Pharmacokinetics, oral bioavailability, and metabolic profile of resveratrol and its dimethylether analog, pterostilbene, in rats (original) (raw)
. Author manuscript; available in PMC: 2012 Sep 1.
Published in final edited form as: Cancer Chemother Pharmacol. 2010 Nov 30;68(3):593–601. doi: 10.1007/s00280-010-1525-4
Abstract
Purpose
Resveratrol (3,5,4′-trihydroxy-_trans_-stilbene) is a naturally occurring polyphenol with a broad range of possible health benefits, including anti-cancer activity. However, the biological activity of resveratrol may be limited by poor absorption and first-pass metabolism: only low plasma concentrations of resveratrol are seen following oral administration, and metabolism to glucuronide and sulfate conjugates is rapid. Methylated polyphenol analogs (such as pterostilbene [3,5-dimethoxy-4′-hydroxy-_trans_-stilbene], the dimethylether analog of resveratrol) may overcome these limitations to pharmacologic efficacy. The present study was designed to compare the bioavailability, pharmacokinetics, and metabolism of resveratrol and pterostilbene following equimolar oral dosing in rats.
Methods
The agents were administered orally via gavage for 14 consecutive days at 50 or 150 mg/kg/day for resveratrol and 56 or 168 mg/kg/day for pterostilbene. Two additional groups were dosed once intravenously with 10 and 11.2 mg/kg for resveratrol and pterostilbene, respectively. Plasma concentrations of agents and metabolites were measured using a high-pressure liquid chromatograph-tandem mass spectrometer system. Noncompartmental analysis was used to derive pharmacokinetic parameters.
Results
Resveratrol and pterostilbene were approximately 20 and 80% bioavailable, respectively. Following oral dosing, plasma levels of pterostilbene and pterostilbene sulfate were markedly greater than were plasma levels of resveratrol and resveratrol sulfate. Although plasma levels of resveratrol glucuronide exceeded those of pterostilbene glucuronide, those differences were smaller than those of the parent drugs and sulfate metabolites.
Conclusions
When administered orally, pterostilbene demonstrates greater bioavailability and total plasma levels of both the parent compound and metabolites than does resveratrol. These differences in agent pharmacokinetics suggest that the in vivo biological activity of equimolar doses of pterostilbene may be greater than that of resveratrol.
Keywords: Resveratrol, Pterostilbene, Pharmacokinetics, Bioavailability, Metabolites, Rat
Introduction
Resveratrol (5-[(E)-2-(4-hydroxyphenyl)ethenyl]benzene-1,3-diol; Fig. 1) is a naturally occurring plant polyphenol that is present in grapes, berries, peanuts, and other foodstuffs. Resveratrol is thought to act as a phytoalexin, protecting plants against pathogens. Since the suggestion in the early 1990s that the apparent cardioprotective effects of red wine (“the French Paradox”) may be mediated by resveratrol, the compound has been studied for a variety of beneficial health effects [4]. In 1997, Jang et al. [17] reported that resveratrol has a number of biological activities that are consistent with cancer chemoprevention and that the agent can inhibit carcinogenesis in experimental animals. Since then, a broad range of desirable health effects have been ascribed to resveratrol; these include anti-cancer, anti-aging, anti-Alzheimer’s, anti-diabetic, anti-viral, neuroprotective, and cardioprotective activities [2–5, 7, 11, 15, 20, 26, 30–35, 37].
Fig. 1.
Chemical structure of resveratrol (a) and pterostilbene (b)
A large number of mechanisms have been identified through which resveratrol may exert cancer chemopreventive and cancer chemotherapeutic effects. Mechanisms of resveratrol anti-cancer activity may include antioxidant activity and radical scavenging; anti-inflammatory effects; modulation of the activity of carcinogen-metabolizing enzymes; inhibition of cell proliferation; induction of apoptosis; inhibition of angiogenesis; chemosensitization; and inhibition of tumor metastasis [7, 18]. However, resveratrol exhibits low oral bioavailability and undergoes rapid first-pass metabolism; glucuronide and sulfate conjugates are the major resveratrol species found in plasma [41, 44]. Pharmacokinetic studies in human volunteers have led to questions concerning whether high oral doses of resveratrol can generate plasma levels of parent drug that are necessary to achieve the chemopreventive and other desirable activities that have been reported in experimental model systems [6]. However, two very recent references reported data suggesting that biologically active concentrations of resveratrol and/or metabolites may be achievable in human subjects on chronic dosing [8, 22].
The relatively poor bioavailability and rapid metabolism of resveratrol are not uncommon among polyphenols. By contrast, methylated polyphenols appear to demonstrate substantially greater intestinal absorption and enhanced hepatic stability [42]. Pterostilbene (4-[(E)-2-(3,5-dime-thoxyphenyl)ethenyl]phenol; Fig. 1), a naturally occurring dimethylether analog of resveratrol, is a phytoalexin that, like resveratrol, is generated by plants in response to microbial infestation or exposure to ultraviolet light [21]. Pterostilbene has also been reported to possess cancer chemopreventive activity and other resveratrol-like health benefits [10, 24, 27–29, 36, 38].
In consideration of the potential limitations that bio-availability and metabolism may impose on the pharmacodynamic activity of resveratrol, congeners and analogs of resveratrol are attracting increasing attention as possible agents for cancer chemoprevention. The present studies were performed to determine the absolute and relative bioavailability of equimolar oral doses of resveratrol and pterostilbene in rats and to characterize their qualitative and quantitative in vivo metabolic profiles.
Experimental methods
Animals and animal husbandry
All animal care and use in this study were performed in accordance with standards set forth in the Guide for Care and Use of Laboratory Animals (National Research Council, 1996), by the U.S. Department of Agriculture through the Animal Welfare Act (7 USC 2131, 1985), and Animal Welfare Standards incorporated in Title 9, Part 3 of the Code of Federal Regulations, 1991.
