Navigating the ribosome’s metastable energy landscape (original) (raw)
. Author manuscript; available in PMC: 2010 Aug 3.
Published in final edited form as: Trends Biochem Sci. 2009 Aug 3;34(8):390–400. doi: 10.1016/j.tibs.2009.04.004
Abstract
The molecular mechanisms by which tRNA molecules enter and transit the ribosome during mRNA translation remains elusive. However, recent genetic, biochemical and structural studies offer important new findings into the ordered sequence of events underpinning the trans-location process that help place the molecular mechanism within reach. In particular, new structural and kinetic insights have been obtained regarding tRNA movements through ‘hybrid state’ configurations. These dynamic views reveal that the macromolecular ribosome particle, like many smaller proteins, has an intrinsic capacity to reversibly sample an ensemble of similarly stable native states. Such perspectives suggest that substrates, factors and environmental cues contribute to translation regulation by helping the dynamic system navigate through a highly complex and metastable energy landscape.
Ribosome-catalyzed protein synthesis
mRNA template-directed synthesis of proteins in the cell (i.e. translation) is catalyzed by the ribosome, a roughly spherical two-subunit, ~2–3-MDa macromolecular assembly (70S in bacteria) comprising more than 60 distinct RNA and protein products (Figure 1a) [1–3]. The assembled ribonuclear protein complex, which possesses enzymatic properties, is a central component of the gene expression mechanism that works cooperatively with exogenous cellular factors and highly conserved GTPases to function as an integration point for controlled expression of the cellular proteome.
Figure 1.

Structural snapshots of functional ribosome complexes reveal distinct conformational degrees of freedom implicated in the translation mechanism. Distinct views of three-dimensional, atomic models of the bacterial ribosome (Protein Data Bank accession codes 2QAL, 2QAM, 1VS9 and 1Q86) are shown to schematically illustrate (with double-headed arrows) mobile structural elements within (a) the 70S particle, (b) the large (50S) ribosomal subunit and (c) the small (30S) ribosomal subunit. Ribosomal proteins are shown in blue and tan in the large and small subunit, respectively. In both subunits rRNA is shown in gray ribbons. Conserved rRNA elements directly contacting tRNA substrates are shown in green. (a) A cross-sectional view of the 70S pre-translocation ribosome reveals the substrate-binding channel formed at the subunit interface. The three tRNA binding sites (E, P and A) are illustrated, and classically configured A- and P-site tRNAs are shown in red. Landmark structural features, including the decoding site (DS), the peptidyl transferase center (PTC), the L1 stalk, the GTPase activating center (GAC), the A-site finger (ASF; H38 of 23S rRNA), the P-site gate (G1338–A1339, A790 of 16S rRNA) and the mRNA track are indicated for structural reference. (b) The isolated 50S subunit interface is shown to schematically illustrate the protein and rRNA components of the L1 stalk and the GAC and their estimated range of motions [26,55,57]. The ASF bridging the large and small subunit head domains, as well as the A- and P-loop structural elements within the PTC that base pair with the 3′-CCA-ends of tRNA, are highlighted in green. (c) The solvent surface of the 30S subunit is shown to illustrate the single-stranded mRNA (red) track, which wraps around the neck domain and contacts the Shine–Dalgarno (SD) sequence at the convergence of the three principal structural domains (head, platform and body). Panels (i–iii) show the identified conformational degrees of freedom in the ribosome that are implicated in the translocation mechanism and their approximate magnitudes. (i) Subunit ratcheting rotates the three 30S domains counterclockwise in a collective fashion; the reverse motion is termed unratcheting. (ii) Head rotation represents a swiveling motion of the 30S head domain around the neck-like feature connecting the principal domains [3]. (iii) Head tilt represents flexion of the neck perpendicular and parallel to the subunit interface [35]. (ii–iii) The head domain is highlighted to indicate that these motions can largely occur in the absence of subunit ratcheting.
Each of the four principal biochemical stages of translation – initiation, elongation, termination and recycling (see Figure I in Box 1) – contributes substantially to the overall regulation of protein synthesis [4–6]. Factormediated elongation reactions consisting of aminoacyl-tRNA (aa-tRNA) selection at the aminoacyl (A) site and directional translocation of A- and peptidyl (P)-site tRNA substrates to the P and exit (E) sites, respectively, represent key determinants of the rate and fidelity of protein synthesis and the observed genetic code. Cyclical elongation processes drive the 70S ribosome particle in a step-wise fashion through the mRNA open reading frame (ORF), where at each step the primary mRNA sequence is decoded in discrete codon increments at the A site through the binding of ~25-kDa, L-shaped aa-tRNA adaptor molecules [7,8]. In this review, recent progress towards understanding the molecular mechanism of elongation will be discussed with an aim to contextualize these findings within the broader context of more than five decades of previous research.
Box 1. Translation and the global architecture of the ribosome.
The process of translation consists of four principal stages: initiation, elongation, termination and recycling (Figure I). In bacteria, translation is initiated by initiation factors (IF-1, -2 and -3), which facilitate subunit association on the mRNA and placement of initiator fMet-tRNA fMet in the ribosomal P site, where its anticodon base pairs with the mRNA start codon [81]. After initiation, aminoacyl-tRNAs (aa-tRNAs) cognate to the mRNA codon presented in the 30S subunit decoding site are delivered to the ribosome in a ternary complex with elongation factor-Tu (EF-Tu) and GTP to the ribosomal A site [8]. Codon-dependent, EF-Tu-catalyzed GTP hydrolysis allows the tRNA to accommodate into the particle. This multistep, 30S-catalyzed decoding reaction is followed immediately by 50S-subunit-catalyzed peptide-bond formation between amino acids linked to the 3′ ends of the adjacently bound tRNAs within the peptidyl transferase center (PTC), completing the process of tRNA selection by generating A-site- bound peptidyl-tRNA. The multistep translocation process facilitated by EF-G-catalyzed GTP hydrolysis (Figure 2) subsequently moves deacylated P-site tRNA, peptidyl A-site tRNA and mRNA with respect to the ribosome bringing the next codon into the 30S decoding site.
Reverse translocation, catalyzed by LepA, which is induced under cellular stress conditions, helps to control the rate and fidelity of the translation process [65]. After translocation and EF-G release, the ribosome is reset for additional cycles of elongation. The dissociation of deacylated tRNA from the E site of the post-translocation complex might occur passively but might also depend on active mechanisms mediated by additional protein factors [82] and/or of subsequent tRNA selection reactions [83]. Each successive elongation cycle extends the nascent peptide chain by a single amino acid, where each mRNA ORF includes N codons. Upon reaching a nonsense stop codon in the mRNA, protein synthesis terminates through the action of release factors (RF-1, -2 and -3) that facilitate water-mediated cleavage of the full-length protein product from P-site-bound peptidyl-tRNA [84].
