Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome (original) (raw)
Abstract
Induced pluripotent stem cells (iPSC) have been generated from somatic cells by introducing reprogramming factors. Integration of foreign genes into the host genome is a technical hurdle for the clinical application. Here, we show that Sendai virus (SeV), an RNA virus and carries no risk of altering host genome, is an efficient solution for generating safe iPSC. Sendai-viral human iPSC expressed pluripotency genes, showed demethylation characteristic of reprogrammed cells. SeV-derived transgenes were decreased during cell division. Moreover, viruses were able to be easily removed by antibody-mediated negative selection utilizing cell surface marker HN that is expressed on SeV-infected cells. Viral-free iPSC differentiated to mature cells of the three embryonic germ layers in vivo and in vitro including beating cardiomyocytes, neurons, bone and pancreatic cells. Our data demonstrated that highly-efficient, non-integrating SeV-based vector system provides a critical solution for reprogramming somatic cells and will accelerate the clinical application.
Keywords: iPS cell, Sendai virus, transgene-free, reprogramming, human
Introduction
After the report of Takahashi and Yamanaka1) induced pluripotent stem (iPS) cells have been generated from somatic cells by transducing reprogramming factors in mammalian including mouse1)–4) and human.5)–9) A major limitation of this technology is the integration of viral transgenes into the host genome that includes the risk of tumorigenicity.3) Several solutions for this problem have been developed with use of adenovirus vectors10) or plasmids11),12) for induction of iPSCs, but the risk of integration still remains as far as DNA-type vectors are used.13) These methods also suffered from the low efficiency of transdution. Alternative methods using a transposon, or Cre/LoxP system require subsequent excision of transgenes from the host genome.14)–16) Use of small molecules that replace the reprogramming genes17) may solve the problem but remain unrealized in human cells. Delivery of recombinant reprogramming proteins was able to generate iPSCs, however, the efficiency was very low and needed repetitive induction.18),19) Considering that all those problems are derived from each of the fact that reprogramming genes have been introduced and present in the host cells as DNA form and/or the low efficiency of protein expression, it is obvious that RNA virus based, and high efficient expression vector such as Sendai virus can bring an ultimate solution for those problem.
We have developed Sendai virus (SeV) vectors that replicate in the form of negative-sense single-stranded RNA in the cytoplasm of infected cells, which do not go through a DNA phase nor integrate into the host genome.20) Since SeV vectors are very efficient for introduction of foreign genes in wide spectrum of host cell species and tissues, moreover, controllable for foreign gene expression,21) SeV vectors have been considered for clinical studies of gene therapy for cystic fibrosis,22),23) critical limb ischemia24) or vaccines for AIDS.25)
In this study, we investigated whether or not Fprotein deficient (ΔF), non-transmissible SeV vectors with low cytotoxity26),27) that allow high-level expression of exogenous genes without integrating into the host genome, could be used to generate human iPSCs.
Material and methods
Cell culture
Human fibroblast BJ from neonatal foreskin (ATCC, USA), and HDF from facial dermis of 36-year femail (Cell Applications, Inc., USA), were maintained in fibroblast growth medium (Cell Applications, Inc., USA) or DMEM (Invitrogen, USA) supplemented with 10% FBS. PA6 feeder cells (RIKEN BRC, Japan) were grown in alpha-MEM (Invitrogen, USA) with 10% FBS. Human iPSCs were maintained on MMC-treated MEF feeder cells (ReproCELL, Japan) in primate ES medium (ReproCELL, Japan) supplemented with 10 ng/ml bFGF (R&D systems, USA). Human iPSCs were passaged by 1mg/ml collagenase IV (Invitrogen, USA).
Generation of SeV vectors
The open reading frames (ORFs) of human OCT3/4 were obtained from NCCIT cDNA and the ORFs of human SOX2, KLF4 and c-Myc genes were amplified from Jurkat cell cDNA by RT-PCR. Those four genes were amplified with Not I-tagged gene-specific forward primer and Not I-tagged gene-specific reverse primer containing SeV-specific transcriptional regulatory signal sequences26) listed in Table 2. The amplified fragment was introduced into F-deficient SeV vector. Recovery and propagation of the SeV/ΔF vectors were carried out as follows. Briefly, 293T cells were transfected with template pSeV/ΔF carrying each transgenes and pCAGGS-plasmids each carrying the T7 RNA polymerase, NP, P, F5R, and L gene. The cells were maintained in DMEM supplemented with 10% heat-inactivated fetal bovine serum and cultured for 1 to 3 days to generate the seed SeV/ΔF vector. Then, the vector was propagated using the LLC-MK2/F7/A cells that were Sendai virus F-expressing LLC-MK2 cells line previously established in MEM containing trypsin (2.5 μg/ml). The vector titers (cell infectious unit (CIU) per ml) of recovered SeV vector were determined by immunostaining using anti-SeV rabbit polyclonal serum described as previously.26)
Table 2.
List of primer sequences for PCR
| For cDNA cloning | ||
|---|---|---|
| Gene | 5′ | 3′ |
| Oct3/4 | CACCATGCTTGGGGCGCCTTCCTTCC | CATCGGAGTTGCTCTCCACCCCGAC |
