Meta-analysis of genome-wide association studies of attention deficit/hyperactivity disorder (original) (raw)
. Author manuscript; available in PMC: 2011 Sep 1.
Published in final edited form as: J Am Acad Child Adolesc Psychiatry. 2010 Aug 1;49(9):884–897. doi: 10.1016/j.jaac.2010.06.008
Abstract
Objective
Although twin and family studies have shown Attention Deficit/Hyperactivity Disorder (ADHD) to be highly heritable, genetic variants influencing the trait at a genome-wide significant level have yet to be identified. As prior genome-wide association scans (GWAS) have not yielded significant results, we conducted a meta-analysis of existing studies to boost statistical power.
Method
We used data from four projects: a) the Children’s Hospital of Philadelphia (CHOP), b) phase I of the International Multicenter ADHD Genetics project (IMAGE), c) phase II of IMAGE (IMAGE II), and d) the Pfizer funded study from the University of California, Los Angeles, Washington University and the Massachusetts General Hospital (PUWMa). The final sample size consisted of 2,064 trios, 896 cases and 2,455 controls. For each study, we imputed HapMap SNPs, computed association test statistics and transformed them to Z-scores, and then combined weighted Z-scores in a meta-analysis.
Results
No genome-wide significant associations were found, although an analysis of candidate genes suggests they may be involved in the disorder.
Conclusions
Given that ADHD is a highly heritable disorder, our negative results suggest that the effects of common ADHD risk variants must, individually, be very small or that other types of variants, e.g. rare ones, account for much of the disorder’s heritability.
Keywords: ADHD, meta-analysis, association, GWAS, genetics
Introduction
Attention Deficit/Hyperactivity Disorder (ADHD) is one of the most common psychiatric phenotypes affecting children and adolescents. It is characterized by an inability to focus, high levels of impulsivity and age-inappropriate hyperactivity, and increased rates of antisocial, anxiety, mood and substance use disorders. With estimates of prevalence ranging from 5–12% 1, 2 and 2–5% in adults 3, 4, ADHD is a substantial public health concern.
Previous work in ADHD has established a strongly heritable component to the phenotype, with genetic factors, across 20 twin studies, explaining approximately 76% of the phenotypic variance 5–7. There appears to be substantial overlap between the two ADHD symptom dimensions hyperactivity/impulsivity and inattention 8. The heritability of ADHD is high regardless of whether it is measured as a dimensional trait or a categorical disorder. Genome-wide linkage and hypothesis-driven candidate gene association studies have failed to unequivocally identify specific genetic variation that predisposes to ADHD, although genome wide levels of significance were derived in a meta-analysis for variants within or close to the dopamine D4 (DRD4) and D5 receptor (DRD5) genes 9. Similarly, genome-wide association studies (GWAS) of ADHD in children have not yet yielded genome-wide significant association to common variation. The first genome-wide association scan of 909 trios from the International Multicenter ADHD Genetics (IMAGE) study did not find any significant associations 10, although an analysis of ADHD-related quantitative traits in this data set revealed (trait-specific) genome-wide significance for two variants, in the CDH13 gene encoding the neural adhesion protein cadherin 13 and the GFOD1 gene encoding a protein of unknown function 11. The other GWAS of childhood ADHD 12, 13, did not reveal any genome-wide significant associations, nor did a small, pooled GWAS in 343 cases with persistent, adult ADHD and 304 controls did not provide statistically significant association findings 14. The recently reported first genome-wide study of copy number variants (CNVs) in approximately 400 trios from Children’s Hospital of Philadelphia did not result in genome-wide significant results, either 15.
To determine whether a combination of the samples from studies in childhood ADHD would be sufficient to identify genes underlying the heritable component of ADHD, we conducted meta-analysis of these genome-wide association scans. While we fail to identify any genome-wide significant association, there is some evidence that true associations are present in the top end of our distribution of results.
Method
Samples
Our total data set comprises four projects: a) the Children’s Hospital of Philadelphia (CHOP), b) phase I of the International Multisite ADHD Genetics Project (IMAGE), c) phase II of IMAGE (IMAGE II), and d) the Pfizer funded study from the University of California, Los Angeles, Washington University and the Massachusetts General Hospital (PUWMa). Each of these datasets has been described elsewhere 15, 10, 12, 13. For this meta-analysis, we attempted to include the same individual-level data as those included in the original publication, but amended the analysis with additional quality control at the SNP and individual level to maintain quality control through the necessary imputation step (described below). We also restricted the analysis to one affected offspring per family (which eliminated 3 individuals from analysis). Additionally, we imputed all missing data, so the results from the imputation are fully complete for all individuals, which slightly changed the results for each subsample from the published results.
The IMAGE trio samples were collected using a common protocol with centralized training and reliability testing of raters and centralized data management. Family members were Caucasians of European origin from countries around Europe including Belgium, Germany, Ireland, the Netherlands, Spain, Switzerland, and the United Kingdom, and Israel. At the IMAGE sites, parents of children were interviewed with the Parental Account of Childhood Symptom (PACS), a semi-structured, standardized, investigator-based interview developed as an instrument to provide an objective measure of child behavior. Both parents and teachers completed the respective versions of the Conners ADHD rating scales and the Strengths and Difficulties Questionnaire. The assessment procedures have been described in more detail elsewhere 16–20. Probands had been referred for assessment of hyperactive, disruptive or disorganized behavior and had been clinically diagnosed as ADHD (or hyperkinetic disorder, the most closely equivalent category in the ICD-10 nomenclature used at some of the clinics). Most probands (N=868) met criteria for DSM-IV combined-type ADHD, additional ones met criteria for DSM-IV inattentive subtype (N=13), hyperactive subtype (N=33) or missed one of the ADHD diagnoses by a single item on the structured interview (N=19). We retained these latter families because, upon review of the medical record and structured interview data, the ADHD diagnosis was confirmed. Exclusion criteria were autism, epilepsy, IQ < 70, brain disorders and any genetic or medical disorder associated with externalizing behaviors that might mimic ADHD. All data were collected with informed consent of parents and with the approval of the site’s Institutional Review Board (IRB) or Ethical Committee.
The IMAGE II ADHD samples included some samples from the original IMAGE project along with samples provided by colleagues at other sites, using similar but not identical methods. In Germany, families were recruited in order of clinical referral in the outpatient clinics in Wuerzburg, Homburg and Trier. Families were of German, Caucasian ancestry. All cases met DSM-IV criteria for ADHD. The index child was aged 6 years or above, further affected siblings were included when aged at least 6 years. All children were assessed by full semi-structured interview (Kiddie-Sads-PL-German Version or Kinder-DIPS) and parent and teacher ADHD DSM-IV based rating scales to ensure pervasiveness of symptoms. Exclusion criteria were IQ below 80, comorbid autistic disorders or somatic disorders (hyperthyroidism, epilepsy, neurological diseases, severe head trauma etc.), primary affective disorders, Tourette-syndrome, psychotic disorders or other severe primary psychiatric disorders, and birth weight below 2000 grams.
