Residual Human Immunodeficiency Virus Type 1 Viremia in Some Patients on Antiretroviral Therapy Is Dominated by a Small Number of Invariant Clones Rarely Found in Circulating CD4+ T Cells (original) (raw)
Abstract
Antiretroviral therapy can reduce human immunodeficiency virus type 1 (HIV-1) viremia to below the detection limit of ultrasensitive clinical assays (50 copies of HIV-1 RNA/ml). However, latent HIV-1 persists in resting CD4+ T cells, and low residual levels of free virus are found in the plasma. Limited characterization of this residual viremia has been done because of the low number of virions per sample. Using intensive sampling, we analyzed residual viremia and compared these viruses to latent proviruses in resting CD4+ T cells in peripheral blood. For each patient, we found some viruses in the plasma that were identical to viruses in resting CD4+ T cells by pol gene sequencing. However, in a majority of patients, the most common viruses in the plasma were rarely found in resting CD4+ T cells even when the resting cell compartment was analyzed with assays that detect replication-competent viruses. Despite the large diversity of pol sequences in resting CD4+ T cells, the residual viremia was dominated by a homogeneous population of viruses with identical pol sequences. In the most extensively studied case, a predominant plasma sequence was also found in analysis of the env gene, and linkage by long-distance reverse transcriptase PCR established that these predominant plasma sequences represented a single predominant plasma virus clone. The predominant plasma clones were released for months to years without evident sequence change. Thus, in some patients on antiretroviral therapy, the major mechanism for residual viremia involves prolonged production of a small number of viral clones without evident evolution, possibly by cells other than circulating CD4+ T cells.
Treatment of human immunodeficiency virus type 1 (HIV-1) infection with highly active antiretroviral therapy (HAART) reduces viremia to below the detection limit of ultrasensitive clinical assays (15, 16, 37). However, HIV-1 persists in resting CD4+ T cells (6, 8, 9, 12, 51) and possibly other reservoirs (4, 58). The latent reservoir in resting CD4+ T cells has a long half-life (11, 41, 44, 47, 56) that will likely preclude virus eradication unless novel approaches (5, 24-28, 42) can purge latently infected cells.
In patients on HAART, HIV-1 persistence is evidenced not only by the latent reservoir in resting CD4+ T cells but also by free virus in the plasma (10, 17, 19, 36, 41, 48, 52). Free virions can be found with special methods, even in patients who do not have clinically detectable viremia (10, 18, 19, 36, 52). Given the short half-life of free virus (20, 49), this residual viremia indicates active virus production. This virus production may reflect low-level ongoing replication that continues despite HAART (7, 10, 13, 14, 18, 21, 33, 48, 56) and/or release of virus from latently infected cells that become activated (19, 22, 34, 48, 55) or from other stable cellular reservoirs (4, 58). The characterization of residual viremia may provide a means for determining the importance of different mechanisms of viral persistence.
Although the presence of free virus can be detected in patients with viral loads below 50 copies/ml by using extremely sensitive reverse transcriptase (RT) PCR assays (10, 18, 36, 52), characterization of this residual viremia has been limited because of the technical difficulties involved in the analysis of extremely low numbers of viral RNA templates. To obtain sufficient numbers of independent plasma virus clones, we carried out intensive sampling in nine patients on HAART and analyzed plasma virus genotypes with a sensitive RT-PCR method. Viral variants in the plasma were compared to viruses in the latent reservoir. The results provided evidence that in some patients on HAART, much of the residual viremia is due to continued production of a small number of viral clones over prolonged periods, without evident sequence change by cells that are not well represented in the circulation. These results have implications for understanding HIV-1 persistence and treatment failure.
MATERIALS AND METHODS
Patient population.
We studied asymptomatic HIV-1-infected adults who had achieved suppression of viremia to <50 copies/ml on a stable HAART regimen for at least 6 months and were willing to make frequent study visits. Patient characteristics and treatment histories have been described previously (34). Volunteers donated 100 ml of blood for initial genotyping of the virus in the plasma and in the cellular reservoir in resting CD4+ T cells. Beginning approximately 1 month thereafter, participants donated blood thrice weekly (Monday, Wednesday, and Friday) for 36 total study visits. To document adherence, plasma concentrations of nonnucleoside RT inhibitors (NNRTIs) and protease inhibitors (PIs) were measured at each visit, as is described elsewhere (34). Thereafter, patients donated 100 ml of blood periodically. The protocol was approved by a Johns Hopkins institutional review board. Informed consent was obtained from all study participants.
Patients maintained viral loads below 50 copies/ml on 95% of the study visits (34). On the other visits, transient low-level viremia (<200 copies/ml) was present. It is likely that the detection of blips in these patients is a direct consequence of frequent sampling used in this study. Our previous analysis suggests that isolated blips under 200 copies/ml occur in many patients on HAART and likely represent biological and statistical variation around mean viral loads slightly below 50 copies/ml rather than clinically significant elevations in viremia (34). No new resistance mutations appeared during or after blips (34). Viruses detected at blip time points were not detectably different from viruses detected between blips (34) and therefore were not distinguished for this analysis.
Plasma HIV-1 RNA quantification.
Viral RNA was quantified using the ultrasensitive Roche Amplicor Monitor system version 1.5 assay (Roche Molecular Systems, Inc., Blanchburg, New Jersey), which has a lower limit of quantification of 50 copies/ml.
Amplification, cloning, and sequencing of the pol and env genes from plasma virus.
To allow consistent amplification and sequencing of the small number of viral genomes present in the plasma of patients with viral loads below 50 copies/ml, plasma virus was first pelleted by ultracentrifugation and then analyzed by limiting dilution RT-PCR, cloning, and sequencing, using a modification of a previously described ultrasensitive genotyping method (19, 34). Briefly, 17 ml of blood was collected using an acid-citrate-dextrose anticoagulant at each visit during the period of intensive sampling. Three milliliters was used for determination of plasma HIV-1 RNA levels, and the remainder was separated on a Ficoll gradient. The resulting plasma (6 to 8 ml) was filtered or spun to remove contaminating cells and then ultracentrifuged at 25,200 × g for 2 h at 4°C to pellet virions. The virions were lysed under denaturing conditions, and viral RNA was isolated using a commercial silica gel membrane binding method (QIAamp viral RNA minikit; QIAGEN, Valencia, CA). The isolated RNA was treated with DNase I to ensure that amplified sequences were derived from viral RNA and not DNA. Viral RNA was then diluted to limit dilution and amplified by a one-step RT-PCR method (Invitrogen Corp., Carlsbad, CA). During the period of intensive sampling, RNA from 6 to 8 ml of plasma was typically distributed into 14 tubes, 7 for amplification of the protease gene and 7 for amplification of the RT gene. For each amplification, one of the seven reactions was run without the addition of SuperScript II RNase H− reverse transcriptase. In the remaining six reactions, the viral RNA was reverse transcribed into cDNA by using Superscript II RNase H− reverse transcriptase and then amplified by nested PCR using high-fidelity Platinum Taq DNA polymerase (Invitrogen Corp., Carlsbad, CA). The primers and PCR conditions are described in Table S1 in the supplemental material. The first PCR product was diluted 1:4 and used in a nested reaction. PCR products were separated on a 2% agarose gel, and bands of the appropriate size were purified using a QIAquick gel extraction kit (QIAGEN, Valencia, CA). Isolated DNA was then cloned using the Zero Blunt TOPO PCR cloning kit (Invitrogen Corp., Carlsbad, CA). Multiple clones from each reaction were sequenced using an ABI Prism 3700 DNA analyzer (Applied Biosystems, Foster City, CA). As is discussed below in “Sequence analysis,” only sequences that could be rigorously shown to be derived from independent templates were used in the analysis.
Amplification and sequencing of latent provirus in resting CD4+ T cells.
Viruses persisting in the resting CD4+ T-cell reservoir were analyzed by multiple complementary methods. Resting CD4+ T cells were purified from peripheral blood mononuclear cells (PBMC) by magnetic bead depletion as previously described (12). As previously described (6), these cells do not produce virus without stimulation and therefore, by definition, harbor latent virus. Most of the sequences were obtained by a novel limiting dilution PCR assay. DNA was purified from resting CD4+ T cells using the PureGene method (Gentra). A segment of the pol gene encompassing all of the protease and the first 219 amino acids of RT was amplified in an initial set of limiting dilution nested PCRs with 12 replicates at each dilution. PCR conditions are given in Table S1 in the supplemental material. The concentration of HIV-1 DNA in each sample was determined from the slope of a plot of the natural log of the fraction of negative wells at each dilution versus the input DNA volume. Cellular DNA was then diluted to 0.2 copies of HIV-1 DNA per well in 96-well plates, and a nested amplification was performed. Based on the Poisson distribution, this approach ensures that positive reactions will have ∼90% probability of being clonal. Products of positive PCRs were separated on a 1.5% agarose gel, purified using a QIAquick gel extraction kit (QIAGEN, Valencia, CA), and directly sequenced. Reaction mixtures containing more than one distinct template were identified by examination of chromatograms and excluded from the analysis. Because of the more robust amplification of proviral DNA, direct sequencing of PCR products was possible, eliminating the vast majority of PCR errors (except for those occurring in the first or second cycle).
Direct sequencing of HIV-1 DNA in resting CD4+ T cells from patients on long-term HAART is likely to capture predominantly the integrated form, since unintegrated forms are generally considered to be labile (1a, 2, 46, 53, 57). In addition to direct sequencing of HIV-1 DNA in resting CD4+ T cells, two additional approaches were used to analyze HIV-1 persisting in the latent reservoir. Resting CD4+ T cells carrying integrated proviruses that are competent for virus production following cellular activation were detected using a recently described assay that involves activation of resting CD4+ T cells in the presence of RT and integrase inhibitors (32). Resting CD4+ T cells carrying replication-competent virus were detected using a well-characterized limiting dilution culture assay (11, 12, 44, 45). Similar populations of viruses were obtained from all three assays (M. Wind-Rotolo, P. K. Lee, R. F. Siliciano, and J. D. Siliciano, unpublished data), with the exception that the last two assays did not detect replication-defective hypermutated sequences.