After termination, deacylated tRNA and mRNA are liberated from the ribosome, and subunits dissociate from each other through the action of ribosome recycling factor (RRF) together with EF-G [85]. Under proliferative growth conditions, synthesis of an average 50-kDa protein can be achieved on the time scale of approximately one minute, where in the absence of attenuated initiation, elongation reactions represent the vast majority of the time required for synthesis.
Figure I.

Schematic structural representation of the four principal phases of translation. This highly simplified diagram shows initiation, elongation, termination and recycling steps, as well as the key translation factors facilitating these reactions, in a manner meant to illustrate the cyclical nature and estimated timescales of translation processes. The range of time scales shown depends strongly on mRNA codon sequence and the availability of substrates and factors, as well as temperature and ionic conditions. The translocation component of the elongation cycle is highlighted in red to indicate the focus on this multistep process in this review.
The mechanistic paradox of substrate–ribosome interactions
The nature of substrate–ribosome interactions, and how tRNAs and mRNA move through the ribosome, have been the subject of investigation for nearly 50 years. During the past decade, cryoelectron microscopy (cryo-EM) and X-ray crystallography efforts have elucidated the global ribosome architecture and the molecular basis of ribosome–substrate interactions underpinning the translocation mechanism at unprecedented resolution [1–3,9,10]. Although these transformative advances provide a physical frame-work for interpreting genetic and biochemical observations amassed since the ribosome’s initial discovery, structural snapshots of the ribosome represent only the most thermo-dynamically stable native state conformations of the particle. Such global and/or local minima on the free energy landscape are likely to represent only a small portion of the many potential conformations of the ribosome transited during the process of translation.
As for each phase of translation, conformational degrees of freedom in both the large and small ribosomal subunits are thought to accompany mRNA transit (Figure 1b,c) [2,10]. Translocation, the directional movement of A-and P-site tRNA and mRNA through the ribosome catalyzed by elongation factor G(EF-G), is a complex, multistep process in which substrates are moved in ~10–30 Å steps through structurally and spatially distinct binding sites within a solvent-accessible channel formed by the interface of large and small subunits (Figure 1a, Figure 2). Here, substrate motions occur via ‘hybrid’ configurations in which tRNAs first move with respect to the large subunit before complete translocation [7,10].
Figure 2.
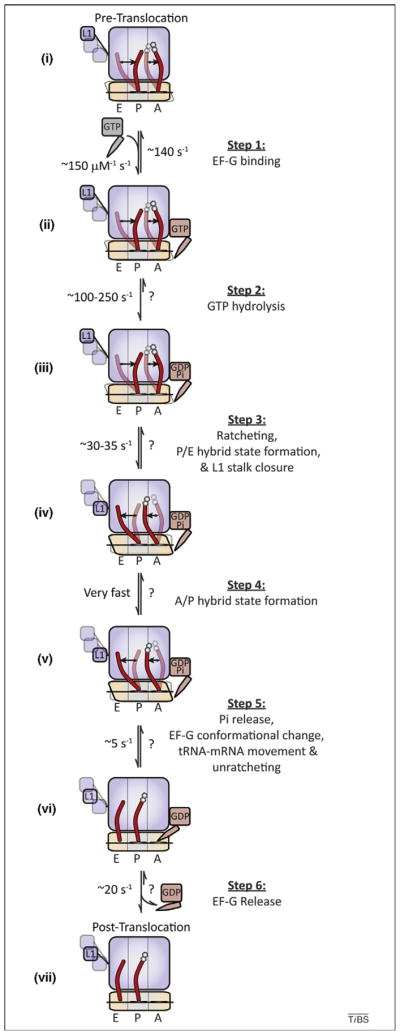
The multistep process of translocation is one of motion, including conformational changes in the ribosome, tRNA and EF-G. A mechanistic model depicting the process of tRNA–mRNA translocation is schematically diagramed to include data from kinetic, structural and smFRET investigations. Dynamic processes and factor- and substrate-induced conformational changes are included where knowledge of such processes is either known or inferred from experimental data. (i) On the dynamic pre-translocation complex, A- and P-site tRNAs (red) spontaneously transition to the A/P and P/E hybrid states [17,41,42]. The 30S subunit exchanges between ratcheted and unratcheted states [53,58,59], and the L1 stalk transitions between open (opaque), intermediate (translucent) and closed (translucent) positions [56,57]. (Step 1) EF-G binds rapidly and reversibly to the dynamic pre-translocation complex (ii). (Step 2) EF-G binding is quickly followed by GTP hydrolysis (iii) [44,45,80]. (Step 3) GTP hydrolysis promotes subunit ratcheting (RSR), P/E-hybrid-state formation and L1 stalk closure (iv), quickly followed by (Step 4) formation of the A/P hybrid state (v). (Step 5) Rate-limiting conformational changes within the ribosome and EF-G, triggered by the direct interaction of EF-G’s tRNA like domains with the 30S decoding site, precipitate movements of both A- and P-site tRNA–mRNA complexes with respect to the 30S subunit and Pi release (perhaps in random order), followed by unratcheting (vi). (Step 6) EF-G(GDP) releases from the post-translocation ribosome complex. In the post-translocation complex the L1 stalk adopts a distinct, partially closed configuration (vii). Rate constants, where indicated, were compiled from previously published models taken under a variety of experimental conditions and must be interpreted with the understanding that global translocation rates are highly sensitive to both temperature and buffer components as well as ribosome pre-translocation complex composition [8,44,45,50,57]; rates that have not been experimentally determined are noted with a ‘?’. Stochastic processes (tRNA and L1 stalk motions, subunit ratcheting/ unratcheting) are presumed to remain dynamic throughout the process, where prior to translocation only the relative populations of potential sub-states change.
In understanding the molecular mechanism of translocation, the balance of two seemingly opposed evolutionary pressures must be considered. Faithful transmission of genetic information requires accurate, processive protein synthesis, which is achieved via stable binding between the ribosome and mRNA and tRNA substrates. However, in competitive growth environments, rapid protein synthesis is required, which necessitates that the ribosome maintains a relatively loose grip on its substrates to ensure expeditious translation [11]. Recent progress, afforded by investigations of the order and timing of events leading to the large-scale repositioning of tRNA within the ribosome, includes structure-determination efforts that have shed light on the dynamic nature of intermediate hybrid states. These findings suggest that conformational plasticities of the system enable ribosome–substrate interactions to change with time.
As foreshadowed by investigations of many small proteins [12–15], the potential for the ribosome to exist in spontaneously interconverting native-state conformations suggests that dynamism contributes to regulated, processive translation. This new framework affords a deeper understanding of both the basic mechanism of protein synthesis and allosteric signaling in translation, where a rugged, metastable energy landscape enables translating particles to integrate and respond to a wide array of regulatory cues.