| CCCGCCGTATGAGTTCTGTGG | GCCGCGGCCGCGTTATCAGTTTGAATGCATGGGAGAGCCCAG | |
| GCCGCGGCCGCACCATGGCGGGACACCTGGCTTC | GCCGCGGCCGCGTTATCAGTTTGAATGCATGGGAGAGCCCAG | |
| Sox2 | CAAAGTCCCGGCCGGGCCGAGGGTCGG | CCCTCCAGTTCGCTGTCCGGCCC |
| GATGTACAACATGATGGAGACGGAGC | GTCACATGTGTGAGAGGGGCAGTG | |
| Klf4 | CCACATTAATGAGGCAGCCACCTGGC | GCAGTGTGGGTCATATCCACTGTCTG |
| GATGGCTGTCAGCGACGCGCTGCTCCC | GTTAAAAATGCCTCTTCATGTGTAAGGCGAG | |
| c-Myc | AACCAGCAGCCTCCCGCGACG | AGGACATTTCTGTTAGAAGGAATCG |
| GATGCCCCTCAACGTTAGCTTCACC | GTTACGCACAAGAGTTCCGTAGCTG | |
| For plasmid constraction of SeV vector | ||
| Notl-_Klf_-4F(5′) | ATTGCGGCCGCGACATGGCTGTCAGCGACGCGCTG | |
| Notl-_Klf_-4R(3′) | ATTGCGGCCGCGATGAACTTTCACCCTAAGTTTTTCTTACTACGGTTAAAAATGCCTCTTCATGTGTAAGGCGAGGTGGTC | |
| Notl-c-Myc F(5′) | ATTGCGGCCGCATGCCCCTCAACGTTAGCTTCAC | |
| Notl-c-Myc R(3′) | ATTGCGGCCGCGATGAACTTTCACCCTAAGTTTTTCTTACTACGGTTACGCACAAGAGTTCCGTAGCTGTTCAAGTTTGTGTTTC | |
| Notl-Sox2 F(5′) | ATTGCGGCCGCATGTACAACATGATGGAGACG | |
| Notl-Sox2 R(3′) | ATTGCGGCCGCGATGAACTTTCACCCTAAGTTTTTCTTACTACGGTCACATGTGTGAGAGGGGCAGTGTGCCGTTAATGGCCGTG | |
| Notl-Oct3/4 F(5′) | ATTGCGGCCGCCCCATGGCGGGACACCTGGCTTC | |
| Notl-Oct3/4 R(3′) | ATTGCGGCCGCGATGAACTTTCACCCTAAGTTTTTCTTACTACGGTCAAAGCGGCAGATGGTCGTTTGGCTGAACACCTTC | |
| For RT-PCR | ||
| Gene | 5′ | 3′ |
| Oct3/4 (SeV-Tg) | CCCGAAAGAGAAAGCGAACCAG (Oct3/4) | AATGTATCGAAGGTGCTCAA (SeV) |
| Sox2 (SeV-Tg) | ACAAGAGAAAAAACATGTATGG (SeV) | ATGCGCTGGTTCACGCCCGCGCCCAGG (Sox2) |
| Klf4 (SeV-Tg) | ACAAGAGAAAAAACATGTATGG (SeV) | CGCGCTGGCAGGGCCGCTGCTCGAC (Klf4) |
| c-Myc (SeV-Tg 18+) | ACAAGAGAAAAAACATGTATGG (SeV) | TCCACATACAGTCCTGGATGATGATG (c-Myc) |
| c-Myc (SeV-Tg HNL) | TAACTGACTAGCAGGCTTGTCG (SeV) | TCCACATACAGTCCTGGATGATGATG (c-Myc) |
| Oct3/4 (3′UTR:endo) | AGTTTGTGCCAGGGTTTTTG | ACTTCACCTTCCCTCCAACC |
| Sox2 (3′UTR:endo) | GGGAAATGGGAGGGGTGCAAAAGAGG | TTGCGTGAGTGTGGATGGGATTGGTG |
| Klf4 (3′UTR:endo) | GACAGTGGATATGACCCACACTGCC | GATAGAAGATCCAGTCACAGACC |
| c-Myc (3′UTR:endo) | ATGTCCTGAGCAATCACCTATG | AAGTTCTTTTATGCCCAAAGTCC |
| Nanog | TACCTCAGCCTCCAGCAGAT | TGCGTCACACCATTGCTATT |
| Tert | TGCCCGGACCTCCATCAGAGCCAG | TCAGTCCAGGATGGTCTTGAAGTCTG |
| TDGF1 | ATGGACTGCAGGAAGATGGCCCGC | TTAATAGTAGCTTTGTATAGAAAGGC |
| Zfp42 | TTGGAGTGCAATGGTGTGAT | TCTGTTCACACAGGCTCCAG |
| Sal4F | AAACCCCAGCACATCAACTC | GTCATTCCCTGGGTGGTTC |
| Dnmt3b | GCAGCGACCAGTCCTCCGACT | AACGTGGGGAAGGCCTGTGC |
| CABRB3 | CTTGACAATCGAGTGGCTGA | TCATCCGTGGTGTAGCCATA |
| CYP26A1 | AACCTGCACGACTCCTCGCACA | AGGATGCGCATGGCGATTCG |
| FOXD3 | GTGAAGCCGCCTTACTCGTAC | CCGAAGCTCTGCATCATGAG |
| β-actin | CAACCGCGAGAAGATGAC | AGGAAGGCTGGAAGAGTG |
| For Bisulfite sequencing | ||
| Gene | 5′ | 3′ |
| Nanog | GGAATTTAAGGTGTATGTATTTTTTATTTT | AACCCACCCTTATAAATTCTCAATTA |
| Oct3/4 (1) | AATAGATTTTGAAGGGGAGTTTAGG | TTCCTCCTTCCTCTAAAAAACTCA |
| For Fingerprinting | ||
| Gene | 5′ | 3′ |
| MCT18 | GAAACTGGCCTCCAAACACTGCCCGCCG | GTCTTGTTGGAGATGCACGTGCCCCTTGC |
| ApoB-100 | ATGGAAACGGAGAAATTATG | CCTTTCTCACTTGGCAAATAC |
| D17S1290 | GCCAACAGAGCAAGACTGTC | GGAAACAGTTAAATGGCCAA |
| For DIG-labeling PCR | ||
| Gene | 5′ | 3′ |
| KLF4 | TGACCCATCCTCCGGAGTCAGTG | GGGGATGGAAGCCGGGAGGAAGCGG |
| c-Myc | TGCCACGTCTCCACACATCAGC | GTTACGCACAAGAGTTCCGTAGCTG |
| Sox2 | GATGTACAACATGATGGAGACGGAGC | GTCACATGTGTGAGAGGGGCAGTG |
| Oct3/4 | CCCGAAAGAGAAAGCGAACCAG | GTCAGTTTGAATGCATGGGAGAGCCCAGAG |
| For cardiomyocytes | ||
| Gene | 5′ | 3′ |
| TnTc | ATGAGCGGGAGAAGGAGCGGCAGAAC | TCAATGGCCAGCACCTTCCTCCTCTC |
| MEF2C | TTTAACACCGCCAGCGCTCTTCACCTTG | TCGTGGCGCGTGTGTTGTGGGTATCTCG |
| MYHCB | CTGGAGGCCGAGCAGAAGCGCAACG | GTCCGCCCGCTCCTCTGCCTCATCC |
| GAPDH | ATCACTGCCACCCAGAAGACT | ACCAGGAAATGAGCTTGACAA |
Induction of pluripotent stem cells
Induction of human iPSCs was similar to previous report5) except using SeV vectors instead of retrovirus. Alternatively, we tested feeder-free protocol: 2 × 105 cells per well were plated and infected with SeV vectors at an MOI of 3 on 12 well plate, cultured in DMEM supplemented with 10% FBS. After ES-like colonies appeared, medium was changed to primate ES medium with bFGF and then growing colonies were harvested with collagenase IV treatment, plated on MEF feeder cells to avoid spontaneous differentiation.
Determining reprogramming efficiency
Reprogramming efficiency was calculated as the number of iPS colonies formed per number of infected cells seeded. iPS colonies were identified based on ES-like morphology, and alkaline phosphatase staining was used to facilitate the identification of iPS colonies.
Alkaline phosphatase (ALP) and immunofluorescence staining
ALP staining was performed with ALP substrate (1 step NBT/BCIP/Pierce) after fixed with 10% neutral buffered formalin solution (Wako, Japan).
Immunofluorescence staining was performed using the following primary antibodies: Nanog, SSEA1, SSEA4, PDX1, SOX17 (R&D systems, USA), SOX2, TH (Chemicon, USA), TRA-1-60 (BD Pharmingen, USA), TRA-1-81, OCT4, βIII-tubulin (2G10) (Santa Cruz Biotechnology, USA), anti-SeV-polyclonal antibody,27) anti-HN-monoclonal antibody IL4.1.28) Samples were analyzed with confocal microscopy (Bio-Rad MRC1024) or FLovel HCD-FL (Flovel, Leica). For FACS, CD34-, CD45-, CD33-PE conjugates and CD66b-FITC conjugate were purchased from BioLegend. TO-PRO3 (Molecular Probes, USA) was used for nuclear staining. 2nd antibodies used here: anti-rabbit IgG, anti-mouse IgG, IgM and ProteinA conjugated with Alexa Fluor 488 (Green) or 568 (Red) were purchased from Molecular Probes.
