Posttranscriptional Crossregulation between Drosha and DGCR8 (original) (raw)
. Author manuscript; available in PMC: 2010 Jan 9.
SUMMARY
The Drosha-DGCR8 complex, also known as Microprocessor, is essential for microRNA (miRNA) maturation. Drosha functions as the catalytic subunit, while DGCR8 (also known as Pasha) recognizes the RNA substrate. Although the action mechanism of this complex has been intensively studied, it remains unclear how Drosha and DGCR8 are regulated and if these proteins have any additional role(s) apart from miRNA processing. Here, we report that Drosha and DGCR8 regulate each other posttranscriptionally. The Drosha-DGCR8 complex cleaves the hairpin structures embedded in the DGCR8 mRNA and thereby destabilizes the mRNA. We further find that DGCR8 stabilizes the Drosha protein via protein-protein interaction. This crossregulation between Drosha and DGCR8 may contribute to the homeostatic control of miRNA biogenesis. Furthermore, microarray analyses suggest that a number of mRNAs may be downregulated in a Microprocessor-dependent, miRNA-independent manner. Our study reveals a previously unsuspected function of Microprocessor in mRNA stability control.
INTRODUCTION
MicroRNAs (miRNAs) are ~22 nt small noncoding RNAs that control gene expression at the posttranscriptional level through translational inhibition and destabilization of their target mRNAs (Ambros et al., 2003; Filipowicz et al., 2008). MiRNAs were initially discovered from C. elegans, as regulatory molecules modulating the developmental timing (Lee et al., 1993). Hundreds of miRNAs have been discovered in most eukaryotic species, and their diverse roles are rapidly being elucidated (Bushati and Cohen, 2007; Jones-Rhoades et al., 2006). Links between miRNA and human diseases such as cancer and neurodegenerative diseases have also been established (Bushati and Cohen, 2007; Hebert et al., 2008; Wang et al., 2008a).
MiRNAs originate from long primary transcripts called primiRNAs (Lee et al., 2002). The hairpin embedded in pri-miRNA is processed in the nucleus by Drosha, a member of ribonuclease III family (RNase III), and converted into precursor miRNA (premiRNA) (Lee et al., 2003). This processing is crucial for the vast majority of miRNAs, although a small subgroup of miRNAs found in short introns can bypass this step (Okamura et al., 2007; Ruby et al., 2007). The pre-miRNA is then exported to the cytoplasm by exportin-5 and turned into ~22 nt miRNA duplex by Dicer, another RNase III protein (Liu et al., 2008). One strand of the miRNA duplex is incorporated into an effector complex known as RNA-induced silencing complex (RISC) (Liu et al., 2008).
Drosha has two RNase III domains and one double-stranded RNA binding domain (dsRBD). The two RNase III domains (RIIIDa and RIIIDb) form an intramolecular dimer and cleave the 3′ and 5′ strands of the stem, respectively (Han et al., 2004). Because the dsRBD of Drosha is insufficient for substrate binding, Drosha needs a partner protein surrogating the RNA recognition function. DGCR8 (also known as Pasha) is the cofactor that interacts with Drosha and forms a functional complex called the “Microprocessor” (Denli et al., 2004; Gregory et al., 2004; Han et al., 2004; Landthaler et al., 2004). DGCR8 contains two dsRBDs and recognizes the unique features of the pri-miRNA, which include the ssRNA segments flanking a stem of appropriate length (Han et al., 2006). DGCR8 anchors at the ssRNA-dsRNA junction and directs Drosha to cleave ~11 bp away from the junction (Han et al., 2006).
Although the mechanism of Microprocessor activity has been intensively interrogated, it remains largely unknown how its components are regulated. In a comparative genomics study, Pedersen et al. predicted a couple of conserved hairpins in DGCR8 mRNA, whose folds are similar to pri-miRNA structure, which implied that the hairpins may be involved in the autoregulation of DGCR8 (Pedersen et al., 2006). A possible link between DGCR8 and heme-mediated signal transduction pathway was also proposed on the basis of the finding that DGCR8 is a heme-binding protein (Faller et al., 2007).
When Microprocessor activity is dysregulated, the miRNA pool is altered, disrupting the normal cellular function. For instance, Dgcr8 knockout mouse embryonic stem (ES) cells do not produce miRNAs with resulting defects in proliferation or differentiation (Wang et al., 2007). Interestingly, in various human cancers, miRNA expression was found to be generally suppressed (Lu et al., 2005), implicating that precise control of miRNA biogenesis may be critical in guarding the cells against malignancy. It was proposed that Drosha processing of certain miRNAs is suppressed in tumor cells and undifferentiated cells (Thomson et al., 2006), although the mechanism of the suppression remains unclear.
In the present study, we show that DGCR8 is negatively regulated by the Drosha-DGCR8 complex through mRNA cleavage. We further report that DGCR8 positively regulates Drosha by protein stabilization. These auto- and crossregulations between Drosha and DGCR8 may help maintain the homeostatic control of miRNA biogenesis.
RESULTS
Drosha Suppresses the Expression of DGCR8
To study the function of Drosha in pri-miRNA processing, we routinely use RNAi technique to deplete Drosha in HeLa cells and analyze RNA by RT-PCR and northern blotting. While performing these RNAi experiments, we observed that the DGCR8 mRNA level increased significantly in Drosha-depleted cells (Figure 1A). Quantitative real time PCR (qRT-PCR) showed ~3 fold increase in the DGCR8 mRNA level under this condition. As with the mRNA level, the DGCR8 protein level also increased 3- to 4-fold after Drosha knockdown, as determined by western blotting (Figure 1B). To exclude the possibility that this is caused by off-target effects, we used a transdominant negative Drosha mutant (TN Drosha) containing point mutations in both RNase III domains. When this mutant protein was overexpressed, Drosha processing of pri-miRNA was effectively blocked (Figure 1C and Figure S1A available online). The DGCR8 mRNA level increased under this condition (Figure 1C, lane 3), whereas it decreased when wild-type Drosha was overexpressed (Figure 1C, lane 2). In accordance with the mRNA level, endogenous DGCR8 protein accumulated after TN Drosha overexpression (Figure 1C, lane 6, and Figure S1A). A similar experiment was carried out with a transdominant negative DGCR8 mutant (Figure S1B). The mutant called mDRBD1&2 DGCR8 (m1&2) contains point mutations in both dsRBDs (Yeom et al., 2006). Expression of this DGCR8 mutant upregulated the endogenous DGCR8 mRNA (Figure S1B, lane 4), suggesting that it is the reduction of the wild-type Drosha-DGCR8 activity that is responsible for the accumulation of DGCR8 mRNA.
Figure 1. Drosha Downregulates DGCR8 mRNA and Protein Expression at the Posttranscriptional Level.

(A) Quantitative real-time PCR (qRT-PCR) after Drosha depletion. Total RNA was prepared from HeLa cells 72 hr after transfection of siRNA against Drosha (siDrosha). siRNA against GFP (siGFP) was used as a control. Three biologically independent experiments were performed for quantification (mean ±SD).