Early insights into the translocation mechanism
As early as the 1960s it was hypothesized that the movement of tRNAs through the ribosome might occur by way of intermediate binding sites [16]. Hybrid tRNA positions on the ribosome were conclusively demonstrated through elegant chemical footprinting and genetic investigations more than two decades later [17]. These studies revealed that the conserved 3′-CCA ends of both A- and P-site tRNAs within the pre-translocation complex, which normally base pair with the A and P loops of 23S rRNA when classically bound (A/A, P/P; Figure 1a) [18,19], can spontaneously move with respect to the large ribosomal subunit before anticodon stem movement on the small subunit (Figure 2, Figure 3a). Hybrid tRNA positions arise spontaneously after peptide-bond formation as a consequence of preferred binding of the nascent peptide within the large subunit P site [17,20]. Thus, A-site tRNA, to which the peptide is linked in the pre-translocation complex, adopts an A/P configuration. Correspondingly, the deacylated 3′-CCA terminus of P-site tRNA adopts a P/E hybrid state by vacating the peptidyltransferase center (PTC) and entering the 50S E site. These early models suggested that tRNAs should predominantly occupy A/P and P/E hybrid state configurations within the pre-translocation ribosome and that EF-G(GTP) specifically operates on this complex.
Figure 3.

Atomic and cartoon models of classical and hybrid tRNA configurations. (a) Cartoon depicting the transitions of A- and P-site tRNAs (red) between the classical state (A/A–P/P), hybrid state 1 (A/P–P/E) and hybrid state 2 (A/A–P/E) observed by smFRET [41]. The Cy3 and Cy5 fluorophores used in smFRET investigations of tRNA motions attached to naturally occurring modified nucleotides (s4U8 on P-site tRNAfMet and acp3U47 on A-site peptidyl-tRNAPhe) are represented as green and red circles, respectively. Also shown are the approximate occupancies of the three configurations, the transition rates and the estimated distance changes observed. (b) Atomic models of the classical (A/A and P/P) and hybrid (A/P and P/E) tRNA configurations observed by cryo-EM [59] embedded in an atomic model of the 70S ribosome. As in (a), the approximate positions of the Cy3 (green) and Cy5 (red) fluorophores used in smFRET investigations are shown near the elbow regions of the A- and P-site tRNAs [41]. The distances shown are the estimated inter-dye displacements between classical and hybrid states [59]. Panel (b) was reproduced, with permission, from Ref. [59].
Conformational degrees of freedom in the ribosome have a direct role in the translocation mechanism
Structural studies later revealed tRNAs bound in classical (A/A and P/P) configurations within the pre-translocation complex [21–23]. Only under certain buffer conditions [24], or when EF-G was bound at the A site in the presence of the non-hydrolyzable GTP analogue, GDPNP [25,26], could the P/E hybrid state tRNA configuration be directly visualized. As foreshadowed by earlier chemical modification studies [17,27], P/E hybrid state formation was shown to be accompanied by global conformational rearrangements of the ribosome particle [25,26]. These motions include an ~20 Å inward flexion of the L1 stalk domain within the 50S E site toward the subunit interface (Figure 1b), as well as ratchet-like subunit rearrangement (RSR), which is defined by an ~8° counterclockwise rotation of the 30S subunit and a ~20 Å lateral displacement of the 30S head domain in the direction of translocation (Figure 1c). The collection of observed conformational rearrangements was speculated to actively participate in the translocation mechanism. In particular, a direct interaction observed between the L1 stalk and the P/E hybrid state tRNA elbow was proposed to stabilize the P/E hybrid tRNA conformation [26]. These studies, later corroborated by chemical footprinting and bulk fluorescence resonance energy transfer (FRET) experiments [28–30], revealed that large-scale conformational remodeling of the ribosome actively participates in the formation of tRNA hybrid states.
In the years that followed, crystallographic and cryo-EM studies continued to shed light on conformational degrees of freedom within the ribosome that might also be relevant to the translocation mechanism [3,31–35] (Figure 1c). Rotations of up to 14° in the small subunit head domain that were attenuated by P-site tRNA and anticodon stem loop (ASL) binding were suggested to accompany and/or facilitate active tRNA repositioning events. Complex head domain motions, involving both rotation and tilt, were also observed upon ASL binding to the isolated 30S subunit, described as ‘domain closure’. These observations suggest the possibility that the conformation of the small subunit might also influence A-site tRNA repositioning during hybrid states formation and translocation. Head domain motions were speculated to regulate the configuration of the P-site gate, a structural element located between the 30S head and shoulder domains and the P and E sites formed by residues G1338–A1339 in 16S rRNA (Figure 1a). This highly conserved RNA element forms stabilizing A-minor interactions with the P-site tRNA anticodon in the classical state in a manner that might potentially block P/E hybrid state formation and translocation [31–33,36]. Conformational degrees of freedom also implicated in the translocation mechanism included local rearrangements in the 30S subunit decoding site (DS) (Figure 1a), as well as in the A-site finger (ASF) and GTPase activating center (GAC) of the 50S subunit (Figure 1b). Collectively, these observations suggest a functional link between ribosome domain motions and the way in which the translating particle interacts with its substrates. Such a link would help to explain the observed relationships between ribo-some conformation and the position of the Shine–Dalgarno (SD)–anti-SD helix [37,38] (Figure 1c), as well as how peptide-bond formation might disrupt the SD interaction [39].
tRNA hybrid states represent translocation intermediates
Peptide-bond formation substantially increases the observed rate of translocation [40]. This observation was believed to reflect the increased propensity of A-site pepti-dyl-tRNA to adopt hybrid state tRNA configurations on the ribosome [17,40–42]. Functional studies of ribosomes mutated in the A and P loops [43] and pre-steady-state kinetic studies of translocation using fluorescently labeled tRNAs [44] later revealed that tRNA hybrid states represent bona fide intermediates in the translocation reaction coordinate. Consistent with this finding, mutations in the large subunit E site, predicted to inhibit P/E hybrid state formation, dramatically decrease trans-location rates [45]. Although unequivocally establishing a role for tRNA hybrid states in the translocation mechanism, these studies were unable to discern the relationship between global remodeling of ribosome architecture, including RSR, and hybrid states formation.
Metastable tRNA hybrid states are spontaneously achieved within the pre-translocation ribosome
The delineation of the order and timing of conformational changes within the ribosome, their relationship to tRNA hybrid states formation, and how both contribute to the translocation reaction coordinate remain crucial to under-standing ribosome regulation. Single-molecule methods have emerged as powerful new means of addressing some of these questions because they are free from ensemble averaging, which masks non-accumulating intermediates and stochastic structural transitions [46,47].
