Myc represses primitive endoderm differentiation in pluripotent stem cells (original) (raw)
. Author manuscript; available in PMC: 2011 Sep 3.
Published in final edited form as: Cell Stem Cell. 2010 Sep 3;7(3):343–354. doi: 10.1016/j.stem.2010.06.023
Abstract
The generation of induced pluripotent stem cells (iPSCs) provides a novel method to facilitate investigations into the mechanisms that control stem cell pluripotency and self-renewal. Myc has previously been shown to be critical for murine embryonic stem cell (mESC) maintenance, while also enhancing directed reprogramming of fibroblasts by effecting widespread changes in gene expression. Despite several studies identifying in vivo target genes, the precise mechanism by which Myc regulates pluripotency remains unknown. Here we report that co-deletion of c- and N-MYC in iPSCs and ESCs results in their spontaneous differentiation to primitive endoderm. We show that Myc sustains pluripotency through repression of the primitive endoderm master regulator GATA6, while also contributing to cell cycle control by regulation of the mir-17-92 miRNA cluster. Our findings demonstrate the indispensable requirement for c- or N-myc in pluripotency beyond proliferative and metabolic control.
Introduction
Myc is widely regarded as being important for stem cell proliferation, but its role in the regulation of pluripotency remains unclear. To determine the role of Myc in mESCs, a multitude of genome-wide chromatin immunoprecipitation analyses (ChIP-Chip, ChIP-Seq) have been performed (Chen et al, 2008; Kidder et al., 2008; Kim et al., 2008; Sridharan et al., 2009). Despite the identification of cell cycle control and metabolic genes as direct targets, lineage specific regulators have not been functionally defined. These data therefore leave open the question of how Myc maintains the self-renewing, pluripotent state.
The establishment of pluripotency has been widely analyzed by reprogramming somatic cells via the introduction of four exogenous factors, Oct4, Sox2, Klf4, and c-myc (Takahashi and Yamanaka, 2006). Although the exogenous introduction of c-myc is not absolutely required for reprogramming, it significantly enhances the efficiency of iPSC generation by causing sweeping changes to gene expression (Nakagawa et al., 2008; Sridharan et al., 2009). These data indicate that c-myc is important in initializing reprogramming but do not address its role in maintenance of the pluripotent state. Dramatic changes in the mode of cell cycle regulation accompany somatic cell reprogramming, a facet of pluripotent cell biology that is widely thought to be under the control of Myc (Singh and Dalton, 2009). However, mechanisms by which Myc controls the cell cycle in pluripotent cells remain undefined.
We previously demonstrated that c-myc promotes self-renewal of mESCs in the absence of leukemia inhibitor factor (LIF), while overexpression of a dominant-negative c-myc promotes differentiation (Cartwright et al., 2005). More recently, enforced expression of Myc has been shown to promote a metastable pluripotent state in mESCs that are otherwise unstable (Hanna et al., 2009). Interpretation of this data however, is confounded by observations that c-myc and N-myc knockout mice develop well past the blastocyst stage of development, and that mESCs derived from these mice self-renew in a manner comparable to wild-type cells (Baudino et al., 2002; Charron et al., 1990). This can be attributed to the functional redundancy between Myc family members and their overlapping expression during early development (Malynn et al., 2000).
In this report, we address this issue by analyzing the effects of simultaneous c- and N-MYC inactivation in pluripotent stem cells. Myc is shown to impact on self-renewal through regulation of the cell cycle regulatory network and to maintain pluripotency by imposing a primitive endoderm differentiation blockade involving the master regulator, GATA6.
Results
Myc is Essential for the Maintenance of Pluripotency and Inhibits Primitive Endoderm Formation
To examine the requirement for Myc in pluripotent stem cells, we generated iPSCs from mouse embryonic fibroblasts containing c-MYC and N-MYC floxed alleles using Oct4, Sox2 and Klf4 retroviruses. Flox miPSCs (c-MYCfl/fl;N-MYCfl/fl) have a mESC-like domed-shaped colony morphology, express markers of pluripotency, are capable of multilineage differentiation in vitro and form teratomas in vivo (Figure S1). After transfection of CreGFP, c-MYCfl/fl;N-MYCfl/fl miPSCs were subjected to FACS and genotyped to confirm deletion of c- and N-MYC (Figure 1A and 1B). Upon plating of cells into mESC medium, GFP+ double knockout (dKO; c-mycΔ/Δ;N-mycΔ/Δ) cells underwent spontaneous differentiation, as determined by loss of alkaline phosphatase staining and by loss of a tightly-packed colony morphology (Figure 1C and 1D). c-MYCfl/fl;N-MYCfl/fl mESCs were used in parallel experiments and generated similar results (Figure S2A, B). Simultaneous loss of c- and N-myc is therefore not compatible with maintenance of pluripotent cells.
Figure 1. Deletion of c- and N-myc in iPSCs results in loss of self-renewal.
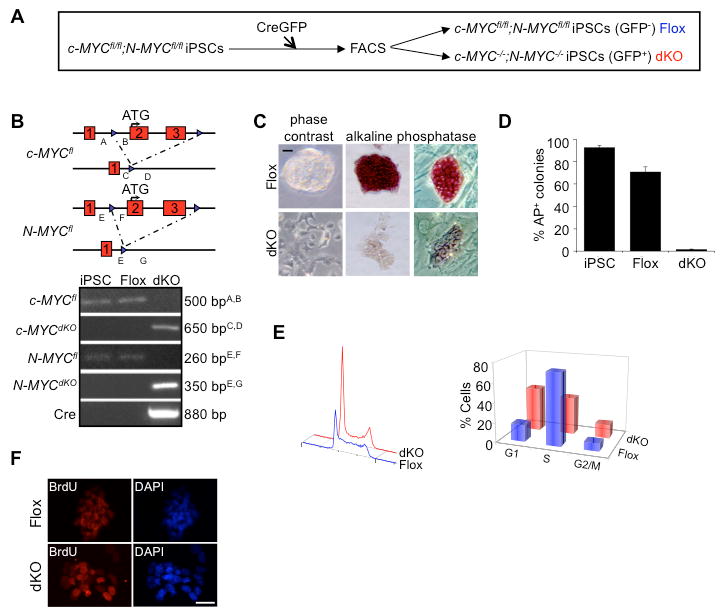
(A) Derivation of double knockout c-myc; N-myc miPSCs by transfection of CreGFP and isolation by FACS. (B) Locus map of targeted alleles and genotype analysis. Genotype analysis was performed on genomic DNA isolated from parental c-MYCfl/fl;N-MYCfl/fl, CreGFP- (Flox) miPSCs, and CreGFP+ (dKO) miPSCs. Amplicon lengths and corresponding primer sets corresponding to their position at the c-MYC and N-MYC loci are indicated. (C) c-MYCfl/fl;N-MYCfl/fl miPSCs transfected with CreGFP were FACS-isolated to separate dKO and Flox cells. GFP- (Flox) and GFP+ (dKO) cells were then plated in mESC medium for 3 days. Left panels; phase contrast images of dKO and Flox cells on gelatin. Middle and right panels; images of dKO and Flox cells following alkaline phosphatase staining on gelatin and mouse embryo fibroblast feeders, respectively. Scale bar, 100μm. (D) Quantitative analysis of alkaline phosphatase staining for wild-type miPSCs, Flox and dKO cells. n>150, for each condition. (E) Left panel; cell cycle profiles of propidium iodide stained Flox and dKO cells obtained by flow cytometric analyses. Right panel; % of Flox and dKO cells in G1-, S- and G2/M-phases of the cell cycle as determined by flow cytometry analysis. (F) Immunostaining demonstrates dKO cells remain proliferative, compared to Flox cells, by BrdU incorporation after labeling for 24 hrs. Scale bar, 100μm. (See also Figure S1).
