Usher syndrome and Leber congenital amaurosis are molecularly linked via a novel isoform of the centrosomal ninein-like protein (original) (raw)
Abstract
Usher syndrome (USH) and Leber congenital amaurosis (LCA) are autosomal recessive disorders resulting in syndromic and non-syndromic forms of blindness. In order to gain insight into the pathogenic mechanisms underlying retinal degeneration, we searched for interacting proteins of USH2A isoform B (USH2AisoB) and the _LCA5_-encoded protein lebercilin. We identified a novel isoform of the centrosomal ninein-like protein, hereby named Nlp isoform B (NlpisoB), as a common interactor. Although we identified the capacity of this protein to bind calcium with one of its three EF-hand domains, the interacton with USH2AisoB did not depend on this. Upon expression in ARPE-19 cells, recombinant NlpisoB, lebercilin and USH2AisoB were all found to co-localize at the centrosomes. Staining of retinal sections with specific antibodies against all three proteins revealed their co-localization at the basal bodies of the photoreceptor-connecting cilia. Based on this subcellular localization and the nature of their previously identified binding partners, we hypothesize that the pathogenic mechanisms for LCA and USH show significant overlap and involve defects in ciliogenesis, cilia maintenance and intraflagellar and/or microtubule-based transport. The direct association of NlpisoB with USH2AisoB and lebercilin indicates that Nlp can be considered as a novel candidate gene for USH, LCA and allied retinal ciliopathies.
INTRODUCTION
Usher syndrome (USH) is the most common cause of combined deaf-blindness, with a prevalence of about one in 20 000 in the Caucasian population (1–3). It is a clinically and genetically heterogeneous disorder for which three different types can be distinguished based on the severity and progression of the hearing loss, the presence or absence of vestibular symptoms and the age of onset of retinitis pigmentosa (RP) (4). To date, six different genetic loci have been identified for USH type I (USH1), three loci for USH type II (USH2) and one locus for USH type III (USH3) (reviewed in 5). Mutations have been identified in the genes encoding myosin VIIa, harmonin, cadherin 23, protocadherin 15 and SANS as the underlying defect in USH1b, -c, -d, -f and -g, respectively (6–11). Defects in USH2A, GPR98 (VLGR1) and whirlin are causative for USH2a, -c and -d, respectively (12–15). USH3 is caused by mutations in the clarin-1 encoding USH3A gene (16).
Recently, we and others have shown that all known USH1 and USH2 proteins are present in a protein network, the Usher interactome, in which harmonin and whirlin play the role of key organizers that interconnect the other USH proteins (17–22). Localization studies in rodents revealed that this interactome is present in the stereocilia and the synaptic region of hair cells in the cochlea and in the synaptic region of photoreceptor cells. Except for harmonin, all known USH1 and USH2 proteins are also present at the region of the connecting cilium of photoreceptor cells, a microtubule-based structure which separates the outer segments from the inner segments (21). This region encompasses the connecting cilium, its basal body and centriole as well as the periciliary region surrounding the connecting cilium. In the region of the connecting cilium, the Usher interactome may be recruited via PDZ (postsynaptic density 95, PSD-95; discs large, Dlg; zonula occludens-1, ZO-1) domain-based interactions with whirlin (19,21,23). Based on the localization of the Usher protein network to the region of the connecting cilium and the identification of ciliary abnormalities in photoreceptor cells and sperm cells of several patients with USH (24), the syndrome can be considered as a (retinal) ciliopathy (25). Recent phenotypic data of patients with different Usher subtypes (USH 1B, 1F, 2A or 2C) also suggest that the primary pathogenic insult does not take place at the photoreceptor synapse (26).
Leber congenital amaurosis (LCA) is another clinically and genetically heterogeneous recessive disorder which has been described as a severe form of RP presenting before the age of 1 year with the absence of photoreceptor function (27). Patients present with profound visual loss, nystagmus, poorly reactive pupils and a markedly diminished or non-detectable electro-retinogram (27,28). Defects in the centrosomal and cilia-associated proteins RPGRIP1, CEP290 and lebercilin (encoded by LCA5) have been identified to be associated with LCA (29–31), implicating a role of these proteins in ciliary processes.
At the moment, mutations in several different genes have been described to be the underlying cause of USH and LCA, but the molecular basis of the pathogenic defects of these syndromes still remains largely elusive. Based on similarities in clinical manifestations and the subcellular localization of proteins involved in USH and a number of proteins involved in LCA, the pathogenic mechanisms underlying both disorders might significantly overlap. In order to gain a better insight into the molecular basis of USH and LCA, we searched for novel interacting partners of the recently identified lebercilin (LCA5) and the intracellular region of USH2A isoform B (USH2AisoB) by using yeast two-hybrid screening.
In this study, we show that both lebercilin and the intracellular region of USH2AisoB directly interact with a novel isoform of the second member of the ninein family, the centrosomal ninein-like protein [Nlp isoform B (NlpisoB)], encoded by the Nlp (KIAA0980) gene. NlpisoB is a novel component of the Usher interactome, connecting USH and LCA at the molecular level, and the Nlp gene can therefore be considered as a novel candidate for USH, LCA, and related disorders.
RESULTS
A novel isoform, isoform B, of the centrosome-associated ninein-like protein interacts with USH2A and lebercilin
Yeast two-hybrid screens of an oligo-d(T) primed human retina cDNA library were performed to identify proteins interacting with lebercilin or with the intracellular region of USH2AisoB. Analysis of positive clones that activated all reporter genes revealed a common interactor for both bait proteins. In total, 11 overlapping clones of a novel splice variant of the centrosomal ninein-like protein (Nlp), hereby named NlpisoB, were identified. By using the intracellular region of USH2AisoB as a bait, two clones for NlpisoB were identified. These encode amino acids 598–1033 and 658–1033, respectively. For lebercilin, four Nlp clones encoding amino acids 598–1033 and five clones encoding amino acids 658–1033 were identified. The transcript encoding NlpisoB lacks exon 17 from the originally described Nlp gene, resulting in in-frame skipping of 349 amino acids after residue 734. The specificity of the interaction in the yeast two-hybrid assay was determined by co-transforming pAD-NlpisoB (amino acids 658–1033) with the pBD-Gal4 vector expressing the non-related p63 protein. No interaction was observed performing this control experiment (data not shown).
Bioinformatic analysis of the NlpisoB protein (SMART database; http://smart.embl-heidelberg.de/) identified three regions that are predicted to form EF-hand domains, potentially Ca2+-binding, which are known to be involved in the interaction with γ-tubulin (32). In addition, the C-terminal region was predicted to form an intermediate filament (IF) domain (amino acids 656–925), likely to be involved in protein–protein interactions (Pfam Home Page; http://pfam.sanger.ac.uk/) (Fig. 1A). The IF domain, however, was predicted with a low significance (_E_-value: 5.2 × 10−1). We analysed the spatial and temporal expression pattern of transcripts encoding NlpisoA and NlpisoB by semi-quantitative RT–PCR, using several human fetal and adult tissues. No major differences in expression were observed for isoforms A and B, indicating that both isoforms function in the same tissues (Fig. 1B). However, differences at the cellular level cannot be excluded.
Figure 1.

Schematic representation of the protein structure of NlpisoA and NlpisoB. (A) The predicted EF hands 1, 2 and 3 are formed by amino acids 11–39, 200–228 and 237–265, respectively. Coiled-coil domains (CC) 1, 2, 3 and 4 are formed by amino acids 384–425, 470–579, 621–699 and 1046–1375, respectively, and the predicted IF domain is formed by amino acids 656–925. (B) Semi-quantitative RT–PCR for transcripts encoding NlpisoA and NlpisoB in human fetal and adult tissues. Shown are the samples that were taken after 35 cycles. The results are representative for the samples taken after 25 and 30 cycles. As a control, RT–PCR analysis of the household gene GAPDH was performed. The transcripts for NlpisoA and NlpisoB show a similar distribution, with the strongest expression in fetal cochlea and adult brain, testis and kidney.
Deletion constructs were made of the intracellular region of USH2AisoB and NlpisoB and tested in yeast two-hybrid analysis to determine the regions that are involved in the interaction. By using these deletion constructs, we were able to show that in USH2A, the interacting region is located in the fragment containing amino acids 5124–5196 and in NlpisoB in the fragment containing the predicted IF domain (amino acids 656–925) (Fig. 2A). In order to further pinpoint the domains involved in the interaction between NlpisoB and lebercilin, parts of the lebercilin protein comprising the amino acids 1–95, 96–305, 306–490 and 491–697 were tested in a yeast two-hybrid assay with different parts of NlpisoB. This revealed that the predicted IF domain of NlpisoB specifically interacts with the fragment encompassing the first two coiled-coil domains of lebercilin (amino acids 96–305) (Fig. 2B). To test whether the interactions of USH2AisoB and lebercilin with Nlp are isoform-specific, a liquid β-galactosidase assay was performed using both NlpisoA and NlpisoB together with lebercilin and the intracellular region of USH2AisoB. A specific interaction was observed between both proteins and NlpisoB, whereas no interaction with NlpisoA could be detected (Fig. 2C and D).
