Protein kinase C isozymes as therapeutic targets for treatment of human cancers (original) (raw)
. Author manuscript; available in PMC: 2009 Jan 1.
Introduction
Protein kinase C (PKC; EC 2.7.11.13) was originally identified as a calcium-activated serine/threonine kinase that was dependent upon phospholipid and diacylglycerol (DAG) for full activity. The PKC gene family comprises 10 structurally related phospholipid-dependent serine/threonine protein kinases whose catalytic activity can be regulated by phosphorylation, interaction with calcium and lipid-derived co-factors, and through specific protein-protein interactions. The PKC enzyme family is divided into three subgroups: the conventional, calcium-dependent cPKCs [alpha (α), beta I (βI), beta II (βII), and gamma (γ)]; the novel nPKCs [delta (δ), epsilon (ε), eta (η) and theta (Θ)]; and the atypical aPKCs [zeta (ζ) and iota (ι), also known as lambda (λ) in mice]. These groupings are based on the presence or absence of functional domains that confer isozyme-specific co-factor and activator requirements. Conventional PKCs are calcium, DAG and phosphatidyl-serine dependent due to the presence of conserved modular C1 and C2 domains within the regulatory region of the enzyme. Novel PKCs are DAG and phosphatidyl-serine dependent but do not require calcium. Due to the unique structure of their N-terminal regulatory region, the atypical PKCs do not require calcium, DAG or phosphatidylserine for activation. Instead, the N-terminal regulatory region of atypical PKCs contains a Phox/Bem1 (PB1) domain that mediates homo- and hetero-typic protein-protein interactions that are critical for atypical PKC activation and localization (Lamark et al., 2003). All of the PKC isozymes are the products of independent genes, except PKCβI and PKCβII which are splice variants of the PKCβ gene that result from the inclusion of alternative final exons encoding the last ∼50 amino acids of the protein (Kubo et al., 1987).
Biochemical and immunologic studies indicate that multiple PKC isozymes are expressed in virtually all cell and tissue types. The expression of individual PKC isozymes is developmentally regulated and is responsive to the differentiation state of the cell. For these reasons, PKC isozymes are thought to fulfill distinct, often non-redundant functions in cells.
Similar activator requirements and substrate specificities in vitro have made identification of physiologically relevant, isozyme-specific substrates and functions difficult. The ability to genetically manipulate the expression level of specific PKC isozymes and to express mutant forms of individual PKC isozymes with altered kinase activity have allowed greater success in identification of PKC isozyme-specific functions. Using these genetic techniques, many laboratories, including our own, have demonstrated isozyme- and cell type-specific roles for PKC isozymes in cellular proliferation, differentiation, apoptosis and cell polarity. The discovery that PKC is a cellular receptor for the tumor-promoting phorbol esters led to an intense interest in the role of individual PKC isozymes in cancer development. Indeed, the expression level and cellular localization of individual PKC isozymes has been shown to be regulated during carcinogenesis in numerous tissue types. However, despite exhaustive investigation, no somatic or germline mutations have been found in the coding regions of any PKCs associated with human cancers or other diseases, and to date, only one PKC isozyme, atypical PKCι, been shown to satisfy the criteria of a human oncogene. This report summarizes work from our laboratory to characterize PKC isozyme-specific functions in human cancer and puts this work in the context of the relevant literature. We have also included a discussion of the identification and characterization of two PKC isozyme-selective inhibitors that are currently being evaluated clinically for their therapeutic potential in the treatment of cancer.
PKC Isozyme-specific Functions in Cancer
The Human Leukemia Model
In initial studies to evaluate PKC isozyme-specific functions, we utilized human leukemia cell lines as a model system. The human chronic myelogenous leukemia (K562) cell line expresses two major conventional PKCs, PKCα and PKCβII, that serve distinct functions in these cells (Hocevar et al., 1992). K562 cells can be induced to cease dividing and to differentiate to megakaryocytes by treatment with PMA (Hocevar et al., 1992). Expression of PKCα, but not PKCβII, correlates with PMA-induced cytostasis and megakaryocytic differentiation and over-expression of PKCα induced cytostasis in K562 cells (Murray et al., 1993). Others groups have reported a similar role for PKCα in leukemia cell differentiation (Nagata et al., 1996; Dieter and Schwende, 2000). Expression of chimeric PKC molecules in which the regulatory and catalytic domains of PKCα and PKCβII are exchanged established that the effects of PKCα on differentiation and cytostasis are mediated by isozyme-specific sequences within the catalytic domain of the PKCα (Walker et al., 1995).
In contrast to PKCα, expression of PKCβII is required for K562 cell proliferation (Murray et al., 1993). PKCβII selectively translocates to the nuclear membrane of these cells where it phosphorylates the nuclear envelope protein, lamin B (Hocevar and Fields, 1991). Further studies revealed that PKCβII is a physiologic mitotic lamin kinase that is required for nuclear envelope breakdown and G2/M phase transition (Goss et al., 1994; Thompson and Fields, 1996). Nuclear translocation and lamin B phosphorylation is specific to the PKCβII isozyme in this cellular context, and selective nuclear translocation and activation of PKCβII is conferred by the extreme carboxyl-terminal 13 amino acids of PKCβII (Gokmen-Polar and Fields, 1998).
PKCι, an atypical PKC, is also highly expressed in K562 cells (Murray and Fields, 1997). Despite a high level of homology with the other atypical PKC, PKCζ (72% homology at the amino acid level), we and others have determined that these isozymes play distinct, non-overlapping roles in most, if not all systems studied (Selbie et al., 1993; Murray and Fields, 1997; Leitges et al., 2001; Soloff et al., 2004). By genetically altering the expression and activity of PKCι, we identified a critical role for PKCι in human K562 leukemia cells (Murray and Fields, 1997; Jamieson et al., 1999). K562 cells are highly resistant to chemotherapeutic drugs, such as taxol, that induce cell death by apoptosis. This resistance is mediated by the chimeric tyrosine kinase oncogene Bcr-Abl, the transforming activity that causes chronic myelogenous leukemia (Bedi et al., 1995). Over-expression or inhibition of expression of PKCι in K562 cells has no significant effect on the proliferative capacity or the sensitivity to PMA-induced cytostasis and megakaryocytic differentiation, indicating that PKCι does not play a critical role in these processes (Murray and Fields, 1997). Rather, PKCι serves to protect K562 cells against chemotherapeutic drug-induced apoptosis (Murray and Fields, 1997). Over-expression of PKCι protects K562 cells against both okadaic acid- and taxol-induced apoptosis, whereas inhibition of PKCι expression sensitizes cells to induction of apoptosis, indicating that PKCι plays a general protective role against apoptotic stimuli (Murray and Fields, 1997). Over-expression of PKCζ has no protective effect against apoptosis, demonstrating that the anti-apoptotic effect is isozyme-specific (Murray and Fields, 1997). Treatment of K562 cells with taxol leads to a strong, sustained activation of PKCι in K562 cells (Jamieson et al., 1999). In contrast, Bcr-Abl-negative, myeloid leukemia (HL60) cells, which are sensitive to taxol-induced apoptosis, exhibit only weak and transient PKCι activation in response to taxol treatment (Jamieson et al., 1999). Mutational analysis of the human PKCι promoter reveals that Bcr-Abl regulates PKCι expression through the MAPK/ERK kinase- (MEK; EC 2.7.12.2) dependent activation of an Elk1 element within the proximal PKCι promoter (Gustafson et al., 2003). Expression of a constitutively active mutant of PKCι (caPKCι) protects K562 cells from taxol-induced apoptosis in the presence of a Bcr-Abl inhibitor (Jamieson et al., 1999). Taken together, these results demonstrated that both Bcr-Abl and PKCι activity are necessary for apoptotic resistance in K562 cells and that PKCι is a critical downstream target of Bcr-Abl that is sufficient to mediate the anti-apoptotic effects of Bcr-Abl.