Male CD ([Crl:CD(SD) IGS BR]; Charles River Laboratories, Portage, MI) rats were received at 6–7 weeks of age and were held in quarantine for 1 week prior to use in the study. During the quarantine period, rats were observed daily for survival and general health status. Prior to randomization into experimental groups, each animal underwent a detailed physical examination to demonstrate its suitability for use as a test animal.
Throughout the study, rats were housed individually in stainless steel cages in a windowless room that was maintained within a temperature range of approximately 18–23°C and a humidity range of approximately 50–80%. Fluorescent lighting in the animal room was provided on a daily cycle of 12 h of light followed by 12 h of darkness. At all times during the quarantine and dosing periods, rats were permitted free access to Certified Rodent Diet #5002 (PMI Nutrition International, Brentwood, MO) and City of Chicago drinking water (administered by an automatic watering system).
Test articles and dosing formulations
Resveratrol (Sochinaz SA, Vionnaz, Switzerland) and pterostilbene (H&Y International Group Ltd., Hangzhou, China) were provided by the Division of Cancer Prevention, National Cancer Institute. The purity of each agent was >99%, as determined by HPLC.
Dosing formulations of resveratrol and pterostilbene were prepared for intravenous administration in a vehicle of DMSO:PEG-300 (15:85; v/v)]; a dosing volume of 2.5 mL/kg body weight was used for intravenous injections. Oral dosing formulations were prepared in a vehicle of 0.5% (w/v) aqueous methylcellulose containing 0.2% (w/v) Tween 80; a dosing volume of 10 mL/kg body weight was used for gavage administration. All vehicle components were purchased from Sigma–Aldrich (St. Louis, MO).
Study design and conduct
At the end of the quarantine period, rats were randomly assigned to one of ten dosing groups using a computerized body weight stratification procedure that produced similar group mean body weight values. Body weights for the animals assigned to the study ranged from 201 to 241 g. The study design is summarized in Table 1.
Table 1.
Experimental design
| Group number | Number of rats | Test article | Route of administration | Number of daily doses | Dose (mg/kg/day) |
|---|---|---|---|---|---|
| 1 | 12 | Resveratrol | Intravenous | 1 | 10 |
| 2 | 12 | Pterostilbene | Intravenous | 1 | 11.2 |
| 3 | 12 | Resveratrol | Oral | 1 | 50 |
| 4 | 12 | Pterostilbene | Oral | 1 | 56 |
| 5 | 12 | Resveratrol | Oral | 14 | 50 |
| 6 | 12 | Pterostilbene | Oral | 14 | 56 |
| 7 | 12 | Resveratrol | Oral | 1 | 150 |
| 8 | 12 | Pterostilbene | Oral | 1 | 168 |
| 9 | 12 | Resveratrol | Oral | 14 | 150 |
| 10 | 12 | Pterostilbene | Oral | 14 | 168 |
Detailed clinical and physical observations, including body weight collection, were performed on each surviving animal once during the quarantine period, and on Days 1, 8, and 14 of the treatment period. Food consumption was measured individually for each study animal in the repeat-dose groups on Days 8 and 14 of the treatment period.
In groups receiving a single dose of resveratrol or pterostilbene, blood samples were collected from three animals per group at 10 time points (oral dose groups) or 11 time points (intravenous dose groups); blood was collected at time 0 (predose), 5 min (iv groups only), 15 min, 30 min and 1, 2, 4, 6, 8, 12, and 24 h after dose administration. Animals were anesthetized with 70% CO2/30% air, and blood samples (approximately 1 mL) were collected via retro-orbital sinus puncture. The same schedule of blood collection was used after the final dose in animals receiving resveratrol or pterostilbene daily for 14 days.
Blood samples were collected in Vacutainer tubes (Fisher Scientific, Pittsburgh, PA) containing ethylenediaminetetraacetic acid (EDTA). Tubes were inverted several times to mix and were then placed on ice until centrifuged to separate plasma within 1 h time. After centrifugation, plasma was transferred into storage tubes (0.5 mL), which were stored frozen (approximately −70°C) until analyzed.
Analytical method
Plasma levels of resveratrol and pterostilbene were measured using a tandem mass spectrometer (API 3000; Applied Biosystems/MDS Sciex, Foster City, CA) equipped with a high performance liquid chromatograph (Agilent 1200; Agilent Technologies, Wilmington, DE). For resveratrol or pterostilbene determination, a 100 μL aliquot of plasma was mixed with 1 mL of acetonitrile (ACN; Sigma–Aldrich, St. Louis, MO). For resveratrol analysis, an internal standard resveratrol-13C6 (Toronto Research Chemicals, Inc., Ontario, Canada) was added at a concentration of 25 ng/mL. After vortex mixing for 1 min, the sample was centrifuged at 7,000 RPM at 4°C for 10 min to remove precipitated proteins; the supernatant was transferred to a clean tube and dried under nitrogen at room temperature (approximately 25°C). After the evaporation was completed, the residue was reconstituted in 100 μL of methanol with 5 min of sonication, added to 400 μL of water, vortex mixed, and centrifuged again. The resulting supernatant was transferred to a sample vial for instrumental analysis. All sample preparations were conducted under yellow light and using opaque plastic ware to avoid light exposure of the agent.
Freshly prepared resveratrol and pterostilbene standard curves were analyzed along with samples on each day of analysis. Instrument calibrators and quality control (QC) samples were prepared by adding 10 μL of a stock resveratrol or pterostilbene solution (in a methanol/water mixture [v/v 50:50] to 100 μL of rat plasma (Bioreclamation Inc., Westbury, NY). Target calibrator concentrations were 5, 10, 20, 50, 100, 200, 500, and 1,000 ng/mL. QC samples were prepared at approximately 12, 400, and 800 ng/mL. Calibrators and QC samples were processed for analysis using the procedure described earlier. The concentration of conjugated metabolites for both agents was estimated using the calibration curve for each parent compound.