Figure 2.

Protein–protein interaction studies. (A) The schematic protein structure of NlpisoB and protein fragments encoded by deletion constructs with the corresponding amino acids are depicted. Yeast two-hybrid analysis showed a specific interaction between the fragment of the intracellular region of USH2AisoB encompassing amino acids 5124–5196 and the predicted IF domain (amino acids 656–925) of NlpisoB. (B) Schematic representation of the protein structure of lebercilin and protein fragments encoded by deletion constructs with the corresponding amino acids. Yeast two-hybrid analysis revealed a specific interaction between the lebercilin domain containing the two N-terminal coiled-coil domains (amino acids 96–305) and the predicted IF domain of NlpisoB (amino acids 656–925). A liquid β-galactosidase assay revealed a specific interaction between NlpisoB and the USH2A_tail (C) and NlpisoB and lebercilin (D). No interaction was observed between USH2A and NlpisoA (C) and lebercilin and NlpisoA (D). aa, amino acids; NA, not assayed.
Co-immunoprecipitations were performed to confirm the interaction between USH2A and NlpisoB. For this purpose, COS-1 cells were co-transfected with constructs encoding HA-tagged NlpisoB and the flag-tagged intracellular region of USH2A (USH2A_tail). From the COS-1 cell lysates we were able to co-immunoprecipitate HA-tagged NlpisoB and flag-tagged USH2A with antibodies against the flag-tag. As a negative control, unrelated flag-tagged STRAD was co-expressed with HA-tagged NlpisoB. As expected, HA-tagged NlpisoB and flag-tagged STRAD did not co-immunoprecipitate (Fig. 3A). The interaction between lebercilin and NlpisoB could be confirmed in a glutathion-S-transferase (GST) pull-down experiment and in co-immunoprecipitation assays. We were able to pull down flag-tagged lebercilin from a COS-1 cell lysate with GST-fused NlpisoB but not with GST alone (Fig. 3B). In addition, we were able to co-immunoprecipitate flag-tagged NlpisoB and HA-tagged lebercilin with antibodies against the HA-tag. As a negative control, unrelated flag-tagged LRRK2 was co-expressed with HA-tagged lebercilin. No LRRK2 was co-immunoprecipitated with lebercilin (Fig. 3C). Also, co-immunoprecipitation assays of endogenous proteins were performed from bovine retinal extracts by using antibodies against lebercilin and Nlp. We were able to co-immunoprecipitate Nlp with an antibody against lebercilin. As a negative control, rabbit IgGs were used (Fig. 3D).
Figure 3.

Co-immunoprecipitation of NlpisoB with the intracellular region of USH2AisoB (USH2A_tail), but not with STRAD. (A) The immunoblot (IB) in the left upper panel shows that HA-NlpisoB (lane 1) co-immunoprecipitated with flag-USH2A_tail, but not with the unrelated protein flag-STRAD (lane 2). Protein input is shown in the left lower panel; the anti-flag immunoprecipitates are shown in the right panel. (B) GST pull-down assays showing that flag-tagged lebercilin was efficiently pulled down by GST-fused NlpisoB, but not by GST alone. The left lane shows 2% input of the protein lysate. (C) Co-immunoprecipitation of lebercilin with NlpisoB, but not with LRRK2. The immunoblot (IB) in the right panel shows that flag-NlpisoB (right lane) co-immunoprecipitated with HA-lebercilin, whereas the unrelated protein flag-LRRK2 (left lane) did not. Protein input is shown in the left panel; the anti-HA immunoprecipitates are shown in the middle panel. (D) Co-immunoprecipitation of endogenous Nlp with lebercilin from bovine retinal extracts, but not with rabbit IgGs. The top panel shows that Nlp was co-immunoprecipitated with lebercilin, indicated by an arrow, whereas it was not with rabbit IgGs. The bottom panel shows that lebercilin was immunoprecipitated with lebercilin-specific antibodies, as indicated by an arrow, but not with rabbit IgGs.
Interaction between USH2A and Nlp is calcium-independent
Because of the predicted presence of three EF-hand domains in the N-terminal half of Nlp, we hypothesized that the interaction between USH2AisoB and NlpisoB might be dependent on the binding of Ca2+ ions to these EF hands, as was shown for the interaction between the EF-hand domain containing proteins centrin and transducin (33). Therefore, we first determined the Ca2+-binding capacity of Nlp. At residue 12 of EF-hand domains, an invariant Glu or Asp provides two oxygens for liganding Ca2+ (bidentate ligand) (34). Based on this, only EF hand 3 would be able to efficiently bind Ca2+ (Fig. 4A). To test this hypothesis, GST-fusion proteins were made of the three predicted EF hands of Nlp. As a positive control, the GST-fused calmodulin 28K subunit was used. We indeed were able to show binding of Ca2+ to Nlp EF hand 3 and not to EF hands 1 and 2 (Fig. 4B). Subsequently, we performed GST pull-down assays in the presence or absence of 2 mm CaCl2 in which HA-tagged full-length NlpisoB was pulled down from a COS-1 cell lysate with GST-USH2A_tail but not with GST alone. Also, no HA-tagged NlpisoB was pulled down with GST-fused NBC3_tail. NBC3 was used as an additional negative control and is a previously described member of the Usher interactome (17). Similar results were obtained for pull-down experiments performed in the presence or absence of calcium, and therefore, there are no indications for calcium-dependence of the interaction (Fig. 4C).
Figure 4.

Analysis of Ca2+-binding properties of Nlp. (A) Multiple protein alignment of the predicted EF-hand domains in Nlp and the consensus sequence for EF-hand domains (SMART database). The invariable D or E at position 12 is boxed and indicated by an arrow and is only present in EF hand 3. (B) A calcium-binding assay showing the specific calcium-binding capacities of Nlp EF hand 3. The 28K subunit of calmodulin was used as a positive control. (C) GST pull-down analysis showing that HA-tagged NlpisoB was efficiently pulled down by GST-fused USH2A_tail in the presence and absence of calcium, but not by GST-NBC3_tail and GST alone. Lanes 1 and 5 show 5% of the input protein lysate.
Nlp co-localizes with USH2AisoB and lebercilin at the photoreceptor cell basal body and centriole
We performed immunohistochemistry for Nlp and USH2AisoB to determine whether both proteins co-localize in the retina. Monoclonal anti-_pan_-centrin antibodies (20H5) were used as a marker for the basal body and connecting cilium. With affinity-purified antibodies against Nlp, the presence of Nlp was detected in the inner segment and at the basal body, shown as a partial co-localization with centrins (Fig. 5B). On retinal cryosections, Nlp and USH2AisoB co-localized in the inner segments and at the region of the connecting cilium (Fig. 5A). In addition, USH2AisoB localized to the outer plexiform layer as was already described (17,19).
Figure 5.

Co-localization of Nlp and USH2A in the retina. (A–A″) Co-immunostaining of USH2A and Nlp in radial cryosections of adult (P20) rat retina by using anti-Nlp antibodies (green signal; A) and anti-USH2A antibodies (red signal; A′) showing co-localization (yellow signal; A″) in the inner segment (IS) and in the region of the connecting cilium (CC). (B) High magnification fluorescence microscopy analysis of double immunofluorescence with anti-Nlp (green) and anti-_pan_-centrin antibodies (red; marker for the ciliary apparatus: connecting cilium, centriole and basal body) in cryosections through the ciliary part of rat photoreceptor cells. Merged images indicate partial co-localization of Nlp with centrins in the centriole and basal body (yellow signal). Pre-embedding immunolabellings of the ciliary region of mouse photoreceptors by antibodies against Nlp (C) and USH2AisoB (D) show a clear staining of the (apical part of) inner segment (IS), the centriole (indicated by an asterisk) and the basal body (BB).
To determine the exact subcellular localization of Nlp in the retina, immunoelectron microscopy was performed. We detected Nlp in the basal body and the centriole of the photoreceptor-connecting cilium as well as in the periciliary region of the apical inner segments of mouse photoreceptor cells (Fig. 5C). Immunoelectron microscopy for USH2AisoB on the retina showed the presence of USH2AisoB in the periciliary region, the connecting cilium, basal body and the centriole of photoreceptor cells (Fig. 5D) (21). Immunohistochemistry on mouse retinal cryosections revealed lebercilin as a component of the photoreceptor-connecting cilium and basal body (29). Thus, our results indicate that Nlp co-localizes with USH2AisoB and also with lebercilin at the photoreceptor cell basal body.
