PPARα mediates the hypolipidemic action of fibrates by antagonizing FoxO1 (original) (raw)
. Author manuscript; available in PMC: 2009 Apr 3.
Published in final edited form as: Am J Physiol Endocrinol Metab. 2006 Sep 19;292(2):E421–E434. doi: 10.1152/ajpendo.00157.2006
Abstract
High-fructose consumption is associated with insulin resistance and diabetic dyslipidemia, but the underlying mechanism is unclear. We show in hamsters that high-fructose feeding stimulated forkhead box O1 (FoxO1) production and promoted its nuclear redistribution in liver, correlating with augmented apolipoprotein C-III (apoC-III) production and impaired triglyceride metabolism. High-fructose feeding upregulated peroxisome proliferator-activated receptor-γ coactivator-1β and sterol regulatory element binding protein-1c expression, accounting for increased fat infiltration in liver. High-fructose-fed hamsters developed hypertriglyceridemia, accompanied by hyperinsulinemia and glucose intolerance. These metabolic aberrations were reversible by fenofibrate, a commonly used anti-hypertriglyceridemia agent that is known to bind and activate peroxisome proliferator-activated receptor-α (PPARα). PPARα physically interacted with, but functionally antagonized, FoxO1 in hepatic apoC-III expression. These data underscore the importance of FoxO1 deregulation in the pathogenesis of hypertriglyceridemia in high-fructose-fed hamsters. Counterregulation of hepatic FoxO1 activity by PPARα constitutes an important mechanism by which fibrates act to curb apoC-III overproduction and ameliorate hypertriglyceridemia.
Keywords: hypertriglyceridemia, forkhead box O1, peroxisome proliferator-activated receptor-α, apolipoprotein C-III, very-low-density lipoprotein-triglyceride, microsomal triglyceride transfer protein
Hypertriglyceridemia is characterized by increased production of very-low-density lipoprotein (VLDL) and/or decreased clearance of triglyceride (TG)-rich particles (4). Due to its proatherogenic profile, hypertriglyceridemia is a major contributing factor for the pathogenesis of atherosclerosis and coronary artery disease in obesity and type 2 diabetes. While the pathophysiology of hypertriglyceridemia is poorly understood, its close association with visceral adiposity and type 2 diabetes implicates insulin resistance as a causative factor in the development of hypertriglyceridemia (5, 12, 22, 29, 42). However, the molecular events that link insulin resistance to the pathogenesis of hypertriglyceridemia remain elusive.
An important player in plasma VLDL-TG metabolism is apolipoprotein C-III (apoC-III). It functions as an inhibitor of lipoprotein lipase and hepatic lipase, playing a pivotal role in the hydrolysis and clearance of TG-rich particles such as VLDL-TG and chylomicrons (27, 36, 51, 53). Elevated plasma apoC-III levels are associated with impaired clearance of TG-rich particles, leading to the accumulation of TG-rich lipoprotein remnants in plasma and the development of hypertriglyceridemia.
Our recent data indicate that forkhead box O1 (FoxO1), a nuclear transcription factor known to be a mediator of insulin signaling, plays an important role in hepatic regulation of apoC-III. FoxO1 stimulates hepatic apoC-III expression, which is counteracted by insulin (2). Elevated FoxO1 production in liver augments hepatic apoC-III expression, resulting in increased plasma TG levels and impaired postprandial fat clearance in mice (2). Transgenic mice expressing a FoxO1 constitutively active allele develop hypertriglyceridemia, as manifested by significantly elevated plasma VLDL-TG levels and retarded postprandial fat clearance (2). In insulin-resistant states, hepatic FoxO1 activity becomes deregulated, culminating in elevated hepatic production along with skewed nuclear redistribution and accounting for its increased _trans_-activation activity in liver (2, 3). Furthermore, anti-sense oligonucleotide-mediated inhibition of FoxO1 activity improves hepatic and peripheral insulin action in high-fat-diet-induced obese mice (47). These data implicate FoxO1 at the interface between impaired insulin action and aberrant apoC-III expression, contributing to the pathogenesis of hypertriglyceridemia in insulin-resistant subjects.
Fibrates are commonly used as hypolipidemic agents for treating hypertriglyceridemia (13, 23, 33). An important function of fibrates is to suppress apoC-III production, contributing to increased VLDL-TG catabolism and decreased plasma TG levels (15, 16, 45, 52). Fibrates bind and activate peroxisome proliferator-activated receptor-α (PPARα), a transcription factor that is abundantly expressed in liver (8, 21, 32, 37, 38). However, it is conceptually difficult to reconcile fibrate-mediated activation of PPARα activity with reduced apoC-III production and improved TG metabolism (24, 25, 28, 44). Based on the findings that FoxO1 mediates the inhibitory effect of insulin on hepatic apoC-III expression and excessive FoxO1 activity may contribute to unrestrained apoC-III production and metabolic aberrations in diabetes (2), we hypothesized that fibrates would suppress FoxO1 expression and reduce apoC-III production, contributing to improved VLDL-TG metabolism.
We tested this hypothesis in high-fructose-fed hamsters. Syrian golden hamsters develop hypertriglyceridemia, accompanied by insulin resistance, in response to high-fructose feeding (10, 14, 30). But the underlying pathogenesis of high-fructose-induced insulin resistance and hypertriglyceridemia remains elusive. To address the function of FoxO1 in hypertriglyceridemia and define the mechanism of fibrate action in TG metabolism, we studied hepatic FoxO1 expression in hamsters fed a high-fructose diet vs. regular chow, with and without fibrate treatment. We show that high-fructose feeding augmented hepatic FoxO1 production and promoted its nuclear localization. This effect, along with impaired insulin action, contributes to apoC-III overproduction and the development of hypertriglyceridemia. High-fructose feeding also resulted in marked induction of hepatic expression of PPARγ coactivator-1β (PGC-1β) (31), a coactivator of sterol regulatory element-binding protein-1c (SREBP-1c), accounting for increased fat infiltration in liver. Fibrates upregulated hepatic production of PPARα, which antagonized FoxO1 activity and corrected FoxO1 deregulation in liver, contributing to enhanced insulin sensitivity and the amelioration of hypertriglyceridemia in high-fructose-fed hamsters. These data underscore the importance of FoxO1 deregulation in the pathogenesis of hypertriglyceridemia and illustrate a novel mechanism by which fibrates act to improve plasma VLDL-TG metabolism.
MATERIALS AND METHODS
Animal studies
Syrian golden hamsters (male, 5-wk old, body weight of 81–90 g) were purchased from Charles River Laboratory (Wilmington, MA). Animals were fed with standard rodent chow and water ad libitum in sterile cages with a 12:12-h light-dark cycle. To induce hypertriglyceridemia, hamsters were fed a high-fructose diet (60% fructose, 6% fat, DYET no. 161506; Dyets, Bethlehem, PA) for 4 wk. For fibrate treatment, high-fructose-induced hamsters were randomly divided into two groups (n = 9), which were treated with once-daily oral gavage of 250-μl fenofibrate (50 mg/kg; Abbott Laboratories, Chicago, IL) or 250-μl saline as control. All hamsters were maintained on a high-fructose diet during a 4-wk treatment. In addition, one group of hamsters fed regular rodent chow was used as normal control. Blood was collected from the tail vein into capillary tubes precoated with potassium-EDTA (Sarstedt, Nümbrecht, Germany) for the preparation of plasma or the determination of blood glucose levels using Glucometer Elite (Bayer, IN). Plasma levels of TG and cholesterol were determined using Thermo Infinity TG and cholesterol reagents (Thermo Electron, Melbourne, Australia). Plasma free fatty acid (FFA) levels were determined using the Wako FFA assay kit (Wako Chemical USA, Richmond, VA). Plasma insulin levels were determined by anti-human insulin ELISA that cross-reacts with hamster insulin (ALPCO, Windham, NH). At the end of study, animals were killed by CO2 inhalation, and liver tissue was frozen in liquid N2. All procedures were approved by the Institutional Animal Care and Use Committee of the Children’s Hospital of Pittsburgh (protocol no. 41-04).
Glucose tolerance test
Hamsters were fasted for 5 h and injected intraperitoneally with 50% dextrose solution (Abbott Laboratories) at 5 g/kg body wt. Blood glucose levels were determined and plotted as a function of time. Area under the curve was calculated using the KaleidaGraph software (Synergy Software, Reading, PA).