Sensitive optical trapping methods have recently enabled the first direct observation of single ribosomes translocating through the mRNA ORF [48]. This landmark achievement provided the first microscopic views of elongation, revealing that multiple rate-limiting, and relatively slow (>25 ms), kinetic events underpin the ribosome’s step-wise movement through the mRNA ORF. These findings are consistent with the notion that rate-limiting conformational events in the ribosome occur both before and during the process of translocation [49,50]. Single-molecule FRET (smFRET) studies of dye-labeled tRNAs on the pre-translocation ribosome complex provided further insights into the elongation mechanism by showing that A- and P-site tRNAs undergo reversible fluctuations between classical states and two distinct hybrid tRNA configurations (Figure 3a) [41,42]. The estimated activation barriers for motion (~70 KJ/mol), as well as the changes in barrier heights observed in response to single-base pair disruptions in the A and P loops (~1–2 KJ/ mol), argued that A- and P-site tRNAs can move independently, or in a coupled fashion, via global conformational changes in the pre-translocation complex. Importantly, these studies suggested that the most stable hybrid state (hybrid state 2 [H2]) corresponded to an isolated P-site tRNA motion in the P/E hybrid state (~15 Å), whereas A-site tRNA remained classically configured (A/A). Motions of A-site tRNA into its hybrid (A/P) position (hybrid state 1 [H1]) were the most transient fluctuations observed (~100 ms lifetime). Here, the estimated distance moved in the A/A–A/P transition was ~8 Å. Together with previous and contemporaneous bulk observations [17,44,51,52], these investigations revealed by direct means that the pre-translocation complex is highly dynamic in nature and suggested that the P/E hybrid state is achieved before A/P-hybrid-state formation during translocation. These smFRET data also led to the speculation that P-site tRNA motions might be related to the RSR observed in the EF-G-bound ribosome.
Coupled and uncoupled motions occur within the pretranslocation ribosome complex
smFRET studies have since provided direct evidence that RSR and the reverse process of ‘unratcheting’ occur spontaneously in the pre-translocation ribosome [53]. However, the observed rates of ratcheting and unratcheting were considerably slower than those of P/E-hybrid-state formation [41]. smFRET experiments employing a distinct labeling strategy and site of fluorophore attachment called the spontaneous and reversible nature of RSR into question [54], showing that peptide-bond formation was necessary to trigger RSR while EF-G was required to reverse this process. These mechanistic distinctions might stem from differences in the biochemical methods, labeling strategies and buffers employed. Alternatively, the structural elements of the ribosome examined by each group might move at distinct time scales.
Further insights into dynamic processes occurring in the pre-translocation ribosome have come from recent smFRET studies measuring L1 stalk motions. In line with conformational changes observed in EF-G-bound ribosome structures containing deacylated P-site tRNA [26,55], spontaneous and reversible changes in the intermolecular distance have been observed between P-site tRNA and the L1 stalk in the pre-translocation complex [56]. smFRET measurements reporting on isolated L1 stalk motions with respect to the body of the 50S subunit revealed additional complexities in the range of stalk conformations accessible [57]. In the latter report, three distinct L1 domain positions were observed in the pre-translocation complex: two configurations consistent with the initial L1 stalk smFRET study and a third, intermediate position specific to complexes containing E-site tRNA. Here, a positive correlation was shown between the rates of L1 stalk motions and RSR formation, suggesting that subunit ratcheting and L1 stalk closure might be coupled. However, the stochastic nature of both processes and an absence of correlation in reverse transitions – unratcheting and L1 stalk opening – leaves open the possibility that certain L1 stalk transitions could be substantially uncoupled from RSR and/or unratcheting. Consistent with previous cryo-EM and smFRET studies of tRNA motions within the pre-translocation complex [26,41], both investigations support the notion that L1 stalk closure might stabilize the P/E hybrid state through direct interactions with the P-site tRNA elbow.
Structures of the metastable tRNA hybrid state revealed by cryo-EM
Through the use of particle-sorting algorithms, independent research groups recently reported that the pre-trans-location complex can occupy at least two clearly distinct conformational sub-states corresponding to classical and hybrid states. These snapshot views confirming the meta-stable nature of the pre-translocation ribosome complex revealed the first molecular descriptions of hybrid tRNA configurations (Figure 3b) [58,59]. Specifically, the globally distinct structures observed corresponded to: (i) an unratcheted, classical pre-translocation complex configuration in which the A- and P-site tRNAs were in contact with the A and P loops, respectively (Figure 1a,b), with the L1 stalk in an open position; and (ii) a configuration in which RSR was observed, where both A- and P-site tRNAs occupied hybrid positions. Although the extent of RSR was estimated to be less than observed in the EF-G-bound complex [26], the L1 stalk was found in a closed configuration, interacting directly with the P/E tRNA elbow.
Surprisingly, the estimated distance between A- and P-site tRNAs in these structures was inconsistent with the intermolecular tRNA–tRNA distance estimated for the P/ E–A/P configuration (H1; ~8 Å) using smFRET (Figure 3a) [41,58,59]. Instead, the hybrid state configuration observed more closely approximated the tRNA–tRNA distances estimated for the P/E–A/A complex (H2; ~15 Å). Yet, occupancy of the A/P hybrid state was convincingly shown by both groups, as the 3′-CCA end of A-site peptidyl-tRNA clearly occupied the large subunit P site. Interestingly, the observed A/P hybrid state was largely facilitated by rotation of the A-site tRNA body. Its elbow region, the site of fluorophore labeling used in the smFRET experiments, moved by less than ~3 Å from its classical position. Indeed, such a small lateral movement would not be likely to result in appreciable FRET changes given the fluorescent labeling strategies employed. Therefore, one plausible explanation for the apparent differences is that the H2 state observed through smFRET is actually a mixture of two states with nearly identical FRET values: one containing classically configured A-site tRNA and the other containing the A/P hybrid state revealed by cryo-EM. This model, which specifies the existence of more than one A/P hybrid configuration, will need to be examined through additional experiments. Alternatively, the hybrid state structures observed might represent a weighted-average structure of H1 and H2 tRNA configurations whose structural features could not be distinguished by the particle-sorting protocols employed. In this case, the observed A-site tRNA position would represent a mixture of A/A and A/P tRNA positions. If RSR amplitudes vary depending on the nature and/or position of A-site substrates, then this model could explain why RSR amplitudes were smaller than those observed in the EF-G-bound complex [26].
Here, the value of parallel cryo-EM and smFRET studies becomes apparent. Although both methods suffer inherent limitations – particle averaging in cryo-EM and limited structural perspective in smFRET – combined efforts might deliver a more complete biological under-standing. Cryo-EM provides an invaluable and proven method for elucidating the structures of intermediate states in translation, whereas smFRET can reveal the dynamic properties of these systems. Particle average phenomena are likely to remain a challenge in cryo-EM structure determination, where the number of potential subpopulations present in the ensemble scales with the number of conformational degrees of freedom in the ribo-some. Averaging considerations are likely to dampen the amplitudes of motion revealed by cryo-EM investigations and must be interpreted in this context. Continued advances in cryo-EM data collection methods combined with improved computational power for data analysis are sure to meet these challenges in the future by allowing larger numbers of particles to be sorted into smaller and smaller subpopulations.