At the Cardiff site, children ages 6 to 16, of British, Caucasian ancestry, were assessed by interviewing parents with the Parent Child and Adolescent Psychiatric Assessment (CAPA)-a semi-structured research diagnostic interview and a telephone interview with the teacher using the Child ADHD Teacher Telephone Interview. All cases met diagnostic criteria for DSM-IV ADHD or ICD-10 hyperkinetic disorder or DSM-III-R ADHD and had IQ test scores above 70. Exclusion criteria were pervasive developmental disorder, Tourette syndrome, psychosis or any neurological conditions. At the Scottish site, children ages 6 to 16, of British, Caucasian ancestry, were assessed by interviewing parents with the CAPA. To confirm pervasiveness, teachers completed the Conners Teacher Rating Scale. All cases met diagnostic criteria for DSM-IV ADHD. Children with an IQ below 70, autistic spectrum disorder, head injury, known chromosomal abnormality, encephalitis or significant medical conditions such as epilepsy were excluded. At the Dutch site, assessment data are available for 112 subjects aged 3–18 years with DSM-IV ADHD and related comorbidities, including mood, anxiety, oppositional defiant (ODD) and conduct (CD) disorders. Most of the sample was collected as part of a sib pair genome-wide linkage study in ADHD 21. Subjects were assessed using the DSM-IV version of the Diagnostic Interview Schedule for Children (DISC-P) with both parents, supplemented by Conners’ Questionnaires (old versions), the Childhood Behavior Checklist (CBCL) and Teacher Report Form (TRF). In addition, we obtained ADHD symptom severity scores from the parents on the SWAN (Strengths and Weaknesses of ADHD symptoms and Normal behaviors).
The IMAGE II control samples (2,653 population controls of European ancestry) were collected for an IRB approved GWAS of schizophrenia and have been described elsewhere 22. Briefly, the control participants were drawn from a US Nationally representative survey panel (of approximately 60,000 adult individuals at any one time, with constant turnover) ascertained via random digit dialing. Participants were screened for psychosis and bipolar disorder, but not ADHD. Control participants were not screened for ADHD. A blood sample was collected via a US national phlebotomy service. Control participants gave written consent for their biological materials to be used for medical research at the discretion of NIMH.
The PUWMa samples were collected independently at three sites using similar but slightly different methods: MGH, Washington University and UCLA. At MGH, 309 families were recruited from clinics at the Massachusetts General Hospital. Screening and recruitment for some subjects (N= 121) occurred prior to the publication of DSM-IV. Initial affection status for those subjects was based on DSM-IIIR criteria but lifetime DSM-IV-TR criteria was asked at follow up, and only those subjects endorsing a life-time DSM-IV-TR diagnosis of ADHD were enrolled. Remaining subjects were screened and assessed according to DSM-IV-TR criteria (N=188). Psychiatric assessments were made with K-SADSE (Epidemiologic Version). Potential probands were excluded if they had major sensorimotor handicaps (deafness, blindness), psychosis/schizophrenia, autism, inadequate command of the English language, or a Full Scale IQ less than 80. At Washington University, 272 families were selected from a population-representative sample identified through birth records of the state of Missouri, for a genetic epidemiologic study of the prevalence and heritability of ADHD. The original sample included 812 complete male and female twin pairs and six individual twins ages 7 to 19 years at the time of interview identified from the Missouri Family Registry from 1996 to 2002. Families were invited into the study if at least one child exhibited three or more inattentive symptoms on a brief screening interview. Parents reported on their children and themselves, and the youths on themselves, using the Missouri Assessment of Genetics Interview for Children (MAGIC), a semi-structured psychiatric interview. DSM-IV diagnoses of ADHD were based upon parental reports (most of the time, maternal). Families were excluded if a parent/guardian reported mental retardation or if the parent/guardian and twins could not speak English. At UCLA, 156 subjects were drawn from 540 children and adolescents ages 5 to 18 years and 519 of their parents ascertained from 370 families with ADHD-affected sibling pairs. Lifetime psychiatric diagnoses were based on semi-structured diagnostic interviews conducted by master’s level clinical psychologists or highly trained interviewers with extensive experience and reliability training in psychiatric diagnoses. Children and adolescents were assessed using the Schedule for Affective Disorders and Schizophrenia for School-Age Children-Present and Lifetime version (K-SADS-PL). Adult parents were assessed using the Schedule for Affective Disorders and Schizophrenia-Lifetime version (SADS-LA-IV), supplemented with the K-SADS Behavioral Disorders module for diagnosis of ADHD and disruptive behavior disorders. Direct interviews were supplemented with parent and teacher versions of the Swanson, Nolan, and Pelham, version IV (SNAP-IV) rating scale, as well as a parent-completed CBCL and Teacher Report Form. Parents also completed current ratings of self and spouse behavior with the ADHD Rating Scale IV. Subjects were excluded from participation if they were positive for any of the following: neurological disorder, head injury resulting in concussion, lifetime diagnoses of schizophrenia or autism, or estimated Full Scale IQ < 70. Subjects on stimulant medication were asked to discontinue use for 24 hours prior to their visit.
The CHOP ADHD trio families were recruited from pediatric and behavioral health clinics in the Philadelphia area 15. Inclusion criteria included families of European descent with an ADHD proband (age 6–18). Exclusionary criteria included prematurity (< 36 weeks), mental retardation, major medical and neurological disorders, pervasive developmental disorder, psychoses and major mood disorders. Diagnostic data were collected using the K-SADS interview.
Table 1 presents the breakdown of samples that were included in the analysis. Three of these samples are proband-parent trio-based samples, with the fourth being a case only sample. To analyze these controls, we selected genomically matched, pre-existing controls, available through the genetics data repository of the National Institute of Mental Health (NIMH). The final sample size consisted of 2,064 trios, 896 cases and 2,455 controls.
Table 1.
| Samples | Cases | Controls | Trios | SNPs |
|---|---|---|---|---|
| CHOP | - | - | 423 | 469,283 |
| IMAGE | - | - | 909 | 438,784 |
| IMAGE II | 896 | 2,455 | - | 294,811 |
| PUWMa | - | - | 732 | 645,995 |
| Total | 896 | 2,455 | 2,064 | 1,206,462a |
Imputation and association analysis
Each of the studies had been conducted on different platforms (Illumina 550K for CHOP, Perlegen 600K for IMAGE, Affymetrix 500K for IMAGE II, and Illumina 1M Duo for PUWMa). From these genome-wide association datasets, we imputed untyped loci across the genome using the HapMap Phase III European CEU and TSI samples as the reference panel 23, 24. The IMAGE II sample had already undergone imputation, which we carried through to this meta-analysis. We had to impute SNPs for the trio samples prior to the meta-analysis. As of writing this, and to the best of our knowledge, none of the imputation programs handle trios and supply posterior probabilities of the offspring’s transmitted and non-transmitted alleles at each imputed locus. To resolve this, we phased the trios using Beagle, which outputs the most likely haplotypes for each parent split by transmitted and non-transmitted haplotypes. We then created case pseudo-control individuals consistent with the Haplotype Relative Risk (HRR) test 25, 26. We then passed these case and pseudo-controls through Beagle 27, 28 to conduct imputation and analyzed the samples using MACH2DAT 29, using a logistic regression model. The P-value from this analysis was then transformed into a Z-score. The P-value represents the probability of observing a test statistic equally or more deviant than the observed test statistic from the analysis. By the same token, we can consider the normal distribution, and identify the critical value for which the ratio of the area under the normal distribution above the critical value plus the area under the normal distribution below the negative critical value to the total area under the normal distribution is equal to the P-value. Considered another way, we are aiming to identify the Z-score that is as deviant as the test statistic from the regression analysis. Conditional on this transformation we can then conduct meta-analysis of Z-scores, which is described in de Bakker and colleagues 30. Any SNP which was imputed with an_r̂_2 of less than 0.6 was excluded from each individual dataset before meta-analysis.
Meta-analysis
To conduct the meta-analysis, we first calculated the computed an association Z-score corresponding to the association test statistic for each sample and then combined Z’s after weighting each by the inverse of the variances of the estimate of the odds ratios from each analysis. We then calculated the P-value from this Z-score. Formula 1 shows the analytic method:
where wi is the weight of each individual study, as defined by the variance of the estimate from the logistic regression, wt is the sum of these weights, and Zi is the Z-score for each study, as defined by the P-value and direction.