(B) Western blot analysis after knockdown. HeLa cell extract was prepared 72 hr after siRNA transfection. The p27, a target of miR-221/222, is used as a control. Quantitative results are shown in the right panel. Three independent experiments were performed for quantification (mean ±SD).
(C) RT-PCR and western blot analysis after overexpression of the transdominant negative (TN) Drosha mutant in HEK293T cells. Drosha (TN) is a mutant lacking the catalytic activity. Wild-type (WT) Drosha was used as a control. Though the juxtaposed lanes are not contiguous, all of them are from a single gel, which is true for all the membranes with dashed lines. The primer set, Ex1-Ex2, amplifies endogenous DGCR8 mRNA.
(D) Nuclear runoff assay after Drosha depletion. The nuclei of miR-30a-inducible HeLa cells were prepared after siRNA transfection and doxycycline treatment. For quantification of the nuclear runoff, the band intensity was determined by phosphoimager and normalized against β-actin levels. Two independent experiments were performed for quantification (mean ±SD).
(E) Turnover rate of DGCR8 mRNA in Drosha-depleted cells. Actinomycin D (ActD) (5 μg/ml) was added to HEK293T cells 40 hr after siRNA transfection. Total RNAs are prepared at the indicated point after ActD treatment. The band intensity from northern blot was measured by Multi Gauge program(Fuji) and normalized against GAPDH mRNA. The relative DGCR8 mRNA levels were determined from three independent experiments (mean ±SD). A representative data is shown in Figure S3A.
(F) RT-PCR analysis of DGCR8 mRNA in subcellular fractions after RNAi in HEK293T cells. The exon and intron of GAPDH were amplified as markers of cytoplasmic RNA and nuclear RNA, respectively.
Drosha Regulates the DGCR8 mRNA Posttranscriptionally in the Nucleus
To unravel the mechanism of this regulation, we first determined the RNA polymerase density on DGCR8 gene by nuclear runoff assay. For this experiment, a HeLa cell line that expresses primiR-30a from the tetracycline-inducible promoter was used. The inducible pri-miR-30a gene was used as a control for a differentially transcribed gene. Polymerase density on DGCR8 did not change after Drosha knockdown (Figure 1D), despite the increase of the steady-state level of the DGCR8 mRNA (Figure S2A), which indicated that the regulation of DGCR8 may occur posttranscriptionally.
To be certain of this result, we performed qRT-PCR to amplify the intronic region of the DGCR8 pre-mRNA using a primer set that bind to the first intron (Figure 1A). Knockdown of Drosha had no effect on the DGCR8 pre-mRNA level (DGCR8 first intron), whereas it dramatically increased the mature DGCR8 mRNA level (DGCR8 CDS). This was also confirmed by RT-PCR with a different primer set that amplifies the first intron (Int1-Int1) (Figure S2B). Thus, the DGCR8 mRNA is likely to be regulated by Drosha after transcription and splicing of the first intron.
To examine whether Drosha affects the stability of the DGCR8 mRNA, we measured the turnover rate of the mRNA after blocking transcription by treating the cells with actinomycin D (ActD) (Figure 1E and Figure S3). HEK293T cells were first transfected with siRNA and then treated with ActD 40 hr later. Total RNA was subsequently prepared on several time points and analyzed by northern blotting. The DGCR8 mRNA prepared from Drosha-depleted cells was more stable than that from control cells (Figure 1E and Figure S3A). The RT-PCR analysis with RNAs prepared from similarly treated cells confirmed that the half-life of DGCR8 mRNA was lengthened upon Drosha depletion (Figure S3B). Pri-miR-21 and GAPDH transcripts served as the positive and negative controls, respectively.
Because the DGCR8 3′ untranslated region (UTR) is predicted to possess several miRNA target sites, it is conceivable that mRNA destabilization is caused by RISC-associated miRNAs. To test this possibility, we depleted Dicer, which is another essential factor for miRNA biogenesis. The reduction of Dicer did not increase the protein level of DGCR8 under this condition, excluding the possibility that the observed suppression of DGCR8 is mediated by RISC-associated miRNA (Figure 1B). Accordingly, the mRNA level of DGCR8 did not increase in Dicer-depleted cells, as determined by RT-PCR(Figure 1F). Effective depletion of Dicer was confirmed by strong induction of the p27 protein that is a known target of miR-221/222 (Figure 1B) (Visone et al., 2007; Galardi et al., 2007; le Sage et al., 2007).
Furthermore, when the cells were separated into nuclear and cytoplasmic fractions after Drosha depletion, the accumulation of the DGCR8 mRNA was observed in both fractions (Figure 1F, lanes 7 and 11). The overexpression of transdominant negative mutant (TN Drosha) also induced the DGCR8 mRNA accumulation in the nucleus, as well as in the cytoplasm (Figure S4). The siRNA against DGCR8 (siDGCR8) reduced DGCR8 mRNA in the cytoplasm (Figure 1F, lane 12), as expected since siRNAs silence their target mRNAs mainly in the cytoplasm (Zeng and Cullen, 2002). The same siRNA increased DGCR8 mRNA in the nucleus (Figure 1F, lane 8), indicating that DGCR8 protein may be involved in the nuclear destruction of its own mRNA. Because Microprocessor acts in the nucleus, these results indicate that Microprocessor may directly target the DGCR8 mRNA.
Hairpin Structures in the DGCR8 mRNA Are Cleaved by Drosha In Vitro
Pedersen et al. reported that vertebrate DGCR8 mRNAs contain two conserved hairpins in the 5′ UTR (hairpin A) and in the coding sequence near the start codon (hairpin B) (Figure 2A) (Pedersen et al., 2006). These structures were detected by an algorithm called EvoFold, which searches for evolutionarily conserved secondary structures (Pedersen et al., 2006). Because these hairpins have similar features to those of pri-miRNAs (Han et al., 2006), we investigated whether the hairpins can be cleaved by Drosha through in vitro processing assay (Lee et al., 2003). The transcript used for this assay contains the 5′ UTR (+69-+350 nt) and the first 217 nt of the coding region (+351-+567 nt) (Figure 2A). When this RNA was incubated with cell extract or immunoprecipitated protein complex, two fragments were generated, demonstrating that the hairpins can be cleaved by Drosha (Figure 2B). The lengths of these products (~60 nt and ~76 nt) were similar to those of typical pre-miRNAs. In order to validate that these fragments were produced by Drosha not by contaminating nucleases, we performed a processing assay using a Drosha mutant, E110aQ (Figure 2C). The E110aQ contains a point mutation (E to Q) at the catalytic site of RIIIDa (Han et al., 2004). Because this mutant can cleave only the 5′ strand of a hairpin, it released larger cleavage fragments as expected (Figure 2C). Pri-miR-16-1 was used as a control. On the basis of these results, we conclude that the hairpin structures in the DGCR8 mRNA are cleaved by Microprocessor. It is noted that the DGCR8 hairpins are cleaved less efficiently than pri-miR-16-1, which is expected if the DGCR8 hairpins have evolved to modulate, rather than to abrogate, DGCR8 expression. Consistent with this idea, pri-miRNAs such as pri-miR-16-1, pri-let-7a-1, and pri-miR-21 were induced more strongly (4- to 10-fold) than DGCR8 mRNA (3- to 4-fold) in Drosha-depleted cells (Figures 1A and 1F).