PKCι mediates resistance to taxol-induced apoptosis downstream of Bcr-Abl through a mechanism involving nuclear factor-κB (NF-κB). Taxol weakly induces IκBα proteolysis and NF-κB nuclear translocation in K562 cells, but potently induces its transcriptional activity (Lu et al., 2001). Inhibition of NF-κB activity (by blocking IκBα degradation) significantly sensitizes cells to taxol-induced apoptosis (Lu et al., 2001). Likewise, expression of antisense PKCι mRNA or kdPKCι in K562 cells sensitizes cells to taxol, and this effect can be overcome by overexpression of NF-κB (Lu et al., 2001). Expression of caPKCι upregulates NF-κB transactivation and rescued cells from apoptosis induced by inhibition of Bcr-Abl (Lu et al., 2001). Taxol potently trans-activates RelA-dependent DNA binding and transcriptional activity (Lu et al., 2001) and this trans-activation can be further upregulated by expression of caPKCι, and inhibited by expression of kdPKCι (Lu et al., 2001). Taken together, these results demonstrate that RelA trans-activation is an important downstream target of the PKCι-mediated Bcr-Abl signaling pathway and is required for resistance to taxol-induced apoptosis in K562 cells.
Colon Cancer Models
Our laboratory has utilized complimentary cell-based and genetic animal models to investigate the functional role of PKC isozymes in intestinal epithelial cell homeostasis and colon carcinogenesis. PKCα mRNA abundance and protein expression is dramatically reduced in early preneoplastic lesions, aberrant crypt foci (ACF), induced by the colon-specific carcinogen azoxymethane (AOM) (Gokmen-Polar et al., 2001). The reduction in PKCα expression is sustained and progressive throughout the carcinogenic process, such that PKCα expression is almost completely lost in AOM-induced colon tumors (Gokmen-Polar et al., 2001). Others have observed a similarly dramatic downregulation of PKCα in intestinal tumors of APCMin/+ mice, a model for familial colon cancer (Oster and Leitges, 2006). In addition, genetic knockout of PKCα increases tumor formation and decreases survival of APCMin/+ mice (Oster and Leitges, 2006). These data demonstrate that, similar to its role in leukemia cells, PKCα does not promote tumorigenesis in the colon but rather functions as a tumor suppressor. This conclusion is further supported by in vitro studies demonstrating a role for PKCα in growth suppression and enhanced differentiation in intestinal epithelial and colon cancer cell lines (Scaglione-Sewell et al., 1998; Guan et al., 2007).
In contrast to PKCα, PKCβII mRNA abundance and protein expression is highly induced in ACF and subsequent colon tumors in AOM-treated mice (Gokmen-Polar et al., 2001). PKCβ expression is also induced in intestinal tumors in APCMin/+ mice (Oster and Leitges, 2006). Transgenic mice that express elevated PKCβII in the colonic epithelium (transgenic PKCβII mice) are more susceptible to colon cancer than their non-transgenic littermates (Murray et al., 1999). On the other hand, PKCβ knockout mice are resistant to AOM-mediated colon carcinogenesis (Liu et al., 2004). Bi-transgenic PKCβ KO/PKCβII mice (which express PKCβII only in the colonic epithelium) exhibit sensitivity to AOM-mediated colon carcinogenesis indistinguishable from non-transgenic mice (Liu et al., 2004) demonstrating that expression of PKCβII in the colonic epithelium is required for colon carcinogenesis.
The cancer-prone phenotype of transgenic PKCβII mice results, at least in part, from PKCβII-mediated hyper-proliferation of the colonic epithelium, as characterized by an increased proliferative index and resultant hypercellularity of the colonic crypts (Murray et al., 1999; Murray et al., 2002). This hyper-proliferative phenotype is caused by PKCβII-mediated activation of several pro-carcinogenic/proliferative signaling pathways in the intestinal epithelium. The Wnt/APC/β-catenin proliferative signaling pathway is a key regulator of colon carcinogenesis in mice and humans (Su et al., 1992; Pennisi, 1998; Takahashi et al., 2000). Transgenic PKCβII mice exhibit reduced GSK-3β (EC 2.7.11.26) activity and increased β-catenin protein expression in the colonic epithelium (Murray et al., 1999). These data indicate that PKCβII induces colonic epithelial hyper-proliferation via activation of the Wnt/APC/β-catenin signaling pathway.
To further investigate the molecular mechanisms underlying PKCβII-mediated carcinogenesis, we generated and analyzed non-transformed rat intestinal epithelial (RIE) cell lines that over-express PKCβII (RIE/PKCβII) (Yu et al., 2003). Unlike parental RIE cells, RIE/PKCβII cells exhibit a highly invasive phenotype (Zhang et al., 2004). PKCβII-induced invasion in RIE cells is blocked by the PKCβ-selective inhibitor LY379196, as well as by expression of a kinase-deficient allele of PKCβII, demonstrating the requirement for PKCβII activity for invasion (Zhang et al., 2004). PKCβII–induced invasion is mediated through a Ras→PKCι/Rac1→MEK signaling pathway, indicating that elevated expression of PKCβII early in colon carcinogenesis may represent a phenocopy of an activated Ras allele (Zhang et al., 2004) and can stimulate the invasive properties associated with Ras-transformed cells.
In addition, PKCβII represses transforming growth factor beta receptor type II (TGF-β RII) expression in transgenic PKCβII mice in vivo and in intestinal epithelial cells in vitro, resulting in reduced sensitivity to TGF-β-mediated growth inhibition (Murray et al., 2002). In contrast, PKCβII induces the expression of pro-carcinogenic cyclooxygenase type 2 (Cox-2) in RIE cells in vitro and in the colonic epithelium of transgenic PKCβII mice in vivo (Yu et al., 2003). Celecoxib, a selective Cox-2 inhibitor, and inhibitor of colon carcinogenesis, restores expression of TGF-β RII in RIE/PKCβII cells in vitro and in the colonic epithelium of transgenic PKCβII mice in vivo (Yu et al., 2003). Thus PKCβII promotes colon cancer, in part, through induction of Cox-2, suppression of TGF-β signaling, and establishment of a TGF-β-resistant, hyper-proliferative state in the colonic epithelium. These studies defined a pro-carcinogenic PKCβII→Cox-2→TGF-β signaling axis within the colonic epithelium, and provided a plausible molecular mechanism by which non-steroidal anti-inflammatory agents such as Celecoxib suppress colon carcinogenesis.
