigns residues 443–450 (sequence DNASYVEI) to RNP1 and 406–411 (sequence LYFNGG) to RNP2. In the DbpA/YxiN subfamily, at the structural equivalent of RNP1 position 1, an acidic residue or a proline is commonly found, in contrast to the conserved basic residue in eukaryotic RRMs. The residue at position 3 is variable, while position 5 is conservative, having either tyrosine or phenylalanine. The residue at the structural equivalent of RNP2 position 2 is also variable; tyrosine and cysteine are the most common amino acids at this position, but serine, arginine, glutamine, and glutamic acid are also found. The features that are most similar between YxiN and the conserved residues that interact with RNA in the eukaryotic RRMs are Tyr447 at position 5 of RNP1 and Tyr407 at position 2 of RNP2. Hence, these residues were targeted for mutagenesis to test whether the YxiN RBD binds RNA in a similar manner. Each residue was mutated to alanine; in addition, Tyr407 was mutated to cysteine, the residue found at this position in E. coli DbpA. All of the mutant proteins bind RNA with essentially wild-type affinity.
Two arguments suggest that the YxiN RBD binds RNA in a manner that differs from the common mode of binding observed in structures of eukaryotic RRMs complexed with nucleic acid binding targets. First, we show here that although YxiN contains aromatic residues at the structurally orthologous positions that stack on ssRNA in the eukaryotic RRMs, their mutation to alanine does not significantly impair specific RNA binding. This can be contrasted to data on the U1A protein–RNA complex. Mutation of the residue of RNP1 that base-stacks with RNA, Phe56, to alanine reduces the binding affinity by 4 orders of magnitude, while mutation to leucine reduces affinity by 3 orders of magnitude, and a relatively conservative change to tyrosine reduces the RNA affinity 50-fold (Nolan et al. 1999). Mutation of the U1A residue of RNP2 that base-stacks with RNA, Tyr13, to phenylalanine reduces the binding affinity 25-fold (Jessen et al. 1991).
Second, a comparison of members of the DbpA protein subfamily indicate that aside from Tyr447 in RNP1, most of the residues on the face of the β-sheet are not conserved, arguing against their involvement in specific binding of RNA in a manner shared by all DbpA proteins. Residues of high conservation localize primarily in the two glycine-, lysine-rich loops (410–415 between β1 and α1; 468–470 between α2 and β4) and in the interior of the protein. The high level of sequence conservation of the glycine-, lysine-rich loops, in combination with their significant level of disorder in the crystal structure in the absence of an RNA ligand, is suggestive of a participation in RNA binding. (Interestingly, the residues that align with Gly411 and Gly423 of YxiN are conserved across both the DbpA and CsdA subfamilies; however, the basic residues between these two glycines that are conserved in DbpA and its homologs are variable in the CsdA homologs.) Additionally, high-affinity binding of RNA by the YxiN RBD requires both the single-strand loop of the H92 hairpin and a ssRNA extension of several nucleotides 5′ to the 5-bp stem of the hairpin (C.M. Diges and O.C. Uhlenbeck, in prep.). Mutagenesis of the RNA confirms sequence-specific dependence on the single-strand loop of the hairpin. However, if this loop were docked to the YxiN RBD in a manner consistent with the ssRNA binding scheme of eukaryotic RRMs, the single-strand extension at the distal end of the duplex stem would be a significant distance away from the protein (Fig. 3). Thus it appears that YxiN has developed an alternative strategy to achieve sequence-specific recognition of hairpin 92 of 23S rRNA.
FIGURE 3.

YxiN RBD with conserved residues highlighted. (Right) Ribbon drawing on YxiN RBD, with residues shown that have conservative substitutions in 36 aligned sequences (Karginov et al. 2005). Conserved external residues, with side chains presented as stick models in red: K412, K413, K415, R417, D420, F/Y447, K468, K470. Internal residues having only conservative substitution (L/I/V), with side chains shown as green stick model surrounded by semitransparent surface: V422, I425, V431, I436, I439, I441, V448, I450, V467. Conserved glycines, shown as magenta spheres at Cα positions: G411, G423, G437, G469. Residues that are included in figure in an approximate conformation that follows continuous electron density but not in final model due to inability to precisely define their conformation: 415–418 and 468–470. Selected residues are labeled. (Left) For reference, an anticodon fragment of tRNA (taken from PDB 1EIY) having the same features of secondary structure as the tight-binding RNA target of the YxiN RBD: a hairpin with a 5-bp stem and a 5-nt loop, plus a single-strand extension.