Activated CD4+ T cells (CD4+ and CD25+) and monocytes (CD14+) were purified to >90% purity by positive selection, using the appropriate monoclonal antibodies conjugated to magnetic beads.
Analysis of tropism.
Coreceptor usage was evaluated using HIV-1 pseudoviruses as previously described (39, 54).
Sequence analysis.
All sequences were screened against local and public databases to rule out contamination. Sequences from each patient were readily distinguishable and showed patient-specific clustering on phylogenetic analysis by multiple methods. Where possible, sequence ambiguities were resolved by direct inspection of chromatograms.
Several steps were taken to ensure that the plasma and cellular sequences obtained accurately reflected independent viral genomes present in vivo. For both plasma and cellular samples, multiple independent PCRs were set up at limiting dilution. In the case of the plasma virus, multiple independent RT-PCRs were set up at each of the 36 visits during the period of intensive sampling. On average, only 26.5% of these reactions were positive. In general, positive reactions were distributed evenly among the visits. By Poisson statistics, an average of 84.2% of the positive reactions represented amplification of single template molecules.
Because multiple clones were sequenced for each positive reaction, we were able to detect reactions containing more than one distinct template and to correct PCR errors. To avoid PCR resampling (29) and to remove PCR-induced sequence errors, the following steps were taken. First, a formal error analysis was carried out. HIV-1 virions produced by transfecting 293T cells with the plasmid standard pNL4-3 were quantitated using the Roche Amplicor assay. Limiting dilutions of these virions were amplified by RT-PCR, cloned, and sequenced using exactly the same protocol used in the analysis of patient plasma samples. After 64 total cycles, errors were detected in 21% of the clones. Sixteen percent had a single mutation in the 546-bp region of RT amplified, while 4% had two errors and 1% had three errors. These results fit the Poisson distribution, with a polymerase error rate of 6.9 × 10−6 mutations/nucleotide/cycle. Next, patient-derived sequences were analyzed. Clones obtained from a single PCR were compared, and an intrareaction consensus was established. Sequences differing from the intrareaction consensus by one, two, or three nucleotides appeared at frequencies consistent with Poisson statistics and the above error rate. These mutations were typically present in a single clone but not in other clones from the same reaction or in other clones from that patient. These were considered to be PCR errors. Thus, the sequencing of multiple clones from a single positive PCR set up at limiting dilution allowed for the correction of PCR-induced errors. Rarely, a single reaction gave rise to sequences that differed at four or more positions. Based on the directly measured error rate, this degree of difference cannot be attributed to PCR error. Typically, these mutations were observed in other clones from the same patient. These sequences were considered to be independent clones. The frequency of reactions containing more than one distinct template was actually less than that predicted by Poisson statistics. For example, an average of 4.5% of the positive reactions contained two distinct clones, less than the 12.7% predicted by the Poisson distribution. As is discussed in Results, this may reflect the presence of predominant plasma sequences in some patients.
Proviral sequences were obtained under limiting dilution conditions with an ∼90% probability of clonality. Direct sequencing of PCR products resulted in a dramatic reduction in PCR errors, since only mutations introduced in the first or second cycle of a 64-cycle amplification are present in a high-enough fraction of the product DNA molecules to be detected. Each positive reaction was sequenced with two different primers, giving overlapping sequence data. Contigs were assembled using CodonCode Aligner, version 1.3.1, and chromatograms were manually examined for the presence of double peaks indicative of two distinct templates per reaction. Such sequences were discarded. Where possible, sequence ambiguities were resolved by direct inspection of chromatograms. Sequences were examined for the presence of G→A hypermutation as previously described (23). Hypermutated sequences were not included in the phylogenetic analysis except in the case of the env gene, where hypermutation is more difficult to distinguish from mutation.
Sequences were analyzed for drug-resistant mutations by using the Los Alamos (http://hiv-web.lanl.gov/content/index) and Stanford (http://hivdb.stanford.edu/) databases. Drug resistance phenotypes were predicted using the HIVdb program developed by R. W. Shafer et al. (http://hivdb.stanford.edu/).
Phylogenetic analysis.
To select an appropriate analytical model of pol gene evolution, a test set consisting of 45 pol sequences (5 randomly selected sequences from each patient) was evaluated using the Akaike information criterion method implemented in ModelTest version 3.7 (40). The selected model was the general time-reversible model with correction for invariant sites and nonuniform evolutionary rates using a gamma distribution (GTR+I+Γ; specific parameters are provided in Table S2 in the supplemental material). Similarly, for the env tree shown in Fig. 5F, the sequences depicted in the figure were used to select a two-parameter model with correction for nonuniform evolutionary rates using a gamma distribution (HKY85+Γ; specific parameters are provided in Table S2 in the supplemental material). Phylogenetic tree estimation was performed using PAUP* version 4b10 (Sinauer Associates, Sunderland, MA), applying the above-described models in a heuristic search using the maximum likelihood criterion. The search started with a random-addition tree and was performed 10 times, with tree-bisection-reconnection branch swapping. Trees were visualized using TreeView version 1.6.6 (35). Key findings were confirmed using other phylogenetic methods, including simple neighbor-joining trees.
FIG. 5.


Phylogenetic analysis of RT and env sequences from free virus and from provirus in various cell types in patient 154. (A) Sampling timeline. (B) Maximum likelihood phylogenetic tree of RT sequences from free virus in the plasma (triangles) and CSF (inverted triangles) and from provirus in resting CD4+ T cells (circles), activated CD4+ T cells (diamonds), monocytes (hexagons), and unfractionated PBMC (squares). Some resting cell sequences (indicated with an I through the circle) were obtained using a special assay that definitively detects integrated HIV-1 DNA that is competent for virion production following cellular activation (32). Sequences with documented R5 tropism are indicated (see Fig. S8 in the supplemental material). Three independent plasma sequences obtained from long amplicons that include both the RT and env genes are indicated. (C) RT genotype of plasma and cellular sequences. Prior to study entry, the patient took AZT monotherapy and then failed a regimen consisting of ddI, stavudine (d4T), and EFV. At 21 months prior to study entry, the patient started a suppressive HAART regimen consisting of AZT, 3TC, tenofovir (TDF), the nucleoside/nucleotide RT inhibitor (NRTI) abacavir (ABC), and the protease inhibitor lopinavir boosted with ritonavir. Colored boxes indicate the mutations conferring resistance to NRTIs (red) and NNRTIs (blue). Note that the top portions of Fig. 2C and D are truncated. However, no RT resistance mutations were present for taxa 1 to 45. (D) Predicted phenotype of each RT sequence in the presence of RT inhibitors used in prior regimens or the regimen taken during the study period. (E) Regions of the HIV-1 genome analyzed to show that the PPS in RT represents a PPC. Colored arrows indicate the locations of nested primers used for amplification of RT (red) and the C2-V4 region of env (blue). These two regions were linked by long-distance RT-PCR (green). Additional sequence information was obtained by amplification of the full-length env gene (orange). Products from this reaction were also used in phenotypic analysis of coreceptor usage. LTR, long terminal repeat. (F) Maximum likelihood phylogenetic tree of sequences of the C2-V4 region of the env gene from free virus in the plasma (triangles) and provirus in resting CD4+ T cells (circles). Three independent plasma sequences obtained from long amplicons that also include the RT gene are indicated. Sequences obtained by amplification of the full-length env gene are indicated by outlined triangles. Colors indicate sampling times, as shown in panel A.
Modeling of the persistence of the PPC.
The persistence of the predominant plasma clone (PPC) was studied using a set of equations similar to Eigen's quasispecies equations, but for discrete generations. Initially, we considered the 546-bp segment of the RT gene analyzed in Fig. 1, 2, 4, and 5. Let _x_0(t) be the fraction of plasma virus that corresponds to the PPC and x_1(t) the fraction of all other plasma virus sequences. We assumed that the PPC replicates with a fitness equal to 1. We did not make any specific assumptions about the fitness of the other individual sequences, but we assumed that as a whole, they reproduce with fitness 1−_s (50).
FIG. 1.

Phylogenetic analysis of RT sequences from free plasma virus and from provirus in resting CD4+ T cells of patient 147. (A) Sampling timeline. After an initial blood draw for baseline analysis of plasma and latent reservoir sequences, the patient began a period of intensive sampling with visits every 2 to 3 days, as indicated by colored vertical lines and symbols. Note that each color represents five or six consecutive study visits. The number of clones obtained at each time point was roughly constant for each patient. A single follow-up sample was obtained 1 month after the intensive sampling period. (B) Maximum likelihood phylogenetic tree of RT sequences from plasma (triangles) and from resting CD4+ T cells (circles). The sequences represent codons 38 to 219 of RT. Clusters of five or more identical sequences are boxed. Individual taxa are numbered from top to bottom, and numbers are shown for taxa that illustrate coclustering of plasma and cellular sequences or release of wild-type virus into the plasma (Table 1). All sequences from patient 147 clustered together, away from those of other study patients and reference sequences (HXB2, JRFL, and SF2). Patient-specific clustering was observed for all patients studied. (C) RT genotype of plasma and cellular sequences. This patient was initially treated with AZT, 3TC, and indinavir for approximately 1 year. Then, 65 months prior to entry, he began a regimen consisting of EFV along with the PIs ritonavir (RTV) and saquinavir (SQV) and various nucleoside/nucleotide RT inhibitors (NRTIs). RTV was given at full dose. Although there were some treatment interruptions early on, the patient maintained good suppression of viremia on these regimens. At 15 months prior to entry, the patient began a regimen of EFV, RTV, and SQV along with the NRTIs 3TC and tenofovir (TDF). This was taken throughout the study. Colored boxes indicate the mutations conferring resistance to NRTIs (red) or NNRTIs (blue). (D) Predicted phenotype of each RT sequence in the presence of RT inhibitors used in prior regimens or the regimen taken during the study period. Predictions were made using a well-characterized, rules-based algorithm developed by R. W. Shafer and colleagues (http://hivdb.stanford.edu/). Colors indicate predicted levels of resistance to the indicated drugs. d4T, stavudine.