Regional association plotting
We highlight the most significant regions from the meta-analysis by use of the SNP annotation and Proxy (SNAP) Search website31. SNAP displays and annotates association results, such that the LD patterns of the genetic variation in the region is included along with the association results and recombination rate.
Results
Genome-wide results
The full distribution of results can be seen in the QQ plot in Figure 1. Under the null hypothesis of no association, these points should fall along the diagonal line. The dotted line plots the 95% confidence interval. The QQ plot and the lambda statistic (1.025) show no appreciable inflation of the test statistic. The lambdas for each individual study are 1.085 for IMAGE II, 1.012 for IMAGE, 0.970 for PUWMa and 1.047 for CHOP, which yields an expected lambda of 1.028 based on the average lambda, weighted by case size. The lambda statistic is the ratio of the observed median chi square test statistic to the theoretical expectation of the median chi square32. This approach capitalizes on the observation that an increase in the variance of the distribution of the test statistic will increase the median of the chi square. This increase in variance can be caused by population stratification, technical bias, or other confounders. The reason that stratification can yield lambda is that the null conditions that the allele frequencies in cases and controls are the same are violated and thus the test statistic is expected to increase. This slightly lower than expected lambda from the meta-analysis is indicative of no particular correlated bias across these studies, enhancing our confidence in the quality of results. These results show that our quality control procedures removed most association signals that could be attributed to either technical artifacts or population stratification, and that there is little evidence for correlated biases across the studies, as correlated biases will yield a further inflation of lambda.
Figure 1.

Quantile-Quantile (QQ) plot of the meta-analysis of four attention-deficit/hyperactivity disorder genome-wide associations studies. Note: The QQ plot shows the distribution of expected p-values based against observer distribution. There is slight inflammation in the distribution of results, as indicated by the lambda of 1.025. The red dashed line represents the 95% confidence interval for the distribution of results.
Table 2 presents the top 50 findings of the meta-analysis. None of our results achieved genome-wide significance, defined as a P-value of 5×10−8, 33, 34. We present the regional association plots for our top three findings across the meta-analysis, using SNAP31 in Figures 2a, 2b, and 2c. Our top hit is rs1464807 (p=1.10E-06), which is in a gene-poor region. The SNP lies 230 kb 5′ to SHFM1. The product of this gene may be involved in proteolysis and the regulation of the cell cycle. According to the NCBI Entrez Gene database (http://www.ncbi.nlm.nih.gov/gene/), mutations in the product of _SHFM1_cause split hand/foot malformation type I. A total of eight SNPs in the region were among the top 50 findings in Table 2, which lends credibility to this association finding. Our second hit, rs177290098 (p=1.68E-06), located in an intergenic region on chromosome 20, appears to be a false positive, as it is sufficiently poorly imputed in the three trio samples to be filtered from inclusion in the meta-analysis (Table 1) and SNPs in the region, which are in linkage disequilibrium (LD) with this SNP, do not show any association signal (Figure 2b). Given the low levels of recombination around this SNP, these surrounding SNPs are more likely to be reflective of the true association evidence in this region rather than the single large association observed. The third best hit, rs7463256, shows strong regional association, indicating that this is not likely to be a technical artifact. This association signal is close to the 5′ end of the CHMP7 gene, with a number of additional SNPs in the top 50 list present located within the gene. CHMP7 is involved in the endosomal sorting pathway. Another 3 genes are present within 100kb in this gene-rich area: TNFRSF10D, which protects against apoptosis, TNFRSF10A, which transduces cell death signals to start apoptosis and LOXL2, which is involved in the biogenesis of connective tissue.
Table 2.
Top 50 hits from the genome-wide attention-deficit/hyperactivity disorder (ADHD) Meta-analysis
| CHR | BPa | SNPb | A1c | A2d | Weighte | Zf | P-valueg | Directionh | Genei |
|---|---|---|---|---|---|---|---|---|---|
| 7 | 96200280 | rs1464807 | T | G | 23.54 | 4.874 | 1.10E-06 | ++++ | . |
| 20 | 58511043 | rs17729098 | T | C | 2.85 | 4.788 | 1.68E-06 | ???+ | . |
| 7 | 96238981 | rs12375086 | T | C | 23.33 | 4.704 | 2.56E-06 | ++++ | . |
| 7 | 96215976 | rs12673393 | A | G | 23.41 | 4.665 | 3.09E-06 | ++++ | . |
| 8 | 23157565 | rs7463256 | T | C | 36.67 | −4.659 | 3.17E-06 | −−−? | CHMP7 |
| 8 | 23160140 | rs2294123 | T | G | 36.67 | 4.608 | 4.07E-06 | +++? | CHMP7 |
| 8 | 23166448 | rs11135712 | T | C | 35.75 | 4.575 | 4.77E-06 | +++? | CHMP7 |