Figure 2. The Hairpins in DGCR8 mRNA Are Cleaved by Drosha In Vitro.

(A) Partial sequences of DGCR8 mRNA of human, mouse, rat, chicken, and zebrafish. There are two hairpin structures, hairpin A in the 5′ UTR and hairpin B in the coding region. A black bar presents a template for in vitro transcription. This figure is adapted from Pedersen et al. (2006).
(B) In vitro processing of the DGCR8 hairpins. Internally labeled hairpins were incubated with total cell extract (TCE) or FLAG-immunopurified (FLAG-IP) Drosha. Cleavage fragments are marked by red or blue triangles.
(C) In vitro processing of the DGCR8 hairpins with a Drosha mutant E110aQ. Pri-miR-16-1 was used as a control.
(D) Cleavage site mapping of the two hairpins. The cleavage sites are indicated with triangles. The color of the triangles shows the origin of the fragments presented in Figure 2B. Size of the triangle reflects the clone frequency such that the frequencies of 99%-75%, 74%-50%, and 49%-25% are shown with large, medium, and small triangles, respectively. The cleavage sites with clone frequencies of under 25% are not presented in this figure. Gray lines under the hairpins represent the binding sites for the probes used for northern blotting.
To clarify the identity of the fragments, we determined the cleavage sites by a directional cloning method (Elbashir et al., 2001). The RNAs were gel purified, ligated with adapters, and reverse transcribed. The cDNAs were then amplified by PCR, cloned, and sequenced. We found that the longer cleavage fragment (indicated with a red triangle) was created from the cleavage at the site A1 of hairpin A, whereas the smaller fragments (marked with a blue triangle) were derived from two different cleavage sites-at the site A2 of hairpin A and the site B1 of hairpin B(Figure 2D). The mapping of the cleavage sites indicated that the fragments have short 3′ overhangs (Figure 2D), which were expected for the products of RNase III-mediated cleavage. The cleavage sites were 6-14 nt away from the ssRNA-dsRNA junction, depending on the conditions that were used for secondary structure prediction. This is consistent with the previous results on Drosha cleavage site selection (Han et al., 2006).
Drosha-Mediated Destabilization of the DGCR8 mRNA Is Conserved in Other Species
The DGCR8 mRNA is highly conserved among vertebrates, including mouse, rat, chicken, and zebrafish (Pedersen et al., 2006)(Figure 2A), so we asked whether the regulatory mechanism of DGCR8 is evolutionary conserved. For this, siRNA targeting mouse Drosha was transfected into NIH 3T3 cells (Figure 3A and Figure S5A) and mouse embryonic stem (mES) cells (Figure S6). As in humans, mouse DGCR8 mRNA and protein accumulated when Drosha was knocked down, indicating evolutionary conservation of the regulation.
Figure 3. Conservation of the Regulation of DGCR8.
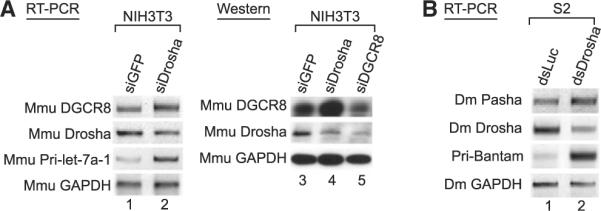
(A) RT-PCR and western blot analysis after mouse Drosha and mouse DGCR8 depletion. Total RNA and cell extract of NIH 3T3 cells were prepared 72 hr siRNA transfection.
(B) RT-PCR analysis after Drosha knockdown in D. melanogaster S2 cells. Total RNA of S2 cells was prepared 7 days after addition of long doublestranded RNAs that target fly Drosha (dsDrosha). Reverse transcription was carried out with random primer. Pri-Bantam was amplified as a positive control.
We also found, using EvoFold, that the 5′ UTR of Drosophila DGCR8/Pasha mRNA contains a hairpin structure, although the sequences themselves are not conserved with vertebrates (Figure S5C). When we knocked-down Drosophila Drosha in S2 cells, Pasha mRNA accumulated (Figure 3B and Figure S5B). This suggests that a similar regulatory mechanism may be present in invertebrates and thus that this regulation may have played a fundamental role throughout animal evolution.
Drosha Cleaves the DGCR8 mRNA In Vivo
To confirm that the Drosha-mediated cleavage event of the DGCR8 mRNA occurs in vivo, we carried out a northern blot assay to detect the cleavage products in the cell. When we used probes that are complementary to hairpin A, we could detect an ~60 nt fragment (Figure 4A). The level of this RNA was reduced by knockdown of Drosha or DGCR8 (Figure 4A, lanes 3 and 4), which is expected for the product of Microprocessor. An ~60 nt RNA was also produced from the plasmid that ectopically expresses the RNA containing both hairpins (Figure 4B, lane 2). This indicated that the ~60 nt band corresponds to the cleavage product generated from hairpin A. When we used probes that are complementary to the stem sequence of hairpin B, we could not detect any band by northern blotting (Figures S6A and S7). This suggested that Drosha may prefer the cleavage site A2 of hairpin A to the other sites for cleavage in vivo (Figure 2D). It is also plausible that the other fragments (the ~76 nt fragment from the site A1 and the ~60 nt fragment from the site B1) may be degraded rapidly in the cell, defying detection by northern blotting.
Figure 4. Drosha Cleaves the DGCR8 mRNA In Vivo.

(A) Northern blot assay to detect the cleavage fragments from the hairpin A in HEK293T cells. Probe is complementary to the 5′ stem region of hairpin A marked as a gray underline in Figure 2D. Small RNAs under 200 nt were enriched after siRNA transfection. As a loading control, tRNA was probed. Protein knockdown was confirmed by western blotting.
(B) Northern blot assay. Total RNAs were prepared from HEK293T cells transfected with either empty vector or the plasmid expressing both hairpins.
(C) Northern blot assay. HEK293T cells were fractionated into nucleus and cytoplasm. The RNAs were extracted from each fraction, dissolved in the same amount of TE, and loaded on the gel. Fractionation efficiency is confirmed by measurement of GAPDH mRNA and pri-miR-21 as the cytoplasmic and nuclear markers, respectively.
Notably, we could not detect any small RNA of ~22 nt, even when small RNA was enriched (Figure 4 and Figure S6A). More-over, we observed that when we carried out subcellular fractionation, the ~60 nt fragment was confined in the nuclear fraction (Figure 4C). This may be because the nuclear export of this ~60 nt fragment is very inefficient or because the fragment gets degraded rapidly in the cytoplasm. Thus, these results suggest that the DGCR8 hairpin may not produce mature miRNA and that the hairpin may function as an RNA instability element rather than as a precursor for miRNA biogenesis.