The energy landscape interpretation of ribosome dynamics
Knowledge that the ribosome can spontaneously adopt distinct native state conformations that are in equilibrium at room temperature necessitates a metastable energy landscape view of ribosome function (Box 2) [60]. This landscape must contain energy wells that represent distinct conformational states that are accessible to the ribo-some as well as the activation energies that govern the rates of transition between them. Similar models have been articulated for protein folding and dynamics for decades [12–15]; by contrast, its application to larger macromolecular systems such as the ribosome remains in its infancy.
Box 2. The free energy landscape view of the translation reaction coordinate.
Distinct structural elements of the ribosome (e.g. L1 stalk, tRNAs, GAC, 30S head), each intrinsically dynamic on multiple length and time scales, participate in the process of translation. Such considerations suggest that translation must proceed via the orchestrated navigation of a multidimensional, hierarchical and rugged free energy landscape. Local minima on the free energy landscape represent macroscopic, mesoscopic and microscopic sub-state conformations distinguished by changes in one or more distinct conformational degrees of freedom within the particle (Figure I). In a regime where each conformational degree of freedom in the ribosome is uncoupled fromall others and the activation barriers for motion are on the order of ~k_B_T, the number of sub-state conformations in equilibrium at room temperature scales with the number of unique mobile elements. In this view, transitions between distinct configurations can occur via multiple structural and kinetic pathways, where each pathway corresponds to a unique sequence of conformational events (Figure I). Here, the number of distinct structural and kinetic pathways transited during navigation of the energy landscape, and their relative probabilities, depends strongly on the barrier heights of each transition and the extent to which conformational degrees of freedom are coupled.
The existence of a distribution of pathways through the energy landscape is analogous to the well-established protein- and RNAfolding funnel models, where the final folded state of a biopolymer can be reached by way of numerous structural pathways consisting of a diverse range of intermediate states [76]. A key distinction between the protein folding and ribosome energy landscape models is that a single, deep global energy minimum does not exist on the translation reaction coordinate. Rather, the energy landscape navigated during translation is likely to consist of a multitude of conformational sub-states, each with relatively similar stabilities. Here, the absolute free energies of sub-state conformations of the ribosome and the barrier heights for structural transitions are anticipated to depend strongly on temperature, ionic conditions in the cellular environment, factor and ligand availability, posttranscriptional and -translational modifications of the ribosome, and small-molecule effectors of translation. Here, uncoupled and/or loosely coupled conformational processes in the ribosome provide a degree of robustness to the system where mutational and/or kinetic traps are avoided as well as an increase the number of regulation mechanisms.
Figure I.

Schematic of ligand-induced modulation of the free-energy landscape. (a) Graph indicating the hierarchical structure of the free energy landscape with the magnitude of activation barriers relative to k_B_T (_k_B is Boltzmann’s constant, T is temperature). Also shown is the remodeling of the landscape before (black) and after (red) ligand binding. (b) Hypothetical graph diagram indicating the existence of multiple pathways connecting states on the energy landscape and the re-weighting of preferred pathways before (black) and after (red) ligand binding.
To encompass the many time and length scales of dynamics expected to occur within the ribosome, the energy landscape model must be hierarchical in nature (see Figure Ia in Box 2). Large-scale changes in the conformation of the ribosome complex, such as the transition between the pre- and post-translocation configurations, represent macroscopic states on the landscape with deep energy basins separated by energy barriers significantly greater than k_B_T (_k_B is Boltzmann’s constant and T is temperature). To account for the spontaneous motions of tRNAs between classical and hybrid states, subunit ratcheting and unratcheting events and L1 stalk dynamics observed through smFRET experiments, macroscopic states must contain numerous mesoscopic states separated by activation energies on the order of k_B_T. The existence of smaller scale motions, such as those of individual protein side chains and unstructured RNA elements, specifies that mesoscopic state in turn consists of a large ensemble of microscopic states with still smaller activation energies (<k_B_T). This basic energetic framework provides a basis for understanding the potential complexity underpinning ribosome conformational processes, where motions across multiple time and length scales can contribute to function.
An important aspect of this, or any energy landscape framework, is that the ground state energies and/or activation barriers to motion can be remodeled in response to changes in the composition of the ribosome complex and changes in solution environment. For example, EF-G accelerates the rate of translocation more than ~50 000-fold over spontaneous translocation [61]. Conversely, binding of deacylated tRNA to the ribosomal E site stimulates reverse translocation, a reaction that occurs at negligible rates in its absence [62,63]. Ligand-modulated remodeling of the ribosome energy landscape might also explain the action of ribosome-targeting antibiotics [15,64], as well as factors such as leader peptidase A (LepA) that drive the translating ribosome complex in a reverse direction [65,66]. Environmental variables, such as temperature or ionic strength, can regulate function by altering the energy landscape in ways that favor or inhibit specific transitions through ground-state stabilization or destabilization. This notion is apparent in both the structural studies of pre-translocation complexes discussed above, where relative populations of the ratcheted (hybrid) state and unratcheted (classical) state subpopulations (~20% ratcheted at ~20 mM MgCl2 [58] and ~70% ratcheted at 3.5 mM MgCl2 [59]) are dramatically different [46].
The energy landscape model is also useful for under-standing how mutations within, or distal to, the ribosome’s active centers can alter function [67]. For example, mutations near the sites of interactions between tRNA and the ribosome can affect the rate and/or fidelity of translocation [68–71] and tRNA selection [35]. Unusual ribosome–substrate interactions can stall [48], promote [72] and alter [73] the translation mechanism. Such observations point to the existence of crucial nodes in the network of interactions within the ribosome that modulate dynamics over both short [74] and long distances [75].
According to this framework, the intrinsically dynamic nature of the ribosome is a central feature of cellular translation control [10,60]. In processive translation reactions, such as elongation, small changes in the activation energies of crucial transitions can have a profound impact at the systems level. The energy landscape view also implies that, much like the funnel model of protein folding [76], multiple structural and kinetic pathways can exist between macroscopic, mesoscopic and microscopic states (see Figure Ib in Box 2). Such complexity has been implicated through smFRET observations demonstrating that transitions between classical and hybrid states can occur through independent and/or coupled processes [41].
