Fluorescence changes reveal kinetic steps of muscarinic receptor–mediated modulation of phosphoinositides and Kv7.2/7.3 K+ channels (original) (raw)
Abstract
G protein–coupled receptors initiate signaling cascades. M1 muscarinic receptor (M1R) activation couples through Gαq to stimulate phospholipase C (PLC), which cleaves phosphatidylinositol 4,5-bisphosphate (PIP2). Depletion of PIP2 closes PIP2-requiring Kv7.2/7.3 potassium channels (M current), thereby increasing neuronal excitability. This modulation of M current is relatively slow (6.4 s to reach within 1/e of the steady-state value). To identify the rate-limiting steps, we investigated the kinetics of each step using pairwise optical interactions likely to represent fluorescence resonance energy transfer for M1R activation, M1R/Gβ interaction, Gαq/Gβ separation, Gαq/PLC interaction, and PIP2 hydrolysis. Electrophysiology was used to monitor channel closure. Time constants for M1R activation (<100 ms) and M1R/Gβ interaction (200 ms) are both fast, suggesting that neither of them is rate limiting during muscarinic suppression of M current. Gαq/Gβ separation and Gαq/PLC interaction have intermediate 1/e times (2.9 and 1.7 s, respectively), and PIP2 hydrolysis (6.7 s) occurs on the timescale of M current suppression. Overexpression of PLC accelerates the rate of M current suppression threefold (to 2.0 s) to become nearly contemporaneous with Gαq/PLC interaction. Evidently, channel release of PIP2 and closure are rapid, and the availability of active PLC limits the rate of M current suppression.
INTRODUCTION
G protein–coupled receptors (GPCRs) comprise the largest receptor family in the human genome, mediate a vast array of cellular processes, and constitute a large fraction of current pharmaceutical targets. GPCR signal transduction pathways use diverse signaling mechanisms and kinetics, and only a few G protein–coupled systems have received much quantitative attention. Recent studies reveal nuances in GPCR-G protein specificity (Kenakin, 1997), G protein heterotrimer stability (Evanko et al., 2005; Digby et al., 2006; Yuan et al., 2007), G protein trafficking among membranes (Chisari et al., 2007; Saini et al., 2007), and spatial organization of GPCRs with G proteins and effectors (Nobles et al., 2005; Dowal et al., 2006). We seek to deepen understanding of GPCR signaling by analyzing the underlying kinetics of the relatively slow modulation of a K+ channel by muscarinic receptors.
Activation of Gq/11-coupled muscarinic acetylcholine receptors in sympathetic neurons attenuates M-type potassium current and thus increases neuronal excitability (Brown and Adams, 1980; Brown, 1983). M current, an outwardly rectifying neuronal potassium current encoded by KCNQ2 and KCNQ3 (Kv7.2 and 7.3) channel subunits (Wang et al., 1998), requires phosphatidylinositol 4,5-bisphosphate (PIP2) to be active (Suh and Hille, 2002; Zhang et al., 2003). Muscarinic modulation of M current acts through a chain of events: Gαq activates phospholipase C-β (PLCβ), which hydrolyzes PIP2 to generate inositol 1,4,5-trisphosphate (IP3) and diacylglycerol. PIP2 is a principal determinant of M current activity, and its depletion induces closure of Kv7.2/7.3 channels (Suh et al., 2006). Signal transduction through these steps from receptor to channel requires 10–15 s to come to completion.
Previously, we formulated a preliminary kinetic model for the steps from activation of the M1 muscarinic acetylcholine receptor (M1R) to closure of Kv7.2/7.3 channels (Suh et al., 2004). We found, however, that many intermediate rate constants were not constrained by empirical measurements. Here, we use optical signals likely to represent fluorescence resonance energy transfer (FRET) to tease apart these steps. We wish to resolve which steps contribute to the relative slowness of this signal. FRET is an optical technique that relies on the close proximity (<100 Å) of two fluorophores to monitor their relative molecular dynamics in intact cells in real time. Changes in FRET can reveal the kinetics of changes in protein conformation (intramolecular) or interaction (intermolecular). FRET has been used to determine the kinetics of signaling of several GPCRs, with a focus on Gi/o- and Gs-coupled systems (Lohse et al., 2007a,b, 2008). Turning our attention to the Gq-coupled M1R, we used FRET experiments to probe the kinetics of receptor activation, G protein activation and rearrangement, PLC activation, and PIP2 hydrolysis. Electrophysiology was used to examine Kv7.2/7.3 channel closure. In this initial report, we emphasize the relative timing of the optical signals without close attention to their amplitude or to full kinetic modeling.
MATERIALS AND METHODS
Constructs
Cerulean, a variant of enhanced cyan fluorescent protein (ECFP), was appended to mouse M1 receptor cDNA (provided by N. Nathanson, University of Washington, Seattle, WA) after Cys460 at the C terminus to generate M1R-Cerulean. To generate the intramolecular fluorescent probe M1R-EYFP-Cerulean, enhanced yellow fluorescent protein (EYFP) replaced a segment between Ala223 and Val358 in the third intracellular loop of the Cerulean-labeled receptor.
cDNAs for other fluorescent probes were obtained through the generosity of other laboratories: mouse Gαq-ECFP (Hughes et al., 2001; Scarlata and Dowal, 2004) from C. Berlot (Geisinger Clinic, Danville, PA); bovine EYFP-Gβ1 and ECFP-Gγ2 (Ruiz-Velasco and Ikeda, 2001) from S. Ikeda (National Institutes of Health, Rockville, MD); rat EYFP-PLCβ1 (Scarlata and Dowal, 2004) from L. Runnels (University of Medicine and Dentistry, Piscataway, NJ); and human pleckstrin homology (PH) domain probes PH(PLCδ1)-ECFP and PH(PLCδ1)-EYFP (van der Wal et al., 2001) from K. Jalink (The Netherlands Cancer Institute, Amsterdam, Netherlands). For some controls we used ECFP-Mem, an ECFP that becomes palmitoylated and localizes mostly to the plasma membrane (Bal et al., 2008), from M. Shapiro (University of Texas Health Sciences, San Antonio, TX). Hereafter, we refer to fluorophores simply as CFP or YFP regardless of whether regular or enhanced fluorescent proteins were used.
Plasmids containing unlabeled human Gαq, Gβ1, and Gγ2 were from the Missouri S&T cDNA Resource Center, human KCNQ2 and rat KCNQ3 were from D. McKinnon (State University of New York, Stony Brook, NY), and bovine GPCR kinase 2 (GRK2) was from M. Bünemann (University of Würzburg, Würzburg, Germany).
Cell culture
All experiments were performed on transiently transfected tsA-201 cells. The 2-ml transfection medium contained 10 µl Lipofectamine-2000 and 0.2–0.8 µg of each cDNA. For better membrane expression of any G protein subunit probe, we always transfected three G protein subunits (α, β, and γ) together. The next day, cells were plated onto poly-l-lysine–coated #0 glass coverslip chips, and fluorescent cells were studied 36–48 h after transfection.
Epifluorescence photometry
To measure fluorescence interactions between CFP and YFP, we made photometric measurements on single cells using an epifluorescence microscope equipped with two photomultipliers in photon-counting mode. The cells were excited by shutter-controlled light from a 75-W xenon arc lamp and measured on an inverted Nikon diaphot microscope using a 40×, 1.3 numerical aperture oil-immersion objective. Excitation light passed through a 0.2 neutral density filter and a cube containing a 440 ± 10 nm bandpass excitation filter and a 465-nm dichroic mirror. This cube excites CFP and not YFP, and transmits light from both CFP and YFP emissions. The entire cell was centered within a circular pinhole at the image plane of the side port of the microscope, and the total light in this circular field of view was pooled and counted. Emitted light was separated by two cubes in series: a 505-nm dichroic mirror with a 480 ± 15 nm bandpass filter deflected light to one photomultiplier tube (“short-wavelength channel”), and a 570-nm dichroic mirror with a 535 ± 12.5 nm bandpass filter deflected light to the other photomultiplier tube (“long-wavelength channel”). Cells were also epi-illuminated with red light, and a CCD camera with video monitor collected undeflected light above 570 nm to visualize the positioning of the single cell within the pinhole.
For slow sampling, the shutter was opened for 24 ms every 100 or 500 ms. For fast sampling, the shutter remained open and the photon counters were activated for 24 ms every 50 ms. Shutter and counters were controlled by an in-house DOS-based program. Solution exchange was accomplished by a theta tube moved laterally by a step-driven motor (Warner Instruments) and was complete within 50 ms. Cells were simultaneously subjected to continuous slow bath flow of Ringer's solution.