To ask whether the hairpins are required for Drosha-dependent regulation, we generated luciferase reporter constructs, one containing the DGCR8 hairpins and the other with the hairpins deleted (Figure S8). The reporter construct possessing the hairpin region is induced by Drosha or DGCR8 knockdown, whereas the reporter lacking the hairpins is not responsive to the knockdown. This indicates that the hairpins are necessary for Microprocessor-dependent gene regulation.
DGCR8 Protein Stabilizes the Drosha Protein
When we depleted DGCR8 by RNAi, we also noticed that there may be an additional layer of regulation between Drosha and DGCR8. Namely, the protein level of the endogenous Drosha was downregulated after DGCR8 knockdown (Figure 1B). This reduction at the Drosha protein level was not accompanied by that of the Drosha mRNA level (Figure 1F and Figure S2B).
To further examine the effect of DGCR8 on Drosha, we ectopically expressed FLAG-tagged DGCR8 and Drosha proteins in HEK293T cells. Drosha protein accumulated significantly when coexpressed with DGCR8 protein (Figure 5A). In addition, the endogenous Drosha protein increased when DGCR8 was over-expressed (Figure 5B). To confirm and extend this initial finding, we examined DGCR8 deletion mutants that cannot bind to Drosha. We formerly showed that the mutants DGCR8[1-483] and DGCR8[1-614] do not interact with Drosha, whereas the mutants DGCR8[276-773] and DGCR8[m1&2] can bind to Drosha (Yeom et al., 2006). When coexpressed with Drosha, the DGCR8 mutants that cannot bind to Drosha failed to upregulate Drosha (Figure 5C, lanes 4 and 5). It is noted that we made a related observation with different DGCR8 mutants (Yeom et al., 2006). Taken together, these results indicate that DGCR8 may stabilize Drosha through direct protein-protein interaction.
Figure 5. Drosha Is Positively Controlled by DGCR8 through Protein-Protein Interaction.
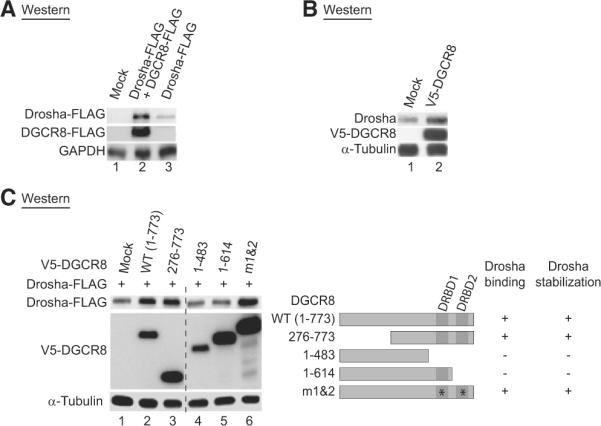
(A) Western blot analysis of the transiently expressed Drosha protein. In HEK293T cells, Drosha-FLAG was transiently transfected with FLAG null vector or DGCR8-FLAG.
(B) Western blot analysis of endogenous Drosha protein after overexpression of DGCR8. V5-tagged DGCR8 was transiently expressed in HeLa cells.
(C) Western blot analysis after transient expression of various DGCR8 mutants. The Drosha-FLAG and V5-DGCR8 mutants were coexpressed in HEK293T cells. Though the juxtaposed lanes are not contiguous, all of them are from a single gel, which is true for all the membranes with dashed lines. DGCR8 mutants were schematized and their Drosha binding abilities are presented on the right.
To further test this hypothesis, we examined Dgcr8 knockout (KO) mouse ES cells (Wang et al., 2007). We compared the protein expression levels in wild-type homozygote (flox/flox), heterozygote (Δ/flox), and KO homozygote (Δ/Δ) lines by western blot assay. In Dgcr8 null ES cells, the Drosha protein levels were significantly lower than that in wild-type cells, whereas they remained unchanged in Dicer KO ES cells (Figure 6A). The Drosha mRNA levels were comparable between wild-type and Dgcr8 null ES cells (Figure 6B). We also examined mouse embryonic fibroblast (MEF) from Dgcr8 KO mouse (Figure S9). Like in ES cells, the Drosha protein was expressed at a lower level (~40%) in the Dgcr8 Δ/Δ MEF compared to the wild-type cells (Figure S9A), even though the mRNA level of Drosha in Δ/Δ MEF remained similar to that in wild-type or Δ/flox MEF (Figure S9B). This result indicates that in the absence of DGCR8, Drosha is downregulated and that DGCR8 may be required to maintain the normal level of Drosha protein.
Figure 6. Expression of Drosha and DGCR8 in Dgcr8 KO Mouse ES Cells.

(A) Western blot analysis of Drosha in mouse ES cells. Δ/flox indicates heterozygote cells, and Δ/Δ indicates homozygous null cells. Two different KO lines (c1 and c2) were used for the experiments (lanes 3 and 4). The Dicer KO ES line (dicer Δ/Δ) was used as a control (Babiarz et al., 2008).
(B) Quantitation of the Drosha mRNA by Affymetrix chip analysis. Three independent experiments were performed (mean ±SD). The methods are described in Wang et al. (2008b).
(C) Western blot analysis of DGCR8 in mouse ES cells. The asterisk represents a nonspecific band which serves as a loading control.
(D) Quantitative real time PCR (qRT-PCR) of the DGCR8 mRNA in Dgcr8 KO mES cell. The relative DGCR8 mRNA levels were determined in wild-type (flox/flox) and heterozygote (Δ/flox). Three independent experiments were performed for quantification (mean ±SD).
Interestingly, we noticed that neither DGCR8 nor Drosha protein levels appeared to be significantly diminished in heterozygote ES or MEF (Figure 6C, lane 2, and Figure S9C, lane 2), although it is expected that only 50% of DGCR8 is expressed in these cells compared to the wild-type cells. To test whether there is compensatory expression from the remaining Dgcr8 allele in the heterozygous background, we performed qRT-PCR for exon 3 of DGCR8, which only measures expression from the wild-type allele (Figure 6D). The DGCR8 mRNA level in heterozygous cells is also ~90% compared to that of the wild-type (Figure 6D). Consistent with the DGCR8 level, the miR-130a level in heterozygous cells is also similar to that of wild-type cells (Figure S9D). This suggests that the reduction of DGCR8 in heterozygous cells resulted in the decrease of Microprocessor activity, which in turn enhanced the expression of DGCR8 protein through the feedback circuit. Thus, the feedback involving Drosha and DGCR8 may play a significant role in vivo in the control of Microprocessor activity and miRNA production.
DISCUSSION
Here, we report two main findings (Figure 7). First, Drosha can act directly to eliminate a specific mRNA. Thus, Drosha plays an additional role in posttranscriptional control apart from miRNA processing. Second, this direct action of Drosha is part of a feedback control system between Drosha and DGCR8, which also includes a protein stabilization component.
