Recognition of Unmodified Histone H3 by the First PHD Finger of Bromodomain-PHD Finger Protein 2 Provides Insights into the Regulation of Histone Acetyltransferases Monocytic Leukemic Zinc-finger Protein (MOZ) and MOZ-related factor (MORF) (original) (raw)
Abstract
MOZ (monocytic leukemic zinc-finger protein) and MORF (MOZ-related factor) are histone acetyltransferases important for HOX gene expression as well as embryo and postnatal development. They form complexes with other regulatory subunits through the scaffold proteins BRPF1/2/3 (bromodomain-PHD (plant homeodomain) finger proteins 1, 2, or 3). BRPF proteins have multiple domains, including two PHD fingers, for potential interactions with histones. Here we show that the first PHD finger of BRPF2 specifically recognizes the N-terminal tail of unmodified histone H3 (unH3) and report the solution structures of this PHD finger both free and in complex with the unH3 peptide. Structural analysis revealed that the unH3 peptide forms a third antiparallel β-strand that pairs with the PHD1 two-stranded antiparallel β-sheet. The binding specificity was determined primarily through the recognition of arginine 2 and lysine 4 of the unH3 by conserved aspartic acids of PHD1 and of threonine 6 of the unH3 by a conserved asparagine. Isothermal titration calorimetry and NMR assays showed that post-translational modifications such as H3R2me2as, H3T3ph, H3K4me, H3K4ac, and H3T6ph antagonized the interaction between histone H3 and PHD1. Furthermore, histone binding by PHD1 was important for BRPF2 to localize to the HOXA9 locus in vivo. PHD1 is highly conserved in yeast NuA3 and other histone acetyltransferase complexes, so the results reported here also shed light on the function and regulation of these complexes.
Keywords: Histone Acetylase, Histone Modification, NMR, Protein Structure, Zinc Finger
Introduction
Histone acetyltransferases (HATs)3 are enzymes that catalyze the transfer of an acetyl group from acetyl-CoA to the ϵ-amino groups of lysine on histones, which results in important regulatory effects in chromatin structure and assembly as well as gene expression. HATs are highly diverse and generally form multiprotein complexes. Different HAT complexes are composed of various unique subunits, and the combination of these subunits contributes to the unique features of each HAT complex (1).
MOZ (monocytic leukemic zinc-finger protein) and MORF (MOZ-related factor), a pair of large HATs of the MYST (Moz, Ybf2/Sas3, Sas2, Tip60) family, are important for hematopoiesis, skeletogenesis, neurogenesis, and other developmental processes, and they also have been implicated in leukemogenic and other tumorigenic progressions (2). A recent study in mice embryos reported that MOZ is required for normal levels of spatially correct Hox gene expression and for correct body segment identity. MOZ is specifically required for the H3K9 acetylation of active Hox gene loci (3). MOZ is also required for the maintenance of hematopoietic stem cells and plays a role in the differentiation of erythroid and myeloid cells (4). By contrast, MORF is required for the maintenance of undifferentiated neuronal progenitors and for adult neural stem cell self-renewal and neuronal differentiation (5). MOZ and MORF genes are often translocated in acute myeloid leukemia cells, producing either fusion proteins with CBP (cAMP-response element-binding protein (CREB)-binding protein)/p300 HATs or TIF2, a member of the p160 family of coactivators. It is thought that aberrant targeting/acetylation by the fusion proteins is responsible for oncogenic transformation (2).
MOZ and MORF form tetrameric complexes with ING5 (inhibitor of growth 5), EAF6 (Esa1-associated factor 6 ortholog), and BRPF1/2/3 (bromodomain-PHD (plant homeodomain) finger protein 1, 2, or 3) (6). BRPF proteins bridge the association of MOZ and MORF with ING5 and EAF6. At the functional level, complex formation with BRPF1 and ING5 powerfully stimulates the activity of the acetyltransferase on nucleosomal histone H3 and enhances the transcriptional potential of MOZ (7).
During zebrafish development, BRPF1 is required for histone acetylation, the maintenance of cranial Hox gene expression, and the proper determination of pharyngeal segmental identities (8). BRPF1 is associated with chromatin in discrete locations in both interphase and mitotic chromosomes of HEK293 cells and zebrafish embryos. Interestingly, in contrast to interphase, BRPF1 and MOZ do not co-localize during mitosis, when MOZ is largely excluded from chromosomes. This suggests that BRPF1 contributes to transcriptional memory throughout mitosis (8). The BRPF2 protein is 49.2 and 49.9% identical to BRPF1 and BRPF3, respectively. The similarity is even higher within the conserved domains. Despite its identification almost 10 years ago, there has been little information about the biological role of BRPF2 in vertebrate systems. Studies have suggested that BRPF2 expression is highly regulated in the embryonic brain and that the BRPF2 gene is associated with schizophrenia and bipolar affective disorder (9). In a population-based screening study of 446 consecutive childhood acute lymphoblastic leukemia cases, it was found that the entire BRPF2 protein was fused to PAX5 (10), a master regulator of B-cell development. These results strongly suggested that BRPF2 plays important roles in developmental and disease processes.
BRPF proteins contain a unique combination of domains typically found in chromatin-associated factors, including two PHD fingers, PHD1 and PHD2, linked by a mononuclear Zn2+ knuckle to form a PZP (PHD/ Zn2+-knuckle/PHD) motif (11), a bromodomain, and a PWWP domain. Bromodomains interact with acetylated lysines on N-terminal tails of histones and other proteins (12), and the PWWP domain of BRPF1 has been shown to bind histone H3K36me3 (13). PHD fingers were shown to recognize methylated or unmethylated lysine 4 of histone H3 (14, 15). However, the role of the PHD fingers in BRPF proteins remains unclear. In this study we classified the BRPF2-PHD1 finger as an unmodified histone H3 binding module and revealed its structural basis. ChIP assays showed that histone binding by PHD1 is important for BRPF2 to localize at the target gene. Sequence alignments revealed that this PHD finger is highly conserved in yeast NuA3-like HAT complexes, and corresponding interactions with histones were also observed in these complexes. Herein, our study sheds light on a conserved mechanism regulating these groups of HATs.
EXPERIMENTAL PROCEDURES
Plasmids Construction
For NMR and isothermal titration calorimetry (ITC) experiments, human BRPF2 PHD1 (Gln-208—Ala-269) and PHD2 (Asn-317—Thr-394) were cloned into a modified pET-28a (+) (Novagen) plasmid, generating a fusion protein with a MGHHHHHHM tag at the N terminus. For the structure determination of the complex of the BRPF2-PHD1 finger with histone H3, the H3 tail (Ala-1—Gly-12) was fused to the N terminus of the PHD1 finger (H3-PHD1 fusion), linked by a six reside linker, GSSGSS. The DNA fragment coding for the H3-PHD1 fusion protein was subcloned into the plasmid pET-22b (+) (Novagen), giving an N-terminal Met and a C-terminal His-tag (-LEHHHHHH). The N-terminal Met was cleaved during expression. For GST (glutathione _S_-transferase) pulldown assays, the BRPF2 PHD1 and PHD2 finger were subcloned into pGEX 4T-1. For transient expression in mammalian cells, the full-length human BRPF2 gene (obtained from RZPD, IMAGE clone ID IRATp970A0587D) was cloned into p3XFLAG-myc-CMV™-24. Mutants were generated by PCR-mediated site-directed mutagenesis. All constructs were verified by DNA sequencing.