Nlp, lebercilin and USH2A co-localize at the centrosome of ARPE-19 cells
Casenghi et al. (32) have shown that Nlp localizes at the mother centriole of the centrosome in cells during interphase. In order to visualize the interaction between the intracellular domain of USH2A and NlpisoB, we fused these proteins at their N-terminus to enhanced cyan fluorescent protein (eCFP) and monomeric red fluorescent protein (mRFP), respectively. In single transfected human retinal pigment epithelium cells (ARPE-19) expressing NlpisoB, this protein shows a centrosomal localization specifically at one centriole, most probably the mother centriole (Fig. 6A). In mitotic cells, a punctate localization in the cell periphery was observed for NlpisoB (data not shown). In single transfected cells, a nuclear localization (data not shown) (17,19) or a centrosomal localization was observed for eCFP-USH2A (Fig. 6B). In cells co-expressing NlpisoB and USH2A, co-localization was observed at both the mother and the daughter centriole of the cell (Fig. 6D–D″). In addition, overexpression assays in ARPE-19 cells were performed by co-expressing N-terminally fused mRFP-NlpisoB and C-terminally fused lebercilin-eYFP. In single transfected cells, lebercilin-eYFP was localized to the centrosome and microtubule network of the cell, as previously described (29) (Fig. 6C). In cells co-expressing NlpisoB and lebercilin-eYFP, co-localization at the centrosome and non-centrosomal microtubule organization centres (MTOCs) was observed (Fig. 6E–E″).
Figure 6.

Centrosomal localization of NlpisoB, USH2A_tail and lebercilin in ARPE-19 cells. When expressed alone, mRFP–NlpisoB (red signal) was localized to the mother centriole of the centrosome (A), eCFP-USH2A_tail (green signal) was localized to the nucleus and the centrosome (indicated by an arrow and in the inlay) (B), whereas eYFP-lebercilin (green signal) was localized at the centrosome and the microtubule network of the cell (C). After co-expression of NlpisoB and USH2A, both proteins were localized at the mother and daughter centriole of the centrosome, confirming the interaction between both proteins (D–D″). Co-expression of NlpisoB and lebercilin showed co-localization of both proteins at the centrosome and non-centrosomal MTOCs (yellow signal; E″). Lebercilin in addition partly localized at the microtubule network (E–E″). Nuclei were stained with DAPI (blue signal).
To determine the subcellular localization of endogenous Nlp and lebercilin, ARPE-19 cells were induced to form primary cilia by serum starvation and subsequently used for immunohistochemical stainings with antibodies directed against Nlp and lebercilin. Both proteins could be detected at the base of the cilium, most probably the basal body (Fig. 7A and B). Interestingly, we detected both lebercilin and Nlp at the midbody during cytokinesis (Fig. 7C and D). After telophase, the mother centrioles which are present at the midbody develop into the basal bodies of the newly formed cells (35). No endogenous USH2A was detected in ARPE-19 cells.
Figure 7.

Localization of endogenously expressed Nlp and lebercilin in ARPE-19 cells by immunocytochemistry using the anti-Nlp and anti-lebercilin antibodies (green signals) and anti-acetylated tubulin antibodies as an axoneme and microtubule marker (red signal). Nlp (A) and lebercilin (B) were present at the basal body of primary cilia and both proteins were found at the midbody region of ARPE-19 cells during telophase (C and D). Nuclei were stained with DAPI (blue signal).
Lebercilin mutations affect the interaction with NlpisoB
Recently, we have identified truncating lebercilin mutations in LCA patients (29). We addressed the biologic relevance of the interaction between lebercilin and NlpisoB by testing the effect of two of these mutations, p.Q279X and p.P493TfsX1, on the association with NlpisoB. In a quantitative yeast two-hybrid interaction assay, the p.Q279X mutation significantly enhanced the interaction. The interaction of NlpisoB and the p.P493TfsX1 mutant was found to be completely abolished (Fig. 8A).
Figure 8.
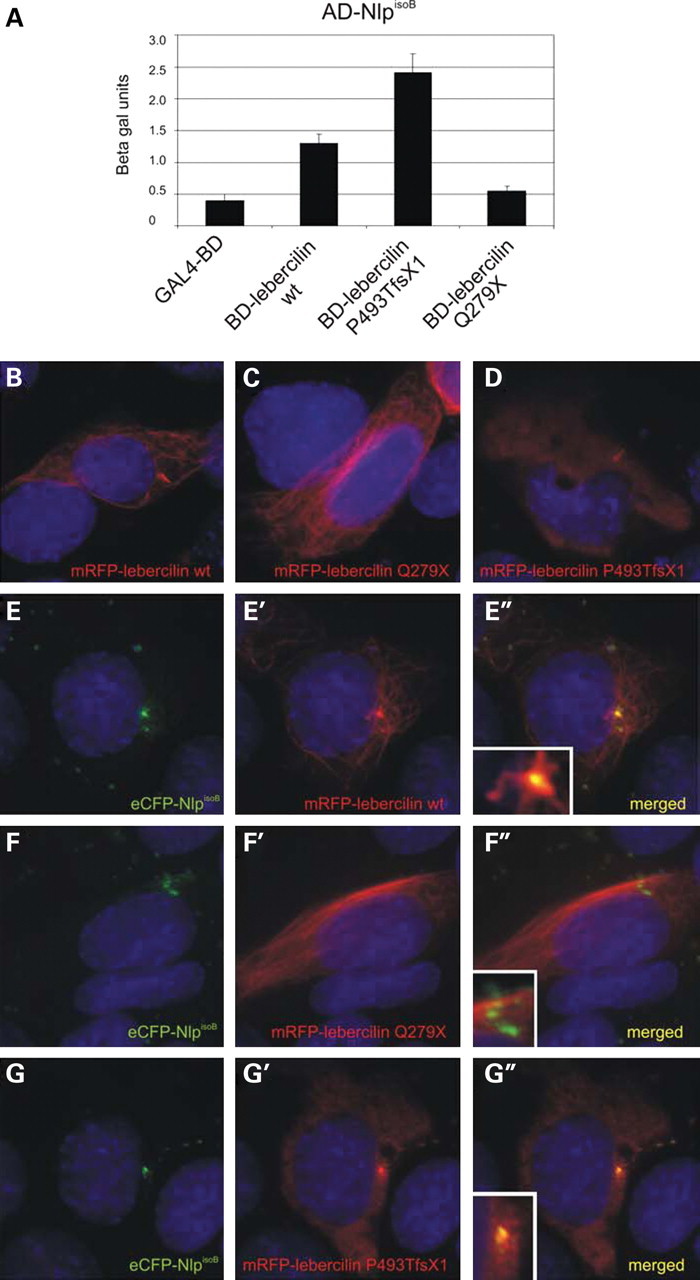
Mutations in lebercilin affect the interaction with NlpisoB. A quantitative liquid β-galactosidase assay shows an enhanced interaction of NlpisoB and lebercilinQ279X when compared with wild-type lebercilin and a reduced interaction of NlpisoB and lebercilinP493TfsX1 (A). When expressed alone in ARPE-19 cells, mRFP–lebercilin wild-type (red signal) was localized to the basal body, the primary cilium and the microtubule network of the cell (B), mRFP-lebercilinQ279X was localized to the basal body and primary cilium (C) and mRFP-lebercilinP493TfsX1 was localized to the microtubule network of the cell (D). Co-expression of NlpisoB and lebercilin showed co-localization of both proteins at the basal body (yellow signal; E″, inset). In addition, lebercilin localized to the primary cilium and the microtubule network in the cell periphery (red signal; E′–E″, inset). Upon co-expression of NlpisoB and lebercilinQ279X, both proteins co-localize at the basal body (yellow signal; F″, inset). The mutant lebercilin was not found in the primary cilium (F′–F″, inset). After co-expression of NlpisoB and lebercilinP493TfsX1, no co-localization of these proteins was observed (G–G″, inset). Nuclei were stained with DAPI (blue signal).
The subcellular localization of the lebercilin mutants was studied upon expression in ciliated ARPE-19 cells. When expressed alone, mRFP-tagged lebercilin localizes to the basal body, the primary cilium and the microtubule network of the cell (Fig. 8B) (29). The mRFP-lebercilinQ279X mutant does not associate with the microtubule network any longer but is present in the primary cilium and its basal body (Fig. 8C). In contrast, mRFP-lebercilinP493TfsX1 only localizes to the microtubule network in the cell periphery but no longer to the cilium and the basal body (Fig. 8D). Co-expression of NlpisoB and lebercilin showed a clear co-localization, specifically at the basal body (Fig. 8E–E″). In the primary cilium and the microtubule network, only lebercilin was present. As indicated by the yeast two-hybrid assay, the interaction between NlpisoB and lebercilinQ279X is enhanced. When co-expressed with NlpisoB, lebercilinQ279X is recruited to the basal body, but is no longer observed in the primary cilium (Fig. 8F–F″). Upon co-expression of NlpisoB and lebercilinP493TfsX1, no co-localization was observed, confirming the loss of interaction between the two proteins (Fig. 8G–G″). These data indicate that truncating mutations in lebercilin severely affect the association with Nlp at the basal body. However, downregulation of both endogenous Nlp and LCA5 by RNAi in ciliated ARPE-19 cells does not result in altered protein localization of Nlp or lebercilin (Supplementary Material, Fig. S2), suggesting that additional binding partners are involved in their localization at the basal body.