As Myc is widely regarded as a critical regulator of the cell cycle and cellular proliferation (Meyer and Penn, 2008), we examined the cell cycle profile and proliferative capacity of dKO cells. Not surprisingly, dKO cells showed a lengthening of G1 and G2/M and a decrease in the percentage of S-phase cells (Figure 1E). Such cell cycle remodeling is typical as mESCs undergo differentiation (Stead et al., 2002; White et al., 2005). dKO cells also had reduced uptake of BrdU relative to Flox cells following a 2 hr pulse (data not shown), but were >90% labeled following a 24 hr BrdU pulse, indicating that they remain proliferative (Figure 1F). This is consistent with the cell cycle changes seen as pluripotent cells differentiate towards endoderm and mesoderm lineages following LIF withdrawal (Stead et al., 2002). c- and N-MYC deletion led to only small changes in apoptosis as determined by Tunel staining (data not shown).
We next set out to determine if there was lineage specific differentiation upon c- and N-MYC deletion. Quantitative reverse transcriptase PCR (qRT-PCR) and immunostaining were performed on Flox and dKO cells cultured in mESC medium (Figure 2A and 2B). Significant increases in transcript levels for primitive endoderm markers were observed (Gata6 and FoxA2), but not for the early mesoderm marker Brachyury and the primitive ectoderm marker Fgf5. Similarly, mESC-derived dKO cells also displayed increases in endoderm marker transcripts (Figure S2C). Immunostaining revealed that the majority of dKO cells were positive for Gata4 and FoxA2 with a decrease in expression in pluripotent stem cell markers Nanog and SSEA1. These results indicate that when cultured under normal maintenance conditions, dKO cells undergo differentiation to primitive endoderm.
Figure 2. Deletion of c- and N-myc in iPSCs results in differentiation to primitive endoderm.
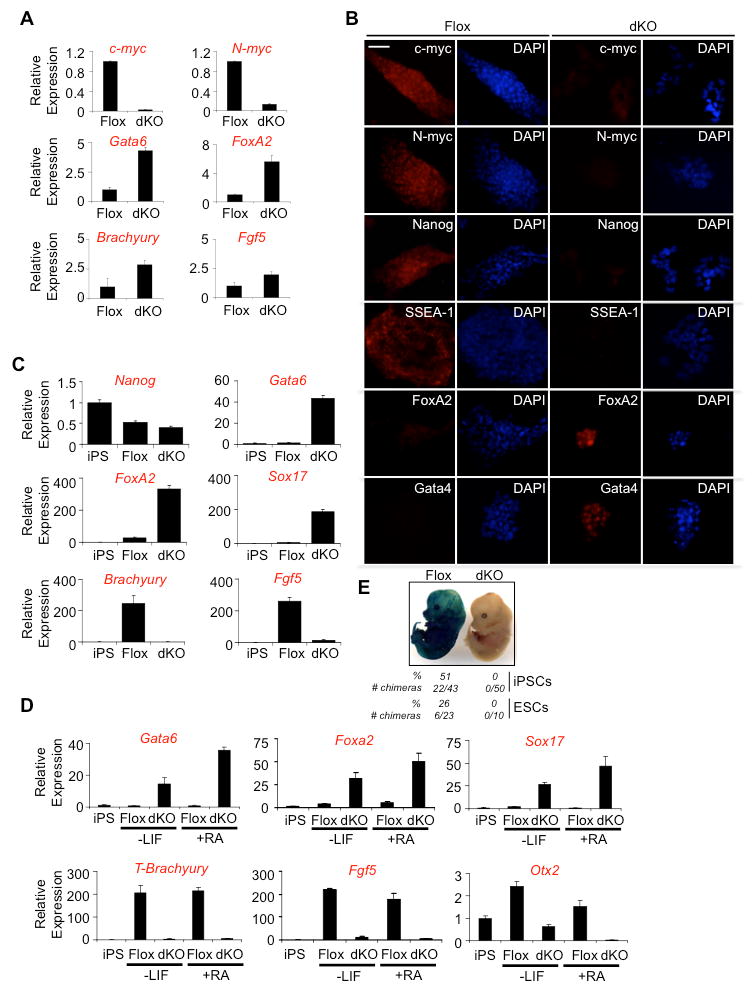
(A) qRT-PCR of c-myc, N-myc, endoderm (Gata6 and Foxa2), mesoderm (Brachyury), and primitive ectoderm (Fgf5) markers indicate endoderm differentiation in dKO cells compared to Flox cells. Cells were cultured in the presence of LIF and experiments were performed in triplicate, normalized to GAPDH and represented as mean ± s.d. (B) Immunostaining for c-myc, N-myc, pluripotency markers, Nanog and SSEA-1 and endoderm markers, FoxA2 and Gata4, reveals the spontaneous differentiation to endoderm following loss of Myc in miPSCs cultured in LIF. Scale bar, 100 μm. (C) qRT-PCR examining Nanog, Gata6, Foxa2, Sox17, Brachyury, and Fgf5 transcripts in miPSCs cultured in LIF (iPS), and during embryoid body differentiation (Flox, dKO) indicates that the loss of Myc predisposes miPSCs to primitive endoderm differentiation. Experiments were performed in triplicate, normalized to GAPDH and represented as mean ± s.d. (D) qRT-PCR analysis of endoderm markers, Gata6, Foxa2, and Sox17; mesendoderm marker, Brachyury; primitive ectoderm marker, Fgf5; and ectoderm marker Otx2, 4 days after LIF removal with and without retinoic acid. iPS represents control miPSCs cultured in the presence of LIF. Triplicate experiments were performed, normalized to GAPDH and represented as mean ± s.d. (E) Flox and dKO miPSCs and mESCs expressing β-galactosidase were injected into blastocyst stage C57BL/6 embryos, transferred into recipient females and allowed to develop until E14.5. LacZ staining was then performed on fixed, whole embryos. The number of blastocysts injected, the number of chimeras generated and the % of chimeras generated are indicated. (See also Figure S2).
To further explore the differentiation potential of dKO cells, in vitro differentiation was performed in suspension culture following FACS in the absence of LIF (Figure 2C). Additionally, differentiation was carried out in adherent culture in the absence of LIF, and in the presence or absence of retinoic acid (Figure 2D). In both cases, marker transcript levels were compared to miPSCs cultured in mESC medium. Regardless of whether dKO cells were cultured in the absence of LIF only, or in the presence of retinoic acid, they consistently up-regulated the primitive endoderm markers Gata6, FoxA2 and Sox17 (Figures 2C and 2D). No up-regulation of the mesoderm marker Brachyury, primitive ectoderm marker Fgf5, or neuroectoderm marker Otx2 was observed under these conditions (Figures 2C and 2D). Flox cells however, did up-regulate these other lineage markers under these conditions, indicating that dKO cells have a more restricted differentiation potential relative to Flox cells. Injection of GFP- (Flox) miPSCs and mESCs into blastocysts resulted in incorporation and broad contribution to embryos, while GFP+ (dKO) cells failed to generate chimeras (Figure 2E). dKO cells therefore fail to retain pluripotency and are predisposed to become primitive endoderm.
Either c- or N-myc is Sufficient to Maintain Pluripotency
Previously, c-MYC null mESCs were found to retain the potential for self-renewal with slight changes in differentiation capacity (Baudino et al., 2002). Since N-myc can functionally compensate for loss of c-myc in early embryonic development (Malynn et al., 2000), this suggested a potential overlapping role with c-myc in pluripotency. To evaluate the ability of different Myc family members to promote pluripotency, c-, N-, or L-mycER were introduced into dKO cells and assayed for their ability to generate alkaline phosphatase positive colonies in the presence or absence of 4-hydroxytamoxifen (4OHT; Figure 3A). c-mycER and N-mycER, but not L-mycER, maintained the pluripotent state as determined by alkaline phosphatase staining (Figures 3B and S3). Both c- and N-mycER were able to maintain dKO cells in an undifferentiated state in the presence of 4OHT for multiple passages (data not shown).