The fluorescence ratio was taken as the ratio of YFP to CFP emission (YFPC/CFPC) during 440-nm illumination after corrections for background fluorescence and bleed-through determined in separate experiments on cells transfected with single fluorophores. The subscript C is a reminder that the excitation light is exciting CFP in both cases. In single-fluorophore control experiments, the fraction of CFP emission that shows up in the long-wavelength channel is 0.17, and the fraction of YFP emission that shows up in the short-wavelength channel is 0.00. Direct excitation of YFP by 440 nm light was small and not corrected for. In principle, any correction would be proportional to YFP expression levels. If LW is the background-corrected number of counts in the long-wavelength channel, and SW is the number in the short-wavelength channel, the corrected fluorescence values are:
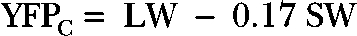 |
(1) |
|---|
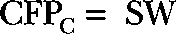 |
(2) |
|---|
The ratio of these quantities, YFPC/CFPC, is often called the FRET ratio (Bünemann et al., 2003; Lohse et al., 2003; Vilardaga et al., 2003; Frank et al., 2005; Hein et al., 2005, 2006), but here we will call it FRETr to indicate that we use a common formula for FRET ratio but have not entirely proven that all the signals represent true FRET.
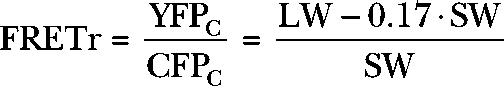 |
(3) |
|---|
For questions of how long it takes for a certain step in the signaling cascade to be changed by agonist addition, it is not important whether FRETr is in fact FRET. Nevertheless, in Results and Discussion we give lines of evidence that our calculated FRETr represents proper FRET. Slow photobleaching occurs during the measurements, but it had negligible effects on the FRETr for the experiment durations and sampling frequencies we used.
For each pair of fluorescent constructs studied we provide three lines of evidence that the baseline ratios and agonist-induced signals calculated by Eq. 3 represent FRET rather than some other optical change. (1) During perfusion of agonist, the CFPC and YFPC values invariably changed in opposite directions with identical time courses. (2) The changes in the calculated FRETr ratio were nearly fully reversed by removing the agonist. (3) When strong illumination at 500 nm was used to bleach the YFP fluorophore, CFPC increased appreciably and the calculated baseline FRETr ratio fell to near zero. This experiment, donor dequenching after acceptor photobleaching, was performed on separate populations of cells under the same transfection conditions used for kinetic FRETr measurements. Bleaching was accomplished by a 5-min illumination without the neutral density filter and using a YFP filter cube containing a 500 ± 10-nm bandpass excitation filter for YFP excitation, a 515-nm dichroic mirror, and a 535 ± 15-nm bandpass emission filter. Control experiments measuring YFP photon counts showed that YFP was bleached with an exponential time constant of ∼60 s with this steady light, and YFP fluorescence was reduced by 94% after 5 min of illumination. In cells expressing membrane-directed CFP-Mem only, CFP was bleached 6.5 ± 1.5% (n = 6) in this time. Control experiments using a presumed non-interacting pair of fluorophores, CFP-Mem and PLC-YFP, showed an average increase in CFPC of 9.0 ± 1.9% (n = 6) after acceptor photobleaching, confirming minimal energy transfer. This value has been corrected for 6.5% CFP bleaching, as have all values reported later for donor dequenching after acceptor photobleaching.
We performed control experiments to test the function of fluorescent constructs. Calcium photometry and electrophysiology confirmed that the M1R-CFP construct coupled appropriately to modulate intracellular Ca2+ and M current with standard kinetics and efficacy. However, the M1R-YFP-CFP construct failed to couple effectively to M current, likely because the YFP insert disrupts association with G proteins. Electrophysiology confirmed the coupling of other fluorescent constructs. To ensure the specificity of FRETr responses to muscarinic agonist oxotremorine-methiodide (oxo-M), we confirmed that coincubation with 10 µM of muscarinic antagonist atropine blocked oxo-M–induced FRETr changes in all construct pairs studied. Atropine alone had no effect on FRETr for most pairs of constructs; when FRETr changes were observed, they were minimal and opposed the direction of oxo-M–induced changes.
Cell selection for photometry
After transfection of fluorescent proteins, the cell population is not uniform. Fewer than 10% of the cells are bright enough to use for photometry, and some of these are too bright. We selected cells for study under 440-nm illumination on the basis of several criteria. The short-wavelength counts had to be in the range of 500–12,000 counts (per 24 ms). The long-wavelength counts had to exceed the value expected from simple CFP bleed-through into the YFP channel. These criteria ensure adequate expression of CFP and YFP. The cell had to be firmly adherent to the substrate. For photometry, confocal microscopy, and patch clamp, we often chose cells that were slightly rounded rather than strongly flattened. They were easier to patch onto with a pipette, and in confocal optical section, they had a clearer vertical region of plasma membrane, permitting us to assess membrane localization of the probes. All such cells responded robustly in patch clamp (current measurement) and photometry to the muscarinic agonist oxo-M. Finally, we did not use cells that had bright fluorescent regions inside the cell.
Confocal fluorescence imaging
To verify membrane expression, cells were imaged using a Leica SP1 confocal microscope with a 63× water or 100× oil-immersion objective. The confocal images shown in several figures were used to determine subcellular localizations of probes, but not for any of the FRETr calculations. Cells pictured in confocal images are different from those on which FRETr calculations were performed. For cyan images, the cells were illuminated with the 457-nm laser line (RSP465 beam splitter), and light from 462 to 551 nm was collected. For yellow images, the cells were illuminated with the 488-nm laser line (RSP500 beam splitter), and light from 523 to 593 nm was collected. Both because the 457-nm line excites CFP inefficiently (it is much weaker than the 488-nm line) and because CFP is intrinsically less bright, the confocal images for CFP required higher gain than those for YFP, in contrast to the epifluorescence photometry experiments using only 440-nm light, where the CFPC counts were larger than the YFPC counts. The confocal images shown are labeled cyan and yellow and represent the raw data with no corrections.
Current recording and analysis
We recorded M currents from voltage-clamped cells in whole cell configuration at room temperature (23°C). Electrodes had resistances of 1–3 MΩ. The whole cell access resistance was 2–5 MΩ, and series-resistance errors were compensated 70%. Fast and slow capacitances were also compensated. M current was measured using a standard deactivation protocol: cells were held at −20 mV, and a 500-ms hyperpolarizing step to −60 mV was applied every 4 s. Data acquisition and analysis used PULSE software in combination with an EPC-9 amplifier (HEKA).
Radioligand binding
tsA cells were grown and transfected in 150-mm cell culture plates. Membranes were prepared using a cell harvester (Brandel) and radioligand binding was assayed as described previously (Chen et al., 2004). Receptor dissociation constants (Kd) were determined by saturation binding assays with the M1R-specific antagonist _N_-methyl-3_H_-scopolamine (3H-NMS), and receptor inhibition constants (Ki) were determined by competition binding experiments including 1 nM 3H-NMS and 0.1 nM to 300 µM oxo-M. Nonspecific binding was determined in the presence of 10 µM atropine. Samples were counted with a Packard Tri-Carb 2200 CA liquid scintillation analyzer (PerkinElmer). Each result reflects two experiments performed in triplicate. Saturation and competition binding curves were fitted with rectangular hyperbolas for one-site binding. Inhibition constants were determined using the Cheng-Prusoff equation.
Solutions and materials
The external Ringer's solution used for photometry and current recording contained (in mM): 160 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 8 glucose, adjusted to pH 7.4 with NaOH. The pipette solution contained (in mM): 175 KCl, 5 MgCl2, 5 HEPES, 0.1 BAPTA, 3 Na2ATP, and 0.1 Na3GTP, adjusted to pH 7.4 with KOH.
Atropine, oxo-M, and poly-l-lysine were from Sigma-Aldrich. DMEM, Lipofectamine-2000, and penicillin/streptomycin were from Invitrogen. Fetal bovine serum was from Gemini Bio-Products. 3H-NMS was from PerkinElmer.