DISCUSSION
In this study, we demonstrate that a novel isoform of the centrosome-associated ninein-like protein, NlpisoB, specifically interacts with lebercilin and the cytoplasmic region of USH2A, thereby linking USH and LCA at the molecular level. Previous analysis of Xenopus laevis Nlp already suggested the presence of two Nlp isoforms, which correspond with human NlpisoA and NlpisoB (36). Nlp is the second member of the ninein protein family and has a 37% sequence identity with ninein (32). Nlp is predominantly present in the mother centriole of the centrosome and in the basal body of primary cilia in cultured cells and is involved in microtubule-nucleation, -anchoring and -outgrowth (32,36,37). However, it remains to be elucidated which isoform of Nlp is present and functions at these subcellular structures. The interaction between USH2AisoB, lebercilin and NlpisoB and their co-localization in the basal bodies, the centrioles and the periciliary compartments of photoreceptor cells show that at least NlpisoB molecules are present there.
Basal bodies and associated centrioles are found at the base of cilia and serve as a nucleation site for the axonemal and cytoplasmic microtubules, respectively (38,39). The photoreceptor cell outer segment is regarded as a highly specialized cilium corresponding to the ciliary shaft of a prototypic cilium (38,39). The connecting cilium then correlates with the short junction between the basal body and the axoneme of a prototypic cilium, the transition zone (38). The presence of Nlp at the basal bodies and centrioles of the ciliary apparatus of photoreceptor cells matches with the previously determined localization of Nlp in the basal body of ciliated cells (36). Based on the knowledge on the Nlp function in mitotic cells (32,36,37), we propose that Nlp functions in the development and maintenance of the connecting cilium and the outer segment and in the establishment of a microtubule network in (the apical part of) the inner segment in differentiated photoreceptor cells.
In addition to the physical interaction of both USH2AisoB and lebercilin with NlpisoB, similarities in clinical manifestations between USH and LCA suggest a significant overlap in the pathogenic mechanisms underlying both disorders. Based on the subcellular localization and the current knowledge on USH2AisoB and lebercilin, these pathogenic mechanisms are likely to include ciliary dysfunction. Post-mortem observation of connecting cilium defects in the retina and the identification of sperm abnormalities in some USH patients (reviewed in 40), as well as the involvement of other cilia-associated proteins in non-syndromic forms of retinal degeneration and USH, contribute to this hypothesis (19,24,29,30,41,42). Two proteins involved in (non-)syndromic RP, RPGR and RP1, and even the LCA-associated proteins, RPGRIP1 and CEP290, have been shown to localize to the connecting cilia of photoreceptor cells, emphasizing the role of impaired cilia function in the pathogenesis of retinal degeneration (30,43–46).
The connecting cilium of photoreceptor cells connects the inner and outer segments. Within the outer segments the actual phototransduction takes place. Since protein synthesis does not occur in the outer segments, the proteins essential for phototransduction and assembly and maintenance of the axonemal and outer segment structure are transported through the connecting cilium between the inner segment and the outer segment, analogous to the intraflagellar transport (IFT) (47,48). In cilia and flagella, IFT complexes are assembled near the basal body and transported along the axoneme by the heterotrimeric kinesin II (KIF3A/3B/KAP3) and the homodimeric KIF17 in an anterograde direction and back to the basal body region by cytoplasmic dynein 2/1b (49–52). In photoreceptor cells, IFT20, IFT52, IFT57 and IFT88 molecules and both kinesin motors are thought to be present in an anterograde transport complex (53–55). Dysfunction of the components of the IFT transport complex lead to mislocalization of structural proteins and proteins involved in the phototransduction and subsequently lead to retinal degeneration (53,56). This is corroborated by the development of both polycystic kidney disease in addition to retinal degeneration in mice after the introduction of a hypermorphic mutation in IFT88 (53,57). Also, photoreceptor-specific silencing of KIF3A leads to mislocalization of opsin and arrestin, proteins involved in phototransduction (56). Similar defects were observed in mice lacking BBS4, a gene involved in the Bardet–Biedl syndrome (58). BBS4 is a member of the recently discovered BBSome, which is required for ciliogenesis and is hypothesized to function in IFT and vesicular transport to the cilium (59). The evaluation of the cellular function of USH proteins in photoreceptors suggests the participation of these proteins in the transport through the connecting cilium (21,48,60,61). Also, there is growing evidence that BBS molecules, IFT proteins and USH proteins participate in microtubule-based vesicular transport through the cytoplasm (21,53,59,62). The molecular links of NlpisoB with the USH2AisoB and lebercilin proteins qualified these as common denominators in the associated retinal protein network, suggesting that common molecular processes are disrupted in the retinas of USH2A and LCA5 patients. We indeed were able to determine that protein truncating mutations in lebercilin affect the interaction with Nlp, but as downregulation of expression of any of these proteins by RNAi had no effect on the localization of the interacting partner, the physiological defect is more complex and remains to be identified. The USH2A-associated protein network, the lebercilin interactome (29) and the Nlp interactors p150glued and Polo-like kinase 1 (Plk1) (32,36,37) provide multiple links with centrosomal and ciliary processes and pathways that are now potentially connected through Nlp.
Recently, we and others showed the presence of the Usher protein network in a periciliary collar-like structure at the apical part of the photoreceptor inner segments, analogous to the periciliary ridge complex of Xenopus photoreceptor cells, and the Usher proteins were hypothesized to function in the transport and docking of vesicles containing proteins for the outer segment (21,23,63). These cargo vesicles originate from the _trans_-Golgi network of the photoreceptor cells and are thought to be transported along microtubules by cytoplasmic dynein through the inner segment to the apical membrane (21,64). Nlp might function in this vesicular transport by the direct association with the dynactin p150glued subunit of the dynein–dynactin motor complex (37), explaining the presence of Nlp in the inner segments of photoreceptor cells. The periciliary region and also the basal body and centriole are thought to serve as a docking site from where ciliary proteins (e.g. IFT proteins and proteins involved in signalling) are further distributed (65). In such a model, Nlp would function as the molecular switch between intracellular and IFT transport. The effect of mutations in lebercilin on its interaction with NlpisoB and on its localization in the cilium is in line with this model.
Interestingly, Nlp is known to be phosphorylated by Plk1 (32,36,37), which is a key regulator in centrosome function. Plk1 also phosphorylates nucleophosmin (66), a centrosome-associated protein that acts in nucleocytoplasmic shuttling (67) and is a member of the previously identified lebercilin interactome (29). In addition, nucleophosmin associates with RPGR-ORF15, a protein involved in X-linked retinal degeneration (68). The Nlp-interacting protein p150glued (37) was found in a protein complex with RPGR-ORF15 and with lebercilin (29,69). The direct or indirect association of Nlp with several protein complexes in the region of the photoreceptor-connecting cilium stresses the importance of this protein in photoreceptor cell function.
In conclusion, our data show that USH and LCA are molecularly linked by the direct association of USH2AisoB and lebercilin with a novel isoform of Nlp, NlpisoB. Co-localization of these proteins at the basal body of photoreceptor cells and the current knowledge on the function of the existing Usher protein network(s) point towards an interdependent function for NlpisoB, USH2AisoB and lebercilin, possibly in cytoplasmic trafficking and ciliary transport of proteins involved in photoreception. We propose that at the basal body, NlpisoB could function as a molecular hinge connecting cytoplasmic transport mechanisms to the IFT transport machinery, suggesting an important role for Nlp in photoreceptor cell function. The central position of Nlp in the protein network indicates that Nlp can be considered as an excellent candidate gene for USH, LCA or other (retinal) ciliopathies. However, no pathogenic mutations have been identified so far.
MATERIAL AND METHODS
Animals and tissues
In this study, mature Wistar rats and C57BL/6J mice housed in standard cages and receiving water and food ad libitum were used. Animal experiments were conducted in accordance with international and institutional guidelines. Bovine retinas were dissected from eye balls obtained from the local slaughterhouse (21).
Plasmids and antibodies
Affinity-purified polyclonal antibodies directed against Nlp were described earlier (32). Monoclonal antibodies recognizing centrins 1–4 (20H5), polyclonal antibodies directed against the cytoplasmic region of USH2A and affinity-purified polyclonal antibodies directed against lebercilin were described previously (17,19,29). The immunohistochemical stainings of Nlp and USH2A were specific. No staining was observed after pre-adsorption of the primary antibodies with the corresponding peptide epitope. In addition, primary Nlp antibodies did not recognize GFP-tagged ninein after western blot analysis (Supplementary data, S1). Anti-flag, anti-HA, anti-acetylated tubulin and anti-gamma tubulin antibodies were purchased from Sigma (Germany). Anti-HA beads were purchased from Roche (Germany). Secondary antibodies for immunohistochemistry and western blot analysis were purchased from Molecular Probes—Invitrogen (USA), Rockland (USA) and Jackson ImmunoResearch Laboratories (USA). cDNA encoding (parts of) the cytoplasmic region of human USH2A (GenBank NP_996816) (amino acids 5064–5202, 5064–5168, 5064–5196, 5124–5196, 5124–5202 and 5169–5202) were cloned in the pDONR201 vector using the Gateway cloning system (Invitrogen) according to manufacturers' instructions. cDNA fragments of the human Nlp gene were amplified by using IMAGE clone IRATp970C1131D (RZPD, Germany) as a template. By using the Gateway technology (Invitrogen), cDNAs encoding human full-length NlpisoB (amino acids 1–1033) (GenBank EU718622), NlpisoA (amino acids 1–1382) (GenBank NM_025176), the predicted EF-hand domains (amino acids 11–39, 200–228 and 237–265), the predicted IF domain (amino acids 656–925) of NlpisoB and deletion constructs for NlpisoB encoding amino acids 599–1033, 656–1033 and 656–825 were cloned in the pDONR201 vector. Lebercilin fragments (GenBank NP_859065) encoding amino acids 1–95, 96–305, 306–490 and 491–697 were amplified by PCR, using pDONR201-lebercilinfl as a template, and cloned in pDONR201 (29).
