Tetraspanin15 regulates cellular trafficking and activity of the ectodomain sheddase ADAM10 (original) (raw)
Abstract
A disintegrin and metalloproteinase10 (ADAM10) has been implicated as a major sheddase responsible for the ectodomain shedding of a number of important surface molecules including the amyloid precursor protein and cadherins. Despite a well-documented role of ADAM10 in health and disease, little is known about the regulation of this protease. To address this issue we conducted a split-ubiquitin yeast two-hybrid screen to identify membrane proteins that interact with ADAM10. The yeast experiments and co-immunoprecipitation studies in mammalian cell lines revealed tetraspanin15 (TSPAN15) to specifically associate with ADAM10. Overexpression of TSPAN15 or RNAi-mediated knockdown of TSPAN15 led to significant changes in the maturation process and surface expression of ADAM10. Expression of an endoplasmic reticulum (ER) retention mutant of TSPAN15 demonstrated an interaction with ADAM10 already in the ER. Pulse-chase experiments confirmed that TSPAN15 accelerates the ER-exit of the ADAM10–TSPAN15 complex and stabilizes the active form of ADAM10 at the cell surface. Importantly, TSPAN15 also showed the ability to mediate the regulation of ADAM10 protease activity exemplified by an increased shedding of N-cadherin and the amyloid precursor protein. In conclusion, our data show that TSPAN15 is a central modulator of ADAM10-mediated ectodomain shedding. Therapeutic manipulation of its expression levels may be an additional approach to specifically regulate the activity of the amyloid precursor protein alpha-secretase ADAM10.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-012-0960-2) contains supplementary material, which is available to authorized users.
Keywords: ADAM10, Tetraspanin, APP, Cadherin
Introduction
Since the discovery of ADAM10 as the key protease involved in the alpha-secretase processing of the amyloid precursor protein (APP) knowledge about the spectrum of other substrates cleaved by this protease and its role in a wide range of physiological functions has increased [1, 2]. ADAM10 is a ubiquitously expressed proteinase of the large family of ADAMs. It is synthesized as an inactive proform in the endoplasmic reticulum (ER) and is activated by the removal of its prodomain by furin or proprotein convertases during transport to the cell surface [3–5]. ADAM10 is highly expressed in the central nervous system (CNS) [6] and is thought to be responsible for the ectodomain shedding of neuronal cell surface proteins such as the ephrins [7, 8], prion protein [9, 10], the neuronal adhesion molecule [11], L1 adhesion molecule [12], N-cadherin [13] and importantly APP [14–16]. The phenotype of classical and conditional knockout mice for ADAM10 underlines the central role of ADAM10 in the development of the brain through the regulation of early cell fate decisions [17, 18]. This can be explained by the role of ADAM10 in the proteolytic processing of the Notch receptor, which mediates lateral inhibition during neurogenesis [19]. The overexpression of ADAM10 in mice suggested a beneficial role of this protease to prevent the excessive accumulation of the amyloid beta peptide in Alzheimer's disease (AD) mouse models [14] through activation of the non-amyloidogenic alpha-secretase pathway. Based on these findings strategies to upregulate ADAM10 expression for the treatment of AD have been suggested. All-trans retinoic acid led to an increased expression of the mature form of ADAM10 and shifted the processing of APP in the direction of the non-amyloidogenic pathway [20–22]. In addition, members of the tetraspanin family have been identified as ADAM modulators [23, 24]. This family is composed of more than 30 members in mammals, spanning the membrane four times and forming a small and a large extracellular loop (LEL) and carrying a conserved Cys–Cys–Gly motif in the LEL domain [25, 26]. Stable protein assembly is most likely mediated through conserved polar amino acid residues in the transmembrane domains [27]. Tetraspanins, defined as “molecular facilitators,” tend to associate with other molecules, such as growth factors and their receptors, signaling enzymes, integrins, proteins of the immunoglobulin superfamily, proteoglycans, complement regulatory proteins and other tetraspanins [28, 29]. Specific cell surface proteins are grouped by tetraspanins. This modulates the formation and stability of functional signaling complexes involved in cell activation, adhesion, motility, differentiation and malignancy [29–31]. The tetraspanin-enriched microdomains (TEMs) are also known to regulate shedding and intramembrane proteolysis [32, 33]. In this context ADAM10 was shown to interact with CD9, CD81, CD82 and TSPAN12 [23, 24]. These interactions may regulate APP alpha-secretase activity through stabilization and by increasing activity of the protease.