Concluding remarks and future perspectives
The many recent advances in structural and functional investigations of the ribosome highlight the potentially dynamic nature of the translating particle and how such dynamism could have a role in modulating both ribosome– substrate interactions and the translation mechanism. Although the metastable energy landscape model foreshadows the existence of many conformational degrees of freedom and sub-states of the ribosome beyond those previously described – global state-1 (unratcheted, classical, and L1 stalk open) and global state-2 (ratcheted, hybrid and L1 stalk closed) [10,56,58,59] – a complete structural description of the many key structural transitions under-pinning the translation process could soon be within reach.
Ultimately, this important challenge might be best addressed by using integrated experimental strategies that employ a combination of cryo-EM, smFRET and computational modeling. Here, smFRET could be used to accelerate the pipeline of structure determination efforts by identifying and stabilizing unique translation intermediates. Computational modeling could be used to provide molecular descriptions of the cryo-EM data and an atomic resolution framework in which to interpret the smFRET findings. Ultimately, the integrated whole could also be used to design new smFRET experiments, where new fluorophore labeling strategies and site-directed mutagenesis could be used to further explore structural and dynamic features of the system or to examine complex allosteric signaling pathways. Here, targeted molecular dynamic simulations and/or long-time-scale simulations [74,77] might provide an important tool for exploring the trajectories between the discrete sub-states observed. Such efforts could be particularly valuable when addressing potentially uncoupled or loosely coupled conformational events wherein numerous structural and kinetic pathways might exist between macroscopic, mesoscopic and microscopic sub-states. Such integrated efforts, when combined with novel experimental platforms probing the ribosome from multiple structural and kinetic perspectives simultaneously, will be crucial for advancing our knowledge of the energy landscape’s dimensionality and for providing unified views of the structural, biochemical and kinetic descriptions of the translation process. On the immediate horizon, multi-color smFRET imaging [78] of multiple elongation cycles and the complete translation process, in combination with integrated optical trap-smFRET investigations [79], will facilitate new discoveries in both the basic mechanism of translation as well as the cellular strategies employed to regulate this key gene expression pathway.
Acknowledgments
We are grateful to members of the Blanchard laboratory for their careful reviews and critical comments during the preparation of this manuscript. The authors also thank Joachim Frank (Howard Hughes Medical Institute, Columbia University) for granting permission to present structural models of tRNA hybrid states determined by cryo-EM. This work was supported by the National Institutes of Health (GM 079238), the National Science Foundation (0644129) and the Human Frontiers in Science Program.
References
- 1.Ramakrishnan V. What we have learned from ribosome structures. Biochem Soc Trans. 2008;36:567–574. doi: 10.1042/BST0360567. [DOI] [PubMed] [Google Scholar]
- 2.Korostelev A, et al. Structural dynamics of the ribosome. Curr Opin Chem Biol. 2008;12:674–683. doi: 10.1016/j.cbpa.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berk V, Cate JH. Insights into protein biosynthesis from structures of bacterial ribosomes. Curr Opin Struct Biol. 2007;17:302–309. doi: 10.1016/j.sbi.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 4.Wilson DN, Nierhaus KH. The weird and wonderful world of bacterial ribosome regulation. Crit Rev Biochem Mol Biol. 2007;42:187– 219. doi: 10.1080/10409230701360843. [DOI] [PubMed] [Google Scholar]
- 5.Herbert TP, Proud CG. Regulation of translational elongation and the cotranslational protein targeting pathway. In: Mathews MB, et al., editors. Translational Control in Biology and Medicine. Cold Spring Harbor Laboratory Press; 2007. pp. 601–624. [Google Scholar]
- 6.Kozak M. Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene. 2005;361:13–37. doi: 10.1016/j.gene.2005.06.037. [DOI] [PubMed] [Google Scholar]
- 7.Shoji S, et al. Ribosomal translocation: one step closer to the molecular mechanism. ACS Chem Biol. 2009;4:93–107. doi: 10.1021/cb8002946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wintermeyer W, et al. Mechanisms of elongation on the ribosome: dynamics of a macromolecular machine. Biochem Soc Trans. 2004;32:733–737. doi: 10.1042/BST0320733. [DOI] [PubMed] [Google Scholar]
- 9.Mitra K, Frank J. Ribosome dynamics: insights from atomic structure modeling into cryo-electron microscopy maps. Annu Rev Biophys Biomol Struct. 2006;35:299–317. doi: 10.1146/annurev.biophys.35.040405.101950. [DOI] [PubMed] [Google Scholar]
- 10.Frank J, et al. The process of mRNA–tRNA translocation. Proc Natl Acad Sci U S A. 2007;104:19671–19678. doi: 10.1073/pnas.0708517104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kurland CG, Ehrenberg M. Growth-optimizing accuracy of gene expression. Annu Rev Biophys Biophys Chem. 1987;16:291–317. doi: 10.1146/annurev.bb.16.060187.001451. [DOI] [PubMed] [Google Scholar]
- 12.Frauenfelder H, et al. The energy landscapes and motions of proteins. Science. 1991;254:1598–1603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- 13.Henzler-Wildman K, Kern D. Dynamic personalities of proteins. Nature. 2007;450:964–972. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- 14.Smock RG, Gierasch LM. Sending signals dynamically. Science. 2009;324:198–203. doi: 10.1126/science.1169377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee GM, Craik CS. Trapping moving targets with small molecules. Science. 2009;324:213–215. doi: 10.1126/science.1169378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bretscher MS. Translocation in protein synthesis: a hybrid structure model. Nature. 1968;218:675–677. doi: 10.1038/218675a0. [DOI] [PubMed] [Google Scholar]
- 17.Moazed D, Noller HF. Intermediate states in the movement of transfer RNA in the ribosome. Nature. 1989;342:142–148. doi: 10.1038/342142a0. [DOI] [PubMed] [Google Scholar]
- 18.Samaha RR, et al. A base pair between tRNA and 23S rRNA in the peptidyl transferase centre of the ribosome. Nature. 1995;377:309– 314. doi: 10.1038/377309a0. [DOI] [PubMed] [Google Scholar]
- 19.Kim DF, Green R. Base-pairing between 23S rRNA and tRNA in the ribosomal A site. Mol Cell. 1999;4:859–864. doi: 10.1016/s1097-2765(00)80395-0. [DOI] [PubMed] [Google Scholar]