In vitro differentiation of human iPSCs
Embryoid bodies were generated from clumps of human iPSCs in suspension culture for 6 days in IMDM with 15% FBS, and then grown in adherent culture on gelatin-coated dish with cytokine cocktails (100 ng/ml SCF, 100 ng/ml Flt3L, 50 ng/ml TPO, 100 ng/ml G-CSF, 20 ng/ml IGF-2, and 100 ng/ml VEGF) to induce lymphoid lineage cells and cardimyocytes. 29) For differentiation to dopaminergic neurons, small clumps of SeV-iPSC were cocultured with PA6 (stromal cells derived from skull bone marrow; RIKEN BRC, Japan) in GMEM (Invitrogen, USA) containing 10% KSR (Invitrogen, USA), 1 × 10−4 M non-essential amino acids and 2-mercaptoethanol for 16 days.30) For induction of definitive endoderm cells and pancreatic cells, small clumps of iPSCs were cultured on feeder cells with 100 ng/ml activin A (R&D Systems, USA) in RPMI1640 (Invitrogen, USA) supplemented with 2% FBS for 4 days, and followed by additional 8 days culture in DMEM/F12 supplemented with N2 and B27, non-essential amino acids, β-mercaptoethanol, 0.5 mg/ml bovine serum albumin, l-glutamine and penicillin/streptomycin.31)
Teratoma formation by human iPSCs
Human iPSCs grown on MEF feeder layers were collected by collagenase IV treatment and injected subcutaneously into SCID mice. Palpable tumors were observed about one month after injection. Tumor samples were collected typically in two months, fixed in 10% formalin, and processed for paraffin embedding and hematoxylin-eosin staining following standard procedures.
Whole-genome expression analysis
For transcriptional analysis, total RNA was isolated from cells cultured in 6-well dishes using RNeasy Mini Kit. Cyanine labeled antisense RNA were amplified using Quick Amp Labeling Kit from Agilent, hybridized with Gene Expression Hybridization Kit on Whole Human Genome Oligo Microarray (one color, 4 × 44K, Agilent, USA) and analyzed by Agilent Microarray Scanner. Data were analyzed by using GeneSpring GX10.0 software (Agilent, USA). Two normalization procedures were applied; first, signal intensities less than 1 were set to 1. Then each chip was normalized to the 50th percentile of the measurements taken from that chip. Each gene was normalized to the median of that gene in the respective controls to enable comparison of relative changes in gene expression levels between different conditions. The microarray data of hES H9 cells32) and hiPSCs5) were retrived from GEO DataSets (GSM194390 and GSE9561, respectively). These analyses were performed at Bio Matrix Research, Inc.
Detection of telomerase activity
Telomerase activity was detected with a TRAPEZE telomerase detection kit (Chemicon, USA). The samples were separated by TBE-based 10% acrylamide non-denaturing gel electrophoresis. The gel was stained with ethidium bromide.
Bisulfite genomic sequencing
Genomic DNA (1 μg) from BJ, HDF and iPSCs were treated with sodium bisulfite using the BisulFast DNA Modification Kit (Toyobo, Japan) according to the manufacturer’s instruction. The promoter regions of Oct4 and Nanog were amplified by PCR using primer sets previously described.5),9) The resultant PCR products were cloned into pGEM-Teasy vector (Promega, USA) and sequenced.
RNA isolation, RT-PCR, and real-time quantitative PCR analysis
Total RNA was isolated using ISOGEN (Nippon Gene, Japan) and cDNA was synthesized using the SuperScript VILO cDNA Synthesis Kit (Invitrogen, USA). Real-time PCR was performed with the ABI Prism 7700 sequence detection system (Applied Biosystems, USA) and SYBR™ Green PCR Master Mix (Applied Biosystems, USA). RT-PCR was performed with cDNAs using gene-specific primers. Primer sequences are listed in Table 2.
DNA fingerprinting
Genomic DNA was isolated from parental fibroblasts, SeV vector-induced iPS clones using the DNeasy kit (Qiagen, Germany). Three variable number of tandem repeats (VNTR) loci, MCT118, D17S1290 and 3’ApoB were amplified by PCR and analysed by 3% agarose gel electrophoresis.
Southern blot analysis
10 μg of genomic DNA was digested with Afl II and Bam HI, or Dra I and Nco I, separated on a 1% agarose gel and blotted onto Hybond N+ membrane (Amersham Biosciences, GE healthcare, USA). Dig-labeled probes were prepared with DIG-High Prime (Roche, Swiss) using full-length PCR products of Oct3/4, Sox2, Klf4 and c-Myc genes, respectively. The membranes were hybridized with the DIG-labeled probes and then incubated with the anti-DIG antibody conjugated with alkaline phosphatase conjugate. Signals were visualized with CDP-Star chemiluminescent substrate (Roche, Swiss) and the resulted chemiluminescence was detected using the LAS-1000 (FUJIFILM, Japan).
Karyotyping
Cell division was blocked in mitotic metaphase using colcemid-spindle formation inhibitor (karyoMax colcemid solution, Gibco Invitrogen, USA). Nuclear membranes were broken after hypotonic treatment. For the chromosome visualization we used G-band standard staining (Giemsa, Wako, Japan).
Results
Expression of exogenous genes in human fibroblasts by SeV vectors
To test the efficiency of the expression of exogenous genes by SeV vectors in human fibroblast cells, BJ from neonatal foreskin, and HDF from adult facial dermis, were infected with green fluorescent protein (GFP)-carrying non-transmissible ΔF/TS-SeV vector.26),27) Almost all the infected cells expressed GFP at low multiplicities of infection (MOI, number of viral particles per cell) of three (Fig. 1A).
Fig. 1.
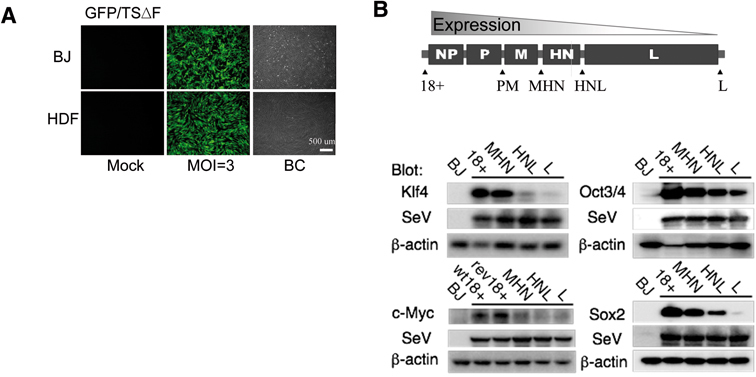
Expression of exogenous genes in human fibroblasts by SeV vectors. A. Efficient induction of GFP cDNA by TSΔF/SeV in BJ and HDF at an MOI of 3. BC: bright contrast. B. Schematic presentation of SeV vector genomes. Reprogramming genes were inserted at 18+, PM, HN, HNL and Leis (L), respectively. The expression levels of inserted genes decreased depending on the inserted site (polar effect: Refs. 21) as shown by Western blotting on day 3 after infection. Anti-SeV blot was performed to confirm equal infection efficiency of the vectors.