Data analysis
Data analysis was performed using IGOR Pro (Wavemetrics). Traces of FRETr or current versus time were fitted with a linear delay to accommodate the time required by preceding steps, followed by a single-exponential component. Fitting was performed with a least-squares criterion to determine delays and time constants (τ) of activation and deactivation. The fitted equations during agonist onset were:
 |
(4a) |
|---|
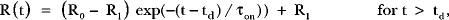 |
(4b) |
|---|
where R is the FRETr, R0 and R1 are the baseline and final values, and td is the time delay. For receptor activation, kon was taken as the slope of 1/τon versus oxo-M concentration, and koff was 1/τoff. Half-maximal effective concentrations (EC50) of agonist were obtained from fits of the Hill equation to graphs of normalized, steady-state amplitude change versus oxo-M concentration. Error for EC50 is reported as the standard deviation of the fit parameter in IGOR, a measure analogous to the SEM. Elsewhere, reported errors are SEM.
Online supplemental material
Fig. S1 has two graphs showing radioligand saturation and competition binding data for receptor constructs expressed in tsA cells. It is available at http://www.jgp.org/cgi/content/full/jgp.200810075/DC1.
RESULTS
M1R activation
We examined receptor activation by measuring intramolecular FRETr in the double-labeled receptor construct, M1R-YFP-CFP (Fig. 1 A). Imaging in a confocal microscope confirmed that the construct localized principally to the plasma membrane (Fig. 1 B). In our epifluorescence photometry apparatus, YFPC (acceptor) fluorescence was large, although the excitation light (440 nm) excited only CFP (donor), as would be expected for an intramolecular FRET interaction with fluorophores in close proximity. The calculated resting FRETr for the receptor construct (0.88) was much larger than the intermolecular FRETr for the other probe combinations we studied here. As evidence that this resting FRETr actually represents FRET between the fluorophores, we found that bleaching the YFP fluorophore with 5 min of 500 nm light increased CFPC counts by 82 ± 4% and decreased the calculated baseline FRETr to 0.02 (n = 7). Washing 10 µM of the muscarinic agonist oxo-M onto cells expressing M1R-YFP-CFP resulted in a rapid increase of acceptor YFPC counts (Fig. 1 C, yellow line) and a decrease of donor CFPC counts (blue line) corresponding to an increase in FRETr (black line). Averaging five agonist exposures in a single cell, Fig. 1 D shows that the FRETr rose 6% above the already high baseline. The rising phase could not be resolved, as it exceeded the 10-Hz sampling frequency. Faster sampling required leaving the shutter open and resulted in excessive bleaching of the construct, which confounded kinetic measurements. The FRETr change was readily reversed upon agonist washout; the falling phase was fitted with a single-exponential time constant of 180 ms. Table I summarizes these and subsequent kinetic measurements.
Figure 1.

Kinetics of M1R activation. (A) Cartoon of the double-labeled M1R construct, M1R-YFP-CFP. (B) Confocal images of three resting cells, only one of which expresses the M1R. The transfected cell shows the distributions of cyan and yellow fluorescence. Bar, 10 µm. (C) FRETr photometry time course for a single cell. The top panel shows corrected CFPC fluorescence (blue trace, left axis) and YFPC fluorescence (yellow trace, right axis), and the bottom panel shows the corrected ratio, YFPC/CFPC (black), for a 5-s exposure to 10 µM oxo-M. Sampling frequency: 1 Hz during baseline and 10 Hz during agonist. (D) Normalized mean time course for five 5-s exposures to oxo-M in a single cell (same cell as C). Black line is a single-exponential fit with τoff = 180 ms.
TABLE I.
Summary of kinetics
| Step | Probes | Resting FRETr % | ΔFRETr % | Delayon s | τon s | Delayoff s | τoff s | EC50 (oxo-M) nM |
|---|---|---|---|---|---|---|---|---|
| M1R activation | M1R-YFP-CFP | 0.88 | +6 | <0.1 | 0.18 | |||
| M1R/Gβ interaction | M1R-CFP | 0.42 ± 0.07 | +33 ± 6 | 0.20 ± 0.03 | 3.7 ± 0.2 | 330 ± 150 | ||
| Gβ1-YFP | ||||||||
| Gαq/Gβ1 separation | Gαq-CFP | 0.15 ± 0.01 | −10 | 2.0 | 5.8 | 35 | ||
| Gβ1-YFP (with GRK2) | 0.26 ± 0.03 | −17 ± 2 | 0.14 ± 0.05 | 2.8 ± 0.3 | 9.9 ± 2.9 | 28 ± 2 | 160 ± 100 | |
| Gαq/PLCβ1 interaction | Gαq-CFP | 0.14 ± 0.03 | +20 ± 2 | 0.38 ± 0.25 | 1.3 ± 0.3 | 0.34 ± 0.14 | 3.6 ± 0.5 | 260 ± 190 |
| PLCβ1-YFP | ||||||||
| PIP2 hydrolysis | PH(PLCδ1)-CFP | 0.14 ± 0.03 | −44 ± 3 | 1.3 ± 0.2 | 5.4 ± 1.6 | 29 ± 2 | 59 ± 7 | 28 ± 14 |
| PH(PLCδ1)-YFP | ||||||||
| Kv7.2/7.3 closure | M current | 1.4 ± 0.3 | 5.0 ± 0.6 | 34 ± 6 | 123 ± 20 | 120 ± 100 | ||
| Kv7.2/7.3 closure with PLCβ | M current | 0.78 ± 0.07 | 1.2 ± 0.1 | 11 ± 7 | 62 ± 22 | |||
| PLCβ1-YFP | ||||||||
| Kv7.2/7.3 closure with PH probes | M current | 2.1 ± 0.1 | 5.7 ± 0.7 | 11 ± 5 | 63 ± 9 | |||
| PH(PLCδ1)-CFP | ||||||||
| PH(PLCδ1)-YFP |
M1R affinity
Because coupling to G proteins was compromised in the M1R-YFP-CFP construct (see Materials and methods), we wanted to verify that its ligand binding was close to that for wild-type M1R. Using a radioactive ligand, we measured saturation (Fig. S1 A) and competition binding curves (Fig. S1 B) for membranes containing wild-type M1R, M1R-CFP, or M1R-YFP-CFP, and for untransfected membranes. Dissociation constants (Kd) for the radioactive M1 receptor ligand 3H-NMS were not significantly different among the three receptor constructs (mean ± SEM): wild-type M1R, 740 ± 580 pM; M1R-CFP, 940 ± 400 pM; and M1R-YFP-CFP, 760 ± 510 pM. The number of binding sites in untransfected membranes was negligible. Oxo-M inhibition constants, which should represent the apparent Kd for oxo-M at M1Rs, were also very similar: wild-type M1R, 9.2 ± 7.4 µM; M1R-CFP, 6.2 ± 1.7 µM; and M1R-YFP-CFP, 4.2 ± 1.0 µM. Thus, ligand binding remained normal in the compromised receptor.
M1R/G protein interaction
Next, we measured coupling kinetics between receptor and G protein using M1R-CFP and Gβ1-YFP constructs (Fig. 2 A). When coexpressed with unlabeled G protein subunits Gαq and Gγ2, these constructs localized primarily to the plasma membrane, with a small intracellular component (Fig. 2 B). Baseline FRETr averaged 0.42. Bleaching the YFP fluorophore with 5 min of 500 nm light increased FCFP by 10.2 ± 0.5% and decreased the baseline FRETr to 0.01 (n = 8). Application of 10 µM oxo-M consistently produced robust increases in YFPC and decreases in CFPC, and the FRETr rose 33% above baseline on average (Fig. 2 C). The rising phase had an average time constant of 200 ms, and the falling phase had an average of 3.7 s. Changes in amplitude were concentration dependent, as shown in the time course of FRETr as the oxo-M concentration was varied from 10 nM to 50 µM (Fig. 2 E). Normalizing responses like these to their maximal effect at 50 µM and averaging over several cells revealed a half-maximal effective concentration (EC50) of 330 nM oxo-M by Hill fit (Fig. 2 F). Apparently, half-maximal interaction between receptors and Gβ requires much less than half-maximal receptor occupancy (compare Fig. S1 B).
Figure 2.

Kinetics of M1R/Gβ1 interaction. (A) Cartoon of M1R-CFP and Gβ1-YFP constructs and cognate G proteins. (B) Confocal images of a pair of cells expressing M1R-CFP and Gβ1-YFP. Bar, 10 µm. (C) FRETr photometry time course for a single cell undergoing a 5-s exposure to 10 µM oxo-M. The top panel shows CFPC fluorescence (blue trace, left axis) and YFPC fluorescence (yellow trace, right axis), and the bottom panel shows the ratio, YFPC/CFPC (black). Sampling frequency: 2 Hz during baseline and 20 Hz during agonist. (D) Mean time course for 5-s exposures to oxo-M in six cells. Note the different time scales for onset and washout. Mean ± SEM (E) FRETr time course for a single cell. Oxo-M was stepped to different concentrations ranging from 10 nM to 50 µM as labeled. (F) FRETr concentration–response curve from steady-state values in E for six cells. Mean ± SEM.