Recombinant Protein Expression and Purification
The recombinant plasmids harboring the respective target genes were transformed into Escherichia coli BL21 (DE3) host cells, and the cells were induced at 16 °C with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside in LB or SV40 media plus 50 μm ZnSO4. Uniformly 15N- and 13C,15N-labeled proteins were prepared by growing bacteria in SV40 media using 15NH4Cl (0.5 g/liter) and [13C6]glucose (2.5 g/liter) as stable isotope sources. The His-tag proteins were purified using chelated nickel and gel filtration columns. The GST fusion proteins were purified using immobilized glutathione and gel filtration columns.
Peptide Synthesis and Purification
The histone H3 N-terminal tail peptides were chemically synthesized using a standard Fmoc (_N_-(9-fluorenyl)methoxycarbonyl) strategy at GL Biochem (Shanghai) Ltd. The synthetic peptides were purified by reverse-phase high pressure liquid chromatography using a C18 column. Pooled fractions of the pure peptides were lyophilized and verified by MALDI-TOF mass spectrometry.
Calf Thymus Histone Binding Assays
GST proteins (20 μg) were bound to glutathione beads (Pharmacia). The beads were then incubated for 1 h at 4 °C with histones (50 μg, Worthington) in buffer containing 20 mm Tris, pH 7.5, 500 mm NaCl, and 0.2% Triton X-100. After six washes, the bound material was resolved on SDS-polyacrylamide gels and stained with Coomassie Brilliant Blue.
Isothermal Titration Calorimetry
The BRPF2 PHD1 and mutants were exchanged into 50 mm Tris-HCl buffer containing 100 mm NaCl, pH 7.5, by gel filtration chromatography. Lyophilized H3 peptides were dissolved in the same buffer, and the pH values were corrected. ITC measurements were carried out with protein and peptide concentrations ranging from 100–200 μm and 2–5 mm, respectively, in a MicroCal VP-ITC instrument at 20 °C. For each peptide, a reference titration of peptide into buffer was subtracted from the experimental data to compensate for the heat of dilution. Binding constants were calculated by fitting the data using the ITC data analysis module Origin 7.0 (OriginLab Corp.).
NMR Sample Preparation and NMR Spectroscopy
Both the purified 15N- and 13C,15N-labeled proteins were dissolved to a final concentration of 0.8 mm in 500 μl of buffer containing 20 mm Bis-Tris, pH 6.7, 150 mm NaCl, and 10% D2O. All NMR experiments were performed at 293 K in a Bruker DMX500 spectrometer.
The following spectra were recorded to obtain backbone and side chain resonance assignments (16): two-dimensional 1H,15N HSQC, three-dimensional triple resonance spectra HNCO, HN(CA)CO, CBCA(CO)NH, CBCANH, HBHA(CO)NH, C(CO)NH-TOCSY H(CCO)NH-TOCSY, two-dimensional 13C,1H HSQC, and three-dimensional HCCH-TOCSY and HCCH-COSY. Three-dimensional 15N-separated and 13C-separated NOESY were acquired with a mixing time of 130 and 110 ms, respectively. All NMR spectra were processed with NMRPipe (17) and analyzed using Sparky3 software (T. D. Goddard and D. G. Kneller, University of California, San Francisco).
Structure Calculation and Validation
The NOE cross-peaks from the three-dimensional 15N- and 13C-separated NOESYs were assigned and converted into distance restraints. Dihedral angle restraints for the φ and ψ angles were derived from N, CO, CA, CB, and HA chemical shifts by using TALOS (18). Protein-zinc restraints were added as described in Ref. 19. The structures were calculated and further refined using the standard simulated annealing protocol for torsion angle dynamics in the CNS1.2 program (20). A number of hydrogen bonds derived from the H/D exchange experiment and from observed NOEs characteristic of α-helices and β-sheets were added in the final rounds of structure refinement. In the final stage, 200 structures were calculated, and the 20 lowest energy conformers were selected to form the final ensemble. The final structures were validated and visualized by the programs Procheck-NMR (21), MOLMOL (22), and PyMOL (Delano Scientific).
Chemical Shift Perturbation
NMR titrations of the PHD with H3 peptides were performed on 15N-labeled proteins. 1H,15N HSQC spectra of free PHD were recorded as controls. Peptide stock solutions in identical buffer were titrated stepwise with a sample dilution of less than 10%. The combined chemical shift perturbation was calculated using the equation,
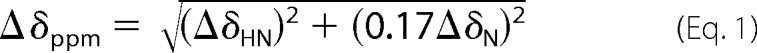
where ΔσHN and ΔσN are the chemical shift variations in the proton and nitrogen dimensions, respectively. Dissociation constants were estimated as described previously (23).
15N Relaxation Measurements
15N relaxation experiments were carried out on a Bruker DMX 500 NMR spectrometer at 293 K using published methods (24). The samples of 15N-labeled PHD1, free or bound to unlabeled unH3 peptide, were dissolved in the same buffer described above. With a 1-s recycle delay, the T1 and T2 were measured with 8 relaxation delays (11.2, 61.4, 142, 242 (run twice), 363, 523, 754, and 1150 ms) and 7 relaxation delays (0, 17.6, 35.2, 52.8 (run twice), 70.4, 106, and 141 ms), respectively. The spectra measuring 1H,15N NOE were acquired with a 2-s relaxation delay followed by a 3-s period of proton saturation. In the absence of proton saturation, the spectra were recorded using a relaxation delay of 5 s. The exponential curve fitting and data analysis were carried out with the program Sparky.
Chromatin Immunoprecipitation Assay (ChIP)
293T cells were maintained in DMEM media supplemented with 10% fetal bovine serum (Invitrogen). Transient transfection was performed with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. ChIP assays were performed as previously described (25). Briefly, 293T cells (2 × l07 cells) were fixed with 1% formaldehyde and then neutralized by adding 0.125 m glycine. Cells were collected and lysed in cell lysis buffer (5 mm PIPES, pH 8.0, 85 mm KCl, 0.5% Nonidet P-40, and protease inhibitor mixture). The nuclei pellet was obtained by centrifugation and was subsequently lysed in nuclei lysis buffer (50 mm Tris-HCl, pH 8.1, 10 mm EDTA, 1% SDS, and protease inhibitor mixture). The nuclei lysate was sonicated to obtain soluble chromatin with an average length of 1000 bp. After 1:10 dilution in dilution buffer (10 mm Tris-HCl, pH 8.1, 2 mm EDTA, 150 mm NaCl, and 1% Triton X-100), chromatin solutions were precleared and then were incubated with or without anti-FLAG antibody (Sigma). Next, the mixture was incubated at 4 °C overnight on a rotating platform. The immunocomplexes were recovered with protein A-Sepharose beads. After extensive washing, the bound DNA fragments were eluted, and the resulting DNA was subjected to PCR using the following primer pairs: HOXA9 forward (CGA CCC ACG GAA ATT ATG AA) and reverse (CGT TGG CCA CAA TTA AAA CA) and actin forward (GAC CTG ACT GAC TAC CTC ATG AAG AT) and reverse (GTC ACA CTT CAT GAT GGA GTT GAA GG). PCR products were separated by electrophoresis on 2% agarose gels and visualized.