We next attempted to test the efficiency of SeV vectors to transduce reprogramming factors for iPSC induction. We cloned the cDNAs for reprogramming factors, Oct3/4, Sox2, Klf4, and c-Myc1) into ΔF/TS-SeV vectors at different sites, designated 18+, HMN, PM, MHN and L, to control the expression level (polar effect: 21) (Fig. 1B). Since the abundance of AT-rich region decreased the expression of target genes in SeV (Stuttering sequence: 23), silent mutations of the codons without replacement of amino acids were also prepared for wt c-Myc (designated rev-c-Myc). Expressions of these genes in human fibroblast cells by these SeV vectors were confirmed by Western blotting (Fig. 1B). Expressions of the inserted genes decreased depending on the location of insertions: insertion at 18+ showed maximum expression while insertion at L lead to minimum expression. Anti-SeV blot showed the equivalent level of infection of each SeV vector (Fig. 1B).
Optimization of SeV transduction for generating human iPSCs
Above results suggest that SeV vector that allows sustained expression of transgenes in a relatively controlled manner would be suitable for introducing reprogramming factors for iPSC formation. We next determined optimum conditions for applying SeV vectors to induce iPSC. Induction of human iPSC was carried out in a similar mannner described previously.5) In brief, 1 × 106 cells of BJ or HDF on 100 mm dish were infected with Oct3/4, Sox2, Klf4 and/or c-Myc by a series of SeV vectors at an MOI of 3. One week after infection, cells were collected and re-plated on mitomycin C-treated MEF feeder cells. The next day, medium was changed to primate ES medium supplemented with 10 ng/ml bFGF. The conditions to generate SeV-iPSC performed were summarized in the column in Fig. 2A. Reprogramming efficiency was calculated as the number of alkaline phosphatase (ALP)-positive, ES-like colonies formed per number of infected cells seeded (Fig. 2B). Typical colonies were shown in Fig. 2C. Reduction in numbers of seeded cells on MEF improved the induction efficiency (Fig. 2A, lanes 2, 3). AT-revised c-Myc also enhanced the efficiency depending on the expression level of c-Myc (rev-c-Myc: lanes 4–6). All genes inserted at HNL showed the maximum efficiency, around 1% (lane 7). The efficiency of reprogramming decreased approximately 100 fold when c-Myc was absent (lane 1), comparable to the result of retroviral induction without c-Myc.33) When Klf4, but not Oct3/4 and Sox2, were moved to the other sites than 18+ or HNL, no colonies were observed (lanes 8, 9), suggesting that the balance of expression among reprogramming factors Oct3/4, Sox2 and Klf4 is a critical factor in efficient induction of reprogramming.
Fig. 2.
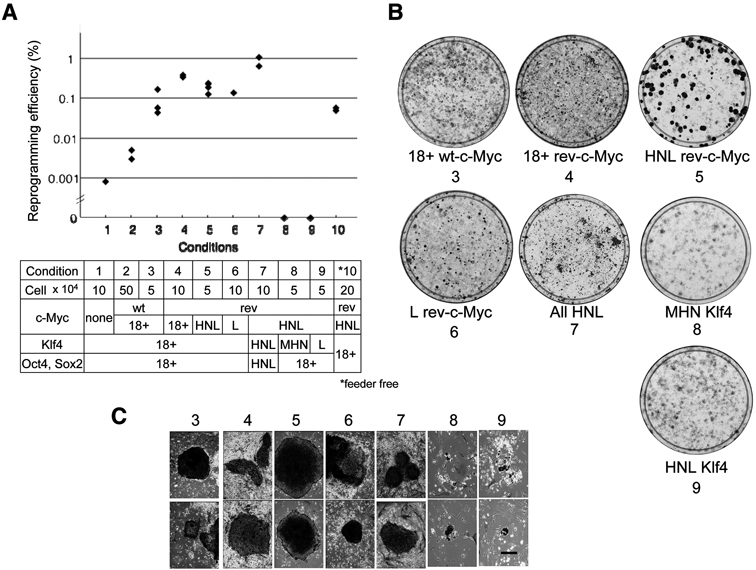
Efficient generation of human iPSCs by non-integrating SeV vectors. A. The reprogramming efficiency with SeV vectors. iPS colonies were determined by ALP positive and ES-like morphology. Lane numbers correlate with the conditions listed in the column under the figure. Each dot represents one experiment. The bars represent the average efficiency for each condition. B. ALP-staining of growing cells on 100 mm dishes. C. Typical ALP-positive colonies (scale bar: 100 μm). Numbering of C and B are correlated with the numbers in the column in A. Colonies in conditions 1,2 and feeder free were similar to those of 3 (data not shown).
We were also able to induce iPSCs by SeV vectors without feeder cells: (Fig. 2A, lane 10). Taken together, the efficiency of iPSC induction by SeV vectors with c-Myc significantly surpassed those of retroviral transduction (0.02%: Ref. 5).
Human iPSCs generated by SeV vectors (SeV-iPSCs) express hES Markers
In order to determine how many ES-like colonies fulfill the more stringent criteria for iPSCs,34) we randomly picked up ES-like colonies and established 20 clones and the results are as summarized in Table 1. To our surprise, ES-like morphology was sufficient to select SeV-generated iPSCs that fulfill typical phenotype of human ES cells. All clones expressed pluripotent markers,34) including endogenous Oct3/4, Sox2, Nanog, GDF3, TDGF1, Zfp42, Sal4F, Dnmt3b, CABRB3, CYP26A1, FOXD3, and telomerase reverse transcriptase (hTERT), at the comparable level to the expression by human embryonic carcinoma cell line, NCCIT (Fig. 3A). SeV-iPSC also expressed surface markers such as SSEA4 and TRA-1-60 and -81 (Fig. 3C), high telomerase activity (Fig. 3B) and proliferated more than six months and over 30 passages (Table 1). DNA fingerprinting analysis8) confirmed their fibroblast origin (Fig. 4A). Taken together, SeV-vector system enables a highly efficient method to generate bona fide iPSC.
Table 1.