Chromatography was performed using a Luna 3_μ_ C18, 30 × 2.0 mm column (Phenomenex, Torrance, CA) maintained at a temperature of 25°C. A flow rate of 0.25 mL/min was used. Mobile phase (MP) A consisted of 5 mM ammonium acetate in water:isopropanol (98:2, v/v); and MP B consisted of methanol:isopropanol (98:2, v/v). The MP gradient was as follows: after injection, initial conditions with MP A at 90% were held for 0.5 min, decreased to 5% in 3.5 min and held constant for 5 min, returning to initial conditions for another 3 min of re-equilibration time. Retention times for the target analytes were: resveratrol and resveratrol-13C6, 5.2 min; resveratrol glucuronide, 4.1 min; resveratrol sulfate, 4.7 min; pterostilbene, 6.3 min; pterostilbene glucuronide, 5.4 min; and pterostilbene sulfate, 5.5 min. Total run time was 12 min.
A turbo ion spray interface was used as the ion source operating in negative ion mode. Acquisition was performed in multiple reaction monitoring mode using the following ions: resveratrol, 227.0 (Q1) → 185.0 (Q3) Dalton; resveratrol-13C6, 233.0 → 191.0; resveratrol glucuronide, 403.0 → 227.0; resveratrol sulfate, 307.0 → 227.0; pterostilbene, 255.1 → 197.5; pterostilbene glucuronide, 431.1 → 255.1; and pterostilbene sulfate, 335.1 → 255.1. Ion spray voltage was −3,000 V; ion source temperature was 450°C; and collision energy was −30, −35, or 45 V.
The selectivity of the method was assessed by analyzing plasma extracts from different lots for the presence of analytical interferences and comparing the results to those obtained from spiking the blank plasma sources with analytes at the lower limit of quantitation (LLOQ; 5 ng/mL). Linearity was assessed using the internal standard (resveratrol) or external standard method (pterostilbene) and up to eight calibrators with analyte concentrations in the 5–1,000 ng/mL range. The curves were built from peak areas using least-squares linear regression with (1/x) weighting factor. The weighting factor was chosen based on goodness-of-fit criteria including coefficient of determination (r2), the back-calculated concentration of individual calibrators, and minimizing intercept value. Precision and accuracy of the method were determined from QC sample results. Within-run precision and accuracy were assessed from the results from a single day, while between-run precision and accuracy were determined from the results from multiple runs.
No significant peaks interfering with the quantitation of the agents were detected in the chromatograms of blank plasma. Calibration curves for both agents were linear from 5 to 1,000 ng/mL. The r2 values were greater than 0.995. The back-calculated concentration of individual calibrators used to determine the calibration curve ranged from 90 to 100% of the true value. The method’s precision (CV%) was less than 10% and within-run accuracy ranged from 92 to 109% for both agents. Between-run accuracy ranged from 95 to 105% and 101 to 103% for resveratrol and pterostilbene, respectively.
Reference standards for glucuronide and sulfate metabolites were not commercially available for pterostilbene. For this reason, calibration standards were not used to quantitate conjugated metabolites of either agent. Specific enzymatic hydrolysis of the conjugated metabolites was attempted, but resulted in the degradation of pterostilbene. As such, this approach was not considered to be viable for metabolite quantitation.
The analytical results obtained for metabolites of both agents are estimates obtained using the corresponding calibration curve for each parent compound; as such, these data carry a larger experimental uncertainty than do those generated for the parent compound.
Pharmacokinetic analysis
Mean plasma concentration–time profiles of resveratrol and pterostilbene in the rats at scheduled (nominal) sampling times were analyzed by noncompartmental pharmacokinetic methods using WinNonlin® Professional Edition software, Version 5.0.1 (Pharsight Corporation, Mountain View, CA). Key pharmacokinetic parameters, including Tmax,, Cmax, AUC0–t, AUC0–inf, and t½, were calculated for both routes of administration. Additional parameters, including C0, CL, and Vss, were calculated for the intravenous route only, and %F was calculated for the oral dosing route only. AUC from time zero to the last measured concentration was estimated by the linear trapezoidal rule up to Cmax, followed by the log trapezoidal rule for the remainder of the curve. AUC extrapolated to infinity is defined as AUC0–inf = AUC0–t + Ct/_λ_z, where _λ_z is the disposition rate constant estimated using log-linear regression during the terminal elimination phase and Ct is the last measureable plasma concentration.
Results
Pharmacokinetic parameters for resveratrol and pterostilbene following a single intravenous administration at equimolar doses are summarized in Table 2. Even though equimolar doses were administered, systemic exposure to pterostilbene was several-fold greater than was exposure to resveratrol based on their respective values for C0 and AUC0–inf.
Table 2.
Summary of pharmacokinetic parameters of resveratrol and pterostilbene following a single intravenous dose
| Chemical | Dose (mg/kg) | C0 (ng/mL) | AUC0–inf (ng*h/mL) | T1/2 (h) | CL (L/h/kg) | Vss (L/kg) |
|---|---|---|---|---|---|---|
| Resveratrol | 10.0 | 3,450 | 906 | NC | 11.0 | NC |
| Pterostilbene | 11.2 | 7,340 | 4,090 | 2.9 | 2.7 | 5.3 |
Pharmacokinetic profiles of resveratrol and pterostilbene after 14 days of oral dosing are depicted in Fig. 2; similar results were seen in animals receiving a single oral dose (data not shown). After administration of equimolar doses, plasma concentrations of pterostilbene were substantially greater than were plasma concentrations of resveratrol at both dose levels evaluated in the study. The bioavailability of both resveratrol and pterostilbene appeared to be largely independent of the dose (over the 3-fold dose range used in the study) and number of doses (1 or 14; Table 3); at both doses and durations of exposure, the bioavailability of pterostilbene was approximately 3- to 4-fold greater than was the bioavailability of resveratrol. This finding is consistent with much greater Cmax and AUC0–inf values for pterostilbene.