| 8 | 23152539 | rs13268919 | A | G | 35.78 | 4.551 | 5.33E-06 | +++? | . |
| 8 | 70971815 | rs12680109 | T | C | 36.13 | −4.502 | 6.73E-06 | −−−? | . |
| 8 | 94154715 | rs1027730 | A | G | 21.6 | 4.497 | 6.89E-06 | ++++ | . |
| 7 | 96215498 | rs12673272 | T | C | 23.54 | 4.462 | 8.14E-06 | ++++ | . |
| 20 | 51017390 | rs10485813 | A | G | 20.41 | 4.46 | 8.20E-06 | ++++ | . |
| 8 | 94152003 | rs16915515 | A | G | 21.91 | −4.456 | 8.37E-06 | −−−− | . |
| 20 | 51015479 | rs16997358 | A | T | 20.41 | 4.436 | 9.17E-06 | ++++ | . |
| 3 | 15321704 | rs7340679 | T | C | 23 | 4.409 | 1.04E-05 | ++?+ | SH3BP5 |
| 20 | 58235038 | rs7271202 | A | G | 39.25 | −4.406 | 1.05E-05 | −−−− | . |
| 8 | 94153512 | rs1384769 | A | G | 22.37 | 4.393 | 1.12E-05 | ++++ | . |
| 15 | 85242738 | rs7176964 | A | T | 17.95 | −4.384 | 1.17E-05 | −−?− | AGBL1 |
| 19 | 54116059 | rs9676447 | T | C | 2.48 | −4.369 | 1.25E-05 | ??−? | NUCB1 |
| 8 | 94150207 | rs1472747 | A | G | 22 | −4.368 | 1.25E-05 | −−−− | . |
| 15 | 89221977 | rs1573643 | T | C | 21.93 | 4.357 | 1.32E-05 | ++?? | FURIN |
| 8 | 2316261015110474 | rs2280934 | A | G | 36.77 | −4.355 | 1.33E-05 | −−−? | CHMP7 |
| 7 | 6 | rs7809589 | T | G | 27.1 | 4.347 | 1.38E-05 | +++? | PRKAG2 |
| 7 | 96243316 | rs17499178 | T | G | 17.29 | 4.342 | 1.41E-05 | ++++ | . |
| 16 | 3307062 | rs17611827 | A | G | 37.06 | −4.336 | 1.45E-05 | −−−− | ZNF75A |
| 7 | 96207549 | rs17167761 | A | C | 23.49 | 4.327 | 1.51E-05 | ++++ | . |
| 6 | 16858880 | rs649266 | T | C | 46.63 | −4.325 | 1.52E-05 | −−−− | ATXN1 |
| 11 | 70829906 | rs736894 | T | C | 39.28 | −4.317 | 1.58E-05 | −−− | DHCR7 |
| 11 | 70836489 | rs3750997 | A | C | 42.89 | −4.312 | 1.62E-05 | −−− | DHCR7 |
| 6 | 16857546 | rs607138 | A | G | 46.97 | −4.303 | 1.69E-05 | −−−− | ATXN1 |
| 3 | 61578705 | rs36051446 | T | C | 47.5 | −4.299 | 1.72E-05 | +−−− | PTPRG |
| 14 | 97563832 | rs8019853 | A | G | 13.85 | −4.285 | 1.83E-05 | −−−− | . |
| 20 | 58232796 | rs2865793 | T | G | 39.22 | 4.276 | 1.91E-05 | ++++ | . |
| 20 | 58234268 | rs6100796 | T | C | 38.79 | −4.262 | 2.03E-05 | −−−− | . |
| 5 | 50381012 | rs4866023 | A | G | 45.29 | 4.261 | 2.03E-05 | ++++ | . |
| 11 | 70832468 | rs12422045 | A | G | 42.85 | 4.261 | 2.03E-05 | ++++ | DHCR7 |
| 11 | 70838711 | rs7928249 | A | G | 42.6 | −4.259 | 2.05E-05 | −−−− | . |
| 16 | 10540666 | rs11074889 | A | G | 7.87 | 4.251 | 2.13E-05 | ?+?? | EMP2 |
| 11 | 70831107 | rs11603330 | A | C | 42.95 | 4.249 | 2.14E-05 | ++++ | DHCR7 |
| 14 | 97566932 | rs12892356 | T | G | 13.64 | −4.243 | 2.21E-05 | −−−− | .NADSYN |
| 11 | 70848651 | rs12800438 | A | G | 42.58 | 4.24 | 2.23E-05 | ++++ | 1 |
| 7 | 96262263 | rs1004561 | A | T | 17.29 | 4.237 | 2.26E-05 | ++++ | . |
| 8 | 75041641 | rs16938747 | C | G | 8.88 | −4.234 | 2.30E-05 | −−?− | TCEB1 |
| 15 | 50953701 | rs2120445 | A | G | 22.04 | −4.231 | 2.33E-05 | ?−−? | . |
| 11 | 70828168 | rs1790324 | T | G | 39.27 | 4.228 | 2.36E-05 | ++++ | DHCR7 |
| 7 | 96235685 | rs1449591 | T | C | 35.29 | 4.225 | 2.39E-05 | ++++ | . |
| 20 | 58247823 | rs1535028 | T | C | 44.04 | 4.216 | 2.48E-05 | ++++ | . |
| 21 | 22305480 | rs9982805 | A | C | 12.43 | −4.206 | 2.60E-05 | −−−− | . |
| 10 | 12630181 | rs4747989 | T | C | 12.39 | 4.194 | 2.74E-05 | ++?? | CAMK1D |
| 11 | 70828433 | rs1630498 | A | C | 39.15 | 4.191 | 2.77E-05 | ++++ | DHCR7 |
Figure 2.



Regional association plots for the top three hits in the attention-deficit/hyperactivity disorder meta-analysis. Note: Three region association plots are shown here. On the x-axis is the base pair position based on the human genome 18 build, with gene regions coded in green. On the left y-axis, the log10(P-value) is reported. On the right y-axis, the recombination rate of cM per Mb is shown. The points are each individual single-nucleotide polymorphism (SNP), color-coded by r2 to the most significant SNP in the region with red indicating high values and white indicating low values.
Candidate region results
We also generated the distribution of results for a previously defined set of ADHD candidate genes selected by the IMAGE team 35ADRA1A, ADRA1B, ADRA2A, ADRA2C, ADRB2, ADRBK2, ARRB1, BDNF, CDH13, CHRNA4, COMT, CSNK1E, DBH, DDC, DRD1, DRD2, DRD3, DRD4, FADS1, FADS2, HES1, HTR1B, HTR1E, HTR2A, HTR2C, HTR3B, MAOA, MAOB, NFIL3, NR4A2, PER1, PER2, SLC18A2, SLC6A1, SLC6A2, SLC6A3, SLC6A4, SLC9A9, SNAP25, STX1A, SYT1, TPH1, and TPH2. These results from the 2752 SNPs in these candidate genes can be seen in Figure 3. The marked deviation of this SNP set from the null expectation (the diagonal line) suggests that some of these variants are associated with ADHD, although none achieve genome-wide significance. Finally, we present the top 50 results from the candidate gene SNPs in Table 3.
Figure 3.
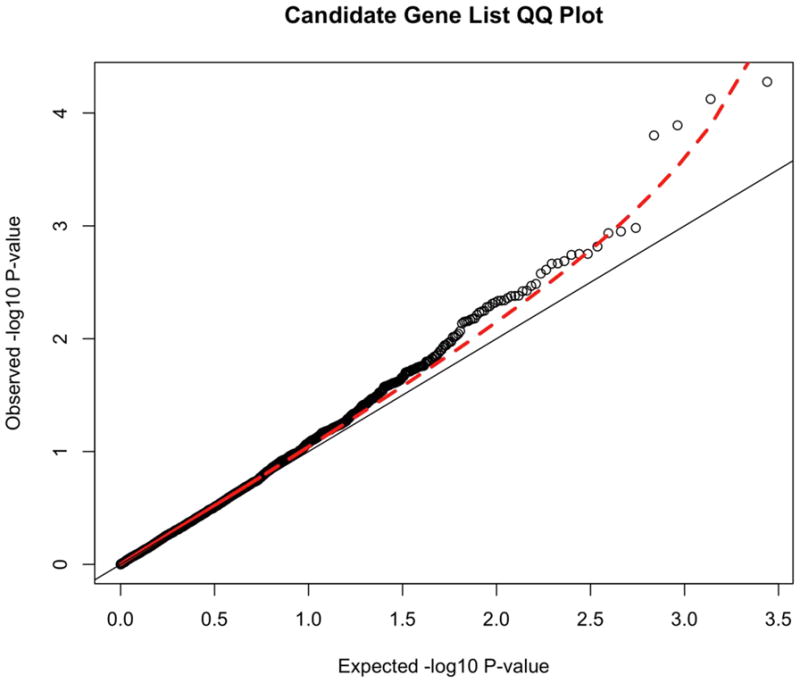
Quantile-Quantile (QQ) plot of candidate genes. Note: The QQ plot shows the distribution of expected p-values based against the observer distribution. The red dashed line represents the 95% confidence interval for the distribution of results. These p-values are uncorrected.
Table 3.