Characterization of established clones
| clone | source | c-Myc | marker expression | epigenetics | pluripotency | SeV(-) established at | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RT-PCR | IH | WB | telo mer ase | bisulte | micro array | EB | terato ma | cardio myocytes | PA6 | pancreas | ||
| XH1 | HDF | 18+ | ✓ | ✓ | ✓ | ✓ | P(30) | |||||
| 7H5 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | –** | –** | –** | *(P20) | ||
| 7H2 | ✓ | ✓ | ||||||||||
| 7H8 | ✓ | ✓ | ||||||||||
| 7H9 | ✓ | ✓ | ||||||||||
| 7H10 | ✓ | ✓ | ||||||||||
| 4BJ1 | BJ | ✓ | ✓ | ✓ | – | – | – | ** | ||||
| B1 | ✓ | ✓ | ✓ | ✓ | ✓ | ± | – | ** | ||||
| 5B1 | ✓ | ✓ | ||||||||||
| 7B6 | ✓ | ✓ | ||||||||||
| 7B1 | ✓ | ✓ | ✓ | |||||||||
| HNLs | HNL | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | P17 |
| HNL1 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | P5 | ||
| HNL2 | ✓ | ✓ | ||||||||||
| HNL4 | ✓ | ✓ | ||||||||||
| HNL5 | ✓ | ✓ | – | ** | ||||||||
| HNL6 | ✓ | ✓ | ||||||||||
| HNLp | ✓ | ✓ | ✓ | *P20 | ||||||||
| T10 | ✓ | ✓ | ||||||||||
| T11 | ✓ | ✓ |
Fig. 3.
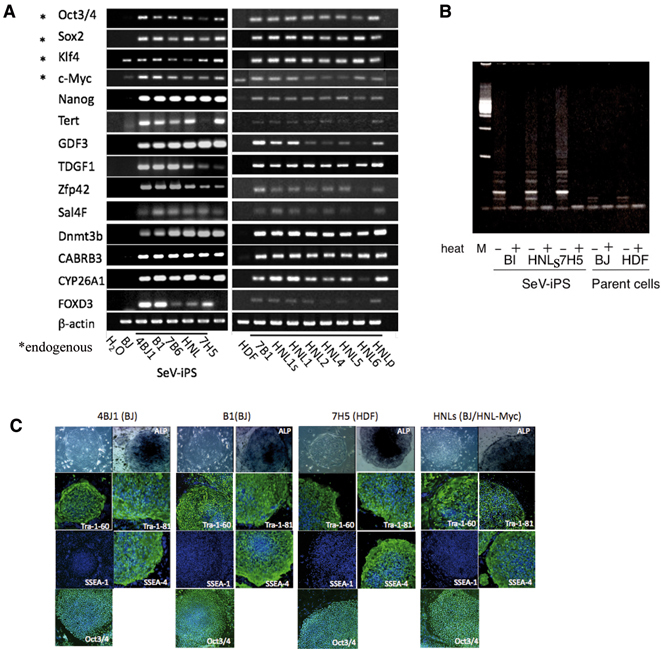
Expression of hES markers and telomerase activity in SeV-iPSC. A. RT-PCR anaysis of human ES cell-marker genes. Primers used for Oct3/4, Sox2, Klf4 and c-Myc were designed to detect the expressions of endogenous genes, but not of transgenes. Cont: PCR without cDNA. B. Telomerase activity of human SeV-iPSC. Telomerase activity was detected by the TRAP method.5) Heat-inactivated samples (+) were used as negative controls. C. Immunofluorescence staining of established clones with human ES cell-markers (Tra-1-60, Tra-1-81, SSEA-4 and Nanog). SeV-iPS colonies were positive for ALP and negative for SSEA-1 as in hES cells. Nuclei were stained with TO-PRO3 (blue).
Fig. 4.
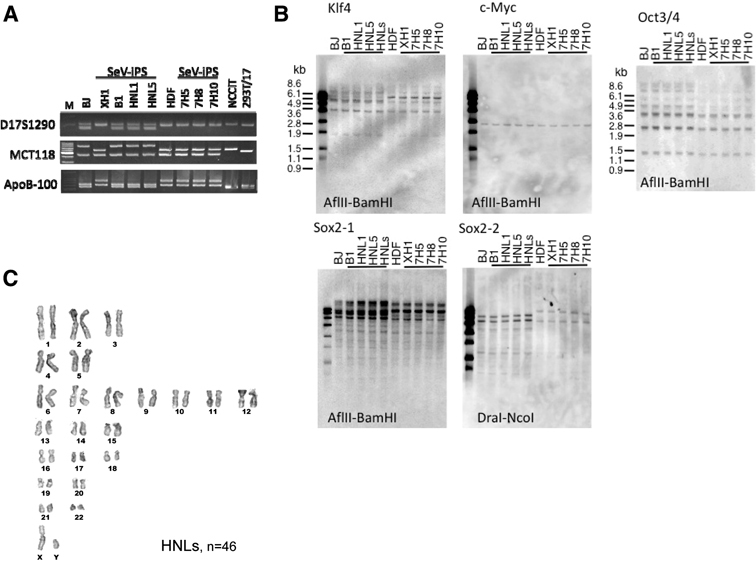
Genomic Southern blot, karyotyping and fingerprinting of SeV-iPSC. A. DNA fingerprinting of SeV-iPS clones. PCR analysis of three variable number of tandem repeats (VNTR) loci of D17S1290, MCT118 and ApoB-100 using genomic DNA from the SeV-iPS clones confirmed that these clones were originated from human fibroblasts BJ or HDF. B1, HNL1 and HNL5 were derived from BJ; XH1, 7H5, 7H8 and 7H10 were from HDF. B. Viral transgenes were not detected from the host genome as analyzed by genomic Southern blot. C. Karyotyping of SeV-iPSC. Viral-free SeV-iPSC HNLs at passage 34 were used for karyotyping.
SeV vectors were diluted and disappeared during cell growth
SeV vectors used here usually replicated constitutively in the cytoplasm of infected cells.26) Thus we next analyzed the level of vector RNA in iPSCs by RT-PCR using SeV-specific primers (Table 2).
Transgenes were expressed as early as three days after transduction (Fig. 1), and continued to be present after iPSCs were established (Fig. 5). However, transgenes and vectors tend to decrease over time. Fig. 5A showed our analyses on three clones that were generated with c-Myc inserted at 18+ site. Transgenes were lost at variable extent among these clones. In 4BJ1 and B1 (BJ-iPSC), Oct3/4 transgene persisted over 20 and 15 passages, whereas, in 7H5 (HDF-iPSC), Sox2 and Klf4 persisted longer. Among clones derived form vectors with c-Myc insertion at HNL, c-myc transgene usually persisted longer than other transgenes (Fig. 5A, right panel). This may be due to the difference of vector replication and growth advantage of c-Myc expressing cells. However, even from those clones derived from HNL-myc vectors, we could obtain two clones that lost all viral genomes (HNLs, HNL1). These clones were free of viral genome and proteins (Fig. 5B, C), carried unaltered host genome (Fig. 4B), and karyotypically normal (Fig. 4C). Moreover, we found that SeV-distributions were heterologous in passaged SeV-iPS colonies (Fig. 5D). Thus, antibody against HN protein, a major protein expressed on the surface of SeV-infected cells,20) enables us to remove HN positive cells from these heterologous populations (Fig. 5D, lower panels). The established SeV-negative clones tested here were listed in Table 1. From all clones, the virus can be removed by anti-HN-antibody-mediated negative selection theoretically, because SeV vectors were decreased in all clones tested (data not shown).
Fig. 5.
