Cellular and genetic diversity in the progression of in situ human breast carcinomas to an invasive phenotype (original) (raw)
Abstract
Intratumor genetic heterogeneity is a key mechanism underlying tumor progression and therapeutic resistance. The prevailing model for explaining intratumor diversity, the clonal evolution model, has recently been challenged by proponents of the cancer stem cell hypothesis. To investigate this issue, we performed combined analyses of markers associated with cellular differentiation states and genotypic alterations in human breast carcinomas and evaluated diversity with ecological and evolutionary methods. Our analyses showed a high degree of genetic heterogeneity both within and between distinct tumor cell populations that were defined based on markers of cellular phenotypes including stem cell–like characteristics. In several tumors, stem cell–like and more-differentiated cancer cell populations were genetically distinct, leading us to question the validity of a simple differentiation hierarchy–based cancer stem cell model. The degree of diversity correlated with clinically relevant breast tumor subtypes and in some tumors was markedly different between the in situ and invasive cell populations. We also found that diversity measures were associated with clinical variables. Our findings highlight the importance of genetic diversity in intratumor heterogeneity and the value of analyzing tumors as distinct populations of cancer cells to more effectively plan treatments.
Introduction
With rare exceptions, human malignancies are thought to originate from a single cell, yet by the time of diagnosis, most tumors display startling heterogeneity in cell morphology, proliferation rates, angiogenic and metastatic potential, and expression of cell surface molecules (1, 2). This heterogeneity is in part caused by epigenetic and morphological plasticity, including variability for stem cell–like and more-differentiated cell characteristics, but there is also strong evidence for the existence of genetically distinct clones within the same tumor. This intratumor clonal heterogeneity has been reported for a wide range of malignancies, ranging from hematopoietic cancers to different types of solid tumors (3–7). Among others, the existence of clonal heterogeneity was documented in breast carcinomas using a variety of molecular and cytological techniques, both within primary tumors (8–10) and between matched primary tumors and distant metastases (9, 11). It is widely hypothesized that intratumor clonal heterogeneity underlies therapeutic resistance (2, 3). Supporting this hypothesis, the extent of the intratumor clonal heterogeneity measured based on FISH and TP53 and CDKN2A mutation data was associated with higher risk of tumor progression in esophageal carcinoma (4).
Despite the importance of intratumor genetic heterogeneity in tumor progression and therapeutic resistance, currently there are no established methods for the quantitative assessment of intratumor diversity at the cellular level that could be used as a biomarker for establishing the prognosis of cancer patients and predicting the risk of therapeutic resistance. Furthermore, methods for the combined analysis of phenotypic and genetic diversity at the single-cell level in situ in tissue sections are also lacking.
Here we report the development of methods that can be used for the quantitative description of intratumor heterogeneity in primary human tumors. We also show the utility of these methods for assessing genetic diversity of stem cell–like and more-differentiated breast cancer cells during progression from in situ to invasive carcinoma. Furthermore, we correlate diversity measures of breast carcinomas with clinical variables such as tumor grade.
Results
Combined measurement of phenotypic and genetic diversity at single-cell resolution.
We previously characterized stem cell–like CD44+ and more-differentiated CD24+ breast cancer cells from multiple tumors and determined that even within the same tumor, the 2 cell populations have distinct molecular and functional properties (10). These discrete characteristics are in part determined by epigenetic programs that might change during tumor progression (10, 12, 13). We also found evidence for genetic divergence between CD44+ and CD24+ breast cancer cells in one short-term primary culture derived from a pleural effusion sample (10).
To further investigate intratumor genetic and phenotypic heterogeneity in relation to stem cell–like and more-differentiated cell characteristics during progression from in situ to invasive breast carcinoma, we performed combined immunofluorescence staining and FISH (iFISH) (14) analyses of 15 invasive breast tumors of different subtypes containing both in situ and invasive components in the same section (Supplemental Table 1; supplemental material available online with this article; doi:10.1172/JCI40724DS1). In iFISH, immunofluorescence staining and FISH are used to define variability for phenotypic traits and copy number alterations, respectively. Six of the tumors were HER2+, 4 luminal A, and 5 basal-like, as defined by immunohistochemical analyses of estrogen and progesterone receptors (ER and PR), HER2, CK5/6, and EGFR (15). Stem cell–like and more-differentiated breast cancer cells were categorized based on positivity for the CD44 and CD24 cell surface markers, respectively.
First, we analyzed the tumors for the expression of CD24 and CD44 by immunohistochemistry to ensure that both cell populations were well represented on the slides to be used for iFISH. We observed high variability for the expression of these 2 markers both among and within tumors. Consistent with our prior studies (16), CD24+ breast cancer cells were infrequently detected in basal-like tumors, whereas the frequency of CD44+ cells was highest in basal-like and lowest in HER2+ cases. Thus, in basal-like tumors, we categorized breast cancer cells as CD44– and CD44+ populations. The frequency of CD24+ and CD44+ tumor cells was also highly variable in different regions of the same tumor (Supplemental Figure 1). To quantitatively assess this variability, we measured topological diversity of the tumor subtypes and histologies by determining the frequency of CD44+ and CD24+ cells in 4 independent quadrants of each tumor. Despite the high variability in the frequencies of these 2 cell types within some tumors (Supplemental Table 2 and Supplemental Figure 2), no significant differences were detected when the ratio of CD24+ to CD44+ cell frequencies was compared across all 15 cases (P = 0.91).