Using a yeast split-ubiquitin based screening system we identified TSPAN15 as a specific binding partner of ADAM10. This interaction occurs already in the ER and triggers the transport of the activated form of ADAM10 to the cell surface. This leads in turn to an increased shedding of known substrates of ADAM10 such as N-cadherin and APP. Our data suggest that an altered expression of members of the tetraspanin family may be exploited as a therapeutic tool to modulate ADAM10-mediated shedding events.
Materials and methods
Antibodies, cells, transfection and plasmids
The following antibodies were used: B42.1, polyclonal rabbit anti-mouse ADAM10 serum (gift of Wim Annaert, Leuven), anti-ADAM10 antibody raised against a peptide corresponding to the C-terminus of murine ADAM10 (used for immunoprecipitation and pulse-chase experiments), anti-actin (IB, Sigma-Aldrich, Hamburg, Germany), MAB946 anti-ADAM10 ectodomain (FACS and IF, R&D Systems, Wiesbaden, Germany), anti-human ADAM10 antibody (11G2) (Gen-Probe, Wiesbaden, Germany), anti-KDEL antibody (Enzo Life Science, Lörrach, Germany), anti-PDI antibody (Santa Cruz Biotechnology, Santa Cruz, USA), 9B11 anti-myc (IP, IB and IF, Cell Signaling Technology, Frankfurt am Main, Germany), B63.3 polyclonal rabbit anti-mouse APP C-terminus (IB, a kind gift of Wim Annaert, Leuven, Belgium), H68.4 anti-transferrin receptor (IB, Life Technologies GmbH, Darmstadt, Germany); FL-335 anti-GAPDH (Santa Cruz Biotechnology, Heidelberg, Germany) and anti N-cadherin (BD Transduction Laboratories, Heidelberg, Germany).
All cells (HeLa, Cos7, N2A, SHSY, HEK) were cultured in DMEM (high glucose) (PAA Laboratories, Cölbe, Germany) with 10 % fetal calf serum (FCS) and 1 % penicillin/streptomycin at 37 °C, 5 % CO2 atmosphere and 95 % relative humidity. Cells were transiently transfected with FuGENE HD (Roche, Mannheim, Germany) according to the manufacturer’s instructions. Mammalian expression vectors were pcDNA3.1Hygro- and pFrog3 derived from pcDNA3 (Invitrogen, Life Technologies, Darmstadt, Germany) [34]. The myc-epitope was inserted terminally of the last amino acid of TSPAN15.
Split-ubiquitin membrane yeast two-hybrid screening
The split-ubiquitin yeast two-hybrid screen was performed according to the manufacturer’s instructions (MoBiTec, Göttingen, Germany). Briefly, murine ADAM10 cDNA was cloned into a yeast expression vector (pTMBV4), C-terminal fused to a Cub-LexA-VP16 cassette and used as a bait protein. Autoactivation of the bait protein was checked on selective media plates lacking leucine (Leu) and histidine (His). Expression of the ADAM10 bait construct was monitored using adequate positive (NubI) and negative (NubG) controls as supplied by the manufacturer. Yeast expressing the ADAM10 bait construct was transfected with a N-terminal NubG-tagged murine brain library (MoBiTec), seeded on selective media plates (-Leu, -Trp, -His) and incubated for 3–5 days at 30 °C. Subsequently, clones were selected and expanded, prey plasmids extracted and analyzed by sequencing (GATC, Konstanz, Germany).