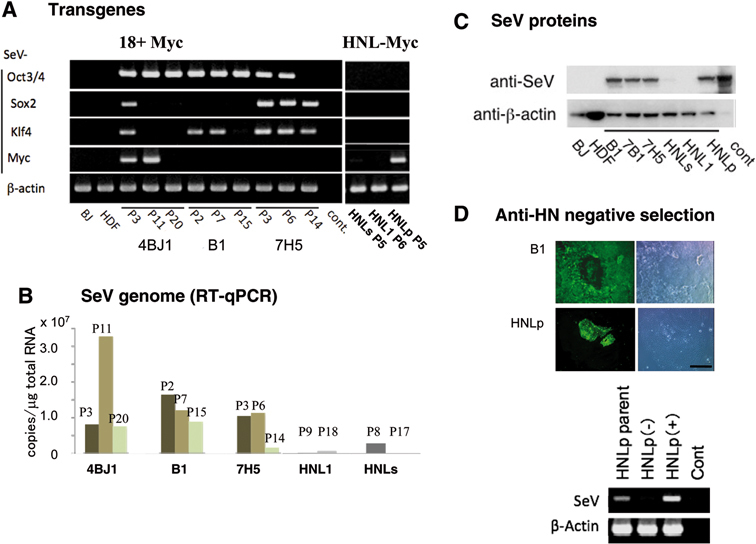
SeV vectors were diluted and lost during cell growth. A. Kinetics of transgene expression determined by RT-PCR using combination of specific primers for SeV and transgenes. cont: no template. B. Kinetics of SeV genome expression during cell growth by real time quantitative PCR. Pn means passage numbers. C. SeV-derived protein expression determined by Western blotting with anti-SeV polyclonal antibody. Passage numbers are correlated with A. cont: positive control from SeV-infected LLC-MK2 cells. Viral proteins in HNLs were slightly existed at this time (P8), but those of HNL1 were completely lost later at P17 as well as HNL1 at P9 (B). D. Anti-SeV-immunostaining revealed that SeV distribution was heterologous in iPS colonies (Upper). SeV could be removed by anti-HN-antibody mediated negative selection using anti-mouse IgG1-conjugated IMag-beads. Anti-HN antibody separated SeV-negative population (−) and SeV-enriched population (+).
We selected these viral-free clones, HNL1 and HNLs, for further studies.
DNA methylation and global gene expression profiles of human SeV-iPSCs
Reprogramming of methylated sites of genome is a signature of pluripotent cells. Thus, we analyzed the methylation states of CpG dinucleotides in the Oct3/4 and Nanog promoter regions by bisulfite genomic sequencing.5),9) The result showed that Oct3/4 and Nanog promoter regions were demethylated in SeV-iPSC contrary to parental fibroblasts (Fig. 6A). We also compared the gene expression profile of our SeV-iPSC with that of human H9 ES cell line and HDF iPSC line that were reported previously.5) Our data of HNL1 is markedly similar to that of H9 ES cell lines (Fig. 6B).
Fig. 6.
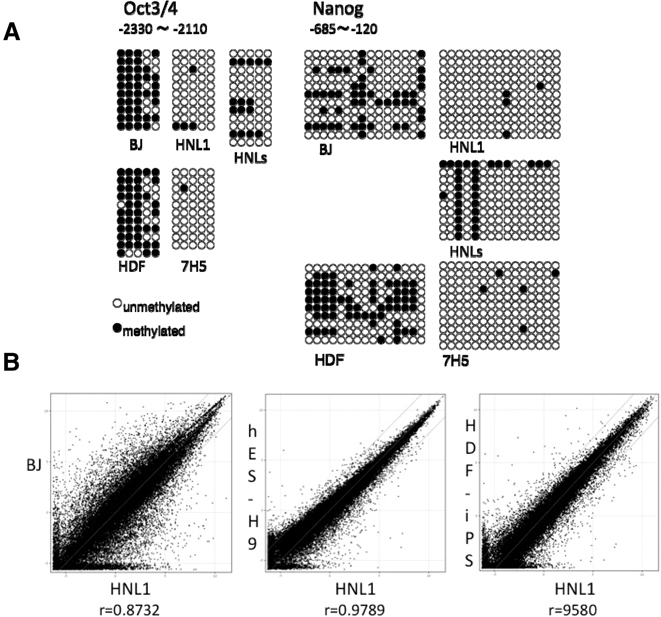
DNA methylation and global gene expression profiles of SeV-iPSC. A. Methylation analysis of Oct3/4 and Nanog promoter regions in SeV-iPSC. B. The global gene-expression patterns were compared between SeV-iPSC (HNL1) and BJ, human ES cells (H9)32) and HDF-iPSC5) with microarrays. The lines indicate the diagonal and 5-fold changes between two samples.
Pluripotency of SeV-iPSCs in vitro and in vivo
Differentiation potential of viral-free SeV-iPS clones were analyzed either by in vitro embryoid-body culture or in vivo teratoma formation according to the methods described previously.29)–31) As shown in Fig. 7A and B, we were able to induce representative mature cell lineages derived from all three germ layers; hematopoietic cells and beating cardiomyocytes for mesoderm lineage, Sox17 or PDX1 positive cells for endoderm lineage, and beta III- and tyrosine hydroxylase-positive neuron for ectodermal linage.
Fig. 7.