FIG. 2.

Phylogenetic analysis of RT sequences from free plasma virus and from provirus in resting CD4+ T cells and monocytes of patient 148. (A) Sampling timeline. (B) Maximum likelihood phylogenetic tree of RT sequences from free virus in the plasma (triangles) and provirus in resting CD4+ T cells (circles) and monocytes (hexagons). Some resting cell sequences (indicated with an I through the circle) were obtained using a special assay that definitively detects integrated HIV-1 DNA that is competent for virion production following cellular activation (32). Clusters of five or more identical sequences are boxed. Individual taxa are numbered from top to bottom, and numbers for taxa with particular properties are shown. (C) RT genotype of plasma and cellular sequences. Prior to study entry, the patient was treated with monotherapy and dual-nucleoside analogue therapy involving AZT, stavudine (d4T), and 3TC for a total of 59 months. The patient then started a three-drug regimen consisting of d4T, 3TC, and indinavir but failed after the protease inhibitor was switched to nelfinavir. Then, 66 months before study entry, the patient began a suppressive regimen consisting of ddI, EFV, ritonavir (RTV), quinavir (SQV). Subsequently, the NNRTI was changed to nevirapine (NVP), and SQV was replaced with lopinavir boosted with RTV (LPR/r). The patient maintained excellent suppression of viremia throughout the study on ddI, NVP, and LPR/r. At approximately day 570, ddI was replaced with tenofovir (TDF) and 3TC. Colored boxes indicate the mutations conferring resistance to nucleoside/nucleotide RT inhibitors. No NNRTI resistance was seen. (D) Predicted phenotype of each RT sequence in the presence of RT inhibitors used in prior regimens or the regimen taken during the study period.
FIG. 4.

Phylogenetic analysis of RT sequences from free plasma virus and from provirus in resting CD4+ T cells of patient 135. (A) Sampling timeline. (B) Maximum likelihood phylogenetic tree of RT sequences from free virus in the plasma (triangles) and provirus in resting CD4+ T cells (circles). (C) RT genotype of plasma and cellular sequences. Prior to study entry, the patient was treated with AZT and stavudine (d4T) monotherapy for a total of 31 months and then with d4T and 3TC for 5 months. At 79 months prior to entry, the patient started a suppressive HAART regimen consisting of AZT, 3TC, ritonavir (RTV) (full dose), and saquinavir (SQV). At day 470, RTV and SQV were replaced with lopinavir boosted with ritonavir, and 2 months later, AZT was replaced with tenofovir. Nucleoside/nucleotide RT inhibitor (NRTI) resistance mutations are indicated by red boxes. No NNRTI resistance was seen. (D) Predicted phenotype of each RT sequence in the presence of RT inhibitors used in prior regimens or the regimen taken during the study period.
We defined m as the mutation rate away from the PPC. More exactly, m is the rate at which the PPC produces mutant sequences that make a substantial contribution to the second pool of sequences. We write m = α_L_μ, where α is the fraction of sites at which a mutation leads to a viable mutant sequence, L is the length of the relevant region of the RT gene (546 sites), and μ is the per-site mutation rate (μ = 3.4 × 10−5 substitutions/site/cycle [30]). The per-site mutation rate μ for HIV-1 is small enough so that we can neglect more deleterious mutants and back mutations. The frequencies _x_0(t) and _x_1(t) then evolve according to
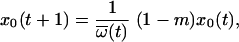 |
(1) |
|---|

where 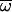 is the average fitness at generation t,
is the average fitness at generation t,
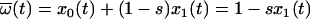 |
(2) |
|---|
The solution to this set of equations for the initial conditions _x_0(0) and _x_1(0) is
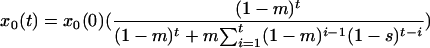 |
(3) |
|---|
and _x_1(t) = 1 − _x_0(t), as can be verified by induction. Using equation 3, we calculated _x_0(t) for values of t corresponding to late time points for patient no. 154 (days 410 and 516). For _x_0(0), we used a value of 0.721, which represents the fraction of plasma sequences consisting of the PPC during the intensive plasma sampling period at the beginning of the study. Our results do not change significantly even in the extreme case where _x_0(t) = 1.0.
Our data set shows that approximately 56% of the sites in the relevant region of RT are mutable (data not shown). However, the fraction α of these mutations that results in viral fitness comparable to that of the PPC is unknown. Therefore, we determined the maximum value of α that would produce a data set consistent with ours given various amounts of selective pressure. For mutations that lead to a fitness advantage (negative s), it is not clear a priori that we can neglect back mutations. Therefore, for all results reported for negative s, we solved the full equations for _x_0(t) and _x_1(t). We added the term 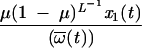 to the expression for _x_0(t + 1) in equation 1 and subtracted the same term from the expression for _x_1(t + 1) numerically and found that the deviations from our approximate solution were negligible.
to the expression for _x_0(t + 1) in equation 1 and subtracted the same term from the expression for _x_1(t + 1) numerically and found that the deviations from our approximate solution were negligible.
Nucleotide sequence accession numbers.
The sequences have been deposited in GenBank. The accession numbers are DQ391352 to DQ392955 (for RT) and DQ391282 to DQ391351 (for env).
RESULTS
Study design.
We studied residual viremia in infected adults with prolonged suppression of viremia to <50 copies/ml on a stable HAART regimen (mean duration of suppression prior to enrollment, 34 months; range, 11 to 79 months). So that a sufficient number of independent viral sequences could be obtained from the plasma, patients provided blood samples every 2 to 3 days for 36 closely spaced visits. The stable latent reservoir in resting CD4+ T cells was sampled at enrollment 1 month before the intensive sampling period and then 1 month after the intensive sampling. To characterize the extremely small number of viral RNA molecules in the plasma of patients with viral loads below 50 copies/ml, virions were pelleted by ultracentrifugation and analyzed by limiting dilution RT-PCR amplification of segments of the pol gene, followed by cloning and sequencing. Importantly, only clones that could be rigorously shown to be derived from independent templates in vivo were used in the analysis (see Materials and Methods). Thus, great care was taken to avoid the PCR resampling problem that complicates many studies of HIV-1 compartmentalization (29). The analysis of multiple clones from each limiting dilution PCR also allowed for the correction of PCR errors as described in Materials and Methods. Control reactions set up without the inclusion of RT enzyme were invariably negative, indicating that the sequences obtained were derived from genomic viral RNA and not DNA. The analysis of proviral DNA from resting CD4+ T cells was also carried out by limiting dilution PCR, with special precautions to avoid PCR resampling and PCR errors (see Materials and Methods).
General characteristics of the residual plasma virus in patients with viral loads below 50 copies/ml.
Analysis of the RT genes of viruses from plasma and resting CD4+ T cells revealed a diverse set of patient-specific sequences. Figure 1 shows a representative phylogenetic tree from patient 147. The tree is complex, with 100 taxa. This diversity is consistent with chronic infection (43). Plasma and cellular sequences were clearly intermingled. For some of the plasma viruses, it was possible to find viruses in the resting CD4+ T-cell compartment with identical RT sequences despite the extensive diversification present (taxa 23, 39, 41, and 87) (Fig. 1B). Viruses with identical RT sequences were obtained from the plasma and resting CD4+ T-cell compartments of all nine patients (Table 1). Some sequences were isolated repeatedly from the plasma of patient 147 (taxa 23 and 29) (Fig. 1B). For each of these two most commonly observed plasma sequences, identical sequences were observed in resting CD4+ T cells. As shown in Fig. 1C and D, some viruses in both the plasma and the resting CD4+ T-cell compartments carried the K103N mutation, which confers high-level resistance to all currently licensed NNRTIs. Interestingly, despite the fact that the patient continued to take efavirenz (EFV) throughout the study with excellent adherence, 34% of the plasma viruses lacked the K103N mutation (taxa 9, 38, 50 to 53, 55, 56, 58, 71, 73, 76, and 82). Release of wild-type viruses in patients on HAART who also had partially resistant viruses was seen in patients 134 and 139 (see Fig. S1 and S2 in the supplemental material, respectively) (Table 1) as well as in patient 135 (described below). The detection of viruses with identical RT sequences in both plasma and resting CD4+ T cells and the continued release of wild-type virus in patients with resistance are consistent with the hypothesis that at least some of the residual viremia reflects release of virus from latently infected resting CD4+ T cells that become activated. However, the source of the residual viremia cannot be definitively established from these observations.
TABLE 1.
Phylogenetic analysis of plasma and resting CD4+ T-cell sequences from patients on HAART
| Patient | No. of independent sequences analyzed | Figure no. for RT treea | Total no. of taxa on RT tree | Taxon no. with identical plasma and resting CD4+ T-cell sequencesb | Release of archival wild-type virus in patients with resistancec | PPS presentd | PPS taxon no. | Frequency of PPS (%)e | |
|---|---|---|---|---|---|---|---|---|---|
| Plasma | Resting CD4+ T cells | ||||||||
| 099 | 97 | 67 | S3 | 90 | 23, 26, 32, 37, 67, 72 | 4, 5, 9-27, 30, 32-35 | No | ||
| 113 | 27 | 24 | S5 | 32 | 2, 27 | NAf | Yes | 24 | 51.9 |
| 134 | 72 | 12 | S1 | 63 | 49 | 2, 12, 30, 35 | No | ||
| 135 | 97 | 151 | 4 | 145 | 115, 139 | 3, 12, 13, 16, 17, 25, 29, 33-35, 67, 68, 84, 90, 91 | Yes | 130 | 70.1 |
| 136 | 53 | 66 | S4 | 62 | 4, 14, 22 | NA | No | ||
| 139 | 46 | 83 | S2 | 64 | 57, 61 | 10, 25 | Yesg | 57 | 37.0 |
| 63 | 34.8 | ||||||||
| 147 | 47 | 88 | 1 | 100 | 23, 39, 41, 87 | 9, 38, 50-53, 55, 56, 71, 73, 76, 82 | No | ||
| 148 | 51 | 139 | 2 | 75 | 35, 68 | Not detected | Yes | 50 | 84.3 |
| 154 | 92 | 241 | 5 | 194 | 46, 57, 94, 140, 163, 173, 185, 192 | NA | Yes | 140 | 66.8 |
Viruses with identical pol sequences dominate the residual viremia in many patients with suppression of viremia to below 50 copies/ml.