Insulin sensitivity index
Insulin sensitivity index (ISI) was calculated using the formula ISI = 2/[(INS × GLU) + 1], where INS is fasting plasma insulin levels and GLU is fasting blood glucose levels, with all values being converted to μmol/1 as described (7). A similar homeostasis model assessment (HOMA) for calculating ISI has been described by Mattews et al. (35). The principle of this formula is as follows: a reduction in insulin sensitivity results in elevated levels of either blood glucose, plasma insulin, or both. ISI is a function of the compound effect of fasting blood glucose and plasma insulin levels. Preclinical and clinical data indicate that ISI correlates inversely with insulin resistance (6, 7, 35).
Hepatic lipid content
Forty milligrams of liver tissue were homogenized in 800 μl of HPLC grade acetone. After incubation with agitation at room temperature overnight, aliquots (5 μl) of acetone-extract lipid suspension were used for the determination of TG concentrations using the Thermo Infinity TG reagent (Thermo Electron). Hepatic lipid content was defined as milligrams of TG per gram of liver tissue.
Western blot analysis
Isolation of nuclear proteins from homogenized liver tissue using the Pierce NE-PER extraction reagents (Pierce, Rockford, IL) has been described (2). Aliquots (40 mg) of liver tissue were homogenized in 400 μl of ice-cold cytosolic extraction reagent-I solution (Pierce), supplemented with 4-μl protease inhibitor cocktail (Pierce). Nuclear fractions were separated from cytoplasm and subjected to immunoblot analysis using antibody against FoxO1, PPARα, SREBP-1c, PGC-1β, and β-actin proteins, as previously described (2). The intensity of protein bands was quantified by densitometry using the National Institutes of Health Image software (NIH, Bethesda, MD), as described (2). Polyclonal rabbit anti-FoxO1 antibody was developed in our laboratory by immunization of rabbits with the glutathione S-transferase-tagged human FoxO1 protein (Genemed Synthesis, San Francisco, CA). Rabbit anti-PGC-1β antibody was provided by Dr. Spiegelman’s laboratory (31). Antibodies against PPARα (ABR Affinity BioReagents, Golden, CO), SREBP-1c (catalog no. sc-13551; Santa Cruz Biotechnology, Santa Cruz, CA), and β-actin (sc-1615, Santa Cruz Biotechnology) were commercially available. To determine plasma apoC-III levels, aliquots of plasma at a fixed quantity of protein (20 μg) were blotted to a piece of nitrocellulose membrane by dot blotting. The membrane was probed with goat anti-apoC-III antibody (1:1,000 dilution; Abcam, Cambridge, MA), followed by incubation with rabbit anti-goat IgG conjugated with peroxidase (1:6,000 dilution; Jackson ImmunoResearch Laboratories, West Grove, PA). ApoC-III protein was detected using the enhanced chemiluminescence detection reagents (GE HealthCare), as described (2). In addition, aliquots of plasma at a fixed concentration of 15-μg proteins per lane were applied to 4–20% polyacrylamide gels. The gels were subjected to Western blot assay using goat anti-apoC-III antibody (Abeam). ApoC-III proteins were visualized by enhanced chemiluminescence detection reagents, and the relative intensities of protein bands were quantified by densitometry, as described (2). For the determination of hepatic microsomal TG transfer protein (MTP) levels by the immunoblot assay, we have generated polyclonal anti-MTP antibody derived against a homologous region of MTP protein between rodents and humans (SYSASVKGHTTGLSLN, corresponding to MTP amino acid residues 14_–_29). After affinity purification by protein-A column chromatography, this MTP antibody (1:1,000 dilution) reacted specifically with both rodent and human MTP proteins on Western blots.
Immunoprecipitation
Immunoprecipitation of FoxO1 from liver tissue by protein-A chromatography has been described (3). Aliquots of liver tissue (20 mg) were homogenized in 800 μl of M-PER buffer supplemented with 8-μl protease inhibitor cocktail (Pierce). After centrifugation at 13,000 rpm for 10 min in a microfuge, aliquots of protein lysates (600 μg) were incubated with 5 μg of rabbit anti-FoxO1 antibody or preimmune serum for 1 h at room temperature. The samples were mixed with 80 μl of 50% protein A slurry and incubated for 1 h at room temperature, followed by washing in 80 μl of 1× PBS containing 0.05% Tween 20 for three cycles, each 30 min. Immunoprecipitated proteins were eluted and analyzed on immunoblots using anti-FoxO1 and anti-PPARα antibodies, respectively.
Immunoprecipitation of FoxO1-Δ256 and PPARα complex was performed. FoxO1-Δ256 is a FoxO1 dominant-negative mutant that contains the amino domain (1–256 amino acid residues), as described (3, 39). HepG2 were transfected with 4 μg of pPPARα plasmid in Vitacell culture medium (American Type Culture Collection) in six-well plates. Four hours later, culture media were renewed with prewarmed media supplemented with adenoviral vector expressing FoxO1-Δ256 at a multiplicity of infection of 100 plaque-forming units/cell, as described (3). After 48-h incubation, cells were harvested and lysed in 100 μl of M-PER buffer supplemented with 1-μl protease inhibitor cocktail (Pierce). Protein lysates were subjected to immunoprecipitation by rabbit anti-FoxO1 antibody or preimmune serum, as described above. Each condition was run in triplicate. Immunoprecipitates were analyzed by immunoblot assay using anti-FoxO1 or anti-PPARα antibody.
Effect of fenofibrate on FoxO1 and PPARα interaction was determined as follows: HepG2 cells were cotransfected with FoxO1- and PPARα-expressing plasmids (2 μg each plasmid). After 24 h of incubation, cells were incubated in the presence and absence of 50 mM fenofibrate in culture medium for 16 h, followed by immunoprecipitation using anti-FoxO1 antibody. Immunoprecipitated proteins were subjected to semiquantitative immunoblot assay by anti-FoxO1 and anti-PPARα antibodies, respectively.
Fast protein liquid chromatography fractionation of lipoproteins
Aliquots (500 μl) of plasma were applied to two head-to-tail linked Tricorn high-performance Superose S-6 10/300GL columns using a fast protein liquid chromatography system (Amersham Biosciences), followed by elution with PBS at a constant flow rate of 0.5 ml/min. Fractions (500 μl) were eluted and assayed for TG and cholesterol concentrations using the Thermo Infinity TG and cholesterol reagents (Thermo Electron).
Immunohistochemistry
Liver tissue from euthanized hamsters was embedded with Histoprep tissue embedding media (Fisher Scientific). Frozen sections were cut (8 μm) and subjected to immunohistochemistry using rabbit anti-FoxO1 antibody (dilution at 1:1,500). The second antibody was Cy3-conjugated goat anti-rabbit IgG (dilution 1:500, Jackson ImmunoResearch Laboratories). The nuclei of hepatocytes were visualized by staining with TO-PRO-3 dye (Molecular Probes, Eugene, OR). Specimens were examined under a laser scanning confocal microscope (Nikon E800, Nikon).
Liver histology
Liver tissue was embedded in the Histoprep tissue embedding media and snap-frozen. Frozen sections (8 μm) were cut for fat staining with Oil red O, as described (19).
Plasmid transfection and luciferase assay
HepG2 cells in six-well microplates were transfected with 1-μg plasmid pHD317 encoding luciferase cDNA under the control of the human APOC3 promoter using the Lipofectamine 2000 (Invitrogen, Carlsbad, CA). In each transfection, 1-μg plasmid pCA35-LacZ was included, and the amount of β-galactosidase (β-gal) activity was used as control for the normalization of transfection efficiency. After 48-h incubation, cells were collected for luciferase and β-gal activity assays as described (2).
FoxO1 promoter-directed luciferase reporter system
The 2-kb mouse FoxO1 promoter [−1,748/+407 nucleotide (nt)], including the 5′-untranslated region, was cloned from Balb/C mouse genomic DNA (BioChain Institute, Hayward, CA) into the TA-cloning vector pCR2.1 (Invitrogen) by PCR using specific primers (for forward reaction, 5′-GATCTGGGGAAAGCTTTAGGAAAAC-3′; and reverse reaction, 5′-CAGAGGGCCATGAAGAGACGAC-3′). After verification by DNA sequencing, the _Kpn_I_-Xho_I DNA fragment harboring FoxO1 promoter was subcloned upstream of the luciferase gene in the pGL3-basic plasmid (Promega, Madison, WI).