Fig. 2.
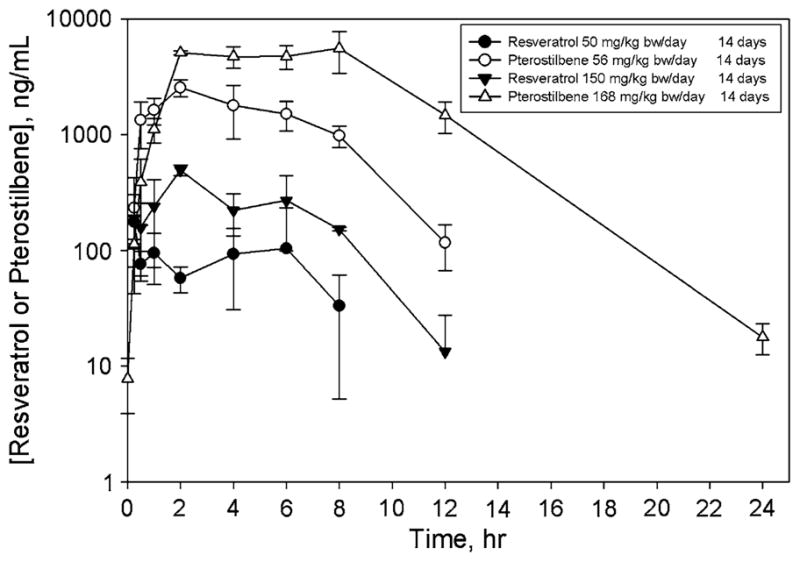
Plasma concentration–time curves for resveratrol and pterostilbene. Animals were orally dosed daily for 14 days, and pharmacokinetic profiles were obtained after the last dose. Symbols represent mean ± SD for n = 3
Table 3.
Summary of pharmacokinetic parameters of resveratrol and pterostilbene following daily oral dosing for 1 and 14 days
| Chemical | Dose (mg/kg) | Number of doses | Tmax (h) | Cmax (ng/mL) | AUC0–inf (ng*h/mL) | T1/2 (h) | %F |
|---|---|---|---|---|---|---|---|
| Resveratrol | 50 | 1 | 1.0 | 76.7 | 1,350 | 11.8 | 29.8 |
| Pterostilbene | 56 | 1 | 2.0 | 2,820 | 13,700 | 1.5 | 66.9 |
| Resveratrol | 50 | 14 | 0.25 | 176 | 902 | 5.5 | 19.9 |
| Pterostilbene | 56 | 14 | 2.0 | 2,550 | 15,000 | 1.6 | 73.2 |
| Resveratrol | 150 | 1 | 1.0 | 847 | 2,580 | 3.6 | 19.0 |
| Pterostilbene | 168 | 1 | 4.0 | 7,880 | 57,700 | 1.9 | 94.2 |
| Resveratrol | 150 | 14 | 2.0 | 494 | 2,370 | 1.8 | 17.5 |
| Pterostilbene | 168 | 14 | 8.0 | 5,560 | 49,600 | 1.9 | 80.8 |
In addition to the plasma levels of the two parent drugs, patterns of metabolism of resveratrol and pterostilbene were also compared. Glucuronide and sulfate conjugates were the primary metabolites of both agents; resveratrol was not detectable after pterostilbene administration. For both agents, sulfate conjugation was generally more extensive than was glucuronidation; an exception to this general pattern was seen with administration of the low dose of resveratrol, after which levels of sulfate and glucuronide conjugates were similar (Tables 4, 5, 6).
Table 4.
Summary of pharmacokinetic parameters of resveratrol glucuronide and pterostilbene glucuronide following a daily oral dosing for 1 and 14 days
| Chemical | Dose (mg/kg) | Number of doses | Tmax (h) | Cmax (ng/mL) | AUC0–inf (ng*h/mL) | T1/2 (h) |
|---|---|---|---|---|---|---|
| Resveratrol | 50 | 1 | 4 | 2,290 | 11,600 | 1.2 |
| Pterostilbene | 56 | 1 | 2 | 516 | 2,380 | 1.4 |
| Resveratrol | 50 | 14 | 1 | 1,590 | 11,600 | 6.6 |
| Pterostilbene | 56 | 14 | 2 | 456 | 2,520 | 1.5 |
| Resveratrol | 150 | 1 | 2 | 3,740 | 22,700 | 3.4 |
| Pterostilbene | 168 | 1 | 2 | 1,400 | 10,500 | 2.3 |
| Resveratrol | 150 | 14 | 2 | 2,620 | 15,700 | 2.2 |
| Pterostilbene | 168 | 14 | 6 | 2,390 | 16,300 | 1.6 |
Table 5.
Summary of pharmacokinetic parameters of resveratrol sulfate and pterostilbene sulfate following a daily oral dosing for 1 and 14 days
| Chemical | Dose (mg/kg) | Number of doses | Tmax (h) | Cmax (ng/mL) | AUC0–inf (ng*h/mL) | T1/2 (h) |
|---|---|---|---|---|---|---|
| Resveratrol | 50 | 1 | 4 | 2,020 | 12,600 | 3.2 |
| Pterostilbene | 56 | 1 | 4 | 1,970,000 | 1,280,000 | 1.3 |
| Resveratrol | 50 | 14 | .25 | 2,770 | 12,200 | .93 |
| Pterostilbene | 56 | 14 | 4 | 2,060,000 | 1,110,000 | 1.5 |
| Resveratrol | 150 | 1 | 1 | 8,710 | 46,000 | 3.2 |
| Pterostilbene | 168 | 1 | 8 | 2,630,000 | 2,450,000 | 1.6 |
| Resveratrol | 150 | 14 | 2 | 10,500 | 56,400 | 1.6 |
| Pterostilbene | 168 | 14 | 6 | 2,050,000 | 2,130,000 | 1.6 |
Table 6.