Figure 7. Model for the Crossregulation between Drosha and DGCR8.

The Drosha protein cleaves the hairpins on the DGCR8 mRNA and destabilizes the mRNA, while the DGCR8 protein positively regulates the Drosha protein via protein-protein interaction. Drosha may be involved in the stability control of other mRNAs.
Regulation of Microprocessor Activity
Most eukaryotic RNase III proteins interact with dsRBD-containing proteins. It seems to be a general theme that RNase III proteins are stabilized by their binding partners. For instance, human Dicer binds to PACT, a protein containing three dsRBDs (Lee et al., 2006). Depletion of PACT results in the reduction of Dicer protein. Similarly, in Drosophila, Dicer-2, and a dsRNA-binding protein, R2D2, interact with each other and depend on each other for stable expression (Liu et al., 2003). In the present study, we show that DGCR8 stabilizes Drosha protein through protein-protein interaction (Figure 5). Although the molecular details of such protein degradation/stabilization remain to be determined, the data from mouse KO cells suggest that protein stability control of RNase III may be significant in vivo.
Drosha and its cofactor, DGCR8, are engaged in a complex regulatory circuit (Figure 7). If Drosha and DGCR8 levels are elevated in the cell, Microprocessor would cleave and destabilize the DGCR8 mRNA, resulting in the reduction of DGCR8. This will in turn reduces the Drosha protein through protein destabilization, lowering the Microprocessor activity. This autoregulatory feedback circuit may help minimize the potentially harmful fluctuation of Microprocessor activity in the cells. In light of this, it is noteworthy that we observe an interesting dosage compensation effect in Dgcr8 heterozygous cells that contains only one copy of the Dgcr8 gene. In heterozygous cells, the DGCR8 level is expected to be 50% of that in wild-type cells. However, the DGCR8 protein level in heterozygous cells is over 80% of the normal level (Figure 6 and Figure S9). Moreover, the miR-130a level in heterozygous cells is similar to that in wild-type MEF cells (Figure S9). Similarly, the Blelloch group previously showed that miRNA levels in Dgcr8 heterozygous ES cells are over 85% of those in wild-type ES cells (Wang et al., 2007). Thus, Microprocessor activity in heterozygous cells may be maintained at over 80% of that in wild-type cells. It was recently reported that Dgcr8 heterozygous mice produce less miRNAs in the brain than does the wild-type (Stark et al., 2008). However, significant changes were observed only for a small subset of miRNAs, while the majority of miRNAs remained largely unaffected. Taken together, these results indicate that the feedback circuit found in our study may be significant in vivo and contribute to tight control of miRNA production in the cell. The evolutionary evidence also suggests that the regulations between Drosha and DGCR8 may play an important role in controlling miRNA biogenesis in vivo. The hairpins in DGCR8 mRNAs are conserved throughout evolution, and similar regulation occurs not only in mammalian cells but also in insect cells (Figure 3). Further work will be required to confirm the physiological significance of this regulation.
Both Drosha and DGCR8 are ubiquitously expressed proteins, but the levels of these proteins vary depending on the cell types (J.H. and V.N.K., unpublished data). It is conceivable that Drosha and DGCR8 are modulated by additional factors in a cell type-specific manner. Previous studies showed that Drosha and DGCR8 are associated with multiple factors (Gregory et al., 2004; Faller et al., 2007; Shiohama et al., 2007). The functional significance of these interactions remains to be determined. Given the general suppression of miRNAs in cancer and stem cells (Lu et al., 2005; Thomson et al., 2006), it would be particularly interesting to investigate how the miRNA processing activities are modulated during tumorigenesis and cell differentiation.
A Function for Drosha as a Regulator of mRNA Stability
RNase III family members vary widely and play diverse roles in RNA metabolism (MacRae and Doudna, 2007). For instance, yeast RNase III protein, Rnt1, functions in the processing of pre-rRNAs, small nuclear RNAs, and small nucleolar RNAs. Rnt1 also cleaves and controls certain specific mRNAs. Dicer functions mainly in small RNA pathways, but it was recently shown that human Dicer can target mRNAs with CNG repeat-containing hairpins (Krol et al., 2007). Drosha is known to play a critical role in miRNA maturation (Lee et al., 2003). Our present study reveals a function of Drosha in mRNA stability control.
The EvoFold program predicted that vertebrate DGCR8 mRNAs contain two highly conserved hairpin structures, both of which can be cleaved by Drosha in vitro (Figure 2). We detected the cleavage product from the first hairpin by northern blotting (Figure 4). In recent deep sequencing studies, small RNAs from the second hairpin of DGCR8 mRNA were detected and named miR-1306 (Friedlander et al., 2008; Morin et al., 2008). From the peripheral blood of a dog, miR-1306-5p was sequenced (Friedlander et al., 2008), while miR-1306-3p was identified in human embryonic stem cells (Morin et al., 2008). Taken together, both hairpins (A and B) are cleaved by Microprocessor in at least certain cell types. But these hairpins seem to be quite inefficient precursors for miRNA biogenesis because the small RNAs were detected only by massive parallel sequencing approach (Friedlander et al., 2008; Morin et al., 2008). So it remains to be determined whether miR-1306 is a functional miRNA, a by-product of regulated mRNA cleavage, or both.
It is noteworthy that over 10% of the known miRNAs are located in the exonic regions of protein-coding or noncoding transcripts (Kim and Kim, 2007; Morin et al., 2008; Rodriguez et al., 2004). Because Drosha processing is expected to destabilize the host transcripts, it would be interesting to investigate whether such exonic miRNA hairpins serve dual roles as RNA instability elements as well as miRNA precursors. It would also be of great interest to search for additional mRNAs that are controlled by Drosha. To begin investigating this possibility, we performed a series of knockdown and microarray experiments. We found that 335 out of 16,309 mRNAs detected in HeLa cells by Affymetrix chip are upregulated over 2-fold in Drosha-depleted cells without significant changes in Dicer-depleted cells (Figure S10A, a; Group I; shown in red). Most mRNAs that are upregulated in Drosha-depleted cells are also upregulated in DGCR8-depleted cells (Figure S10A, b, and Figure S10B, a; Group II; shown in dark blue). Out of the 335 mRNAs, 104 mRNAs are elevated over 2-fold in DGCR8-depleted cells without changing significantly in Ago2-depleted cells (Figure S10A, b; Group II; shown in dark blue). These genes do not show signifi-cant fold changes in Ago2 knockdown cells (Figure S10B, b). Thus, in HeLa cells, ~100 genes are Microprocessor dependent and RISC independent. Although it is possible that some of these genes are regulated indirectly through unknown mechanism(s), our result implies that there may be a significant number of genes that are directly controlled by Microprocessor.