Diet is a major risk factor for colon cancer development both in humans and mice. We and others have shown that a diet high in colon cancer-promotive ω-6 fatty acids increases membrane-associated PKCβII (Davidson et al., 2000; Murray et al., 2002; Jalili et al., 2003). In contrast, chemopreventive ω-3 fatty acids, which inhibit AOM-induced colon carcinogenesis in vivo (Reddy and Maeura, 1984; Reddy and Maruyama, 1986; Nelson et al., 1988; Reddy and Sugie, 1988; Deschner et al., 1990; Reddy et al., 1991), can directly inhibit PKCβII activity in vitro (Holian and Nelson, 1992; Davidson et al., 2000; Seung Kim et al., 2001). Furthermore, we have shown that chemopreventive ω-3 fatty acids inhibit colonic PKCβII activity in vivo and block PKCβII-mediated hyper-proliferation and enhanced colon carcinogenesis in transgenic PKCβII mice (Murray et al., 2002). Chemopreventive ω-3 fatty acids also inhibit PKCβII-mediated induction of Cox-2 protein expression and repression of TGF-β RII expression in RIE/PKCβII cells (Murray et al., 2002). Thus, our studies demonstrate that PKCβII is not only an important colon cancer gene, but also an important target for cancer risk modulation by chemopreventive dietary fatty acids in vivo (Murray et al., 2002). PKCβII drives key aspects of colon carcinogenesis through activation of several major signaling pathways involved in proliferation, invasion and resistance to TGFβ-mediated growth inhibition (Figure 1). PKCβII is an important mediator of environmental, particularly dietary, effects on colon cancer and is therefore, is an ideal target for chemopreventive drug therapy.
Figure 1. PKCβII activates multiple pro-carcinogenic signal transduction pathways.
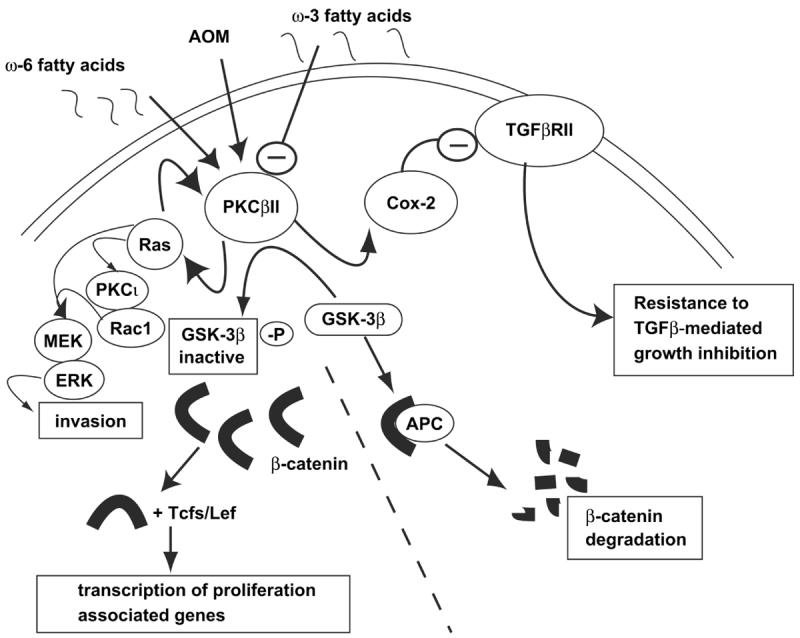
PKCβII activates the Wnt/APC/β-catenin proliferative signaling pathway by inactivating GSK-3β and increasing the levels of β-catenin (Murray et al., 1999). PKCβII also induces resistance to the growth inhibitory effects of TGFβ via a PKCβII→Cox-2→TGFβRII signaling pathway in vitro and in vivo (Murray et al., 2002; Yu et al., 2003). Finally, PKCβII induces Ras activation thereby inducing an invasive phenotype in vitro (Zhang et al., 2004).
Like PKCβII, PKCι expression is elevated in human colon cancer and AOM-induced colon tumors in mice (Murray et al., 2004). PKCι expression is also induced in intestinal tumors in APCMin/+ mice compared to normal intestinal epithelium (Oster and Leitges, 2006), suggesting that PKCι may play an important role in colon carcinogenesis. To test this hypothesis, we generated transgenic mice that express either a constitutively active PKCι allele (caPKCι) or a dominant negative, kinase-deficient PKCι allele (kdPKCι) in the colonic epithelium. Expression of either PKCι mutant has no demonstrable effect on proliferation or differentiation of the colonic epithelium (Murray et al., 2004). However, expression of caPKCι in the colon increases susceptibility to AOM-mediated ACF, while expression of kdPKCι significantly inhibits AOM-induced ACF formation (Murray et al., 2004). caPKCι mice also exhibit an increase in the number of AOM-induced colon tumors, and an increase in progression of these tumors from benign adenoma to malignant intramucosal carcinoma (Murray et al., 2004).
Ras signaling is thought to play an important role in colon carcinogenesis. Oncogenic Ras mutations are detected in ∼30% of colon cancers (Slattery et al., 2001; Takayama et al., 2001) and atypical PKCs have been implicated in oncogenic Ras signaling (Uberall et al., 1999; Kampfer et al., 2001). Therefore, we investigated the role of PKCι in oncogenic _Ras_-mediated transformation of RIE cells. We found that oncogenic Ras activates PKCι activity in RIE cells and that expression of kdPKCι inhibits oncogenic _Ras_-mediated invasion and anchorage-independent growth (Murray et al., 2004). Rac1 is activated by oncogenic Ras and is required for _Ras_-mediated transformation (Qiu et al., 1995; Murray et al., 2004). kdPKCι blocks oncogenic _Ras_-mediated Rac1 activation, and expression of a constitutively active Rac1 allele, RacV12, overcomes dnPKCι-mediated inhibition of invasion (Murray et al., 2004). Thus, oncogenic _Ras_-mediated cellular invasion is dependent on both Rac1 and PKCι. Interestingly, expression of either wildtype PKCι or caPKCι in the absence of oncogenic Ras fails to induce invasion or alter anchorage-dependent or independent growth, indicating that PKCι is necessary for _Ra_s-mediated invasion, but is not sufficient to induce transformation in the absence of oncogenic Ras (Murray et al., 2004). Finally, in an in vivo model of _K-ras_-mediated colon carcinogenesis, the K-rasLA2 mouse, expression of kdPKCι in the colonic epithelium inhibits _K-ras_-mediated ACF formation (Murray et al., 2004). Thus, PKCι is required for oncogenic _Ras_-mediated transformation of the intestinal epithelium in vitro and in vivo.