RNA isolation and RT-PCR
Total RNA preparation from HepG2 cells for RT-PCR analysis are as described (3). The primers for PPARα are 5′-AAGGGCTTCTTTCGGCGAAC-3′ (corresponding to a conserved region of mouse and human PPARα cDNA 367–386 nt) and 5′-TGACCTTGTTCATGTTGAAGTTCTTCA-3′ (mouse and human PPARα cDNA 644–670 nt). Primers for FoxO1 and β-actin mRNA have been described (3). All primers were obtained commercially from Integrated DNA Technologies (Coralville, IA).
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed as described (2). HepG2 cells (1 × 106 cells) were transfected with pFoxO1 (2 μg) alone or in combination with pPPARα (2 μg). Both pFoxO1 and pPPARα plasmids express the respective transgene under the control of the cytomegalovirus promoter, as derived from the pCR3.1 vector (Invitrogen). They were cloned from C56 BL/6J mouse liver RNA by RT-PCR using specific primers flanking FoxO1 (forward 5′-TCCTAGGCACGAACTCGGAG-3′ and reverse 5′-TTAGCCTGACACCCAGCTGT-3′) and PPARα coding regions (forward 5′-ATCACAGCTTAGCGCTCTGT-3′ and reverse 5′-TCAGTACATGTCTCTGTAGATC-3′). To compensate for the difference in the amount of DNA used for transfection, a control pCA35-LacZ plasmid was included to ensure the same amount of DNA (4 μg) in each transfection. After 48-h incubation, cells were cross-linked with 1% formaldehyde, followed by sonication in a Microson 100-W Ultrasonicator (Structure Probe, West Chester, PA) at 10% of maximum power for 60 pulses, 2 s for each pulse. After centrifugation at 13,000 rpm for 10 min, the supernatant was incubated with 8 μg of polyclonal rabbit anti-FoxO1 antibody, followed by immunoprecipitation using the ChIP kit (Upstate Biotechnology, Lake Placid, NY). As controls, aliquots (1 × 106) of pFoxO1-transfected or pFoxO1/pPPARα-cotransfected HepG2 cells were treated identically for the preparation of cell lysates, which were immunoprecipitated with 8 μg of preimmune rabbit IgG (Genemed Synthesis). The immunoprecipitated DNA was analyzed by PCR using 20-nt primers that flank −675/+1 nt of the APOC3 promoter, as described (2). PCR products were analyzed on 1% agarose gels, and the relative intensity of DNA bands was quantified by densitometry using the NIH Image software, as described (2).
To analyze the potential association between PPARα and APOC3 promoter, HepG2 cells were transfected with 4 μg of pPPARα DNA, followed by ChIP assay using monoclonal anti-PPARα antibody (8 μg, ABR Affinity BioReagents) or 8 μg of preimmune rabbit IgG (Genemed Synthesis). Immunoprecipitates were analyzed by PCR using the APOC3 promoter-specific primers, as described above.
Statistics
Statistical analyses of data were performed by ANOVA using StatView software (Abacus Concepts). Pairwise comparisons were performed to study the significance between different conditions. Data were expressed as means ± SE. P values <0.05 are considered statistically significant.
RESULTS
Effect of fibrates on metabolism in high-fructose-fed hamsters
Fibrates are commonly used oral agents for treating hypertriglyceridemia in subjects with obesity and type 2 diabetes, but their mechanisms of action are not completely understood. To define the molecular mechanism by which fibrates improve TG metabolism, we treated high-fructose-induced hyperlipidemic hamsters with either an oral dose of fenofibrate or saline for 4 wk, using one group of hamsters fed on regular chow as control. High-fructose feeding did not result in significant alterations in blood glucose levels (Fig. 1_A_). However, all hamsters fed on high-fructose diet had significantly higher plasma insulin levels (Fig. 1_B_), which is indicative of insulin resistance.
Fig. 1.

Effect of fenofibrate on glucose metabolism in high-fructose-fed hamsters. A: blood glucose levels. B: plasma insulin levels. C: intraperitoneal glucose tolerance. D: area under the curve (AUC) of blood glucose profiles in response to glucose tolerance. After normalizing to regular chow-treated hamsters, the relative AUC values were plotted. E: insulin sensitivity indexes (ISI). F: body weight. Blood glucose and plasma insulin levels were determined after 16-h fasting. Data were obtained from hamsters fed on regular chow (regular, n = 8), high-fructose diet (fructose, n = 9), and high-fructose plus fenofibrate treatment (fibrate, n = 9) after 4-wk treatment with fenofibrate. NS, not significant. *P < 0.05 by ANOVA.
To corroborate this finding, we performed a glucose tolerance test. Hamsters fed high-fructose diet were associated with impaired abilities to tolerate intraperitoneally infused glucose (Fig. 1, C and D). But in response to a 4-wk treatment with fenofibrate, this impaired glucose tolerance along with elevated plasma insulin levels in high-fructose-fed hamsters were restored to normal. Based on fasting blood glucose and plasma insulin levels, we calculated ISI using a HOMA as described (7). As shown in Fig. 1_E_, high-fructose-fed hamsters were associated with markedly reduced ISI, which was reversed to normal after 4-wk treatment with fenofibrate. Fibrate treatment also resulted in a small reduction in body weight, but the mean body weight of fibrate-treated hamsters was not significantly different from that of regular chow-fed controls (Fig. 1_F_).
To determine the effect of high-fructose diet on lipid metabolism, we determined plasma FFA, TG, and total cholesterol levels. High-fructose-fed hamsters are associated with significantly increased plasma FFA (Fig. 2_A_), TG (Fig. 2B), and cholesterol levels (Fig. 2_C_), compared with hamsters fed regular chow. However, these aberrations in lipid metabolism were reversed to near-normal levels after 4 wk of treatment with fenofibrate.
Fig. 2.

Effect of fenofibrate on plasma lipid metabolism. A: plasma free fatty acid (FFA) levels. B: plasma triglyceride (TG) levels. C: plasma cholesterol levels. Data were obtained following 4-wk treatment with fenofibrate. Aliquots (500 μl) of plasma pooled from killed hamsters in individual groups were subjected to gel filtration column chromatography through 2 consecutive Tricom high-performance Superose S-6 10/300GL columns in a fast protein liquid chromatography (FPLC) system. Seventy fractions were collected for the determination of TG (D) and cholesterol (E) concentrations. Plasma apolipoprotein C-III (apoC-III) levels were determined by dot blot (F) and Western blot (G) assays. Dot blot assay seemed to result in an overestimation of plasma apoC-III levels. H: hepatic microsomal TG transfer protein (MTP) levels were determined by semiquantitative Western blot assay. LDL, lipoprotein lipase; IDL, intermediate-density lipoprotein; HDL, high-density lipoprotein; VLDL, very-low-density lipoprotein. *P < 0.05.
Effect of fibrates on lipoprotein metabolism
To study the effect of fibrates on lipoprotein metabolism, we subjected plasma pooled from hamsters in individual groups to gel filtration chromatography for the fractionation of lipoproteins. In accordance with their elevated plasma TG levels, high-fructose-fed hamsters exhibited significantly increased VLDL-TG levels (Fig. 2D). The fractional concentrations of cholesterol in VLDL and HDL were also markedly increased in response to high-fructose feeding (Fig. 2_E_). After fenofibrate treatment, these elevated VLDL-TG levels were significantly ameliorated, but not restored to normal (Fig. 2_D_). However, fenofibrate treatment reversed elevated VLDL- and HDL-cholesterol levels to normal (Fig. 2_E_).
To gain insight into the molecular basis of fibrate-mediated beneficial effects on VLDL-TG metabolism, we determined the expression levels of apoC-III and MTP, two functions that catalyze the rate-limiting steps in plasma VLDL-TG metabolism (26, 51). Plasma apoC-III levels were determined by both dot blot and Western blot assays. These two approaches consistently show that plasma apoC-III levels were significantly increased in response to high-fructose feeding and were reduced to normal levels after 4-wk fenofibrate treatment (Fig. 2, F and G). Likewise, hepatic MTP production was markedly induced in response to high-fructose feeding, as detected by Western blot analysis (Fig. 2_H_). Fenofibrate treatment suppressed excessive MTP production to normal levels in high-fructose-fed hamsters.
Hepatic lipid content in high-fructose-fed hamsters in response to fibrate treatment
To investigate the metabolic consequence associated with high-fructose feeding, liver tissue from euthanized hamsters was histologically examined after staining with Oil Red O. As shown in Fig. 3, A–C, significantly higher levels of lipid droplets were detected in livers of high-fructose-fed hamsters. This observed increase in hepatic lipid deposition was confirmed by the quantification of overall hepatic fat content. Hamsters fed a high-fructose diet exhibited significantly higher levels of intrahepatic TG content (10.7 ± 0.8 vs. 6.3 ± 0.3 mg/g liver tissue in control diet-fed hamsters, P < 0.001 by ANOVA; Fig. 3_D_). However, in response to fenofibrate treatment, this increased intrahepatic deposition in high-fructose-fed hamsters was reduced to normal, as revealed by both histological examination and quantitative assay of fat content in liver.