The phylogenetic relationship between plasma and resting CD4+ T-cell sequences observed for patient 147 was also seen in patients 134, 099, and 136 (see Fig. S1, S3, and S4 in the supplemental material, respectively). However, in five of nine patients studied, phylogenetic analysis of plasma and resting CD4+ T-cell sequences produced a strikingly different pattern. In each of these patients, the majority of viruses found in the plasma were identical in sequence throughout the region analyzed. The same patient-specific RT sequence was identified repeatedly in multiple independent samples from multiple time points. This modal sequence constituted a single taxon of a complex patient-specific phylogenetic tree. Importantly, on this branch, there were few, if any, resting CD4+ T-cell sequences.
An example is shown in Fig. 2. In this patient (148), analysis of resting CD4+ T-cell and plasma sequences over a 2-year period produced a complex phylogenetic tree with 75 taxa. Thus, despite the relatively high degree of conservation in RT, considerable diversification took place. A total of 139 independent resting CD4+ T-cell sequences obtained by limiting dilution PCR constituted 70 of the 75 taxa. These included archival wild-type sequences and sequences capturing the progressive accumulation of zidovudine (AZT) and lamivudine (3TC) resistance mutations during prior nonsuppressive therapy (Fig. 2C and D).
In sharp contrast to the diverse nature of the resting CD4+ T-cell sequences, 51 independent plasma sequences constituted only five taxa (taxa 27, 35, 50, 51, and 68) (Fig. 2B). Of these 51 sequences, 43 (84.3%) were identical (taxon 50). None of the 139 independent cellular sequences matched this sequence.
We defined a predominant plasma sequence (PPS) as a single sequence representing more than 50% of a large sample of independent plasma sequences from a given patient. Several explanations for the presence of a PPS were considered. The study was designed to avoid PCR resampling which can give the artifactual appearance of a dominant population (29). Taxon 50 sequences were detected in multiple independent limiting dilution PCRs at the time of enrollment and at days 26, 28, 30, 33, 35, 37, 40, 42, 44, 49, 51, and 89. Therefore, the results cannot be explained by PCR resampling. Nor can they be attributed to the use of any particular method of sequence processing and phylogenetic tree construction. Although rigorous methods were used in the analysis presented above, the presence of a PPS was apparent regardless of the analytical methods used.
To further rule out PCR artifacts and to determine whether viruses with identical RT sequences were identical in other genes as well, a similar analysis was carried out for the HIV-1 protease gene. Full-length protease was amplified from the plasma separately at each time point in separate reactions, using a different primer set. For patient 148, the protease tree showed a comparable pattern in which 47 of 65 independent plasma sequences (72%) were identical, constituting a single taxon (taxon 47) of a complex tree with 75 taxa (Fig. 3). No cellular sequences matched. Thus, the finding that residual viremia was dominated by a PPS was not unique to particular viral genes, amplicons, or primers.
FIG. 3.

Phylogenetic analysis of protease sequences from free plasma virus and from provirus in resting CD4+ T cells of patient 148. (A) Sampling timeline. (B) Maximum likelihood phylogenetic tree of protease sequences from plasma (triangles) and from resting CD4+ T cells (circles). Note that protease and RT sequences were obtained from independent PCRs. Therefore, different numbers of positive reactions were obtained, and these represented different viral genomes present in vivo. Thus, the shape of the tree is not identical to that of the RT tree shown in Fig. 2B. Nevertheless, there is a PPS that constitutes most of the residual viremia. (C) Protease genotype of plasma and cellular sequences. See the legend to Fig. 2 for treatment history. Colored boxes indicate the mutations conferring resistance to PIs. (D) Predicted phenotype of each protease sequence in the presence of PIs used in prior regimens or the regimen taken during the study period. IDV, indinavir; NFV, nelfinavir; SQV, saquinavir; RTV, ritonavir; APV, amprenavir.
The presence of a PPS is common in patients with suppression of viremia to below 50 copies/ml.
The presence of a PPS was not unique to patient 148. The same phenomenon was clearly present in patients 135 and 154 (Fig. 4 and 5 ) and to a lesser extent in patients 139 and 113 (see Fig. S2 and S5 in the supplemental material). Smaller plasma-unique taxa were also seen in patients 099 and 136 (see Fig. S3 and S4 in the supplemental material).
For patient 135, 68 of 97 independent plasma sequences (70%) were identical (taxon 150) (Fig. 4). This PPS was isolated in multiple independent PCRs from 24 different blood samples taken over a 6-month period but not at later time points (Fig. 4A and B). As with patient 148, no viruses with this sequence were found in resting CD4+ T cells despite extensive sampling (151 independent clones). Together, the plasma and cellular sequences produced a tree with 145 taxa, but the vast majority of the plasma viruses comprised a single taxon on this tree.
Patient 154 provided the most dramatic example of this phenomenon (Fig. 5). Extensive sampling of the plasma and cellular compartments produced a tree with 194 taxa. Of 241 independent plasma sequences, 161 (66.8%) were identical and constituted a single taxon on this tree (taxon 140). This PPS was isolated repeatedly throughout the study, even at the last time point almost 2 years after initial detection. Extensive sampling of the resting CD4+ T-cell compartment (92 independent sequences) yielded only a single matching sequence, which was obtained on day 410 using a special assay for integrated virus (see below).
In the case of patient 139 (see Fig. S2 in the supplemental material), there were actually two predominant plasma sequences that together represented 72% of the plasma sequences (taxa 57 and 63). Of 83 cellular sequences, only one matched one of these commonly detected plasma sequences.
The PPS represents a predominant plasma clone.
Although a PPS was detected in the analysis of both RT and protease genes, it remained possible that these sequences were from a family of closely related viruses identical in pol but different elsewhere in the genome. The 546-bp region of the RT gene sequenced represents only 5.6% of the HIV-1 genome. Therefore, to determine whether viruses represented by the PPS are identical throughout the genome, we carried out separate limiting dilution amplifications of the env gene, which is located at the opposite end of the genome (Fig. 5E). The env gene undergoes extensive diversification (43), and thus, analysis of env provides a rigorous test of the hypothesis that the PPS reflects a single viral clone. As expected, there was more divergence in env than in protease or RT (Fig. 5F). Nevertheless, of 45 independent plasma sequences obtained from patient 154 by limiting dilution RT-PCR, 23 (51.1%) were identical, constituting a single taxon on the tree with 35 taxa (taxon 10). The same PPS in env was obtained with three different primer sets, one that amplifies the C2-V4 region of env, one that amplifies full-length env, and one that amplifies a long 5-kb segment of the genome linking RT and env. Thus, the detection of a PPS was not due to the use of any particular primer set. The env tree was generated with sequences stripped of gaps created by insertions and deletions and thus does not capture the extraordinary length and sequence polymorphism present, particularly in the V4 region. Importantly, the sequences comprising taxon 10 were absolutely identical to one another before gap stripping, all sharing a uniquely short V4 loop (see Fig. S6 in the supplemental material). Thus, the phenomenon of a PPS can be reproduced by independent amplification of an entirely separate, highly variable region of the genome and appears to reflect the presence of a predominant plasma clone.
To determine the relationship of the PPS observed in the RT gene and the PPS observed in the env gene, plasma virus from patient 154 was amplified with primers that would produce a single 5-kb amplicon containing the relevant regions of both RT and env (Fig. 5E). Three of five RT-PCRs were positive, and products from each positive reaction were cloned and sequenced (Fig. 5B and E). The RT regions of these clones were identical in sequence to the PPS of the RT tree (taxon 140) (Fig. 5B) while the env regions were identical to the PPS of the env tree (taxon 10) (Fig. 5E), establishing linkage between the two PPSs. Most importantly, full-length sequencing of all three independent 5-kb clones gave exactly the same sequence throughout. Thus, the PPS in RT and the PPS in env are linked by 5 kb of identical sequence representing over 50% of the viral genome. In addition, six of the taxon 10 sequences were obtained by amplification of the full-length env gene. All six were identical throughout the region sequenced, which extended into the nef gene. Thus, as is indicated in Fig. 5E, a region comprising over 65% of the HIV-1 genome is identical in the most commonly isolated viruses from the plasma. These results strongly suggest that the PPSs detected by sequencing of the RT or env gene represent viruses that are identical throughout the entire genome and that constitute a single predominant plasma clone (PPC).
Resistance and neutralization profiles of the PPC.
One possible explanation for the presence of a PPC is that it is a uniquely fit virus. To address this issue, we examined the RT and protease genotypes which are critical determinants of viral fitness in patients on potent HAART regimens. For patient 148, the PPC (taxon 50) (Fig. 2B) had a single T215Y mutation in RT, which may confer a low level of resistance to the didanosine (ddI) component of the active regimen (Fig. 2C and D). Phylogenetic analysis suggested that this virus was derived from an ancestral sequence that also had the M184V mutation that confers resistance to 3TC. Interestingly, many of the patient's other viruses had multiple mutations which are predicted to confer higher levels of resistance to the active regimen. The PPC in this patient did not carry resistance mutations in protease (taxon 47) (Fig. 3). The PPC in patient 135 (Fig. 4) also had a T215Y mutation conferring low-level resistance to the active regimen, and in patient 139, both dominant taxa had a V108I mutation, which may confer low-level resistance to EFV. However, in the case of patients 154 (Fig. 5) and 113 (see Fig. S5 in the supplemental material), the PPC did not have significant resistance in either RT or protease. Thus, this phenomenon is not clearly related to drug resistance.