PKCι is a Human Lung Cancer Oncogene
In addition to its promotive role in colon cancer, PKCι has also been implicated as a downstream effector of oncogenic K-ras in lung cancer (Regala et al., 2005a). PKCι, but not PKCζ, is highly expressed in non-small cell lung cancer (NSCLC) cells (Regala et al., 2005a). Consistent with our observations in human leukemic and intestinal epithelial cell lines, expression of kdPKCι has no effect on adherent growth or survival in NSCLC cells (Regala et al., 2005a). However, expression of kdPKCι in NSCLC cells blocks transformed (anchorage-independent) growth in vitro and tumorigenicity in vivo (Regala et al., 2005a). Likewise, we have shown that Rac1 is a critical downstream target of PKCι-dependent transformation of NSCLC cells in vitro and in vivo (Regala et al., 2005a). Expression of the PB1 domain of PKCι blocks PKCι-dependent Rac1 activation and inhibits the transformed growth of NSCLC cells in vitro (Regala et al., 2005a) demonstrating the importance of the PB1 domain in PKCι-dependent transformation of NSCLC cells. Thus, PKCι plays a critical role in the transformed growth, invasion and survival of cancer cells through several oncogenic signaling pathways (Figure 2).
Figure 2. PKCι is an oncogene that activates multiple signaling pathways critical for transformed growth, invasion and survival of human cancer cells.
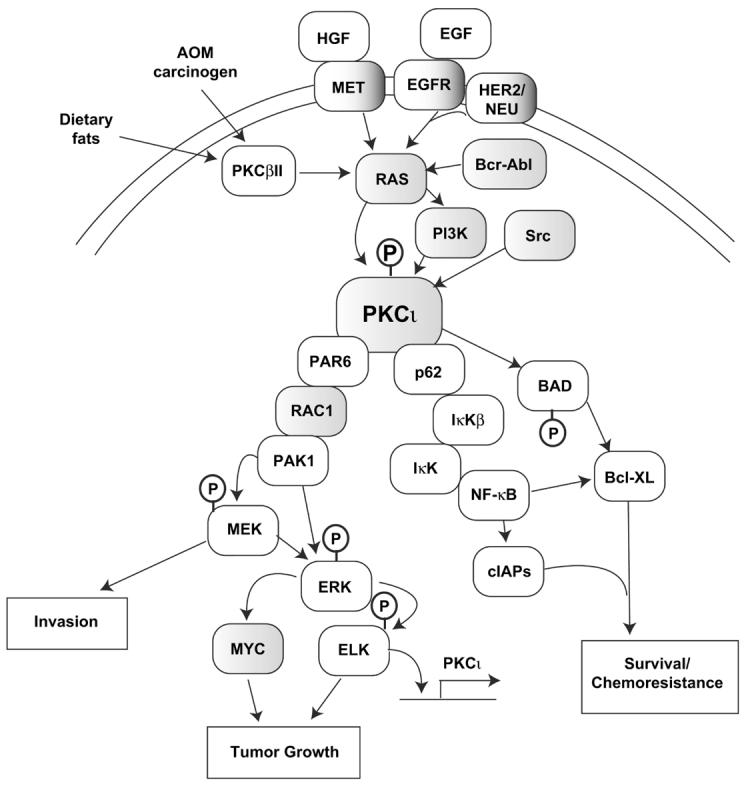
PKCι resides in several major signaling pathways implicated in human cancer. Many components of these pathways, including PKCι itself are oncogenically activated, often by multiple mechanisms (i.e. gene amplification and somatic mutation), in human tumors (indicated by shaded boxes). Arrows indicate flow through signaling pathways; touching boxes indicate direct binding of signaling components. Phosphorylation events are indicated by circled Ps.
Our finding that PKCι signaling is required for transformed growth of human lung cancer cells led us to investigate the functional significance of PKCι in human lung cancers. We observed that PKCι (but not PKCζ) is highly over-expressed at both the mRNA and protein level in the majority (∼70%) of primary NSCLC tumors (Regala et al., 2005b). Furthermore, the level of PKCι expression in tumors is highly predictive of poor patient survival independent of tumor stage (Regala et al., 2005b). Thus, patients with early stage lung cancer expressing high PKCι levels are significantly more likely to die from their disease than those with low PKCι expression. Given the prevalence of PKCι over-expression in NSCLC tumors, we investigated the mechanisms responsible for elevated PKCι expression in NSCLC. The PKCι gene resides at chromosome 3q26, one of the most frequently amplified chromosomal regions in NSCLC, particularly in lung squamous cell carcinoma (LSCC) (Brass et al., 1996). Therefore, we assessed PKCι gene copy number in LSCC tumors. We determined that the PKCι gene is frequently amplified in LSSC tumors (∼70%) and that gene amplification drives PKCι expression in these tumors (Regala et al., 2005b). Disruption of PKCι signaling by expression of dnPKCι inhibits the transformed phenotype in human NSCLC cells with PKCι gene amplification demonstrating that PKCι is a critical target of amplification of chromosomal region 3q26 in lung cancer (Regala et al., 2005b). These data demonstrate that PKCι is a human oncogene in NSCLC. More recently, PKCι has also been identified as a potential oncogene in ovarian cancer indicating that chromosomal instability and genomic amplification of PKCι may be a common feature of numerous tumor types (Zhang et al., 2006). Genomic amplification is not the only mechanism by which PKCι is over-expressed in NSCLC, however, since PKCι is also highly expressed in lung adenocarcinoma tumors which do not harbor PKCι gene amplification (Regala et al., 2005b). We are currently investigating mechanisms of oncogenic PKCι activation of PKCι in lung adenocarcinomas. These data are the first to demonstrate that any PKC isozyme is a bonafide human oncogene that is a specific target of tumor-specific genetic alteration and activation.
PKC Isozymes as Targets for Chemotherapy
Multiple PKC isozymes have been the target for therapeutic drug development for treatment of cancer. The first isozyme-selective PKC inhibitor to reach the clinic as a potential cancer therapeutic targeted PKCα. A 20-base antisense oligonucleotide directed against PKCα (LY900003/Aprinocarsen) inhibits PKCα expression and exhibits anti-tumor activity in various human cancer cell xenografts in nude mice (Dean et al., 1996; Geiger et al., 1998). However, no clinical benefit was observed in phase II clinical trials using Aprinocarsen as a single agent against ovarian cancer (Advani et al., 2004) or high grade glioma (Grossman et al., 2005). Likewise, a large phase III clinical trial using Aprinocarsen in combination with gemcitabine and cispatin to treat advanced stage non-small cell lung cancer observed no survival benefit or difference in response rate (Paz-Ares et al., 2006). Therefore, despite encouraging pre-clinical data, PKCα has not yet been shown to be a successful target of cancer therapeutics. It is unclear whether the failure of Aprinocarsen in the clinical setting is due to an inability of the drug to sufficiently inhibit PKCα in vivo or relates to the relative importance of PKCα as a tumor promoting gene in the target cancers. In this regard, our studies and others in leukemia and colon cancer indicate that PKCα suppresses tumorigenesis, rather than promoting it, calling into question the rationale for PKCα inhibitors in cancer therapy.