Fig. 3.

Hepatic lipid content. Hamsters were killed after 4 wk of fibrate treatment. Liver tissues of hamsters treated with regular chow (A), high-fructose diet (B), and high-fructose diet plus fenofibrate (C) were embedded with Histoprep tissue embedding media. Frozen sections (8 μm) were cut and stained with Oil red O, followed by counterstaining with hematoxylin. D: hepatic lipid content in different groups of hamsters was also determined using a quantitative hepatic TG assay. Bar = 50 μm. *P < 0.001.
To investigate the molecular mechanism underlying increased fat accumulation in livers of high-fructose-fed hamsters, we determined the expression levels of two key transcription factors in hepatic lipid metabolism, SREBP-1c and its coactivator PGC-1β (31). Hepatic expression of both SREBP-1c (Fig. 4_A_) and PGC-1β (Fig. 4_B_) was significantly upregulated, coinciding with increased intrahepatic fat infiltration in high-fructose-fed hamsters. After fenofibrate treatment, hepatic SREBP-1c and PGC-1β expression levels were suppressed to normal, correlating with the reversal of hepatic fat content in fenofibrate-treated hamsters (Fig. 3).
Fig. 4.

Hepatic gene expression. Sterol regulatory element binding protein-1c (SREBP-1c; A), peroxisome proliferator-activated receptor-γ coactivator-1β (PGC-1β; B), forkhead box O1 (FoxO1; C), and peroxisome proliferator-activated receptor-α (PPARα; D) protein levels in livers of euthanized hamsters were determined by semiquantitative immunoblot assays. *P < 0.05.
Hepatic expression of FoxO1 in hyperlipidemic hamsters in response to fibrate treatment
To account for the mechanism of fenofibrate-mediated beneficial effect on hepatic TG metabolism, we determined hepatic FoxO1 abundance in high-fructose-fed hamsters treated with fenofibrate. High-fructose feeding resulted in a more than fourfold increase in hepatic FoxO1 protein levels, which were reduced to normal after a 4-wk treatment with fenofibrate (Fig. 4_C_).
As control, we studied hepatic expression profiles of PPARα, the nuclear receptor of fibrates. As shown in Fig. 4_D_, hepatic PPARα protein levels remained at basal levels in high-fructose-fed hamsters. However, in response to a 4-wk treatment with fenofibrate, hepatic PPARα protein levels were elevated about twofold, which correlated inversely with hepatic FoxO1 expression in fenofibrate-treated hamsters (Fig. 4_C_).
Our laboratory has previously shown that hepatic FoxO1 activity becomes deregulated, culminating in increased nuclear localization in livers with impaired insulin action (2, 3). To determine the potential alterations in hepatic FoxO1 subcellular localization, we performed immunohistochemistry on liver tissue of killed hamsters using anti-FoxO1 antibody. Compared with regular chow-fed hamsters (Fig. 5, A–C), FoxO1 was predominantly localized within the nucleus of hepatocytes in high-fructose-fed hamsters (Fig. 5, D–F). This skewed subcellular distribution of FoxO1 was reversed to normal in response to fenofibrate treatment (Fig. 5, G–I).
Fig. 5.

FoxO1 immunohistochemistry. Liver tissue of hamsters fed regular chow (A, B, and C), high-fructose diet (D, E, and F), and high-fructose diet plus fenofibrate treatment (G, H, and I) was embedded with the Histoprep tissue embedding media. Frozen sections (8 μm) were cut and incubated with rabbit anti-FoxO1 antibody (A, D, and G), followed by immunostaining with Cy3-conjugated goat anti-rabbit IgG (red). The nuclei of hepatocytes (B, E, and H) were stained with the TO-PRO-3 dye (blue). Bar =10 μm.
Functional interplay between FoxO1 and PPARα in hepatic apoC-III expression
To study whether fenofibrate-mediated upregulation of PPARα expression in liver contributes to the inhibition of hepatic FoxO1 activity, we determined the potential interaction between FoxO1 and PPARα proteins in liver. Liver tissue (20 mg) from fibrate-treated hamsters was homogenized and subjected to immunoprecipitation using rabbit anti-FoxO1 antibody or preimmune serum, followed by immunoblot analysis. As shown in Fig. 6_A_, we detected PPARα protein in the product that was immunoprecipitated by anti-FoxO1 antibody, implicating a potential interaction between FoxO1 and PPARα proteins in liver.
Fig. 6.
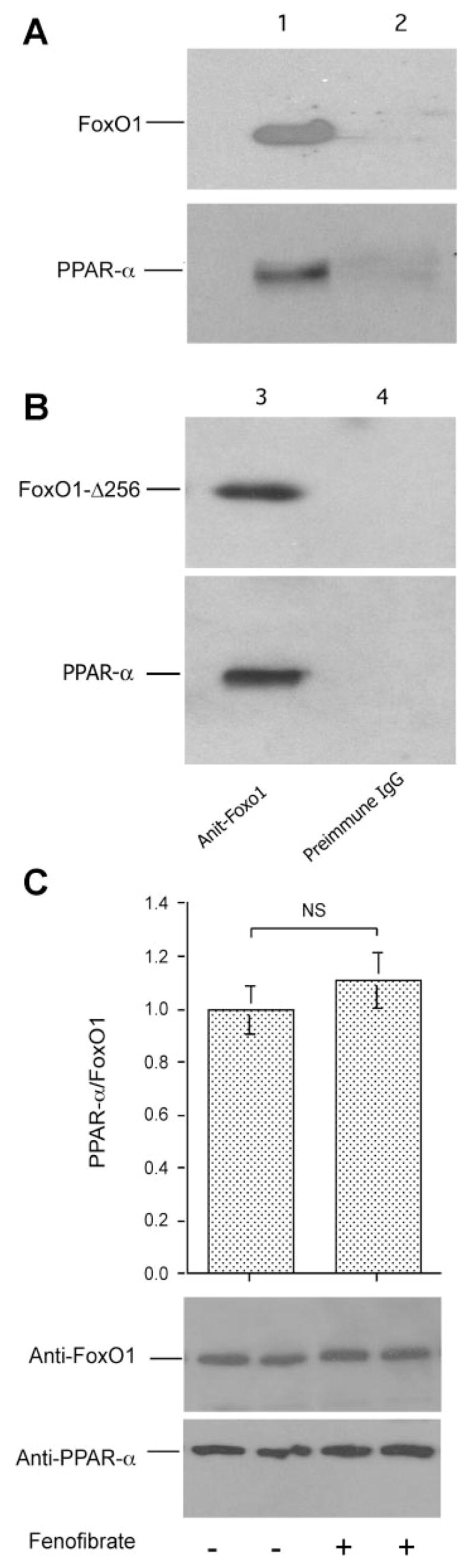
FoxO1 and PPARα interaction. A: immunoblots. Liver tissue (20 mg) of fenofibrate-treated high-fructose-fed hamsters was homogenized and subjected to immunoprecipitation using rabbit anti-FoxO1 antibody (lane 1) or rabbit preimmune serum (lane 2). Immunoprecipitates were analyzed by immunoblot assay using anti-FoxO1 and anti-PPARα antibodies, respectively. B: immunoblots. HepG2 cells were transfected with pPPARα and transduced with adenoviral vector expressing FoxO1-Δ256. After 48-h incubation, cells were subjected to immunoprecipitation by rabbit anti-FoxO1 antibody (lane 3) or preimmune serum (lane 4), followed by immunoblot analysis using anti-FoxO1 or anti-PPARα antibody. C: effect of fenofibrate on FoxO1 and PPARα interaction. HepG2 cells expressing FoxO1 and PPARα were cultured in the presence and absence of 50 mM fenofibrate for 16 h, followed by immunoprecipitation using anti-FoxO1 antibody. Immunoprecipitated proteins were subjected to semiquantitative immunoblot assay. The amounts of PPARα were normalized to FoxO1 and compared between mock and fenofibrate treatment groups. Data were from 3 independent experiments.