Top 50 results from Candidate Genes
| CHR | BPa | SNPb | A1c | A2d | Weighte | Z-scoref | P-valueg | Directionh | Genei |
|---|---|---|---|---|---|---|---|---|---|
| 16 | 81819599 | rs8045365 | A | G | 11.03 | −4.043 | 5.28E-05 | −−−− | CDH13 |
| 3 | 11028517 | rs6778281 | A | C | 20.97 | 3.959 | 7.52E-05 | ++++ | SLC6A1 |
| 16 | 81822613 | rs12917991 | A | C | 10.97 | 3.829 | 0.0001286 | ++++ | CDH13 |
| 16 | 81813774 | rs8045006 | A | C | 15.84 | 3.778 | 0.000158 | ++++ | CDH13 |
| 16 | 81280799 | rs13334902 | T | C | 24.53 | −3.279 | 0.001043 | −−−+ | CDH13 |
| 16 | 81828083 | rs7184058 | A | G | 22.73 | 3.258 | 0.001121 | +++? | CDH13 |
| 16 | 81298757 | rs10514564 | T | G | 34.52 | 3.248 | 0.001161 | ++++ | CDH13 |
| 16 | 81277337 | rs8060937 | T | C | 23.89 | −3.17 | 0.001526 | −−−− | CDH13 |
| 16 | 81273739 | rs8048202 | A | G | 25 | −3.126 | 0.001771 | −−−+ | CDH13 |
| 16 | 81254805 | rs12444015 | T | C | 29.01 | −3.125 | 0.001775 | −−−+ | CDH13 |
| 16 | 81384008 | rs17192646 | C | G | 21.29 | −3.12 | 0.001808 | −−−− | CDH13 |
| 16 | 81283227 | rs13336758 | A | G | 24.32 | −3.082 | 0.002054 | −−−+ | CDH13 |
| 3 | 11041876 | rs9822125 | A | T | 20.06 | −3.068 | 0.002152 | −−−− | SLC6A1 |
| 16 | 81271375 | rs4081995 | T | C | 23.59 | 3.066 | 0.002166 | +++− | CDH13 |
| 16 | 81267473 | rs8054295 | T | C | 18.54 | −3.029 | 0.002451 | −−−− | CDH13 |
| 8 | 26773734 | rs17426222 | T | C | 31.51 | 3.006 | 0.002645 | ++++ | ADRA1A |
| 16 | 81469875 | rs17674039 | T | C | 24.23 | −2.941 | 0.003268 | −−−+ | CDH13 |
| 16 | 81281593 | rs12932203 | A | G | 24.63 | 2.929 | 0.0034 | +++− | CDH13 |
| 3 | 11018019 | rs1919075 | A | G | 33.96 | −2.899 | 0.003749 | −−−− | SLC6A1FIGNL1(+17.31kb) |
| 7 | 50502889 | rs11575522 | T | C | 9.84 | −2.895 | 0.003795 | −−+− | DDCFIGNL1(+12.78kb) |
| 7 | 50498363 | rs11575543 | A | G | 9.68 | −2.866 | 0.004158 | −−+− | DDC |
| 16 | 81370055 | rs17276565 | T | G | 23.12 | 2.864 | 0.004178 | ++++ | CDH13FIGNL1(+18.08kb) |
| 7 | 50503662 | rs11575510 | A | G | 9.68 | −2.864 | 0.004188 | −−+− | DDC |
| 16 | 81468190 | rs7192135 | A | C | 23.78 | 2.851 | 0.004362 | +++− | CDH13FIGNL1(+16.24kb) |
| 7 | 50501821 | rs11575527 | C | G | 9.73 | −2.836 | 0.004569 | −−+− | DDC |
| 16 | 81264849 | rs9936363 | T | C | 18.08 | −2.834 | 0.004598 | −−−− | CDH13 |
| 7 | 50575188 | rs11575302 | A | G | 10.21 | −2.832 | 0.004619 | −−+− | DDC |
| 7 | 50503581 | rs11575511 | T | C | 9.68 | −2.821 | 0.004783 | −−+− | FIGNL1(+18kb) DDC |
| 3 | 11025563 | rs11710097 | A | G | 19.22 | −2.814 | 0.004896 | −−−− | SLC6A1FIGNL1(+19.96kb) |
| 7 | 50505546 | rs11575494 | A | G | 9.67 | −2.797 | 0.005165 | −−+− | DDCFIGNL1(+13.91kb) |
| 7 | 50499488 | rs11575535 | T | C | 9.72 | −2.791 | 0.005248 | −−+− | DDC |
| 16 | 81497693 | rs16958840 | C | G | 25.06 | 2.767 | 0.005651 | +++− | CDH13FIGNL1(+19.21kb) |
| 7 | 50504792 | rs10499693 | A | G | 9.71 | −2.763 | 0.005729 | −−+− | DDC |
| 3 | 11025518 | rs17466478 | T | C | 14.51 | −2.756 | 0.00586 | −−?? | SLC6A1 |
| 7 | 50563965 | rs11575340 | T | G | 9.71 | −2.74 | 0.00615 | −−+− | DDC |
| 16 | 81349970 | rs1870847 | T | C | 29.09 | −2.715 | 0.006625 | −−−− | CDH13FIGNL1(+19.1kb) |
| 7 | 50504681 | rs11575501 | A | G | 9.66 | 2.713 | 0.006677 | ++−+ | DDC |
| 12 | 70629554 | rs11179003 | T | C | 12.45 | 2.707 | 0.006791 | ++++ | TPH2 |
| 16 | 81854762 | rs16960006 | T | G | 19.76 | 2.698 | 0.006982 | +++− | CDH13FIGNL1(+13.8kb) |
| 7 | 50499379 | rs11575537 | T | C | 9.69 | −2.696 | 0.007019 | −−+− | DDC |
| 3 | 11038545 | rs11712912 | A | G | 23.37 | 2.693 | 0.007086 | ++++ | SLC6A1 |
| 16 | 81495401 | rs17674776 | T | C | 25.11 | 2.681 | 0.007334 | +++− | CDH13 |
| 16 | 81490402 | rs16958834 | T | C | 25.15 | −2.629 | 0.008564 | −−−+ | CDH13 |
| 16 | 81491933 | rs9646331 | C | G | 25.06 | −2.61 | 0.009053 | −−−+ | CDH13 |
| 3 | 11024133 | rs1710891 | T | C | 35.51 | 2.599 | 0.009352 | ++++ | SLC6A1 |
| 3 | 11015439 | rs9990174 | T | G | 33.21 | −2.589 | 0.009613 | −−−− | SLC6A1 |
| 16 | 81275531 | rs8055389 | A | G | 24.72 | −2.587 | 0.009683 | −−−+ | CDH13 |
| 22 | 24309234 | rs713921 | A | G | 5.95 | 2.555 | 0.01063 | +??+ | ADRBK2 |
| 16 | 81483050 | rs16958826 | T | G | 30.61 | 2.552 | 0.01071 | +++− | CDH13 |
| 22 | 24308356 | rs738344 | A | G | 5.94 | 2.542 | 0.01101 | +??+ | ADRBK2 |
Discussion
We have presented the first meta-analysis of genome-wide association datasets for childhood ADHD, including a total of 2,064 trios, 896 cases and 2,455 controls. Genome-wide significant effects still elude detection for this disorder, suggesting that the effect sizes for the common variants influencing risk for ADHD are likely to be very small. However, these results include a number of promising regions, for which replication is an essential next step. Moreover, our analysis of candidate genes suggests that some of these loci are worth studying further.
The most strongly implicated region is on chromosome 7, where 8 SNPs feature among the top 50 association findings. The chromosome 7 region is gene-poor. The gene most near to the findings is the SHFM1_gene, which still more than 200 kb away from the association findings and is not an obvious candidate for ADHD. According to Entrez, the product of_SHFM1 may be involved in proteolysis and the regulation of the cell cycle. Mutations in the gene cause split hand/foot malformation, indicating that it functions from an early time point in embryonic development. SHFM1 is also expressed in the brain, which points to a possibly yet unknown function for this gene in the context of ADHD. Genetic variation in the same region was previously found to be associated with major depressive disorder and bipolar disorder at p-values of 10−4 in genome-wide association studies 36, 29. In addition to the finding on chromosome 7, a region on chromosome 20 contains the second best hit in our study. It includes four other SNPs from the top 50 list. This region does not lie close to known genes, though.