Alternative modes of sequence-specific RNA binding by protein domains with the RRM fold are found in NMR structures of complexes of an RNA binding fragment of nucleolin with its target RNAs, the nucleolin recognition elements (NREs) (Allain et al. 2000a,b; Johansson et al. 2004), and also in the crystal structure of the complex between Thermus thermophilus phenylalanyl tRNA synthetase and its tRNA (PDB ID 1EIY) (Goldgur et al. 1997). The nucleolin fragment includes the two amino-terminal RRM domains and the polypeptide linker between them. This fragment binds both a NRE derived from in vitro selection (Allain et al. 2000a,b) and a naturally occurring sequence (b2NRE) (Johansson et al. 2004) with high affinity and sequence specificity. A substantial fraction of the binding interactions of the RNA are with amino acid residues of the polypeptide linker between the RRM domains. The aromatic residues Phe17 of RNP2 and Tyr58 of RNP1 in the RRM-1 of nucleolin do not interact with the RNA through parallel stacking of planar rings of the amino acids and RNA and thus differ from the consensus binding mode seen in cases where a single RRM binds a segment of ssRNA. Those parts of the nucleolin fragment that interact directly with RNA only become well-ordered in the protein–RNA complex; they are disordered in the absence of ligand.
In the synthetase–tRNA complex, the anticodon stem-loop binds a domain of the synthetase that has an RRM fold. The anticodon loop interfaces to the loop between β-strands 2 and 3, as well as to other residues on the β-sheet. Translation of this general scheme to the YxiN RBD suggests one candidate binding interface. Figure 3 shows the YxiN RBD, highlighting the conserved external (red), glycine (magenta spheres), and conservative substitution internal (green) residues, and for comparison, a fragment of RNA with a stem-loop and single-strand extension similar to that of the H92 hairpin. It would be feasible for the loop of the hairpin, in which mutations impair binding, to bind at the base of the protein, possibly interfacing to the glycine, lysine-rich loops. It would then be sterically feasible for the ssRNA extension at the distal end of the duplex to bind the other extremity of the protein. Such a binding scheme would allow significant interactions with both the loop and the ssRNA extension by a single RBD protomer. It would juxtapose the single-strand loop of the hairpin to a region of the YxiN RBD where the highly conserved external amino acid residues are localized. This would also suggest that those parts of the YxiN RBD that are most poorly ordered in the structure in the absence of RNA may be extensively involved in specific RNA recognition. Whether RNA is bound in this manner or in some alternative manner can only be answered by structural work on protein–RNA complexes.
MATERIALS AND METHODS
Plasmids for expression of recombinant YxiN(404–479)
The subcloning of the coding sequence for wild-type YxiN residues 404–479 into the pTWIN1 vector of an intein-based expression system (New England Biolabs) has been described (Karginov et al. 2005). Expression vectors for proteins with mutations Y407C, Y407A, and Y447A were constructed from the parent expression vector using suitable primers and the QuikChange mutagenesis system (Stratagene). Mutations were confirmed by DNA sequencing of the protein coding regions of the expression plasmids.
Protein expression and purification
Wild-type and mutant YxiN(404–479) fragment proteins were expressed and purified using the protocol described previously (Karginov et al. 2005). Expression of seleno-L-methionine (SeMet)-labeled YxiN(404–479) was carried out in the methionine auxotroph E. coli strain B834(DE3). The medium for protein expression was 2× M9 minimal salts enriched with 0.4% glucose, supplemented with 2 mM MgSO4, 25 μg mL−1 FeSO4·7H2O, 1 μg mL−1 riboflavin, 1μg mL−1 thiamine, and 100 μg mL−1 ampicillin. All amino acids were added at a concentration of 40 μg/mL, with SeMet (Sigma) substituted for methionine (Yu et al. 1998). Cells were grown at 37°C in this expression medium to a cell density at which A600 = 0.6, at which point protein expression was induced by adding 0.4 mM IPTG; the cells were incubated for an additional 4 h in a shaker at 25°C and then harvested. The subsequent purification protocol for the SeMet-labeled protein was identical to that used for the native protein. To prevent oxidation of SeMet, 1 mM DTT was added to all solutions during all purification steps.