Co-immunoprecipitations
Cells were lysed with buffer (120 mM NaCl, 50 mM Tris-HCl, complete proteinase inhibitor cocktail, pH 7.4) containing 0.5 % NP-40 (Sigma-Aldrich, Hamburg, Germany) on ice; 60 μl of lysate was removed as input control. Residual lysate amounts were incubated with primary antibody overnight at 4 °C. Then 50 μl of Protein G agarose beads (Thermo Scientific, Bonn, Germany) were washed twice with lysis buffer and added to the lysate-antibody complexes for 30 min at 4 °C. After the final washing steps, the beads were heated in 2× sample loading buffer to elute immunoprecipitated proteins, which were then loaded on 10 % SDS-PAGE gels [35, 36].
TSPAN15 knockdown
N2A cells were seeded in six-well culture dish plates and transfected with 140 pmol of either murine TSPAN15 (Silencer® siRNA no. s88814 and s88815) or control siRNA using Lipofectamin2000 (all Invitrogen, Life Technologies) according to the manufacturer’s instructions. The procedure was repeated after 24 h, and cells were harvested 48 h post first transfection.
Immunoblotting
Cell lysates were prepared as described previously [13]. Proteins were separated on 10 % SDS-PAGE gels or 4–12 % gradient BIS/Tris NuPAGE® gels (Invitrogen, Darmstadt, Germany) and transferred to nitrocellulose (Whatman, GE Healthcare, Munich, Germany) or PVDF (Roth, Karlsruhe, Germany) membranes by tank blotting for 2 h at 4 °C. After antibody incubations and washing Western blots were developed using the ECL Advanced Detection System (GE Healthcare, Freiburg, Germany) and detected using a chemoluminescence detection system LAS4000 (GE Healthcare, München, Germany). Band intensities were quantified using Image J calculation software (http://rsbweb.nih.gov/ij/).
Flow cytometry
Cells were washed with PBS and detached by applying Accutase (PAA Laboratories). Thereafter, all steps were performed on ice; 5 × 105 cells per sample were incubated with 1 μl anti-ADAM10 antibody (MAB946, R&D System, Wiesbaden, Germany) for 1 h. Cells were washed with buffer (PBS containing 1 % BSA and 0.01 % NaN3) twice and incubated with 1 μl of an IgG2b goat anti-rat Alexa-488 labeled secondary antibody for 1 h. Afterwards cells were washed three times using PBS. The addition of 0.5 μl propidium-iodide was used to control for dead cells during measurements.
Surface biotinylation
Biotinylation of surface proteins was performed as described previously [37]. In brief, cells were washed with ice-cold PBS-CM (0.1 mM CaCl2, 1 mM MgCl2 in PBS) and then covered with a solution containing 1 mg/ml biotin/PBS-CM (+biotin) or PBS-CM (-biotin) for 30 min. After incubation for 10 min with quenching buffer (50 mM Tris-HCl/PBS-CM, pH 8.0), cells were washed three times with PBS-CM, collected and lysed with pulldown buffer [50 mM Tris-HCl, 150 mM NaCl, 1 % (w/v) Triton X-100, 0.1 % (w/v) SDS, complete proteinase inhibitor cocktail (Roche, Mannheim, Germany)] using the standard procedure. Then 10 % of cell lysate was removed as expression control (total) and prepared for SDS-PAGE. Residual lysate amounts were incubated with streptavidin-coupled Agarose-Beads (Thermo Scientific) for 1 h. These fractions (bound) were washed four times with pulldown buffer and finally prepared for SDS-PAGE and immunoblotting.
Determination of sAPPα in cell culture supernatants by ELISA
One-day post transfection, media of cultured cells was renewed (2 ml per 6-well), and supernatants were collected after 24 h. ELISA kits for sAPPα (mouse, rat; product no. JP27415) were obtained from IBL International (Hamburg, Germany) and performed according to the manufacturer’s instructions.