A Small Molecule PKCβ Inhibitor Shows Promise in Some Forms of Human Cancer
Enzastaurin (LY317615) is a macrocyclic bisindolylmaleimide compound that exhibits PKCβ-selective inhibition in an ATP-competitive manner (Ishii et al., 1996; Jirousek et al., 1996). Enzastaurin is one of a series of PKCβ-selective inhibitors produced and characterized by Lilly Research Laboratories. LY379196 (Jirousek et al., 1996), a closely related compound to Enzastaurin, blocks PKCβII-mediated cellular invasion (Zhang et al., 2004) and inhibition of TGF-β RII expression (Murray et al., 2002) in rat intestinal epithelial cells and similar results have been obtained with Enzastaurin (unpublished observations). In addition, Enzastaurin blocks PKCβII-mediated gene expression in human colon cancer cells (Liu et al., 2004). Preclinical studies in human tumor xenografts in nude mice have shown that oral administration of Enzastaurin inhibits tumor cell proliferation, induces apoptosis, and decreases plasma VEGF levels and intratumoral vessel formation (Keyes et al., 2004; Graff et al., 2005). A phase I clinical trial demonstrated that Enzastaurin is well tolerated with few toxic side effects (Carducci et al., 2006). Phase II clinical trials of Enzastaurin have shown promising results in treatment of diffuse large B-cell lymphomas and in high grade gliomas (Fine, 2005; Robertson et al., 2007). A phase III clinical trial of Enzastaurin for treatment of recurrent glioblastoma is currently ongoing.
Therapeutic Targeting of Oncogenic PKCι Signaling
The fact that PKCι is a bonafide oncogene in human lung cancer led us to explore whether PKCι is a tractable drug target. Rather than targeting the catalytic domain of PKCι, we chose to target the PB1 domain, which plays an important role in oncogenic PKCι signaling (Regala et al., 2005a). A novel fluorescence resonance energy transfer (FRET) based assay was developed and used to identify compounds in a commercial compound library that disrupt the PB1-PB1 domain interaction between PKCι and its binding partner Par6 (Stallings-Mann et al., 2006). The most potent compounds identified in this screen were the gold salts aurothioglucose (ATG) and aurothiomalate (ATM). These compounds are FDA approved for clinical use for the treatment of rheumatoid arthritis, and ATM is still clinically available for this indication. These compounds effectively inhibit PKCι/Par6 interactions in vitro with an IC50 of ∼1μM, block PKCι-dependent signaling to Rac1, and inhibit the transformed growth of NSCLC cells (Stallings-Mann et al., 2006). Inhibition of transformed growth is relieved by expression of RacV12, consistent with a mechanism of action at the level of the interaction between PKCι and Par6, which couples PKCι to Rac1 (Stallings-Mann et al., 2006). ATM is a highly selective inhibitor of PB1-PB1 domain interactions between PKCι and the two adaptor molecules Par6 and p62, having no appreciable inhibitory effect on other PB1-PB1 domain interactions including p62-p62, p62-NBR1, and MEKK3-MEK5 interactions (Erdogan et al., 2006). Structural analysis, site-directed mutagenesis and molecular modeling studies demonstrate that ATM targets a unique cysteine residue, Cys69, within the PB1 domain of PKCι to exert its inhibitory effect on Par6 binding (Erdogan et al., 2006). Cys69 resides within the OPCA motif of the PKCι PB1 domain within the binding interface between PKCι and Par6 where it interacts with Arg28 on Par6. Molecular modeling predicts that a cysteinyl-aurothiomalate adduct at C69 protrudes into the binding cleft normally occupied by Par6, providing a plausible structural explanation for ATM-mediated inhibition of Par6 binding. Mutation of Cys69 of PKCι to isoleucine or valine, residues frequently found at this position in other PB1 domains, has little or no effect on the affinity of the resultant PKCι mutant for Par6 but confers resistance to ATM-mediated inhibition of Par6 binding. Finally, expression of the PKCι C69I mutant NSCLC cells has no effect on transformed growth, but confers resistance to the inhibitory effects of ATM on transformed growth (Erdogan et al., 2006). Thus, ATM inhibits cellular transformation by selectively targeting Cys69 within the PB1 domain of PKCι and is a potent and effective anti-tumor agent in preclinical models of NSCLC. We are currently conducting a phase I dose escalation clinical trial of ATM in patients with advanced stage NSCLC to establish a tolerated dosing regimen of this compound.
Summary
PKC isozymes play specific, non-redundant functional roles in numerous cellular processes, including proliferation, differentiation, cellular invasion and apoptosis. We have determined that PKCβII and PKCι are both critical pro-carcinogenic genes involved in multiple human cancers. PKCι is a bonafide human oncogene, the first PKC isozyme that can be so classified. Both PKCβII and PKCι are legitimate therapeutic targets to which novel targeted therapy has been successfully developed and is being evaluated clinically.
Acknowledgements
We would like to thank all the members of the Fields laboratory for their contributions to the work described in this review. We would also like to acknowledge the work of the many investigators whose combined work has made critical contributions to our understanding of PKC functions in cancer. Space limitations necessitated a relatively narrow focus in this review. We apologize to colleagues whose contributions were not cited due to these constraints. The work described here was supported by grants from the National Cancer Institute to APF (R01 CA081436 and R01CA056869), and to NRM (R01CA094122). Additional funding was provided by the American Lung Association/LUNGevity Lung Cancer Discovery Award (LCD-22766-N), the James and Esther King Biomedical Research Program, and the V Foundation for Cancer Research to APF.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Advani R, Peethambaram P, Lum BL, Fisher GA, Hartmann L, Long HJ, Halsey J, Holmlund JT, Dorr A, Sikic BI. A Phase II trial of aprinocarsen, an antisense oligonucleotide inhibitor of protein kinase C alpha, administered as a 21-day infusion to patients with advanced ovarian carcinoma. Cancer. 2004;100:321–326. doi: 10.1002/cncr.11909. [DOI] [PubMed] [Google Scholar]