Next, we performed interphase iFISH analyses using 3 BAC probes localized to different chromosomal regions for each tumor. BAC clones were selected for each tumor subtype corresponding to commonly gained regions based on our previous SNP array studies (17) (Supplemental Table 3). Variability for chromosome 8q24 copy number was evaluated in all tumors, since this locus is often altered in all breast tumor subtypes. Basal-like tumors were also analyzed for chromosome 12p13 and 10p13, luminal A tumors for 11q13 and 16p13, and HER2+ tumors for 17q21 (ERBB2) and 1q32. Each pair of probes (BAC and corresponding centromeric probe) was evaluated individually using serial sections in each tumor, and the ratio of BAC to centromeric probe was determined and used for further calculations. Visual inspection of the iFISH images demonstrated variable copy numbers in different areas of some tumors (Figures 1 and 2, Supplemental Figure 3). In a luminal A tumor (tumor 10), clear evidence of clonal evolution during the in situ–to–invasive breast carcinoma transition was detected, as a CD24+CD44+ subclone with high 11q13 copy number gain in ductal carcinoma in situ (DCIS) became the dominant clone in the invasive areas (Figure 2). Interestingly, this 11q13 BAC includes the CCND1 gene encoding for cyclin D1; thus, amplification of this locus might explain the significantly higher rate of cellular proliferation (defined by the percentage of cells positive for the Ki67 marker) seen in the invasive (30% Ki67+ cells) compared with the in situ (2% Ki67+ cells) areas of this tumor (Supplemental Table 1).
Figure 1. Cellular and genetic diversity in breast cancer defined by iFISH analysis.

A representative example (tumor 2) of HER2+ invasive ductal breast carcinoma with adjacent DCIS displaying a high degree of diversity for the expression of HER2, CD44, and CD24 and for copy number gain of ERBB2 and 8q24 based on immunohistochemical staining and iFISH, respectively. CD24 showed membrano-cytoplasmic expression in invasive tumor cells but apical membranous expression in DCIS. In iFISH, blue corresponds to CD24 or CD44 immunofluorescence; ERBB2 and 8q24-specific probes are red; and centromeric probes (chromosomes 17 and 8 for ERBB2 and 8q24, respectively) are green. Faint green and yellow are background autofluorescence. Scale bars: 10 μm; original magnification, ×400 (immunohistochemistry) and ×600 (iFISH).
Figure 2. Clonal evolution during in situ to invasive breast carcinoma progression detected by iFISH.

iFISH analyses using 11q13/CCDN1 (red) and chromosome 11 centromeric probe (green) in a luminal A subtype breast cancer (tumor 10). In the invasive areas, both CD44+ and CD24+ tumor cells (blue) display high-level amplification, whereas in adjacent DCIS, this is restricted to a subset of CD24+CD44+ tumor cells (dotted line), with the majority of the tumor demonstrating normal copy number for this locus. iFISH analysis of adjacent sections using 8q24 (red) and chromosome 8 centromeric probe (green) demonstrates normal (2n) copy numbers for 8q24 in both DCIS and invasive areas. Faint green and yellow are background autofluorescence. Yellow spots and lines are autofluorescent collagen fibers. Scale bars: 10 μm; original magnification, ×600.
Tumors are composed of populations of cancer cells with distinct properties.
To obtain a quantitative measure of genetic heterogeneity in distinct tumor cell populations, we recorded copy number data for both BAC and centromeric probes in 100 individual CD24+ or CD44+ tumor cells in both invasive and in situ areas (a total of 400 individual cancer cells/tumor were evaluated) (Supplemental Table 4). Because we used 4-μm sections for iFISH (cutting nuclei approximately in half), it is possible that some chromosomal regions may not be well represented in the section and thus would not be detected by FISH. However, this sampling bias is expected to be the same in all cell populations analyzed. To ensure that the observed tumor cell diversity was not due to technical variability stemming from FISH procedures, we also determined BAC and centromeric probe counts in 100 normal stromal cells adjacent to tumors on the same slide for each probe as control (Supplemental Table 5 and Supplemental Figure 4).