Figure 3. Conditional activation of c- or N-myc, but not L-myc, is sufficient to maintain pluripotency in dKO cells.
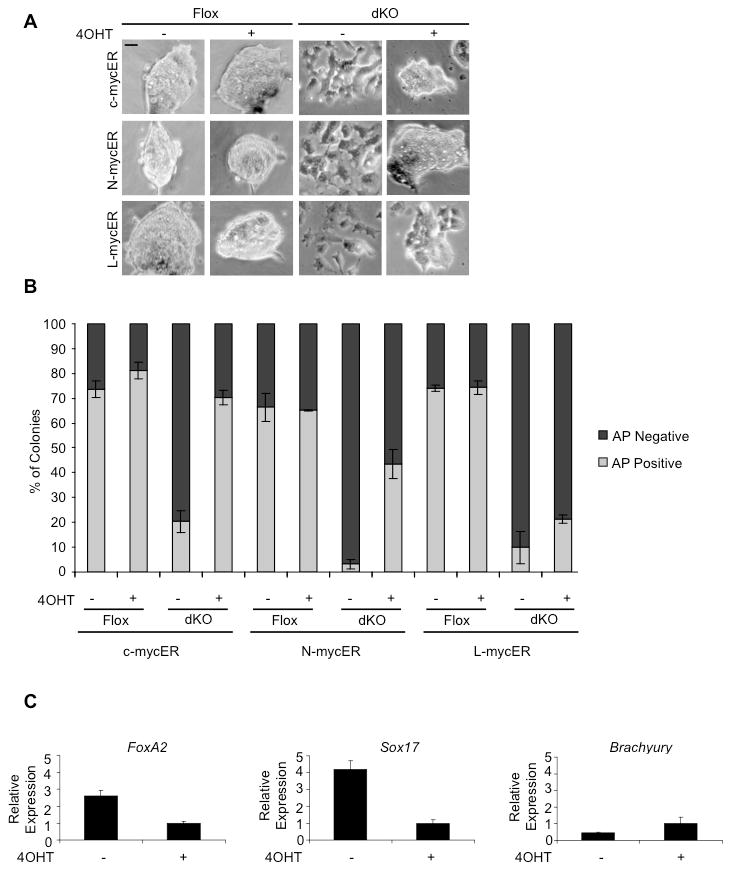
(A) Flox and dKO cell morphology after transfection with c-mycER, N-mycER or L-mycER in the presence or absence of 4OHT for 3 days. Scale bar, 100 μm. (B) Quantitation of alkaline phosphatase staining (Figure S3) showing the % positive colonies versus the % negative colonies. n>300 for each condition. Error bars represent mean ± s.d. from triplicate experiments. (C) Activation of 4OHT-inducible N-mycER inhibits the activation of FoxA2 and Sox17 transcript as determined by qRT-PCR. Experiments were performed in triplicate, normalized to GAPDH and represented as mean ± s.d. The data are representative of multiple experiments where either c- or N-mycER expressing cell lines were used. (See also Figure S3).
To further confirm that Myc blocks primitive endoderm differentiation, CreGFP transfected dKO cells (GFP+) were plated for 3 days in mESC media, +/-4OHT. qRT-PCR analysis showed that activation of N-mycER strongly suppressed the up-regulation of endoderm markers FoxA2 and Sox17 (Figure 3C). These data demonstrate that either c- or N-myc, but not L-myc, is sufficient to maintain pluripotency and explain how single knockout c- or N-myc mESCs are able to self-renew in a comparable manner to wild-type cells (see Baudino et al., 2002; Charron et al., 1990).
Myc targets the mir-17-92 cluster which impacts on the pluripotent cell cycle
Our data show that following deletion of c- and N-MYC, pluripotent cells remodel their cell cycle and differentiate into primitive endoderm. To understand mechanisms of Myc-mediated cell cycle regulation and endoderm repression, we set out to identify in vivo targets for c-myc by ChIP-Chip analysis. Previous studies (Kidder et al., 2008; Kim et al., 2008) identified in vivo Myc targets in mESCs but these did not provide any insight into our observations relating to loss of pluripotency in dKO cells. Since the potential existed that critical in vivo targets may have been missed by previous studies we generated new tools that could expose new Myc target genes. Our approach was to generate c-mycΔ/Δ mESC lines expressing epitope-tagged versions of c-myc (c-mycΔ/6×9e10 or c-mycΔ/3×HA). Myc tagged with multiple epitopes, combined with use of monoclonal antibodies, would then allow for the identification of targets in ChIP assays. Cell lines were selected that expressed levels of epitope-tagged Myc that were at, or below, wild-type levels (Figure S4A). Many previously identified Myc target genes such as SET, EZH2 and Mybbp1a were identified by this approach (Table S1 and Figure S4B).
Similar to the situation in cancer cells (O'Donnell et al., 2005), the mir-17-92 cluster was also bound by c-myc in mESCs (Figure 4A and Table S1). The mir-17-92 cluster was next confirmed as a target by ChIP followed by qPCR (Figure 4B). Since the six miRNAs within this cluster (mir-17, mir-18a, mir-19a, mir-20a, mir-19b-1 and mir-92a-1) are co-regulated through the activity of a common promoter (O'Donnell et al., 2005), responses of the mir-17-92 cluster to changes in Myc activity were evaluated by assaying one of the transcripts- mir-20a. Deletion of c- and N-MYC in miPSCs resulted in a >5-fold decrease in mir-20a transcript levels (Figure 4C). Furthermore, when c-mycER was activated by addition of 4OHT in mESCs, there was a significant increase in mir-20a transcripts above existing levels (Figure 4D). These data confirm that the mir-17-92 cluster is directly regulated by c-myc in pluripotent cells.
Figure 4. c-myc regulates the miR-17-92 cluster to control the cell cycle.
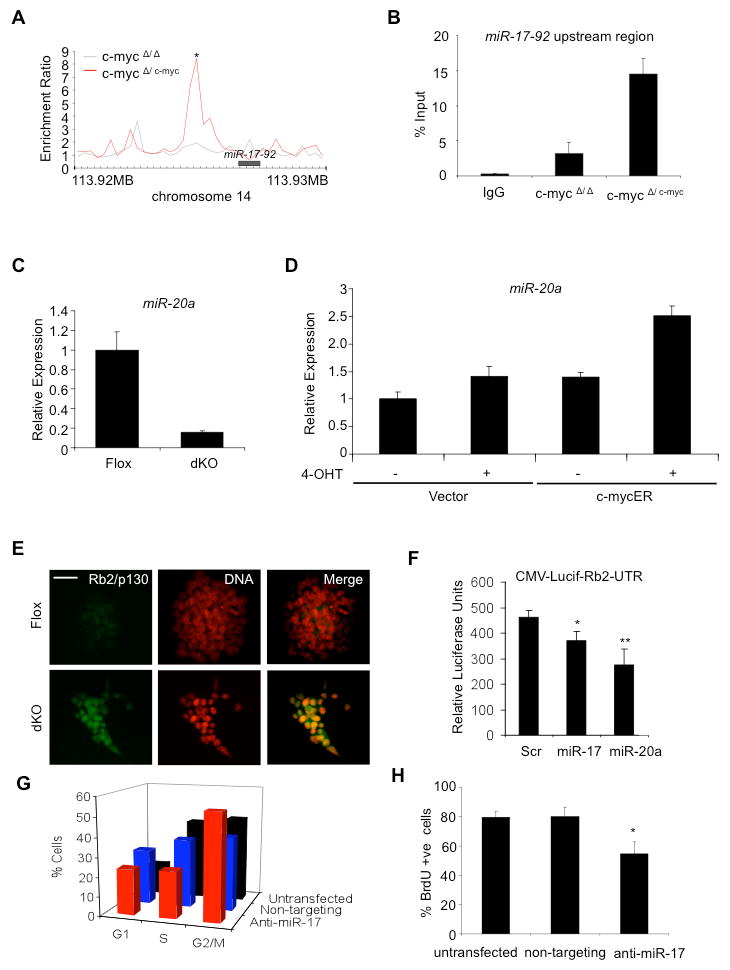
(A) c-myc6×9e10 binds to the upstream regulatory region of the miR-17-92 cluster in ChIP-Chip assays; *p<0.001. (B) Independent validation of ChIP-Chip analysis with ChIP-qPCR using the AB2.1 c-mycΔ/6×9e10 cell line. In control samples, ChIP-qPCR was carried out using chromatin immunoprecipitated by the 9e10 antibody with the c-mycΔ/Δ cell line and control IgG with the c-mycΔ/6×9e10 cell line. (C) miR-20a transcript is down-regulated upon deletion of c- and N-MYC in miPSCs. (D) Activation of c-mycER with 4OHT in mESCs increases miR-20a transcript over basal levels. Experiments were performed in triplicate, normalized to GAPDH and represented as mean ± s.d. (E) Increased expression of the mir-17-92 target, Rb2/p130 upon deletion of Myc in dKO cells. Flox and dKO cells were immunostained with an antibody for Rb2/p130. DNA, DAPI staining. Scale bar, 100 μm. (F) Myc targets miR-17 and miR-20a regulate Rb2. miPSCs were transfected with a firefly luciferase reporter containing a Rb2-UTR under a CMV promoter and either a scrambled control, miR-17 precursor, or a miR-20a precursor. Luciferase assays were performed in triplicate and normalized to a Renilla luciferase control. Data are representative of multiple experiments, *p<0.05; **p<0.01. (G) Cell cycle profiles (flow cytometry) comparing untransfected wild-type miPSCs (black bars), cells transfected with a non-targeting control (blue bars) or, cells transfected with an antisense oligonucleotide against miR-17 (red bars). % of cells in G1-, S-, and G2/M-phases are shown 48 hours after transfection. Similar results were obtained in mESCs (data not shown). (H) Immunostaining monitoring the incorporation of BrdU after labeling for 24 hrs demonstrates reduced cellular proliferation of cells upon transfection of antisense oligonucleotide inhibitors against miR-17. Assay was performed in triplicate and is represented as mean ± s.d., *p<0.05. (See also Figure S4).