- 20.Odom OW, et al. Movement of tRNA but not the nascent peptide during peptide bond formation on ribosomes. Biochemistry. 1990;29:10734–10744. doi: 10.1021/bi00500a004. [DOI] [PubMed] [Google Scholar]
- 21.Stark H, et al. Arrangement of tRNAs in pre- and posttranslocational ribosomes revealed by electron cryomicroscopy. Cell. 1997;88:19–28. doi: 10.1016/s0092-8674(00)81854-1. [DOI] [PubMed] [Google Scholar]
- 22.Agrawal RK, et al. Visualization of tRNA movements on the Escherichia coli 70S ribosome during the elongation cycle. J Cell Biol. 2000;150:447–460. doi: 10.1083/jcb.150.3.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yusupov MM, et al. Crystal structure of the ribosome at 5.5 Å resolution. Science. 2001;292:883–896. doi: 10.1126/science.1060089. [DOI] [PubMed] [Google Scholar]
- 24.Agrawal RK, et al. Effect of buffer conditions on the position of tRNA on the 70S ribosome as visualized by cryoelectron microscopy. J Biol Chem. 1999;274:8723–8729. doi: 10.1074/jbc.274.13.8723. [DOI] [PubMed] [Google Scholar]
- 25.Frank J, Agrawal RK. A ratchet-like inter-subunit reorganization of the ribosome during translocation. Nature. 2000;406:318–322. doi: 10.1038/35018597. [DOI] [PubMed] [Google Scholar]
- 26.Valle M, et al. Locking and unlocking of ribosomal motions. Cell. 2003;114:123–134. doi: 10.1016/s0092-8674(03)00476-8. [DOI] [PubMed] [Google Scholar]
- 27.Moazed D, Noller HF. Transfer RNA shields specific nucleotides in 16S ribosomal RNA from attack by chemical probes. Cell. 1986;47:985–994. doi: 10.1016/0092-8674(86)90813-5. [DOI] [PubMed] [Google Scholar]
- 28.Spiegel PC, et al. Elongation factor G stabilizes the hybrid-state conformation of the 70S ribosome. RNA. 2007;13:1473–1482. doi: 10.1261/rna.601507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ermolenko DN, et al. Observation of intersubunit movement of the ribosome in solution using FRET. J Mol Biol. 2007;370:530–540. doi: 10.1016/j.jmb.2007.04.042. [DOI] [PubMed] [Google Scholar]
- 30.Ermolenko DN, et al. The antibiotic viomycin traps the ribosome in an intermediate state of translocation. Nat Struct Mol Biol. 2007;14:493–497. doi: 10.1038/nsmb1243. [DOI] [PubMed] [Google Scholar]
- 31.Korostelev A, et al. Crystal structure of a 70S ribosome–tRNA complex reveals functional interactions and rearrangements. Cell. 2006;126:1065–1077. doi: 10.1016/j.cell.2006.08.032. [DOI] [PubMed] [Google Scholar]
- 32.Selmer M, et al. Structure of the 70S ribosome complexed with mRNA and tRNA. Science. 2006;313:1935–1942. doi: 10.1126/science.1131127. [DOI] [PubMed] [Google Scholar]
- 33.Berk V, et al. Structural basis for mRNA and tRNA positioning on the ribosome. Proc Natl Acad Sci U S A. 2006;103:15830–15834. doi: 10.1073/pnas.0607541103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schuwirth BS, et al. Structures of the bacterial ribosome at 3.5 Å resolution. Science. 2005;310:827–834. doi: 10.1126/science.1117230. [DOI] [PubMed] [Google Scholar]
- 35.Ogle JM, Ramakrishnan V. Structural insights into translational fidelity. Annu Rev Biochem. 2005;74:129–177. doi: 10.1146/annurev.biochem.74.061903.155440. [DOI] [PubMed] [Google Scholar]
- 36.Abdi NM, Fredrick K. Contribution of 16S rRNA nucleotides forming the 30S subunit A and P sites to translation in Escherichia coli. RNA. 2005;11:1624–1632. doi: 10.1261/rna.2118105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jenner L, et al. Messenger RNA conformations in the ribosomal E site revealed by X-ray crystallography. EMBO Rep. 2007;8:846–850. doi: 10.1038/sj.embor.7401044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Korostelev A, et al. Interactions and dynamics of the Shine Dalgarno helix in the 70S ribosome. Proc Natl Acad Sci U S A. 2007;104:16840–16843. doi: 10.1073/pnas.0707850104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Uemura S, et al. Peptide bond formation destabilizes Shine– Dalgarno interaction on the ribosome. Nature. 2007;446:454–457. doi: 10.1038/nature05625. [DOI] [PubMed] [Google Scholar]
- 40.Semenkov YP, et al. Energetic contribution of tRNA hybrid state formation to translocation catalysis on the ribosome. Nat Struct Biol. 2000;7:1027–1031. doi: 10.1038/80938. [DOI] [PubMed] [Google Scholar]
- 41.Munro JB, et al. Identification of two distinct hybrid state intermediates on the ribosome. Mol Cell. 2007;25:505–517. doi: 10.1016/j.molcel.2007.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blanchard SC, et al. tRNA dynamics on the ribosome during translation. Proc Natl Acad Sci U S A. 2004;101:12893–12898. doi: 10.1073/pnas.0403884101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dorner S, et al. The hybrid state of tRNA binding is an authentic translation elongation intermediate. Nat Struct Mol Biol. 2006;13:234– 241. doi: 10.1038/nsmb1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pan D, et al. Kinetically competent intermediates in the translocation step of protein synthesis. Mol Cell. 2007;25:519–529. doi: 10.1016/j.molcel.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Walker SE, et al. Role of hybrid tRNA-binding states in ribosomal translocation. Proc Natl Acad Sci U S A. 2008;105:9192– 9197. doi: 10.1073/pnas.0710146105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marshall RA, et al. Translation at the single-molecule level. Annu Rev Biochem. 2008;77:177–203. doi: 10.1146/annurev.biochem.77.070606.101431. [DOI] [PubMed] [Google Scholar]
- 47.Blanchard SC. Single-molecule observations of ribosome function. Curr Opin Struct Biol. 2009;19:103–109. doi: 10.1016/j.sbi.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wen JD, et al. Following translation by single ribosomes one codon at a time. Nature. 2008;452:598–603. doi: 10.1038/nature06716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peske F, et al. Conformational changes of the small ribosomal subunit during elongation factor G-dependent tRNA–mRNA translocation. J Mol Biol. 2004;343:1183–1194. doi: 10.1016/j.jmb.2004.08.097. [DOI] [PubMed] [Google Scholar]
- 50.Savelsbergh A, et al. An elongation factor G-induced ribosome rearrangement precedes tRNA–mRNA translocation. Mol Cell. 2003;11:1517–1523. doi: 10.1016/s1097-2765(03)00230-2. [DOI] [PubMed] [Google Scholar]
- 51.Sharma D, et al. EF-G-independent reactivity of a pre-translocation- state ribosome complex with the aminoacyl tRNA substrate puromycin supports an intermediate (hybrid) state of tRNA binding. RNA. 2004;10:102–113. doi: 10.1261/rna.5148704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Semenkov Y, et al. Puromycin reaction for the A site-bound peptidyl-tRNA. FEBS Lett. 1992;296:207–210. doi: 10.1016/0014-5793(92)80380-y. [DOI] [PubMed] [Google Scholar]