Copy number ratios of the 8q24 BAC and chromosome 8 centromeric probes depicted using box plots demonstrated substantial variability across cell and tumor types (Figure 3A). Importantly, CD24+ and CD44+ tumor cells displayed discordant copy number ratios both within the same histology as well as between the in situ and invasive areas of some tumors. To further explore the distribution of copy number ratios within each cell type and tumor, we used histograms and kernel density estimates (18) (Figure 3B). The latter is a nonparametric way of estimating the probability density function of a random variable, providing a method to estimate the density function of the population from data obtained for 100 cells in each cell type, with minimal assumptions. Visualization of cancer cell population diversity using these approaches further highlighted the pronounced genetic heterogeneity within and between populations of CD24+ and CD44+ cells in the same histology as well as between in situ and invasive components. Similar observations were made using all other BAC probes (Supplemental Figures 5–10). Thus, despite the uniform expression of CD24 or CD44 in a subset of tumor cells, these 2 cell populations are genetically highly heterogeneous and as a consequence of this, they are likely to display variability for biological and functional traits including tumor-initiating potential and response to therapeutic agents.
Figure 3. Diversity for 8q24 copy number gain in breast tumors.

In basal-like tumors, CD24+ corresponds to CD44– cells, because no CD24 expression is seen in these cases. (A) Box plots depicting the distribution of 8q24 copy number gain defined as the ratio of signal observed for the 8q24-specific and centromeric probes in 100 individual cells in each of the 4 indicated tumor cell subpopulations. The filled circle represents the mean; boxes show the 25th to 75th percentiles; the horizontal lines inside the boxes represent the median; whiskers extend to the 10th and 90th percentiles; and outlying black circles are individual data points outside the 10th and 90th percentiles. Differences are seen between cell populations and also progression stages both for median copy number gain and for range of distribution. (B) Histograms and kernel density estimates depicting the distribution of cells with the indicated copy number ratio. Differences are seen between DCIS and invasive areas of the same tumor (e.g., tumor 4 [T4]) and also between CD24+ and CD44+ cells within the same compartment (e.g., T13A).
Numerical indices of tumor cell diversity.
To express the observed genetic diversity as a numerical value that can potentially be a clinically useful biomarker predicting the risk of progression or response to treatment, we applied diversity measures from the ecology and evolution sciences (19) to our copy number data. These diversity measures estimate the number and distribution of species in a certain geographical area or environmental niche. In our context, a species is a cancer cell population defined by a unique value of the iFISH measurement specifying the ratio of gene-specific BAC and centromeric probes. Hence, a region of a tumor containing cancer cells with 3 different copy number ratios is interpreted to contain 3 distinct “species.” We used the Shannon index as a measure of diversity
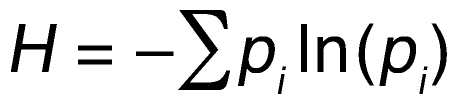
(Equation 1)
where pi is the frequency of species i in the tumor sample. This index is borrowed from information theory, where it specifies the information content of a message, and can be used to summarize the diversity of a population by a single number. An alternative measure of diversity, Simpson’s index, was also used. For discussion of the relative advantages of Shannon index versus Simpson’s index, see Methods.
We calculated the Shannon diversity index for 8q24 copy number gain in both CD24+ and CD44+ cell populations in the invasive and in situ areas of each tumor. A scatter plot of these Shannon indices suggested 2 distinct diversity groups (Figure 4A). We exploited this pattern by identifying 2 clusters for each probe and then testing whether these clusters are distinct by using the parametric bootstrap method (20) (Table 1). In the HER2+ and luminal A tumor subtypes, the diversity of 8q24 copy number as measured by the Shannon index fell into 2 significantly distinct groups; cell populations in one group had a lower diversity index than those in the other group. Whereas the 2 groups were equally large for luminal A tumors, the group with lower diversity contained fewer samples in HER2+ tumors. Interestingly, the Shannon index of the low-diversity group of luminal A tumors was essentially the same as that of normal cells (Figure 4A and Supplemental Figure 4). Basal-like tumors formed a single group with diversity measures similar to the group with high diversity in the other 2 subtypes (Figure 4A). To ensure that the observed tumor cell diversity was not due to technical issues associated with iFISH, we also defined the diversity indices of normal stromal cells adjacent to tumors on the same slide and found low and non-variable diversity for each of the chromosomal regions analyzed (Supplemental Figure 4). These data suggest that the Shannon index might be used as a clinically useful biomarker that further refines breast tumor subtypes according to their diversity.
Figure 4. Diversity for 8q24 copy number gain in breast tumors defined by Shannon index and Whittaker plots.

In basal-like tumors, CD24+ corresponds to CD44– cells, because no CD24 expression is seen in these cases. (A) The Shannon index, H, indicating diversity within tumor cell subpopulations and tumors. For each tumor, 100 different cells for each of the 4 different types (IDC CD24+, IDC CD44+, DCIS CD24+, and DCIS CD44+) were analyzed, and their Shannon indices are depicted in dark blue, dark red, light blue, and light red, respectively. Higher score indicates higher diversity. Basal-like tumors are all uniformly highly diverse for 8q24, whereas a subset of HER2+ and luminal A tumors show a lower degree of diversity. (B) Whittaker plots (rank-abundance plots) depicting the abundance of unique cancer cells.
Table 1 .