A region on chromosome 8, featuring five SNPs in the top list, also seems interesting. The genes in this region play a role in immunity and cardiovascular processes, but the CHMP7 gene, implicated most stringently by our findings, also shows expression in the brain and has a function in endosomal sorting and vesicular transport 28. Mutations in a related gene, CHMP2B, lead to frontotemporal dementia, disinhibition and executive dysfunction37. A second (small) region on chromosome 8 implicated through four association findings in the top 50 does not contain any genes, but lies less than 100 kb 5′ to C8ORF83, a gene of unknown function.
In addition to those, a region on chromosome 11 contains 8 of the 50 top findings. This region contains the 5′ ends of two genes, DHCR7 and NADSYN1. The former encodes 7-dehydrocholesterol reductase, and mutations in this gene cause Smith-Lemli-Opitz syndrome. In adults, the ubiquitously transcribed DHCR7 is most abundant in adrenal gland, liver, testis, and brain. NADSYN1 encodes NAD synthase 1, which catalyzes the final step in the biosynthesis of NAD from nicotinic acid adenine dinucleotide (NaAD). Nicotinamide adenine dinucleotide (NAD) is a coenzyme in metabolic redox reactions, a precursor for several cell signaling molecules, and a substrate for protein posttranslational modifications. Replication efforts will be necessary to be certain that these putative associations are actually related to ADHD.
Clearly, this sample still lacks power to be able to unequivocally identify genome-wide significant associations of common genetic variants (SNPs) with ADHD. There are a number of possible explanations for why this lack of power still persists. First off, it may be that the true effect sizes of individual genetic variants for ADHD are extremely small, accounting for less than 0.51% of the variance. As such our findings remain in line with other psychiatric phenotypes. Recent work on psychiatric phenotypes such as schizophrenia and bipolar disorder have identified bona fide associations, but the combined sample size required to do is two to three times as large as the results presented in this study. As a result, further genome-wide association studies and replication efforts are essential. We note that at similar sample sizes for schizophrenia and bipolar disorder, there were no unequivocal associations, either. Essentially, this meta-analysis imposes a ceiling on the effect size for common variation in the sample. We have 98% power to detect an effect accounting for 0.5% of the variance of the phenotype. The power to detect is 48% when we decrease the effect size to 0.25% of the variance and drops to 2% power when we consider an effect size of 0.1% of the phenotypic variance explained. These power calculations are all conducted assuming an alpha level of 5e-8, consistent with the genome-wide significance threshold, and are based on the post-cleaning sample size.
Beyond the question of effect size, there are other possible explanations for our negative findings. Although each data collection site provided diagnoses of ADHD based on structured, systematic interviews, there may be variability in measurement across data collection sites. Such variability would induce additional phenotypic (and probably genotypic) heterogeneity. Alternatively, a similar problem would arise if differences in local referral patterns lead to increased heterogeneity between the datasets. Additionally, it may be that much of the genetic variation that predisposes to ADHD is extremely rare and deleterious, rather than common and conferring modest risk. A number of studies in other psychiatric disorders recently suggested that a significant (though undefined) proportion of cases may be caused by such rare variants, in this case CNVs 38. A first study in ADHD by members of this group also pointed to a role of such rare variants in this disorder 15. We also note that we may lose power from incomplete tagging of the putative causal variation. The expected non-centrality parameter for a SNP in LD with a functional variant is equal to the product of the r2 and the non-centrality parameter at the causal locus39. Furthermore, gene by environment interaction may also yield a reduction in power, as might gene by gene interaction. Although much of our sample is drawn from populations of western European descent, there is likely a great deal of environmental variability between the countries of origin that are included in this study, in terms of diet, exposure to toxins, and so forth. This variation may interact and influence the genetic mechanisms which predispose to ADHD and as a consequence hurt power to detect association. In all likelihood each of these possible explanations may partially contribute to the difficulty in finding ADHD susceptibility genes through GWAS.
This work should be viewed in the context of some limitations. Although we have conducted the largest genetic study of ADHD ever, our sample size is still relatively small compared with successful GWAS of complex disorders. Because creating this large sample required a collaborative effort among many sites, differences in ascertainment approaches and clinical assessments could have added noise to our sample and reduced power. Moreover, although using repository controls is a cost effective strategy, because our control sample was not screened for ADHD, our power was less than it would have been for a screened sample.
In conclusion, we amassed a sizeable genome-wide association dataset for meta-analysis consisting of 2,960 individual childhood ADHD cases plus parental or independent control samples. Such numbers are indicative of a strong commitment by the ADHD genetics community to share data and collaborate in the name of the identification of significant associations to gain biological insight. Additional collaborative efforts should return true associations that will start to unravel the complex biological underpinnings of ADHD.
Acknowledgments
This project was supported by the following grants: US National of Institute of Health Grants R13MH059126, R01MH62873, U01MH085518 and R01MH081803 to S.V. Faraone, U01MH085515 to M. Daly K23MH066275-01 to J. Elia; and R01MH58277 to S. Smalley; Institutional Development Award to the Center for Applied Genomics from the Children’s Hospital of Philadelphia to H. Hakonarson. Affymetrix Power Award, 2007 to B. Franke; NHMRC (Australia) and Sidney Sax Public Health Fellowship (443036) to S.E. Medland; Wellcome Trust, UK for sample collection to L Kent. UMC Utrecht Genvlag Grant and Internal Grant of Radboud University, Nijmegen Medical Centre to J. Buitelaar, the Deutsche Forschungsgemeinschaft (KFO 125, SFB 581, GRK 1156 to K.P. Lesch, ME 1923/5-1, ME 1923/5-3, GRK 1389 to C. Freitag and J. Meyer, SCHA 542/10-3 to H. Schäfer) and the Bundesministerium für Bildung und Forschung (BMBF 01GV0605 to K.P. Lesch). Additional analytical support was from Foundation for the NIH and R01MH080403 (PI Sullivan). All computational work was conducted on the Genetic Cluster Computer (the Netherlands) which is funded by an NWO Medium Investment grant (480-05-003, PI Posthuma), the Faculty of Psychology and Education of VU University (Amsterdam), and by the Dutch Brain Foundation (PI Ophoff) and is hosted by the Dutch National Computing and Networking Services.
The Psychiatric GWAS Consortium: ADHD Subgroup consists of: Writing team: Benjamin M Neale, PhD (team leader), Sarah E. Medland, PhD, Stephan Ripke, MD, Philip Asherson, MRCPsych, PhD, Barbara Franke, PhD, Klaus-Peter Lesch, MD, Stephen V. Faraone, PhD; Data Analysis team: Sarah E. Medland, PhD (team leader), Stephan Ripke, MD, Thuy Trang Nguyen, Dipl. Math. oec, Helmut Schäfer, PhD, Peter Holmans, PhD, Mark Daly, PhD, Benjamin M Neale, PhD (chair); Data Collection Team: Hans-Christoph Steinhausen, MD, PhD, DMSc, Klaus-Peter Lesch, MD, Christine Freitag, MD, MA; Andreas Reif, MD, Tobias J. Renner, MD, Marcel Romanos, MD, Jasmin Romanos, MD, Susanne Walitza, MD, Andreas Warnke, MD, PhD, Thuy Trang Nguyen, Dipl. Math. oec, Helmut Schäfer, PhD, Jobst Meyer, PhD, Haukur Palmason, PhD, Jan Buitelaar, MD, Barbara Franke, PhD, Alejandro Arias Vasquez, PhD, Nanda Lambregts-Rommelse, PhD, Michael Gill, Mb BCh BAO, MD, MRCPsych, FTCD, Richard J.L. Anney, PhD, Kate Langely, PhD, Michael O’Donovan, FRCPsych, PhD, Nigel Williams, PhD, Peter Holmans, PhD, Michael Owen, PhD FRCPsych, Anita Thapar, MD, Lindsey Kent, MD, PhD, Joseph Sergeant, PhD, Herbert Roeyers, MD, PhD, Philip Asherson, MRCPsych, PhD, Eric Mick, Sc.D, Joseph Biederman, MD, Alysa Doyle, PhD, Susan Smalley, PhD, Sandra Loo, PhD, Hakon Hakonarson, MD, PhD, Josephine Elia, MD, Alexandre Todorov, PhD, Ana Miranda, MD, Fernando Mulas, M.D., Ph.D., Richard P. Ebstein, PhD, Aribert Rothenberger, MD, PhD, Tobias Banaschewski, MD, PhD, Robert D. Oades, PhD, Edmund Sonuga-Barke, PhD, James McGough, MD, Laura Nisenbaum, PhD, Stephen V. Faraone, PhD (Chair); Molecular Genetics team: Frank Middleton, PhD, Xiaolan Hu, PhD, Stan Nelson, MD; Project Management team: Anita Thapar, MD, James McGough, MD, Eric Mick, Sc.D, Benjamin M Neale, PhD, Stephen V. Faraone, PhD (Chair); Statistical experts: Benjamin M Neale, PhD Mark Daly, PhD.