Gel mobility shift assay of rna binding
RNA oligoribonucleotides were purchased from Dharmacon, Inc., or synthesized by runoff T7 transcription as described (Tsu et al. 2001). Assays of the protein-induced shift of RNA gel mobility were carried out as described previously for the wild-type protein (Karginov et al. 2005). Briefly, a trace amount (0.4 nM) of 5′-32P-labeled RNA was titrated with increasing concentrations of proteins in 50 mM HEPES (pH 7.5), 50 or 200 mM KCl, 5 mM MgCl2, 100 μM DTT, 70 μM poly(A), 5% (v/v) glycerol, and 0.1% (v/v) Tween 20. Reactions were incubated for 10 min to reach equilibrium and 10-μL aliquots were run on 5% native acrylamide gels in 1/3× TBE (= 30 mM Tris-borate, 0.33 mM EDTA at pH ∼8.3). The amount of bound and unbound RNA in each lane was quantified with a PhosphorImager, and the fraction of bound material versus protein concentration was fit with a noncooperative binding curve to parameterize a dissociation constant (Kd).
Crystallization and crystal freezing
Wild-type and SeMet-labeled YxiN(404–479) protein were crystallized at 18°C by vapor diffusion in sitting or hanging drops using protein at an initial concentration of 10 mg/mL and a precipitant of 3.2–3.6 M (NH4)2SO4, buffered to pH in the range 9–10 with 0.1 M glycine. Crystals grew as tetragonal bipyramids with maximum dimension ∼0.05–0.10 mm in 1–2 wk. Crystals were transferred to a cryoprotectant solution of 1% (w/v) glucose, 3.2 M (NH4)2SO4, 0.1 M glycine (pH 9.5) and frozen in liquid nitrogen.
Data collection
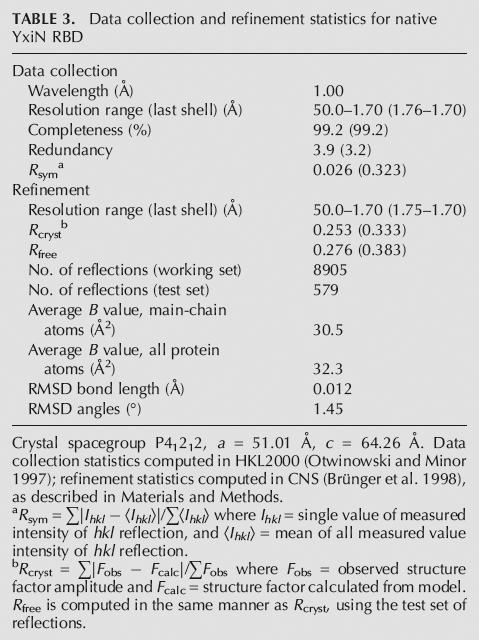
Initial multiwavelength anomalous dispersion (MAD) diffraction data from SeMet-labeled crystals were collected to 2.0 Å resolution at three wavelengths on beamline 4.2.2 of the Advanced Light Source (ALS); these data were used to locate selenium sites and for initial phasing and model building. A subsequent three-wavelength MAD data set was collected to 1.7 Å resolution on beamline 1.5 of the Stanford Synchrotron Radiation Laboratory (SSRL); these data were used for final phasing. Native diffraction data were collected to 1.7 Å resolution on ALS beamline 5.0.1 with radiation of λ = 1.00 Å; these data were used for model refinement. All data were indexed, integrated, and scaled with HKL2000 (Otwinowski and Minor 1997). Unit cell and symmetry parameters and data-collection statistics for crystals of native and SeMet-labeled protein are summarized in Tables 2 and 3.
TABLE 2.
Data collection and phasing statistics for SeMet-YxiN-RBD

TABLE 3.