We also examined the fitness of the PPC with respect to autologous neutralizing antibodies. Full-length env sequences representing the PPC of patient 154 (Fig. 5E and F) were tested for susceptibility to neutralization by antibodies in contemporaneous autologous plasma. The PPC was more easily neutralized than other variants from the same patient (1). Taken together, these results suggest that PPC does not have an obvious fitness advantage with respect to selection by antiretroviral drugs or neutralizing antibodies.
The PPC is underrepresented among integrated proviruses in resting CD4+ T cells from the blood and among replication-competent viruses in resting CD4+ T cells from the blood.
As discussed above, extensive analysis of HIV-1 DNA sequences in resting CD4+ T cells in the circulation demonstrated that these sequences rarely matched the PPC (Fig. 2, 3, 4, and 5; also see Fig. S2 and S5 in the supplemental material). Sampling of the CD4+ T-cell compartment was sufficient to detect taxa composed of large numbers of identical independent sequences from resting CD4+ T cells (Fig. 2 and 3; also see Fig. S2 and S3 in the supplemental material), but these did not contribute measurably to the residual viremia. These results raise the possibility that the circulating pool of resting CD4+ T cells might not be the source of the PPC. However, most of the cellular sequences used in the above-described analyses were obtained by sequencing of HIV-1 DNA in purified resting CD4+ T cells. This type of analysis does not distinguish integrated versus unintegrated HIV-1 DNA or replication-competent versus defective HIV-1 DNA. The latent reservoir in resting CD4+ T cells consists of a small pool of cells carrying stably integrated, replication-competent viral genomes (6, 9). It is not possible to assess both integration status and replication competence in a single assay. However, using separate assays, we showed that the PPC is underrepresented among integrated proviruses in resting CD4+ T cells and among replication-competent viruses in resting CD4+ T cells.
With respect to integration status, we used a recently described assay that is specific for integrated proviruses (32). This assay detects viruses released from resting CD4+ T cells after activation in the presence of RT and integrase inhibitors. Resting CD4+ T cells carrying incomplete reverse transcripts or full-length unintegrated HIV-1 DNA cannot produce virus in this system (32). Figure 6A shows RT sequences from patient 154 at a single time point (day 410), using three different methods: direct sequencing of plasma virus, direct sequencing of HIV-1 DNA in resting CD4+ T cells, and sequencing of viruses released from resting CD4+ T cells following activation in the presence of RT and integrase inhibitors. Resting cell sequences obtained by either method were phylogenetically intermingled and largely distinct from the plasma sequences. At this time point, 17 of 21 plasma sequences (81%) were identical to one another and to the PPC detected at other time points. Only 1 of 22 cellular sequences was identical to the PPC. This sequence was detected with the assay that is specific for integrated provirus. Nevertheless, most of the integrated proviruses were clearly distinct from the PPC. Similar observations were made at day 550 (Fig. 4B). Similar results were obtained with patient 139 (see Fig. S2 in the supplemental material). For patient 139, 1 of 83 independent resting CD4+ T-cell sequences matched 1 of the 2 predominant plasma sequences (taxon 57). This cellular sequence was obtained with the assay that detects integrated HIV-1 in resting CD4+ T cells. Taken together, these results suggest that sequences matching the PPC can be detected in resting CD4+ T cells, but there remains a very strong lack of proportionality—the sequences commonly found in the plasma are rarely found in resting CD4+ T cells.
FIG. 6.

The PPC is underrepresented among integrated proviruses in resting CD4+ T cells from the blood and among replication-competent viruses in resting CD4+ T cells from the blood. (A) Comparison of plasma and resting CD4+ T-cell sequences from patient 154 at day 410. Resting cell sequences were analyzed by direct sequencing or by a method that detects only integrated proviruses (32). Plasma sequences matching the PPC in Fig. 5B are indicated. (B) Comparison of plasma sequences and replication-competent virus in resting CD4+ T cells from patient 154 at day 634. Plasma sequences matching the PPC in Fig. 5B are indicated.
We also considered the possibility that resting CD4+ T cells harboring replication-competent HIV-1 were the source of the PPC but escaped detection in the direct sequencing studies because they represent only a small subset of all the resting CD4+ T cells that carry HIV-1 DNA. To address this possibility, we used a well-established limiting dilution virus culture assay (11, 12, 45) to isolate multiple clones of replication-competent HIV-1 from purified resting CD4+ T cells from patient 154. RT sequences from these clones were phylogenetically intermingled with RT sequences obtained by direct sequencing of proviral DNA (data not shown). As is shown in Fig. 6B, none of the 34 independent clones of replication-competent virus isolated from resting CD4+ T cells matched the PPC, which accounted for 3 of the 6 plasma clones at this time point.
Other potential sources of the PPC.
We carried out additional experiments to determine the cellular source of the PPC. We analyzed purified activated CD4+ T cells from the peripheral blood. As shown in Fig. 5B, activated cell sequences were intermingled with and, in some cases, identical to resting CD4+ T-cell sequences but were absent from the tree branch containing the PPC. The same was true of sequences detected in monocyte-enriched PBMC preparations (Fig. 2 and 5). To rule out other circulating cell types as the source of the PPC, we also examined HIV-1 sequences in unfractionated PBMC from patient 154. None of the 89 independent sequences fell on the branch containing the PPC (Fig. 5B). Analysis of free virus in the cerebrospinal fluid (CSF) of patient 154 yielded a single clone that did not cluster with the PPC. However, one of four CSF clones from patient 139 clustered with one of the two commonly isolated plasma sequences (taxon 63) (see Fig. S2 in the supplemental material). In patient 136, three CSF sequences did not cluster with the small plasma-unique taxon (see Fig. S4 in the supplemental material). To evaluate the tropism of the PPC, full-length env clones with the predominant plasma env sequence from patient 154 (taxon 10) (Fig. 5F) were tested in functional assays with coreceptor-transfected target cells (see Fig. S7 in the supplemental material). Clear R5 tropism was documented. Thus, the source of the PPC in this patient is likely to be a cell capable of expressing CCR5. Because of the relative inefficiency of the 5-kb RT-PCR, we could not link env sequences to the predominant RT sequences in other patients, and thus, it remains possible that the PPC in other patients is an X4 or dual-tropic virus. Taken together, these results suggest that the homogeneous nonevolving PPC that constitutes the bulk of the residual viremia in some patients is not derived from infected cells that are well represented in the circulation.
Ongoing viral replication as a potential source of the PPC.
Elegant studies by Shankarappa and colleagues (43) have shown that HIV-1 replication leads to progressive increases in divergence from the most recent common ancestor and in the diversity of quasispecies present in a given individual. In this context, the persistence of a PPC for months to years without evolution is surprising. We therefore studied the mechanism of persistence of the PPC by modeling the degree of sequence change that would be expected if the PPC was maintained by ongoing cycles of viral replication. We used discrete-time equations similar to Eigen's quasispecies equations in order to predict the fraction of plasma viruses that would retain the same sequence after t generations. The only input parameters necessary for this model are the initial fraction of plasma sequences represented by the PPC, the generation time, and the mutation rate. The last two values were obtained from accepted measurements in the literature (30, 31). The expressions we developed depend on α, the fraction of sites at which a mutation leads to a viable variant (mutations at 1 − α sites are lethal to the virus), and s, the average fitness cost to variants in this region (see Materials and Methods), expressed as a fraction of the fitness of the PPC (i.e., s = 0.01 reflects a 1% fitness cost compared to the PPC). We then determined the maximal value of α that would be consistent with the observed persistence of the PPC in the setting of ongoing viral replication, using longitudinal data from patient 154.
We first considered the possibility that the PPC sits atop a peak in the fitness landscape of viral quasispecies. If the PPC sits on a moderately advantageous local fitness maximum, such that all immediate mutants carry at least some moderately deleterious fitness cost (s ≥ 0.03), then the model predicts that the observed pattern could be explained by ongoing replication since a wide range of values for α are consistent with the observed persistence of the PPC (Table 2). In this setting, the majority of the plasma virus would be composed of the PPC because it out-competes all other possible mutants. However, as discussed above, the PPC does not have any obvious fitness advantage, at least with respect to selection by antiretroviral drugs or neutralizing antibodies.
TABLE 2.