Yeast two-hybrid analysis
A GAL4-based yeast two-hybrid system (HybriZAP, Stratagene, USA) was used to identify proteins that interact with the cytoplasmic region of USH2AisoB (amino acids 5064–5202) and proteins that interact with lebercilin (amino acids 1–697), using methods previously described (70) with minor variations. Yeast strain PJ69-4α (71) was used as a host, which carried the HIS3 (histidine), ADE2 (adenine), MEL1 (α-galactosidase) and LacZ (β-galactosidase) reporter genes. The DNA-binding domain fused to the human USH2A cytoplasmic region (pBD-USH2A_tail) and to the full-length lebercilin (pBD-lebercilinfl) was used as a bait for screening a human oligo-d(T) primed retina cDNA library containing 1.9 × 106 primary clones fused to the activation domain (pAD). In total, 1.1 × 107 clones (USH2A) and 5.0 × 107 clones (lebercilin) were plated on amino acid dropout plates lacking Trp, Leu and His (SD -WLH plates), containing 1 mm 3-aminotriazol, and selected for growth. Clones were then patched on medium additionally lacking adenine (SD-WLHA plates) and selected for growth and α-galactosidase activity by the activation of the MEL1 reporter gene. The latter was done using 20 µg/ml of the chromogenic substrate 5-bromo-4-chloro-3-indolyl α-d-galactopyranoside (X-α-Gal) in the dropout plates and selecting the yeast cells that developed a blue-green colour due to hydrolysis of X-α-Gal by the secreted α-galactosidase enzyme. Further selection of positive clones was based on β-galactosidase activity by the activation of the LacZ reporter gene, which was detected by a filter lift assay as previously described (70).
Liquid β-galactosidase assay
Three independent clones of PJ69-4α yeast cells co-transformed with pAD-NlpisoA full length or NlpisoB full length and pBD-USH2A_tail or pBD-lebercilin full length were cultured in selective medium lacking Trp and Leu (SD-WL). After two overnight incubation steps at 30°C, the optical density of the cultures was determined at a wavelength of 600 nm. Cell lysis and subsequent colourimetric reactions were performed by using the Yeast β-galactosidase assay kit (Pierce, USA) according to the manufacturer's instructions. Absorbance was measured at a wavelength of 420 nm.
Expression analysis
The expression of NlpisoA and NlpisoB was examined by performing semi-quantitative RT–PCR analysis on RNA from human fetal and adult tissues as described before (72). Primers used for the detection of the transcripts encoding NlpisoA are 5′-GAGGGGGAGACCAAAATAGC-3′ and 5′-TCTGAATGGTCACACGATGC-3′. For detection of NlpisoB, the following primers were used: 5′-ACCTGCAGCAGATCAGACTG-3′ and 5′-TGATTTGGTCACTCTGCTG-3′. Samples were taken after 25, 30 and 35 cycles.
Calcium-binding assay
GST-fusion proteins of the predicted Nlp EF hands 1 (amino acids 11–39), 2 (amino acids 200–228) and 3 (amino acids 237–265) were produced by transforming BL21-DE3 cells with pDEST15-Nlp EF hands 1, 2 and 3. Pre-cleared lysates were separated on pre-casted 4–12% NuPage gradient gels (Invitrogen) and subsequently blotted onto nitrocellulose membranes. The membranes were incubated for 10 minutes at room temperature with 10–20 µCi/l 45CaCl2 (New England Nuclear, USA) in 10 mm imidazole, pH 6.8, and 60 mm KCl. The blots were washed twice for 5 minutes with 50% ethanol, dried and subsequently exposed to a radiation sensitive film (Kodak, Germany) (73).
GST pull-down assay
In order to produce GST-fusion proteins, BL21-DE3 cells were transformed with pDEST15-USH2A_tail (amino acids 5064–5202), pDEST15-NBC3_tail (amino acids 1119–1214) and pDEST15-NlpisoB (amino acids 1–1033). Cells were induced at 30°C for 4 h with 0.5 mm IPTG and subsequently lysed with STE buffer (1% Sarkosyl, 1% Triton X-100) supplemented with protease inhibitor cocktail (Roche). Lysates were pre-cleared and incubated at 4°C for 16 h with glutathione Sepharose 4B beads (Amersham Biosciences, USA). Beads with bound fusion proteins were washed twice with lysis buffer and three times with TBS with 1% Triton-X-100 and 2 mm DTT. During each washing step, samples were incubated on a rotating wheel at 4°C for 5 min. The amount of bound GST-fusion protein was verified on a 10% SDS–PAGE gel stained with Gelcode Blue Stain Reagent (Pierce). HA-tagged NlpisoB, 3×flag-tagged lebercilin, 3×flag-tagged USH2A_tail and 3×flag-tagged NBC3_tail were produced by transfecting COS-1 cells with, respectively, pcDNA3-HA-NlpisoB, p3×flag-lebercilin, p3×flag-USH2A_tail and p3×flag-NBC3_tail using Nucleofector kit V (Amaxa, USA) and program A-24 according to the manufacturer's instructions. The pre-cleared supernatants were incubated overnight at 4°C in the presence or absence of 2 mm CaCl2 with equal amounts of blocked beads with GST or beads with GST-fusion proteins. After three washes with lysis buffer, the beads were boiled in 1× SDS loading buffer. Protein complexes were resolved on 4–12% NuPage gradient gels (Invitrogen). For western blot analysis, proteins were electrophoretically transferred onto nitrocellulose membranes, blocked with 5% non-fat dry milk (Biorad, Germany) in PBST (0.1% Tween) and analysed with the appropriate primary and secondary antibodies in 0.5% milk in PBST. Bands were visualized using the Odyssey Infrared Imaging System (LI-COR, USA). Tagged molecules were detected by anti-HA or anti-flag mono- or polyclonal antibodies. As secondary antibodies IRDye800 goat-anti-mouse IgG (Rockland) and Alexa Fluor 680 goat-anti-rabbit IgG were used (Molecular Probes, USA).
Co-immunoprecipitation from COS-1 cells
HA-tagged NlpisoB and HA-tagged lebercilin were expressed by using the mammalian expression vector pcDNA3-HA/DEST. Flag-tagged intracellular region of USH2A, flag-tagged NlpisoB and flag-tagged LRRK2 were expressed by using p3xflag-CMV/DEST from the Gateway cloning system (Invitrogen). Both plasmids contain a CMV promoter. COS-1 cells were (co-)transfected by using Effectene (Qiagen, Germany) according to the manufacturer's instructions. Thirty hours after transfection, cells were washed with PBS and subsequently lysed on ice in lysis buffer (50 mm Tris–HCl pH 7.5, 150 mm NaCl and 0.5% Triton X-100) supplemented with complete protease inhibitor cocktail (Roche). HA-tagged NlpisoB and lebercilin were immunoprecipitated from cleared lysates overnight at 4°C by using rat monoclonal anti-HA beads (Roche), whereas flag-tagged USH2A_tail, NlpisoB and LRRK2 were immunoprecipitated by using polyclonal anti-flag antibodies (Sigma) and Protein A/G PLUS-Sepharose (Santa Cruz Biotechnology, USA). After four washes in lysis buffer, the protein complexes were resolved on 4–12% NuPage gradient gels (Invitrogen) and subsequently analysed on immunoblots as described for the GST pull-down assay. Bands were visualized by using the Odyssey Infrared Imaging System (LI-COR). Tagged molecules were detected by anti-HA or anti-flag mono- or polyclonal antibodies. As secondary antibodies IRDye800 goat-anti-mouse IgG (Rockland) and Alexa Fluor 680 goat-anti-rabbit IgG were used (Molecular Probes).
Co-immunoprecipitations from bovine retinal extracts
For immunoprecipitations, bovine retinas from a local slaughterhouse were used. Retinas were lysed by sonification (two times, 30 s) in lysis buffer (50 mm Tris–HCl pH 7.4, 150 mm NaCl, 0.5% Nonidet P-40 and 1 mm natrium-orthovanadate) supplemented with complete protease inhibitor cocktail (Roche). Lysates were pre-cleared and incubated for 16 h at 4°C with mouse monoclonal anti-rabbit IgGs, rabbit polyclonal Nlp antibodies or polyclonal rabbit lebercilin antibodies. Protein-antibody complexes were coupled to Protein A/G PLUS-Sepharose beads (Santa Cruz) for 2 h at 4°C. After incubations, the beads were pelleted and washed three times with lysis buffer. Beads were boiled and proteins were resolved on 4–12% NuPage gradient gels (Invitrogen) and subsequently analysed on immunoblots as described for GST pull-down. Bands were visualized using the Odyssey Infrared Imaging System (LI-COR).