The mir-17-92 miRNA cluster has previously been shown to control the cell cycle and facilitate cancer cell proliferation (Mendell, 2008; Yu et al., 2008) by directly blocking the expression of various cell cycle control genes such as E2F1 (O'Donnell et al., 2005), cyclin D1 (Yu et al., 2008), p21 (Fontana et al., 2008), and Rb2/p130 (Wang et al., 2008). These mir-17-92 targets are not expressed in pluripotent cells but are up-regulated during differentiation, coinciding with changes in cell cycle regulation (White et al., 2005). As anticipated, we found that upon deletion of c- and N-MYC in miPSCs there was a significant increase in the expression of Rb2/p130 (Figure 4E). Next, the ability of mir-17-92 cluster members to regulate the 3′ untranslated region of Rb2 was tested using a luciferase reporter assay. Transfection of miRNA precursors for either mir-17 or mir-20a significantly reduced Rb2-luciferase reporter activity (Figure 4F). The magnitude of Rb2 luciferase activity suppression is comparable to that reported previously (Wang et al., 2008; see Discussion). miRNAs in the mir-17-92 cluster therefore regulate cell cycle regulators such as Rb2 in pluripotent cells.
To obtain further evidence that Myc-regulated miRNAs in the mir-17-92 cluster have functional relevance to the cell cycle in pluripotent cells, miPSCs were transfected with a specific oligonucleotide inhibitor targeting mir-17, another member of the cluster. Inhibition of miR-17 in miPSCs decreased the percentage of S-phase cells (41% to 23%) and increased the percentage of G1 cells (16% to 24%) and G2/M (44% to 55%; Figure 4G). Only small changes in cell cycle distribution were observed following transfection with a non-targeting siRNA control. Similar results were obtained in parallel experiments with mESCs (data not shown). Incorporation of BrdU in miPSCs was also reduced following a 24 h pulse with introduction of antisense oligonucleotide inhibitors (Figure 4H), indicative of a cell cycle arrest or, a reduction in the rate of cell division. These effects are consistent with the known ability of Myc to regulate the miR-17-92 cluster in tumor cells (Aguda et al., 2009).
Taken together, these data indicate that Myc expression in pluripotent cells promotes the expression of the mir-17-92 cluster, which serves to block the expression of cell cycle regulators, such as Rb2/p130. Inhibiting the activity of miRNAs within this cluster has a significant effect on cell cycle structure, most notably a dramatic reduction in the % of S-phase cells. We therefore establish a functional link between Myc, the miR-17-92 cluster and cell cycle control in pluripotent cells (see Discussion).
Myc Inhibits Primitive Endoderm Specification by Repressing GATA6 Expression
Upon further examination of Myc-bound targets identified by ChIP-Chip in mESCs, GATA6 was also identified as a potential target (Figure 5A). As described above, Gata6 transcript was significantly up-regulated in dKO cells (Figure 2A), consistent with the possibility that Myc regulates GATA6 at the transcriptional level. Previous studies have in fact shown that GATA6 over-expression is sufficient to drive primitive endoderm differentiation of mESCs (Fujikura et al., 2002). Together, these observations suggest that Myc can inhibit primitive endoderm differentiation through regulation of GATA6. To confirm GATA6 as a Myc target, we performed scanning ChIP-qPCR analysis over the region of GATA6 corresponding to that performed in the ChIP-Chip analysis (Figure 5B). Scanning ChIP analysis confirmed that Myc binds to the GATA6 gene at several sites, including those in the 5′ upstream region and within intron 1 (corresponding to peaks 2, 5, 7 and 8).
Figure 5. c-myc binds and represses the primitive endoderm master regulator, GATA6 and suppresses primitive endoderm differentiation.