- Bedi A, Barber JP, Bedi GC, el-Deiry WS, Sidransky D, Vala MS, Akhtar AJ, Hilton J, Jones RJ. BCR-ABL-mediated inhibition of apoptosis with delay of G2/M transition after DNA damage: a mechanism of resistance to multiple anticancer agents. Blood. 1995;86:1148–1158. [PubMed] [Google Scholar]
- Brass N, Ukena I, Remberger K, Mack U, Sybrecht GW, Meese EU. DNA amplification on chromosome 3q26.1-q26.3 in squamous cell carcinoma of the lung detected by reverse chromosome painting. Eur J Cancer. 1996;32A:1205–1208. doi: 10.1016/0959-8049(96)00016-0. [DOI] [PubMed] [Google Scholar]
- Carducci MA, Musib L, Kies MS, Pili R, Truong M, Brahmer JR, Cole P, Sullivan R, Riddle J, Schmidt J, Enas N, Sinha V, Thornton DE, Herbst RS. Phase I dose escalation and pharmacokinetic study of enzastaurin, an oral protein kinase C beta inhibitor, in patients with advanced cancer. J Clin Oncol. 2006;24:4092–4099. doi: 10.1200/JCO.2005.05.3447. [DOI] [PubMed] [Google Scholar]
- Davidson LA, Brown RE, Chang WC, Morris JS, Wang N, Carroll RJ, Turner ND, Lupton JR, Chapkin RS. Morphodensitometric analysis of protein kinase C beta(II) expression in rat colon: modulation by diet and relation to in situ cell proliferation and apoptosis. Carcinogenesis. 2000;21:1513–1519. [PubMed] [Google Scholar]
- Dean N, McKay R, Miraglia L, Howard R, Cooper S, Giddings J, Nicklin P, Meister L, Ziel R, Geiger T, Muller M, Fabbro D. Inhibition of growth of human tumor cell lines in nude mice by an antisense of oligonucleotide inhibitor of protein kinase C-alpha expression. Cancer Res. 1996;56:3499–3507. [PubMed] [Google Scholar]
- Deschner EE, Lytle JS, Wong G, Ruperto JF, Newmark HL. The effect of dietary omega-3 fatty acids (fish oil) on azoxymethanol-induced focal areas of dysplasia and colon tumor incidence. Cancer. 1990;66:2350–2356. doi: 10.1002/1097-0142(19901201)66:11<2350::aid-cncr2820661117>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Dieter P, Schwende H. Protein kinase C-alpha and -beta play antagonistic roles in the differentiation process of THP-1 cells. Cell Signal. 2000;12:297–302. doi: 10.1016/s0898-6568(00)00069-3. [DOI] [PubMed] [Google Scholar]
- Erdogan E, Lamark T, Stallings-Mann M, Lee J, Pellechia M, Thompson EA, Johansen T, Fields AP. Aurothiomalate Inhibits Transformed Growth by Targeting the PB1 Domain of Protein Kinase C{iota} J Biol Chem. 2006;281:28450–28459. doi: 10.1074/jbc.M606054200. [DOI] [PubMed] [Google Scholar]
- Fine HA, Kim L, Royce C, Draper D, Haggarty I, Ellinzano H, Albert P, Kinney P, Musib L, Thorton D.Results from phase II trial of Enzastaurin (LY317615) in patients with recurrent high grade gliomas Journal of Clinical Oncology ASCO Annual Meeting Proceedings 20052316S, Part I of II (June 1 Supplement):1504 [Google Scholar]
- Geiger T, Muller M, Dean NM, Fabbro D. Antitumor activity of a PKC-alpha antisense oligonucleotide in combination with standard chemotherapeutic agents against various human tumors transplanted into nude mice. Anticancer Drug Des. 1998;13:35–45. [PubMed] [Google Scholar]
- Gokmen-Polar Y, Fields AP. Mapping of a molecular determinant for protein kinase C betaII isozyme function. J Biol Chem. 1998;273:20261–20266. doi: 10.1074/jbc.273.32.20261. [DOI] [PubMed] [Google Scholar]
- Gokmen-Polar Y, Murray NR, Velasco MA, Gatalica Z, Fields AP. Elevated protein kinase C betaII is an early promotive event in colon carcinogenesis. Cancer Res. 2001;61:1375–1381. [PubMed] [Google Scholar]
- Goss VL, Hocevar BA, Thompson LJ, Stratton CA, Burns DJ, Fields AP. Identification of nuclear beta II protein kinase C as a mitotic lamin kinase. J Biol Chem. 1994;269:19074–19080. [PubMed] [Google Scholar]
- Graff JR, McNulty AM, Hanna KR, Konicek BW, Lynch RL, Bailey SN, Banks C, Capen A, Goode R, Lewis JE, Sams L, Huss KL, Campbell RM, Iversen PW, Neubauer BL, Brown TJ, Musib L, Geeganage S, Thornton D. The protein kinase Cbeta-selective inhibitor, Enzastaurin ( LY317615.HCl), suppresses signaling through the AKT pathway, induces apoptosis, and suppresses growth of human colon cancer and glioblastoma xenografts. Cancer Res. 2005;65:7462–7469. doi: 10.1158/0008-5472.CAN-05-0071. [DOI] [PubMed] [Google Scholar]
- Grossman SA, Alavi JB, Supko JG, Carson KA, Priet R, Dorr FA, Grundy JS, Holmlund JT. Efficacy and toxicity of the antisense oligonucleotide aprinocarsen directed against protein kinase C-alpha delivered as a 21-day continuous intravenous infusion in patients with recurrent high-grade astrocytomas. Neuro Oncol. 2005;7:32–40. doi: 10.1215/S1152851703000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan L, Song K, Pysz MA, Curry KJ, Hizli AA, Danielpour D, Black AR, Black JD. Protein kinase C-mediated down-regulation of cyclin D1 involves activation of the translational repressor 4E-BP1 via a phosphoinositide 3-kinase/Akt-independent, protein phosphatase 2A-dependent mechanism in intestinal epithelial cells. J Biol Chem. 2007;282:14213–14225. doi: 10.1074/jbc.M610513200. [DOI] [PubMed] [Google Scholar]
- Gustafson WC, Ray S, Jamieson L, Thompson EA, Brasier AR, Fields AP. BCR-ABL regulates protein kinase Ciota transcription via an ELK1 site in the PKCiota promoter. J Biol Chem. 2003 doi: 10.1074/jbc.M312840200. [DOI] [PubMed] [Google Scholar]
- Hocevar BA, Fields AP. Selective translocation of beta II-protein kinase C to the nucleus of human promyelocytic (HL60) leukemia cells. J Biol Chem. 1991;266:28–33. [PubMed] [Google Scholar]
- Hocevar BA, Morrow DM, Tykocinski ML, Fields AP. Protein kinase C isotypes in human erythroleukemia cell proliferation and differentiation. J Cell Sci. 1992;101(Pt 3):671–679. doi: 10.1242/jcs.101.3.671. [DOI] [PubMed] [Google Scholar]
- Holian O, Nelson R. Action of long-chain fatty acids on protein kinase C activity: comparison of omega-6 and omega-3 fatty acids. Anticancer Res. 1992;12:975–980. [PubMed] [Google Scholar]