Data collection and refinement statistics for native YxiN RBD
Model building and refinement
Selenium atoms were located, experimental phases were computed, and a partial model was built automatically using the program package SOLVE/RESOLVE (Terwilliger and Berendzen 1999). Manual model building was effected with the modeling program O (Jones 1978). Final experimental phase computations, map computations, and model refinement were carried out using the program CNS (Brünger et al. 1998). Three selenium sites, corresponding to the three SeMet residues in the labeled YxiN RBD, were located by the automated search algorithms of SOLVE from an anomalous difference Patterson computed from the peak wavelength data. Final MAD phases had an overall figure of merit of 0.713–1.7 Å resolution; the figure of merit was generally >0.80 to ∼2.1 Å and declined beyond that resolution. Experimental phases were improved by solvent flipping applied to the experimental electron density map to yield an electron density map that was readily interpretable for much of the molecule (Fig. 1A). Despite the clarity of the electron density maps for the majority of the molecule, three segments of the polypeptide remained problematic throughout model building and refinement. The first segment begins at residue 410 with a loop rich in glycine and lysine and extends through Gly423 in the middle of the first helix. Although residues 410–413 could be modeled into electron density with little ambiguity, the remainder of the loop and the beginning of the helix, starting at Ala418, remained ambiguous. Difference Fourier maps gave electron density consistent with multiple conformations for this segment (Fig. 4), and the refinement statistics improved significantly when residues 418–423 of helix 1 were modeled in two conformations, each having 50% occupancy. However, residues 414–417 could not be placed in electron density reliably and are absent from the final model. The inability to decipher the apparent multiple conformations of this region presumably contributes to the uncharacteristically high values of the crystallographic and free _R_-factors in the refinement. Similarly, residues 468–470, which are in a loop connecting helix 2 and β-strand 4, and the carboxy-terminal residue 479 could not be modeled reliably. Forty-eight solvent molecules and 2 sulfate ions were apparent in the electron density maps and were included in the final model. Coordinates have been deposited with the Protein Data Bank (ID 2G0C).
FIGURE 4.

Electron density maps in region of helix α1, which displays conformational heterogeneity. Stick models for part of helix in unique conformation (residues 424–427, top of figures) colored green. Segment of helix built in two conformations (residues 418–423): alternate conformation 1 (AC1), green; AC2, red. The break point in the alternate conformations is residue Gly423. (A) Fo − Fc map computed with only AC2 included in calculation of model phases, contoured at 2.5σ. (B) Fo − Fc map computed with only AC1 included in calculation of model phases, contoured at 2.5σ. (C) 2_Fo_ − Fc simulated annealing omit map, contoured at 1.2σ. For clarity, orientation of the model in C differs slightly from that of A and B. Figure made with Pymol (http://pymol.sourceforge.net/index.php).
ACKNOWLEDGMENTS
This work was supported by grants GM71696 to DBM and GM60268 to OCU from the NIH. M.T.O. was supported by fellowships from The Alfred Benzon Foundation and Diagnostic Systems Laboratories, Inc. Parts of this research were carried out at the Stanford Synchrotron Radiation Laboratory (SSRL), a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Science. The SSRL Structural Molecular Biology Program is supported by the Department of Energy and by the NIH. Parts of this research were carried out at the Advanced Light Source of the Lawrence Berkeley Laboratory.
Footnotes
REFERENCES
- Allain F.H., Bouvet P., Dieckmann T., Feigon J. Molecular basis of sequence-specific recognition of pre-ribosomal RNA by nucleolin. EMBO J. 2000a;19:6870–6881. doi: 10.1093/emboj/19.24.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allain F.H., Gilbert D.E., Bouvet P., Feigon J. Solution structure of the two N-terminal RNA-binding domains of nucleolin and NMR study of the interaction with its RNA target. J. Mol. Biol. 2000b;303:227–241. doi: 10.1006/jmbi.2000.4118. [DOI] [PubMed] [Google Scholar]