Cluster analysis of diversity indices

In most tumors, the 4 distinct cell populations (i.e., CD24+ and CD44+ cells in DCIS and invasive areas) had similar diversity scores for each of the 3 BAC probes analyzed; however, in some cases, CD24+ and CD44+ cells displayed divergent scores in the same histology or between DCIS and invasive regions (Figure 4A and Supplemental Figures 5–10). Interestingly, in all but 1 tumor with deviating scores, the invasive areas (both CD24+ and CD44+ cells) showed higher diversity potentially due to the larger number of tumor cells in invasive compared with in situ tumors and their exposure to more varied environmental conditions (e.g., interaction with various stromal cells that cannot occur in DCIS, because the stroma and tumor epithelial cells are physically separated from each other by the myoepithelial cell layer and the basement membrane).
To further define the abundance of unique cancer cells in the tumor samples, we used rank-abundance plots (also called Whittaker plots) (21) as graphical measures of diversity (Figure 4B and Supplemental Figures 5–10). In these graphs, species are plotted in sequence from the most to least abundant on the horizontal axis, and their frequencies are indicated on the vertical axis; hence, a steep slope corresponds to a population dominated by a few abundant species. These data again indicated that luminal A tumors are composed of a few dominant cancer cell populations, whereas basal-like and HER2+ cases more frequently contain a wider array of less abundant tumor cell types.
Associations between diversity indices and histopathologic characteristics of tumors.
To further investigate differences in diversity among breast tumor subtypes, the distribution of the Shannon index for 8q24 copy number gain was explored by a heatmap (Figure 5A) and pairwise scatter plots (Supplemental Figure 11). The heatmap suggested that luminal A and basal-like tumors were mostly characterized by higher diversity in the invasive and in situ components, respectively, whereas the HER2+ subtype was not uniquely characterized by either category. CD44+ cells were more diverse within invasive relative to in situ areas of the tumors, whereas CD24+ cells showed higher diversity in in situ compared with invasive components. These observations were confirmed by the dendrogram depicting the hierarchical clustering of the tumors. The dendrogram displaying the clustering of the diversity of distinct cell populations and invasive and DCIS areas across tumors revealed a strong cluster of the areas (i.e., invasive and DCIS) and weaker subclusters of the cell populations (i.e., CD24+ and CD44+ cells). Pairwise scatter plots did not reveal any further associations that were not obvious from the heatmap and dendrograms (Supplemental Figure 11).
Figure 5. Diversity for different chromosomal probes in breast tumor subtypes and their association with histopathologic features.

(A) Hierarchical clustering of tumor samples based on the Shannon index for the 8q24 probe. Heatmap and dendrograms displaying relatedness of cell types and tumor samples based on their Shannon indices. Red and yellow correspond to high and low diversity, respectively, whereas white represents median levels. Tumor names are colored according to subtype: red, basal-like; pink, HER2+; and blue, luminal A. The color key indicates the correlation between diversity and colors. (B) Differences in diversity for different chromosomal regions in the same tumor. Histograms of copy number ratios in 4 distinct cell types for 3 different chromosomal probes are depicted in 2 individual tumors.
Next, we used a hierarchical model with the copy number ratio as the outcome; tumor subtype, histology, and cell type as the covariates; and the tumor as a random effect (see Methods and Supplemental Table 6). This model allowed us to determine the joint effects of the covariates on the distribution of the copy number ratio. We identified significant differences between CD44– and CD44+ cells in the DCIS portion of basal-like tumors (P = 0.001) for 8q24 copy number gain and between CD24+ and CD44+ cells in the invasive ductal carcinoma (IDC) portion of HER2+ tumors (P = 0.002) for 1q32 copy number gain. These differences might indicate the divergent evolution of the 2 distinct cell populations at different stages of tumor progression.
We found that tumor cell populations differed not only in copy number gain and diversity with regard to a single probe across CD24+ and CD44+ cell populations in invasive and in situ areas and tumor subtypes (Figures 3 and 4) but also with regard to different probes in a single tumor (Figure 5B). The relative presence and diversity of copy number gains for different chromosomes may be used for mapping the evolutionary history of tumors. Interestingly, in a HER2+ tumor, the abundance of tumor cells with 8q24 copy number gain was lower in invasive compared with DCIS areas, whereas the opposite was observed for 17q21/ERBB2 gain (Figure 5B).
To determine whether the diversity of each tumor with regard to 8q24 copy number gain correlates with histopathologic features of the tumors (e.g., tumor grade, nuclear pleomorphism, extent of intra- and peri-tumoral DCIS, necrosis, and proliferation rate), we analyzed associations using a rank-sum test (Supplemental Table 7). We found that several variables were highly associated (Supplemental Table 7). The most significant correlations (Table 2) were detected between DCIS CD24+ cell diversity as measured by the Shannon index and the extent of intra- and peritumoral DCIS, DCIS necrosis, and extensive intraductal component (EIC). These findings imply that larger tumor cell population size and hypoxia might increase intratumor genetic diversity. Once the P values were corrected for multiple testing, none of the associations were statistically significant due to small sample size. Hence, the associations can only be interpreted as suggestive evidence, and confirmation in a larger sample set is needed.
