Study of active controlled tocilizumab monotherapy for rheumatoid arthritis patients with an inadequate response to methotrexate (SATORI): significant reduction in disease activity and serum vascular endothelial growth factor by IL-6 receptor inhibition therapy (original) (raw)
Abstract
We investigated the clinical efficacy and safety of tocilizumab (a humanized anti-IL-6 receptor antibody) monotherapy in active rheumatoid arthritis (RA) patients with an inadequate response to low dose methotrexate (MTX). In a multicenter, double-blind, randomized, controlled trial, 125 patients were allocated to receive either tocilizumab 8 mg/kg every 4 weeks plus MTX placebo (tocilizumab group) or tocilizumab placebo plus MTX 8 mg/week (control group) for 24 weeks. The clinical responses were measured using the American College of Rheumatology (ACR) criteria and the Disease Activity Score in 28 joints. Serum vascular endothelial growth factor (VEGF) levels were also monitored. At week 24, 25.0% in the control group and 80.3% in the tocilizumab group achieved ACR20 response. The tocilizumab group showed superior ACR response criteria over control at all time points. Additionally, serum VEGF levels were significantly decreased by tocilizumab treatment. The overall incidences of adverse events (AEs) were 72 and 92% (serious AEs: 4.7 and 6.6%; serious infections: 1.6 and 3.3%) in the control and the tocilizumab groups, respectively. All serious adverse events improved by adequate treatment. Tocilizumab monotherapy was well tolerated and provided an excellent clinical benefit in active RA patients with an inadequate response to low dose MTX.
Keywords: Clinical trial, Interleukin-6, Rheumatoid arthritis, Tocilizumab, Vascular endothelial growth factor
Introduction
Rheumatoid arthritis (RA) is a common autoimmune disease characterized by persistent synovitis and progressive destruction of cartilage and bone in multiple joints [1]. The affected joints exhibit hyperplasia of inflamed synovium infiltrated with a range of immunocompetent cells, which forms pannus tissue and invade cartilage and bone [2]. Angiogenesis is a characteristic histological feature of rheumatoid synovium for which vascular endothelial growth factor (VEGF) is responsible [3]. In addition, patients with RA show systemic inflammatory manifestations such as fever, fatigue, anemia, and laboratory findings, including elevated erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP), hyper-γ-globulinemia and emergence of various types of autoantibodies. These abnormalities can be explained, at least partly, by deregulated overproduction of interleukin (IL)-6, a pro-inflammatory cytokine, although the etiological causes are not fully understood.
Recently biologics targeting T cells, B cells, as well as pro-inflammatory cytokines, such as tumor necrosis factor (TNF)-α, IL-1, have been successfully used in the treatment of RA. However, these therapies are not always effective. In addition, adverse reactions are also the problem for those treatments. Therefore, we need further new therapeutic agents that have a new mechanism of action and higher efficacy.
Tocilizumab is a humanized anti-human IL-6 receptor (IL-6R) monoclonal antibody that inhibits IL-6 binding to IL-6R [4]. This antibody was humanized by grafting the complementarity-determining regions from a murine anti-IL-6R antibody into human IgG1, thereby creating a functioning antigen-binding site in a reshaped human antibody and reducing the antigenicity of the antibody in human. We have demonstrated that treatment with tocilizumab improves the signs and symptoms and prevents joint damage of RA [5–8]. The objective of this clinical trial was to investigate the safety and efficacy of tocilizumab monotherapy in active RA patients with an inadequate response to MTX, an anchor drug for RA treatment, at a dose of 8 mg/week, which is approved for adult RA patients in Japan. In addition, tocilizumab effect on serum VEGF levels was also examined.