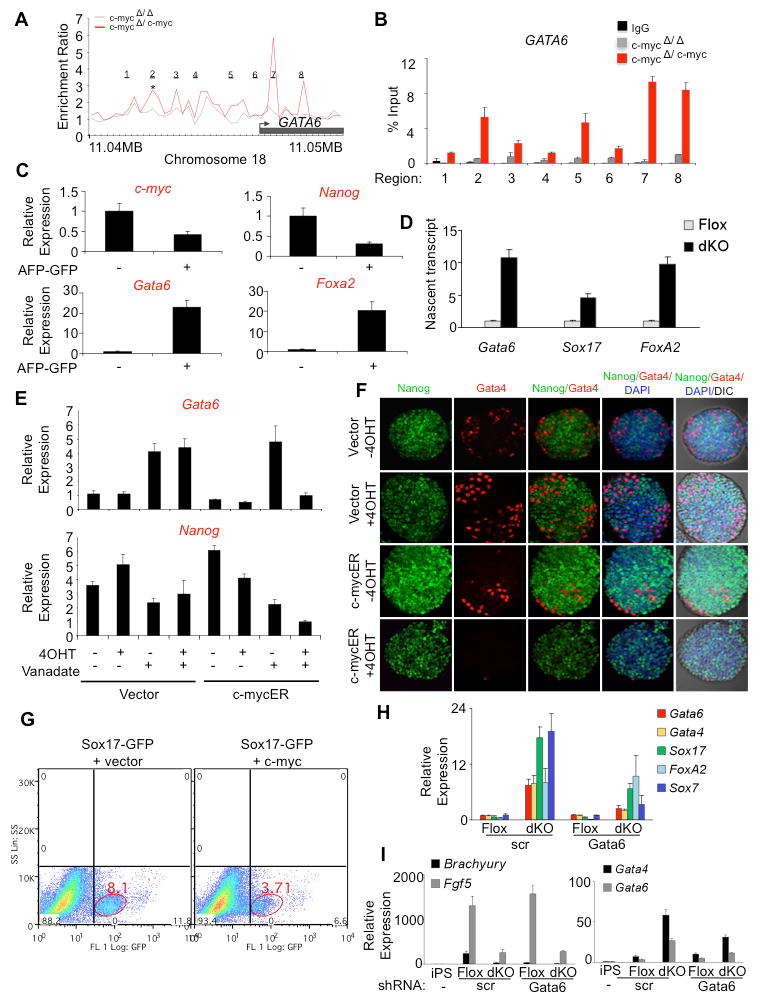
(A) GATA6 is a Myc-bound target identified from ChIP-Chip assays; *p<0.001. (B) Independent validation of GATA6 as a Myc-bound target by ChIP-qPCR using 8 primer sets to scan the GATA6 region corresponding to the ChIP-Chip analysis shown in (A). Primer set 2 corresponds to the statistically significant region identified by DNA Analytics software (Agilent) in the GATA6 promoter shown in (A) to bind c-myc. Primer sets 5, 7 and 8 also represent regions of significant enrichment and primer sets 1, 3, 4 and 6 correspond to regions not bound by c-myc. (C) c-myc transcript is down-regulated in primitive endoderm. AFP-GFP mESCs were aggregated in the presence of LIF then cultured for 3 days. GFP positive and negative cells were isolated by FACS and analyzed by qRT-PCR. (D) Transcription of GATA6, SOX17 and FOXA2 increases in dKO cells. Flox (GFP-) or dKO (GFP+) cells were isolated by FACS, cultured for 3 days, and nuclear run-on assays performed by labeling nascent nuclear transcripts with biotin-16-UTP. After the isolation of biotinylated transcripts on streptavidin beads, qRT-PCR analysis was performed in triplicate. Gata6, Sox17 and FoxA2 values were normalized to GAPDH and represented as mean ± s.d. (E) Induction of Gata6 transcript by sodium orthovanadate is blocked by activation of c-mycER. mESCs were aggregated in the presence of orthovanadate for 24 hr, in the presence or absence of 4OHT. qRT-PCR analysis was performed in triplicate and values normalized to GAPDH and represented as mean ± s.d. Data are representative of multiple experiments. (F) mESCs carrying a c-mycER transgene or vector alone, were aggregated for 3 days in the presence of LIF, in the presence or absence of 4OHT. Embryoid bodies were probed with antibodies for Nanog or the endoderm marker Gata4 and stained with DAPI. Embryoid bodies were analyzed by confocal microscopy and DIC optics. (G) Sox17-GFP mESCs were transfected with vector alone or with a c-myc expression construct. Cells were then aggregated in the absence of LIF for 3 days. Flow cytometry was used to determine the effect of endoderm differentiation by evaluating the % of GFP+ cells. (H) GATA6 is required for endoderm formation following loss of c- and N-Myc. c-MYCfl/fl;N-MYCfl/fl miPSCs were transfected with CreGFP and Gata6 shRNA or scrambled (scr) shRNA construct. GFP positive and negative cells were FACS sorted, plated and analyzed after 3 days by qRT-PCR. Experiments were performed in triplicate, normalized to GAPDH and represented as mean ± s.d. (I) Myc-deleted and Gata6 knockdown cells have restricted differentiation potential. c-MYCfl/fl;N-MYCfl/fl miPSCs were transfected with CreGFP and Gata6 shRNA or scrambled (scr) shRNA construct. Control miPSCs (iPS) were cultured in LIF, and GFP positive (dKO) and negative cells (Flox) were FACS sorted, aggregated in the absence of LIF to induce differentiation and analyzed after 4 days by qRT-PCR. Experiments were performed in triplicate, normalized to GAPDH and represented as mean ± s.d. (See also Figure S5).
To further evaluate the relationship between GATA6 and c-myc during differentiation of pluripotent stem cells to primitive endoderm, we utilized mESCs that express GFP under the control of the alpha-fetoprotein (AFP) promoter (Hamazaki et al., 2004). Upon aggregation of these cells in mESC medium (+LIF), primitive endoderm forms as the outer layer while the inner cells retain pluripotent properties. After aggregation for three days, cells were subjected to FACS followed by qRT-PCR to evaluate marker transcripts (Figure 5C). As expected, Gata6 and FoxA2 mRNAs were elevated in the GFP+ endoderm population, while Nanog and c-myc transcripts were elevated in the GFP- population. These data confirm that c-myc is down-regulated as mESCs transition to primitive endoderm. To directly confirm that GATA6 transcription is up-regulated as dKO cells differentiate into primitive endoderm, nuclear run-on assays were performed. qRT-PCR analysis of RNA isolated from labeled nuclei showed that rates of GATA6 transcription increase ∼11-fold in dKO cells, relative to Flox cells (Figure 5D). Levels of FoxA2 and Sox17 nascent transcripts also increased in dKO cells, consistent with the activation of an endoderm transcriptional program following the activation of GATA6. These results indicate that Myc represses endoderm formation by blocking GATA6 transcription.
Previous studies have shown that sodium orthovanadate activates Grb2/Mek and induces Nanog down-regulation and GATA6 up-regulation, leading to primitive endoderm differentiation (Hamazaki et al., 2006). To determine if c-myc can block the up-regulation of GATA6 induced by sodium orthovanadate, c-mycER mESCs or control mESCs (vector alone) were aggregated in suspension (+LIF), in the presence or absence of sodium orthovanadate, with or without 4OHT for 24 hr (Figure 5E). qRT-PCR analysis shows that activation of c-mycER was able to block Gata6 up-regulation induced by sodium orthovanadate. Nanog mRNA was still down-regulated when c-mycER was activated in the presence of sodium orthovanadate. These data indicate that c-myc is able to inhibit GATA6 expression independently of Nanog.
To investigate the effects of Myc expression on primitive endoderm formation we activated c-mycER during embryoid body differentiation (+LIF, 3d) and examined expression of the Gata6 target, GATA4 in the primitive endoderm outer layer, which accounts for ∼3-5% of cells in the embryoid body (Hamazaki et al., 2004; Figure 5F). Immunostaining followed by confocal microscopy revealed that activated c-myc (+4OHT) almost completely eliminated the expression of Gata4 in cells on the surface of embryoid bodies. We then evaluated the effects of Myc on endoderm formation in a Sox17-GFP reporter mESC line (Borowiak et al., 2009). Sox17-GFP cells were transfected with a c-myc expression construct or vector alone, aggregated for 24 hr and then analyzed by flow cytometry (Figure 5G). Under these conditions over-expression of c-myc reduced the number of GFP+ endoderm cells by over 50%. Altogether, these data demonstrate that c-myc inhibits differentiation to primitive endoderm.
To establish if Myc prevents primitive endoderm formation by repressing GATA6, we knocked down Gata6 transcript using shRNA (Izumi et al., 2007) in dKO miPSCs. qRT-PCR analysis indicates that knockdown of Gata6 mRNA blocks the up-regulation of the endoderm markers Gata4, Sox17 and Sox7 (Figure 5H). FoxA2 was not affected and so may not be directly under the control of GATA6. These data show that c-myc blocks primitive endoderm formation by repressing the expression of GATA6.
Even though knockdown of Gata6 in dKO cells blocks normal activation of endoderm genes, pluripotency markers such as alkaline phosphatase, Nanog and SSEA1 are not maintained suggesting that differentiation into other lineages may occur under these conditions (Figure S5). However, marker analysis for mesendoderm/early mesoderm, definitive endoderm, ectoderm and trophoblast lineages following LIF withdrawal does not support this possibility (Figure 5I, S5 and data not shown). Although dKO-Gata6 knockdown cells are no longer pluripotent, they fail to differentiate into any definable lineage and appear to have lost developmental potential.
Discussion
We have characterized two mechanisms by which Myc contributes to self-renewal and maintenance of the pluripotent state. The first function involves maintenance of a characteristic mode of cell cycle control whereby Myc regulates the mir-17-92 miRNA cluster. The second and perhaps most important function defined by these studies involves repression of the master endoderm regulator gene, GATA6 (Morrisey et al., 1998; Koutsourakis et al., 1999).