Table 2 .
Associations between diversity and histopathologic variables

Discussion
Tumors are highly heterogeneous and dynamic (2). The cell populations, both normal and cancer, composing the tumor continuously evolve, posing a major challenge for effective cancer treatment. Thus, to understand the clinical behavior of tumors, it is essential to define the various cancer cell populations they contain and to determine how these populations behave together as a whole.
Targeting cancer cells with specific mutations, currently the most favored approach for cancer treatment, inevitably selects for resistant clones. Often these resistant cells already exist in the primary tumor at the time of diagnosis (22–24) but go undetected due to the limited sensitivity of the methods currently used. Examples include the emergence of MET-amplified tumors in lung cancer patients resistant to EGFR inhibitors (25) and the relapse of chronic myelogenous leukemia (CML) following imatinib mesylate treatment due to mutations in BCR-ABL that confer resistance (26). Thus, there is a pressing need for the development and application of techniques that allow the quantitative definition of intratumor diversity at the single-cell level in archived clinical samples.
Here we have applied a new approach for the analysis of intratumor diversity based on FISH and immunofluorescence staining for selected markers specific for the phenotype of interest in combination with ecological and evolutionary methods for data analysis. Because both of these experimental methods are routinely used in diagnostic pathology laboratories and both data collection and its mathematical evaluation can fairly easily be automated, our approach can directly be translated into clinical practice.
Using breast cancer as an example, we have shown that application of ecological and evolutionary methods to genetic and phenotypic data collected on populations of individual cancer cells demonstrated a high degree of heterogeneity for chromosomal alterations in cancer cells homogeneous for markers associated with stem cell–like and more-differentiated epithelial cell traits and between in situ and invasive areas of the same tumors. These findings are inconsistent with the hypothesis that CD24+ more-differentiated luminal epithelial breast cancer cells are the direct progeny of CD44+ stem cell–like breast cancer cells. Thus, a simple hierarchical differentiation–based cancer stem cell model (27) does not appear to be applicable for breast carcinomas. Furthermore, our results also show that both stem cell–like and more-differentiated cancer cell populations evolve during tumor progression. Even if CD24 and CD44 are not specific markers for stem cell–like and more-differentiated breast cancer cells, the degree of genetic diversity we observed within tumors is inconsistent with a strictly cellular differentiation status–based tumor progression model. Furthermore, recent data indicate that even homogeneous-appearing cell populations can have heterogeneous responses to physiologic stimuli due to cell-to-cell variability in protein levels and signaling pathways (28). Thus, the clinical behavior, including therapeutic resistance and recurrence, of the tumor is determined by the combined behavior of the tumor as a whole; it must be regarded as a complex system composed of highly variable individual cells.
Intratumor heterogeneity is not limited to markers associated with differentiation states but is likely to be a general feature of all measurable characteristics, including gene expression, mutation, and epigenetic modification patterns. Indeed, prior studies analyzing the expression of various genes including estrogen receptor, cytokeratins, and HER2 in DCIS and invasive breast carcinomas have demonstrated a high degree of diversity in a subset of tumors (29). The intratumor heterogeneity of several of these markers that are used for the categorization of breast tumors into major subtypes (e.g., luminal, HER2+, and basal-like) reflects the imperfection of such classification schemes.
Intratumor heterogeneity has also been reported for mutations in tumor suppressor (TP53) and oncogenes (RAS, PIK3CA) in breast and other tumor types (3, 4, 7, 30), demonstrating a continuous selection process within tumors that drives their evolution. Because several of these mutant genes are targets of new molecular-based cancer therapy, assessing their heterogeneity within tumors prior to treatment is important for the design of more effective combinatorial treatment approaches.
Another interesting question pertains to the underlying mechanisms that maintain and promote intratumor heterogeneity. At this point, we can only speculate on these, since there is limited experimental evidence, especially in human tumors. With rare exceptions, tumors initiate from a single transformed cell, and multiple rounds of clonal expansion are required to produce a clinically symptomatic tumor. During this expansion phase, genetic and epigenetic instability, which is a common feature of most tumors, produces a wide range of tumor cell variants with favorable traits that natural selection acts on. In solid tumors, spatial restrictions lead to the generation of separate niches in different parts of the tumor that favor the outgrowth of cancer cells with different characteristics. This spatial restriction might also promote cooperation among tumor cell populations, as has been demonstrated in colon cancer based on mathematical modeling supported by some experimental observations (5). Due to the importance of these issues for effective cancer treatment, further studies are required to better understand the sources of intratumor diversity and their consequences.
In summary, in this study we have demonstrated the power of analyzing tumors as ecosystems and suggest that quantitative measures of intratumor diversity might be clinically useful biomarkers predicting prognosis and response to treatment.
Methods
Tissue specimens and cell cultures.