Analysis of the maximum fraction of mutable sites consistent with the observed persistence of the PPC in patient 154
| Parameter | Value for study daya: | |
|---|---|---|
| 410 | 516 | |
| Time since first detection of PPCb (days) | 380 | 486 |
| No. of viral generationsc | 190 | 243 |
| No. of taxon 140 sequences/total no. of plasma sequences (%) | 19/23 (83) | 8/8 (100) |
| 95% Confidence intervald | 0.64 | 0.68 |
| α Maximum value ate: | ||
| s = 0.03 | 0.6 | 0.53 |
| s = 0 | 0.13 | 0.09 |
| s = −0.03 | 0.0034 | 0.0006 |
We next considered neutral mutations (s = 0). Because of the error-prone nature of reverse transcriptase, mutations are inevitably introduced during HIV-1 replication, and those mutations that are neutral will have a chance to persist. According to this simple model, the observed persistence of a PPC can be explained by ongoing viral replication only if a very small fraction of the sites are neutral. For example, in patient 154, the persistence of the PPC at day 516 can be explained by replication only if at most 9% of the sites within the 546-bp region of the RT gene analyzed can undergo neutral mutations (Table 2). This result is independent of the fitness of all other mutants at nonneutral sites (viable or nonviable). We observed mutations at 56% of the sites in the region analyzed. On average, mutations at 20% of all sites within this region of the genome lead to synonymous changes. Thus, unless there are unappreciated RNA structural features or major codon usage effects at over half of these synonymous sites, the data are inconsistent with ongoing replication according to this model. As discussed above, linkage to env sequences suggests that predominant plasma RT sequences reflect viral clones identical throughout the genome. If the entire 9,719-bp genome is considered, then by this model, ongoing replication can explain the observed persistence of the PPC only if <0.5% of the sites in the HIV-1 genome can undergo neutral mutation (data not shown). Even more striking is the fact that if mutation at even one site in the region of RT analyzed leads to a moderate fitness advantage (s ≤ −0.03), then generation and subsequent replication of the more fit mutant would lead to significantly greater viral sequence divergence than we observed. In this case, the data are inconsistent with replication as the mechanism of persistence of the PPC. As noted above, this result is independent of the fitness of any other possible mutant. Taken together, these results suggest that the observed persistence of the PPC can be explained by viral replication only if the PPC sits atop a fitness maximum.
DISCUSSION
We have shown that in some HIV-1-infected individuals with suppression of viremia to below 50 copies/ml on HAART, much of the residual viremia consists of only one or two PPCs that are released into the plasma over the course of months to years without apparent evolution. The latent reservoir in resting CD4+ T cells is one potential source for residual viremia in patients on HAART. However, although the plasma of all patients studied contained viruses similar to those found in resting CD4+ T cells, more than half of our small patient population had a PPC that was distinct from most of the viruses found in resting CD4+ T cells in the circulation.
Given that the PPC constitutes most of the residual viremia in many patients with viral loads below 50 copies/ml, it is interesting to consider why this phenomenon has not been previously described. A major reason is that few studies have examined plasma virus in patients with plasma HIV-1 RNA levels below 50 copies/ml (10, 19, 22, 34, 36, 38, 52). In patients who are measurably viremic, most of the virus is produced by recently infected cells, generally thought to be activated CD4+ T cells (3, 20, 49). The large amount of virus produced by these cells is likely to completely obscure the PPC. Thus, the PPC is predominant only when the active replication is largely suppressed by HAART. When the viral load is below 50 copies/ml, only a small number of viral sequences can be obtained from each blood sample, even if amplification can be achieved with perfect efficiency. A PPC cannot be identified without obtaining a sufficiently large number of independent clones to provide a clear picture of the plasma virus population. Because we had used intensive sampling to obtain large numbers of independent viral clones from patients with undetectable viral loads, the existence of a PPC in some patients became readily apparent. Thus, the present study is the first in which a sufficient number of independent sequences have been collected from patients with viral loads below 50 copies/ml to identify a PPC. Interestingly, in a careful study of pediatric patients whose viral loads had risen transiently into the detectable range, Tobin et al. noted that some patients had multiple identical sequences in the analysis of both the RT and env genes (48). Although linkage between the RT and env sequences was not established and the viruses represented by these sequences could have had differences outside the small region sequenced, it is possible that these sequences also represent PPCs.
Several technical aspects of the study may have also contributed to the identification of PPCs. The use of limiting dilution PCR techniques helped to ensure that each reported sequence represented an independent viral template present in vivo while simultaneously avoiding PCR errors which give the false impression of evolutionary diversity. Although labor intensive, these approaches greatly facilitated the detection of a PPC by allowing a clearer picture of the viral diversity present in vivo.
A particularly interesting question is why there are only one or two PPCs in each patient. This may reflect the fact that some rare event is involved in the process, occurring one or two times in some patients and not at all in other patients. Another possibility is that the critical infection event occurs more commonly but that expansion of the infected cell population rarely reaches a point where the viruses released from these cells dominate the plasma population.
A critical unresolved issue is the source of the PPC. Low-level ongoing replication of the virus in CD4+ T cells is frequently considered a source of residual viremia in patients on HAART (7, 10, 13, 14, 18, 21, 33, 48, 56). Although ongoing replication remains a possibility, a simple quantitative analysis of the likelihood that mutations would arise during this ongoing replication suggested that the persistence of the PPC could be explained by ongoing replication only if the PPC had a unique fitness advantage. In patients on HAART, potent selective pressure operates on the genes encoding the viral proteins targeted by the drugs. Analysis of the RT and protease genes failed to reveal mutations that would allow the PPC a unique advantage, and in some cases, the PPC had no known resistance mutations. Ongoing replication would also be expected to generate mutations at sites that are neutral, and the persistence of the PPC can be explained only if there are very few neutral sites. Given the number of sites at which synonymous mutations occur, the absence of synonymous changes can be explained only if there are unrecognized constraints operating at the RNA level.
The other frequently mentioned source of residual viremia for patients on HAART is release of virus from the stable reservoir in resting CD4+ T cells as these cells become activated (19, 22, 34, 48, 55). Interestingly, our results strongly suggest that some of the residual viremia results from this mechanism. In every patient studied, we could find identical sequences in both the plasma and resting CD4+ T-cell compartments. Thus, our approach allows us to see instances in which the latent reservoir may be contributing to residual viremia. However, the PPCs were absent or strikingly underrepresented in resting CD4+ T cells from the blood. Although Tobin et al. suggested that clonal expansion of CD4+ T cells might explain the repeated detection of identical sequences (48), our data raise the possibility that the source of the PPC may not be CD4+ T cells or, in fact, any other cell type in the blood. Resolving this issue will of course require the direct identification of the cells that produce the PPC. This may prove very difficult. We calculate that as few as several hundred cells distributed throughout the body could produce the PPC observed in these patients (data not shown).
An important caveat is that our analysis of the latent reservoir relied primarily on direct sequencing of HIV-1 DNA in resting CD4+ T cells from patients with undetectable viral loads. While most of this HIV-1 DNA is likely to be integrated, not all of it is replication competent. For this reason, we used two additional assays. One of these specifically detected integrated proviruses that were competent for virus production following cellular activation. The other assay detected replication-competent viruses persisting in resting CD4+ T cells. For both assays, the viral sequences detected in the resting CD4+ T cells remained largely distinct from the PPC. Thus, the underrepresentation of the PPC in the latent reservoir in resting CD4+ T cells is apparent regardless of how the reservoir is assayed. Since all of the studies were done on peripheral blood CD4+ T cells, there remains the possibility that the PPC is produced by a subset of CD4+ T cells that does not enter the circulation.
One alternative hypothesis that is consistent with all of the data is that a rare infection event establishes an integrated viral genome in a cell that has proliferative capacity, for example, a stem cell in the monocyte-macrophage lineage. This infected cell proliferates, copying the viral genome without introducing errors and generating an expanded set of progeny cells that release virus. According to this hypothesis, the relevant cell type must be capable of both clonal expansion after infection and low-level virus production. The nature of the PPC suggests that infection of this cell type is inefficient and/or that only rarely does clonal expansion of this cell type reach the level at which viruses produced by the cell and its progeny predominate in the plasma. Importantly, viruses produced by this cell type could infect CD4+ T cells, which explains the rare instances of a PPC in a CD4+ T cell.
The presence of a PPC in patients on HAART is of considerable clinical significance. Although the amount of virus produced is very low, this virus may be a factor in virologic failure in the setting of poor adherence and in rebound viremia in the case of treatment interruption. This is because the PPC is being continuously released into the plasma and comprises the bulk of the residual viremia. An additional issue is related to the problem of HIV-1 eradication. There is general agreement that latent reservoir in resting CD4+ T cells must be eliminated before HIV-1 infection can be cured, and several groups have described strategies for targeting this reservoir (5, 24-28, 42). However, if the source of the PPC turns out not to be a CD4+ T cell, then eradication may also be dependent upon the development of another strategy for elimination of the cells that produce the PPC.
Supplementary Material
[Supplemental material]
Acknowledgments
This study was supported by the Johns Hopkins University School of Medicine General Clinical Research, grant number M01-RR00052, from the National Center for Research Resources, NIH; by NIH grants K08AI060367 (to R.E.N.) and AI43222 and AI51178 (to R.F.S.); and by a grant from the Doris Duke Charitable Foundation. C.O.W. was supported by NIH grant AI 065960.
This work is dedicated to the memory of Ann M. Siliciano.
We thank Devin Dressman and Bert Vogelstein for suggestions on digital PCR.