Co-localization analyses in ARPE-19 cells
To determine the cellular localization of the cytoplasmic region of human USH2A, full-length lebercilin and full-length NlpisoB in ARPE-19 cells, cDNAs encoding the region of USH2A in pDEST501 were cloned by using the Gateway cloning technology (Invitrogen), resulting in N-terminally fused eCFP-USH2A. Nlp was cloned in pDEST733, resulting in N-terminally fused mRFP-NlpisoB. Lebercilin was cloned in pDEST504, resulting in C-terminally fused lebercilin-eYFP. ARPE-19 cells were co-transfected with pDEST733-NlpisoB and pDEST501-USH2A_tail or pDEST504-lebercilin by using Effectene (Qiagen) according to the manufacturer's instructions. Twenty hours after transfection, cells were washed with PBS, fixed with 4% paraformaldehyde and mounted with Vectashield containing DAPI (Vector Laboratories, Inc., UK). Images were taken with an Axioplan2 Imaging fluorescence microscope (Zeiss, Germany) equipped with a DC350FX camera (Zeiss) and processed using Adobe Photoshop (Adobe Systems, USA).
Double immunofluorescence labelling of rat retinas
Unfixed eyes of 20-day-old (P20) Wistar rats were isolated and frozen in melting isopentane. Cryosections were made at a thickness of 7 µm and treated with 0.01% Tween-20 in PBS, followed by a blocking step with a blocking solution (0.1% ovalbumin, 0.5% fish gelatin in PBS). Antibodies diluted in blocking solution were incubated overnight at 4°C. Secondary antibodies were also diluted in the blocking solution and incubated in the dark for 1 h. Sections were embedded with Prolong Gold Anti-fade (Molecular Probes). Pictures were made with an Axioskop2 Mot plus fluorescence microscope (Zeiss) equipped with an AxioCam MRC5 camera (Zeiss). Images were processed using Axiovision 4.3 (Zeiss) and Adobe Photoshop (Adobe Systems).
Pre-embedding immunoelectron microscopy
Immunoelectron microscopy was performed on isolated mouse eyes as previously described (21). In short, vibratome sections through mouse retina were stained by primary antibodies against Nlp and visualized by appropriate secondary antibodies (Vectastain ABC-Kit, Vector, UK). After fixation with 0.5% OsO4, specimens were embedded in araldite and ultrathin sections were analysed with an FEI Tecnai 12 Biotwin transmission electron microscope (FEI, The Netherlands).
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
FUNDING
This work was supported by grants from the British Retinitis Pigmentosa Society (to H.K. and R.R.), the DFG (to U.W.), Forschung contra Blindheit—Initative Usher Syndrome (to H.K., T.M. and U.W.), ProRetina Deutschland (to U.W. and R.R.), the FAUN-Stiftung, Nürnberg (U.W.), the Heinsius Houbolt Foundation (to H.K.), The Netherlands Organisation for Scientific Research (VIDI 917.86.396 to R.R.), the European Commission IP ‘EVI-GenoRet’ LSHG-CT-2005-512036, the Foundation for Retinal Research, the Algemene Nederlandse Vereniging ter Voorkoming van Blindheid, the Stichting Blindenhulp, the Stichting OOG and the Rotterdamse Vereniging Blindenbelangen (to F.P.M.C. and R.R.).
Supplementary Material
[Supplementary Data]
ACKNOWLEDGEMENTS
The authors would like to thank Jaap Oostrik and Saskia van der Velde-Visser and Elisabeth Sehn for technical assistance, Dr Pascal Duijf for providing us with the pBD-p63 vector and Dr Erich Nigg for providing us with the antibody against Nlp.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Marazita M.L., Ploughman L.M., Rawlings B., Remington E., Arnos K.S., Nance W.E. Genetic epidemiological studies of early-onset deafness in the U.S. school-age population. Am. J. Med. Genet. 1993;46:486–491. doi: 10.1002/ajmg.1320460504. [DOI] [PubMed] [Google Scholar]
- 2.Spandau U.H., Rohrschneider K. Prevalence and geographical distribution of Usher syndrome in Germany. Graefes Arch. Clin. Exp. Ophthalmol. 2002;240:495–498. doi: 10.1007/s00417-002-0485-8. [DOI] [PubMed] [Google Scholar]
- 3.Rosenberg T., Haim M., Hauch A.M., Parving A. The prevalence of Usher syndrome and other retinal dystrophy-hearing impairment associations. Clin. Genet. 1997;51:314–321. doi: 10.1111/j.1399-0004.1997.tb02480.x. [DOI] [PubMed] [Google Scholar]
- 4.Smith R.J., Berlin C.I., Hejtmancik J.F., Keats B.J., Kimberling W.J., Lewis R.A., Moller C.G., Pelias M.Z., Tranebjaerg L. Clinical diagnosis of the Usher syndromes. Usher Syndrome Consortium. Am. J. Med. Genet. 1994;50:32–38. doi: 10.1002/ajmg.1320500107. [DOI] [PubMed] [Google Scholar]
- 5.Kremer H., van Wijk E., Maerker T., Wolfrum U., Roepman R. Usher syndrome: molecular links of pathogenesis, proteins and pathways. Hum. Mol. Genet. 2006;15 (Spec no. 2):R262–R270. doi: 10.1093/hmg/ddl205. [DOI] [PubMed] [Google Scholar]
- 6.Weil D., Blanchard S., Kaplan J., Guilford P., Gibson F., Walsh J., Mburu P., Varela A., Levilliers J., Weston M.D. Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature. 1995;374:60–61. doi: 10.1038/374060a0. [DOI] [PubMed] [Google Scholar]
- 7.Verpy E., Leibovici M., Zwaenepoel I., Liu X.Z., Gal A., Salem N., Mansour A., Blanchard S., Kobayashi I., Keats B.J., Slim R., Petit C. A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies Usher syndrome type 1C. Nat. Genet. 2000;26:51–55. doi: 10.1038/79171. [DOI] [PubMed] [Google Scholar]
- 8.Bork J.M., Peters L.M., Riazuddin S., Bernstein S.L., Ahmed Z.M., Ness S.L., Polomeno R., Ramesh A., Schloss M., Srisailpathy C.R., et al. Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am. J. Hum. Genet. 2001;68:26–37. doi: 10.1086/316954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahmed Z.M., Riazuddin S., Bernstein S.L., Ahmed Z., Khan S., Griffith A.J., Morell R.J., Friedman T.B., Riazuddin S., Wilcox E.R. Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am. J. Hum. Genet. 2001;69:25–34. doi: 10.1086/321277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weil D., El Amraoui A., Masmoudi S., Mustapha M., Kikkawa Y., Laine S., Delmaghani S., Adato A., Nadifi S., Zina Z.B., et al. Usher syndrome type I G (USH1G) is caused by mutations in the gene encoding SANS, a protein that associates with the USH1C protein, harmonin. Hum. Mol. Genet. 2003;12:463–471. doi: 10.1093/hmg/ddg051. [DOI] [PubMed] [Google Scholar]
- 11.Bolz H., von Brederlow B., Ramirez A., Bryda E.C., Kutsche K., Nothwang H.G., Seeliger M., del C.-S., Vila M.C., Molina O.P., Gal A., Kubisch C. Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nat. Genet. 2001;27:108–112. doi: 10.1038/83667. [DOI] [PubMed] [Google Scholar]
- 12.van Wijk E., Pennings R.J., te Brinke H., Claassen A., Yntema H.G., Hoefsloot L.H., Cremers F.P.M., Cremers C.W.R.J., Kremer H. Identification of 51 novel exons of the Usher syndrome type 2A (USH2A) gene that encode multiple conserved functional domains and that are mutated in patients with Usher syndrome type II. Am. J. Hum. Genet. 2004;74:738–744. doi: 10.1086/383096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eudy J.D., Weston M.D., Yao S., Hoover D.M., Rehm H.L., Ma-Edmonds M., Yan D., Ahmad I., Cheng J.J., Ayuso C., et al. Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science. 1998;280:1753–1757. doi: 10.1126/science.280.5370.1753. [DOI] [PubMed] [Google Scholar]
- 14.Weston M.D., Luijendijk M.W., Humphrey K.D., Moller C., Kimberling W.J. Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II. Am. J. Hum. Genet. 2004;74:357–366. doi: 10.1086/381685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ebermann I., Scholl H.P., Charbel I.P., Becirovic E., Lamprecht J., Jurklies B., Millan J.M., Aller E., Mitter D., Bolz H. A novel gene for Usher syndrome type 2: mutations in the long isoform of whirlin are associated with retinitis pigmentosa and sensorineural hearing loss. Hum. Genet. 2007;121:203–211. doi: 10.1007/s00439-006-0304-0. [DOI] [PubMed] [Google Scholar]