Fifteen cases of invasive breast tumors with DCIS component were selected from files from 2005–2007 in the Department of Pathology, Seoul National University Bundang Hospital, according to protocol B-0909-083-002, approved by the Institutional Review Board of Seoul National University Bundang Hospital. The use of these tissue samples for experiments was approved by the Institutional Review Board of Dana-Farber Cancer Institute under protocol 98-229. All tissue samples were deidentified; thus, patient consent was not required. Of the 15 cases, 2 were in the same patient as collision tumors. The T47D human breast cancer cell line was obtained from ATCC, cultured and treated with colcemid, harvested, and used for metaphase chromosome spread preparations according to standard protocols (31).
BAC probes.
BAC clones were obtained from Invitrogen and labeled with SpectrumOrange or SpectrumGreen using a Nick Translation Kit (Abbott Molecular) according to the manufacturer’s recommendation, whereas a PathVysion HER-2 DNA probe kit (Abbott Molecular) was used for ERBB2. Each BAC probe was tested in metaphase FISH analyses using the T47D breast cancer cell line to confirm their proper chromosomal localization (data not shown).
Immunohistochemical and iFISH analyses.
iFISH analysis was performed essentially as previously described, with minor modifications (14), using monoclonal antibodies against CD44 and CD24 (CD44, clone 156-3C11, 1:200 and CD24, clone SN3b, 1:100; LabVision). FISH signal enumeration was performed using a fluorescence microscope with single- and triple-band pass filters in intact, non-overlapping nuclei to avoid variability in scoring due to the inaccurate recognition of cell boundaries and probe hybridization. Frequency of cells and FISH signal were counted by a single pathologist to minimize variability due to inter-observer bias. However, to ensure reproducibility, some slides were counted by other individuals, with essentially the same results.
Diversity measures.
The Shannon index, or information entropy, describes the information content of a message and can be used to estimate diversity in a biological sample. One shortcoming of the Shannon index is that it confounds species richness and evenness. To overcome this problem, we additionally applied Simpson’s index to our data and tested our conclusions for robustness to the diversity measure used.
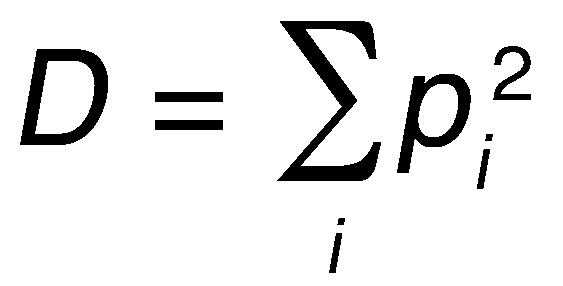
(Equation 2)
where pi is the frequency of species i in the tumor sample. Simpson’s index measures the probability that two individuals randomly sampled from a population belong to the same species. A high value indicates low diversity. We used (1 - D), a number between 0 and 1 that increases with increasing diversity of the sample, as an indicator of diversity, representing the probability that 2 randomly sampled individuals belong to different species. As compared to the Shannon index, Simpson’s index has the advantage of having a clear biological and probabilistic interpretation as well as returning a number between 0 and 1, while the Shannon index can take any non-negative value. The disadvantage of Simpson’s index is that it is dominated by the most abundant species in the population. When comparing our data analyzed with the Shannon versus Simpson’s index, we found very few qualitative differences (Table 1 and Supplemental Figures 5–10 and 12).
Statistics.
We used box plots, histograms, and kernel density estimates to present and visually compare the distribution of copy number ratios for CD24+ and CD44+ cancer cell populations in invasive and in situ areas. The bin sizes and the bandwidth for the Gaussian kernel of the density estimate were automatically chosen using Scott’s algorithm (32). Shannon and Simpson’s indices are estimated by substituting the sample proportions in the definitions, and their sampling variability was assessed by forming bootstrap samples within each tumor, cell population (CD24+ and CD44+), and histology (invasive and in situ). Since scatter plots of both Shannon and Simpson’s indices suggested the presence of 2 distinct groups, the _k_-means algorithm with Euclidian distance was used to form 2 clusters (33). Because this algorithm always identifies a given number of clusters (k = 2 in our case), we also tested whether the resulting clusters are statistically distinct. For this purpose, we used the parametric bootstrap method, representing the null hypothesis with a single normal distribution and the alternative as a mixture of 2 normal distributions with different location and scale parameters (20). The intratumor dependence of the observations (the possibility that 2 observations from the same area within each tumor are not probabilistically independent) is taken into account in the clustering algorithm by scaling the Euclidian distance by (1 – ρ), where ρ, the intratumor correlation between copy number ratios, was estimated using the method of Shrout and Fleiss (34).