Patients and methods
Patients
Eligible patients were between 20 and 75 years old, fulfilled the American college of Rheumatology (ACR; formerly, the American Rheumatism Association) 1987 revised criteria for the classification of RA [9], with disease duration of more than 6 months. All candidates were treated with MTX 8 mg/week for at least 8 weeks until enrolment. They all had ≥6 tender joints (of 49 evaluated), ≥6 swollen joints (of 46 evaluated), ESR of ≥30 mm/h or CRP of ≥10 mg/l at enrolment. An inadequate response to MTX was defined as the presence of active disease, as described above. Patients were not allowed to have received prior anti-TNF agents or leflunomide (within 12 weeks prior to the first dose), plasma exchange therapy or surgical treatments (within 4 weeks prior to the first dose), DMARDs other than MTX or immunosuppressants (within 2 weeks prior to the first dose). Oral corticosteroids (prednisolone, ≤10 mg/day) were allowed if the dosage had not been changed within 2 weeks. Eligible patients had white blood cell counts ≥3.5 × 109/l, lymphocyte counts ≥0.5 × 109/l and platelet count of at least the lower limit of normal as defined by the respective local laboratory used. Patients were excluded if they had functional class IV using Steinbrocker’s criteria [10], aspartate transaminase (AST), alanine transaminase (ALT) and serum creatinine ≥1.5-fold the upper limit of normal, were HBs antigen and/or HCV antibody positive, had pulmonary fibrosis or active pulmonary disease, a history of serious adverse drug reaction to MTX, concomitant pleural effusion, ascites, varicella infection, or were excessive users of alcohol on a regular basis. Patients were also excluded if they had significant cardiac, blood, respiratory system, neurologic, endocrine, renal, hepatic, or gastrointestinal disease, or had an active infection requiring medication within 4 weeks before the first dose or medical history of a serious allergic reaction. Sexually active premenopausal women were required to have a negative urine pregnancy test at the entry to the study and to use effective contraception during the study period.
Study protocol
This study was conducted at 25 sites in Japan. The study protocol was approved by the Ministry of Health, Labor and Welfare of Japan, and by the local ethical committee. Patients gave their written informed consent. This trial was registered with http://www.clinicaltrials.gov (NCT00144521). The first patient was enrolled on January 27, 2004, and the last patient exited the study on February 15, 2005.
Patients were randomly assigned to receive either tocilizumab therapy or MTX therapy as a control: tocilizumab 8 mg/kg every 4 weeks plus MTX placebo (tocilizumab group) or tocilizumab placebo plus MTX 8 mg/week (control group) for 24 weeks. The randomization was done by registering the patients to the patient registration center utilizing a centralized allocation method. The dosage of tocilizumab used in this study was chosen according to a previous dose finding study [7]. The dose of MTX was the maximum dose allowed in Japan (see Discussion). Oral corticosteroids less than 10 mg prednisolone per day were allowed, and the dose could not be increased during the study. Intra-articular injections of corticosteroid (only one joint at one treatment) and hyaluronate preparations were allowed. Use of one nonsteroidal anti-inflammatory drug (NSAID), including switching to another NSAID, was allowed. DMARDs, intravenous or intramuscular corticosteroids, plasmapheresis and surgical treatment were not allowed. Patients who received three or more doses of tocilizumab or tocilizumab placebo were able to join an open-label extension study of tocilizumab.
The clinical responses were measured using the ACR criteria as well as the Disease Activity Score in 28 joints (DAS28) and the European League Against Rheumatism (EULAR) criteria based on DAS28. Remission was defined according to EULAR definition of a DAS28 <2.6 [11]. Serum VEGF levels were also monitored. Safety was assessed through recording of adverse events, physical examinations, and standard laboratory tests.
Statistical analysis
We determined that a sample size of 57 patients per treatment group was required to provide 90% power for detecting a significant (P < 0.05) difference in ACR20 response between the control group and the tocilizumab group by use of the two-side chi-square test, where ACR20 response rates in the population were assumed to be 35 and 65% in the control group and the tocilizumab group, respectively. We decided to recruit 60 patients per treatment group to allow for anticipated withdrawals. The primary end point was the ACR20 response at week 24 with the last observation carried forward (LOCF) method, using an intent-to-treat (ITT) analysis. The incidences of clinical improvements were analyzed by the chi-square test.
All statistical analyses were two-sided and P values less than 0.05 were considered significant. All patients receiving at least one dose of tocilizumab or tocilizumab placebo, and at least 4 weeks of MTX or MTX placebo administration were included in the clinical efficacy analysis.