We thank the patients and the family members who provided data for this project.
Disclosure: Dr. Buitelaar has, in the past three years, served as a consultant to, member of the advisory board, and/or served on the speakers’ bureau for Janssen Cilag BV, Eli Lilly and Co., Bristol-Myers Squibb, Organon/Shering Plough, UCB, Shire, Medice, Servier, and Servier. Dr. Faraone has, in the past year, received consulting fees and has served on advisory boards for Eli Lilly and Co., Ortho-McNeil, and Shire Development, and has received research support from Eli Lilly and Co., Pfizer, Shire, and the National Institutes of Health. In previous years, Dr. Faraone has received consulting fees, served on advisory boards, or been a speaker for Shire, McNeil, Janssen, Novartis, Pfizer, and Eli Lilly and Co. In previous years he has received research support from Eli Lilly and Co., Shire, Pfizer and the National Institutes of Health. Dr. Banaschewski has served on the Advisory Board or as a consultant for Dsitin, Eli Lilly and Co. Medice, Novartis, Pfizer, Shire, UCB, and Viforpharma. He received conference attendance support or served on the speakers’ bureau for Eli Lilly and Co., Janssen McNeil, Medice, Novartis, and UCB. He is or has been involved in clinical trials conducted by Eli Lilly and Co., Shire, and Novartis. The present work is unrelated to the above grants and relationships. Dr. M. Romanos, in the past three years, has served on the speakers’ bureau for Janssen-Cilag. In previous years he has served on the speakers’ bureau for MEDICE. Dr. Freitag, in the past three years, has served on the speakers’ bureau for Eli-Lilly and Co., Shire, Novartis, and Janssen-Cilag. Dr. Mick receives research support from Ortho-McNeil Janssen Scientific Affairs, Pfizer, Shire Pharmaceuticals, and has been an advisory board member for Shire Pharmaceuticals. Dr. Biederman receives research support from Alza, AstraZeneca, Bristol Myers Squibb, Eli Lilly and Co., Janssen Pharmaceuticals Inc., McNeil, Merck, Organon, Otsuka, Shire, the National Institute of Mental Health, and the National Institute of Child Health and Human Development. In 2009, Dr. Biederman served on the speakers’ bureau for Fundacion Areces, Medice Pharmaceuticals, and the Spanish Child Psychiatry Association. In previous years, Dr. Biederman has received research support, consultation fees, or speaker’s fees for/from Abbott, AstraZeneca, Celltech, Cephalon, Eli Lilly and Co., Esai, Forest, Glaxo, Gliatech, Janssen, McNeil, the National Alliance for Research on Schizophrenia and Depression, the National Institute on Drug Abuse, New River, Novartis, Noven, Neurosearch, Pfizer, Pharmacia, The Prechter Foundation, Shire, The Stanley Foundation, UCB Pharma, Inc. and Wyeth. Dr. Sergeant has served on the advisory boards for Eli Lilly and Co. and Shire. He has received research grant support from Eli Lilly and Co. and served on the speakers’ bureau for Shire, Eli Lilly and Co., Janssen-Cilag, and Novartis. Dr. Oades has received research support from UCB GmbH, Janssen-Cilag, and Shire. Dr. Schäfer has served as a consultant for Daiichi Sankyo, served on the speakers’ bureau for the European School of Oncology, and receives research grants from the German Federal Government, and the German Research Foundation. Drs. Neale, Medland, Ripke, Asherson, Franke, Lesch, Nguyen, Holmans, Daly, Steinhausen, Reif, Renner, J. Romanos, Walitza, Warnke, Meyer, Palmason, Vasquez, Lambregts-Rommelse, Gill, Anney, Langely, O’Donovan, Williams, Owen, Thapar, Kent, Roeyers, Doyle, Smalley, Loo, Hakonarson, Elia, Todorov, Miranda, Mulas, Ebstein, Rothenberger, Sonuga-Barke, McGough, Nisenbaum, Middleton, Hu, and Nelson report no biomedical financial interests or potential conflicts of interest.
Footnotes
This article represents one of several articles published in the August and September issues of the Journal of the American Academy of Child and Adolescent Psychiatry that explores the intersection of genetics and mental health disorders in children and adolescents. The editors invite the reader to investigate the additional articles on this burgeoning area of developmental psychopathology.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Faraone SV, Sergeant J, Gillberg C, Biederman J. The Worldwide Prevalence of ADHD: Is it an American Condition? World Psychiatry. 2003;2(2):104–113. [PMC free article] [PubMed] [Google Scholar]
- 2.Polanczyk G, de Lima MS, Horta BL, Biederman J, Rohde LA. The Worldwide Prevalence of ADHD: A Systematic Review and Metaregression Analysis. Am J Psychiatry Jun. 2007;164(6):942–948. doi: 10.1176/ajp.2007.164.6.942. [DOI] [PubMed] [Google Scholar]
- 3.Kessler RC, Adler L, Barkley R, et al. The prevalence and correlates of adult ADHD in the United States: Results from the national comorbidity survey replication. American Journal of Psychiatry. 2006;163(4):716–723. doi: 10.1176/appi.ajp.163.4.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Faraone SV, Biederman J. What is the prevalence of adult ADHD? Results of a population screen of 966 adults. Journal of Attention Disorders. 2005;9(2):384–391. doi: 10.1177/1087054705281478. [DOI] [PubMed] [Google Scholar]
- 5.Freitag CM, Rohde LA, Lempp T, Romanos M. Phenotypic and measurement influences on heritability estimates in childhood ADHD. Eur Child Adolesc Psychiatry Mar. 2010;19(3):311–323. doi: 10.1007/s00787-010-0097-5. [DOI] [PubMed] [Google Scholar]
- 6.Faraone SV, Perlis RH, Doyle AE, et al. Molecular genetics of attention-deficit/hyperactivity disorder. Biol Psychiatry. 2005 Jun 1;57(11):1313–1323. doi: 10.1016/j.biopsych.2004.11.024. [DOI] [PubMed] [Google Scholar]
- 7.Faraone SV, Mick E. Molecular Genetics of Attention Deficit Hyperactivity Disorder. Psychiatric Clinics of North America. 2010;33(1):159–180. doi: 10.1016/j.psc.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McLoughlin G, Ronald A, Kuntsi J, Asherson P, Plomin R. Genetic support for the dual nature of attention deficit hyperactivity disorder: substantial genetic overlap between the inattentive and hyperactive-impulsive components. J Abnorm Child Psychol Dec. 2007;35(6):999–1008. doi: 10.1007/s10802-007-9149-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li D, Sham PC, Owen MJ, He L. Meta-analysis shows significant association between dopamine system genes and attention deficit hyperactivity disorder (ADHD) Hum Mol Genet. 2006 Jul 15;15(14):2276–2284. doi: 10.1093/hmg/ddl152. [DOI] [PubMed] [Google Scholar]