To corroborate this finding, we determined molecular association between PPARα and FoxO1-Δ256, a truncated version of FoxO1 that consists of its DNA-binding domain (amino acids 156–256) plus additional 155 amino acids at the NH2-terminus of FoxO1 (3). HepG2 cells were transduced by adenoviral vector expressing FoxO1-Δ256 in the presence of PPARα expression. After 48-h incubation, cells were subjected to immunoprecipitation by anti-FoxO1 antibody, followed by immunoblot assay. As shown in Fig. 6_B_, a specific protein band corresponding to PPARα was coimmunoprecipitated together with FoxO1-Δ256.
To address whether FoxO1-PPARα interaction is enhanced by fenofibrate, HepG2 cells were transfected with FoxO1 and PPARα expressing plasmids. After 24-h incubation to allow FoxO1 and PPARα production, cells were incubated in the presence and absence of 50 mM fenofibrate in culture medium for 16 h, followed by immunoprecipitation using anti-FoxO1 antibody. Using semiquantitative immunoblot assay, we detected similar levels of PPARα protein in the immunoprecipitated complex of fenofibrate- and mock-treated cells (Fig. 6_C_). These results suggest that fibrates did not significantly enhance FoxO1 and PPARα interaction in HepG2 cells.
To address the functional interplay between FoxO1 and PPARα in VLDL-TG metabolism, we studied the effect of FoxO1 on hepatic apoC-III production in the presence and absence of PPARα production in HepG2 cells using a predefined human APOC3 promoter-directed luciferase reporter system (2). Our laboratory has previously shown that FoxO1 stimulates human apoC-III expression via direct binding to its target site in the APOC3 promoter (2). As shown in Fig. 7_A_, such an APOC3 promoter-directed luciferase expression system in pHD317 was transfected to HepG2 cells that were pretransfected with vectors expressing FoxO1 or PPARα alone or both in combination. Consistent with our previous observations, FoxO1 markedly stimulated APOC3 promoter activity, as evidenced by significantly increased luciferase activity in response to elevated FoxO1 production in HepG2 cells (Fig. 7, B and C). However, this stimulatory effect of FoxO1 on hepatic apoC-III expression was abolished in the presence of elevated PPARα production (Fig. 7, B and C). These results revealed a novel mechanism by which PPARα counteracts the effect of FoxO1 on hepatic apoC-III expression in modulating plasma VLDL-TG metabolism.
Fig. 7.

PPARα counteracts FoxO1 in apoC-III expression. A: the human APOC3 promoter-directed luciferase system in pHD317. The nucleotide sequences corresponding to the insulin-responsive element (IRE) in pHD317 and its mutant version in pHD334 were underlined. B: APOC3 promoter activity. HepG2 cells were transfected with pHD317 together with plasmids expressing FoxO1 or PPARα alone or both in combination, using pCA35-LacZ as control for the normalization of transfection efficiency. After 48-h incubation, cells were collected for the determination of luciferase and β-galactosidase activities. The ratios between luciferase and β-galactosidase activities were compared between different conditions. Results were obtained from 5 independent experiments. *P < 0.001. C: RT-PCR products of FoxO1, PPARα, and β-actin mRNA. A subset of HepG2 cells treated identically as described in B was used for the preparation of total RNA, which was analyzed by RT-PCR using specific primers corresponding to FoxO1, PPARα, and β-actin cDNA. The RT-PCR products were resolved on 1.0% agarose gels and visualized under UV light after staining with ethidium bromide. D: the mutant APOC3 promoter activity. HepG2 cells were transfected with pHD334 expressing the mutant APOC3 promoter-directed luciferase expression system in the presence of FoxO1 or PPARα alone or both in combination, using pCA35-LacZ as control. Each condition was run in triplicate. Cells were subjected to luciferase and β-galactosidase activity assay after 24-h incubation. E: effect of PPARα on FoxO1 promoter activity. The FoxO1 promoter-directed luciferase expression system pFoxO-luc was cotransfected with pPPARα or control pGL3-basic plasmid (2 μg per plasmid) in 6-well plates using pCA35-LacZ (1 μg) as control. After 24-h incubation, cells were collected for the determination of luciferase and β-galactosidase activities. Data were from 3 independent experiments. #P < 0.05. F: effect of PPARα on FoxO1 mRNA expression. A subset of HepG2 cells from experiment in E was used for the preparation of total RNA, which was analyzed by RT-PCR using specific primers corresponding to FoxO1, PPARα, and β-actin cDNA.
To strengthen the above findings, we performed a similar study using a mutant version of the APOC3 promoter. As a result of mutations in the insulin-responsive element (IRE), this mutant APOC3 promoter no longer responds to FoxO1 (2). We transfected this mutant promoter-directed luciferase expression system into HepG2 cells in the presence of FoxO1 or PPARα expression alone or both in combination. As shown in Fig. 7_D_, mutations in the IRE abolished FoxO1-mediated stimulation of APOC3 promoter activity and also abrogated PPARα effect on ApoC-IH expression in HepG2 cells. Furthermore, elevated PPARα production alone did not result in significant changes in the mutant APOC3 promoter activity.
To address whether PPARα suppresses FoxO1 expression at the transcriptional level, we cloned the 2-kb FoxO1 promoter using FoxO1 promoter-specific primers. After verification by DNA sequencing, the FoxO1 promoter was subcloned into a pGL3-basic luciferase reporter system. This FoxO1 promoter-directed luciferase expression system was transfected into HepG2 cells, together with pPPARα or control pGL3-basic plasmid, followed by analysis of luciferase activity after 24 h of incubation. As shown in Fig. 7_E_, PPARα production resulted in a slight induction of FoxO1 promoter activity. In addition, we determined endogenous FoxO1 mRNA levels using semiquantitative RT-PCR assay in PPARα expressing and control cells. No significant difference in FoxO1 mRNA levels was detected in HepG2 cells that were transduced with either PPARα or control plasmid (Fig. 7_F_). These data indicate that PPARα did not directly suppress hepatic FoxO1 expression.
Mechanism of PPARα-mediated antagonism of FoxO1 activity
To define the mechanism of PPARα-mediated counter-regulation of FoxO1 activity, we postulated that PPARα, when associated with FoxO1, interferes with FoxO1 binding to target promoter DNA, resulting in inhibition of gene expression. To test this hypothesis, we determined the molecular interaction between FoxO1 and APOC3 promoter DNA in FoxO1-expressing HepG2 cells in the presence and absence of PPARα production. HepG2 cells were transfected with FoxO1-expressing plasmid alone or in combination with PPARα-expressing plasmid. After 48-h incubation, cells were subjected to ChIP assay. Consistent with previous observations (2), a specific DNA fragment corresponding to the human APOC3 promoter DNA was immunoprecipitated by rabbit anti-FoxO1 antibody in FoxO1-expressing HepG2 cells (Fig. 8_A_). Using the same assay, we detected a similar APOC3 promoter-specific DNA in HepG2 cells expressing both FoxO1 and PPARα (Fig. 8_A_). But the relative amount of APOC3 promoter activity in FoxO1- and PPARα-coexpressing cells was more than fivefold lower than in cells expressing FoxO1 alone, as determined by semiquantitative PCR assay (Fig. 8_B_). As control, no APOC3 promoter-specific DNA was detected when rabbit preimmune IgG was used in the ChIP assay (Fig. 8_A_). FoxO1 stimulates hepatic apoC-III expression via specific binding to its target site (CCAAACA) in the APOC3 promoter DNA (2). These data suggest that this association between FoxO1 and APOC3 promoter DNA was significantly reduced in response to PPARα production, which correlated with the inhibition of APOC3 promoter activity in PPARα-expressing HepG2 cells (Fig. 7, B and C).
Fig. 8.

PPARα interferes with FoxO1 binding to apoC-III promoter. A: chromatin immunoprecipitation (ChIP) analysis of FoxO1-APOC3 promoter DNA interaction in the absence and presence of PPARα production. HepG2 cells were transfected with pFoxO1 alone (lanes 1 and 2) or cotransfected with both pFoxO1 and pPPARα plasmids (lanes 3 and 4) in 6-well dishes. After 48-h incubation, cells were subjected to ChIP assay using preimmune IgG (lanes 1 and 3) or rabbit anti-FoxO1 antibody (lanes 2 and 4). Immunoprecipitates were analyzed by PCR using 20-nucleotide (nt) primers flanking −675/+1 nt region of human APOC3 promoter DNA. The PCR products were analyzed on 1% agarose gels and visualized under UV light after ethidium bromide staining. B: quantification of PCR products. The relative amounts of PCR products corresponding to the specific APOC3 promoter DNA were quantified by densitometry. Results were obtained from 3 independent experiments. *P < 0.001.