- Bateman A., Birney E., Durbin R., Eddy S.R., Howe K.L., Sonnhammer E.L. The Pfam protein families database. Nucleic Acids Res. 2000;28:263–266. doi: 10.1093/nar/28.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S., et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Caruthers J.M., McKay D.B. Helicase structure and mechanism. Curr. Opin. Struct. Biol. 2002;12:123–133. doi: 10.1016/s0959-440x(02)00298-1. [DOI] [PubMed] [Google Scholar]
- Caruthers J.M., Johnson E.R., McKay D.B. Crystal structure of yeast initiation factor 4A, a DEAD-box RNA helicase. Proc. Natl. Acad. Sci. 2000;97:13080–13085. doi: 10.1073/pnas.97.24.13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H., Coller J., Parker R., Song H. Crystal structure and functional analysis of DEAD-box protein Dhh1p. RNA. 2005;11:1258–1270. doi: 10.1261/rna.2920905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deo R.C., Bonanno J.B., Sonenberg N., Burley S.K. Recognition of polyadenylate RNA by the poly(A)-binding protein. Cell. 1999;98:835–845. doi: 10.1016/s0092-8674(00)81517-2. [DOI] [PubMed] [Google Scholar]
- Ding J., Hayashi M.K., Zhang Y., Manche L., Krainer A.R., Xu R.M. Crystal structure of the two-RRM domain of hnRNP A1 (UP1) complexed with single-stranded telomeric DNA. Genes & Dev. 1999;13:1102–1115. doi: 10.1101/gad.13.9.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller-Pace F.V., Nicol S.M., Reid A.D., Lane D.P. DbpA: A DEAD box protein specifically activated by 23s rRNA. EMBO J. 1993;12:3619–3626. doi: 10.1002/j.1460-2075.1993.tb06035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldgur Y., Mosyak L., Reshetnikova L., Ankilova V., Lavrik O., Khodyreva S., Safro M. The crystal structure of phenylalanyl-tRNA synthetase from Thermus thermophilus complexed with cognate tRNAPhe. Structure. 1997;5:59–68. doi: 10.1016/s0969-2126(97)00166-4. [DOI] [PubMed] [Google Scholar]
- Gorbalenya A.E., Koonin E.V. Helicases: Amino acid sequence comparisons and structure-function relationships. Curr. Opin. Struct. Biol. 1993;3:419–429. [Google Scholar]
- Handa N., Nureki O., Kurimoto K., Kim I., Sakamoto H., Shimura Y., Muto Y., Yokoyama S. Structural basis for recognition of the tra mRNA precursor by the Sex-lethal protein. Nature. 1999;398:579–585. doi: 10.1038/19242. [DOI] [PubMed] [Google Scholar]
- Jaramillo M., Dever T.E., Merrick W.C., Sonenberg N. RNA unwinding in translation: Assembly of helicase complex intermediates comprising eukaryotic initiation factors eIF-4F and eIF-4B. Mol. Cell. Biol. 1991;11:5992–5997. doi: 10.1128/mcb.11.12.5992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen T.H., Oubridge C., Teo C.H., Pritchard C., Nagai K. Identification of molecular contacts between the U1 A small nuclear ribonucleoprotein and U1 RNA. EMBO J. 1991;10:3447–3456. doi: 10.1002/j.1460-2075.1991.tb04909.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson C., Finger L.D., Trantirek L., Mueller T.D., Kim S., Laird-Offringa I.A., Feigon J. Solution structure of the complex formed by the two N-terminal RNA-binding domains of nucleolin and a pre-rRNA target. J. Mol. Biol. 2004;337:799–816. doi: 10.1016/j.jmb.2004.01.056. [DOI] [PubMed] [Google Scholar]
- Jones A. A graphics model building and refinement system for macromolecules. J. Appl. Crystallogr. 1978;11:268–272. [Google Scholar]
- Karginov F.V., Caruthers J.M., Hu Y., McKay D.B., Uhlenbeck O.C. YxiN is a modular protein combining a DExD/H core and a specific RNA binding domain. J. Biol. Chem. 2005;280:35499–35505. doi: 10.1074/jbc.M506815200. [DOI] [PubMed] [Google Scholar]
- Kossen K., Uhlenbeck O.C. Cloning and biochemical characterization of Bacillus subtilis YxiN, a DEAD protein specifically activated by 23S rRNA: Delineation of a novel sub-family of bacterial DEAD proteins. Nucleic Acids Res. 1999;27:3811–3820. doi: 10.1093/nar/27.19.3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kossen K., Karginov F.V., Uhlenbeck O.C. The carboxy-terminal domain of the DExDH protein YxiN is sufficient to confer specificity for 23S rRNA. J. Mol. Biol. 2002;324:625–636. doi: 10.1016/s0022-2836(02)01140-3. [DOI] [PubMed] [Google Scholar]
- Kraulis P. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 1991;24:946–950. [Google Scholar]