Relative AUC0–inf values after oral administration for resveratrol, pterostilbene, and their glucuronide and sulfate metabolites
| Chemical | Dose (mg/kg) | Number of doses | AUC0–inf glucuronide/parent | AUC0–inf sulfate/parent | AUC0–inf sulfate/glucuronide |
|---|---|---|---|---|---|
| Resveratrol | 50 | 1 | 8.6 | 9.3 | 1.1 |
| 150 | 1 | 8.8 | 18 | 2.0 | |
| 50 | 14 | 13 | 14 | 1.1 | |
| 150 | 14 | 6.6 | 24 | 3.6 | |
| Pterostilbene | 56 | 1 | 0.18 | 937 | 5,330 |
| 168 | 1 | 0.18 | 425 | 2,330 | |
| 56 | 14 | 0.17 | 740 | 4,410 | |
| 168 | 14 | 0.33 | 430 | 1,310 |
Comparisons of AUC0–inf and Cmax values for sulfate conjugates demonstrated that exposure to pterostilbene sulfate was markedly higher than was exposure to resveratrol sulfate (Table 5). For both agents, these values appeared to be independent of the duration of dosing; however, these values did increase with dose level. In the case of resveratrol, glucuronide/parent AUC0–inf was similar after a single dose, but appeared to decrease with dose level after multiple doses. In contrast, the ratio for sulfate/parent increased with dose (Table 6) after both single and multiple doses. Exposure to the sulfate metabolite (relative to the parent or glucuronide) increased with dose for resveratrol, but decreased for pterostilbene (Table 6).
Comparisons of the AUC0–inf for Phase II metabolites after administration of the low doses of each agent demonstrated that exposure to resveratrol glucuronide was considerably greater than was exposure to pterostilbene glucuronide (Table 4). By contrast, at the high dose, a larger increase in levels of pterostilbene glucuronide resulted in the AUC0–inf for pterostilbene glucuronide approaching that of resveratrol glucuronide.
Discussion
Pharmacokinetic profiles of resveratrol and pterostilbene were compared in male rats following a single intravenous dose, a single oral (gavage) dose, and daily oral (gavage) doses administered over 14 consecutive days. Following administration at equimolar doses, systemic exposure to pterostilbene was substantially greater than was systemic exposure to resveratrol: pterostilbene demonstrated markedly higher Cmax and AUC0–inf values, and its oral bioavailability was several-fold greater than the oral bio-availability of resveratrol.
Following intravenous dosing, total body clearance of resveratrol (11 L/h/kg) exceeded that of pterostilbene (2.7 L/h/kg). Clearance of resveratrol exceeded the rate of hepatic blood flow, while clearance of pterostilbene approached that value (4 L/h/kg). Pterostilbene exhibited a Vss value (5.3 L/kg) that was greater than total body water (~0.7 L/kg), suggesting extensive tissue distribution. Insufficient concentration–time data were available to fully characterize the T1/2 and Vss for resveratrol following intravenous dosing. However, unchanged resveratrol has been shown to be retained in tissues and to be the main form retained [1]. Higher clearance could account at least partially for the lower plasma levels and exposure (AUC0–inf) to resveratrol in comparison with pterostilbene.
Low plasma and tissue levels of resveratrol have been reported following oral administration to both experimental animals and humans [6, 41, 44]; reported in vivo plasma levels are significantly lower than are the concentrations of resveratrol that have commonly been used in static, metabolism-, and elimination-free in vitro systems [4, 6]. Plasma concentrations of Phase II metabolites of both resveratrol and pterostilbene are much higher than are the concentrations of the respective parent compounds. It has been proposed that these conjugates could serve as storage pools for the parent drugs, as has been demonstrated for estrone sulfate [40, 41]. The reported enterohepatic recirculation of resveratrol is generally consistent with this hypothesis [25]. An important open question, however, is whether these conjugates have any significant biological activity on their own; one recent publication has presented data demonstrating the in vitro biological activity of resveratrol sulfate metabolites [9, 16].
Comparisons of pharmacokinetic parameters after a single oral dose and 14 consecutive daily oral doses provided no clear evidence for autoinduction, autoinhibition, or saturation-limited elimination of either resveratrol or pterostilbene under the experimental conditions used. However, a trend of increasing F% with increasing dose was observed in animals exposed to pterostilbene.
While the present study was in progress, a report appeared describing the pharmacokinetics of pterostilbene [23] and comparing it to that of resveratrol in a different publication [12]. Reported clearance values following intravenous dosing with pterostilbene (5 mg/kg) or resveratrol (10 mg/kg) were 2.2 and 20.0 L/h/kg. These values compare to our CL values of 2.7 and 11.0 L/h/kg following intravenous administration of pterostilbene (11.2 mg/kg) and resveratrol (10 mg/kg). The F% values reported in those studies after oral (gavage) dosing with pterostilbene (10 mg/kg) or resveratrol (50 mg/kg) were 12.5 and 38.8%, respectively. These values contrast to our results of 66.9 and 29.8% for F% following administration of pterostilbene at 56 mg/kg or resveratrol at 50 mg/kg, respectively. While intravenous CL values were not dissimilar between the studies, the published results for F% of pterostilbene were surprising in two respects. First, the data from the two published reports suggest that the bioavailability of pterostilbene is lower than that of resveratrol, which is unexpected. Secondly, their reported bioavailability of pterostilbene is considerably lower than was found in our study. This could be explained if the doses used in our study had resulted in plasma drug concentrations in the saturating range. This is possible in view of our data where there was a trend of increasing F% value with increasing (tripling) dose. However, it is not possible to draw any definite conclusions on this matter, as the previously reported data involved different drug doses and were based on comparisons of nonequimolar drug administration protocols. In the present study, doses of resveratrol and pterostilbene were equimolar and were selected to allow adequate sensitivity over the sampling time for poorly bioavailable resveratrol. In addition, we have also systematically examined metabolic profiles, as agent metabolism appears to be the single most important factor impacting the bioavailability of polyphenols.