- 16.Joensuu T., Hamalainen R., Yuan B., Johnson C., Tegelberg S., Gasparini P., Zelante L., Pirvola U., Pakarinen L., Lehesjoki A.E., de la Chapelle A., Sankila E.M. Mutations in a novel gene with transmembrane domains underlie Usher syndrome type 3. Am. J. Hum. Genet. 2001;69:673–684. doi: 10.1086/323610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reiners J., van Wijk E., Maerker T., Zimmermann U., Jurgens K., te Brinke H., Overlack N., Roepman R., Knipper M., Kremer H., Wolfrum U. Scaffold protein harmonin (USH1C) provides molecular links between Usher syndrome type 1 and type 2. Hum. Mol. Genet. 2005;14:3933–3943. doi: 10.1093/hmg/ddi417. [DOI] [PubMed] [Google Scholar]
- 18.Adato A., Michel V., Kikkawa Y., Reiners J., Alagramam K.N., Weil D., Yonekawa H., Wolfrum U., El Amraoui A., Petit C. Interactions in the network of Usher syndrome type 1 proteins. Hum. Mol. Genet. 2005;14:347–356. doi: 10.1093/hmg/ddi031. [DOI] [PubMed] [Google Scholar]
- 19.van Wijk E., van der Zwaag B., Peters T., Zimmermann U., te Brinke H., Kersten F.F.J., Maerker T., Aller E., Hoefsloot L.H., Cremers C.W.R.J., et al. The DFNB31 gene product whirlin connects to the Usher protein network in the cochlea and retina by direct association with USH2A and VLGR1. Hum. Mol. Genet. 2006;15:751–765. doi: 10.1093/hmg/ddi490. [DOI] [PubMed] [Google Scholar]
- 20.Boeda B., El Amraoui A., Bahloul A., Goodyear R., Daviet L., Blanchard S., Perfettini I., Fath K.R., Shorte S., Reiners J., et al. Myosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle. EMBO J. 2002;21:6689–6699. doi: 10.1093/emboj/cdf689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maerker T., van Wijk E., Overlack N., Kersten F.F.J., McGee J., Goldmann T., Sehn E., Roepman R., Walsh E.J., Kremer H., Wolfrum U. A novel Usher protein network at the periciliary reloading point between molecular transport machineries in vertebrate photoreceptor cells. Hum. Mol. Genet. 2008;17:71–86. doi: 10.1093/hmg/ddm285. [DOI] [PubMed] [Google Scholar]
- 22.Adato A., Lefevre G., Delprat B., Michel V., Michalski N., Chardenoux S., Weil D., El Amraoui A., Petit C. Usherin, the defective protein in Usher syndrome type IIA, is likely to be a component of interstereocilia ankle links in the inner ear sensory cells. Hum. Mol. Genet. 2005;14:3921–3932. doi: 10.1093/hmg/ddi416. [DOI] [PubMed] [Google Scholar]
- 23.Liu X., Bulgakov O.V., Darrow K.N., Pawlyk B., Adamian M., Liberman M.C., Li T. Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells. Proc. Natl Acad. Sci. USA. 2007;104:4413–4418. doi: 10.1073/pnas.0610950104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hunter D.G., Fishman G.A., Mehta R.S., Kretzer F.L. Abnormal sperm and photoreceptor axonemes in Usher's syndrome. Arch. Ophthalmol. 1986;104:385–389. doi: 10.1001/archopht.1986.01050150085033. [DOI] [PubMed] [Google Scholar]
- 25.Adams N.A., Awadein A., Toma H.S. The retinal ciliopathies. Ophthal. Genet. 2007;28:113–125. doi: 10.1080/13816810701537424. [DOI] [PubMed] [Google Scholar]
- 26.Jacobson S.G., Cideciyan A.V., Aleman T.S., Sumaroka A., Roman A.J., Gardner L.M., Prosser H.M., Mishra M., Bech-Hansen N.T., Herrera W., et al. Usher syndromes due to MYO7A, PCDH15, USH2A or GPR98 mutations share retinal disease mechanism. Hum. Mol. Genet. 2008;17:2405–2415. doi: 10.1093/hmg/ddn140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leber T. Uber retinitis pigmentosa und angebore amaurose. Graefes Arch. Clin. Exp. Ophthalmol. 1869;15:1–25. [Google Scholar]
- 28.Franceschetti A., Dieterle P. Diagnostic and prognostic importance of the electroretinogram in tapetoretinal degeneration with reduction of the visual field and hemeralopia. Confin. Neurol. 1954;14:184–186. [PubMed] [Google Scholar]
- 29.den Hollander A.I., Koenekoop R.K., Mohamed M.D., Arts H.H., Boldt K., Towns K.V., Sedmak T., Beer M., Nagel-Wolfrum K., McKibbin M., et al. Mutations in LCA5, encoding the ciliary protein lebercilin, cause Leber congenital amaurosis. Nat. Genet. 2007;39:889–895. doi: 10.1038/ng2066. [DOI] [PubMed] [Google Scholar]
- 30.den Hollander A.I., Koenekoop R.K., Yzer S., Lopez I., Arends M.L., Voesenek K.E., Zonneveld M.N., Strom T.M., Meitinger T., Brunner H.G., et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am. J. Hum. Genet. 2006;79:556–561. doi: 10.1086/507318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arts H.H., Cremers F.P.M., Knoers N.V.A.M., Roepman R. Focus on molecules: RPGRIP1. Exp. Eye Res. 2008 doi: 10.1016/j.exer.2008.03.019. In press. [DOI] [PubMed] [Google Scholar]
- 32.Casenghi M., Meraldi P., Weinhart U., Duncan P.I., Korner R., Nigg E.A. Polo-like kinase 1 regulates Nlp, a centrosome protein involved in microtubule nucleation. Dev. Cell. 2003;5:113–125. doi: 10.1016/s1534-5807(03)00193-x. [DOI] [PubMed] [Google Scholar]
- 33.Trojan P., Krauss N., Choe H.W., Giessl A., Pulvermuller A., Wolfrum U. Centrins in retinal photoreceptor cells: Regulators in the connecting cilium. Prog. Retin. Eye Res. 2008;27:237–259. doi: 10.1016/j.preteyeres.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 34.Grabarek Z. Structure of a trapped intermediate of calmodulin: calcium regulation of EF-hand proteins from a new perspective. J. Mol. Biol. 2005;346:1351–1366. doi: 10.1016/j.jmb.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 35.Cohen E., Binet S., Meininger V. Ciliogenesis and centriole formation in the mouse embryonic nervous system. An ultrastructural analysis. Biol. Cell. 1988;62:165–169. [PubMed] [Google Scholar]
- 36.Rapley J., Baxter J.E., Blot J., Wattam S.L., Casenghi M., Meraldi P., Nigg E.A., Fry A.M. Coordinate regulation of the mother centriole component nlp by nek2 and plk1 protein kinases. Mol. Cell Biol. 2005;25:1309–1324. doi: 10.1128/MCB.25.4.1309-1324.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Casenghi M., Barr F.A., Nigg E.A. Phosphorylation of Nlp by Plk1 negatively regulates its dynein-dynactin-dependent targeting to the centrosome. J. Cell Sci. 2005;118:5101–5108. doi: 10.1242/jcs.02622. [DOI] [PubMed] [Google Scholar]
- 38.Roepman R., Wolfrum U. Protein networks and complexes in photoreceptor cilia. Subcell. Biochem. 2007;43:209–235. doi: 10.1007/978-1-4020-5943-8_10. [DOI] [PubMed] [Google Scholar]
- 39.Besharse J.C., Horst C.J. In: Ciliary and Flagellar Membranes. Bloodgood R.A., editor. New York: Plenum; 1990. pp. 389–417. [Google Scholar]
- 40.Reiners J., Nagel-Wolfrum K., Jurgens K., Maerker T., Wolfrum U. Molecular basis of human Usher syndrome: deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease. Exp. Eye Res. 2006;83:97–119. doi: 10.1016/j.exer.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 41.Barrong S.D., Chaitin M.H., Fliesler S.J., Possin D.E., Jacobson S.G., Milam A.H. Ultrastructure of connecting cilia in different forms of retinitis pigmentosa. Arch. Ophthalmol. 1992;110:706–710. doi: 10.1001/archopht.1992.01080170128040. [DOI] [PubMed] [Google Scholar]
- 42.Dryja T.P., Adams S.M., Grimsby J.L., McGee T.L., Hong D.H., Li T., Andreasson S., Berson E.L. Null RPGRIP1 alleles in patients with Leber congenital amaurosis. Am. J. Hum. Genet. 2001;68:1295–1298. doi: 10.1086/320113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Q., Zhou J., Daiger S.P., Farber D.B., Heckenlively J.R., Smith J.E., Sullivan L.S., Zuo J., Milam A.H., Pierce E.A. Identification and subcellular localization of the RP1 protein in human and mouse photoreceptors. Invest. Ophthalmol. Vis. Sci. 2002;43:22–32. [PMC free article] [PubMed] [Google Scholar]