The cell cycle profiles of pluripotent stem cells have a characteristic short G1, with a large percentage of cells in S-phase (Stead et al., 2002; Savatier et al., 1996). These cell cycle profiles are attributed to hyper-phosphorylation of Rb and a lack of regulated E2F-dependent transcription. Upon differentiation, E2F and Rb family members serve to impose the Restriction-point near the G1/S transition leading to the remodeling of the cell cycle (Savatier et al., 1996; Stead et al., 2002; White et al., 2005). The change in cell cycle structure observed upon loss of Myc - lengthening of G1 and a reduction in the percentage of S-phase cells, indicate the establishment of a checkpoint in late G1. How this dramatic switch in cell cycle regulation occurs has not been previously defined at the molecular level. However, it has been recognized for some time that changes in Myc activity could potentially orchestrate many of the cell cycle changes seen during early mESC differentiation (Cartwright et al., 2005).
The down-regulation of mir-17-92 and up-regulation of cell cycle regulators, such as Rb2/p130, upon deletion of c- and N-MYC establishes one way by which Myc impacts on the cell cycle in pluripotent cells. Based on previous work, it is likely that the mir-17-92 cluster targets other key cell cycle regulatory molecules such as E2F1, p21 and cyclin D1 (see O'Donnell et al., 2005; Cloonan et al., 2008). These are all thought to play key roles in remodeling the cell cycle as pluripotent cells transition towards differentiating lineages (Stead et al., 2002). Although mir-17-92 has not been implicated in cell cycle control of ESCs or iPSCs previously, other miRNAs are thought to influence self-renewal, and connections between miRNAs and Myc have been established in the context of reprogramming (see Judson et al., 2009). Even though ESCs proliferate rapidly with an unusual cell cycle structure, the significance of this in relation to pluripotency remains unclear. For example, inhibition of cyclin-dependent kinases slows down the cell cycle but does not impact on pluripotency (Stead et al., 2002). Although Myc seems to be critical for maintaining rapid rates of division and the unusual cell cycle structure of pluripotent cells, it remains to be determined if this is directly linked to the maintenance of pluripotency or to other aspects of early embryonic development, such as embryonic growth control.
Many cell cycle control genes have been previously identified as being Myc targets in pluripotent cells but our work represents the first mechanistic characterization of how Myc intersects with the cell cycle machinery. Other genes identified as being Myc targets (see Kidder et al., 2008 and Kim et al., 2008) are likely to be part of an orchestrated transcriptional program that establishes and maintains the pluripotent cell cycle in conjunction with the mir-17-92 cluster. Although Myc has been proposed to regulate miRNAs in the context of cell reprogramming and pluripotency (Judson et al., 2009), mir-17-92 has not been previously implicated in this process. This underpins the need to understand in greater detail the relationship between Myc, miRNAs and the establishment/maintenance of pluripotency.
During reprogramming, Myc operates during the first few days to repress fibroblast-specific genes (Sridharan et al., 2009). We speculate that during the early reprogramming stage, Myc may also serve to initiate remodeling of the cell cycle to one more reminiscent of a pluripotent cell. This is supported by observations that partially reprogrammed cells exhibit a cell cycle profile that is intermediate between fibroblasts and pluripotent cells (Singh and Dalton, 2009) and that cell cycle control is rate-limiting for reprogramming (Edel et al., 2010). This suggests that remodeling of the cell cycle is an early event prior to, and perhaps required for, the establishment of pluripotency. The exact relationship between cell cycle regulation and establishment/maintenance of the pluripotent state still needs to be resolved. Since Myc is implicated in both processes, this is an area that requires further study.
Myc is Critical for Repression of Primitive Endoderm
In conjunction with cell cycle control, we show that Myc is required for maintenance of pluripotency by directly repressing the expression of GATA6, a master regulator of primitive endoderm formation (Morrisey et al., 1998; Koutsourakis et al., 1999). Other factors that have been implicated in Gata6 regulation, such as Nanog and Polycomb repressive complexes (Boyer et al., 2006; Singh et al., 2007), may cooperate with Myc to repress GATA6. Transcriptional control seems to be a key element of this regulation although the possibility that Myc regulates GATA6 expression at additional levels cannot be ruled out. For example, Myc-regulated miRNAs could potentially impact on Gata6 translation and mRNA stability. We are unaware however, of any Myc-regulated miRNAs that could potentially target Gata6.
Following loss of c- and N-myc, iPSCs preferentially differentiate towards primitive endoderm, even under conditions that would normally favor specification of other lineages such as following retinoic acid treatment or LIF withdrawal. Therefore, besides not being able to maintain dKO cells in a pluripotent state, they also lose the potential for differentiation into non-endoderm lineages. The most likely explanation for this can be accounted for by de-repression of GATA6.
dKO cells also lose pluripotency following knockdown of Gata6, but fail to correctly activate the endoderm transcriptional program. These cells however, show no signs of differentiation towards other lineages under these conditions and so appear to lack full developmental potential. This suggests that c- and N- Myc are required for differentiation of the embryonic germ layers and/or that L-myc is sufficient for endoderm differentiation. Surprisingly, Gata6 knockdown also diminished Brachyury expression. As Gata6 is expressed in mesodermal lineages such as cardiac cells, it may play a role in regulating such mesoderm markers as Brachyury. Moreover, primitive endoderm has been implicated in patterning of embryonic germ layer derivatives, and loss of Gata6 and hence primitive endoderm cell types may impact mesoderm formation.
The identification of GATA6 as a Myc target gene was somewhat of a surprise since several other ChIP-Chip studies (Kidder et al., 2008 and Kim et al., 2008) failed to detect this connection. The predisposition of dKO cells to differentiate towards primitive endoderm suggests however, that key elements of the endoderm transcriptional program are targeted by Myc in pluripotent cells. Hence, we focused on GATA6 because of its well-known function as a primitive endoderm master regulator. It is unclear why previous studies may have overlooked the Myc-GATA6 connection but it is clear that ChIP-Chip studies are not exhaustive in their target identification, as demonstrated by differences in target genes identified by these studies (see Kidder et al., 2008; Kim et al., 2008). From a technical standpoint, Myc may weakly or transiently bind GATA6 in comparison to other loci and therefore be easily overlooked. Technically, our studies varied from other studies and used epitope-tagged Myc and corresponding monoclonal antibodies that may have allowed for the identification of target genes, such as GATA6, that would otherwise have been undetected. In addition to GATA6, our ChIP-Chip screen identified HES1 as another target of Myc in pluripotent cells. HES1, like GATA6, plays an important role in primitive endoderm development (Thomas and Beddington, 1996) and its repression by Myc may also sustain pluripotency. Further work is required to establish if Myc also plays a role in establishing differentiation blockades on pathways other than primitive endoderm.
Experimental Procedures
Cell Culture and blastocyst injections
Mouse embryonic fibroblasts isolated from c-MYCfl/fl;N-MYCfl/fl embryos were used in the generation of iPSCs as described elsewhere (de Alboran et al., 2001; Knoepfler et al., 2002; Takahashi and Yamanaka, 2006). Mouse ESCs (wild-type AB2.1, AB2.1c-mycΔ/Δ, Baudino et al., 2002), AFP-GFP (Hamazaki et al., 2004), Sox17-GFP (Borowiak et al., 2009) and miPSCs were cultured in LIF on gelatin-coated dishes. Differentiation was performed in adherent or suspension culture in medium without LIF. Differentiation to endoderm was performed by aggregation of cells in mESC medium, in the presence or absence of sodium orthovanadate (50 μM) and/or 4OHT (100 nM). The miRNA inhibitors, anti-miR-17 and anti-miR-20a (Ambion), miRNA precursors, pre-miR-17 and pre-miR-20a (Ambion), and non-targeting control were transfected into c-MYCfl/fl;N-MYCfl/fl miPSCs and mESCs with Lipofectamine 2000 reagent (Invitrogen). Gata6 shRNA constructs were cotransfected with pCAGCreGFPiNeo using Lipofectamine2000 reagent (Invitrogen) into c-MYCfl/fl;N-MYCfl/fl miPSCs. LacZ+ miPSCs were generated by transfection with a plasmid expressing the LacZ gene from the constitutive CAGi promoter, followed by selection with puromycin. Blastocyst injections and embryo analysis were performed as described previously (Cartwright et al., 2005).