Results
Characteristics of the patients
One-hundred and twenty-seven patients were enrolled in the study (Fig. 1). Two patients randomized to the control group withdrew before dosing (gall stone and patient’s request). A total of 125 patients (64 in the control group and 61 in the tocilizumab group) received study drugs. Thirty-three patients in the control group and 54 patients in the tocilizumab group completed 24-week treatment. Withdrawal occurred in 31 patients in the control group and seven patients in the tocilizumab group. The reported reasons for withdrawal are shown in Fig. 1.
Fig. 1.

Randomization, reasons for withdrawal, and numbers of patients who completed the trial. Tocilizumab humanized anti-interleukin-6 receptor antibody
Demographics and baseline disease characteristics did not differ between the two groups (Table 1). Mean disease duration was 8.6 years. Patients had active RA, indicated by DAS28 score of 6.1 and CRP of 31 mg/l at baseline after using of MTX 8 mg/week for at least 8 weeks.
Table 1.
Patient demographics and clinical characteristics at baseline
| Control group (n = 64) | Tocilizumab group (n = 61) | |
|---|---|---|
| Demographics | ||
| Age (years) | 50.8 ± 12.2 | 52.6 ± 10.6 |
| Male:female ratio | 16:48 | 6:55 |
| Clinical characteristics | ||
| RA duration (years) | 8.7 ± 7.1 | 8.5 ± 8.4 |
| No. of failed DMARDs, mean (range) | 3.6 (1–8) | 3.3 (1–8) |
| Functional classa, I/II/III/IV | 7:50:7:0 | 2:49:10:0 |
| RA Stagea, I/II/III/IV | 3🔞17:26 | 1:20:22:18 |
| Tender joint count, 0–49 scale | 14.2 ± 8.6 | 13.8 ± 7.5 |
| Swollen joint count, 0–46 scale | 12.7 ± 7.5 | 12.4 ± 5.9 |
| ESR (mm/h) | 51.9 ± 24.0 | 51.9 ± 27.7 |
| CRP (mg/l) | 32 ± 26 | 30 ± 20 |
| DAS28 | 6.2 ± 0.9 | 6.1 ± 0.9 |
| VEGF (pg/ml) | 730.8 ± 445.6 | 711.3 ± 417.4 |
Clinical efficacy
The primary end point of the study, an ACR20 response at week 24 was 25.0% in the control group compared with 80.3% in the tocilizumab group, indicating the superiority of tocilizumab treatment (P < 0.001). The ACR50 and ACR70 response rates in the tocilizumab group were higher than those in the control group at all time points from week 4 onward (Fig. 2a). At the last observation, the ACR50 response rate was 10.9 and 49.2%, and the ACR70 response rate was 6.3 and 29.5% in the control group and the tocilizumab group, respectively. Additionally, the tocilizumab group showed a greater reduction in DAS28 than the control group did (Fig. 2b). At the last observation, the incidence of “Good” was 3.2 and 65.5%, and that of “Good or moderate” was 39.7 and 96.6% in the control group and the tocilizumab group, respectively. Similarly, remission was observed in 1.6% of the control group and 43.1% of the tocilizumab group at the last observation, and tocilizumab treatment achieved significantly higher remission rates than MTX treatment (P < 0.001).
Fig. 2.
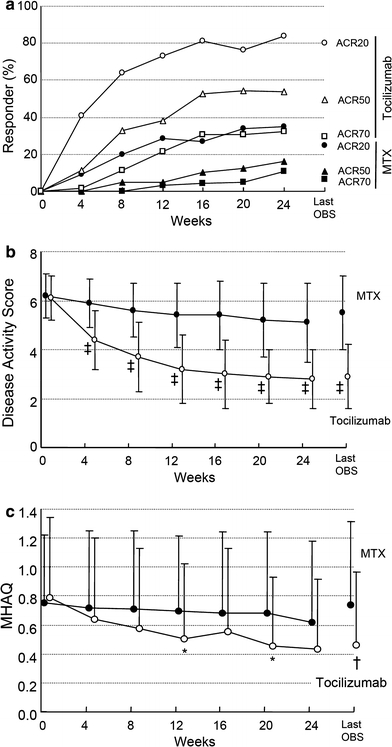
Percentage of responders according to the American College of Rheumatology (ACR) improvement criteria and the Disease Activity Score in 28 joints (DAS28) as well as mean change in Modified Health Assessment Questionnaire (MHAQ) scores. Percentage of responders according to the ACR improvement criteria (a) and the DAS28 (b) according to the ITT analysis over 24 weeks. Mean change in MHAQ scores from baseline to week 24 (c). Last OBS = last observation. *P < 0.05; †P < 0.01; ‡P < 0.001 versus MTX by paired _t_-test
Tocilizumab treatment also significantly improved MHAQ scores compared to MTX treatment (Fig. 2c). A decrease of ≥0.22 units in MHAQ scores represents significant clinical improvement and the minimum clinical important difference [12]. Such improvement was seen in 34% in the control group and 67% in the tocilizumab group at the last observation, demonstrating significant improvement in the tocilizumab group compared to the control group (P < 0.001).