RESULTS
BRPF2-PHD1 Preferentially Binds to Histone H3K4me0
BRPF proteins contain two PHD fingers, PHD1 and PHD2 (Fig. 1A), and PHD fingers have been shown to bind to methylated and unmethylated histones (14, 15). To investigate the function of the PHD fingers of BRPFs, we assayed the interactions between BRPF2 PHD fingers and calf thymus histones using GST pulldown experiments. We found that GST-PHD1, but not GST-PHD2 or GST alone, interacted primarily with histone H3 (Fig. 1B). A sequence alignment search using BLAST in the Protein Data Bank revealed that BRPF2-PHD1 has significant sequence similarity to AIRE-PHD1 and BHC80-PHD (Fig. 5A), two well established H3K4me0 binding modules, whereas a search using PHD2 as the bait returned no target. To investigate whether PHD1 of BRPF2 also binds H3K4me0, the interaction of BRPF2-PHD1 with several histone H3 peptides was studied using ITC. As shown in Fig. 1C and Table 1, BRPF2-PHD1 bound to H3(1–12)K4me0, with a dissociation constant (KD) of ∼190 μm, whereas monomethylated H3K4 peptide significantly reduced the binding affinity, and trimethylated H3K4 peptide completely abolished binding. ITC assays also revealed that the interaction between BRPF2-PHD1 and H3K4me0 was primarily driven by a markedly exothermic binding enthalpy, with Δ_H_ = −4.4 kcal mol−1 and T_Δ_S = 0.59 kcal mol−1. NMR titrations confirmed the interaction between BRPF2-PHD1 and H3K4me0, as a discrete set of chemical shift changes was observed on the addition of H3K4me0 to BRPF2-PHD1 (Fig. 1D and supplemental Fig. S1_A_). The NMR titrations also gave a similar KD of ∼140 μm (supplemental Fig. S1_B_). Taken together, our study established BRPF2-PHD1 as a histone H3-binding module, and this interaction was antagonized by the methylation of H3K4.
FIGURE 1.

BRPF2-PHD1 finger preferentially binds to histone H3K4me0. A, shown is a diagram of BRPF2 domain architecture. B, GST-PHD1, but not GST-PHD2 or GST alone, interacts with histones, visualized by Coomassie staining. C, ITC measurements of the binding of the BRPF2-PHD1 finger to unmodified AND mono- and trimethylated H3K4 peptides (residues 1–12) are shown. D, six superimposed 1H,15N HSQC spectra of PHD1 (0.36 mm) collected during titration of H3K4me0 peptide are color-coded according to the ligand. Inset, PHD1 molal ratio.
FIGURE 5.

Comparison of histone recognition by the different PHD fingers. A, shown is sequence alignment of the PHD fingers of human BRPFs, AIRE, and BHC80. Secondary structure elements of free BRPF2-PHD1 and BHC80-PHD are shown. The numbering refers to the human BRPF2. The same symbols from Fig. 3A are used. B–G, selected PHD-H3 complexes are shown in schematic mode, and the side chains of H3R2, H3K4, and H3T6 (H3K14 for panel G) of histone H3 and their specific interactions with the different PHD fingers are explicitly shown. For DNMT3A-ADD, only the PHD-fold portion is shown for clarity.
TABLE 1.
Dissociation constants of the interactions between BRPF2 PHD1 and H3-tail peptides determined by ITC and NMR titration experiments
| Method | Protein | Peptide | KD value |
|---|---|---|---|
| μ_m_ | |||
| NMR | WTa | unH3(1–12) | 134 ± 5.6 |
| ITC | WT | unH3(1–12) | 192 ± 9.7 |
| ITC | WT | H3(1–12)K4me1 | 552 ± 72 |
| ITC | WT | H3(1–12)K4me3 | Not detected |
| ITC | WT | H3(1–12)K4ac | Not detected |
| ITC | WT | H3(1–12)K9ac | 252 ± 41 |
| ITC | WT | H3(1–12)R2me2a | 694 ± 100 |
| ITC | D212A | unH3(1–12) | 362 ± 84 |
| ITC | D214A | unH3(1–12) | 300 ± 19 |
| ITC | D212A/D214A | unH3(1–12) | Not detected |
| ITC | N229A | unH3(1–12) | Not detected |
| ITC | D235A | unH3(1–12) | Not detected |
| NMR | WT | unH3(1–7) | 176 |
| NMR | WT | H3(1–7)T3ph | Not detected |
| NMR | WT | H3(1–7)T6ph | 1241 |
| ITC | WT | H3(30–44)K36me0 | Not detected |
| ITC | WT | H3(30–44)K36me3 | Not detected |
| NMR | WT | H3(30–44)K36me3 | Not detected |
Structure Determination of BRPF2 PHD1, Both Free and Complexed to Histone H3 Peptide
To understand the molecular basis of histone H3 recognition by BRPF2-PHD1, we solved the solution structure of BRPF2-PHD1 (Gln-208—Ala-269), both free and complexed to histone H3 peptide (Ala-1—Gly-12). To avoid cysteine oxidation, a mutant was produced in which the two non-zinc-chelating cysteines (Cys-218 and Cys-225) were changed to serines (supplemental Fig. S2). 1H NMR spectra and CD spectra of the native and mutant PHD1 fingers were highly similar, suggesting that these two fragments adopted the same three-dimensional structure (supplemental Fig. S3). Other in vitro studies were carried out on the mutant PHD1 finger. To determine the structure of the complex, we expressed a fusion protein with the histone H3 peptide fused to the N terminus of BRPF2-PHD1 (see “Experimental Procedures”). The PHD1 residues in 1H,15N HSQC spectrum of the fusion protein superimposed well with that of BRPF2-PHD1 bound to the H3K4me0 peptide, indicating that the structure of the fusion protein could represent that found in the complex (supplemental Fig. S4). We determined these structures using classical heteronuclear multidimensional NMR experiments (16). The final 20 solution structures with the lowest energy of free PHD1 and H3-PHD1 fusion protein are shown in Fig. 2, and the structural statistics are given in Table 2. The structural coordinates of human BRPF2-PHD1, both free and fused to unH3(1–12), have been deposited into the Protein Data Bank with accession codes 2KU3 and 2L43, respectively.
FIGURE 2.

Solution structures of BRPF2-PHD1 finger, both free and fused to unH3(1–12). Shown are the 20 lowest energy structures of the BRPF2-PHD1 finger, both free (A) and fused to unH3 (C). Ribbon representations of the lowest-energy structure of the BRPF2-PHD1 finger, both free (B) and fused to unH3 (D) are shown. Pink spheres represent Zn2+ atoms. Histone peptide and linker are shown in yellow and pale cyan, respectively.
TABLE 2.