EXPERIMENTAL PROCEDURES
RNA Interference
Thirty two nanomolar of siRNA duplex were transfected into HeLa, HEK293T, or NIH 3T3 cells with Lipofectamine2000 transfection reagent (Invitrogen) according to the manufacturer's instructions. siRNAs were purchased from Samchully Pharmaceuticals. Target sequences of siDrosha, siDGCR8, and siDicer are 5′-AACGAGUAGGCUUCGUGACUU-3′,5′-AACAUCGGACAAGA GUGUGAU-3′, and 5′-UGCUUGAAGCAGCUCUGGA-3′, respectively. A target sequence of mouse siDrosha, siDGCR8, and siDicer are 5′-AGAUCACCGU CUCUAGAAA-3′,5′- AACAAUUUGGAGCUAGAUGAA-3′ and 5′, ACACAGCA GUUGUCCUAAA-3′.
Fifteen micrograms of long dsRNAs targeted on Dm Drosha was added directly to the media of S2 cells grown in 6 well plates. Cells were treated with dsRNA three times in total on days 0, 2, and 4. The cells were then harvested on day 7.
Long Double-Stranded RNA Production for Fly Gene Knockdown
To generate long double-stranded RNAs, each strand of target mRNA is transcribed by MEGASCRIPT T7 transcription kit (Ambion). Then single-stranded RNAs were annealed by incubation at 65° C for 30 min followed by slow cooling to room temperature. DNA templates for transcription are ~700 bp in length, and they were amplified by PCR. All PCR primers contain T7 RNA polymerase binding site 5′-GAATTAATACGACTCACTATAGGGAGA-3′. Primer sequences after T7 polymerase binding site are the following: Dm Drosha, 5′-GGTGTTC CCTCTCACAACTATTCTTC-3′ (forward) and 5′-GGGCGTTCATCTTCTTCA CATAGTC-3′ (reverse); and Luciferase, 5′- TTGTCAGACACATTTCCGAAA A-3′ (forward) and 5′-TCTAGCTAAGCCGGATCAGCTG-3′ (reverse).
RNA Extraction
HeLa, HEK293T, NIH 3T3, and mES cells were lysed directly in tissue culture plates with Trizol (Invitrogen) reagent. RNA was prepared according to the manufacturer's protocol. Total RNA of S2 cells and Dgcr8 knockout MEF cells was prepared from cell pellets and frozen cell pellets, respectively, with Trizol reagent.
RT-PCR
DNase I-treated total RNAs were used for the first-strand cDNA synthesis. HeLa, HEK293T, NIH 3T3, and DGCR8 KO MEF cDNAs were synthesized with M-MuLV reverse transcriptase (Fermentas) and Oligo-dT primers (Invitrogen) according to the manufacturer's protocols. Reverse transcription of S2 total RNA was carried out with Superscript II (Invitrogen) and random primer (Invitrogen). Dgcr8 knockout mES total RNA was reverse transcribed with Superscript II (Invitrogen) and Oligo dT (Invitrogen). Reverse transcription of fly RNA was carried out with Superscript II and random primer (Invitrogen). Primer sequences are given in Figures S11 and S12.
Quantitative Real-Time PCR
The comparative Ct method with SYBR Green was conducted with the 7300 Real-Time PCR System (Applied Biosystems). Human b-actin mRNA was measured as a control in HeLa cells. In _Dgcr_8 knockout mES cells, mouse b-actin mRNA was used as an endogenous control (Wang et al., 2007), whereas in A3-1 mES cells, mouse GAPDH mRNA was used for normalization. Mouse DGCR8 primers were designed to bind to the exon 3 that is deleted in KO cells. Primer sequences are given in Figures S11 and S13.
Western Blot Analysis
HeLa, HEK293T, or NIH 3T3 cells were collected at 72 hr after siRNA transfection. The cells were dispersed by pipetting in lysis buffer (10 mM Tris at pH 7.4, 1 mM ethylenediaminetetraacetic acid [EDTA] at pH 8.0, 500 mM NaCl, and 0.5% Triton X-100) and incubated for 30 min on ice. Dgcr8 knockout mES cells were collected at subconfluent density 48 hr after plating. Cell pellets were rinsed with phosphate-buffered saline (PBS) then lysed in EBC buffer (50 mM Tris at pH 8.0, 120 mM NaCl, 1% NP-40, and 1× Roche complete inhibitor cocktail). Cells were dispersed by pipetting and incubated at 4°C for at least 45 min while rocking. Thirty to eighty micrograms of protein samples were analyzed on 6%-10% SDS-polyacrylamide gels and transferred to Hybond-C Extra membrane (Amersham) or Immuno-Blot PVDF membrane (Biorad). Primary antibodies used in this study are rabbit anti-Drosha antibody (Upstate Biotechnologies and abCam plc.), rabbit anti-DGCR8 antibody raised against recombinant DGCR8 protein prepared in E. coli (Han et al., 2004), rabbit anti-alpha Tubulin antibody (Abfrontier co., Ltd.), mouse anti-alpha Tubulin (Sigma), goat anti-b-actin (Santa Cruz Biotechnology), mouse anti-GAPDH antibody (Santa Cruz Biotechnology), and mouse anti-p27 antibody (BD Biosciences). For mouse Drosha detection, we use the antibody from abCam.
Nuclear Runoff
Thirty two nanomolar of siGFP and siDrosha were transfected into HeLa cell line that expresses pri-miR-30a from the tetracycline-inducible promoter. Fourteen hours after transfection, cells were split into two dishes. Thirty hours after cell splitting, 3 μg/ml of doxycycline was added to culture media. Nuclei were harvested 39 hr after doxycycline treatment. Nuclear runoff assay was carried out as previously (Kim et al., 2006) with full-length DNA fragment of each gene.
Cloning and Sequencing of Cleavage Products Generated from DGCR8 Hairpins
Hairpins of DGCR8 were transcribed in vitro and processed with immunoprecipitated Drosha-FLAG at 37°C for 60 min. The ~60 nt and ~76 nt bands were gel purified and ligated to the 3′ adaptor. The ligated product was gel purified and ligated to the 5′ adaptor. The 3′ and 5′ adapters used for the cloning are 5′-pUUUaaccgcgaattccagidT-3′ and 5′-acggaattcctcactAAA-3′ (upper case, RNA; lower case, DNA; p, phosphate; idT, inverted deoxythymidine), respectively. For reverse transcription, a primer complementary to the 3′ adaptor (3A-reverse) was used. The sequence of 3A-reverse is 5′-ACTGGAATTCGCGGT TAAA-3′. For determination of the 3′ cleavage site of hairpin A, forward primer (′-GATTTCCAATAATTGAGGCAGTG-3′) and 3A-reverse primer were used for PCR amplification. The PCR product was subcloned into pGEM-T-easy (Promega). For mapping the 3′ end of the 76 nt and 60 nt fragments, 20 and 13 clones were sequenced, respectively. For determination of the 5′cleavage site of hairpin A, forward primer which contains same sequence with 5′ adaptor (5A-forward) (5′-ACGGAATTCCTCACTAAA-3′) and reverse primer (5′-ATTGCTCTTTTCATTAATGTAG-3′) were used for PCR amplification. The PCR product was subcloned into pGEM-T-easy (Promega). For mapping the 5′end of the 76 nt and 60 nt fragments, 16 and 12 clones were sequenced, respectively.