G protein separation
We looked for interactions within G protein heterotrimers by measuring FRETr changes between Gαq-CFP and Gβ1-YFP (Fig. 3 A). The resting FRETr ratio averaged 0.15 and always decreased after receptor activation with 10 µM oxo-M. However, on-kinetics varied widely across cells (τon from 0.8 to 10 s) and were obscured by poor signal-to-noise ratios. Averaging records from 10 cells, we found a 10% reduction in FRETr with a mean τon of 2.0 s and a τoff of 35 s after a 5.8-s delay (Fig. 3 D, open circles). The delay presumably reflects the time taken by preceding steps.
Figure 3.

Kinetics of Gαq/Gβ1 separation. All cells coexpress M1R, Gαq-CFP, Gβ1-YFP, Gγ2, and GRK2, except GRK2 is absent in one part of D. (A) Cartoon of Gαq-CFP and Gβ1-YFP constructs and cognate G proteins. (B) Confocal images of a group of cells expressing Gαq-CFP and Gβ1-YFP in the presence of GRK2. Bar, 10 µm. (C) FRETr photometry time course for a single cell undergoing a 10-s exposure to 10 µM oxo-M. The top panel shows CFPC fluorescence (blue trace, left axis) and YFPC fluorescence (yellow trace, right axis), and the bottom panel shows the ratio, YFPC/CFPC (black). Sampling frequency: 2 Hz during baseline and 10 Hz during agonist. (D) Mean time course for 10-s exposures to oxo-M in 10 cells in the absence (open circles) and 8 cells in the presence (closed circles) of GRK2. Note the different time scales for onset and washout. Mean ± SEM. For clarity in display, points were pooled in 500-ms bins for onset and 4-s bins for washout. (E) FRETr time course for a single cell. Oxo-M was stepped to different concentrations ranging from 1 nM to 10 µM as labeled. For clarity in display, trace is smoothed. (F) FRETr concentration–response curve from steady-state values in E for six cells. Mean ± SEM.
Experiments in the laboratory of Moritz Bünemann (Schliefenbaum, J., A.K. Kreile, M.J. Lohse, and M. Bünemann. 2008. Biophysical Society Meeting. Abstr. 1977) suggested that GRK2 could increase the amplitude of G protein FRET changes. In addition to binding GPCRs, GRK2 also binds both Gαq and Gβγ. The binding sites for these subunits are separated by 80–100 Å, as deduced from the crystal structure (Lodowski et al., 2003). Selecting transfected cells with primarily plasma membrane fluorescence (Fig. 3 B), we found that GRK2 increased the resting FRETr and the agonist-induced loss of FRETr relative to the new baseline (Fig. 3 C). Resting FRETr averaged 0.26 and decreased 17% with 10 µM oxo-M (Fig. 3 D, closed circles). The kinetics were largely unchanged but more statistically robust compared with cells not transfected with GRK2. The average τon was 2.8 s after a 140-ms delay, and the average τoff was 28 s after a 10-s delay. Serial concentration–response experiments (Fig. 3 E) gave an EC50 of 160 nM oxo-M (Fig. 3 F), similar to that for receptor–Gβ interaction. Bleaching the YFP fluorophore with 5 min of 500 nm light increased CFPC by 18 ± 2% and decreased the baseline FRETr to 0.02 (n = 8).
G protein/PLC interaction
To examine the kinetics of PLC activation, we measured FRETr between Gαq-CFP and PLCβ1-YFP (Fig. 4 A). These probes, when coexpressed with M1R and unlabeled G protein subunits Gβ1 and Gγ2, localized primarily to the plasma membrane (Fig. 4 B). Some intracellular fluorescence could be seen in the cyan channel. Baseline FRETr averaged 0.14. Bleaching the YFP fluorophore with 5 min of 500 nm light increased CFPC by 12.1 ± 0.9% and decreased the baseline FRETr to 0.01 (n = 4). Application of 10 µM oxo-M produced opposing changes in YFP and CFP fluorescence, and a reliable increase in FRETr averaging 20% above baseline (Fig. 4 C). Fitting with single exponentials yielded mean time constants of 1.3 s after a 380-ms delay for the rising phase and 3.6 s after a 340-ms delay for the falling phase. Changes in the FRETr amplitude were concentration dependent (Fig. 4 E), with an EC50 of 260 nM oxo-M (Fig. 4 F), similar to that for the two preceding steps.
Figure 4.

Kinetics of Gαq/PLCβ1 interaction. (A) Cartoon of Gαq-CFP, PLCβ1-YFP, and cognate G proteins. (B) Confocal images of a pair of cells coexpressing Gαq-CFP and PLCβ1-YFP. Bar, 10 µm. (C) FRETr photometry time course for a single cell undergoing a 5-s exposure to 10 µM oxo-M. The top panel shows CFPC fluorescence (blue trace, left axis) and YFPC fluorescence (yellow trace, right axis), and the bottom panel shows the ratio, YFPC/CFPC (black). Sampling frequency: 2 Hz during baseline and 20 Hz during agonist. (D) Mean time course for 5-s exposures to oxo-M in 10 cells. Mean ± SEM. Note the different time scales for onset and washout. For clarity in display, points from fast sampling were pooled in 200-ms bins. (E) FRETr concentration–response time course for a single cell. Oxo-M from 10 nM to 50 µM as labeled. For clarity in display, trace is smoothed. (F) FRETr concentration–response curve from steady-state values in E for four cells. Mean ± SEM.
PIP2 hydrolysis
To examine changes in PIP2 concentration after PLC activation, we used the PIP2-binding PH(PLCδ1) translocation probe (Fig. 5 A). This probe binds the phosphoinositol headgroup of PIP2 and IP3 within cells and translocates from the membrane to the cytosol when PIP2 is hydrolyzed to IP3 (Stauffer et al., 1998). We measured FRETr between coexpressed PH-CFP and PH-YFP (van der Wal et al., 2001). At rest, the PH probes were primarily localized to the plasma membrane where some of them were in sufficiently close proximity to allow optical interaction to occur (baseline FRETr averaged 0.14). Bleaching the YFP fluorophore with 5 min of 500 nm light increased CFPC by 24 ± 4% and decreased the baseline FRETr to 0.02 (n = 9). Upon application of 10 µM oxo-M, translocation of fluorescence to the cytosol was evident in most cells. It was accompanied by opposing large changes in YFPC and CFPC, and a dramatic drop in the FRETr as the probe molecules leave the membrane. The effect was reversible upon washout (Fig. 5 B). Fig. 5 C shows a robust decrease in FRETr with 10 µM oxo-M, averaging 44% (Fig. 5 D). The FRETr decayed after a 1.3-s delay with a time constant of 5.4 s. Recovery after washout had a 29-s latency and a time constant of 59 s. Decreases in the FRETr amplitude were concentration dependent (Fig. 5 E) with an EC50 of 28 nM (Fig. 5 F), meaning that when compared with the EC50 of other steps (Table I), a very small receptor occupation and a small PLC activation suffice for extensive cleavage of PIP2.
Figure 5.

Kinetics of PIP2 hydrolysis. (A) Cartoon of PH(PLCδ1)-CFP, PH(PLCδ1)-YFP, Kv7.2/7.3 channels, and PIP2. PLC hydrolyzes PIP2 to send PH probes to the cytosol. (B) Confocal images of three cells expressing PH-CFP and PH-YFP. Bar, 10 µm. (C) FRETr photometry time course for a single cell undergoing a 20-s exposure to 10 mM oxo-M. The top panel shows CFPC fluorescence (blue trace, left axis) and YFPC fluorescence (yellow trace, right axis), and the bottom panel shows the ratio, YFPC/CFPC (black). Sampling frequency: 2 Hz throughout. (D) Mean time course for 20-s exposures to oxo-M in 22 cells. Mean ± SEM. Note the different time scales for onset and washout. For clarity in display, points were pooled in 1-s bins for onset and 10-s bins for washout. (E) FRETr concentration–response time course for a single cell. Oxo-M from 1 nM to 10 µM as labeled. (F) FRETr concentration–response curve from steady-state values in E for 12 cells. Mean ± SEM.
Channel closure
Whole cell voltage clamp was used to measure currents from cells expressing M1R and M channel subunits Kv7.2 and Kv7.3. We began with cells not transfected with additional G protein subunits or PLC. M current at −20 mV was almost completely suppressed by 10 µM oxo-M applied for 20 s (Fig. 6 A). On average, suppression of M current had a delay of 1.4 s and a τon of 5.0 s. Washout was followed by a 34-s delay and recovery with a τoff of 123 s (Fig. 6 C). Current suppression was concentration dependent (Fig. 6 D) with an apparent EC50 of 120 nM oxo-M (Fig. 6 E).