Quantitative RT-PCR, Luciferase Assays and Nuclear Run-on Assays
RNA was isolated using the RNeasy Mini Kit (Qiagen), and miRNAs were isolated using the MirVana miRNA isolation kit (Ambion). qRT-PCR for mRNA and miRNA transcripts was performed using Taqman Assays (Applied Biosystems). Luciferase assays were performed using the Dual Luciferase Reporter Kit (Promega) according to instructions and analyzed on a Synergy 2 plate reader (BioTek). Nuclear run-on assays were performed by labeling RNA from isolated nuclei with biotin-16-UTP, essentially as described by Zhang et al., (2005). Biotinylated RNA was purified with streptavidin magnetic beads then reversed transcribed into cDNA. qRT-PCR transcript analysis was performed as described above.
Alkaline Phosphatase Staining, Cell Cycle Analysis and Immunostaining
Alkaline phosphatase staining was carried out using a Leukocyte Alkaline Phosphatase staining kit (Sigma). Cell cycle analysis was performed by flow cytometry after fixing the cells in 70% ethanol and staining using propidium iodide (50 μg/ml), RNase A (200 μg/ml), BSA (100 μg/ml) in PBS for 30 min at 37°C. Immunostaining was performed by fixing cells in 4% paraformaldehyde, blocking in 10% Donkey Serum/PBS, and incubating with the following antibodies in blocking solution, Nanog (CosmoBio); SSEA1 (Developmental Studies Hybridoma Bank); FoxA2 (Upstate); Gata4, Oct4, p130/Rb2, c-myc (Santa Cruz Biotechnology); N-myc (Chemicon); or BrdU (Abcam) overnight.
Chromatin Immunoprecipitation with Microarray Assays
AB2.1c-mycΔ/Δ (c-myc null) cell lines with stable expression of 3×HA or 6×9e10-epitope-tagged c-myc were used for ChIP-Chip assays (see Boyer et al., 2005). Cell lines were selected that expressed c-myc3×HA/6×9e10 at levels equivalent to, or below, that of endogenous c-myc in the parental cell line AB2.1 to eliminate over-expression effects (Figure S4A). The negative control cell line (AB2.1c-mycΔ/Δ) was generated by transfection with empty pCAGiPuro vector. Immunoprecipitations were carried out with AB2.1c-mycΔ/3×HA, AB2.1c-mycΔ/6×9e10 and AB2.1c-mycΔ/Δ cell lines using affinity-purified anti-HA, anti-9e10 monoclonal antibodies (Sigma) or control IgG. Hybridization was carried out with a Mouse Expanded Promoter ChIP-on-Chip Set (Whitehead Institute, Agilent Technologies), and validated using qPCR with SYBR Green (Bio-Rad). Targets were identified using DNA Analytics software (Agilent). Probes used to identify genomic targets were determined in a replicate if the p-value was below a pre-determined cut-off (at least p<0.05). ChIP-Chip data are expressed as an enrichment ratio of immunoprecipitated target and input DNA, calculated using DNA Analytics. Targets obtained using the AB2.1c-mycΔ/Δ cell line were subtracted from those detected with the AB2.1Δ/6×9e10 or AB2.1Δ/3×HA cell lines and used to formulate a consensus Myc target list (Table S1). Experiments were performed in triplicate for each cell line. ChIP-qPCR data are represented as enrichment of the immunoprecipitated target relative to input DNA (Aparicio et al., 2004).
Highlights.
- c- and N-myc perform redundant functions that are essential for maintenance of pluripotency
- Myc blocks primitive endoderm specification by repressing GATA6 transcription
- Myc controls the pluripotent cell cycle by regulation of the miR17-92 cluster
- Myc controls multiple aspects of pluripotent stem cell maintenance
Supplementary Material
01
02
Acknowledgments
This work was supported by grants to SD from the National Institute of Child Health and Human Development (HD049647) and the National Institute for General Medical Sciences (GM75334). We thank Paul Knoepfler for kindly providing c-mycfl/fl;N-mycfl/fl mice and c-mycfl/fl;N-mycfl/fl mESCs, John Cleveland for AB2.1 mESCs, Malgorzata Borowiak and Douglas Melton for the Sox17-GFP mESCs, Naohiro Terada for AFP-GFP mESCs, Shin-Ichi Nishikawa for Gata6 shRNA constructs, and Xinmin Li for the Rb2-Luciferase Reporter. Thanks to Juan Cui and Ying Xu for assistance with bioinformatics analysis, Julie Nelson for assistance with FACS sorting, David Reynolds and Tamas Nagy for assistance with teratoma analysis, David Martin and Helen Zhang for assistance with blastocyst injections.
Footnotes
Supplemental Data: The Supplemental Data include 5 figures and 1 table.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguda BD, Kim Y, Piper-Hunter MG, Friedman A, Marsh CB. MicroRNA regulation of a cancer network: consequences of the feedback loop involving miR-17-92, E2F, and Myc. Proc Natl Acad Sci USA. 2008;105:19678–19683. doi: 10.1073/pnas.0811166106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio O, Geisberg JV, Struhl K. Chromatin immunoprecipitation for determining the association of proteins with specific genomic sequences in vivo. Current Protocols in Cell Biology. 2004;17:1–23. doi: 10.1002/0471143030.cb1707s23. [DOI] [PubMed] [Google Scholar]
- Baudino TA, McKay C, Pendevile-Samain H, Nilsson JA, Maclean KH, White EL, Davis AC, Ihle JN, Cleveland JL. c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev. 2002;16:2530–43. doi: 10.1101/gad.1024602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borowiak M, Maehr R, Chen S, Chen AE, Tang W, Fox JL, Schreiber SL, Melton DA. Small molecules efficiently direct differentiation of mouse and human embryonic stem cells. Cell Stem Cell. 2009;4:348–358. doi: 10.1016/j.stem.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, Jenner RG, Gifford DK, Melton DA, Jaenisch R, Young RA. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer L, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, Bell GW, Otte AP, Vidal M, Gifford DK, Young RA, Jaenisch R. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- Cartwright P, McLean C, Sheppard A, Rivett D, Jones K, Dalton S. LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development. 2005;132:885–896. doi: 10.1242/dev.01670. [DOI] [PubMed] [Google Scholar]
- Charron J, Malynn BA, Robertson EJ, Goff SP, Alt FW. High-frequency disruption of the N-myc gene in embryonic stem and pre-B cell lines by homologous recombination. Mol Cell Biol. 1990;10:1799–804. doi: 10.1128/mcb.10.4.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, Loh YH, Yeo HC, Yeo ZX, Narang V, Govindarajan KR, Leong B, Shahab A, Ruan Y, Bourque G, Sung WK, Clarke ND, Wei CL, Ng HH. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- Cloonan N, Brown NK, Steptoe AL, Wani S, Chan WL, Forrest AR, Kolle G, Gabrielli B, Grimmond S. The mir-17-5p microRNA is a key regulator of the G1/S phase cell cycle transition. Genome Biol. 2008;9:R127. doi: 10.1186/gb-2008-9-8-r127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Alboran IM, O'Hagen RC, Gartner F, Malynn B, Davidson L, Rickert R, Rajewsky K, DePinho RA, Alt FW. Analysis of C-MYC function in Normal Cells via Conditional Gene-Targeted Mutation. Immunity. 2001;14:45–55. doi: 10.1016/s1074-7613(01)00088-7. [DOI] [PubMed] [Google Scholar]