Structural statistics for the BRPF2-PHD1 finger, both free and fused to unH3(1–12) peptide
None of the structures exhibited distance violations of >0.5 Å or dihedral angle violations of >5. r.m.s.d., root mean square deviation.
| BRPF2-PHD1 finger | Free | Fusion |
|---|---|---|
| NMR restraints | ||
| NOE distance restraints | 968 | 1201 |
| Intra-residue | 435 | 464 |
| Sequential (|i − j | = 1) | 231 |
| Medium range (|i − j | < 5) | 97 |
| Long range (|i − j | ≥ 5) | 205 |
| Intermolecular | 74 | |
| Hydrogen bonds | 16 | 28 |
| Dihedral angle restraints | 56 | 68 |
| Protein-zinc restraints | 31 | 31 |
| Lennard-Jones potential energy (kcal mol−1) | −236.5 ± 8.5 | −303.2 ± 10.0 |
| Mean r.m.s.d. from idealized covalent geometry | ||
| Bonds (Å) | 0.0033 ± 0.00018 | 0.0032 ± 0.00016 |
| Angles (degree) | 0.6937 ± 0.0466 | 0.6730 ± 0.0098 |
| Improper (degree) | 0.2491 ± 0.0089 | 0.2491 ± 0.0095 |
| Mean r.m.s.d. from experimental restraints | ||
| Distance (Å) | 0.0138 ± 0.0003 | 0.0137 ± 0.0002 |
| cdih (degree) | 0.3192 ± 0.0367 | 0.3083 ± 0.0203 |
| Structural r.m.s.d. to the mean coordinatesa (Å) | ||
| Backbone atoms | 0.61 | 0.73 |
| Heavy atoms | 1.12 | 1.36 |
| Ramachandran plot analysis (%)a | ||
| Residues in most favored regions | 74.0 | 81.6 |
| Residues in additional allowed regions | 24.9 | 16.4 |
| Residues in generously allowed regions | 0.7 | 1.5 |
| Residues in disallowed regions | 0.4 | 0.5 |
Structural Basis for H3K4me0 Recognition by BRPF2-PHD1 Finger
The BRPF2-PHD1 finger folded into a compact globular domain consisting of extended loops, with a short two-stranded antiparallel β-sheet (β1, Ile-231—Phe-233; β2, Ala-240—His-242) and two α-helices (α1, Gln-243—Tyr-246; α2, Arg-259—Ala-266) as the sole elements of secondary structure (Figs. 2 and 5A). Similar to other PHD finger structures, the two Zn2+ ions, located at opposite ends of the β-sheet, were coordinated by conserved residues in a cross-brace manner (Figs. 2 and 5A). The first zinc-binding site was composed of residues Cys-217, Cys-220, His-242, and Cys-245, and the second zinc-coordinating site was composed of Cys-234, Cys-237, Cys-258, and Cys-261.
The H3K4me0 peptide interacted with the PHD finger through an antiparallel β-sheet pairing. The peptide, adopting an extended β-strand-like conformation (residues Thr-3—Ala-7), was positioned on the surface of the finger, forming a third β-strand antiparallel to the Ser-228—Phe-233 segment of the β-sheet in the protein. A comparison of the PHD finger structures, both free and bound to H3K4me0 peptide, revealed that the PHD finger constitutes a stable preformed binding platform, as the root mean square deviation was only 0.994 Å for the backbone of residues Val-230—Ala-266 between the free and bound states (supplemental Fig. S5). However, an apparent conformation change occurred in the loop (loop 1) connecting the first Zn2+-chelating Cys pair and the β1 segment. Residues Ser-228—Val-230 were induced to a β-conformation to pair with the histone peptide. Consistently, extensive chemical shift perturbations were observed in this region (supplemental Fig. S1_A_).
The substrate specificity of the BRPF2-PHD1 finger was determined primarily through the recognition of the H3 N terminus, H3R2, H3K4, and H3T6. Charge complementarities and hydrophilic interactions contributed greatly to the peptide binding (Fig. 3, C and D), consistent with the thermodynamic data measured using ITC. First, two main chain carbonyl oxygen atoms (Pro-252 and Gly-254) formed a hydrogen bond “pincer” that recognized the N terminus of H3. Second, the side chain of H3A1 was inserted into a hydrophobic pocket formed by Leu-232, Trp-256, Pro-252, and Ile-251, and the methyl group of the H3T3 side chain was inserted into an adjacent hydrophobic pocket formed by Val-230, Leu-232, and Ile-251 (Fig. 3E). Third, the guanidinium moiety of H3R2 formed branched polar interactions with the backbone carbonyl of Cys-234 and the carboxyl group of Asp-235. The interaction between H3R2 and Asp-235 was confirmed by mutagenesis; mutation of Asp-235 to Ala abolished BRPF2-PHD1 binding to the H3K4me0 peptide (Table 1 and supplemental Fig. S6). Fourth, the ϵ-amino group of H3K4 established a network of electrostatic interactions with the carboxyl groups of Asp-212 and Asp-214. Mutagenesis experiments showed that mutants D212A or D214A had reduced binding affinity, whereas the double mutant D212A/D214A abolished the binding (Table 1 and supplemental Fig. S6). Molecular recognition of unmethylated lysine was facilitated primarily through the electrostatic interactions between this Asp pair and the ϵ-amino group of H3K4 and steric exclusion of methyl groups. Finally, the oxygen atom in the hydroxyl group of the H3T6 side chain formed a hydrogen bond with the amino proton in the side chain of residue Asn-229. Mutation of Asn-229 to Ala also abolished BRPF2-PHD1 binding to the histone H3 tail (Table 1 and supplemental Fig. S6). Significantly, residue Asn-229 is strongly conserved in the BRPF-related protein family (Fig. 3A). Residues beyond H3A7 were less structured and had limited interactions with the PHD1 finger.
FIGURE 3.

Details of the interaction of the BRPF2-PHD1 finger with the linked unH3. A, sequence alignment of PZPMs in scaffold proteins of NuA3-like histone acetyltransferase complex are shown. PHD1 zinc binding residues are indicated by blue stars or blue circles; PHD2 zinc binding residues are indicated by blue stars for blue circles; potential mononuclear Zn2+ knuckle zinc-binding residues are indicated by black boxes, and residues mutated for interaction assays in this study are indicated by red triangles. Numbering refers to the human BRPF2. The prefix Hs indicates Homo sapiens; Dr, Danio rerio; Dm, Drosophila melanogaster; Ce, C. elegans; Sc, Saccharomyces cerevisiae. The sequence alignment was performed using ClustalW, and the panel was generated by ESPript 2.2. B, shown is a ribbon representation of the complex highlighting the secondary structural elements. C, shown is electrostatic potential (isocontour value of ±76 kT/e) surface representation of the BRPF2-PHD1 bound to the H3K4me0 peptide (yellow). D, key protein-peptide polar interactions are shown in stick mode. E, H3A1- and H3T3-interacting neighborhood hydrophobic pockets are shown. D and E, the peptide and protein residues are color-coded by atom type with carbon atoms in yellow and cyan, respectively. The orientation of the peptide is the same as that in B.
Dynamic Properties of BRPF2-PHD1 Finger
To investigate the dynamic properties of the BRPF2-PHD1 finger upon binding to the histone H3K4me0 peptide, we recorded the longitudinal relaxation times T1, the transverse relaxation times T2, and the heteronuclear {1H}–15N NOE values of PHD1, both free and bound to the peptide. The steady-state heteronuclear {1H}–15N NOE, a sensitive indicator of internal motions on a subnanosecond timescale, revealed that most residues had similar 1H,15N NOE values of ∼0.7 in free and bound states. However, two regions (Leu-210–Ala-215 and Ile-251—Gln-255) had low 1H,15N NOE values of 0.34 and 0.41, respectively, in free PHD1 and high 1H,15N NOE values of 0.54 and 0.62, respectively, upon binding to the peptide (Fig. 4C). Correspondingly, these two regions had larger T2 values (135 and 136 ms, respectively) than average (95 ms) in free PHD1, whereas they had similar T2 values as other residues (74 and 66 ms, respectively, versus 66 ms) when bound to peptide (Fig. 4B). Structurally, these two regions had key interactions with the histone peptide (Fig. 4D). Taken together, we concluded that substrate binding resulted in increased domain rigidity in these two regions.
FIGURE 4.