Footnotes
REFERENCES
- 1.Bailey, J. R., K. G. Lassen, H. C. Yang, T. C. Quinn, S. C. Ray, J. N. Blanson, and R. F. Siliciano. 2006. Neutralizing antibodies do not mediate suppression of human immunodeficiency virus type 1 in elite suppressors or selection of plasma virus variants in patients on highly active antiretroviral therapy. J. Virol. 80**:**4758-4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1a.Blankson, J. N., D. Finzi, T. C. Pierson, B. P. Sabundayo, K. Chadwich, J. B. Margolick, T. C. Quinn, and R. F. Siliciano. 2000. Biphasic decay of latently infected CD4+ T cells in acute HIV-1 infection. J. Infect. Dis. 182**:**1636-1642. [DOI] [PubMed] [Google Scholar]
- 2.Bukrinsky, M. I., T. L. Stanwick, M. P. Dempsey, and M. Stevenson. 1991. Quiescent T lymphocytes as an inducible virus reservoir in HIV-1 infection. Science 254**:**423-427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cavert, W., D. W. Notermans, K. Staskus, S. W. Wietgrefe, M. Zupancic, K. Gebhard, K. Henry, S.-C. Zhang, R. Mills, H. McDade, J. Goudsmit, S. A. Danner, and A. T. Haase. 1997. Kinetics of response in lymphoid tissue to antiretroviral therapy of HIV-1 infection. Science 276**:**960-964. [DOI] [PubMed] [Google Scholar]
- 4.Chun, T. W., R. T. Davey, Jr., M. Ostrowski, J. J. Shawn, D. Engel, J. I. Mullins, and A. S. Fauci. 2000. Relationship between pre-existing viral reservoirs and the re-emergence of plasma viremia after discontinuation of highly active anti-retroviral therapy. Nat. Med. 6**:**757-761. [DOI] [PubMed] [Google Scholar]
- 5.Chun, T. W., D. Engel, S. B. Mizell, C. W. Hallahan, M. Fischette, S. Park, R. T. Davey, Jr., M. Dybul, J. A. Kovacs, J. A. Metcalf, J. M. Mican, M. M. Berrey, L. Corey, H. C. Lane, and A. S. Fauci. 1999. Effect of interleukin-2 on the pool of latently infected, resting CD4+ T cells in HIV-1-infected patients receiving highly active anti-retroviral therapy. Nat. Med. 5**:**651-655. [DOI] [PubMed] [Google Scholar]
- 6.Chun, T. W., D. Finzi, J. Margolick, K. Chadwick, D. Schwartz, and R. F. Siliciano. 1995. In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat. Med. 1**:**1284-1290. [DOI] [PubMed] [Google Scholar]
- 7.Chun, T. W., D. C. Nickle, J. S. Justement, D. Large, A. Semerjian, M. E. Curlin, M. A. O'Shea, C. W. Hallahan, M. Daucher, D. J. Ward, S. Moir, J. I. Mullins, C. Kovacs, and A. S. Fauci. 2005. HIV-infected individuals receiving effective antiviral therapy for extended periods of time continually replenish their viral reservoir. J. Clin. Investig. 115**:**3250-3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chun, T. W., L. Stuyver, S. B. Mizell, L. A. Ehler, J. M. Mican, M. Baseler, A. L. Lloyd, M. A. Nowak, and A. S. Fauci. 1997. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 94**:**13193-13197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chun, T.-W., L. Carruth, D. Finzi, X. Shen, J. A. Digiuseppe, H. Taylor, M. Hermankova, K. Chadwick, J. Margolick, T. C. Quinn, Y.-H. Kuo, R. Brookmeyer, M. A. Zeiger, P. Barditch-Crovo, and R. F. Siliciano. 1997. Quantitation of latent tissue reservoirs and total body load in HIV-1 infection. Nature 387**:**183-188. [DOI] [PubMed] [Google Scholar]
- 10.Dornadula, G., H. Zhang, B. VanUitert, J. Stern, L. Livornese, Jr., M. J. Ingerman, J. Witek, R. J. Kedanis, J. Natkin, J. DeSimone, and R. J. Pomerantz. 1999. Residual HIV-1 RNA in blood plasma of patients taking suppressive highly active antiretroviral therapy. JAMA 282**:**1627-1632. [DOI] [PubMed] [Google Scholar]
- 11.Finzi, D., J. Blankson, J. D. Siliciano, J. B. Margolick, K. Chadwick, T. Pierson, K. Smith, J. Lisziewicz, F. Lori, C. Flexner, T. C. Quinn, R. E. Chaisson, E. Rosenberg, B. Walker, S. Gange, J. Gallant, and R. F. Siliciano. 1999. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 5**:**512-517. [DOI] [PubMed] [Google Scholar]
- 12.Finzi, D., M. Hermankova, T. Pierson, L. M. Carruth, C. Buck, R. E. Chaisson, T. C. Quinn, K. Chadwick, J. Margolick, R. Brookmeyer, J. Gallant, M. Markowitz, D. D. Ho, D. Richman, and R. F. Siliciano. 1997. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278**:**1295-1300. [DOI] [PubMed] [Google Scholar]
- 13.Frenkel, L. M., Y. Wang, G. H. Learn, J. L. McKernan, G. M. Ellis, K. M. Mohan, S. E. Holte, S. M. De Vange, D. M. Pawluk, A. J. Melvin, P. F. Lewis, L. M. Heath, I. A. Beck, M. Mahalanabis, W. E. Naugler, N. H. Tobin, and J. I. Mullins. 2003. Multiple viral genetic analyses detect low-level human immunodeficiency virus type 1 replication during effective highly active antiretroviral therapy. J. Virol. 77**:**5721-5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Furtado, M. R., D. S. Callaway, J. P. Phair, K. J. Kunstman, J. L. Stanton, C. A. Macken, A. S. Perelson, and S. M. Wolinsky. 1999. Persistence of HIV-1 transcription in peripheral-blood mononuclear cells in patients receiving potent antiretroviral therapy. N. Engl. J. Med. 340**:**1614-1622. [DOI] [PubMed] [Google Scholar]
- 15.Gulick, R. M., J. W. Mellors, D. Havlir, J. J. Eron, C. Gonzalez, D. McMahon, D. D. Richman, F. T. Valentine, L. Jonas, A. Meibohm, E. A. Emini, and J. A. Chodakewitz. 1997. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N. Engl. J. Med. 337**:**734-739. [DOI] [PubMed] [Google Scholar]
- 16.Hammer, S. M., K. E. Squires, M. D. Hughes, J. M. Grimes, L. M. Demeter, J. S. Currier, J. J. Eron, Jr., J. E. Feinberg, H. H. Balfour, Jr., L. R. Deyton, J. A. Chodakewitz, and M. A. Fischl. 1997. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. AIDS Clinical Trials Group 320 Study Team. N. Engl. J. Med. 337**:**725-733. [DOI] [PubMed] [Google Scholar]
- 17.Havlir, D. V., R. Bassett, D. Levitan, P. Gilbert, P. Tebas, A. C. Collier, M. S. Hirsch, C. Ignacio, J. Condra, H. F. Gunthard, D. D. Richman, and J. K. Wong. 2001. Prevalence and predictive value of intermittent viremia with combination HIV therapy. JAMA 286**:**171-179. [DOI] [PubMed] [Google Scholar]
- 18.Havlir, D. V., M. C. Strain, M. Clerici, C. Ignacio, D. Trabattoni, P. Ferrante, and J. K. Wong. 2003. Productive infection maintains a dynamic steady state of residual viremia in human immunodeficiency virus type 1-infected persons treated with suppressive antiretroviral therapy for five years. J. Virol. 77**:**11212-11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hermankova, M., S. C. Ray, C. Ruff, M. Powell-Davis, R. Ingersoll, R. T. D'Aquila, T. C. Quinn, J. D. Siliciano, R. F. Siliciano, and D. Persaud. 2001. HIV-1 drug resistance profiles in children and adults with viral load of <50 copies/ml receiving combination therapy. JAMA 286**:**196-207. [DOI] [PubMed] [Google Scholar]
- 20.Ho, D. D., A. U. Neumann, A. S. Perelson, W. Chen, J. M. Leonard, and M. Markowitz. 1995. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373**:**123-126. [DOI] [PubMed] [Google Scholar]
- 21.Imamichi, H., K. A. Crandall, V. Natarajan, M. K. Jiang, R. L. Dewar, S. Berg, A. Gaddam, M. Bosche, J. A. Metcalf, R. T. Davey, Jr., and H. C. Lane. 2001. Human immunodeficiency virus type 1 quasi species that rebound after discontinuation of highly active antiretroviral therapy are similar to the viral quasi species present before initiation of therapy. J. Infect. Dis. 183**:**36-50. [DOI] [PubMed] [Google Scholar]
- 22.Kieffer, T. L., M. M. Finucane, R. E. Nettles, T. C. Quinn, K. W. Broman, S. C. Ray, D. Persaud, and R. F. Siliciano. 2004. Genotypic analysis of HIV-1 drug resistance at the limit of detection: virus production without evolution in treated adults with undetectable HIV loads. J. Infect. Dis. 189**:**1452-1465. [DOI] [PubMed] [Google Scholar]
- 23.Kieffer, T. L., P. Kwon, R. E. Nettles, Y. Han, S. C. Ray, and R. F. Siliciano. 2005. G→A hypermutation in protease and reverse transcriptase regions of human immunodeficiency virus type 1 residing in resting CD4+ T cells in vivo. J. Virol. 79**:**1975-1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korin, Y. D., D. G. Brooks, S. Brown, A. Korotzer, and J. A. Zack. 2002. Effects of prostratin on T-cell activation and human immunodeficiency virus latency. J. Virol. 76**:**8118-8123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kulkosky, J., D. M. Culnan, J. Roman, G. Dornadula, M. Schnell, M. R. Boyd, and R. J. Pomerantz. 2001. Prostratin: activation of latent HIV-1 expression suggests a potential inductive adjuvant therapy for HAART. Blood 98**:**3006-3015. [DOI] [PubMed] [Google Scholar]
- 26.Kulkosky, J., G. Nunnari, M. Otero, S. Calarota, G. Dornadula, H. Zhang, A. Malin, J. Sullivan, Y. Xu, J. DeSimone, T. Babinchak, J. Stern, W. Cavert, A. Haase, and R. J. Pomerantz. 2002. Intensification and stimulation therapy for human immunodeficiency virus type 1 reservoirs in infected persons receiving virally suppressive highly active antiretroviral therapy. J. Infect. Dis. 186**:**1403-1411. [DOI] [PubMed] [Google Scholar]