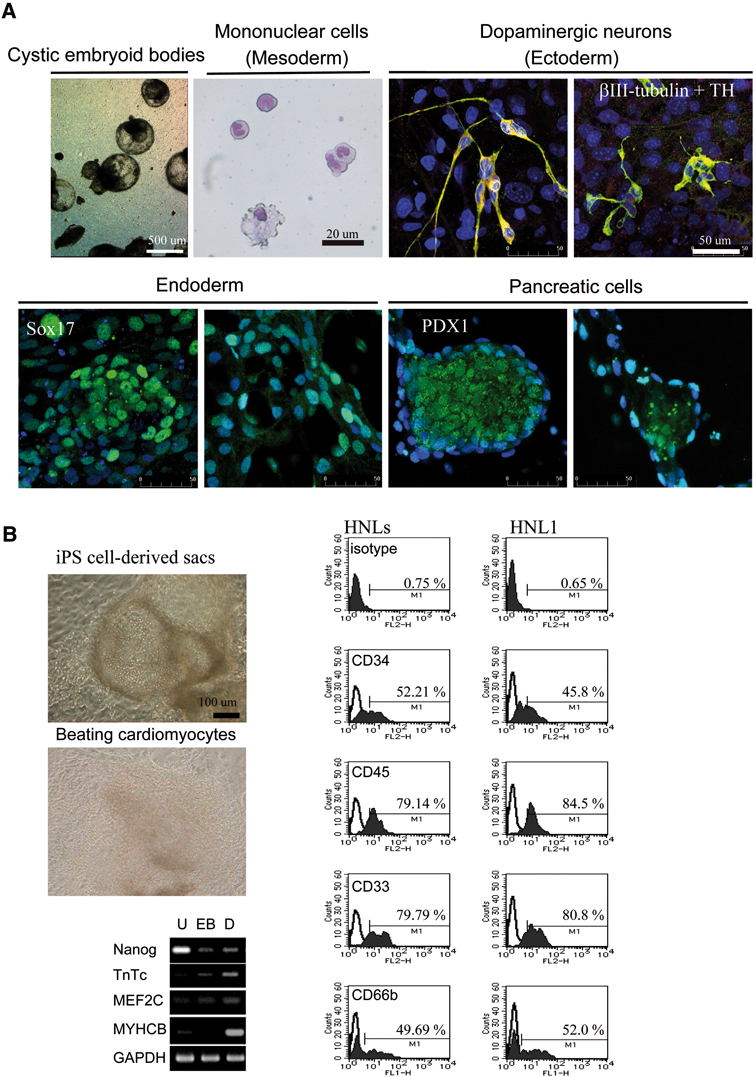
In vitro differentiation of transgenes-free SeV-iPSC. A.In vitro differentiation of mononuclear cells (mesoderm: shown with Wright-Giemsa staining) via embryoid bodies,29) putative dopaminergic neurons co-expressing tyrosine hydroxylase (TH)(ectoderm),30) definitive endoderm (Sox17), and pancreatic cells (PDX1).31)B.In vitro differentiation of cardiomyocytes and mononuclear cells from SeV-iPSC via embryo bodies using cytokine cocktails (SCF, Flt3L, TPO, G-CSF, IGF-2, and VEGF).29) In the adherent culture, hematopoietic sac-like structures filled with mononuclear cells (left, upper) and beating colonies (left, middle) emerged.29) FACS analysis shows differentiation of SeV-iPSC to mononuclear cells (neutrophil, monocyte, and macrophage), expressing CD34, CD45, CD33, and neutrophil-specific marker CD66b (right). RT-PCR analysis shows that pluripotent marker (Nanog) was decreased and various cardiomyocytes-specific differentiation markers (TnTc, MEF2C, MYHCB) were increased after differentiation (left, lower). U: undifferentiated; EB: embryoid body; D: differentiated.
We further tested the pluripotency of SeV-iPSC by teratoma formation. Viral-free-iPSCs, HNL1 and HNLs from neonatal BJ and XH1 from adult HDF cells were injected subcutaneously into the flanks of SCID mice. Histological examination of the teratomas revealed the presence of a set of representative tissues that were originated from the three embryonic germ layers, including neural epithelium, muscle, cartilage, bone, and glandular structures (Fig. 8). Remained expression of transgenes seemed to affect pluripotency (Table 1). From the results of in vitro and in vivo differentiation experiments, viral-free SeV-iPSC showed pluripotency.
Fig. 8.
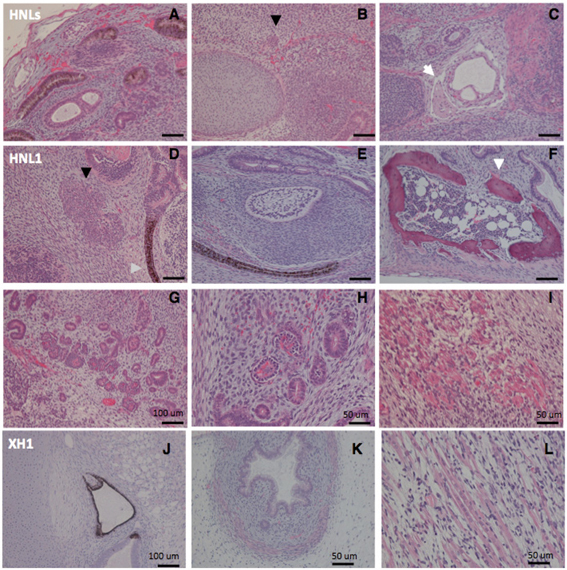
In vivo pluripotency of transgenes-free SeV-iPSC. Hematoxylin and eosin staining of teratoma sections of SeV-iPS clones (6 weeks post-injection into SCID mice). Tissues were differentiated from Tg-free human neonatal fibroblast BJ-derived HNLs (A to C), HNL1 (D to I), and adult fibroblast HDF-derived XH1 (J to L) containing multiple tissues derived from three germ layers: glandular structures (A, G, K), cartilage (B, J), bone (C, F, white arrows) and bone marrow-like structure (F), epithelium (J, D), transitional epithelium (E), population of secreting-like cells (B, D, indicated by black arrows), muscle (C, I, K, L), and glomerulus of kidney-like tissue (H).
Discussion
SeV based vectors have been used as a safe method for gene therapy,22)–25) as SeV per se has no risk of being integrated in the host genome. In this study, we further showed that SeV is an ideal vector for generating human iPSC that fulfills the latest criterion for iPSC. Firstly, as emphasized repeatedly, SeV vector allows expression of transgenes without risk of modification of host genome. Secondly, the efficiency of iPSC generation by gene transduction with SeV vectors is significantly higher than that by other methods especially without any transfection drugs. Finally, it is easy to select iPSC that depleted viral genome from the cytoplasm. Hence, resulting viral-free iPSCs become genetically intact and carries the same genome DNA as the original cells. We emphasize that the method described here has a significant advantage over presently available methods for its safety, efficiency and convenience. Nonetheless, iPS research is entering the new stage by development of methods for epigenetic reprogramming without genetic modification.
Acknowledgments
We thank Drs. Shinya Yamanaka and Makio Iwashima for critical reading of the manuscript, S. Seino, T. Kanaya, K. Washizawa, T, Fujikawa, M. Oshima, Y. Ohara and Y. Ushimaru for technical assistants, H. Iwasaki and Y. Yonemitsu for technical suggestion.