After careful examination, resveratrol was not detectable following dosing with pterostilbene. Therefore, pterostilbene does not appear to act as a prodrug for resveratrol. Phase II metabolites (sulfate and glucuronide conjugates) were the major metabolites of both drugs and were present in the plasma at levels that were substantially greater than levels of the parent compounds. The gastrointestinal tract, liver, and kidneys possess high levels of Phase II metabolizing activities [14, 39]; resveratrol has been reported to undergo rapid and extensive glucuronidation and sulfation, and the gastrointestinal tract may be the major site for its metabolism [13, 19].
In general, the values for sulfate AUC0–inf were substantially greater than the corresponding values for the glucuronide. At both dose levels, the AUC0–inf for pterostilbene sulfate was several orders of magnitude greater than the AUC0–inf for either pterostilbene glucuronide or pterostilbene itself. Differences for resveratrol were smaller: the AUC0–inf for resveratrol sulfate was from 9- to 24-fold higher than the AUC0–inf for resveratrol itself, but was only 2- to 4-fold greater than levels of resveratrol glucuronide following high dose exposure, and comparable to levels of resveratrol glucuronide (1.0–and 1.1-fold) at the low dose of resveratrol. Conversely, the AUC0–inf for pterostilbene glucuronide was much lower than the AUC0–inf for both the parent drug and its sulfate.
A dose-dependent decrease in resveratrol glucuronide/resveratrol with concomitant increases in resveratrol sulfate/resveratrol and its sulfate/glucuronide AUC0–inf ratios suggest a metabolic shift from glucuronidation to sulfation of resveratrol, possibly due to the saturation of the glucuronide pathway and/or differences in the respective values for their Km and Vmax values. A recent study in rats suggested that the specific conjugation pathway of resveratrol may be dose-dependent, with glucuronidation being the first step followed by sulfation, which may occur only after a certain level of resveratrol is reached [43]. On the other hand, a decrease in the sulfate/parent AUC0–inf ratio with dose may suggest saturation of the pterostilbene sulfation pathway under these conditions. Possible contributions of drug transporters to the pharmacokinetic profiles of resveratrol and pterostilbene cannot be discounted as has been proposed for resveratrol [40].
In summary, different exposure profiles of resveratrol, pterostilbene, and their glucuronide and sulfate conjugates are evident in the results of this study. Pterostilbene plasma levels and exposures were much higher than those of equimolar doses of resveratrol, regardless of dose or route of administration. Exposure to pterostilbene and pterostilbene sulfate was markedly greater than to resveratrol and its sulfate metabolite. On the other hand, exposure to resveratrol glucuronide was greater than to pterostilbene glucuronide. These differences stem from differences in absorption and metabolism of these two drugs.
In consideration of the apparent pharmacokinetic advantages of pterostilbene, studies to compare the efficacy and toxicity of pterostilbene and resveratrol are warranted to determine whether its improved pharmacokinetics translate into greater cancer chemopreventive and other pharmacologic activities.
Acknowledgments
These studies were supported by contract number N01-CN-43304 from the National Cancer Institute, Department of Health and Human Services. The authors thank Leigh Ann Senoussi for assistance in preparing the manuscript.
Abbreviations
EDTA
Ethylenediaminetetraacetic acid
QC
Quality control
MP
Mobile phase
LLOQ
Lowest limit of quantitation
Tmax
Time to maximum plasma concentration
Cmax
Peak plasma concentration
AUC
Area under the curve
t½
Elimination half-life
CL
Clearance
Vss
Apparent volume of distribution
F%
Percent bioavailability
Contributor Information
Izet M. Kapetanovic, Division of Cancer Prevention, National Cancer Institute, Bethesda, MD 20892, USA
Miguel Muzzio, Email: mmuzzio@iitri.org, Life Sciences Group, IIT Research Institute, 10 West 35th Street, Chicago, IL 60616, USA.
Zhihua Huang, Life Sciences Group, IIT Research Institute, 10 West 35th Street, Chicago, IL 60616, USA.
Thomas N. Thompson, R&D Services Pharma Consulting, Omaha, NE 68154, USA
David L. McCormick, Life Sciences Group, IIT Research Institute, 10 West 35th Street, Chicago, IL 60616, USA
References
- 1.Abd El-Mohsen M, Bayele H, Kuhnle G, Gibson G, Debnam E, Kaila Srai S, Rice-Evans C, Spencer JP. Distribution of [3H]trans-resveratrol in rat tissues following oral administration. Br J Nutr. 2006;96:62–70. doi: 10.1079/bjn20061810. [DOI] [PubMed] [Google Scholar]
- 2.Athar M, Back JH, Kopelovich L, Bickers DR, Kim AL. Multiple molecular targets of resveratrol: anti-carcinogenic mechanisms. Arch Biochem Biophys. 2009;486:95–102. doi: 10.1016/j.abb.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Athar M, Back JH, Tang X, Kim KH, Kopelovich L, Bickers DR, Kim AL. Resveratrol: a review of preclinical studies for human cancer prevention. Toxicol Appl Pharmacol. 2007;224:274–283. doi: 10.1016/j.taap.2006.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov. 2006;5:493–506. doi: 10.1038/nrd2060. [DOI] [PubMed] [Google Scholar]