Correlations between clinical data and tumor diversity indices were explored only for the 8q24 copy number gain, because only this probe was analyzed in all 15 tumors. Rank correlations between the Shannon index for each of the cell population–tumor area combinations and various clinical parameters were estimated and tested for statistical significance using the rank-sum test with an exact reference distribution. P values were adjusted for multiple comparisons, where noted, using the resampling-based min-P algorithm (35). The intra-class correlation of the cell types from the same patient (4 each) was estimated to be 0.01 (36). In all subsequent analyses, the cell types from the same patient were treated as independent. Additionally, we used a heatmap to represent the joint distribution of the 4 Shannon indices for each tumor, along with dendrograms showing how the columns and the rows of the heatmap are hierarchically clustered. The joint distribution of Shannon indices were further explored using pairwise scatter plots for each bivariate subset.
The distribution of the copy number ratios across various tumor types, cell types, and histologies were compared using a hierarchical model, with cells recognized as nested in tumors. A variance components model was used for the covariance structure, and the estimates were obtained using restricted maximum likelihood (37). PROC MIXED of SAS 9.2 was used for this purpose. Kruskal-Wallis test was used to compare the distribution of diversity indices across groups defined by clinical variables. All P values were adjusted for multiple testing using simulation-based resampling. Two-sided P values less than 0.005 were considered significant.
Supplementary Material
Supplemental data
Supplemental Excel file 1
Supplemental Excel file 2
Acknowledgments
We greatly appreciate the help of Stanislawa Weremowicz (Brigham and Women’s Hospital, Boston, Massachusetts, USA) with the optimization of FISH protocols and the evaluation of metaphase FISH analyses. We thank Ian Krop, Massimo Loda, William Kaelin, William Hahn, and members of the Polyak laboratory for their critical reading of the manuscript and stimulating discussions. This work was supported by the Breast Cancer Research Foundation (F. Michor). K. Polyak is a Leon Levy Foundation Young Investigator.
Footnotes
Conflict of interest: K. Polyak receives research support from Novartis and is a consultant to Novartis Oncology and Xceed Molecular.
Citation for this article: J Clin Invest. 2010;120(2):636–644. doi:10.1172/JCI40724.
See the related Commentary beginning on page 401.
References
- 1.Heppner GH, Loveless SE, Miller FR, Mahoney KH, Fulton AM. Mammary tumor heterogeneity. Symp Fundam Cancer Res. 1983;36:209–221. [PubMed] [Google Scholar]
- 2.Heppner GH, Miller BE. Tumor heterogeneity: biological implications and therapeutic consequences. Cancer Metastasis Rev. 1983;2(1):5–23. doi: 10.1007/BF00046903. [DOI] [PubMed] [Google Scholar]
- 3.Merlo LM, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nat Rev Cancer. 2006;6(12):924–935. doi: 10.1038/nrc2013. [DOI] [PubMed] [Google Scholar]
- 4.Maley CC, et al. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet. 2006;38(4):468–473. doi: 10.1038/ng1768. [DOI] [PubMed] [Google Scholar]
- 5.Gonzalez-Garcia I, Sole RV, Costa J. Metapopulation dynamics and spatial heterogeneity in cancer. Proc Natl Acad Sci U S A. 2002;99(20):13085–13089. doi: 10.1073/pnas.202139299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campbell LL, Polyak K. Breast tumor heterogeneity: cancer stem cells or clonal evolution? Cell Cycle. 2007;6(19):2332–2338. doi: 10.4161/cc.6.19.4914. [DOI] [PubMed] [Google Scholar]
- 7.Konishi N, et al. Intratumor cellular heterogeneity and alterations in ras oncogene and p53 tumor suppressor gene in human prostate carcinoma. . Am J Pathol. 1995;147(4):1112–1122. [PMC free article] [PubMed] [Google Scholar]
- 8.Teixeira MR, et al. Cytogenetic abnormalities in an in situ ductal carcinoma and five prophylactically removed breasts from members of a family with hereditary breast cancer. Breast Cancer Res Treat. 1996;38(2):177–182. doi: 10.1007/BF01806672. [DOI] [PubMed] [Google Scholar]
- 9.Torres L, Ribeiro FR, Pandis N, Andersen JA, Heim S, Teixeira MR. Intratumor genomic heterogeneity in breast cancer with clonal divergence between primary carcinomas and lymph node metastases. Breast Cancer Res Treat. 2007;102(2):143–155. doi: 10.1007/s10549-006-9317-6. [DOI] [PubMed] [Google Scholar]
- 10.Shipitsin M, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11(3):259–273. doi: 10.1016/j.ccr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 11.Klein CA, et al. Genetic heterogeneity of single disseminated tumour cells in minimal residual cancer. Lancet. 2002;360(9334):683–689. doi: 10.1016/S0140-6736(02)09838-0. [DOI] [PubMed] [Google Scholar]