Figure 6.

Kinetics of Kv7.2/7.3 channel closure. (A) Time course for current from a single voltage–clamped cell undergoing a 20-s exposure to 10 µM oxo-M. Current is steady-state measured at −20 mV. Sampling frequency: 0.25 Hz during baseline and 200 Hz during agonist. (B) Individual current traces corresponding to points in A. Voltage was stepped from −20 to −60 mV for 500 ms every 4 s. (C) Time course for normalized mean current in five cells. Note the different time scales for onset and washout. For clarity in display, points from fast sampling were pooled in 1-s bins and points from slow sampling were pooled in 20-s bins. (D) M current concentration–response time course for a single cell. Oxo-M from 1 nM to 10 μM as labeled. (E) M current concentration–response curve from steady-state values for eight cells. Mean ± SEM.
Because our optical measurements required the overexpression of additional fluorescent signaling components, we tested the effect of overexpression of these proteins on the kinetics of M current suppression. Whereas transfecting G proteins (α, β, and γ together) did not alter M current suppression (unpublished data), coexpressing PLC or PH probes with receptor and channel subunits did (Table I). Overexpression of PLC-YFP reduced the delay in current suppression from 1.4 to 0.78 s and shortened the time constant from 5.0 to 1.2 s. Recovery upon washout of agonist was also accelerated, reducing the delay from 34 to 11 s and the time constant from 123 to 62 s (Fig. 7 A). On the other hand, overexpression of PH probes slowed current suppression in a concentration-dependent fashion. Cells with low to moderate expression of PH probes (those with CFPC < 8,000 per 24-ms sampling period) had an average delay of 2.1 s and a time constant of 5.7 s for current suppression, and a delay of 11 s and a time constant of 63 s for recovery (Fig. 7 B). In cells with high expression of PH probes, oxo-M failed to suppress M current fully (not depicted).
Figure 7.

PLC speeds and PH probes slow M current suppression. (A and B) Normalized mean M current at −20 mV from six cells expressing either transfected PLCβ1-YFP (open circles, A) or low levels of PH(PLCδ1)-CFP and PH(PLCδ1)-YFP (open circles, B). For comparison, control M current from five cells lacking exogenous PLC or PH probes (black circles). Note the different time scales for onset and washout.
DISCUSSION
The reaction times summarized in Table I fall into a satisfying sequence that agrees with our understanding of GPCR signaling pathways. Receptor binding and G protein interaction occur in <0.5 s and have minimal delays. Alterations of the Gα/Gβγ complex and interactions with PLC occur within a couple of seconds with sub-second delays. And the depletion of PIP2 and closure of channels take ∼5 s and start after a >1-s delay. We will consider the steps individually. It will be apparent that at present we do not know which of several biochemical steps each fluorescent protein pair reports, so we list major possibilities. First, however, we review the evidence that the FRETr values calculated with Eq. 3 are FRET due to resonance transfer of energy from CFP (donor) to nearby YFP (acceptor).
Relation of FRETr to FRET
With each pair of fluorophores that we studied, there were significant resting YFPC counts (corrected for background and CFP bleed-through), even though the excitation light excited only CFP. Energy is being transferred from CFP to YFP. The calculated mean resting FRETr values were 0.14–0.88 (Table I). In addition, photobleaching the YFP fluorophore with 500 nm light always increased resting CFPC, with the increase in CFPC being largest for pairs that had the largest resting FRETr. These criteria show that the resting FRETr values reflect FRET. Less evident is whether the changes of FRETr during stimulation also reflect FRET changes. It would be ideal to show that photobleaching of YFP increases CFPC more (or less) during the oxo-M–activated state than at rest. However, the small size of the signals, the long time it takes to bleach, the irreversibility of bleaching, and the profound cellular changes that occur if agonist is applied for more than a few seconds do not facilitate doing this experiment. Instead, a clear indicator of FRET changes is the consistent reciprocal time course of CFPC and YFPC during agonist application. Consider Fig. 5, where we know there has to be a FRET decrease because the PH domain probes translocate away from the membrane during receptor activation. Because of their proximity decrease, YFPC dims, and, as for photobleaching of YFP, CFPC brightens. The time courses are exactly reciprocal and fully reversible. This is true of all five FRET pairs we studied. The Gβ1-YFP fluorescence provides a nice demonstration that the intensity changes are not intrinsic to the single probe, but rather to the pair of molecules studied. This probe is paired with M1R-CFP in Fig. 2 and with Gαq-CFP in Fig. 3. The changes in YFPC take 0.2 s when partnered with M1R and 2.0–3.0 s and go in the opposite direction when partnered with Gβ1.
M1R activation is fast
The fast increase in intramolecular FRETr within M1R-YFP-CFP upon the addition of 10 µM oxo-M was finished by 100 ms and ought to reflect some receptor conformational change after agonist binding. We refer to this step as M1R activation. Due to constraints from bleaching and perfusion speed, we were able to determine only a lower limit for the rate of receptor activation. Using kinetic data for 1 and 10 µM oxo-M, and taking the slope of 1/τon versus [oxo-M], we estimate a kon value of 5.0 × 106 M−1s−1. Because this step was very rapid and the receptor construct possibly does not bind G proteins, it is unlikely to be affected by steps downstream in the signaling cascade. For receptor deactivation, we obtained a koff value of 5.6 s−1.
Our results fall within the range of FRET-based activation kinetics measured with other receptor types. Reported time constants for receptor activation are ∼40 ms for the Gi-coupled α2A-adrenergic receptor with 10 µM norepinephrine (Vilardaga et al., 2003), ∼60 ms for the Gs-coupled β1-adrenergic receptor with 10 µM norepinephrine (Rochais et al., 2007), 66 ms for the Gs-coupled adenosine A2A receptor with 1 mM adenosine (Hoffmann et al., 2005), and ∼1 s for the Gs- and Gq-coupled parathyroid receptor with 1 µM parathyroid hormone (Vilardaga et al., 2003). The only deactivation time constant reported so far is ∼2 s for a FlAsH-labeled α2A receptor (Hein et al., 2005). Our estimate of deactivation is 10-fold faster. The four above-mentioned receptor constructs showed decreases in intra-receptor FRET with agonist, unlike ours, implying that the M1R C terminus might move closer to the insertion point in the third intracellular loop, whereas in the other receptors it might move away. However, because in our construct insertion of YFP into the third intracellular loop was compensated by removal of 134 residues of the normal receptor sequence (most of the loop), it may be unwise to try to infer the directions of relative movements of domains of unmodified receptors.
Ligand binding is normal in M1R fluorescent constructs
To rule out altered ligand binding in our modified M1 receptors, we measured dissociation constants for 3H-NMS binding and inhibition constants for oxo-M. Our results for the inhibition constant of oxo-M (4–9 µM) are in the range of reported values: 8.1 µM in Chinese hamster ovary cells in the presence of 0.5 mM GTP (Jakubik et al., 1997), 2.2 ± 0.2 µM for muscarinic receptors in rat cerebral cortex, and 9.0 ± 4.9 µM for M1-M4 subtypes in a mixture of tissues (Sharif et al., 1995). If we take 4 µM as the apparent dissociation constant for oxo-M and 6 s−1 as koff, the predicted kon (=koff/Kd) for the M1R would be 1.5 × 106 M−1s−1. Dissociation constants for 3H-NMS binding to three versions of M1 receptors were internally consistent (580–670 pM) but were higher than those reported in the literature: 145 pM in Chinese hamster ovary cells (Jakubik et al., 1995), 120 pM in human neuroblastoma NB-OK1 cells (Waelbroeck et al., 1990), and 260 pM (Cortés and Palacios, 1986) or 300 pM in rat brain tissue (Ehlert and Tran, 1990).
Signaling to G proteins is not rate limiting
The change in FRETr between M1R-CFP and Gβ1-YFP had a time constant of only 200 ms, ∼30-fold faster than that for M current suppression. Overexpressing G proteins did not accelerate M current suppression. Collectively, these data indicate that signaling to G proteins is not rate limiting for suppression of M current, and that the pool of endogenous G proteins suffices to keep up with the exogenously expressed M1 receptors.