Backbone NMR relaxation data for BRPF2-PHD1, both free and bound to unH3 peptide. 15N longitudinal (T1) (A) and transversal (T2) (B) relaxation times and heteronuclear {1H}-15N NOEs (C) are represented for residues of PHD in its free form and bound to the unlabeled unH3(1–12) peptide. D, shown is a schematic representation of the BRPF2-PHD1-unH3 complex with cyan and yellow representing BRPF2-PHD1 and unH3, respectively. Residues displaying an increase of the heteronuclear NOE values upon peptide binding are represented in stick form (dirty violet).
Structural Comparison with Other Histone H3 Binding PHD Fingers
Studies have suggested that the large family of PHD fingers can be separated into subsets based on their specificity toward different histone modifications. At least five subsets have been identified: (i) H3K4me0 (AIRE, BHC80, and DNMT3L/3A) (15, 26–28), (ii) H3K4me3 (BPTF, ING/YNG, TAF3, etc.) (29–31), (iii) H3K9me3 (ICBP90, Lid2, and SMCX) (32–34), (iv) H3K36me3 (CHD3 and Ecm5) (14, 35), and (v) H3K14ac and H4K16ac (DPF3b) (36). However, only groups i, ii, and v currently have structural information available. Sequence alignments and structural comparisons revealed that the BRPF2-PHD1 finger is most closely related to BHC80-PHD and AIRE-PHD1 (Fig. 5A–D), both of which also recognize H3K4me0. In addition, the PHD portion of the ADD domain of DNMT3L/3A, despite less sequence similarity, also binds histone H3K4me0 (Fig. 5E). Notably, all these domains share a conserved H3K4me0 recognition mode. In these complexes the unmodified N-terminal tail of H3 forms a third antiparallel β-strand that pairs with the protein two-stranded antiparallel β-sheet via an induced fit mechanism. This binding mode is also similar for H3K4me3 binding PHD fingers (e.g. PHD3 of JARID1A; Fig. 5F). The primary difference between H3K4me0 and K4me3 recognition stems from two distinct molecular mechanisms. For H3K4me0, the unmodified lysine 4 is recognized by the carboxylate of the conserved aspartate residues (Fig. 5, B–E). For K4me3, the trimethylated lysine 4 is embraced by an aromatic cage via π-cation interactions (Fig. 5F). Subtle differences also exist in K4me0 recognition. The unmodified H3K4 is recognized by a pair of aspartic acids in the BRPF2-PHD1 finger and DNMT3A-ADD, by one Asp and one Asn in the AIRE-PHD1 finger, and one Asp in the BHC80-PHD finger. In BHC80-PHD, the β-carbon from His-487 further restricts the lysine-binding site. The interactions between the residues flanking H3K4 and PHD fingers also vary. First, the guanidinium moiety of H3R2 has key electrostatic interactions with a conserved aspartic acid in BRPF2-PHD1, BHC80-PHD, AIRE-PHD1, and JARID1A-PHD3, whereas the side chain of H3R2 is fully exposed to solvent in DNMT3A/3L-ADD. Second, H3T6 interacts with the conserved Asn-229 in BRPF2-PHD1. In AIRE and DNMT3A, the hydroxyl group of the H3T6 side chain interacts with the backbone of a Gly, whereas in BHC80 and JARID1A, the H3T6 side chain does not form a hydrogen bond with the PHD finger. Third, in BHC80 and AIRE, H3R8 and H3K9 also contribute to the complex formation.
Recently, the structural mechanism of acetylated histone binding by the double PHD fingers of DPF3b was reported. The H3 residues flanking H3K14ac form an extended conformation bound in an elongated groove nearly perpendicular to the β-sheet of PHD1. This binding mode is totally different from those of H3K4me0 and H3K4me3 recognition (Fig. 5G).
Cross-talk of Modifications on the Histone H3 N-terminal Tail
Histones undergo several different post-translational modifications that regulate a variety of physiological processes. These covalent modifications show substantial cross-regulation, providing a wealth of regulatory potential. To investigate whether such a mechanism exists for the interaction between BRPF2-PHD1 and H3K4me0, we carried out ITC and NMR assays of BRPF2-PHD1 with various histone peptides (Table 1 and supplemental Fig. S7 and S8).
Generally, histone H3K4 methylation is a hallmark of actively transcribed genes. As described above, monomethylated H3K4 peptide significantly reduced binding to BRPF2-PHD1, whereas trimethylated H3K4 peptide completely abolished it (Fig. 1C).
H3K4ac is a recently discovered histone post-translational modification (37). It was detected at low levels in humans, mice, and Tetrahymena thermophila, and its biological functions remain unclear. Our ITC assay showed that acetylated H3K4 peptide completely abolished binding to BRPF2-PHD1 (supplemental Fig. S7). This is most likely due to the loss of the positive charge on H3K4ac, preventing the formation of the critical ion pair with Asp-212 and Asp-214, and the bulky nature of the acetamide moiety.
H3K9ac is a histone modification involved in gene activation. MOZ is specifically required for the H3K9 acetylation of transcriptionally active Hox gene loci (3). Recently, it has been reported that CHD4-PHD2 recognizes H3K4me0, and interestingly, this interaction is facilitated by the acetylation or methylation of H3K9 (38). To test whether this interesting phenomenon also occurs in BRPF2-PHD1, we studied its interaction with H3K9ac using ITC (supplemental Fig. S7). However, the measured KD was ∼250 μm, compared with ∼190 μm for H3K4me0. This indicated that acetylation of H3K9 slightly impaired, rather than abolished, the interaction between histone H3 and BRPF2-PHD1. This was consistent with our structural analysis, as H3K9 had few interactions with the protein.
Asymmetric dimethylation of H3R2 (H3R2me2a) is generally distributed throughout regions of silenced chromatin. This modification counteracts the active transcriptional state by blocking the function of the H3K4 methyltransferase complexes COMPASS in yeast and ASH2/MLL in humans (39). Previously, H3R2me2a was shown to strongly reduce the binding affinity of AIRE-PHD1 to H3K4me0 (26) and of the TAF3-PHD finger to H3K4me3 (40), whereas only a limited effect was observed for the ING2- or BPTF-PHD fingers (30). This suggested that H3R2me2a plays distinct cross-talk roles for different proteins. To evaluate the effect of H3R2me2a on BRPF2-PHD1, we assayed the interaction between the two using ITC. As shown in supplemental Fig. S7, methylation of H3R2 seriously impaired BRPF2-PHD1 histone binding, with a KD value of ∼700 μm versus 190 μm for H3K4me0. This was consistent with our structural analysis, as H3R2 had key electrostatic interactions with the conserved Asp-235 in BRPF2, and it was consistent with the transcriptional activation role of BRPF proteins.
Recently, it was reported that phosphorylation of histone H3 threonine 3 (H3T3ph) by Haspin is required for recruiting Aurora B protein kinase, an enzyme essential for accurate chromosome inheritance, to chromosomes that are poised to segregate into daughter cells (41, 42). In addition, phosphorylation at threonine 6 (H3T6ph) by PKCβI plays a key role in preventing LSD1 from demethylating H3K4 but not from demethylating H3K9 during AR-dependent gene activation (43). A hallmark of mitosis is the appearance of high levels of histone phosphorylation, and BRPF1 has been suggested to contribute to transcriptional memory throughout mitosis (8). These clues led us to evaluate the cross-talk of threonine phosphorylation. As shown in supplemental Fig. S8, the phosphorylation of H3T3 completely abolished the interaction between BRPF2-PHD1 and the tail of histone H3, whereas phosphorylation of H3T6 strongly reduced this interaction (KD of ∼1.2 mm versus ∼170 μm). Structurally, the methyl group of the H3T3 side chain sits in a hydrophobic pocket of PHD1, and the hydroxyl group hydrogen bonds with the backbone carbonyl of H3A1 (Fig. 3, D and E). Phosphorylated H3T3 may have caused large steric hindrance and, hence, abolished the interaction. By contrast, the H3T6-Asn-229 interaction occurs in a more open space (Fig. 3, C and D). Phosphorylated H3T6 may lose the hydrogen bond with Asn-229, but it can adjust to avoid serious steric hindrance.