- Ishii H, Jirousek MR, Koya D, Takagi C, Xia P, Clermont A. Amelioraton of vascular dysfunctions in diabetic rats by an oral PKC beta inhibitor. Science. 1996;272:699–700. doi: 10.1126/science.272.5262.728. [DOI] [PubMed] [Google Scholar]
- Jalili T, Manning J, Kim S. Increased translocation of cardiac protein kinase C beta2 accompanies mild cardiac hypertrophy in rats fed saturated fat. J Nutr. 2003;133:358–361. doi: 10.1093/jn/133.2.358. [DOI] [PubMed] [Google Scholar]
- Jamieson L, Carpenter L, Biden TJ, Fields AP. Protein kinase Ciota activity is necessary for Bcr-Abl-mediated resistance to drug-induced apoptosis. J Biol Chem. 1999;274:3927–3930. doi: 10.1074/jbc.274.7.3927. [DOI] [PubMed] [Google Scholar]
- Jirousek MR, Gillig JR, Gonzalez CM, Heath WF, McDonald JH, 3rd, Neel DA, Rito CJ, Singh U, Stramm LE, Melikian-Badalian A, Baevsky M, Ballas LM, Hall SE, Winneroski LL, Faul MM. (S)-13-[(dimethylamino)methyl]-10,11,14,15-tetrahydro-4,9:16, 21-dimetheno-1H, 13H-dibenzo[e,k]pyrrolo[3,4-h][1,4,13]oxadiazacyclohexadecene-1,3(2H)-d ione ( LY333531) and related analogues: isozyme selective inhibitors of protein kinase C beta. J Med Chem. 1996;39:2664–2671. doi: 10.1021/jm950588y. [DOI] [PubMed] [Google Scholar]
- Kampfer S, Windegger M, Hochholdinger F, Schwaiger W, Pestell RG, Baier G, Grunicke HH, Uberall F. Protein kinase C isoforms involved in the transcriptional activation of cyclin D1 by transforming Ha-Ras. J Biol Chem. 2001;276:42834–42842. doi: 10.1074/jbc.M102047200. [DOI] [PubMed] [Google Scholar]
- Keyes KA, Mann L, Sherman M, Galbreath E, Schirtzinger L, Ballard D, Chen YF, Iversen P, Teicher BA. LY317615 decreases plasma VEGF levels in human tumor xenograft-bearing mice. Cancer Chemother Pharmacol. 2004;53:133–140. doi: 10.1007/s00280-003-0713-x. [DOI] [PubMed] [Google Scholar]
- Kubo K, Ohno S, Suzuki K. Primary structures of human protein kinase C beta I and beta II differ only in their C-terminal sequences. FEBS Lett. 1987;223:138–142. doi: 10.1016/0014-5793(87)80524-0. [DOI] [PubMed] [Google Scholar]
- Lamark T, Perander M, Outzen H, Kristiansen K, Overvatn A, Michaelsen E, Bjorkoy G, Johansen T. Interaction codes within the family of mammalian Phox and Bem1p domain-containing proteins. J Biol Chem. 2003;278:34568–34581. doi: 10.1074/jbc.M303221200. [DOI] [PubMed] [Google Scholar]
- Leitges M, Sanz L, Martin P, Duran A, Braun U, Garcia JF, Camacho F, Diaz-Meco MT, Rennert PD, Moscat J. Targeted disruption of the zetaPKC gene results in the impairment of the NF-kappaB pathway. Mol Cell. 2001;8:771–780. doi: 10.1016/s1097-2765(01)00361-6. [DOI] [PubMed] [Google Scholar]
- Liu Y, Su W, Thompson EA, Leitges M, Murray NR, Fields AP. Protein kinase CbII regulates its own expression in rat intestinal epithelial cells and the colonic epithelium in vivo. J Biol Chem. 2004 doi: 10.1074/jbc.M407701200. [DOI] [PubMed] [Google Scholar]
- Lu Y, Jamieson L, Brasier AR, Fields AP. NF-kappaB/RelA transactivation is required for atypical protein kinase C iota-mediated cell survival. Oncogene. 2001;20:4777–4792. doi: 10.1038/sj.onc.1204607. [DOI] [PubMed] [Google Scholar]
- Murray NR, Baumgardner GP, Burns DJ, Fields AP. Protein kinase C isotypes in human erythroleukemia (K562) cell proliferation and differentiation. Evidence that beta II protein kinase C is required for proliferation. J Biol Chem. 1993;268:15847–15853. [PubMed] [Google Scholar]
- Murray NR, Davidson LA, Chapkin RS, Clay Gustafson W, Schattenberg DG, Fields AP. Overexpression of protein kinase C betaII induces colonic hyperproliferation and increased sensitivity to colon carcinogenesis. J Cell Biol. 1999;145:699–711. doi: 10.1083/jcb.145.4.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray NR, Fields AP. Atypical protein kinase C iota protects human leukemia cells against drug-induced apoptosis. J Biol Chem. 1997;272:27521–27524. doi: 10.1074/jbc.272.44.27521. [DOI] [PubMed] [Google Scholar]
- Murray NR, Jamieson L, Yu W, Zhang J, Gokmen-Polar Y, Sier D, Anastasiadis P, Gatalica Z, Thompson EA, Fields AP. Protein kinase C{iota} is required for Ras transformation and colon carcinogenesis in vivo. J Cell Biol. 2004;164:797–802. doi: 10.1083/jcb.200311011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray NR, Weems C, Chen L, Leon J, Yu W, Davidson LA, Jamieson L, Chapkin RS, Thompson EA, Fields AP. Protein kinase C betaII and TGFbetaRII in omega-3 fatty acid-mediated inhibition of colon carcinogenesis. J Cell Biol. 2002;157:915–920. doi: 10.1083/jcb.200201127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata KI, Okano Y, Nozawa Y. Protein kinase C isozymes in human megakaryoblastic leukemia cell line, MEG-01: possible involvement of the isozymes in the differentiation process of MEG-01 cells. Br J Haematol. 1996;93:762–771. doi: 10.1046/j.1365-2141.1996.d01-1714.x. [DOI] [PubMed] [Google Scholar]
- Nelson RL, Tanure JC, Andrianopoulos G, Souza G, Lands WE. A comparison of dietary fish oil and corn oil in experimental colorectal carcinogenesis. Nutr Cancer. 1988;11:215–220. doi: 10.1080/01635588809513990. [DOI] [PubMed] [Google Scholar]
- Oster H, Leitges M. Protein kinase C alpha but not PKCzeta suppresses intestinal tumor formation in ApcMin/+ mice. Cancer Res. 2006;66:6955–6963. doi: 10.1158/0008-5472.CAN-06-0268. [DOI] [PubMed] [Google Scholar]
- Paz-Ares L, Douillard JY, Koralewski P, Manegold C, Smit EF, Reyes JM, Chang GC, John WJ, Peterson PM, Obasaju CK, Lahn M, Gandara DR. Phase III study of gemcitabine and cisplatin with or without aprinocarsen, a protein kinase C-alpha antisense oligonucleotide, in patients with advanced-stage non-small-cell lung cancer. J Clin Oncol. 2006;24:1428–1434. doi: 10.1200/JCO.2005.04.3299. [DOI] [PubMed] [Google Scholar]
- Pennisi E.How a growth control path takes a wrong turn to cancer Science 19982811438–1439., 1441. [DOI] [PubMed] [Google Scholar]
- Qiu RG, Chen J, Kirn D, McCormick F, Symons M. An essential role for Rac in Ras transformation. Nature. 1995;374:457–459. doi: 10.1038/374457a0. [DOI] [PubMed] [Google Scholar]