- 5.Bishayee A. Cancer prevention and treatment with resveratrol: from rodent studies to clinical trials. Cancer Prev Res. 2009;2:409–418. doi: 10.1158/1940-6207.CAPR-08-0160. [DOI] [PubMed] [Google Scholar]
- 6.Boocock DJ, Faust GES, Patel KR, Schinas AM, Brown VA, Ducharme MP, Booth TD, Crowell JA, Perloff M, Gescher AJ, Steward WP, Brenner DE. Phase I dose escalation pharmacokinetic study in healthy volunteers of resveratrol, a potential cancer chemopreventive agent. Cancer Epidemiol Biomarkers Prev. 2007;16:1246–1252. doi: 10.1158/1055-9965.EPI-07-0022. [DOI] [PubMed] [Google Scholar]
- 7.Brisdelli F, D’Andrea G, Bozzi A. Resveratrol: a natural polyphenol with multiple chemopreventive properties (Review) Curr Drug Metab. 2009;10:530–546. doi: 10.2174/138920009789375423. [DOI] [PubMed] [Google Scholar]
- 8.Brown VA, Patel KR, Viskaduraki M, Crowell JA, Perloff M, Booth TD, Vasilinin G, Sen A, Schinas A, Piccirilli G, Brown K, Steward W, Gescher AJ, Brenner DE. Repeat dose study of the cancer chemopreventive agent resveratrol in healthy volunteers: safety, pharmacokinetics and effect on the insulin-like growth factor axis. Cancer Res. 2010 doi: 10.1158/0008-5472.CAN-10-2364. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calamini B, Ratia K, Malkowski MG, Cuendet M, Pezzuto JM, Santarsiero BD, Mesecar AD. Pleiotropic mechanisms facilitated by resveratrol and its metabolites. Biochem J. 2010;429(2):273–282. doi: 10.1042/BJ20091857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chakraborty A, Gupta N, Ghosh K, Roy P. In vitro evaluation of the cytotoxic, anti-proliferative and anti-oxidant properties of pterostilbene isolated from Pterocarpus marsupium. Toxicol Vitr. 2010;24:1215–1228. doi: 10.1016/j.tiv.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 11.Das DK. Resveratrol in cardioprotection: a therapeutic promise of alternative medicine. Mol Interv. 2006;6:36–47. doi: 10.1124/mi.6.1.7. [DOI] [PubMed] [Google Scholar]
- 12.Das S, Lin HS, Ho PC, Ng KY. The impact of aqueous solubility and dose on the pharmacokinetic profiles of resveratrol. Pharm Res. 2008;25:2593–2600. doi: 10.1007/s11095-008-9677-1. [DOI] [PubMed] [Google Scholar]
- 13.De Santi C, Pietrabissa A, Spisni R, Mosca F, Pacifici GM. Sulphation of resveratrol, a natural product present in grapes and wine, in the human liver and duodenum. Xenobiotica. 2000;30:609–617. doi: 10.1080/004982500406435. [DOI] [PubMed] [Google Scholar]
- 14.Gamage N, Barnett A, Hempel N, Duggleby RG, Windmill KF, Martin JL, McManus ME. Human sulfotransferases and their role in chemical metabolism. Toxicol Sci. 2006;90:5–22. doi: 10.1093/toxsci/kfj061. [DOI] [PubMed] [Google Scholar]
- 15.Goswami SK, Das DK. Resveratrol and chemoprevention. Cancer Lett. 2009;284:1–6. doi: 10.1016/j.canlet.2009.01.041. [DOI] [PubMed] [Google Scholar]
- 16.Hoshino J, Park EJ, Kondratyuk TP, Marler L, Pezzuto JM, van Breemen RB, Mo S, Li Y, Cushman M. Selective synthesis and biological evaluation of sulfate-conjugated resveratrol metabolites. J Med Chem. 2010;53:5033–5043. doi: 10.1021/jm100274c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CW, Fong HH, Farnsworth NR, Kinghorn AD, Mehta RG, Moon RC, Pezzuto JM. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science. 1997;275:218–220. doi: 10.1126/science.275.5297.218. [DOI] [PubMed] [Google Scholar]
- 18.Kraft TE, Parisotto D, Schempp C, Efferth T. Fighting cancer with red wine? Molecular mechanisms of resveratrol. Crit Rev Food Sci Nutr. 2009;49:782–799. doi: 10.1080/10408390802248627. [DOI] [PubMed] [Google Scholar]
- 19.Kuhnle G, Spencer JP, Chowrimootoo G, Schroeter H, Debnam ES, Srai SK, Rice-Evans C, Hahn U. Resveratrol is absorbed in the small intestine as resveratrol glucuronide. Biochem Biophys Res Commun. 2000;272:212–217. doi: 10.1006/bbrc.2000.2750. [DOI] [PubMed] [Google Scholar]
- 20.Kundu JK, Surh YJ. Cancer chemopreventive and therapeutic potential of resveratrol: mechanistic perspectives. Cancer Lett. 2008;269:243–261. doi: 10.1016/j.canlet.2008.03.057. [DOI] [PubMed] [Google Scholar]
- 21.Langcake P. Disease resistance of Vitis spp. and the production of the stress metabolites resveratrol, epsilon-viniferin, alpha-viniferin and pterostilbene. Physiol Plant Pathol. 1981;18:213–226. [Google Scholar]
- 22.la Porte C, Voduc N, Zhang G, Seguin I, Tardiff D, Singhal N, Cameron DW. Steady-State pharmacokinetics and tolerability of trans-resveratrol 2000 mg twice daily with food, quercetin and alcohol (ethanol) in healthy human subjects. Clin Pharmacokinet. 2010;49(7):449–454. doi: 10.2165/11531820-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 23.Lin H-S, Yue B-D, Ho PC. Determination of pterostilbene in rat plasma by a simple HPLC-UV method and its application in pre-clinical pharmacokinetic study. Biomed Chromatogr. 2009;23:1308–1315. doi: 10.1002/bmc.1254. [DOI] [PubMed] [Google Scholar]