BRPF proteins share high sequence similarity with human JADEs and yeast yNto1 in the PHD1 region (see “Discussion” and Fig. 3A), and it has previously been reported that the PHD1 fingers of JADE1 and yNto1 can bind both H3K4me0 and H3K36me3 (35, 44). We examined the H3K36me3 binding ability of BRPF2-PHD1 using ITC and NMR titration. However, no interactions were observed between BRPF2-PHD1 and H3K36me3 (supplemental Fig. S9).
Taken together, the post-translational modifications H3R2me2as, H3T3ph, H3K4me, H3K4ac, and H3T6ph can antagonize the interaction between BRPF2-PHD1 and histone H3, establishing this PHD finger as an unmodified histone H3 (unH3)-recognizing domain.
BRPF2-PHD1 Is Important for Target Gene Binding in Vivo
To test the contribution of unH3 binding by PHD1 to BRPF2 function in vivo, we transfected plasmids containing wild-type full-length FLAG-tagged BRPF2 or full-length BRPF2 that contained either N229A/D235A or D212A/D214A point mutations, which abolish the interaction of the PHD1 finger with histone H3, into 293T cells, along with an empty vector as the control. All three constructs expressed well in 293T cells, and the expression levels were verified by Western blots with an anti-FLAG antibody (Fig. 6A). Afterward, we performed ChIP assays using anti-FLAG antibodies to examine their enrichment on the HOXA9 locus. HOXA9 was selected based on the facts that it is a target of both MOZ (4) and BRPF1 (13) and that it's highly expressed in 293T cells (data not shown). The PCR primers were designed to amplify the region that included the 5′-UTR and start codon of HOXA9. As shown in Fig. 6B, BRPF2 significantly bound to the HOXA9 locus compared with the β-actin gene (lane 10). Moreover, the binding of the two mutant lines was weaker than the wild-type line (lanes 11 and 12 compared with lane 10). Mutations in PHD2, the bromodomain, and the PWWP domain exhibited only slight effects (supplemental Fig. S12). Green fluorescence microscopy showed that wild-type BRPF2 formed speckles in the nucleus in interphase cells (supplemental Fig. S13). Mutations in PHD1 and the PWWP domain resulted in a partial loss of speckles, whereas mutations in PHD2 and the bromodomain had limited effects (supplemental Fig. S13). These results suggested that the binding of unH3 by BRPF2-PHD1 is important, although not solely responsible, for BRPF2 localization at target genes.
FIGURE 6.

Histone binding by PHD1 is important for BRPF2 to bind to the HOXA9 locus in vivo. A, 293T cells were transiently transfected with an empty vector, wild-type full-length FLAG-tagged BRPF2, or full-length BRPF2 that had either N229A/D235A or D212A/D214A point mutations. A Western blot with anti-FLAG antibodies indicated that all three constructs were expressed at high levels. Ctrl, control. B, ChIP assays of the HOXA9 gene from the transfected cells in A are shown. Ab, antibody.
DISCUSSION
Interaction between BRPF2-PHD1 and unH3 Is Conserved in Yeast NuA3-like HAT Complexes
The BRPF proteins and the corresponding HAT complexes are conserved from humans to Caenorhabditis elegans (45). BRPF proteins contain two PHD fingers, PHD1 and PHD2, that are linked by a mononuclear Zn2+ knuckle to form PZPMs (11). Besides BRPF proteins, PZPMs are also found in human JADE1/2/3 and yeast Nto1, scaffold proteins of the human HBO1 HAT complex and yeast NuA3 HAT complex, respectively. These two complexes have similar subunit compositions as the MOZ/MORF complexes (Table S1 and supplemental Fig. S10). Sequence alignments showed that BRPFs, JADEs and yNto1 are highly homologous in the PZPM region (Fig. 3A). Particularly the residues involved in unH3 recognition were highly conserved, and it was previously reported that yNto1 and hJADE1 PHD1 can also bind unH3 (35, 44). The molecular role of BRPF2-PHD2 remains unclear. Although PHD1 and PHD2 are oppositely charged (pI ∼ 4.5 for PHD1 versus ∼9.1 for PHD2), NMR titrations revealed little, if any, interdomain interactions between the isolated domains (supplemental Fig. S11).
Comparison between NuA3-like and NuA4-like HAT complexes further revealed that the PZPM is specific for the NuA3-like complex, whereas ING proteins and EAF6 proteins are present in both NuA3-like and NuA4-like HAT complexes (supplemental Table S1 and supplemental Fig. S10). Moreover, the NuA3-like HATs primarily use histone H3 as a substrate in vivo, whereas the NuA4-like HATs primarily use histone H2A/H4. Whether the PZPMs (especially PHD1 and its interaction with unH3) have a role in distinguishing the substrate specificity remains an open question. In brief, we suggest that the PZPMs and the interaction of PHD1 with unH3 are specific and conserved for NuA3-like HAT complexes.
Potential Roles of Histone Binding Domains in Regulating MOZ/MORF
MOZ/MORF are HATs important for different developmental programs and have been implicated in leukemogenic and other tumorigenic processes. MOZ especially is required for normal levels of H3K9 acetylation and gene expression at a large number of Hox loci (3). There are multiple histone binding domains in the MOZ/MORF complexes, including the BRPF PHD1 finger binding H3K4me0 (KD ∼ 190 μm), the BRPF PWWP domain binding H3K36me3 (K_D_∼ 2.7 mm) (13), the BRPF bromodomain binding acetylated histones (KD typically in mm range), and the ING5 PHD finger binding H3K4me3 (_KD_∼ 2.4 μm) (46). Interestingly, MOZ itself is also able to bind H3K4me3 (47). The presence of multiple histone binding domains in a single complex raises the following questions. Why are so many histone binding domains needed, and how do they cooperate with each other to function? In the past, histone post-translational modifications, which lead to the recruitment of protein complexes that regulate transcription, were simply divided into either activating- or repressing-type modifications. Histone modification “readers” consequently play the role of either activating or repressing target gene expression. However, further exploration questions the one modification-one reader-one function mode (48–50). In fact, there is no clear correlation between a single epigenetic signature and its functional readout, as the same epigenetic modification can be recognized by various readers and consequently result in various effects, depending on the cellular context. For example, H3K4me0 recognition can mediate gene activation or gene repression, as observed for AIRE-PHD1 (51) and BHC80-PHD (15), respectively. In addition, recognition of H3K4me0 by the ADD domain of DNMT3L has been shown to be connected to de novo DNA methylation (27). It seems likely that multiple readers are required to cooperate to read out histone code combinatorially. However, the cooperation mechanism is poorly understood.