The mean serum VEGF levels showed a marked decrease in the tocilizumab group (Fig. 3). Mean changes from baseline were −74.0 pg/ml in the control group and −346.9 pg/ml in the tocilizumab group at week 24 (P < 0.001).
Fig. 3.

Change from baseline in serum levels of VEGF. Values are the mean for each group at each time point. VEGF was measured at three points (0, 12 and 24 weeks) over the study period. Last OBS = last observation. ‡P < 0.001 versus MTX by paired _t_-test
Safety
A total of 104 adverse events occurred in 46 of 64 patients (71.9%) in the control group and 211 adverse events occurred in 56 of 61 patients (91.8%) in the tocilizumab group. Most of the adverse events were mild or moderate. Table 2 shows frequent adverse events observed in at least 5% of the patients. Nasopharyngitis was the most common adverse event in the both groups (the control group 10.9%, the tocilizumab group 18.0%).
Table 2.
Adverse events observed in at least 5% of patients
| Control group (n = 64) | Tocilizumab group (n = 61) | |
|---|---|---|
| Nasopharyngitis | 7 (10.9) | 11 (18.0) |
| Stomatitis | 0 | 7 (11.5) |
| Hyperlipidemia | 1 (1.6) | 4 (6.6) |
| Headache | 2 (3.1) | 4 (6.6) |
| Rash | 2 (3.1) | 4 (6.6) |
| Diarrhea | 1 (1.6) | 4 (6.6) |
| Upper respiratory tract inflammation | 4 (6.3) | 3 (4.9) |
Serious adverse events were reported in 4.7% (3 of 64 patients) and 6.6% (4 of 61 patients) in the control group and tocilizumab group, respectively. In the control group, these consisted of one event each of pneumonia, spinal compression fracture and femoral neck fracture, among which a causal relationship with the study drug could not be ruled out for pneumonia. In the tocilizumab group, the serious adverse events consisted of one event each of pneumonia, infectious arthritis, colonic polyp and headache, among which a causal relationship with the investigational product could not be ruled out for pneumonia and infectious arthritis. All serious adverse events improved with appropriate treatments.
Tuberculosis was not observed in this study. No tuberculosis screening or prophylactic use of any anti-tuberculosis drugs was done for this study.
Drug-related infusion reactions were reported for eight events in seven patients (11.5%) of the tocilizumab group: two with pruritus, and one each with headache, flushing, rash, arthralgia, abnormal feeling and increased blood pressure. The severity of arthralgia was moderate while all other infusion reactions were mild, and all patients continued the study.
Laboratory findings
Laboratory test abnormalities were reported in 23 and 56% of patients in the control and the tocilizumab groups, respectively. In the tocilizumab group, lipid metabolism-related changes such as an increase in total cholesterol (TC), triglycerides (TG), and low-density lipoprotein cholesterol (LDLC) were common. Increases in TC, TG, and LDLC under non-fasting were reported in 36, 20, and 28% of the patients, respectively. Most of them were grade 1 according to the National Cancer Institute Common Toxicity Criteria (NCI-CTC). Values increased until week 4 and thereafter remained constant. High-density lipoprotein cholesterol (HDLC) values were also raised in the tocilizumab group. Therefore, the atherogenic index, calculated by (TC-HDLC)/HDLC, did not change at all throughout the study period of 24 weeks.