- Edel MJ, Menchon C, Menendez S, Consiglio A, Raya A, Izpisua Belmonte JC. Rem2 GTPase maintains survival of human embryonic stem cells as well as enhancing reprogramming by regulating p53 and cyclin D1. Genes Dev. 2010;24:561–573. doi: 10.1101/gad.1876710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana L, Fiori ME, Albini S, Cifaldi L, Giovanni S, Forloni M, Boldrini R, Donfrancesco A, Federici V, Giacomini P, Peschle C, Fruci D. Antagomir-17-5p abolishes the growth of therapy-resistant neuroblastoma through p21 and BIM. PLoS ONE. 2008;21:e2236. doi: 10.1371/journal.pone.0002236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank SR, Schroeder M, Fernandez P, Taubert S, Amati B. Binding of c-Myc to chromatin mediates mitogen-induced acetylation of histone H4 and gene activation. Genes Dev. 2001;15:2069–2082. doi: 10.1101/gad.906601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikura J, Yamato E, Yonemura S, Hosoda K, Masui S, Nakao K, Miyazaki JJ, Niwa H. Differentiation of embryonic stem cells is induced by GATA factors. Genes Dev. 2002;16:784–789. doi: 10.1101/gad.968802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna J, Markoulaki S, Mitalipova M, Cheng AW, Cassady JP, Staerk J, Carey BW, Lengner CJ, Foremen R, Love J, Gao Q, Kim J, Jaenisch R. Metastable pluripotent states in NOD-mouse-derived ESCs. Cell Stem Cell. 2009;4:513–524. doi: 10.1016/j.stem.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamazaki T, Kehoe SM, Nakano T, Terada N. The Grb2/Mek pathway represses Nanog in murine embryonic stem cells. Mol Cell Biol. 2006;26:7539–7549. doi: 10.1128/MCB.00508-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamazaki T, Oka M, Yamanaka S, Terada N. Aggregation of embryonic stem cells induces Nanog repression and primitive endoderm differentiation. J Cell Sci. 2004;117:5681–5686. doi: 10.1242/jcs.01489. [DOI] [PubMed] [Google Scholar]
- Ivanovska I, Ball AS, Diaz RL, Magnus JF, Kibukawa M, Schelter JM, Kobayashi SV, Lim L, Burchard J, Jackson AL, Linsley PS, Cleary MA. MicroRNAs in the miR-106b family regulate p21/CDKN1A and promote cell cycle progression. Mol Cell Biol. 2008;28:2167–2174. doi: 10.1128/MCB.01977-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi N, Era T, Akimaru H, Yasunaga M, Nishikawa S. Dissecting the molecular hierarchy for mesendoderm differentiation through a combination of embryonic stem cell culture and RNA interference. Stem Cells. 2007;25:1664–1674. doi: 10.1634/stemcells.2006-0681. [DOI] [PubMed] [Google Scholar]
- Judson RL, Babiarz JE, Venere M, Blelloch R. Embryonic stem cell-specific microRNAs promote induced pluripotency. Nat Biotechnol. 2009;27:459–461. doi: 10.1038/nbt.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidder B, Yang J, Palmer S. Stat3 and c-Myc genome-wide promoter occupancy in embryonic stem cells. PLoS ONE. 2008;3:e3932. doi: 10.1371/journal.pone.0003932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Chu J, Shen X, Wang J, Orkin SH. An extended transcriptional network for pluripotency of embryonic stem cells. Cell. 2008;132:1049–1061. doi: 10.1016/j.cell.2008.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoepfler PS, Cheng PF, Eisenman RN. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev. 2002;16:2699–2712. doi: 10.1101/gad.1021202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsourakis M, Langeveld A, Patient R, Beddington R, Grosveld F. The transcription factor GATA6 is essential for early extraembryonic development. Development. 1999;126:723–732. [PubMed] [Google Scholar]
- Malynn BA, de Alboran IM, O'Hagen RC, Bronson R, Davidson L, DePinho RA, Alt FW. N-myc can functionally replace c-myc in murine development, cellular growth, and differentiation. Genes Dev. 2000;14:1390–1399. [PMC free article] [PubMed] [Google Scholar]
- Mendell JT. miRiad roles for the miR-17-92 cluster in development and disease. Cell. 2008;133:217–222. doi: 10.1016/j.cell.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer. 2008;8:976–990. doi: 10.1038/nrc2231. [DOI] [PubMed] [Google Scholar]
- Morrisey EE, Tang Z, Sigrist K, Lu MM, Jiang F, Ip HS, Parmacek MS. GATA6 regulates HNF4 and is required for differentiation of visceral endoderm in the mouse embryo. Genes Dev. 1998;12:3579–3590. doi: 10.1101/gad.12.22.3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa M, Koyanagi M, Tanabe K, Takahashi K, Ichisaka T, Aoki T, Okita K, Mocjiduki Y, Takizawa N, Yamanaka S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol. 2008;26:101–106. doi: 10.1038/nbt1374. [DOI] [PubMed] [Google Scholar]
- O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;9:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- Savatier P, Lapillonne H, van Grunsven LA, Rudkin BB, Samarut J. Withdrawal of differentiation inhibitory activity/leukemia inhibitory factor up-regulates D-type cyclins and cyclin-dependent kinase inhibitors in mouse embryonic stem cells. Oncogene. 1996;12:309–322. [PubMed] [Google Scholar]
- Singh AM, Hamazaki T, Hankowski KE, Terada N. A heterogeneous expression pattern for Nanog in embryonic stem cells. Stem Cells. 2007;25:2534–2542. doi: 10.1634/stemcells.2007-0126. [DOI] [PubMed] [Google Scholar]
- Singh A, Dalton S. The cell cycle and Myc intersect with mechanisms that regulate pluripotency and reprogramming. Cell Stem Cell. 2009;5:141–149. doi: 10.1016/j.stem.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridharan R, Tchieu J, Mason MJ, Yachechko R, Kuoy E, Horvath S, Zhou S, Zhou Q, Plath K. Role of the murine reprogramming factors in the induction of pluripotency. Cell. 2009;136:364–377. doi: 10.1016/j.cell.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stead E, White J, Faast R, Conn S, Goldstone S, Rathjen J, Dhingra U, Rathjen P, Walker D, Dalton S. Pluripotent cell division cycles are driven by ectopic Cdk2, cyclin A/E and E2F activities. Oncogene. 2002;21:8320–8333. doi: 10.1038/sj.onc.1206015. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Thomas P, Beddington R. Anterior primitive endoderm may be responsible for patterning the anterior neural plate in the mouse embryo. Curr Biol. 1996;6:1487–1496. doi: 10.1016/s0960-9822(96)00753-1. [DOI] [PubMed] [Google Scholar]
- Wang Q, Li YC, Wang J, Kong J, Qi Y, Quigg RJ, Li X. miR-17-92 cluster accelerates adipocyte differentiation by negatively regulating tumor-suppressor Rb2/p130. Proc Natl Acad Sci U S A. 2008;105:2889–2894. doi: 10.1073/pnas.0800178105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White J, et al. Developmental activation of the Rb-E2F pathway and establishment of cell cycle-regulated cyclin-dependent kinase activity during embryonic stem cell differentiation. Mol Biol Cell. 2005;16:2018–2027. doi: 10.1091/mbc.E04-12-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Wang C, Wang M, Li Z, Casimiro MC, Liu M, Wu K, Whittle J, Ju X, Hyslop T, McCue P, Pestell RG. A cyclin D1/microRNA 17/20 regulatory feedback loop in control of breast cancer cell proliferation. J Cell Biol. 2008;182:509–517. doi: 10.1083/jcb.200801079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang MX, Ou H, Shen YH, Wang J, Coselli J, Wang XL. Regulation of endothelial nitric oxide synthase by small RNA. Proc Natl Acad Sci USA. 2005;102:16967–16972. doi: 10.1073/pnas.0503853102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01
02