It has been proposed that histone modifiers, such as histone acetyltransferase complexes, function in a stepwise manner (48). First, they receive and interpret the signals (e.g. histone modification patterns), which are site-specific and are recruited to the target sites. Then they are further stabilized on the chromatin template to allow the core proteins (e.g. MOZ/MORF) to work efficiently with their substrates. For primary recruitment, multivalent interactions may bring about composite substrate specificity, which is greater than the intrinsic specificity of any of the discrete binding interactions (49). As for MOZ/MORF complexes, BRPF-PHD1 plays potential roles in regulating their preference for histone H3. For the secondary stabilization, multiple modest interactions may drastically improve the substrate affinity through an increase in the probability of rebinding to one of the effector modules in the same complex (48). In the case of the MOZ/MORF complex, we propose that the PHD1 fingers of BRPF proteins are required to strengthen the binding of MOZ/MORF to their substrates. In vitro histone acetyltransferase assays showed that the HAT activity of MORF toward H3 peptide increased when BRPF1 was added. Dimethylation of H3K4, but not H3K9, antagonized this increase in HAT activity (46), consistent with the histone binding affinity of PHD1. Our ChIP assays also showed that disrupting the interaction between BRPF2-PHD1 and histone did not abolish its targeting to the HOXA9 locus (Fig. 6B). One explanation is that the interaction between BRPF-PHD1 and unH3 is not involved in the recruitment of the MOZ/MORF complex to the target sites; rather, it functions to strengthen the binding to chromatin.
In addition, HAT complexes are regulated dynamically because global and local remodeling of chromatin architecture occurs throughout the cell cycle and in response to external stimuli. It should be noted that all the interactions between histones and the corresponding domains in MOZ/MORF complexes are relatively weak. The presence of multiple histone binding domains in one complex offers a good solution to balance the requirements between dynamic regulation and stabilizing a specific complex at a locus after initial recruitment. Weak multivalent interactions are more susceptible to competition due to fast individual on/off rates and thus are more suited to the permitting dynamics of a highly valent chromatin substrate. In conclusion, combinations of multiple histone-binding domains most likely greatly increase the substrate specificity, affinity, and dynamics of HAT complexes (48, 49).
BRPF2-PHD1 Binding H3K4me0 versus ING5-PHD Binding H3K4me3
There are two histone binding PHD fingers in the MOZ/MORF complex, i.e. BRPF2-PHD1 binding H3K4me0 and ING5-PHD binding H3K4me3. It seems puzzling how they collaborate to function. Generally, high levels of H3K4me3 are detected in the promoter regions of active genes (52, 53). However, no information about the genomic localization of H3K4me0 is currently available. It is possible that H3K4me0 and H3K4me3 coexist in the same nucleosome or adjacent nucleosomes, as the H3K4 methylation level is relatively low in certain genomic regions (52, 53). We propose that BRPF2-PHD1 and ING5-PHD can bind their substrates in the same nucleosome or adjacent nucleosomes. Also, ING5-PHD may play a regulatory role in response to signaling cascades, which means it can be present or absent in the MOZ/MORF complex. Indeed, for HBO1, another NuA3-like HAT, two complex architectures are present, one with ING4/5 and the other without ING4/5 (44).
In summary, our study reveals the molecular role of the BRPF2-PHD1 as an unH3 binding module and provides details of the structural basis for this interaction. We also find that unH3 binding by PHD1 is important for BRPF2 to localize at target genes in vivo. Moreover, this PHD finger is highly conserved in yeast NuA3-like HAT complexes, shedding light on a conserved mechanism regulating such HATs. However, the precise molecular mechanism by which BRPF2 regulates MOZ/MORF HATs and gene expression needs to be further studied.
Supplementary Material
Supplemental Data
Acknowledgments
We thank Prof. Xiang-Jiao Yang (McGill University) for helpful discussions and critical editing of this manuscript. We also thank Dr. A. Kwan (University of Sydney) for kindly helping with the structural calculations.
*
This work was supported by Chinese National Natural Science Foundation Grant 30830031, National Basic Research Program of China (973 Program) Grants 2011CB966302 and 2011CB911104, and Chinese National High Technology R&D Program Grant 2006AA02A315.
The atomic coordinates and structure factors (codes 2KU3 and 2L43) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
3
The abbreviations used are:
HAT
histone acetyltransferase
unH3
unmodified histone H3 tail
PZPM
PHD/ Zn2+-knuckle/PHD motif
BRPF
bromodomain-PHD finger protein
HSQC
heteronuclear single quantum correlation
ITC
isothermal titration calorimetry
ING5
inhibitor of growth 5
EAF6
Esa1-associated factor 6
Bis-Tris
2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
PHD
plant homeodomain
AIRE
autoimmune regulation
TOCSY
total correlation spectroscopy
cidh
dihedral angle restraint.
REFERENCES
- 1.Lee K. K., Workman J. L. (2007) Nat. Rev. Mol. Cell Biol. 8, 284–295 [DOI] [PubMed] [Google Scholar]
- 2.Yang X. J., Ullah M. (2007) Oncogene 26, 5408–5419 [DOI] [PubMed] [Google Scholar]
- 3.Voss A. K., Collin C., Dixon M. P., Thomas T. (2009) Dev. Cell 17, 674–686 [DOI] [PubMed] [Google Scholar]
- 4.Katsumoto T., Aikawa Y., Iwama A., Ueda S., Ichikawa H., Ochiya T., Kitabayashi I. (2006) Genes Dev. 20, 1321–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomas T., Voss A. K. (2004) Front. Biosci. 9, 24–31 [DOI] [PubMed] [Google Scholar]
- 6.Doyon Y., Cayrou C., Ullah M., Landry A. J., Côté V., Selleck W., Lane W. S., Tan S., Yang X. J., Côté J. (2006) Mol. Cell 21, 51–64 [DOI] [PubMed] [Google Scholar]
- 7.Ullah M., Pelletier N., Xiao L., Zhao S. P., Wang K., Degerny C., Tahmasebi S., Cayrou C., Doyon Y., Goh S. L., Champagne N., Côté J., Yang X. J. (2008) Mol. Cell. Biol. 28, 6828–6843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laue K., Daujat S., Crump J. G., Plaster N., Roehl H. H., Kimmel C. B., Schneider R., Hammerschmidt M. (2008) Development 135, 1935–1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Severinsen J. E., Bjarkam C. R., Kiaer-Larsen S., Olsen I. M., Nielsen M. M., Blechingberg J., Nielsen A. L., Holm I. E., Foldager L., Young B. D., Muir W. J., Blackwood D. H., Corydon T. J., Mors O., Børglum A. D. (2006) Mol. Psychiatry 11, 1126–1138 [DOI] [PubMed] [Google Scholar]
- 10.Nebral K., Denk D., Attarbaschi A., König M., Mann G., Haas O. A., Strehl S. (2009) Leukemia 23, 134–143 [DOI] [PubMed] [Google Scholar]
- 11.Perry J. (2006) BMC Genomics 7, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeng L., Zhou M. M. (2002) FEBS Lett. 513, 124–128 [DOI] [PubMed] [Google Scholar]