- 44.Liu Q., Zuo J., Pierce E.A. The retinitis pigmentosa 1 protein is a photoreceptor microtubule-associated protein. J. Neurosci. 2004;24:6427–6436. doi: 10.1523/JNEUROSCI.1335-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iannaccone A., Breuer D.K., Wang X.F., Kuo S.F., Normando E.M., Filippova E., Baldi A., Hiriyanna S., MacDonald C.B., Baldi F., et al. Clinical and immunohistochemical evidence for an X linked retinitis pigmentosa syndrome with recurrent infections and hearing loss in association with an RPGR mutation. J. Med. Genet. 2003;40:e118. doi: 10.1136/jmg.40.11.e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hong D.H., Yue G., Adamian M., Li T. Retinitis pigmentosa GTPase regulator (RPGRr)-interacting protein is stably associated with the photoreceptor ciliary axoneme and anchors RPGR to the connecting cilium. J. Biol. Chem. 2001;276:12091–12099. doi: 10.1074/jbc.M009351200. [DOI] [PubMed] [Google Scholar]
- 47.Rosenbaum J.L., Witman G.B. Intraflagellar transport. Nat. Rev. Mol. Cell Biol. 2002;3:813–825. doi: 10.1038/nrm952. [DOI] [PubMed] [Google Scholar]
- 48.Wolfrum U., Schmitt A. Rhodopsin transport in the membrane of the connecting cilium of mammalian photoreceptor cells. Cell Motil. Cytoskeleton. 2000;46:95–107. doi: 10.1002/1097-0169(200006)46:2<95::AID-CM2>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 49.Pazour G.J., Dickert B.L., Witman G.B. The DHC1b (DHC2) isoform of cytoplasmic dynein is required for flagellar assembly. J.Cell Biol. 1999;144:473–481. doi: 10.1083/jcb.144.3.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pedersen L.B., Miller M.S., Geimer S., Leitch J.M., Rosenbaum J.L., Cole D.G. Chlamydomonas IFT172 is encoded by FLA11, interacts with CrEB1, and regulates IFT at the flagellar tip. Curr. Biol. 2005;15:262–266. doi: 10.1016/j.cub.2005.01.037. [DOI] [PubMed] [Google Scholar]
- 51.Kozminski K.G., Beech P.L., Rosenbaum J.L. The Chlamydomonas kinesin-like protein FLA10 is involved in motility associated with the flagellar membrane. J. Cell Biol. 1995;131:1517–1527. doi: 10.1083/jcb.131.6.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Evans J.E., Snow J.J., Gunnarson A.L., Ou G., Stahlberg H., McDonald K.L., Scholey J.M. Functional modulation of IFT kinesins extends the sensory repertoire of ciliated neurons in Caenorhabditis elegans. J. Cell Biol. 2006;172:663–669. doi: 10.1083/jcb.200509115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pazour G.J., Baker S.A., Deane J.A., Cole D.G., Dickert B.L., Rosenbaum J.L., Witman G.B., Besharse J.C. The intraflagellar transport protein, IFT88, is essential for vertebrate photoreceptor assembly and maintenance. J. Cell Biol. 2002;157:103–113. doi: 10.1083/jcb.200107108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baker S.A., Freeman K., Luby-Phelps K., Pazour G.J., Besharse J.C. IFT20 links kinesin II with a mammalian intraflagellar transport complex that is conserved in motile flagella and sensory cilia. J. Biol. Chem. 2003;278:34211–34218. doi: 10.1074/jbc.M300156200. [DOI] [PubMed] [Google Scholar]
- 55.Insinna C., Pathak N., Perkins B., Drummond I., Besharse J.C. The homodimeric kinesin, Kif17, is essential for vertebrate photoreceptor sensory outer segment development. Dev. Biol. 2008;316:160–170. doi: 10.1016/j.ydbio.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marszalek J.R., Liu X., Roberts E.A., Chui D., Marth J.D., Williams D.S., Goldstein L.S. Genetic evidence for selective transport of opsin and arrestin by kinesin-II in mammalian photoreceptors. Cell. 2000;102:175–187. doi: 10.1016/s0092-8674(00)00023-4. [DOI] [PubMed] [Google Scholar]
- 57.Pazour G.J., Dickert B.L., Vucica Y., Seeley E.S., Rosenbaum J.L., Witman G.B., Cole D.G. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J. Cell Biol. 2000;151:709–718. doi: 10.1083/jcb.151.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abd-El-Barr M.M., Sykoudis K., Andrabi S., Eichers E.R., Pennesi M.E., Tan P.L., Wilson J.H., Katsanis N., Lupski J.R., Wu S.M. Impaired photoreceptor protein transport and synaptic transmission in a mouse model of Bardet–Biedl syndrome. Vision Res. 2007;47:3394–3407. doi: 10.1016/j.visres.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nachury M.V., Loktev A.V., Zhang Q., Westlake C.J., Peranen J., Merdes A., Slusarski D.C., Scheller R.H., Bazan J.F., Sheffield V.C., Jackson P.K. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129:1201–1213. doi: 10.1016/j.cell.2007.03.053. [DOI] [PubMed] [Google Scholar]
- 60.Overlack N., Maerker T., Latz M., Nagel-Wolfrum K., Wolfrum U. SANS (USH1G) expression in developing and mature mammalian retina. Vision Res. 2008;48:400–412. doi: 10.1016/j.visres.2007.08.021. [DOI] [PubMed] [Google Scholar]
- 61.Liu X., Udovichenko I.P., Brown S.D., Steel K.P., Williams D.S. Myosin VIIa participates in opsin transport through the photoreceptor cilium. J. Neurosci. 1999;19:6267–6274. doi: 10.1523/JNEUROSCI.19-15-06267.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Follit J.A., Tuft R.A., Fogarty K.E., Pazour G.J. The intraflagellar transport protein IFT20 is associated with the Golgi complex and is required for cilia assembly. Mol. Biol. Cell. 2006;17:3781–3792. doi: 10.1091/mbc.E06-02-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Papermaster D.S. The birth and death of photoreceptors: the Friedenwald Lecture. Invest Ophthalmol. Vis. Sci. 2002;43:1300–1309. [PubMed] [Google Scholar]
- 64.Tai A.W., Chuang J.Z., Bode C., Wolfrum U., Sung C.H. Rhodopsin's carboxy-terminal cytoplasmic tail acts as a membrane receptor for cytoplasmic dynein by binding to the dynein light chain Tctex-1. Cell. 1999;97:877–887. doi: 10.1016/s0092-8674(00)80800-4. [DOI] [PubMed] [Google Scholar]
- 65.Reiter J.F., Mostov K. Vesicle transport, cilium formation, and membrane specialization: the origins of a sensory organelle. Proc. Natl Acad. Sci. USA. 2006;103:18383–18384. doi: 10.1073/pnas.0609324103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang H., Shi X., Paddon H., Hampong M., Dai W., Pelech S. B23/nucleophosmin serine 4 phosphorylation mediates mitotic functions of polo-like kinase 1. J. Biol. Chem. 2004;279:35726–35734. doi: 10.1074/jbc.M403264200. [DOI] [PubMed] [Google Scholar]
- 67.Shinmura K., Tarapore P., Tokuyama Y., George K.R., Fukasawa K. Characterization of centrosomal association of nucleophosmin/B23 linked to Crm1 activity. FEBS Lett. 2005;579:6621–6634. doi: 10.1016/j.febslet.2005.10.057. [DOI] [PubMed] [Google Scholar]
- 68.Shu X., Fry A.M., Tulloch B., Manson F.D., Crabb J.W., Khanna H., Faragher A.J., Lennon A., He S., Trojan P., et al. RPGR ORF15 isoform co-localizes with RPGRIP1 at centrioles and basal bodies and interacts with nucleophosmin. Hum. Mol. Genet. 2005;14:1183–1197. doi: 10.1093/hmg/ddi129. [DOI] [PubMed] [Google Scholar]
- 69.Khanna H., Hurd T.W., Lillo C., Shu X., Parapuram S.K., He S., Akimoto M., Wright A.F., Margolis B., Williams D.S., Swaroop A. RPGR-ORF15, which is mutated in retinitis pigmentosa, associates with SMC1, SMC3, and microtubule transport proteins. J. Biol. Chem. 2005;280:33580–33587. doi: 10.1074/jbc.M505827200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Roepman R., Schick D., Ferreira P.A. Isolation of retinal proteins that interact with retinitis pigmentosa GTPase regulator by interaction trap screen in yeast. Methods Enzymol. 2000;316:688–704. doi: 10.1016/s0076-6879(00)16757-6. [DOI] [PubMed] [Google Scholar]
- 71.James P., Halladay J., Craig E.A. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics. 1996;144:1425–1436. doi: 10.1093/genetics/144.4.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Luijendijk M.W., van de Pol T.J., van Duijnhoven G., den Hollander A.I., ten Caat J., van Limpt V., Brunner H.G., Kremer H., Cremers F.P.M. Cloning, characterization, and mRNA expression analysis of novel human fetal cochlear cDNAs. Genomics. 2003;82:480–490. doi: 10.1016/s0888-7543(03)00150-2. [DOI] [PubMed] [Google Scholar]
- 73.Maruyama K., Mikawa T., Ebashi S. Detection of calcium-binding proteins by 45Ca autoradiography on nitrocellulose membrane after sodium dodecyl sulfate gel electrophoresis. J. Biochem. (Tokyo) 1984;95:511–519. doi: 10.1093/oxfordjournals.jbchem.a134633. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
[Supplementary Data]