Abbreviations:
MOI
multiplicities of infection
iPSC
induced pluripotent stem cell
EB
embryoid body
IH
immunohistochemistry
SeV
Sendai virus
SeV-iPSC
iPS cell generated by SeV vectors
References
- 1).Takahashi, K. and Yamanaka, S. (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblat cultures by defined factors. Cell 126, 663–676 [DOI] [PubMed] [Google Scholar]
- 2).Maherali, N., Sridharan, R., Xie, W., Utikal, J., Eminli, S., Arnold, K.et al. (2007) Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell 1, 55–70 [DOI] [PubMed] [Google Scholar]
- 3).Okita, K., Ichisaka, T. and Yamanaka, S. (2007) Generation of germ-line-competent induced pluripotent stem cells. Nature 448, 313–317 [DOI] [PubMed] [Google Scholar]
- 4).Wernig, M., Meissner, A., Foreman, R., Brambrink, T., Ku, M., Hochedlinger, K.et al. (2007) In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature 448, 318–324 [DOI] [PubMed] [Google Scholar]
- 5).Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K.et al. (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 [DOI] [PubMed] [Google Scholar]
- 6).Yu, J., Vodyanik, M.A., Smuga-Otto, K., Antosiewicz-Bourget, J., Frane, J.L., Tian, S.et al. (2007) Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917–1920 [DOI] [PubMed] [Google Scholar]
- 7).Lowry, W.E., Richter, L., Yachechko, R., Pyle, A.D., Tchieu, J., Sridharan, R.et al. (2008) Generation of human induced pluripotent stem cells from dermal fibroblats. Proc. Natl. Acad. Sci. USA 105, 2883–2888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8).Park, I.H., Zhao, R., West, J.A., Yabuuchi, A., Huo, H., Ince, T.A.et al. (2008) Reprogramming of human somatic cells to pluripotency with defined factors. Nature 451, 141–146 [DOI] [PubMed] [Google Scholar]
- 9).Masaki, H., Ishikawa, T., Takahashi, S., Okumura, M., Sakai, N., Haga, M.et al. (2008) Heterogeneity of pluripotent marker gene expression in colonies generated in human iPS cell induction culture. Stem Cell Res. 1, 105–115 [DOI] [PubMed] [Google Scholar]
- 10).Stadtfeld, M., Nagaya, M., Utikal, J., Weir, G. and Hochedlinger, K. (2008) Induced pluripotent stem cells generated without viral integration. Science 322, 945–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11).Okita, K., Nakagawa, M., Hyenjong, H., Ichisaka, T. and Yamanaka, S. (2008) Generation of mouse induced pluripotent stem cells without viral vectors. Science 322, 949–953 [DOI] [PubMed] [Google Scholar]
- 12).Yu, J., Hu, K., Smuga-Otto, K., Tian, S., Stewart, R., Slukvin, I.I.et al. (2009) Human induced pluripotent stem cells free of vector and transgene sequences. Science 324, 797–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).Harui, A., Suzuki, S., Kochanek, S. and Mitani, K. (1999) Frequency and stability of chromosomal integration of adenovirus vectors. J. Virol. 73, 6141–6146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14).Woltjen, K., Michael, I.P., Mohseni, P., Desai, R., Mileikovsky, M., Hämäläinen, Ret al. (2009) piggy-Bac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 458, 766–770(advance online publication Mar. 1, 2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15).Kaji, K., Norrby, K., Paca, A., Mileikovsky, M., Mohseni, P. and Woltjen, K. (2009) Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature 458, 771–775(advance online publication Mar. 1, 2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16).Soldner, F., Hockemeyer, D., Beard, C., Gao, Q., Bell, G.W., Cook, E.G.et al. (2009) Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 136, 964–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17).Huangfu, D., Osafune, K., Maehr, R., Guo, W., Eijkelenboom, A., Chen, S.et al. (2008) Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat. Biotechnol. 26, 1269–1275 [DOI] [PubMed] [Google Scholar]
- 18).Zhou, H., Wu, S., Joo, J.Y., Zhu, S., Han, D.W., Lin, T.et al. (2009) Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell 4, 381–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19).Kim, D., Kim, C.H., Moon, J.I., Chung, Y.G., Chang, M.Y., Han, B.S.et al. (2009) Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell 4, 472–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20).Lamb, R. A. and Kolakofsky, D. (2001) Paramyxoviridae: the viruses and their replication. Lippincott Williams & Wilkins, New York, pp. 1177–1204 [Google Scholar]
- 21).Tokusumi, T., Iida, A., Hirata, T., Kato, A., Nagai, Y. and Hasegawa, M. (2002) Recombinant Sendai viruses expressing different levels of a foreign reporter gene. Virus Res. 86, 33–38 [DOI] [PubMed] [Google Scholar]
- 22).Yonemitsu, Y., Kitson, C., Ferrari, S., Farley, R., Griesenbach, U., Judd, D.et al. (2000) Efficient gene transfer to airway epithelium using recombinant Sendai virus. Nat. Biotechnol. 18, 970–973 [DOI] [PubMed] [Google Scholar]
- 23).Ferrari, S., Griesenbach, U., Iida, A., Farley, R., Wright, A.M., Zhu, J.et al. (2007) Sendai virus-mediated CFTR gene transfer to the airway epithelium. Gene Ther. 14, 1371–1379 [DOI] [PubMed] [Google Scholar]
- 24).Masaki, I., Yonemitsu, Y., Komori, K., Ueno, H., Nakashima, Y., Nakagawa, K.et al. (2001) Recombinant Sendai virus-mediated gene transfer to vasculature: a new class of efficient gene transfer vector to the vascular system. FASEB J. 15, 1294–1296 [DOI] [PubMed] [Google Scholar]
- 25).Takeda, A., Igarashi, H., Kawada, M., Tsukamoto, T., Yamamoto, H., Inoue, M.et al. (2008) Evaluation of the immunogenicity of replication-competent V-knocked-out and replication-defective F-deleted Sendai virus vector-based vaccines in macaques. Vaccine 26, 6839–6843 [DOI] [PubMed] [Google Scholar]
- 26).Li, H.-O., Zhu, Y.-F., Asakawa, M., Kuma, H., Hirata, T., Ueda, Y.et al. (2000) A cytoplasmic RNA vector derived from nontransmissible Sendai virus with efficient gene transfer and expression. J. Virol. 74, 6564–9569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27).Inoue, M., Tokusumi, Y., Ban, H., Kanaya, T., Tokusumi, T., Nagai, Y.et al. (2003) Nontransmissible virus-like particle formation by F-deficient Sendai virus is temperature sensitive and reduced by mutations in M and HN proteins. J. Virol. 77, 3238–3246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28).Miura, N., Uchida, T. and Okada, Y. (1982) HVJ (Sendai virus)-induced envelope fusion and cell fusion are blocked by monoclonal anti-HN protein antibody that does not inhibit hemagglutination activity of HVJ. Exp. Cell Res. 141, 409–420 [DOI] [PubMed] [Google Scholar]
- 29).Saeki, K., Saeki, K., Nakahara, M., Matsuyama, S., Nakamura, N., Yogiashi, Y.et al. (2009) A feeder-free and efficient production of functional neutrophils from human embryonic stem cells. Stem Cells 27, 59–67 [DOI] [PubMed] [Google Scholar]
- 30).Kawasaki, H., Mizuseki, K., Nishikawa, S., Kaneko, S., Kuwana, Y., Nakanishi, S.et al. (2000) Induction of midbrain dopaminergic neurons from ES cells by stromal cell-derived inducing activity. Neuron 28, 31–40 [DOI] [PubMed] [Google Scholar]
- 31).D’Amour, K.A., Agulnick, A.D., Eliazer, S., Kelly, O.G., Kroon, E. and Baetge, E.E. (2005) Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat. Biotechnol. 23, 1534–1541 [DOI] [PubMed] [Google Scholar]
- 32).Tesar, P.J., Chenoweth, J.G., Brook, F.A., Davies, T.J., Evans, E.P., Mack, D.L.et al. (2007) New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature 448, 196–199 [DOI] [PubMed] [Google Scholar]
- 33).Nakagawa, M., Koyanagi, M., Tanabe, K., Takahashi, K., Ichisaka, T., Aoi, T.et al. (2008) Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 26, 101–106 [DOI] [PubMed] [Google Scholar]
- 34).Adewumi, O., Aflatoonian, B., Ahrlund-Richter, L., Amit, M., Andrews, P.W., Beighton, G.et al. (2007) Characterization of human embryonic stem cell lines by the International Stem Cell Initiative. Nat. Biotechnol. 25, 803–816 [DOI] [PubMed] [Google Scholar]