For determination of the 3′ cleavage site of hairpin B, forward primer (5′CAAACGTCCAGTGGTGCAGAG-3′) and 3A-reverse primer were used for PCR amplification. The PCR product was subcloned into pGEM-T-easy (Promega), and 16 clones were sequenced. To determine the 5′ cleavage site of hairpin B, 5A-forward primer and reverse primer (5′- CCACCAGAGCCAACGTCCATTACC-3′) were used for PCR amplification. The PCR product was subcloned into pGEM-T-easy (Promega), and 11 clones were sequenced.
Plasmid Construction
So that the template for in vitro transcription of DGCR8 hairpins could be produced, the partial 5′ UTR and coding regions of human DGCR8 were amplified from HeLa cDNA by PCR (forward primer, 5′-CCTCAGGTAGAAGAAG AAAGG-3′, and reverse primer, 5′-GAAGCTCCGTAGAAGTTGAAG-3′). PCR product was subcloned into pGEM-T easy vector (Promega) and sequenced. For generation of a mammalian expression vector, the clone was inserted into pcDNA3 vector.
Northern Blot Analysis of Small RNAs
Twenty five micrograms of HEK293T small RNA enriched by mirVana (Ambion) and 50 mg of total HEK293T RNA were resolved on 12.5% urea-polyacrylamide gel. Nuclear and cytoplasmic RNA were loaded on 12.5% urea-polyacryl-amide gel with the same volume ratio of prepared RNA after subcellular fractionation of HEK293T cells grown in a 10 cm dish. Then RNA was transferred to a Zetaprobe-GT-membrane (Bio-Rad). Oligonucleotides complementary to the 5′ UTR of DGCR8 were end labeled with T4 polynucleotide kinase (Takara) and used as probes. The sequence of probe in Figure 4 is 5′-TGCCTCAATT ATTGGAAATC-3′.
Northern Blot Analysis of mRNAs
Total RNA was isolated from transfected HEK293T cells with TRIzol reagent (Invitrogen), separated on denaturing formaldehyde agarose gels (15 mg per well), and blotted onto Zeta-probe membranes (BioRad). 32P-labeled probes were generated by the Prime-It Random Primer Labeling Kit (Stratagene) with plasmid DNA fragments as the templates. After incubating the membrane for 30 min at 68°C in prehybridization solution (Express hybridization solution, Clontech), hybridizations were carried out for 80 min at 68°C. Membranes were washed twice for 45 min at room temperature in washing solution I (2× sodium chloride-sodium citrate [SSC], 0.05% sodium dodecyl sulfate [SDS]) and then twice for 30 min at 50° C in washing solution II (0.1× SSC, 1% SDS).
Subcellular Fractionation
HEK293T cells were grown in a 10 cm dish and they were collected in 450 μl of ice-cold buffer A (10 mM HEPES at pH 7.9, 10 mM KCl, 1 mM dithiothreitol [DTT], and 0.1 mM EDTA at pH 8.0). Cells were dispersed by pipetting and incubated for 25 min on ice. Then 5 μl of 10% NP-40 was added, and cells were incubated for 2 min on ice. The nuclei were precipitated by centrifugation at 5000 rpm for 3 min in 4°C. The supernatant was taken as the cytoplasmic fraction.
Supplementary Material
PIIS
ACKNOWLEDGMENTS
We are grateful to Dr. Seogang Hyun for help with S2 transfection, to Dr. Chirlmin Joo for critical reading of this manuscript, and to members of our laboratory for helpful discussion. We also thank Dr. Young-Joon Kim for kindly providing S2 cell. This work was supported by the Creative Research Initiatives Program (R16-2007-073-01000-0) (V.N.K.) and the BK21 Research Fellowships (J.H. and S.C.K.) from the Ministry of Education, Science, and Technology of Korea. J.S.P. was supported by the Danish Medical Research Council (271-06-0711). C.B. was supported through a University of California, San Francisco, Urology Developmental Award and the Prostate Cancer Foundation. R.B. is a Pew Scholar and was supported by National Institutes of Health grants (K08 NS48118, RO1 NS057221), the Prostate Cancer Foundation, and Stem Cell Research Foundation. J.H., S.C.K., Y.K.K., K.H.Y., and W.Y.Y. performed the experiments and analyzed the data. C.B. and R.B. carried out experiments with Dgcr8 KO mES cells, generated Dgcr8 KO MEF, and helped with manuscript preparation. J.S.P. performed bioinformatics analyses with D.H. and helped with editing of paper. J.H. and V.N.K. designed the study and wrote the paper.
Footnotes
REFERENCES
- Ambros V, Bartel B, Bartel DP, Burge CB, Carrington JC, Chen X, Dreyfuss G, Eddy SR, Griffiths-Jones S, Marshall M, et al. A uniform system for microRNA annotation. RNA. 2003;9:277–279. doi: 10.1261/rna.2183803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babiarz JE, Ruby JG, Wang Y, Bartel DP, Blelloch R. Mouse ES cells express endogenous shRNAs, siRNAs, and other Microprocessor-independent, Dicer-dependent small RNAs. Genes Dev. 2008;22:2773–2785. doi: 10.1101/gad.1705308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushati N, Cohen SM. microRNA functions. Annu. Rev. Cell Dev. Biol. 2007;23:175–205. doi: 10.1146/annurev.cellbio.23.090506.123406. [DOI] [PubMed] [Google Scholar]
- Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ. Processing of primary microRNAs by the Microprocessor complex. Nature. 2004;432:231–235. doi: 10.1038/nature03049. [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001;15:188–200. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faller M, Matsunaga M, Yin S, Loo JA, Guo F. Heme is involved in microRNA processing. Nat. Struct. Mol. Biol. 2007;14:23–29. doi: 10.1038/nsmb1182. [DOI] [PubMed] [Google Scholar]
- Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat. Rev. Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- Friedlander MR, Chen W, Adamidi C, Maaskola J, Einspanier R, Knespel S, Rajewsky N. Discovering microRNAs from deep sequencing data using miRDeep. Nat. Biotechnol. 2008;26:407–415. doi: 10.1038/nbt1394. [DOI] [PubMed] [Google Scholar]
- Galardi S, Mercatelli N, Giorda E, Massalini S, Frajese GV, Ciafre SA, Farace MG. miR-221 and miR-222 expression affects the proliferation potential of human prostate carcinoma cell lines by targeting p27Kip1. J. Biol. Chem. 2007;282:23716–23724. doi: 10.1074/jbc.M701805200. [DOI] [PubMed] [Google Scholar]
- Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, Shiekhattar R. The Microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432:235–240. doi: 10.1038/nature03120. [DOI] [PubMed] [Google Scholar]
- Han J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004;18:3016–3027. doi: 10.1101/gad.1262504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Lee Y, Yeom KH, Nam JW, Heo I, Rhee JK, Sohn SY, Cho Y, Zhang BT, Kim VN. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell. 2006;125:887–901. doi: 10.1016/j.cell.2006.03.043. [DOI] [PubMed] [Google Scholar]