- Lorković Z.J., Barta A. Genome analysis: RNA recognition motif (RRM) and K homology (KH) domain RNA-binding proteins from the flowering plant Arabidopsis thaliana . Nucleic Acids Res. 2002;30:623–635. doi: 10.1093/nar/30.3.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maris C., Dominguez C., Allain F.H. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 2005;272:2118–2131. doi: 10.1111/j.1742-4658.2005.04653.x. [DOI] [PubMed] [Google Scholar]
- Merritt E.A., Bacon D.J. Raster3D: Photorealistic molecular graphics. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- Nagai K., Oubridge C., Jessen T.H., Li J., Evans P.R. Crystal structure of the RNA-binding domain of the U1 small nuclear ribonucleoprotein A. Nature. 1990;348:515–520. doi: 10.1038/348515a0. [DOI] [PubMed] [Google Scholar]
- Nicol S.M., Fuller-Pace F.V. The “DEAD box” protein DbpA interacts specifically with the peptidyltransferase center in 23S rRNA. Proc. Natl. Acad. Sci. 1995;92:11681–11685. doi: 10.1073/pnas.92.25.11681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan S.J., Shiels J.C., Tuite J.B., Cecere K.L., Baranger A.M. Recognition of an essential adenine at a protein–RNA interface: Comparison of the contributions of hydrogen bonds and a stacking interaction. J. Am. Chem. Soc. 1999;121:8951–8952. [Google Scholar]
- Otwinowski Z., Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Oubridge C., Ito N., Evans P.R., Teo C.H., Nagai K. Crystal structure at 1.92 Å resolution of the RNA-binding domain of the U1A spliceosomal protein complexed with an RNA hairpin. Nature. 1994;372:432–438. doi: 10.1038/372432a0. [DOI] [PubMed] [Google Scholar]
- Pause A., Sonenberg N. Mutational analysis of a DEAD box RNA helicase: The mammalian translation initiation factor eIF-4A. EMBO J. 1992;11:2643–2654. doi: 10.1002/j.1460-2075.1992.tb05330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price S.R., Evans P.R., Nagai K. Crystal structure of the spliceosomal U2B″-U2A′ protein complex bound to a fragment of U2 small nuclear RNA. Nature. 1998;394:645–650. doi: 10.1038/29234. [DOI] [PubMed] [Google Scholar]
- Rocak S., Linder P. DEAD-box proteins: The driving forces behind RNA metabolism. Nat. Rev. Mol. Cell Biol. 2004;5:232–241. doi: 10.1038/nrm1335. [DOI] [PubMed] [Google Scholar]
- Rogers G.W., Jr., Richter N.J., Merrick W.C. Biochemical and kinetic characterization of the RNA helicase activity of eukaryotic initiation factor 4A. J. Biol. Chem. 1999;274:12236–12244. doi: 10.1074/jbc.274.18.12236. [DOI] [PubMed] [Google Scholar]
- Shi H., Cordin O., Minder C.M., Linder P., Xu R.M. Crystal structure of the human ATP-dependent splicing and export factor UAP56. Proc. Natl. Acad. Sci. 2004;101:17628–17633. doi: 10.1073/pnas.0408172101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman E., Edwalds-Gilbert G., Lin R.J. DExD/H-box proteins and their partners: Helping RNA helicases unwind. Gene. 2003;312:1–16. doi: 10.1016/s0378-1119(03)00626-7. [DOI] [PubMed] [Google Scholar]
- Staley J.P., Guthrie C. Mechanical devices of the spliceosome: Motors, clocks, springs, and things. Cell. 1998;92:315–326. doi: 10.1016/s0092-8674(00)80925-3. [DOI] [PubMed] [Google Scholar]
- Story R.M., Li H., Abelson J.N. Crystal structure of a DEAD box protein from the hyperthermophile Methanococcus jannaschii . Proc. Natl. Acad. Sci. 2001;98:1465–1470. doi: 10.1073/pnas.98.4.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger T.C., Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr. D Biol Crystallogr. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsu C.A., Kossen K., Uhlenbeck O.C. The Escherichia coli DEAD protein DbpA recognizes a small RNA hairpin in 23S rRNA. RNA. 2001;7:702–709. doi: 10.1017/s1355838201010135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker J.E., Saraste M., Runswick M.J., Gay N.J. Distantly related sequences in the α- and β-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982;1:945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Tanaka Hall T.M. Structural basis for recognition of AU-rich element RNA by the HuD protein. Nat. Struct. Biol. 2001;8:141–145. doi: 10.1038/84131. [DOI] [PubMed] [Google Scholar]
- Washington University Genome Sequencing Center. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science. 1998;282:2012–2018. doi: 10.1126/science.282.5396.2012. [DOI] [PubMed] [Google Scholar]
- Yu R.C., Hanson P.I., Jahn R., Brünger A.T. Structure of the ATP-dependent oligomerization domain of N-ethylmaleimide sensitive factor complexed with ATP. Nat. Struct. Biol. 1998;5:803–811. doi: 10.1038/1843. [DOI] [PubMed] [Google Scholar]