To investigate whether PPARα alone binds to APOC3 promoter DNA, we subjected pPPARα-expressing HepG2 cells to ChIP assay using anti-PPARα antibody, followed by PCR analysis using the same set of APOC3 promoter-specific primers, as described (Fig. 8_A_). No specific PCR DNA was produced. Although PPARα is known to bind specifically to a highly conserved DNA sequence that comprises 2-hexamer repeats with one nucleotide in between (AGGTCAXAGGTCA) in target promoters (11), sequence analysis did not reveal the presence of such a consensus PPARα binding site within a 5-kb DNA region of the APOC3 promoter (−5,000/+1 nt). These data were consistent with the lack of significant alterations in APOC3 promoter activity in response to PPARα production in HepG2 cells (Fig. 7, B and C), suggesting that PPARα modulates hepatic apoC-III expression by counteracting the stimulatory effect of FoxO1 on APOC3 promoter activity (2).
DISCUSSION
The goal of this study is to determine the role of FoxO1 in the pathogenesis of hypertriglyceridemia and address whether FoxO1 mediates the beneficial effect of fibrates on VLDL-TG metabolism in high-fructose-fed hamsters. We show that high-fructose feeding resulted in marked elevations in plasma FFA, TG, and total cholesterol levels, accompanied by significantly elevated plasma insulin levels and impaired glucose tolerance. Interestingly, fasting blood glucose levels and body weight remained unchanged in high-fructose-fed hamsters. These data indicate that hamsters fed a high-fructose diet, as opposed to regular chow, developed insulin resistance and hypertriglyceridemia in the absence of fasting hyperglycemia and excessive body weight gain. Thus high-fructose-fed hamsters represent a nonobese model of insulin resistance with altered TG metabolism.
Our present studies indicate that hepatic FoxO1 activity becomes deregulated, culminating in markedly elevated production along with increased nuclear localization in livers of high-fructose-fed hamsters. Similar observations have been made in insulin-resistant livers of high-fat-induced obese mice (unpublished data) and diabetic db/db mice (2, 3). These data unveil an association between hepatic FoxO1 deregulation and impaired hepatic metabolism. Indeed, FoxO1 gain of function, as a result of adenovirus-mediated hepatic FoxO1 production or transgenic expression of its constitutively active allele in liver, is associated with impaired glucose metabolism in mice (40, 46). Conversely, FoxO1 loss of function, caused by hepatic expression of its dominant-negative allele in insulin-resistant liver, is associated with significantly improved fasting glycemia and amelioration of hyperinsulinemia in diabetic db/db mice (3). FoxO1 haplo insufficiency is sufficient to protect mice from developing high-fat-induced insulin resistance (41) and rescue the diabetic phenotype in insulin receptor substrate 2-deficient diabetic mice (43). Furthermore, targeted FoxO1 inhibition by anti-sense approach improved hepatic metabolism and peripheral insulin action in high-fat-diet-induced obesity (47). Although high-fructose feeding is associated with hypertriglyceridemia, the underlying mechanism remains elusive. Our present data suggest that aberrant hepatic FoxO1 function is a contributing factor for metabolic abnormalities in high-fructose-fed hamsters.
Consistent with this interpretation is the revelation that plasma apoC-III levels were significantly elevated, correlating with increased hepatic FoxO1 activity in high-fructose-fed hamsters. We have previously shown that FoxO1 mediates the inhibitory effect of insulin on hepatic apoC-III expression, and FoxO1 deregulation links loss of insulin inhibition to unrestrained apoC-III production, contributing to the pathogenesis of hypertriglyceridemia (2). This predicts that a selective inhibition of FoxO1 activity will suppress apoC-III production in high-fructose-fed hamsters. Indeed, we show that hepatic FoxO1 deregulation was corrected in high-fructose-fed hamsters in response to fenofibrate treatment. This effect parallels the normalization of plasma apoC-III levels and amelioration of hypertriglyceridemia in fenofibrate-treated hamsters. Thus modification of FoxO1 transcriptional activity represents an important mechanism by which fibrates curb apoC-III overproduction and improve TG metabolism in subjects with hypertriglyceridemia.
In addition to FoxO1, we observed that multiple factors, such as PGC-lβ and SREBP-1c, contribute to high-fructose-induced hypertriglyceridemia. A coactivator of SREBP-1c, PGC-lβ has been shown to bind and augment SREBP-1c transcription activity in stimulating lipogenesis, contributing to elevated hepatic VLDL-TG production and secretion and the development of hyperlipidemia in high-fat-induced obese mice (31). Likewise, we detected a significant increase in hepatic fat content in high-fructose-fed hamsters, which correlated with augmented hepatic PGC-lβ and SREBP-1c expression. These observations, along with the induction of hepatic MTP production, help explain previous observations that high-fructose-fed hamsters are associated with inappropriately increased hepatic VLDL-TG production (10). Increased fat infiltration in organs other than adipose tissue, such as liver and skeletal muscle, is closely associated with insulin resistance (9, 17, 29, 48–50, 54). Our present findings provide important insights into the mechanism of fructose-induced insulin resistance, suggesting that an increased intrahepatic lipid deposition plays a causative role in the development of insulin resistance, as reflected in hyperinsulinemia and glucose intolerance in high-fructose-induced hamsters. In keeping with this conclusion, we show that fenofibrate treatment suppressed hepatic PGC-lβ and SREBP-1c expression to basal levels, resulting in the normalization of hepatic fat content in high-fructose-fed hamsters. These data suggest that fenofibrate-mediated suppression of PGC-lβ and SREBP-1c expression also contributes to the reversal of insulin resistance and amelioration of hypertriglyceridemia in high-fructose-fed hamsters.
Although it is known that hepatic PPARα activity is upregulated by fibrates (8, 13, 21), the molecular link between upregulated PPARα activity and improved TG metabolism remains undefined. We show in high-fructose-fed hamsters that PPARα and FoxO1 are differentially regulated by fibrates. Hepatic PPARα levels were significantly increased, accompanied by a corresponding decrease in hepatic FoxO1 levels in fenofibrate-treated hyperlipidemic hamsters. Although inappropriately increased FoxO1 activity in liver is associated with abnormal hepatic metabolism (1, 34, 46), these observations raise the possibility that fenofibrate-mediated activation of PPARα activity counteracts the effect of FoxO1 on hepatic apoC-III production, contributing to improved VLDL-TG metabolism in high-fructose-fed hamsters. Implicit in this assumption are recent studies by Dowell et al. (20), who showed that PPARγ, another member of the PPAR nuclear receptor subfamily, interacts with FoxO1 and antagonizes its function in kidney epithelial cells. To underpin this hypothesis, we show that PPARα associated with FoxO1 interfered with FoxO1 binding to APOC3 promoter and diminished the ability of FoxO1 to stimulate hepatic apoC-III expression. It follows that PPARα contributes to improved VLDL-TG metabolism in response to fibrate treatment by counterbalancing the effect of FoxO1 on hepatic apoC-III expression. We would like to acknowledge the limitation of coimmunoprecipitation used in our studies, as this assay does not allow the distinction of a direct interaction between FoxO1 and PPARα from indirect association via a third partner. Likewise, FoxO1-Δ256 used in this study contains FoxO1 DNA binding domain (amino acids 156_–_256) plus 155 amino acid residues of FoxO1 NH2-terminal domain. It remains unknown whether PPARα counteracted FoxO1 via specific interaction with FoxO1 DNA binding domain. Further studies are needed to characterize the underlying mechanism by which PPARα antagonizes FoxO1 function in improving hypertriglyceridemia in animal models with altered TG metabolism.
In addition to its activation of PPARα (21, 32, 37, 38), fenofibrate has been shown to display pleiotropic effects on the expression of genes in lipid metabolism. Fibrates upregulate lipoprotein lipase expression (18, 21). This effect, along with the suppression of apoC-III overproduction, accounts for enhanced clearance of lipoprotein remnants. Furthermore, fibrates are shown to enhance carnitine palmitoyltransferase I expression, accounting for increased fatty acid oxidation in liver and muscle and contributing to improved insulin sensitivity (37).
Increased consumption of fructose, a major ingredient of corn syrup used in soft drinks, is associated with the development of insulin resistance and dyslipidemia, but the underlying mechanism is unknown. Our studies highlight the importance of hepatic FoxO1 deregulation along with augmented PGC-lβ production in the pathogenesis of high-fructose-induced insulin resistance and hypertriglyceridemia and, furthermore, elucidate a novel molecular antagonism of PPARα against FoxO1 in mediating the effect of fibrates on the reversal of hypertriglyceridemia. These data provide evidence that selective inhibition of FoxO1 activity in liver can be explored as a distinctive mechanism for better management of hypertriglyceridemia.