- Reddy BS, Burill C, Rigotty J. Effect of diets high in omega-3 and omega-6 fatty acids on initiation and postinitiation stages of colon carcinogenesis. Cancer Res. 1991;51:487–491. [PubMed] [Google Scholar]
- Reddy BS, Maeura Y. Tumor promotion by dietary fat in azoxymethane-induced colon carcinogenesis in female F344 rats: influence of amount and source of dietary fat. J Natl Cancer Inst. 1984;72:745–750. [PubMed] [Google Scholar]
- Reddy BS, Maruyama H. Effect of dietary fish oil on azoxymethane-induced colon carcinogenesis in male F344 rats. Cancer Res. 1986;46:3367–3370. [PubMed] [Google Scholar]
- Reddy BS, Sugie S. Effect of different levels of omega-3 and omega-6 fatty acids on azoxymethane-induced colon carcinogenesis in F344 rats. Cancer Res. 1988;48:6642–6647. [PubMed] [Google Scholar]
- Regala RP, Weems C, Jamieson L, Copland JA, Thompson EA, Fields AP. Atypical protein kinase Ciota plays a critical role in human lung cancer cell growth and tumorigenicity. J Biol Chem. 2005a;280:31109–31115. doi: 10.1074/jbc.M505402200. [DOI] [PubMed] [Google Scholar]
- Regala RP, Weems C, Jamieson L, Khoor A, Edell ES, Lohse CM, Fields AP. Atypical protein kinase C iota is an oncogene in human non-small cell lung cancer. Cancer Res. 2005b;65:8905–8911. doi: 10.1158/0008-5472.CAN-05-2372. [DOI] [PubMed] [Google Scholar]
- Robertson MJ, Kahl BS, Vose JM, de Vos S, Laughlin M, Flynn PJ, Rowland K, Cruz JC, Goldberg SL, Musib L, Darstein C, Enas N, Kutok JL, Aster JC, Neuberg D, Savage KJ, LaCasce A, Thornton D, Slapak CA, Shipp MA. Phase II study of enzastaurin, a protein kinase C beta inhibitor, in patients with relapsed or refractory diffuse large B-cell lymphoma. J Clin Oncol. 2007;25:1741–1746. doi: 10.1200/JCO.2006.09.3146. [DOI] [PubMed] [Google Scholar]
- Scaglione-Sewell B, Abraham C, Bissonnette M, Skarosi SF, Hart J, Davidson NO, Wali RK, Davis BH, Sitrin M, Brasitus TA. Decreased PKC-alpha expression increases cellular proliferation, decreases differentiation, and enhances the transformed phenotype of CaCo-2 cells. Cancer Res. 1998;58:1074–1081. [PubMed] [Google Scholar]
- Selbie LA, Schmitz-Peiffer C, Sheng Y, Biden TJ. Molecular cloning and characterization of PKC iota, an atypical isoform of protein kinase C derived from insulin-secreting cells. J Biol Chem. 1993;268:24296–24302. [PubMed] [Google Scholar]
- Seung Kim HF, Weeber EJ, Sweatt JD, Stoll AL, Marangell LB. Inhibitory effects of omega-3 fatty acids on protein kinase C activity in vitro. Mol Psychiatry. 2001;6:246–248. doi: 10.1038/sj.mp.4000837. [DOI] [PubMed] [Google Scholar]
- Slattery ML, Anderson K, Curtin K, Ma K, Schaffer D, Edwards S, Samowitz W. Lifestyle factors and Ki-ras mutations in colon cancer tumors. Mutat Res. 2001;483:73–81. doi: 10.1016/s0027-5107(01)00228-7. [DOI] [PubMed] [Google Scholar]
- Soloff RS, Katayama C, Lin MY, Feramisco JR, Hedrick SM. Targeted deletion of protein kinase C lambda reveals a distribution of functions between the two atypical protein kinase C isoforms. J Immunol. 2004;173:3250–3260. doi: 10.4049/jimmunol.173.5.3250. [DOI] [PubMed] [Google Scholar]
- Stallings-Mann M, Jamieson L, Regala RP, Weems C, Murray NR, Fields AP. A novel small-molecule inhibitor of protein kinase Ciota blocks transformed growth of non-small-cell lung cancer cells. Cancer Res. 2006;66:1767–1774. doi: 10.1158/0008-5472.CAN-05-3405. [DOI] [PubMed] [Google Scholar]
- Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, Gould KA, Dove WF. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 1992;256:668–670. doi: 10.1126/science.1350108. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Nakatsugi S, Sugimura T, Wakabayashi K. Frequent mutations of the beta-catenin gene in mouse colon tumors induced by azoxymethane. Carcinogenesis. 2000;21:1117–1120. [PubMed] [Google Scholar]
- Takayama T, Ohi M, Hayashi T, Miyanishi K, Nobuoka A, Nakajima T, Satoh T, Takimoto R, Kato J, Sakamaki S, Niitsu Y. Analysis of K-ras, APC, and beta-catenin in aberrant crypt foci in sporadic adenoma, cancer, and familial adenomatous polyposis. Gastroenterology. 2001;121:599–611. doi: 10.1053/gast.2001.27203. [DOI] [PubMed] [Google Scholar]
- Thompson LJ, Fields AP. betaII protein kinase C is required for the G2/M phase transition of cell cycle. J Biol Chem. 1996;271:15045–15053. doi: 10.1074/jbc.271.25.15045. [DOI] [PubMed] [Google Scholar]
- Uberall F, Hellbert K, Kampfer S, Maly K, Villunger A, Spitaler M, Mwanjewe J, Baier-Bitterlich G, Baier G, Grunicke HH. Evidence that atypical protein kinase C-lambda and atypical protein kinase C-zeta participate in Ras-mediated reorganization of the F-actin cytoskeleton. J Cell Biol. 1999;144:413–425. doi: 10.1083/jcb.144.3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker SD, Murray NR, Burns DJ, Fields AP. Protein kinase C chimeras: catalytic domains of alpha and beta II protein kinase C contain determinants for isotype-specific function. Proc Natl Acad Sci U S A. 1995;92:9156–9160. doi: 10.1073/pnas.92.20.9156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Murray NR, Weems C, Chen L, Guo H, Ethridge R, Ceci JD, Evers BM, Thompson EA, Fields AP. Role of cyclooxygenase 2 in protein kinase C beta II-mediated colon carcinogenesis. J Biol Chem. 2003;278:11167–11174. doi: 10.1074/jbc.M211424200. [DOI] [PubMed] [Google Scholar]
- Zhang J, Anastasiadis PZ, Liu Y, Thompson EA, Fields AP. Protein kinase C (PKC) betaII induces cell invasion through a Ras/Mek-, PKC iota/Rac 1-dependent signaling pathway. J Biol Chem. 2004;279:22118–22123. doi: 10.1074/jbc.M400774200. [DOI] [PubMed] [Google Scholar]
- Zhang L, Huang J, Yang N, Liang S, Barchetti A, Giannakakis A, Cadungog MG, O’Brien-Jenkins A, Massobrio M, Roby KF, Katsaros D, Gimotty P, Butzow R, Weber BL, Coukos G. Integrative genomic analysis of protein kinase C (PKC) family identifies PKCiota as a biomarker and potential oncogene in ovarian carcinoma. Cancer Res. 2006;66:4627–4635. doi: 10.1158/0008-5472.CAN-05-4527. [DOI] [PubMed] [Google Scholar]