Increases in AST, ALT, and total bilirubin were reported in 8, 11, and 0% of the patients in the control group, and 3, 5, and 2% in the tocilizumab group, respectively. These values did not continue to be increased, but became stable at week 16 in the tocilizumab group (Fig. 4). All the patients with these abnormalities in the tocilizumab group were NCI-CTC grade 1 except for 1 patient with grade 2 increase in total bilirubin. There was no patient who withdrew from this study for the reason of liver functional abnormality.
Fig. 4.
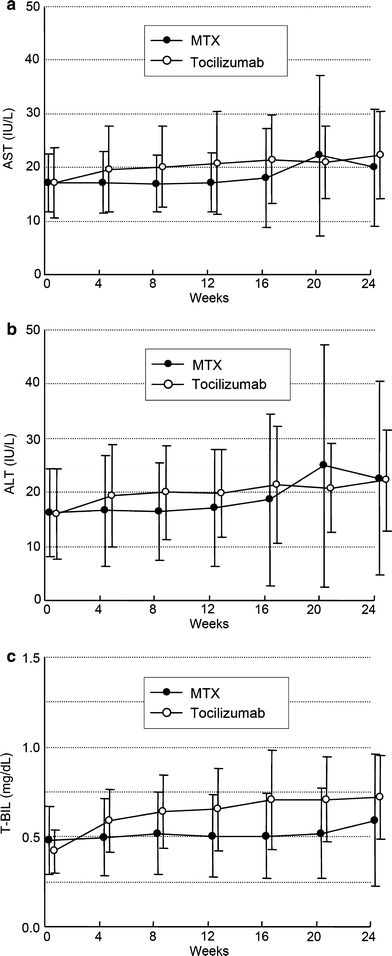
Change from baseline in serum levels of aspartate transaminase (AST), alanine transaminase (ALT), total bilirubin (T-BIL). Values are the mean for each group at each time point
Anti-tocilizumab antibodies were not detected in this study.
Discussion
This study was a multi-center, randomized, blinded, double-dummy trial of tocilizumab in active RA patients who had an inadequate response to low dose MTX treatment in Japan. The results of this study confirmed the excellent efficacy of tocilizumab monotherapy for signs and symptoms as shown in previous studies [7, 8]. Since MTX is an anchor drug in RA treatment, it is noteworthy that tocilizumab treatment is a very efficacious treatment for the patients with an inadequate response to MTX. In addition, switching MTX therapy to tocilizumab monotherapy was safe and effective.
The dose of MTX prior to enrollment was 10–25 mg/week in CHARISMA study, which was conducted in Europe [13], while all patients in this study were treated with MTX 8 mg/week. The dosage 8 mg of MTX/week in this trial is the maximum dosage approved in Japan. The Japanese government recommends 6–8 mg/week of MTX based on the evidence from the Japanese clinical trials of MTX for RA [14, 15]. The MTX dosage used in Japan is lower than that used in Western populations treated in the EU or US. The average body weight of the patients in this study was 54 kg, and much lower than those of EU and US patients. Additionally, all patients were given folic acid in the CHARISMA study, in contrast to only 51% of the patients in this study. Considering these factors, the differences in the clinical efficacy of MTX between the CHARISMA study and this study might not be so large as expected when looking at the difference in the MTX dose.
Maini et al. demonstrated in the CHARISMA study, that adding MTX to tocilizumab increased the efficacy in terms of ACR50 and ACR70 response rates, although there was no difference in ACR20 response rates between the tocilizumab 8 mg/kg monotherapy and the tocilizumab 8 mg/kg plus MTX. Thus, the combination with MTX may be a therapeutic option, if the toxicity is not increased. In this study, however, even monotherapy with tocilizumab 8 mg/kg induced DAS28 remission at 6 months in about 40% of patients. Furthermore, since anti-tocilizumab antibodies are not detected without use of MTX, the combination with MTX is not required to suppress the emergence of anti-tocilizumab antibodies as long as 8 mg/kg of tocilizumab is used. Therefore, tocilizumab will be useful for the patients who do not tolerate MTX.