- 53.Cornish PV, et al. Spontaneous intersubunit rotation in single ribosomes. Mol Cell. 2008;30:578–588. doi: 10.1016/j.molcel.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marshall RA, et al. Irreversible chemical steps control intersubunit dynamics during translation. Proc Natl Acad Sci U S A. 2008;105:15364–15369. doi: 10.1073/pnas.0805299105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Connell SR, et al. Structural basis for interaction of the ribosome with the switch regions of GTP-bound elongation factors. Mol Cell. 2007;25:751–764. doi: 10.1016/j.molcel.2007.01.027. [DOI] [PubMed] [Google Scholar]
- 56.Fei J, et al. Coupling of ribosomal L1 stalk and tRNA dynamics during translation elongation. Mol Cell. 2008;30:348–359. doi: 10.1016/j.molcel.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 57.Cornish PV, et al. Following movement of the L1 stalk between three functional states in single ribosomes. Proc Natl Acad Sci U S A. 2009;106:2571–2576. doi: 10.1073/pnas.0813180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Julian P, et al. Structure of ratcheted ribosomes with tRNAs in hybrid states. Proc Natl Acad Sci U S A. 2008;105:16924–16927. doi: 10.1073/pnas.0809587105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Agirrezabala X, et al. Visualization of the hybrid state of tRNA binding promoted by spontaneous ratcheting of the ribosome. Mol Cell. 2008;32:190–197. doi: 10.1016/j.molcel.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Munro JB, et al. A new view of protein synthesis: mapping the free energy landscape of the ribosome using single-molecule FRET. Biopolymers. 2008;89:565–577. doi: 10.1002/bip.20961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Katunin VI, et al. Coupling of GTP hydrolysis by elongation factor G to translocation and factor recycling on the ribosome. Biochemistry. 2002;41:12806–12812. doi: 10.1021/bi0264871. [DOI] [PubMed] [Google Scholar]
- 62.Shoji S, et al. Reverse translocation of tRNA in the ribosome. Mol Cell. 2006;24:931–942. doi: 10.1016/j.molcel.2006.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Konevega AL, et al. Spontaneous reverse movement of mRNA-bound tRNA through the ribosome. Nat Struct Mol Biol. 2007;14:318– 324. doi: 10.1038/nsmb1221. [DOI] [PubMed] [Google Scholar]
- 64.Tenson T, Mankin A. Antibiotics and the ribosome. Mol Microbiol. 2006;59:1664–1677. doi: 10.1111/j.1365-2958.2006.05063.x. [DOI] [PubMed] [Google Scholar]
- 65.Qin Y, et al. The highly conserved LepA is a ribosomal elongation factor that back-translocates the ribosome. Cell. 2006;127:721– 733. doi: 10.1016/j.cell.2006.09.037. [DOI] [PubMed] [Google Scholar]
- 66.Connell SR, et al. A new tRNA intermediate revealed on the ribosome during EF4-mediated back-translocation. Nat Struct Mol Biol. 2008;15:910–915. doi: 10.1038/nsmb.1469. [DOI] [PubMed] [Google Scholar]
- 67.Kurland CG, et al. Limitations of Translational Accuracy. American Society for Microbiology Press; 1996. [Google Scholar]
- 68.Garcia-Ortega L, et al. Precise alignment of peptidyl tRNA by the decoding center is essential for EF-G-dependent translocation. Mol Cell. 2008;32:292–299. doi: 10.1016/j.molcel.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lancaster LE, et al. Colicin E3 cleavage of 16S rRNA impairs decoding and accelerates tRNA translocation on Escherichia coli ribosomes. Mol Microbiol. 2008;69:390–401. doi: 10.1111/j.1365-2958.2008.06283.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pan D, et al. Rapid ribosomal translocation depends on the conserved 18–55 base pair in P-site transfer RNA. Nat Struct Mol Biol. 2006;13:354–359. doi: 10.1038/nsmb1074. [DOI] [PubMed] [Google Scholar]
- 71.Komoda T, et al. The A-site finger in 23 S rRNA acts as a functional attenuator for translocation. J Biol Chem. 2006;281:32303– 32309. doi: 10.1074/jbc.M607058200. [DOI] [PubMed] [Google Scholar]
- 72.Kieft JS. Viral IRES RNA structures and ribosome interactions. Trends Biochem Sci. 2008;33:274–283. doi: 10.1016/j.tibs.2008.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zaher HS, Green R. Quality control by the ribosome following peptide bond formation. Nature. 2009;457:161–166. doi: 10.1038/nature07582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vaiana AC, Sanbonmatsu KY. Stochastic gating and drug-ribosome interactions. J Mol Biol. 2009;386:648–661. doi: 10.1016/j.jmb.2008.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Di Giacco V, et al. Shine–Dalgarno interaction prevents incorporation of noncognate amino acids at the codon following the AUG. Proc Natl Acad Sci U S A. 2008;105:10715–10720. doi: 10.1073/pnas.0801974105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Onuchic JN, Wolynes PG. Theory of protein folding. Curr Opin Struct Biol. 2004;14:70–75. doi: 10.1016/j.sbi.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 77.Sanbonmatsu KY, et al. Simulating movement of tRNA into the ribosome during decoding. Proc Natl Acad Sci U S A. 2005;102:15854– 15859. doi: 10.1073/pnas.0503456102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hohng S, et al. Single-molecule three-color FRET. Biophys J. 2004;87:1328–1337. doi: 10.1529/biophysj.104.043935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lang MJ, et al. Simultaneous, coincident optical trapping and single-molecule fluorescence. Nat Methods. 2004;1:133–139. doi: 10.1038/nmeth714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wilden B, et al. Role and timing of GTP binding and hydrolysis during EF-G-dependent tRNA translocation on the ribosome. Proc Natl Acad Sci U S A. 2006;103:13670–13675. doi: 10.1073/pnas.0606099103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Marintchev A, Wagner G. Translation initiation: structures, mechanisms and evolution. Q Rev Biophys. 2004;37:197–284. doi: 10.1017/S0033583505004026. [DOI] [PubMed] [Google Scholar]
- 82.Xu J, et al. Molecular localization of a ribosome-dependent ATPase on Escherichia coli ribosomes. Nucleic Acids Res. 2006;34:1158–1165. doi: 10.1093/nar/gkj508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dinos G, et al. Deacylated tRNA is released from the E site upon A site occupation but before GTP hydrolyzed by EF-Tu. Nucleic Acids Res. 2005;33:5291–5296. doi: 10.1093/nar/gki833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Petry S, et al. The termination of translation. Curr Opin Struct Biol. 2008;18:70–77. doi: 10.1016/j.sbi.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 85.Peske F, et al. Sequence of steps in ribosome recycling as defined by kinetic analysis. Mol Cell. 2005;18:403–412. doi: 10.1016/j.molcel.2005.04.009. [DOI] [PubMed] [Google Scholar]