- 12.Bloushtain-Qimron N, et al. Cell type-specific DNA methylation patterns in the human breast. Proc Natl Acad Sci U S A. 2008;105(37):14076–14081. doi: 10.1073/pnas.0805206105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bloushtain-Qimron N, Yao J, Shipitsin M, Maruyama R, Polyak K. Epigenetic patterns of embryonic and adult stem cells. Cell Cycle. 2009;8(6):809–817. doi: 10.4161/cc.8.6.7938. [DOI] [PubMed] [Google Scholar]
- 14.Hu M, et al. Regulation of in situ to invasive breast carcinoma transition. Cancer Cell. 2008;13(5):394–406. doi: 10.1016/j.ccr.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carey LA, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA. 2006;295(21):2492–2502. doi: 10.1001/jama.295.21.2492. [DOI] [PubMed] [Google Scholar]
- 16.Park SY, Lee HE, Li H, Shipitsin M, Gelman R, Polyak K. Heterogeneity for stem cell-realted markers according to tumor subtype and progression stage in breast cancer. Clin Cancer Res. doi: 10.1158/1078-0432.CCR-09-1532. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nikolsky Y, et al. Genome-wide functional synergy between amplified and mutated genes in human breast cancer. Cancer Res. 2008;68(22):9532–9540. doi: 10.1158/0008-5472.CAN-08-3082. [DOI] [PubMed] [Google Scholar]
- Scott DW.Multivariate Density Estimation: Theory, Practice, and Visualization . New York, NY: John Wiley & Sons; 1992. [Google Scholar]
- Magurran AE.Measuring Biological Diversity . Malden, MA: Blackwell; 2004. [Google Scholar]
- 20.McLachlan GJ. On bootstrapping the likelihood ratio test statistic for the number of components in a normal mixture. Appl Stat. 1987;36:318–324. doi: 10.2307/2347790. [DOI] [Google Scholar]
- Krebs CJ.Ecological Methodology . Menlo Park, CA: Benjamin/Cummings; 1999. [Google Scholar]
- 22.Mullighan CG, et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008;322(5906):1377–1380. doi: 10.1126/science.1164266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hofmann WK, et al. Presence of the BCR-ABL mutation Glu255Lys prior to STI571 (imatinib) treatment in patients with Ph+ acute lymphoblastic leukemia. Blood. 2003;102(2):659–661. doi: 10.1182/blood-2002-06-1756. [DOI] [PubMed] [Google Scholar]
- 24.Roche-Lestienne C, et al. Several types of mutations of the Abl gene can be found in chronic myeloid leukemia patients resistant to STI571, and they can pre-exist to the onset of treatment. Blood. 2002;100(3):1014–1018. doi: 10.1182/blood.V100.3.1014. [DOI] [PubMed] [Google Scholar]
- 25.Engelman JA, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 26.Deininger M. Resistance to imatinib: mechanisms and management. J Natl Compr Canc Netw. 2005;3(6):757–768. doi: 10.6004/jnccn.2005.0045. [DOI] [PubMed] [Google Scholar]
- 27.Dick JE. Stem cell concepts renew cancer research. Blood. 2008;112(13):4793–4807. doi: 10.1182/blood-2008-08-077941. [DOI] [PubMed] [Google Scholar]
- 28.Spencer SL, Gaudet S, Albeck JG, Burke JM, Sorger PK. Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature. 2009;459(7245):428–432. doi: 10.1038/nature08012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Allred DC, et al. Ductal carcinoma in situ and the emergence of diversity during breast cancer evolution. Clin Cancer Res. 2008;14(2):370–378. doi: 10.1158/1078-0432.CCR-07-1127. [DOI] [PubMed] [Google Scholar]
- 30.Giaretti W, Monaco R, Pujic N, Rapallo A, Nigro S, Geido E. Intratumor heterogeneity of K-ras2 mutations in colorectal adenocarcinomas: association with degree of DNA aneuploidy. Am J Pathol. 1996;149(1):237–245. [PMC free article] [PubMed] [Google Scholar]
- 31.Ney PA, et al. Purification of the human NF-E2 complex: cDNA cloning of the hematopoietic cell-specific subunit and evidence for an associated partner. Mol Cell Biol. 1993;13(9):5604–5612. doi: 10.1128/mcb.13.9.5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scott DW. On optimal and data-based histograms. Biometrika. 1979;66:605–610. doi: 10.1093/biomet/66.3.605. [DOI] [Google Scholar]
- 33.Hartigan JA, Wong M. A K-means clustering algorithm. Appl Stat. 1979;28:100–108. doi: 10.2307/2346830. [DOI] [Google Scholar]
- 34.Shrout PE, Fleiss JL. Intraclass correlation: uses in assessing rater reliability. Psychol Bull. 1979;86(2):420–428. doi: 10.1037/0033-2909.86.2.420. [DOI] [PubMed] [Google Scholar]
- Westfall PH, Young SS.Resampling-Based Multiple Testing: Examples and Methods for P-Value Adjustment . New York, NY: John Wiley & Sons; 1993. [Google Scholar]
- 36.Gonen M, Panageas KS, Larson SM. Statistical issues in analysis of diagnostic imaging experiments with multiple observations per patient. Radiology. 2001;221(3):763–767. doi: 10.1148/radiol.2212010280. [DOI] [PubMed] [Google Scholar]
- Searle S, Casella G, McCulloch G.Variance Components . New York, NY: John Wiley & Sons; 1992 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental data
Supplemental Excel file 1
Supplemental Excel file 2