- Hebert SS, Horre K, Nicolai L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN, Kauppinen S, Delacourte A, De Strooper B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer's disease correlates with increased BACE1/beta-secretase expression. Proc. Natl. Acad. Sci. USA. 2008;105:6415–6420. doi: 10.1073/pnas.0710263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones-Rhoades MW, Bartel DP, Bartel B. MicroRNAS and their regulatory roles in plants. Annu. Rev. Plant Biol. 2006;57:19–53. doi: 10.1146/annurev.arplant.57.032905.105218. [DOI] [PubMed] [Google Scholar]
- Kim HK, Lee YS, Sivaprasad U, Malhotra A, Dutta A. Muscle-specific microRNA miR-206 promotes muscle differentiation. J. Cell Biol. 2006;174:677–687. doi: 10.1083/jcb.200603008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YK, Kim VN. Processing of intronic microRNAs. EMBO J. 2007;26:775–783. doi: 10.1038/sj.emboj.7601512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krol J, Fiszer A, Mykowska A, Sobczak K, de Mezer M, Krzyzosiak WJ. Ribonuclease dicer cleaves triplet repeat hairpins into shorter repeats that silence specific targets. Mol. Cell. 2007;25:575–586. doi: 10.1016/j.molcel.2007.01.031. [DOI] [PubMed] [Google Scholar]
- Landthaler M, Yalcin A, Tuschl T. The human DiGeorge syndrome critical region gene 8 and Its D. melanogaster homolog are required for miRNA biogenesis. Curr. Biol. 2004;14:2162–2167. doi: 10.1016/j.cub.2004.11.001. [DOI] [PubMed] [Google Scholar]
- le Sage C, Nagel R, Egan DA, Schrier M, Mesman E, Mangiola A, Anile C, Maira G, Mercatelli N, Ciafre SA, et al. Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 2007;26:3699–3708. doi: 10.1038/sj.emboj.7601790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RC, Feinbaum RL, Ambros V. The C. elegans hetero-chronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- Lee Y, Jeon K, Lee JT, Kim S, Kim VN. MicroRNA maturation: stepwise processing and subcellular localization. EMBO J. 2002;21:4663–4670. doi: 10.1093/emboj/cdf476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Rad-mark O, Kim S, et al. The nuclear RNase III Drosha initiates micro-RNA processing. Nature. 2003;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- Lee Y, Hur I, Park SY, Kim YK, Suh MR, Kim VN. The role of PACT in the RNA silencing pathway. EMBO J. 2006;25:522–532. doi: 10.1038/sj.emboj.7600942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Rand TA, Kalidas S, Du F, Kim HE, Smith DP, Wang X. R2D2, a bridge between the initiation and effector steps of the Drosophila RNAi pathway. Science. 2003;301:1921–1925. doi: 10.1126/science.1088710. [DOI] [PubMed] [Google Scholar]
- Liu X, Fortin K, Mourelatos Z. MicroRNAs: biogenesis and molecular functions. Brain Pathol. 2008;18:113–121. doi: 10.1111/j.1750-3639.2007.00121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- MacRae IJ, Doudna JA. Ribonuclease revisited: structural insights into ribonuclease III family enzymes. Curr. Opin. Struct. Biol. 2007;17:138–145. doi: 10.1016/j.sbi.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Morin RD, O'Connor MD, Griffith M, Kuchenbauer F, Delaney A, Prabhu AL, Zhao Y, McDonald H, Zeng T, Hirst M, et al. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res. 2008;18:610–621. doi: 10.1101/gr.7179508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura K, Hagen JW, Duan H, Tyler DM, Lai EC. The mirtron pathway generates microRNA-class regulatory RNAs in Drosophila. Cell. 2007;130:89–100. doi: 10.1016/j.cell.2007.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen JS, Bejerano G, Siepel A, Rosenbloom K, Lindblad-Toh K, Lander ES, Kent J, Miller W, Haussler D. Identification and classification of conserved RNA secondary structures in the human genome. PLoS Comput. Biol. 2006;2:e33. doi: 10.1371/journal.pcbi.0020033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004;14:1902–1910. doi: 10.1101/gr.2722704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby JG, Jan CH, Bartel DP. Intronic microRNA precursors that bypass Drosha processing. Nature. 2007;448:83–86. doi: 10.1038/nature05983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiohama A, Sasaki T, Noda S, Minoshima S, Shimizu N. Nucleolar localization of DGCR8 and identification of eleven DGCR8-associated proteins. Exp. Cell Res. 2007;313:4196–4207. doi: 10.1016/j.yexcr.2007.07.020. [DOI] [PubMed] [Google Scholar]
- Stark KL, Xu B, Bagchi A, Lai WS, Liu H, Hsu R, Wan X, Pavlidis P, Mills AA, Karayiorgou M, et al. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat. Genet. 2008;40:751–760. doi: 10.1038/ng.138. [DOI] [PubMed] [Google Scholar]
- Thomson JM, Newman M, Parker JS, Morin-Kensicki EM, Wright T, Hammond SM. Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes Dev. 2006;20:2202–2207. doi: 10.1101/gad.1444406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visone R, Russo L, Pallante P, De Martino I, Ferraro A, Leone V, Bor-bone E, Petrocca F, Alder H, Croce CM, et al. MicroRNAs (miR)-221 and miR-222, both overexpressed in human thyroid papillary carcinomas, regulate p27Kip1 protein levels and cell cycle. Endocr. Relat. Cancer. 2007;14:791–798. doi: 10.1677/ERC-07-0129. [DOI] [PubMed] [Google Scholar]
- Wang WX, Rajeev BW, Stromberg AJ, Ren N, Tang G, Huang Q, Rigoutsos I, Nelson PT. The expression of microRNA miR-107 decreases early in Alzheimer's disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J. Neurosci. 2008a;28:1213–1223. doi: 10.1523/JNEUROSCI.5065-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Medvid R, Melton C, Jaenisch R, Blelloch R. DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nat. Genet. 2007;39:380–385. doi: 10.1038/ng1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Baskerville S, Shenoy A, Babiarz JE, Baehner L, Blelloch R. Embryonic stem cell specific microRNAs regulate the G1/S transition and promote rapid proliferation. Nat. Genet. 2008b;40:1478–1483. doi: 10.1038/ng.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeom KH, Lee Y, Han J, Suh MR, Kim VN. Characterization of DGCR8/Pasha, the essential cofactor for Drosha in primary miRNA processing. Nucleic Acids Res. 2006;34:4622–4629. doi: 10.1093/nar/gkl458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Cullen BR. RNA interference in human cells is restricted to the cytoplasm. RNA. 2002;8:855–860. doi: 10.1017/s1355838202020071. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PIIS