The FRETr increase observed between M1R-CFP and Gβ1-YFP likely represents either increased association between the two proteins or a conformational change within a preformed complex. Because the kinetics are slower than those of M1R-YFP-CFP and faster than those of Gαq-CFP/Gβ1-YFP, the events represented probably occur between receptor activation and G protein activation. The large resting FRETr (0.42) suggests that some significant fraction of receptors is pre-coupled to G proteins. There is no optical sign of dissociation of Gβγ from receptors upon activation because we see a stable elevation in the FRETr between receptor and Gβ constructs throughout the application of agonist. These results are consistent with the observation that M1R activation increases receptor affinity for G proteins (Potter et al., 1988). Recovery of this signal (τoff = 3.7 s) may reflect partial receptor/G protein dissociation.
Our receptor/G protein kinetics are in the same range as those reported for other receptors and G proteins. Bioluminescence resonance energy transfer between the Gs-coupled β2 adrenergic receptor and Gβ1 or Gγ2 increased with a t1/2 of ∼300 ms and recovered within a few seconds using 10 µM isoproterenol (Galés et al., 2005). FRET between the α2A adrenergic receptor and Gγ2 subunits increased with a t1/2 of 86 ms and recovered with a t1/2 of 13 s using 100 µM norepinephrine in the presence of only endogenous Gαi (Hein et al., 2005). In that study, coexpressing Gαi accelerated the on-kinetics to 44 ms, so that they overlapped with receptor activation. FRET between the A2A adenosine receptor and Gγ2 increased with τon = 50 ms (1 mM adenosine) and recovered with τoff = 15 s (100 µM adenosine), and the β1 adrenergic receptor and Gγ2 had τon = 58 ms (1 mM norepinephrine) and τoff = 8 s (Hein et al., 2006).
G proteins rearrange or dissociate and slowly reset
Traditionally the Gα/Gβγ complex is said to dissociate upon activation by GTP. Indeed, the decrease in FRETr we see between Gαq-CFP and Gβ1-YFP would be consistent with such dissociation upon receptor activation or with some other rearrangement among the G proteins that increases the distance between the fluorophores. Recovery may reflect relaxation or reassociation of the G protein subunits. GRK2 increased the resting FRETr and improved the signal-to-noise ratio for changes in Gαq/Gβ1 FRETr. It may have increased the resting value by recruiting more Gβ1 (acceptors) to the cell surface. In addition, it may have bound one or both G protein subunits after separation, thus increasing the distance between the fluorophores considerably or increasing the fraction of subunits that are dissociated after activation (compare Schliefenbaum, J., A.K. Kreile, M.J. Lohse, and M. Bünemann. 2008. Biophysical Society Meeting. Abstr. 1977).
Our kinetic measurements of G protein subunit rearrangement are similar to those reported for other GPCRs. We found a FRETr decrease with τon = ∼3 s and delay plus τoff = ∼40 s. In our protocols, all of our measurements are on cells that coexpressed exogenous Gα, β, and γ subunits. For comparison, Bünemann et al. (2003) found an increase in FRET between Gαi and Gβ1 with α2A adrenergic receptor activation, with a t1/2 for onset of 1 s and a t1/2 for washout of 38 s with 1 µM norepinephrine. The same laboratory reported a decrease in FRET for Gαs/Gγ2 interaction with τon = 500 ms and τoff = 37 s for A2A adenosine receptor activation with 1 mM adenosine, and τon = 440 ms and τoff = 15 s for β1 adrenergic receptor activation with 100 µM norepinephrine (Hein et al., 2006). The off-kinetics we measured are consistent with these. Although the increase in FRET between G protein subunits seen for α2A receptors does not suggest G protein dissociation, the decrease in FRETr we see with M1Rs could be explained either by subunit rearrangement or by dissociation.
For each example discussed above, recovery from G protein dissociation or rearrangement as measured by recovery of Gα/Gβ or Gα/Gγ FRETr takes longer (∼15–40 s) than classically discussed G protein cycles. Are we overlooking some events? For example, some Gβγ subunits (including β1 but excluding γ2) visit intracellular membranes after G protein activation and then would have to return to the plasma membrane to reassociate (Chisari et al., 2007; Saini et al., 2007). Additionally, in several published receptor–G protein FRET experiments already described, it seems that G protein takes as long as 8–15 s to dissociate from the receptor, suggesting a continued activation. Because in our work τoff for receptor–G protein interaction is only ∼4 s, we return to the idea of slow GTPase. Hydrolysis of Gαq-GTP in vitro is supposed to be extremely slow without and accelerated almost 1,000-fold in the presence of PLCβ1 (0.013 s−1 vs. 9–12 s−1) (Mukhopadhyay and Ross, 1999). If we had expressed an excess of G proteins compared with PLC, the free G proteins would have an exceedingly slow GTPase rate and would have to wait to partner with a free PLC to be able to complete GTP hydrolysis. This would slow overall deactivation of Gα subunits and delay subsequent steps, such as rearrangement or reassembly of G protein subunits. We regard the widely observed slow recovery of G proteins as a puzzle that still needs further conceptual explanation.
PLC activation is fast when PLC is abundant
Interaction between Gαq-CFP and PLC-YFP (delay plus τon = 1.7 s) followed quickly after G protein activation. This step likely reflects G protein/PLC binding or conformational changes associated with PLC activation. Coexpression of RGS2 occludes this FRETr change (unpublished data), indicating that activation of Gαq by GTP is a prerequisite. Recovery from the FRETr increase may reflect GTPase activity or G protein/PLC unbinding. The interpretation of this step is complicated by the fact that we must transfect PLC to measure its activation kinetics—this step might be slower in the presence of only endogenous PLC.
Our data are consistent with the “fast activation” of PLCβ1 observed in vitro by Biddlecome et al. (1996). Using a vesicle preparation including M1R, Gαq, and PLCβ1 and measuring IP3 production, they observed both fast (<2 s) and slow (12 s) activation of PLC. Fast activation occurred when GTP was added to vesicles preincubated with agonist, and slow activation occurred when agonist was added to vesicles preincubated with GTP, suggesting that guanine nucleotide exchange occurred rapidly and receptor/Gαq interaction was rate limiting for PLC activation. Our data suggest that receptor/Gβ1 interaction is not rate limiting for PLC activation. Biddlecome et al. (1996) postulated that agonist exposure could induce the formation of receptor-Gαq-PLC complexes, which would exhibit accelerated activation over multiple GTPase cycles. In agreement, we see an increase of M1R/Gβ FRETr and an increase of Gαq/PLC FRETr. It is possible that overexpressing PLC promotes the formation of such complexes, permitting faster activation of PLC without agonist preincubation. Dowal et al. (2006) demonstrated baseline association between Gαq and PLCβ1 using FRET in PC12 and HEK293 cells, but did not observe an increase in Gαq/PLC association upon the addition of cholinergic agonists. Two differences may explain this discrepancy between our studies: first, lower receptor expression levels in their cells may have failed to produce an observable response; second, the response may have been rapid enough to escape their lower sampling frequency (every 15 s).
PIP2 hydrolysis is rate limiting
PIP2 hydrolysis, as indicated by intermolecular FRETr with PH domain probes, had similar on-kinetics (6–7 s combined delay and τon) to M current suppression. Because Gαq interacts with PLC in <2 s, the rate-limiting step for channel closing must be the gradual depletion of PIP2 after PLC activation. Thus, we found that overexpression of PLC speeded M current suppression more than threefold, giving an on-rate nearly identical to that for interaction between Gαq-CFP and PLC-YFP. With abundant PLC, the sum of the delay and the τon for suppression of M current becomes only ∼2 s. In that short time PLC is activated, PIP2 unbinds from channel subunits, PIP2 is hydrolyzed, and channels close.
Comparison of steady-state concentration–response data from each step suggests that PIP2 hydrolysis comes to completion at agonist concentrations that activate receptors, G proteins, and PLC only partially. Evidently activating a fraction of G proteins and PLC can, given enough time, lead to hydrolysis of a large proportion of available PIP2. This suggests that PLC molecules undergo multiple activation cycles while receptors remain active, and that reduction of PIP2 levels is cumulative during agonist exposure. The normal excess of receptors, G proteins, and PLC permits much brisker physiological responses at higher agonist concentrations.
Consistent with PIP2 hydrolysis being rate limiting, expression of PH domain probes slowed M current suppression in a manner that depended on the PH probe expression level. This slowing probably reflects buffering of PIP2 by the PH probes, which would reduce the availability of free PIP2 and slow its access to PLC (Várnai and Balla, 1998; Gamper et al., 2004). This would imply that the amount of the PH probe expressed approaches or exceeds the size of the usual free PIP2 pool. If there normally is a metabolic set point for the level of free PIP2 in the plasma membrane, sequestering of PIP2 by PH domain probes for 24 h would induce a compensatory rise in the total membrane PIP2 (free and bound). In agreement, cells with high PH probe expression had markedly slower declines in PH domain FRETr with agonist. They were discarded from kinetic analysis. M current suppression was complete in the presence of PH probes, but was slowed by 1.4 s relative to cells not expressing PH probes. Accordingly, the reported time constant for PIP2 hydrolysis may be overestimated by up to 1.4 s.