Acknowledgments
We thank Drs. Steve Ringquist, Fiona D’Souza, and Robert O’Doherty for critical proofreading of this manuscript. We also thank Dr. Bruce Spiegelman for providing anti-PGC-lβ antibody. This study has been orally presented at the 65th American Diabetes Association Scientific Session (2005), San Diego, CA.
GRANTS
This study was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK-066301.
References
- 1.Accili D, Arden KC. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell. 2004;117:421–426. doi: 10.1016/s0092-8674(04)00452-0. [DOI] [PubMed] [Google Scholar]
- 2.Altomonte J, Cong L, Harbaran S, Richter A, Xu J, Meseck M, Dong HH. Foxo1 mediates insulin action on ApoC-III and triglyceride metabolism. J Clin Invest. 2004;114:1493–1503. doi: 10.1172/JCI19992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Altomonte J, Richter A, Harbaran S, Suriawinata j, Nakae J, Thung SN, Meseck M, Accili D, Dong H. Inhibition of Foxo1 function is associated with improved fasting glycemia in diabetic mice. Am J Physiol Endocrinol Metab. 2003;285:E718–E728. doi: 10.1152/ajpendo.00156.2003. [DOI] [PubMed] [Google Scholar]
- 4.Avramoglu RK, Qiu W, Adeli K. Mechanisms of metabolic dyslipidemia in insulin resistant states: deregulation of hepatic and intestinal lipoprotein secretion. Front Biosci. 2003;8:d464–d476. doi: 10.2741/1022. [DOI] [PubMed] [Google Scholar]
- 5.Bard JM, Charles MA, Juhan-Vague I, Vague P, Andre P, Safar M, Fruchart JC, Eschwege E. Accumulation of triglyceride-rich lipoprotein in subjects with abdominal obesity. Arterioscler Thromb Vase Biol. 2001;21:407–414. doi: 10.1161/01.atv.21.3.407. [DOI] [PubMed] [Google Scholar]
- 6.Belfiore F, Iannello S. Insulin resistance in obesity: metabolic mechanisms and measurement methods. Mol Genet Metab. 1998;65:121–128. doi: 10.1006/mgme.1998.2727. [DOI] [PubMed] [Google Scholar]
- 7.Belfiore F, Iannello S, Volpicelli G. Insulin sensitivity indices calculated from basal and OGTT-induced insulin, glucose, and FFA levels. Mol Genet Metab. 1998;63:134–141. doi: 10.1006/mgme.1997.2658. [DOI] [PubMed] [Google Scholar]
- 8.Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002;53:409–435. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- 9.Bugianesi E, Zannoni C, Vanni E, Marzocchi R, Marchesini G. Non-alcoholic fatty liver and insulin resistance: a cause-effect relationship? Dig Liver Dis. 2004;36:165–173. doi: 10.1016/j.dld.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 10.Carpentier A, Taghibiglou C, Leung N, Szeto L, Van Iderstine SC, Uffelman KD, Buckingham R, Adeli K, Lewis GF. Ameliorated hepatic insulin resistance is associated with normalization of microsomal triglyceride transfer protein expression and reduction in very low density lipoprotein assembly and secretion in the fructose-fed hamster. J Biol Chem. 2002;277:28795–28802. doi: 10.1074/jbc.M204568200. [DOI] [PubMed] [Google Scholar]
- 11.Castelein H, Declercq PE, Baes M. DNA binding preferences of PPAR alpha/RXR alpha heterodimers. Biochem Biophys Res Commun. 1997;233:91–95. doi: 10.1006/bbrc.1997.6395. [DOI] [PubMed] [Google Scholar]
- 12.Chan DC, Watts GF, Barrett PH, Mamo JCL, Redgrave TG. Markers of triglyceride-rich lipoprotein remnant metabolism in visceral obesity. Clin Chem. 2002;48:278–283. [PubMed] [Google Scholar]
- 13.Chapman MJ. Fibrates in 2003: therapeutic action in atherogenic dyslipidaemia and future perspectives. Atherosclerosis. 2003;171:1–13. doi: 10.1016/s0021-9150(03)00156-4. [DOI] [PubMed] [Google Scholar]
- 14.Chong T, Naples M, Federico L, Taylor D, Smith GJ, Cheung RC, Adeli K. Effect of rosuvastatin on hepatic production of apolipoprotein B-containing lipoproteins in an animal model of insulin resistance and metabolic dyslipidemia. Atherosclerosis. 2006;185:21–31. doi: 10.1016/j.atherosclerosis.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 15.Clavey V, Copin C, Mariotte MC, Bauge E, Chinetti G, Fruchart J, Fruchart JC, Dallongeville J, Staels B. Cell culture conditions determine apolipoprotein CIII secretion and regulation by fibrates in human hepatoma HepG2 cells. Cell Physiol Biochem. 1999;9:139–149. doi: 10.1159/000016311. [DOI] [PubMed] [Google Scholar]
- 16.Dana SL, Hoener PA, Bilakovics JM, Crombie DL, Ogilvie KM, Kauffman RF, Mukherjee R, Paterniti JR., Jr Peroxisome proliferator- activated receptor subtype-specific regulation of hepatic and peripheral gene expression in the Zucker diabetic fatty rat. Metabolism. 2001;50:963–971. doi: 10.1053/meta.2001.24870. [DOI] [PubMed] [Google Scholar]
- 17.Day CP. Pathogenesis of steatohepatitis. Best Pract Res Clin Gastroenterol. 2002;16:663–678. doi: 10.1053/bega.2002.0333. [DOI] [PubMed] [Google Scholar]
- 18.de Man FH, de Beer F, van der Laarse A, Jansen H, Leuven JA, Souverijn JH, Vroom TF, Schoormans SC, Fruchart JC, Havekes LM, Smelt AH. The hypolipidemic action of bezafibrate therapy in hypertriglyceridemia is mediated by upregulation of lipoprotein lipase: no effects on VLDL substrate affinity to lipolysis or LDL receptor binding. Atherosclerosis. 2000;153:363–371. doi: 10.1016/s0021-9150(00)00409-3. [DOI] [PubMed] [Google Scholar]
- 19.Dong H, Altomonte J, Morral N, Meseck M, Thung SN, Woo SL. Basal insulin gene expression significantly improves conventional insulin therapy in type 1 diabetic rats. Diabetes. 2002;51:130–138. doi: 10.2337/diabetes.51.1.130. [DOI] [PubMed] [Google Scholar]
- 20.Dowell P, Otto TC, Adi S, Lane MD. Convergence of peroxisome proliferator-activated receptor gamma and Foxo1 signaling pathways. J Biol Chem. 2003;278:45485–45491. doi: 10.1074/jbc.M309069200. [DOI] [PubMed] [Google Scholar]
- 21.Frederiksen KS, Wulf EM, Wassermann K, Sauerberg P, Fleckner J. Identification of hepatic transcriptional changes in insulin-resistant rats treated with peroxisome proliferator activated receptor-alpha agonists. J Mol Endocrinol. 2003;30:317–329. doi: 10.1677/jme.0.0300317. [DOI] [PubMed] [Google Scholar]
- 22.Ginsberg HN, Huang LS. The insulin resistance syndrome: impact on lipoprotein metabolism and atherothrombosis. J Cardiovasc Risk. 2000;7:325–331. doi: 10.1177/204748730000700505. [DOI] [PubMed] [Google Scholar]
- 23.Gotto AM., Jr Management of dyslipidemia. Am J Med. 2002;122:10S–18S. doi: 10.1016/s0002-9343(02)01085-9. [DOI] [PubMed] [Google Scholar]
- 24.Guo Q, Wang PR, Milot DP, Ippolito MC, Hernandez M, Burton CA, Wright SD, Chao Y. Regulation of lipid metabolism and gene expression by fenofibrate in hamsters. Biochim Biophys Acta. 2001;1533:220–232. doi: 10.1016/s1388-1981(01)00156-1. [DOI] [PubMed] [Google Scholar]
- 25.Haubenwallner S, Essenburg AD, Barnett BC, Pape ME, DeMattos RB, Krause BR, Minton LL, Auerbach BJ, Newton RS, Leff T, Bisgaier CL. Hypolipidemic activity of select fibrates correlates to changes in hepatic apolipoprotein C-III expression: a potential physiologic basis for their mode of action. J Lipid Res. 1995;36:2541–2551. [PubMed] [Google Scholar]