- 24.Mannal PW, Alosi JA, Schneider JG, McDonald DE, McFadden DW. Pterostilbene inhibits pancreatic cancer in vitro. J Gastrointest Surg. 2010;14:873–879. doi: 10.1007/s11605-010-1164-4. [DOI] [PubMed] [Google Scholar]
- 25.Marier JF, Vachon P, Gritsas A, Zhang J, Moreau JP, Ducharme MP. Metabolism and disposition of resveratrol in rats: extent of absorption, glucuronidation, and enterohepatic recirculation evidenced by a linked-rat model. J Pharmacol Exp Ther. 2002;302:369–373. doi: 10.1124/jpet.102.033340. [DOI] [PubMed] [Google Scholar]
- 26.Marques FZ, Markus MA, Morris BJ. Resveratrol: cellular actions of a potent natural chemical that confers a diversity of health benefits. Int J Biochem Cell Biol. 2009;41:2125–2128. doi: 10.1016/j.biocel.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 27.Pan Z, Agarwal AK, Xu T, Feng Q, Baerson SR, Duke SO, Rimando AM. Identification of molecular pathways affected by pterostilbene, a natural dimethylether analog of resveratrol. BMC Med Genomics. 2008;1:7. doi: 10.1186/1755-8794-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paul S, DeCastro A, Lee HJ, Smolarek AK, So JY, Simi B, Wang CX, Zhou R, Rimando AM, Suh N. Dietary intake of pterostilbene, a constituent of blueberries, inhibits the {beta}-catenin/p65 downstream signaling pathway and colon carcinogenesis in rats. Carcinogenesis. 2010;31:1272–1278. doi: 10.1093/carcin/bgq004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paul S, Rimando AM, Lee HJ, Ji Y, Reddy BS, Suh N. Anti-inflammatory action of pterostilbene is mediated through the p38 mitogen-activated protein kinase pathway in colon cancer cells. Cancer Prev Res. 2009;2:650–657. doi: 10.1158/1940-6207.CAPR-08-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pervaiz S, Holme AL. Resveratrol: its biologic targets and functional activity. Antioxid Redox Signal. 2009;11:2851–2897. doi: 10.1089/ars.2008.2412. [DOI] [PubMed] [Google Scholar]
- 31.Pezzuto JM. Resveratrol as an inhibitor of carcinogenesis. Pharm Biol. 2008;46:443–573. [Google Scholar]
- 32.Pirola L, Frojdo S. Resveratrol: one molecule, many targets. IUBMB Life. 2008;60:323–332. doi: 10.1002/iub.47. [DOI] [PubMed] [Google Scholar]
- 33.Rimando AM, Cuendet M, Desmarchelier C, Mehta RG, Pezzuto JM, Duke SO. Cancer chemopreventive and antioxidant activities of pterostilbene, a naturally occurring analogue of resveratrol. J Agric Food Chem. 2002;50:3453–3457. doi: 10.1021/jf0116855. [DOI] [PubMed] [Google Scholar]
- 34.Rimando AM, Suh N. Biological/chemopreventive activity of stilbenes and their effect on colon cancer. Planta Med. 2008;74:1635–1643. doi: 10.1055/s-0028-1088301. [DOI] [PubMed] [Google Scholar]
- 35.Saiko P, Szakmary A, Jaeger W, Szekeres T. Resveratrol and its analogs: defense against cancer, coronary disease and neurodegenerative maladies or just a fad? Mutat Res Rev Mutat Res. 2008;658:68–94. doi: 10.1016/j.mrrev.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 36.Schmidlin L, Poutaraud A, Claudel P, Mestre P, Prado E, Santos-Rosa M, Wiedemann-Merdinoglu S, Karst F, Merdinoglu D, Hugueney P. A stress-inducible resveratrol O-methyl-transferase involved in the biosynthesis of pterostilbene in grapevine. Plant Physiol. 2008;148:1630–1639. doi: 10.1104/pp.108.126003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shankar S, Singh G, Srivastava RK. Chemoprevention by resveratrol: molecular mechanisms and therapeutic potential. Front Biosci. 2007;12:4839–4854. doi: 10.2741/2432. [DOI] [PubMed] [Google Scholar]
- 38.Suh N, Paul S, Hao X, Simi B, Xiao H, Rimando AM, Reddy BS. Pterostilbene, an active constituent of blueberries, suppresses aberrant crypt foci formation in the azoxymethane-induced colon carcinogenesis model in rats. Clin Cancer Res. 2007;13:350–355. doi: 10.1158/1078-0432.CCR-06-1528. [DOI] [PubMed] [Google Scholar]
- 39.Tukey RH, Strassburg CP. Human UDP-glucuronosyl-transferases: metabolism, expression, and disease. Annu Rev Pharmacol Toxicol. 2000;40:581–616. doi: 10.1146/annurev.pharmtox.40.1.581. [DOI] [PubMed] [Google Scholar]
- 40.van de Wetering K, Burkon A, Feddema W, Bot A, de Jonge H, Somoza V, Borst P. Intestinal breast cancer resistance protein (BCRP)/Bcrp1 and multidrug resistance protein 3 (MRP3)/Mrp3 are involved in the pharmacokinetics of resveratrol. Mol Pharmacol. 2009;75:876–885. doi: 10.1124/mol.108.052019. [DOI] [PubMed] [Google Scholar]
- 41.Walle T, Hsieh F, DeLegge MH, Oatis JE, Jr, Walle UK. High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab Dispos. 2004;32:1377–1382. doi: 10.1124/dmd.104.000885. [DOI] [PubMed] [Google Scholar]
- 42.Wen X, Walle T. Methylated flavonoids have greatly improved intestinal absorption and metabolic stability. Drug Metab Dispos. 2006;34:1786–1792. doi: 10.1124/dmd.106.011122. [DOI] [PubMed] [Google Scholar]
- 43.Wenzel E, Soldo T, Erbersdobler H, Somoza V. Bioactivity and metabolism of trans-resveratrol orally administered to Wistar rats. Mol Nutr Food Res. 2005;49:482–494. doi: 10.1002/mnfr.200500003. [DOI] [PubMed] [Google Scholar]
- 44.Wenzel E, Somoza V. Metabolism and bioavailability of trans-resveratrol. Mol Nutr Food Res. 2005;49:472–481. doi: 10.1002/mnfr.200500010. [DOI] [PubMed] [Google Scholar]