Unexpectedly, recovery from suppression of M current was accelerated in cells transfected with PLC or PH probes. As a working hypothesis, we can suggest that chronic reduction in levels of free PIP2 (by enhanced hydrolysis or buffering, respectively) produces positive feedback on PIP2 synthesis via up-regulation of PI 4-kinase and/or PIP 5-kinase. For the case of PLC overexpression, we provide two additional concepts. Accelerated recovery may be partially explained by PLC's function as a GTPase accelerating protein for Gαq (Biddlecome et al., 1996)—when PLC is overexpressed, G protein activity (and downstream events) may be shut off more quickly. In addition, PLC overexpression may speed M current recovery in a calcium-dependent fashion. That is, enhanced IP3 production could increase the calcium signal and potentiate the calcium-dependent PI 4-kinase, accelerating PIP2 resynthesis (Gamper and Shapiro, 2007).
Pre-coupled or collision-coupled?
The mechanism of coupling among G protein–coupled signaling molecules is an important determinant of signaling kinetics and efficacy. Our baseline FRETr data and photobleaching results indicate some baseline proximity between M1R and Gβ1, Gαq and Gβ1, and Gαq and PLCβ1. In the case of both M1R/Gβ1 and Gαq/PLCβ1, FRETr increases substantially upon muscarinic activation, indicating that not every copy of these proteins is paired/active before stimulation.
The fast activation of PLC that we observe is consistent with the preformation of stable receptor/G protein/PLC complexes (Biddlecome et al., 1996), but may also be explained by an increase in the collisional frequency between Gαq and PLC or by potentiation of G protein activity when PLC is overexpressed. If complexes are formed, the fraction of one protein that enters the complex would be sensitive to the expression level of any other partner protein(s). Additionally, RGS4 binds to activated Gαq as well as to Gβγ and PLCβ1 (Dowal et al., 2001), and may be involved in a signaling complex. Given the relatively low apparent affinity (640 nM) interaction between PLCβ1 and Gαq(GDP) (Dowal et al., 2006), formation of ternary complexes may require scaffold proteins or PLC in excess of endogenous levels. PLC overexpression might tilt the balance in favor of forming complexes. Results from a recent kinetic model for G protein–coupled signaling additionally suggest that PLCβ1 potentiates G protein activity by stabilizing receptor–G protein interaction and by increasing GDP/GTP exchange (Turcotte et al., 2008). Alternatively, PLC overexpression may simply provide a higher concentration of targets for diffusing Gαq and increase the frequency of collision between Gαq and PLC.
Although observing FRETr in the resting condition is a positive indicator for proximity of the components, further experiments are needed to distinguish between these paradigms using multiple approaches. It would be instructive to constrain protein interaction dynamics, for instance by limiting diffusion in the membrane or linking proteins, and comparing activation kinetics and the mobility of possible partners with those in unconstrained systems. Additionally, developing quantitative models and comparing kinetic results with those collected in other systems will provide an important check on our data and potentially shed light on the underlying structure of the system.
These experiments show an orderly temporal progression of the receptor-mediated signaling cascade, and they demonstrate that the rate-limiting step for channel closure is the consumption of PIP2 by PLC. They supply the background material needed to develop a more quantitative model of the steps of the overall signaling pathway.
Acknowledgments
We thank Ken Mackie, Matthew Wallace, Sharona Gordon, and Mika Munari for assistance with molecular biology; Greg Martin for support in the Keck Imaging Center; Björn Falkenburger for many discussions and help in fitting the data; Björn Falkenburger, Peter Detwiler, and Ken Mackie for feedback on the manuscript; and Lea Miller for administrative assistance. We are grateful to Catherine Berlot, Moritz Bünemann, Stephen Ikeda, Kees Jalink, Loren Runnels, and Mark Shapiro for gifts of cDNA.
This work was supported by National Institutes of Health grants NS08174, GM08391 (to B. Hille), T32 GM07108 (to J.B. Jensen), and T32 GM07270 (to J.S. Lyssand).
Edward N. Pugh Jr. served as editor.
Footnotes
Abbreviations used in this paper: ECFP, enhanced cyan fluorescent protein; EYFP; enhanced yellow fluorescent protein; FRET, fluorescence resonance energy transfer; GPCR, G protein–coupled receptor; GRK2, GPCR kinase 2; 3H-NMS, _N_-methyl-3_H_-scopolamine; IP3, inositol 1,4,5-trisphosphate; M1R, M1 muscarinic receptor; oxo-M, oxotremorine-methiodide; PH, pleckstrin homology; PIP2, phosphatidylinositol 4,5-bisphosphate; PLCβ, phospholipase C-β.
References
- Bal M., Zaika O., Martin P., Shapiro M.S. 2008. Calmodulin binding to M-type K+ channels assayed by TIRF/FRET in living cells.J. Physiol. 586:2307–2320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biddlecome G.H., Berstein G., Ross E.M. 1996. Regulation of phospholipase C-β1 by Gq and m1 muscarinic cholinergic receptor. Steady-state balance of receptor-mediated activation and GTPase-activating protein-promoted deactivation.J. Biol. Chem. 271:7999–8007 [DOI] [PubMed] [Google Scholar]
- Brown D.A. 1983. Slow cholinergic excitation—a mechanism for increasing neuronal excitability.Trends Neurosci. 6:302–307 [Google Scholar]
- Brown D.A., Adams P.R. 1980. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone.Nature. 283:673–676 [DOI] [PubMed] [Google Scholar]
- Bünemann M., Frank M., Lohse M.J. 2003. Gi protein activation in intact cells involves subunit rearrangement rather than dissociation.Proc. Natl. Acad. Sci. USA. 100:16077–16082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Rogge G., Hague C., Alewood D., Colless B., Lewis R.J., Minneman K.P. 2004. Subtype-selective noncompetitive or competitive inhibition of human α1-adrenergic receptors by ρ-TIA.J. Biol. Chem. 279:35326–35333 [DOI] [PubMed] [Google Scholar]
- Chisari M., Saini D.K., Kalyanaraman V., Gautam N. 2007. Shuttling of G protein subunits between the plasma membrane and intracellular membranes.J. Biol. Chem. 282:24092–24098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortés R., Palacios J.M. 1986. Muscarinic cholinergic receptor subtypes in the rat brain. I. Quantitative autoradiographic studies.Brain Res. 362:227–238 [DOI] [PubMed] [Google Scholar]
- Digby G.J., Lober R.M., Sethi P.R., Lambert N.A. 2006. Some G protein heterotrimers physically dissociate in living cells.Proc. Natl. Acad. Sci. USA. 103:17789–17794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowal L., Elliott J., Popov S., Wilkie T.M., Scarlata S. 2001. Determination of the contact energies between a regulator of G protein signaling and G protein subunits and phospholipase Cβ1.Biochemistry. 40:414–421 [DOI] [PubMed] [Google Scholar]
- Dowal L., Provitera P., Scarlata S. 2006. Stable association between Gαq and phospholipase Cβ1 in living cells.J. Biol. Chem. 281:23999–24014 [DOI] [PubMed] [Google Scholar]
- Ehlert F.J., Tran L.P. 1990. Regional distribution of M1, M2 and non-M1, non-M2 subtypes of muscarinic binding sites in rat brain.J. Pharmacol. Exp. Ther. 255:1148–1157 [PubMed] [Google Scholar]
- Evanko D.S., Thiyagarajan M.M., Takida S., Wedegaertner P.B. 2005. Loss of association between activated Gαq and Gβγ disrupts receptor-dependent and receptor-independent signaling.Cell. Signal. 17:1218–1228 [DOI] [PubMed] [Google Scholar]
- Frank M., Thumer L., Lohse M.J., Bünemann M. 2005. G Protein activation without subunit dissociation depends on a Gai-specific region.J. Biol. Chem. 280:24584–24590 [DOI] [PubMed] [Google Scholar]