- 10.Neale BM, Lasky-Su J, Anney R, et al. Genome-wide association scan of attention deficit hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet. 2008 Dec 5;147B(8):1337–1344. doi: 10.1002/ajmg.b.30866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lasky-Su J, Anney RJ, Neale BM, et al. Genome-wide association scan of the time to onset of attention deficit hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet. 2008 Dec 5;147B(8):1355–1358. doi: 10.1002/ajmg.b.30869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mick E, Todorov A, Smalley S, et al. Family-Based Genome-Wide Association Scan of Attention-Deficit/Hyperactivity Disorder. Journal of American Academy of Child and Adolescent Psychiatry. 2010;49(9):xxx–xxx. doi: 10.1016/j.jaac.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Consortium II. Case Control Genome-wide Association of Attention Deficit/Hyperactivity Disorder. J of Amer Academy of Child and Adolescent Psychiatry. 2010;49(9):xxx–xxx. doi: 10.1016/j.jaac.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lesch KP, Timmesfeld N, Renner TJ, et al. Molecular genetics of adult ADHD: converging evidence from genome-wide association and extended pedigree linkage studies. J Neural Transm Nov. 2008;115(11):1573–1585. doi: 10.1007/s00702-008-0119-3. [DOI] [PubMed] [Google Scholar]
- 15.Elia J, Gai X, Xie HM, et al. Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol Psychiatry. 2009 Jun 23; doi: 10.1038/mp.2009.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neale BM, Lasky-Su J, Anney R, et al. Genome-wide association scan of attention deficit hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet. 2008 Nov 3;147B(8):1337–1344. doi: 10.1002/ajmg.b.30866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anney RJ, Hawi Z, Sheehan K, et al. Parent of origin effects in attention/deficit hyperactivity disorder (ADHD): Analysis of data from the international multicenter ADHD genetics (IMAGE) program. Am J Med Genet B Neuropsychiatr Genet. 2007 Dec 28;147B(8):1495–1500. doi: 10.1002/ajmg.b.30659. [DOI] [PubMed] [Google Scholar]
- 18.Lasky-Su J, Neale BM, Franke B, et al. Genome-wide association scan of quantitative traits for attention deficit hyperactivity disorder identifies novel associations and confirms candidate gene associations. Am J Med Genet B Neuropsychiatr Genet. 2008 Sep 26;147B(8):1345–1354. doi: 10.1002/ajmg.b.30867. [DOI] [PubMed] [Google Scholar]
- 19.Lasky-Su J, Anney RJ, Neale BM, et al. Genome-wide association scan of the time to onset of attention deficit hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet. 2008 Oct 20;147B(8):1355–1358. doi: 10.1002/ajmg.b.30869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anney RJ, Lasky-Su J, O’Dushlaine C, et al. Conduct disorder and ADHD: Evaluation of conduct problems as a categorical and quantitative trait in the international multicentre ADHD genetics study. Am J Med Genet B Neuropsychiatr Genet. 2008 Oct 24;147B(8):1369–1378. doi: 10.1002/ajmg.b.30871. [DOI] [PubMed] [Google Scholar]
- 21.Bakker SC, van der Meulen EM, Buitelaar JK, et al. A whole-genome scan in 164 Dutch sib pairs with attention-deficit/hyperactivity disorder: suggestive evidence for linkage on chromosomes 7p and 15q. Am J Hum Genet. 2003;72(5):1251–1260. doi: 10.1086/375143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Donovan MC, Craddock N, Norton N, et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat Genet. 2008 Jul 30; doi: 10.1038/ng.201. [DOI] [PubMed] [Google Scholar]
- 23.Thorisson GA, Smith AV, Krishnan L, Stein LD. The International HapMap Project Web site. Genome Res Nov. 2005;15(11):1592–1593. doi: 10.1101/gr.4413105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.The International HapMap Project. Nature. 2003 Dec 18;426(6968):789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 25.Knapp M, Seuchter SA, Baur MP. The haplotype-relative-risk (HRR) method for analysis of association in nuclear families. Am J Hum Genet Jun. 1993;52(6):1085–1093. [PMC free article] [PubMed] [Google Scholar]
- 26.Terwilliger JD, Ott J. A haplotype-based ‘haplotype relative risk’ approach to detecting allelic associations. Hum Hered. 1992;42(6):337–346. doi: 10.1159/000154096. [DOI] [PubMed] [Google Scholar]
- 27.Browning BL, Browning SR. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet Feb. 2009;84(2):210–223. doi: 10.1016/j.ajhg.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet Nov. 2007;81(5):1084–1097. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moskvina V, Craddock N, Holmans P, et al. Gene-wide analyses of genome-wide association data sets: evidence for multiple common risk alleles for schizophrenia and bipolar disorder and for overlap in genetic risk. Mol Psychiatry Mar. 2009;14(3):252–260. doi: 10.1038/mp.2008.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Bakker PI, Ferreira MA, Jia X, Neale BM, Raychaudhuri S, Voight BF. Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum Mol Genet. 2008 Oct 15;17(R2):R122–128. doi: 10.1093/hmg/ddn288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson AD, Handsaker RE, Pulit SL, Nizzari MM, O’Donnell CJ, de Bakker PI. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008 Dec 15;24(24):2938–2939. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55(4):997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 33.Pe’er I, Yelensky R, Altshuler D, Daly MJ. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet Epidemiol May. 2008;32(4):381–385. doi: 10.1002/gepi.20303. [DOI] [PubMed] [Google Scholar]
- 34.Dudbridge F, Gusnanto A. Estimation of significance thresholds for genomewide association scans. Genet Epidemiol Apr. 2008;32(3):227–234. doi: 10.1002/gepi.20297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brookes K, Xu X, Chen W, et al. The analysis of 51 genes in DSM-IV combined type attention deficit hyperactivity disorder: association signals in DRD4, DAT1 and 16 other genes. Mol Psychiatry. 2006 Aug 8; doi: 10.1038/sj.mp.4001869. [DOI] [PubMed] [Google Scholar]
- 36.Konneker T, Barnes T, Furberg H, Losh M, Bulik CM, Sullivan PF. A searchable database of genetic evidence for psychiatric disorders. Am J Med Genet B Neuropsychiatr Genet. 2008 Sep 5;147B(6):671–675. doi: 10.1002/ajmg.b.30802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Urwin H, Ghazi-Noori S, Collinge J, Isaacs A. The role of CHMP2B in frontotemporal dementia. Biochem Soc Trans Feb. 2009;37(Pt 1):208–212. doi: 10.1042/BST0370208. [DOI] [PubMed] [Google Scholar]
- 38.Weiss LA, Shen Y, Korn JM, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008 Feb 14;358(7):667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 39.Sham PC, Zhao JH, Cherny SS, Hewitt JK. Variance-Components QTL linkage analysis of selected and non-normal samples: conditioning on trait values. Genetic Epidemiology. 2000;19(Suppl 1):S22–28. doi: 10.1002/1098-2272(2000)19:1+<::AID-GEPI4>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]