This double-blind study of tocilizumab also confirms the previous finding that IL-6 receptor inhibition improves serum VEGF levels of RA patients [16]. Serum VEGF levels markedly decreased during tocilizumab therapy compared to the treatment with MTX. VEGF is produced by macrophages, fibroblasts surrounding microvessels, vascular smooth muscle cells, synovial lining cells in synovium [17], neutrophils in synovial fluid [18], and peripheral blood mononuclear cells [19] from patients with RA. VEGF is a potent angiogenic factor and thought to be responsible for the angiogenesis necessary to oxygenate the hypertrophic synovial tissues of the affected joints of RA patients [20, 21]. VEGF also induces vascular permeability and mediates inflammation [22–24]. Therefore, the decrease in VEGF may be an important part of the mechanism how IL-6 receptor inhibition with tocilizumab exerts its therapeutic efficacy in RA. Since serum VEGF levels correlate with disease activity scores and radiographic progression in RA patients [16, 25], the dramatic decrease in VEGF also underlines the efficacy of tocilizumab. The normalization of VEGF by blockade of IL-6 function alone indicates that IL-6 is essential for the VEGF production in this disease.
Tocilizumab monotherapy was generally well tolerated. There was no specific type of infection related to tocilizumab therapy. There is no indication for an increased risk of reactivation of latent tuberculosis, which is often a problem in anti-TNF therapy [26]. In this study patients did not receive prophylactic medication nor were they screened for latent or active tuberculosis at the time of screening.
The increase in TC observed is in concordance with observations in previous studies of tocilizumab [7, 8]. This may be secondary to the improvement in inflammation. Furthermore, there was no cardiovascular adverse event related to the increase in TC. However, further investigation will be required to evaluate whether or not tocilizumab might increase the risk for developing ischemic heart diseases.
The mean value elevations of liver functional tests (AST, ALT and total bilirubin) were seen in the tocilizumab group as well as in the control group, but they were within normal range. Liver functional tests abnormalities were more frequently observed in the control group than in the tocilizumab group. Moreover, most of them were grade 1 according to the NCI-CTC. These abnormalities were clinically not significant and no hepatitis was observed. Therefore, tocilizumab monotherapy appears to be as tolerable as MTX in terms of liver function.
Conclusion
This study demonstrates that tocilizumab monotherapy in patients with active RA who had an inadequate response to low dose MTX treatment in Japan has an excellent efficacy with a positive benefit-risk ratio.
Acknowledgments
The authors wish to thank the SATORI investigators for the treatment and Takahiro Kakehi, B·Sc., Yuji Kimura, M.Sc., Kenichi Yamada, B·Sc. and Karsten Kissel, MD for their valuable assistance with the design and analysis of the study and preparation of this manuscript. This work was financially supported by Chugai Pharmaceutical Co., Ltd., Tokyo, Japan. The SATORI investigators were as follows: Tatsuya Atsumi, M.D. (Hokkaido University, Hokkaido, Japan); Takeshi Sasaki, M.D. (Tohoku University, Miyagi, Japan); Takayuki Sumida, M.D. (University of Tsukuba, Ibaraki, Japan); Masahiro Iwamoto, M.D. (Jichi Medical University, Tochigi, Japan); Tsutomu Takeuchi, M.D. (Saitama Medical Center/School, Saitama, Japan); Nobuyuki Miyasaka, M.D. (Tokyo Medical and Dental Unversity, Tokyo, Japan); Michito Hirakata, M.D. (Keio University, Tokyo, Japan); Kazuhiko Yamamoto, M.D. (University of Tokyo, Tokyo, Japan); Akio Yamada, M.D. (Jikei University, Tokyo, Japan); Shigeto Tohma, M.D. (Sagamihara National Hospital, Kanagawa, Japan); Hirahito Endo, M.D. (Kitasato University, Kanagawa, Japan); Yoshiaki Ishigatsubo, M.D. (Yokohama City University, Kanagawa, Japan); Takeshi Kuroda, M.D. (Niigata University, Niigata, Japan); Shogo Banno, M.D., Yoshihito Hayami, M.D. (Nagoya City University, Aichi, Japan); Masao Tanaka, M.D. (Kyoto University, Kyoto, Japan); Tadao Miyake, M.D. (Osaka General Medical Center, Osaka, Japan); Masaki Suemura, M.D. (Nissay Hospital, Osaka, Japan); Shiro Ohshima, M.D. (Osaka University, Osaka, Japan); Masato Matsushita, M.D. (Osaka Second Police Hospital, Osaka, Japan); Yukihiko Saeki, M.D. (Osaka Minami Medical Center, Osaka, Japan); Hiroshi Uda, M.D. (Sakai-Onshinkai Hospital, Osaka, Japan); Kiyoshi Takasugi, M.D. (Dogo Spa Hospital, Ehime, Japan); Takahiko Horiuchi, M.D. (Kyushu University, Fukuoka, Japan); Kazuyoshi Saito, M.D. (University of Occupational and Environmental Health, Fukuoka, Japan); Katsumi Eguchi, M.D. (Nagasaki University, Nagasaki, Japan).