- 26.Hussain MM, Shi J, Dreizen P. Microsomal triglyceride transfer protein and its role in apoB-lipoprotein assembly. J Lipid Res. 2003;44:22–32. doi: 10.1194/jlr.r200014-jlr200. [DOI] [PubMed] [Google Scholar]
- 27.Kinnunen PK, Ehnholm C. Effect of serum and C apolipoproteins from very low density lipoproteins on human post-heparin plasma hepatic lipase. FEBS Lett. 1976;65:354–357. doi: 10.1016/0014-5793(76)80145-7. [DOI] [PubMed] [Google Scholar]
- 28.Lee CH, Olson P, Evans RM. Lipid metabolism, metabolic diseases, and peroxisome proliferator-activated receptors. Endocrinology. 2003;144:2201–2207. doi: 10.1210/en.2003-0288. [DOI] [PubMed] [Google Scholar]
- 29.Lewis GF, Carpentier A, Adeli K, Giacca A. Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr Rev. 2002;23:201–229. doi: 10.1210/edrv.23.2.0461. [DOI] [PubMed] [Google Scholar]
- 30.Lewis GF, Uffelman K, Naples M, Szeto L, Haidari M, Adeli K. Intestinal lipoprotein overproduction, a newly recognized component of insulin resistance, is ameliorated by the insulin sensitizer rosiglitazone: studies in the fructose-fed Syrian golden hamster. Endocrinology. 2005;146:247–255. doi: 10.1210/en.2004-1143. [DOI] [PubMed] [Google Scholar]
- 31.Lin J, Yang R, Tarr PT, Wu PH, Handschin C, Li S, Yang W, Pei L, Uldry M, Tontonoz P, Newgard CB, Spiegelman BM. Hyperlipidemic effects of dietary saturated fats mediated through PGC-1beta coactivation of SREBP. Cell. 2005;120:261–273. doi: 10.1016/j.cell.2004.11.043. [DOI] [PubMed] [Google Scholar]
- 32.Liu PC, Huber R, Stow MD, Schlingmann KL, Collier P, Liao B, Link J, Burn TC, Hollis G, Young PR, Mukherjee R. Induction of endogenous genes by peroxisome proliferator activated receptor alpha ligands in a human kidney cell line and in vivo. J Steroid Biochem Mol Biol. 2003;85:71–79. doi: 10.1016/s0960-0760(03)00135-3. [DOI] [PubMed] [Google Scholar]
- 33.Maki KC. Fibrates for treatment of the metabolic syndrome. Curr Atheroscler Rep. 2004;6:45–51. doi: 10.1007/s11883-004-0115-5. [DOI] [PubMed] [Google Scholar]
- 34.Matsumoto M, Han S, Kitamura T, Accili D. Dual role of transcription factor FoxO1 in controlling hepatic insulin sensitivity and lipid metabolism. J Clin Invest. 2006;116:2464–2472. doi: 10.1172/JCI27047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 36.McConathy WJ, Gesquiere JC, Bass H, Tartar A, Fruchart JC. Inhibition of lipoprotein lipase activity by synthetic peptides of apolipoprotein C-III. J Lipid Res. 1992;33:995–1003. [PubMed] [Google Scholar]
- 37.Minnich A, Tian N, Byan L, Bilder G. A potent PPARα agonist stimulates mitochondrial fatty acid β-oxidation in liver and skeletal muscle. Am J Physiol Endocrinol Metab. 2001;280:E270–E279. doi: 10.1152/ajpendo.2001.280.2.E270. [DOI] [PubMed] [Google Scholar]
- 38.Nagai Y, Nishio Y, Nakamura T, Maegawa H, Kikkawa R, Kashiwagi A. Amelioration of high fructose-induced metabolic derangements by activation of PPARα. Am J Physiol Endocrinol Metab. 2002;282:E1180–E1190. doi: 10.1152/ajpendo.00471.2001. [DOI] [PubMed] [Google Scholar]
- 39.Nakae J, Barr V, Accili D. Differential regulation of gene expression by insulin and IGF-1 receptors correlates with phosphorylation of a single amino acid residue in the forkhead transcription factor FKHR. EMBO J. 2000;19:989–996. doi: 10.1093/emboj/19.5.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakae J, Biggs WH, 3rd, Kitamura T, Cavenee WK, Wright CV, Arden KC, Accili D. Regulation of insulin action and pancreatic β-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1. Nat Genet. 2002;32:245–253. doi: 10.1038/ng890. [DOI] [PubMed] [Google Scholar]
- 41.Nakae J, Kitamura T, Kitamura Y, Biggs WH, 3rd, Arden KC, Accili D. The forkhead transcription factor Foxo1 regulates adipocyte differentiation. Dev Cell. 2003;4:119–129. doi: 10.1016/s1534-5807(02)00401-x. [DOI] [PubMed] [Google Scholar]
- 42.Nieves DJ, Cnop M, Retzlaff B, Walden CE, Brunzell JD, Knopp RH, Kahn SE. The atherogenic lipoprotein profile associated with obesity and insulin resistance is largely attributable to intra-abdominal fat. Diabetes. 2003;52:172–179. doi: 10.2337/diabetes.52.1.172. [DOI] [PubMed] [Google Scholar]
- 43.Okamoto H, Nakae J, Kitamura T, Park BC, Dragatsis I, Accili D. Transgenic rescue of insulin receptor-deficient mice. J Clin Invest. 2004;114:214–223. doi: 10.1172/JCI21645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Packard CJ. Overview of fenofibrate. Eur Heart J. 1998;19 Suppl A:A62–A65. [PubMed] [Google Scholar]
- 45.Peters JM, Hennuyer N, Staels B, Fruchart JC, Fievet C, Gonzalez FJ, Auwerx J. Alterations in lipoprotein metabolism in peroxisome proliferator-activated receptor alpha-deficient mice. J Biol Chem. 1997;272:27307–27312. doi: 10.1074/jbc.272.43.27307. [DOI] [PubMed] [Google Scholar]
- 46.Qu S, Altomonte J, Perdomo G, He J, Fan Y, Kamagate A, Meseck M, Dong HH. Aberrant forkhead box O1 function is associated with impaired hepatic metabolism. Endocrinology. 2006;147:5641–5652. doi: 10.1210/en.2006-0541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Samuel VT, Choi CS, Phillips TG, Romanelli AJ, Geisler JG, Bhanot S, McKay R, Monia B, Shutter JR, Lindberg RA, Shulman GI, Veniant MM. Targeting Foxo1 in mice using antisense oligonucleotide improves hepatic and peripheral insulin action. Diabetes. 2006;55:2042–2050. doi: 10.2337/db05-0705. [DOI] [PubMed] [Google Scholar]
- 48.Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, Befroy D, Romanelli AJ, Shulman GI. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem. 2004;279:32345–32353. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- 49.Scheen AJ. Pathophysiology of type 2 diabetes. Acta Clin Belg. 2003;58:335–341. doi: 10.1179/acb.2003.58.6.001. [DOI] [PubMed] [Google Scholar]
- 50.Seppala-Lindroos A, Vehkavaara S, Hakkinen AM, Goto T, Westerbacka J, Sovijarvi A, Halavaara J, Yki-Jarvinen H. Fat accumulation in the liver is associated with defects in insulin suppression of glucose production and serum free fatty acids independent of obesity in normal men. J Clin Endocrinol Metab. 2002;87:3023–3028. doi: 10.1210/jcem.87.7.8638. [DOI] [PubMed] [Google Scholar]
- 51.Shachter NS. Apolipoproteins C-1 and C-III as important modulators of lipoprotein metabolism. Curr Opin Lipidol. 2001;12:297–304. doi: 10.1097/00041433-200106000-00009. [DOI] [PubMed] [Google Scholar]
- 52.Staels B, Vu-Dac N, Kosykh VA, Saladin R, Fruchart JC, Dallongeville J, Auwerx J. Fibrates downregulate apolipoprotein C-III expression independent of induction of peroxisomal acyl coenzyme A oxidase. A potential mechanism for the hypolipidemic action of fibrates. J Clin Invest. 1995;95:741–750. doi: 10.1172/JCI117717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang C, McConathy WJ, Kloer HJ, Alaupovic P. Modulation of lipoprotein lipase activity by apolipoproteins: effect of apolipoprotein C-III. J Clin Invest. 1985;75:384–390. doi: 10.1172/JCI111711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Youssef W, McCullough AJ. Diabetes mellitus, obesity, and hepatic steatosis. Semin Gastrointest Dis. 2002;13:17–30. [PubMed] [Google Scholar]