- Galés C., Rebois R.V., Hogue M., Trieu P., Breit A., Hébert T.E., Bouvier M. 2005. Real-time monitoring of receptor and G-protein interactions in living cells.Nat. Methods. 2:177–184 [DOI] [PubMed] [Google Scholar]
- Gamper N., Shapiro M.S. 2007. Target-specific PIP2 signalling: how might it work? J. Physiol. 582:967–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N., Reznikov V., Yamada Y., Yang J., Shapiro M.S. 2004. Phosphatidylinositol [correction] 4,5-bisphosphate signals underlie receptor-specific Gq/11-mediated modulation of N-type Ca2+ channels.J. Neurosci. 24:10980–10992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein P., Frank M., Hoffmann C., Lohse M.J., Bünemann M. 2005. Dynamics of receptor/G protein coupling in living cells.EMBO J. 24:4106–4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein P., Rochais F., Hoffmann C., Dorsch S., Nikolaev V.O., Engelhardt S., Berlot C.H., Lohse M.J., Bünemann M. 2006. Gs activation is time-limiting in initiating receptor-mediated signaling.J. Biol. Chem. 281:33345–33351 [DOI] [PubMed] [Google Scholar]
- Hoffmann C., Gaietta G., Bünemann M., Adams S.R., Oberdorff-Maass S., Behr B., Vilardaga J.P., Tsien R.Y., Ellisman M.H., Lohse M.J. 2005. A FlAsH-based FRET approach to determine G protein-coupled receptor activation in living cells.Nat. Methods. 2:171–176 [DOI] [PubMed] [Google Scholar]
- Hughes T.E., Zhang H., Logothetis D.E., Berlot C.H. 2001. Visualization of a functional Gαq-green fluorescent protein fusion in living cells. Association with the plasma membrane is disrupted by mutational activation and by elimination of palmitoylation sites, but not by activation mediated by receptors or AlF4−.J. Biol. Chem. 276:4227–4235 [DOI] [PubMed] [Google Scholar]
- Jakubik J., Bacakova L., el-Fakahany E.E., Tucek S. 1995. Subtype selectivity of the positive allosteric action of alcuronium at cloned M1-M5 muscarinic acetylcholine receptors.J. Pharmacol. Exp. Ther. 274:1077–1083 [PubMed] [Google Scholar]
- Jakubik J., Bacakova L., El-Fakahany E.E., Tucek S. 1997. Positive cooperativity of acetylcholine and other agonists with allosteric ligands on muscarinic acetylcholine receptors.Mol. Pharmacol. 52:172–179 [DOI] [PubMed] [Google Scholar]
- Kenakin T. 1997. Agonist-specific receptor conformations.Trends Pharmacol. Sci. 18:416–417 [DOI] [PubMed] [Google Scholar]
- Lodowski D.T., Pitcher J.A., Capel W.D., Lefkowitz R.J., Tesmer J.J. 2003. Keeping G proteins at bay: a complex between G protein-coupled receptor kinase 2 and Gβγ.Science. 300:1256–1262 [DOI] [PubMed] [Google Scholar]
- Lohse M.J., Vilardaga J.P., Bünemann M. 2003. Direct optical recording of intrinsic efficacy at a G protein-coupled receptor.Life Sci. 74:397–404 [DOI] [PubMed] [Google Scholar]
- Lohse M.J., Bünemann M., Hoffmann C., Vilardaga J.P., Nikolaev V.O. 2007a. Monitoring receptor signaling by intramolecular FRET.Curr. Opin. Pharmacol. 7:547–553 [DOI] [PubMed] [Google Scholar]
- Lohse M.J., Hoffmann C., Nikolaev V.O., Vilardaga J.P., Bünemann M. 2007b. Kinetic analysis of G protein-coupled receptor signaling using fluorescence resonance energy transfer in living cells.Adv. Protein Chem. 74:167–188 [DOI] [PubMed] [Google Scholar]
- Lohse M.J., Nikolaev V.O., Hein P., Hoffmann C., Vilardaga J.P., Bünemann M. 2008. Optical techniques to analyze real-time activation and signaling of G-protein-coupled receptors.Trends Pharmacol. Sci. 29:159–165 [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S., Ross E.M. 1999. Rapid GTP binding and hydrolysis by Gq promoted by receptor and GTPase-activating proteins.Proc. Natl. Acad. Sci. USA. 96:9539–9544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobles M., Benians A., Tinker A. 2005. Heterotrimeric G proteins precouple with G protein-coupled receptors in living cells.Proc. Natl. Acad. Sci. USA. 102:18706–18711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter L.T., Ferrendelli C.A., Hanchett H.E. 1988. Two affinity states of M1 muscarine receptors.Cell. Mol. Neurobiol. 8:181–191 [DOI] [PubMed] [Google Scholar]
- Rochais F., Vilardaga J.P., Nikolaev V.O., Bünemann M., Lohse M.J., Engelhardt S. 2007. Real-time optical recording of β1-adrenergic receptor activation reveals supersensitivity of the Arg389 variant to carvedilol.J. Clin. Invest. 117:229–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Velasco V., Ikeda S.R. 2001. Functional expression and FRET analysis of green fluorescent proteins fused to G-protein subunits in rat sympathetic neurons.J. Physiol. 537:679–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini D.K., Kalyanaraman V., Chisari M., Gautam N. 2007. A family of G protein βγ subunits translocate reversibly from the plasma membrane to endomembranes on receptor activation.J. Biol. Chem. 282:24099–24108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarlata S., Dowal L. 2004. The use of green fluorescent proteins to view association between phospholipase Cβ and G protein subunits in cells.Methods Mol. Biol. 237:223–232 [DOI] [PubMed] [Google Scholar]
- Sharif N.A., Williams G.W., DeSantis L.M. 1995. Affinities of muscarinic drugs for [3H]N-methylscopolamine (NMS) and [3H]oxotremorine (OXO) binding to a mixture of M1-M4 muscarinic receptors: use of NMS/OXO-M ratios to group compounds into potential agonist, partial agonist, and antagonist classes.Neurochem. Res. 20:669–674 [DOI] [PubMed] [Google Scholar]
- Stauffer T.P., Ahn S., Meyer T. 1998. Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells.Curr. Biol. 8:343–346 [DOI] [PubMed] [Google Scholar]
- Suh B.C., Hille B. 2002. Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis.Neuron. 35:507–520 [DOI] [PubMed] [Google Scholar]
- Suh B.C., Horowitz L.F., Hirdes W., Mackie K., Hille B. 2004. Regulation of KCNQ2/KCNQ3 current by G protein cycling: the kinetics of receptor-mediated signaling by Gq.J. Gen. Physiol. 123:663–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh B.C., Inoue T., Meyer T., Hille B. 2006. Rapid chemically induced changes of PtdIns(4,5)P2 gate KCNQ ion channels.Science. 314:1454–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcotte M., Tang W., Ross E.M. 2008. Coordinate regulation of G protein signaling via dynamic interactions of receptor and GAP.PLOS Comput. Biol. 4:e1000148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Wal J., Habets R., Várnai P., Balla T., Jalink K. 2001. Monitoring agonist-induced phospholipase C activation in live cells by fluorescence resonance energy transfer.J. Biol. Chem. 276:15337–15344 [DOI] [PubMed] [Google Scholar]
- Várnai P., Balla T. 1998. Visualization of phosphoinositides that bind pleckstrin homology domains: calcium- and agonist-induced dynamic changes and relationship to myo-[3H]inositol-labeled phosphoinositide pools.J. Cell Biol. 143:501–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilardaga J.P., Bünemann M., Krasel C., Castro M., Lohse M.J. 2003. Measurement of the millisecond activation switch of G protein-coupled receptors in living cells.Nat. Biotechnol. 21:807–812 [DOI] [PubMed] [Google Scholar]
- Waelbroeck M., Tastenoy M., Camus J., Christophe J. 1990. Binding of selective antagonists to four muscarinic receptors (M1 to M4) in rat forebrain.Mol. Pharmacol. 38:267–273 [PubMed] [Google Scholar]
- Wang H.S., Pan Z., Shi W., Brown B.S., Wymore R.S., Cohen I.S., Dixon J.E., McKinnon D. 1998. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel.Science. 282:1890–1893 [DOI] [PubMed] [Google Scholar]
- Yuan C., Sato M., Lanier S.M., Smrcka A.V. 2007. Signaling by a non-dissociated complex of G Protein βγ and α subunits stimulated by a receptor-independent activator of G protein signaling, AGS8.J. Biol. Chem. 282:19938–19947 [DOI] [PubMed] [Google Scholar]
- Zhang H., Craciun L.C., Mirshahi T., Rohács T., Lopes C.M., Jin T., Logothetis D.E. 2003. PIP2 activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents.Neuron. 37:963–975 [DOI] [PubMed] [Google Scholar]