- 27.Lehrman, G., I. B. Hogue, S. Palmer, C. Jennings, C. A. Spina, A. Wiegand, A. L. Landay, R. W. Coombs, D. D. Richman, J. W. Mellors, J. M. Coffin, R. J. Bosch, and D. M. Margolis. 2005. Depletion of latent HIV-1 infection in vivo: a proof-of-concept study. Lancet 366**:**549-555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin, X., D. Irwin, S. Kanazawa, L. Huang, J. Romeo, T. S. Yen, and B. M. Peterlin. 2003. Transcriptional profiles of latent human immunodeficiency virus in infected individuals: effects of Tat on the host and reservoir. J. Virol. 77**:**8227-8236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu, S. L., A. G. Rodrigo, R. Shankarappa, G. H. Learn, L. Hsu, O. Davidov, L. P. Zhao, and J. I. Mullins. 1996. HIV quasispecies and resampling. Science 273**:**415-416. [DOI] [PubMed] [Google Scholar]
- 30.Mansky, L. M., and H. M. Temin. 1995. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 69**:**5087-5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Markowitz, M., M. Louie, A. Hurley, E. Sun, M. Di Mascio, A. S. Perelson, and D. D. Ho. 2003. A novel antiviral intervention results in more accurate assessment of human immunodeficiency virus type 1 replication dynamics and T-cell decay in vivo. J. Virol. 77**:**5037-5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Monie, D., R. P. Simmons, R. E. Nettles, T. L. Kieffer, Y. Zhou, H. Zhang, S. Karmon, R. Ingersoll, K. Chadwick, H. Zhang, J. B. Margolick, T. C. Quinn, S. C. Ray, M. Wind-Rotolo, M. Miller, D. Persaud, and R. F. Siliciano. 2005. A novel assay allows genotyping of the latent reservoir for human immunodeficiency virus type 1 in the resting CD4+ T cells of viremic patients. J. Virol. 79**:**5185-5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Natarajan, V., M. Bosche, J. A. Metcalf, D. J. Ward, H. C. Lane, and J. A. Kovacs. 1999. HIV-1 replication in patients with undetectable plasma virus receiving HAART. Highly active antiretroviral therapy. Lancet 353**:**119-120. [DOI] [PubMed] [Google Scholar]
- 34.Nettles, R. E., T. L. Kieffer, P. Kwon, D. Monie, Y. Han, T. Parsons, J. Cofrancesco, Jr., J. E. Gallant, T. C. Quinn, B. Jackson, C. Flexner, K. Carson, S. Ray, D. Persaud, and R. F. Siliciano. 2005. Intermittent HIV-1 viremia (Blips) and drug resistance in patients receiving HAART. JAMA 293**:**817-829. [DOI] [PubMed] [Google Scholar]
- 35.Page, R. D. 1996. TreeView: an application to display phylogenetic trees on personal computers. Comput. Appl. Biosci. 12**:**357-358. [DOI] [PubMed] [Google Scholar]
- 36.Palmer, S., A. P. Wiegand, F. Maldarelli, H. Bazmi, J. M. Mican, M. Polis, R. L. Dewar, A. Planta, S. Liu, J. A. Metcalf, J. W. Mellors, and J. M. Coffin. 2003. New real-time reverse transcriptase-initiated PCR assay with single-copy sensitivity for human immunodeficiency virus type 1 RNA in plasma. J. Clin. Microbiol. 41**:**4531-4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perelson, A. S., P. Essunger, Y. Cao, M. Vesanen, A. Hurley, K. Saksela, M. Markowitz, and D. D. Ho. 1997. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature 387**:**188-191. [DOI] [PubMed] [Google Scholar]
- 38.Persaud, D., G. K. Siberry, A. Ahonkhai, J. Kajdas, D. Monie, N. Hutton, D. C. Watson, T. C. Quinn, S. C. Ray, and R. F. Siliciano. 2004. Continued production of drug-sensitive human immunodeficiency virus type 1 in children on combination antiretroviral therapy who have undetectable viral loads. J. Virol. 78**:**968-979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pierson, T., T. L. Hoffman, J. Blankson, D. Finzi, K. Chadwich, J. B. Margolick, C. Buck, J. D. Siliciano, R. W. Doms, and R. F. Siliciano. 2000. Characterization of chemokine receptor utilization of viruses in the latent reservoir for HIV-1. J. Virol. 74**:**7824-7833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Posada, D., and K. A. Crandall. 1998. MODELTEST: testing the model of DNA substitution. Bioinformatics 14**:**817-818. [DOI] [PubMed] [Google Scholar]
- 41.Ramratnam, B., J. E. Mittler, L. Zhang, D. Boden, A. Hurley, F. Fang, C. A. Macken, A. S. Perelson, M. Markowitz, and D. D. Ho. 2000. The decay of the latent reservoir of replication-competent HIV-1 is inversely correlated with the extent of residual viral replication during prolonged anti-retroviral therapy. Nat. Med. 6**:**82-85. [DOI] [PubMed] [Google Scholar]
- 42.Scripture-Adams, D. D., D. G. Brooks, Y. D. Korin, and J. A. Zack. 2002. Interleukin-7 induces expression of latent human immunodeficiency virus type 1 with minimal effects on T-cell phenotype. J. Virol. 76**:**13077-13082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shankarappa, R., J. B. Margolick, S. J. Gange, A. G. Rodrigo, D. Upchurch, H. Farzadegan, P. Gupta, C. R. Rinaldo, G. H. Learn, X. He, X. L. Huang, and J. I. Mullins. 1999. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J. Virol. 73**:**10489-10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Siliciano, J. D., J. Kajdas, D. Finzi, T. C. Quinn, K. Chadwick, J. B. Margolick, C. Kovacs, S. J. Gange, and R. F. Siliciano. 2003. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 9**:**727-728. [DOI] [PubMed] [Google Scholar]
- 45.Siliciano, J. D., and R. F. Siliciano. 2005. Enhanced culture assay for detection and quantitation of latently infected, resting CD4+ T-cells carrying replication-competent virus in HIV-1-infected individuals. Methods Mol. Biol. 304**:**3-15. [DOI] [PubMed] [Google Scholar]
- 46.Spina, C. A., J. C. Guatelli, and D. D. Richman. 1995. Establishment of a stable, inducible form of human immunodeficiency virus type 1 DNA in quiescent CD4 lymphocytes in vitro. J. Virol. 69**:**2977-2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Strain, M. C., H. F. Gunthard, D. V. Havlir, C. C. Ignacio, D. M. Smith, A. J. Leigh-Brown, T. R. Macaranas, R. Y. Lam, O. A. Daly, M. Fischer, M. Opravil, H. Levine, L. Bacheler, C. A. Spina, D. D. Richman, and J. K. Wong. 2003. Heterogeneous clearance rates of long-lived lymphocytes infected with HIV: intrinsic stability predicts lifelong persistence. Proc. Natl. Acad. Sci. USA 100**:**4819-4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tobin, N. H., G. H. Learn, S. E. Holte, Y. Wang, A. J. Melvin, J. L. McKernan, D. M. Pawluk, K. M. Mohan, P. F. Lewis, J. I. Mullins, and L. M. Frenkel. 2005. Evidence that low-level viremias during effective highly active antiretroviral therapy result from two processes: expression of archival virus and replication of virus. J. Virol. 79**:**9625-9634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wei, X., S. K. Ghosh, M. E. Taylor, V. A. Johnson, E. A. Emini, P. Deutsch, J. D. Lifson, S. Bonhoeffer, M. A. Nowak, B. H. Hahn, and G. M. Shaw. 1995. Viral dynamics in human immunodeficiency virus type 1 infection. Nature 373**:**117-122. [DOI] [PubMed] [Google Scholar]
- 50.Wilke, C. O. 2003. Probability of fixation of an advantageous mutant in a viral quasispecies. Genetics 163**:**467-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wong, J. K., M. Hezareh, H. F. Gunthard, D. V. Havlir, C. C. Ignacio, C. A. Spina, and D. D. Richman. 1997. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 278**:**1291-1295. [DOI] [PubMed] [Google Scholar]
- 52.Yerly, S., L. Kaiser, T. V. Perneger, R. W. Cone, M. Opravil, J. P. Chave, H. Furrer, B. Hirschel, and L. Perrin. 2000. Time of initiation of antiretroviral therapy: impact on HIV-1 viraemia. The Swiss HIV Cohort Study. AIDS 14**:**243-249. [DOI] [PubMed] [Google Scholar]
- 53.Zack, J. A., S. J. Arrigo, S. R. Weitsman, A. S. Go, A. Haislip, and I. S. Y. Chen. 1990. HIV-1 entry into quiescent primary lymphocytes: molecular analysis reveals a labile, latent viral structure. Cell 61**:**213-222. [DOI] [PubMed] [Google Scholar]
- 54.Zhang, H., Y. Zhou, C. Alcock, T. Kiefer, D. Monie, J. Siliciano, Q. Li, P. Pham, J. Cofrancesco, D. Persaud, and R. F. Siliciano. 2004. Novel single-cell-level phenotypic assay for residual drug susceptibility and reduced replication capacity of drug-resistant human immunodeficiency virus type 1. J. Virol. 78**:**1718-1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang, L., C. Chung, B.-S. Hu, T. He, Y. Guo, A. J. Kim, E. Skulsky, X. Jin, A. Hurley, B. Ramratnam, M. Markowitz, and D. D. Ho. 2000. Genetic characterization of rebounding HIV-1 after cessation of highly active antiretroviral therapy. J. Clin. Investig. 106**:**839-845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang, L., B. Ramratnam, K. Tenner-Racz, Y. He, M. Vesanen, S. Lewin, A. Talal, P. Racz, A. S. Perelson, B. Korber, M. Markowitz, and D. D. Ho. 1999. Quantifying residual HIV-1 replication in patients receiving combination antiretroviral therapy. N. Engl. J. Med. 340**:**1605-1613. [DOI] [PubMed] [Google Scholar]
- 57.Zhou, Y., H. Zhang, J. D. Siliciano, and R. F. Siliciano. 2005. Kinetics of human immunodeficiency virus type 1 decay following entry into resting CD4+ T cells. J. Virol. 79**:**2199-2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu, T., D. Muthui, S. Holte, D. Nickle, F. Feng, S. Brodie, Y. Hwangbo, J. I. Mullins, and L. Corey. 2002. Evidence for human immunodeficiency virus type 1 replication in vivo in CD14+ monocytes and its potential role as a source of virus in patients on highly active antiretroviral therapy. J. Virol. 76**:**707-716. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
[Supplemental material]