- 13.Vezzoli A., Bonadies N., Allen M. D., Freund S. M., Santiveri C. M., Kvinlaug B. T., Huntly B. J., Göttgens B., Bycroft M. (2010) Nat. Struct. Mol. Biol. 17, 617–619 [DOI] [PubMed] [Google Scholar]
- 14.Shi X., Hong T., Walter K. L., Ewalt M., Michishita E., Hung T., Carney D., Peña P., Lan F., Kaadige M. R., Lacoste N., Cayrou C., Davrazou F., Saha A., Cairns B. R., Ayer D. E., Kutateladze T. G., Shi Y., Côté J., Chua K. F., Gozani O. (2006) Nature 442, 96–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lan F., Collins R. E., De Cegli R., Alpatov R., Horton J. R., Shi X., Gozani O., Cheng X., Shi Y. (2007) Nature 448, 718–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clore G. M., Gronenborn A. M. (1994) Methods Enzymol. 239, 349–363 [DOI] [PubMed] [Google Scholar]
- 17.Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 18.Cornilescu G., Delaglio F., Bax A. (1999) J. Biomol. NMR 13, 289–302 [DOI] [PubMed] [Google Scholar]
- 19.Simonson T., Calimet N. (2002) Proteins 49, 37–48 [DOI] [PubMed] [Google Scholar]
- 20.Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Acta Crystallogr. D. Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 21.Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., Thornton J. M. (1996) J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 22.Koradi R., Billeter M., Wuthrich K. (1996) J. Mol. Graph. 14, 51–55, 29–32 [DOI] [PubMed] [Google Scholar]
- 23.Shen W., Xu C., Huang W., Zhang J., Carlson J. E., Tu X., Wu J., Shi Y. (2007) Biochemistry 46, 2100–2110 [DOI] [PubMed] [Google Scholar]
- 24.Farrow N. A., Muhandiram R., Singer A. U., Pascal S. M., Kay C. M., Gish G., Shoelson S. E., Pawson T., Forman-Kay J. D., Kay L. E. (1994) Biochemistry 33, 5984–6003 [DOI] [PubMed] [Google Scholar]
- 25.Xie W., Jin L., Mei Y., Wu M. (2009) J. Cell Mol. Med. 13, 1719–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chignola F., Gaetani M., Rebane A., Org T., Mollica L., Zucchelli C., Spitaleri A., Mannella V., Peterson P., Musco G. (2009) Nucleic Acids Res. 37, 2951–2961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ooi S. K., Qiu C., Bernstein E., Li K., Jia D., Yang Z., Erdjument-Bromage H., Tempst P., Lin S. P., Allis C. D., Cheng X., Bestor T. H. (2007) Nature 448, 714–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Otani J., Nankumo T., Arita K., Inamoto S., Ariyoshi M., Shirakawa M. (2009) EMBO Rep. 10, 1235–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H., Ilin S., Wang W., Duncan E. M., Wysocka J., Allis C. D., Patel D. J. (2006) Nature 442, 91–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Ingen H., van Schaik F. M., Wienk H., Ballering J., Rehmann H., Dechesne A. C., Kruijzer J. A., Liskamp R. M., Timmers H. T., Boelens R. (2008) Structure 16, 1245–1256 [DOI] [PubMed] [Google Scholar]
- 31.Taverna S. D., Ilin S., Rogers R. S., Tanny J. C., Lavender H., Li H., Baker L., Boyle J., Blair L. P., Chait B. T., Patel D. J., Aitchison J. D., Tackett A. J., Allis C. D. (2006) Mol. Cell 24, 785–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karagianni P., Amazit L., Qin J., Wong J. (2008) Mol. Cell. Biol. 28, 705–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li F., Huarte M., Zaratiegui M., Vaughn M. W., Shi Y., Martienssen R., Cande W. Z. (2008) Cell 135, 272–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iwase S., Lan F., Bayliss P., de la Torre-Ubieta L., Huarte M., Qi H. H., Whetstine J. R., Bonni A., Roberts T. M., Shi Y. (2007) Cell 128, 1077–1088 [DOI] [PubMed] [Google Scholar]
- 35.Shi X., Kachirskaia I., Walter K. L., Kuo J. H., Lake A., Davrazou F., Chan S. M., Martin D. G., Fingerman I. M., Briggs S. D., Howe L., Utz P. J., Kutateladze T. G., Lugovskoy A. A., Bedford M. T., Gozani O. (2007) J. Biol. Chem. 282, 2450–2455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zeng L., Zhang Q., Li S., Plotnikov A. N., Walsh M. J., Zhou M. M. (2010) Nature 466, 258–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garcia B. A., Hake S. B., Diaz R. L., Kauer M., Morris S. A., Recht J., Shabanowitz J., Mishra N., Strahl B. D., Allis C. D., Hunt D. F. (2007) J. Biol. Chem. 282, 7641–7655 [DOI] [PubMed] [Google Scholar]
- 38.Musselman C. A., Mansfield R. E., Garske A. L., Davrazou F., Kwan A. H., Oliver S. S., O'Leary H., Denu J. M., Mackay J. P., Kutateladze T. G. (2009) Biochem. J. 423, 179–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kirmizis A., Santos-Rosa H., Penkett C. J., Singer M. A., Vermeulen M., Mann M., Bähler J., Green R. D., Kouzarides T. (2007) Nature 449, 928–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vermeulen M., Mulder K. W., Denissov S., Pijnappel W. W., van Schaik F. M., Varier R. A., Baltissen M. P., Stunnenberg H. G., Mann M., Timmers H. T. (2007) Cell 131, 58–69 [DOI] [PubMed] [Google Scholar]
- 41.Wang F., Dai J., Daum J. R., Niedzialkowska E., Banerjee B., Stukenberg P. T., Gorbsky G. J., Higgins J. M. (2010) Science 330, 231–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kelly A. E., Ghenoiu C., Xue J. Z., Zierhut C., Kimura H., Funabiki H. (2010) Science 330, 235–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Metzger E., Imhof A., Patel D., Kahl P., Hoffmeyer K., Friedrichs N., Müller J. M., Greschik H., Kirfel J., Ji S., Kunowska N., Beisenherz-Huss C., Günther T., Buettner R., Schüle R. (2010) Nature 464, 792–796 [DOI] [PubMed] [Google Scholar]
- 44.Saksouk N., Avvakumov N., Champagne K. S., Hung T., Doyon Y., Cayrou C., Paquet E., Ullah M., Landry A. J., Côté V., Yang X. J., Gozani O., Kutateladze T. G., Côté J. (2009) Mol. Cell 33, 257–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O'Meara M. M., Zhang F., Hobert O. (2010) Genetics 186, 1497–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Champagne K. S., Saksouk N., Peña P. V., Johnson K., Ullah M., Yang X. J., Côté J., Kutateladze T. G. (2008) Proteins 72, 1371–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paggetti J., Largeot A., Aucagne R., Jacquel A., Lagrange B., Yang X. J., Solary E., Bastie J. N., Delva L. (2010) Oncogene 29, 5019–5031 [DOI] [PubMed] [Google Scholar]
- 48.Ruthenburg A. J., Allis C. D., Wysocka J. (2007) Mol. Cell 25, 15–30 [DOI] [PubMed] [Google Scholar]
- 49.Ruthenburg A. J., Li H., Patel D. J., Allis C. D. (2007) Nat. Rev. Mol. Cell Biol. 8, 983–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berger S. L. (2007) Nature 447, 407–412 [DOI] [PubMed] [Google Scholar]
- 51.Org T., Chignola F., Hetényi C., Gaetani M., Rebane A., Liiv I., Maran U., Mollica L., Bottomley M. J., Musco G., Peterson P. (2008) EMBO Rep. 9, 370–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barski A., Cuddapah S., Cui K., Roh T. Y., Schones D. E., Wang Z., Wei G., Chepelev I., Zhao K. (2007) Cell 129, 823–837 [DOI] [PubMed] [Google Scholar]
- 53.Bernstein B. E., Kamal M., Lindblad-Toh K., Bekiranov S., Bailey D. K., Huebert D. J., McMahon S., Karlsson E. K., Kulbokas E. J., 3rd, Gingeras T. R., Schreiber S. L., Lander E. S. (2005) Cell 120, 169–181 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data