Quantitative RT-PCR
RNA was extracted from treated N2A cells using the NucleoSpin RNAII Kit (Macherey-Nagel, Düren, Germany). RNA (2 μg) was reversely transcribed with the RevertAid™ First Strand cDNA Synthesis Kit (Fermentas, St. Leon Rot, Germany) using a oligo(dT)18 primer according to the manufacturer's protocol. ADAM10 (Mm00545742_m1, Applied Biosystems, Foster City, CA, USA), TSPAN15 (Mm01150417_m1, Applied Biosystems) and GAPDH (Mm99999915_g1, Applied Biosystems) expression were determined by real-time PCR analysis of 0.5 μl cDNA on a 7900HT Fast Real-time PCR System (Applied Biosystems) in 10-μl reaction volume in 384-well plates in duplicate. Measurements of ADAM9, ADAM17 and TSPAN12 were performed on a LightCycler480 (Roche Diagnosics, Rotkreuz, Switzerland) under conditions as described above with the Universal Probe Library System (UPL, Roche) using the following primer/probes combinations: ADAM9: tccggcagtgagtacaagaa (for.), gcattgaagctttccacaca (rev.), actgggaa (UPL no. 48); ADAM17: ctttggtgcctttcgtcct (for.), gagcaaagaatcaagcttctcaa (rev.), ggaagcag (UPL no. 38); TSPAN12: aactggcttgcggtgtgt (for.); tcaaagtaaccatatctgaccactg (rev.); gagcagga (UPL no. 15). The expression levels of the genes were depicted as the percentage of GAPDH expression using the Δ_C_ t for calculation. The actual PCR efficiency of each assay was determined previously by serial dilution of standards and these values were used for calculation. The t tests were performed on the mean Δ_C_ t values of the technical duplicate measurements.
Pulse-chase labeling
N2A cells were transfected with expression constructs for either EGFP or TSPAN15-myc; 24-h post transfection, cells were starved in DMEM High Glucose 1x (Invitrogen) lacking l-methionine and l-cysteine for 3 h. Cells were then pulsed for 1 h using 200 μCi/ml 35S cysteine/methionine (Hartmann Analytic GmbH, Braunschweig, Germany) and subsequently chased in DMEM (10 % FCS, 1 % penicillin/streptomycin). Cells were harvested 0, 2, 6, 18 and 30 h after pulse and lysed, and ADAM10 was isolated by immunoprecipitation using a C-terminal anti-murine ADAM10 antibody. Immunoprecipitates were separated by SDS-PAGE and analyzed by fluorographics using the Enhance system (Perkin Elmer, Rodgau, Germany).
Immunofluorescence analysis
Cos7 and SHSY cells were grown on coverslips in six wells; 36 h post transfection, cells were washed three times with ice-cold PBS, fixed with 4 % (w/v) paraformaldehyde in PBS and permeabilized with 0.2 % saponin, which was added in all subsequent incubation steps, and 0.12 % glycine in PBS. Non-specific binding sites have been blocked by pre-incubation with 10 % (v/v) FCS in PBS for 1 h, which was also used as diluent for primary antibodies. Coverslips were then incubated with solutions of primary antibodies for 1 h (MAB946, 1:500; 9B11, 1:1,000; KDEL, 1:300, 11G2, 1:500, PDI 1:300). Afterwards, cells were washed five times with 0.2 % saponin/PBS and then incubated for 1 h with Alexa®-488 and -594 labeled secondary antibodies. Again, cells were washed five times with washing buffer plus two additional washing steps in ddH2O. Coverslips were mounted with mowiol containing 0.1 % DAPI (4-,6-diamidino-2-phenylindole; Sigma-Aldrich, Hamburg, Germany) for nucleus staining. Confocal pictures were acquired with an Olympus FV1000 confocal laser scanning microscope (Hamburg, Germany).