Conflict of interest statement NN has served as a consultant to and received honoraria from Chugai Pharmaceutical, the manufacturer of tocilizumab. NN also works as a scientific advisory board of Hoffmann-La Roche. TK holds a patent for tocilizumab. The other authors have no competing interests.
References
- 1.Harris ED Jr. Rheumatoid arthritis: pathophysiology and implications for therapy. N Engl J Med. 1990;322:1277–89. [DOI] [PubMed]
- 2.Cush JJ, Lipsky PE. Phenotypic analysis of synovial tissue and peripheral blood lymphocytes isolated from patients with rheumatoid arthritis. Arthritis Rheum. 1988;31:1230–8. [DOI] [PubMed]
- 3.Paleolog EM. Angiogenesis: a critical process in the pathogenesis of RA—a role for VEGF? Br J Rheumatol. 1996;35:917–9. [DOI] [PubMed]
- 4.Sato K, Tsuchiya M, Saldanha J, Koishihara Y, Ohsugi Y, Kishimoto T, et al. Reshaping a human antibody to inhibit the interleukin 6-dependent tumor cell growth. Cancer Res. 1993;53:851–6. [PubMed]
- 5.Choy EHS, Isenberg DA, Garrood T, Farrow S, Ioannou Y, Bird H, et al. Therapeutic benefit of blocking interleukin–6 activity with an anti-interleukin-6 receptor monoclonal antibody in rheumatoid arthritis. Arthritis Rheum. 2002;46:3143–50. [DOI] [PubMed]
- 6.Nishimoto N, Yoshizaki K, Maeda K, Kuritani T, Deguchi H, Sato B, et al. Toxicity, pharmacokinetics, and dose-finding study of repetitive treatment with the humanized anti-interleukin 6 receptor antibody MRA in rheumatoid arthritis. Phase I/II clinical study. J Rheumatol. 2003;30:1426–35. [PubMed]
- 7.Nishimoto N, Yoshizaki K, Miyasaka N, Yamamoto K, Kawai S, Takeuchi T, et al. Treatment of rheumatoid arthritis with humanized anti-interleukin-6 receptor antibody: a multicenter, double-blind, placebo-controlled trial. Arthritis Rheum. 2004;50:1761–9. [DOI] [PubMed]
- 8.Nishimoto N, Hashimoto J, Miyasaka N, Yamamoto K, Kawai S, Takeuchi T, et al. Study of active controlled monotherapy used for rheumatoid arthritis, an IL-6 inhibitor (SAMURAI): evidence of clinical and radiographic benefit from an X-ray reader-blinded randomized controlled trial of tocilizumab. Ann Rheum Dis. 2007;66:1162–7. [DOI] [PMC free article] [PubMed]
- 9.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–24. [DOI] [PubMed]
- 10.Hochberg MC, Chang RW, Dwosh I, Lindsey S, Pincus T, Wolfe F. The American College of Rheumatology 1991 revised criteria for the classification of global functional status in rheumatoid arthritis. Arthritis Rheum. 1992;35:498–502. [DOI] [PubMed]
- 11.Fransen J, Creemers MC, Van Riel PL. Remission in rheumatoid arthritis: agreement of the disease activity score (DAS28) with the ARA preliminary remission criteria. Rheumatology. 2004;43:1252–5. [DOI] [PubMed]
- 12.Kosinski M, Zhao SZ, Dedhiya S, Osterhaus JT, Ware JE Jr. Determining minimally important changes in generic and disease-specific health-related quality of life questionnaires in clinical trials of rheumatoid arthritis. Arthritis Rheum. 2000;43:1478–87. [DOI] [PubMed]
- 13.Maini RN, Taylor PC, Szechinski J, Pavelka K, Broll J, Balint G, et al. Double-blind randomized controlled clinical trial of the interleukin-6 receptor antagonist, tocilizumab, in European patients with rheumatoid arthritis who had an incomplete response to methotrexate. Arthritis Rheum. 2006;54:2817–29. [DOI] [PubMed]
- 14.Kashiwazaki S, Ichikawa Y, Sugawara S, Nagaya I, Kawai S, Hakota M, et al. Determination of the clinical optimal dose of L-377 (methotrexate capsule) for the treatment of rheumatoid arthritis (in Japanese). Ensyo. 1996;16:437–58.