Statistical analysis
Statistical significance was performed as unpaired Student’s t test using Microsoft Excel software. Error bars indicate the ± standard deviation. p values: *p < 0.05, **p < 0.01, ***p < 0.005.
Results
ADAM10-TSPAN15 interaction in yeast
To unravel putative modulators of ADAM10-mediated ectodomain shedding, we used a split-ubiquitin yeast screening assay to identify new interacting proteins [38]. Mouse brain cDNAs encoding for N-terminal tagged proteins were used as prey and murine (m) ADAM10 as bait. Close proximity of bait and prey proteins led to the reconstitution of ubiquitin, which finally led to the transcriptional activation of the histidine 3 gene (Fig. 1a), allowing yeast clones to grow on selective media plates. TSPAN15 was identified as an interacting protein. Specific interaction with ADAM10 was verified after reexpression in yeast, allowing the growth on selective media plates (Fig. 1b). To validate this interaction in mammalian cells we expressed a C-terminal myc-tagged mouse TSPAN15 and wild-type mouse ADAM10 in HeLa cells and co-precipitated both proteins with anti-myc and anti-ADAM10 antibodies, respectively. ADAM10 could be found in the precipitate after the pulldown of TSPAN15 (Fig. 1c-I), and vice versa TSPAN15 was found in the precipitate after ADAM10 pulldown (Fig. 1c-II). The band intensities of the pro and mature form of ADAM10 of the lysate and the co-immunoprecipitated fractions were quantified and revealed that only minor amounts of mature ADAM10 were co-precipitated, indicating a strong interaction between the proform of ADAM10 and TSPAN15 (Fig. 1c-III). Based on results in two independent systems these data provide strong evidence that ADAM10 and TSPAN15 physically interact.
Fig. 1.

TSPAN15, a new ADAM10 interaction partner. a-I, II: Split-ubiquitin yeast two-hybrid system (modified from [50]). ADAM10 C-terminally fused to the C-terminal part of ubiquitin (Cub) and an artificial transcription factor (LexA-VP16) coexpressed in yeast together with a N-terminal NubG-tagged murine brain library. The close proximity between the ADAM10 bait protein and an interaction partner leads to the reconstitution of “split ubiquitin” to ubiquitin, which is recognized by cellular ubiquitin proteases, which in turn release the artificial transcription factor from the membrane. This enables yeast to grow on selective media [without leucine (-leu), tryptophane (-trp), histidine (-his)] plates. C-terminal part of ubiquitin (Cub, C), N-terminal part of ubiquitin (NubG, N), LexA (L). b ADAM10 bait protein coexpressed with the identified TSPAN15 prey protein and controls in NMY51 yeast. Transfection of both bait and prey protein is verified on Leu/Trp-lacking selective media plates, and interaction of ADAM10 and TSPAN15 is monitored under selective pressure on Leu/Trp/His-lacking media plates in comparison to controls (“+” and “−”). **c-**I: Mammalian expression constructs of murine ADAM10 and murine TSPAN15-myc were transiently coexpressed in HeLa cells. TSPAN15-myc (35–37 kDa) was precipitated using an anti-myc antibody, and coprecipitation of ADAM10 was detected with an anti-ADAM10 antibody. II: ADAM10 and TSPAN15-myc were transiently coexpressed in HeLa cells, and ADAM10 was precipitated using an anti-ADAM10 antibody. Coprecipitation was analyzed by Western blot using an anti-myc antibody. Single transfections of TSPAN15-myc and ADAM10 served as specificity controls for the antibodies used for immunoprecipitation. III: Quantification of band intensities of pro and mature form of ADAM10 in lysates and CoIP fractions. Ratios of band intensities of pro/(pro + mature) forms of ADAM10 were calculated (%, n = 5). Student’s t test was performed (***p < 0.005). [pro(p) ADAM10, 95 kDa, mature(m) ADAM10, 75 kDa]. Abbreviations: untransfected (Ø), vector (mock) transfected (V), murine ADAM10 (A10), murine TSPAN15-myc (T15), asterisk marks immunoglobulin signals
TSPAN15 expression leads to increased maturation of ADAM10
To address the question if this interaction has any impact on the function of ADAM10, we heterologously expressed ADAM10 and C-terminal myc-tagged TSPAN15 in Cos7 cells. Expression of ADAM10 led to an increased ADAM10 proform, whereas only a minor increase of the mature form of ADAM10 was detectable. Interestingly, coexpression of TSPAN15 and ADAM10 led to a striking increase of the mature form of ADAM10, while the signal of the proform remained unaltered (Fig. 2a). Accordingly, expression of TSPAN15 in the murine neuroblastoma cell line N2A significantly influenced endogenous ADAM10 protein, showing decreased pro-ADAM10, but drastically increased mature ADAM10 (Fig. 2b).
Fig. 2.

TSPAN15-myc influences ADAM10 maturation. a Murine ADAM10 transiently coexpressed with either EGFP or murine TSPAN15-myc in Cos7 cells. After immunoblotting ADAM10 was detected using an ADAM10 specific C-terminal antibody (B42.1) and TSPAN15-myc was detected using an anti-myc antibody. b TSPAN15-myc or EGFP was transiently expressed in N2A cells. After lysis proteins were immunoblotted, and endogenous ADAM10 and TSPAN15-myc were detected. c Murine ADAM17 or murine ADAM10 was transiently expressed either with EGFP or TSPAN15-myc in HeLa cells. Presence of ADAM17 [pro(p) ADAM17, closed arrowhead, 120 kDa; putative mature ADAM17, _open arrowhead_] was analyzed using a C-terminal-specific antibody, and TSPAN15-myc and ADAM10 expressions were monitored as mentioned above. Actin served as protein loading control. Asterisks mark unspecific antibody binding
To exclude any transcriptional influence on the ADAM10 expression level due to TSPAN15-myc overexpression, qRT-PCR experiments were performed. TSPAN15-myc expression had no influence on ADAM10 mRNA expression and vice versa (Suppl. Fig. 1). In order to analyze if TSPAN15 acts specifically on ADAM10, we also coexpressed the related protease ADAM17. These experiments revealed no effect on the maturation of ADAM17, whereas the effect of the generation of the active mature form of ADAM10 could be reproduced in HeLa cells. Interestingly, higher levels of TSPAN15 expression could be detected when cotransfected with ADAM10 in contrast to ADAM17 co-transfections (Fig. 2c). Taken together, our data revealed TSPAN15 as a specific ADAM10 binding partner leading to an increased maturation and apparent stabilization of this protease.
TSPAN15 expression increases ADAM10 cell surface levels
The effect of TSPAN15 expression on the maturation state of ADAM10 suggested that trafficking of the protease from the endoplasmic reticulum (ER) through the secretory route to the cell surface is also accelerated upon coexpression of TSPAN15. To analyze if at steady state the expression of TSPAN15 causes a change in the subcellular localization of ADAM10, we expressed the protease alone or in combination with TSPAN15-myc in Cos7 cells (Fig. 3a). Whereas ADAM10 overexpression led to intracellular staining mainly within the ER (Fig. 3a-I) and Golgi structures (not shown), the simultaneous expression of TSPAN15-myc (Fig. 3a-II) dramatically redistributed ADAM10 from an intracellular localization to an apparent localization at the cell surface. In contrast, the mainly intracellular localization of ADAM17 was not changed upon co-expression of TSPAN15 (Fig. 3a-III, IV). We also analyzed a possible effect of TSPAN15 on the localization of ADAM10 in the human system. Expression of a human C-terminal EGFP-tagged variant of TSPAN15 led to a redistribution of endogenous human ADAM10 to the cell surface compared to untransfected cells (Fig. 3a-V, VI), highlighting TSPAN15 as a species-conserved regulator of ADAM10 trafficking. The increased cell surface expression of ADAM10 could also be confirmed by biochemical analysis. Surface biotinylation of N2A cells showed that the endogenous level of ADAM10 at the cell surface increased 2.3 fold as compared to control cells (Fig. 3b; Suppl. Fig. 2). Additionally, analysis by flow cytometry demonstrated a 1.8-fold increase of ADAM10 surface expression after TSPAN15 expression and a 2.9 fold significantly increased expression after the expression of both TSPAN15 and ADAM10 (Fig. 3c-I, II). Thus, these data indicate that TSPAN15 is an essential regulator of the cellular trafficking of ADAM10.