- 15.Ito S, Gross WL, Reinhold-Keller E, Gause A, Aries P, Ruther W, et al. Rheumatology in Japan, Germany, and Egypt: a comparison of medical practices. Acta Medica et Biologica. 2006;54:51–8.
- 16.Nakahara H, Song J, Sugimoto M, Hagihara K, Kishimoto T, Yoshizaki K, et al. Anti-interleukin-6 receptor antibody therapy reduces vascular endothelial growth factor production in rheumatoid arthritis. Arthritis Rheum. 2003;48:1521–9. [DOI] [PubMed]
- 17.Nagashima M, Yoshino S, Ishiwata T, Asano G. Role of vascular endothelial growth factor in angiogenesis of rheumatoid arthritis. J Rheumatol. 1995;22:1624–30. [PubMed]
- 18.Kasama T, Kobayashi K, Yajima N, Shiozawa F, Yoda Y, Takeuchi HT, et al. Expression of vascular endothelial growth factor by synovial fluid neutrophils in rheumatoid arthritis (RA). Clin Exp Immunol. 2000;121:533–8. [DOI] [PMC free article] [PubMed]
- 19.Bottomley MJ, Webb NJA, Watson CJ, Holt PJL, Freemont AJ, Brenchley PEC. Peripheral blood mononuclear cells from patients with rheumatoid arthritis spontaneously secrete vascular endothelial growth factor (VEGF): specific up-regulation by tumor necrosis factor-alpha (TNF-α) in synovial fluid. Clin Exp Immunol. 1999;117:171–6. [DOI] [PMC free article] [PubMed]
- 20.Folkman J. Angiogenesis in cancer, vascular, rheumatoid, and other disease. Nat Med. 1995;1:27–31. [DOI] [PubMed]
- 21.Colville-Nash PR, Scott DL. Angiogenesis and rheumatoid arthritis: pathogenic and therapeutic implications. Ann Rheum Dis. 1992;51:919–25. [DOI] [PMC free article] [PubMed]
- 22.Keck PJ, Hauser SD, Krivi G, Sanzo K, Warren T, Feder J, et al. Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science. 1989;246:1309–12. [DOI] [PubMed]
- 23.Connolly DT, Heuvelman DM, Nelson R, Olander JV, Eppley BL, Delfino JJ, et al. Tumor vascular permeability factor stimulates endothelial cell growth and angiogenesis. J Clin Invest. 1989;84:1470–8. [DOI] [PMC free article] [PubMed]
- 24.Koch AE, Harlow LA, Haines GK, Amento EP, Unemori EN, Wong WL, et al. Vascular endothelial growth factor: a cytokine modulating endothelial function in rheumatoid arthritis. J Immunol. 1994;152:4149–56. [PubMed]
- 25.Ballara S, Taylor PC, Reusch P, Marme D, Feldmann M, Maini RN, et al. Raised serum vascular endothelial growth factor levels are associated with destructive change in inflammatory arthritis. Arthritis Rheum. 2001;44:2055–64. [DOI] [PubMed]
- 26.Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med. 2001;345:1098–104. [DOI] [PubMed]