Fig. 3.
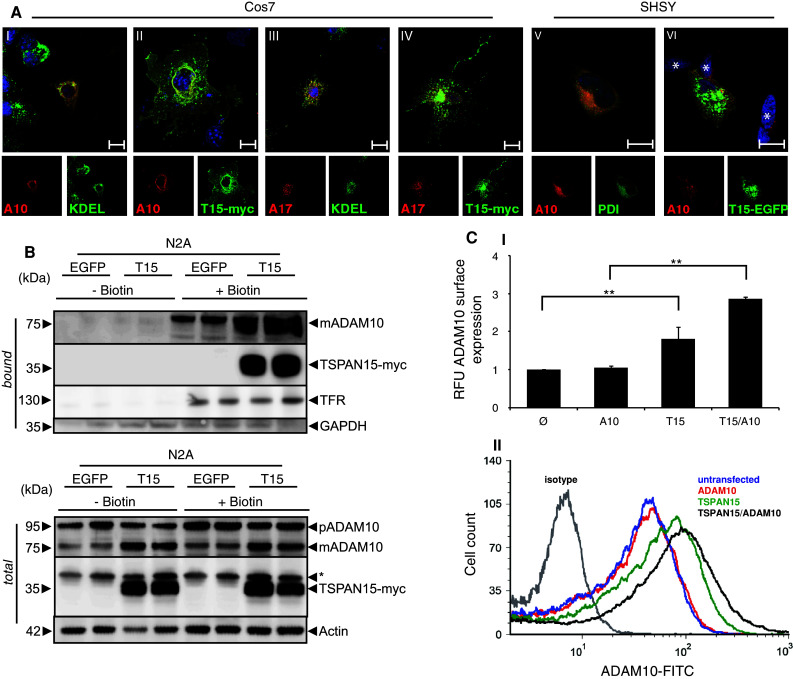
TSPAN15-myc influences ADAM10 localization. **a-**I, II: Murine ADAM10 and ADAM10/TSPAN15-myc expressed in Cos7 cells. III, IV: Murine ADAM17 and ADAM17/TSPAN15-myc expressed in Cos7 cells. Confocal immunofluorescence pictures were taken using an anti-KDEL antibody, an anti-ADAM10 antibody, and an anti-ADAM17 antibody, respectively. Scale bar 100 μm. V, VI: SHSY cells were transiently transfected with a C-terminal EGFP-tagged variant of human TSPAN15. Endogenous ADAM10 was stained using an anti-ADAM10 antibody (11G2), and an anti-PDI antibody was used as ER marker. Asterisks mark untransfected cells. Scale bar 10 μm. b N2A cells were biotinylated after transfection with either EGFP or murine TSPAN15-myc. Following cell lysis total protein samples were taken, and after precipitation of biotin-labeled proteins immunoblotting of total lysates and bound fractions was performed. TSPAN15-myc and ADAM10 were detected. The detection of the transferrin receptor (TFR) was included as a control for biotinylated surface proteins, and antibodies against the intracellular glycerin-aldehyd-3-phosphate dehydrogenase (GAPDH) were used as a negative control. c-I: N2A cells were transfected with ADAM10, TSPAN15-myc and ADAM10/TSPAN15-myc; ADAM10 cell surface expression was determined through FACS analysis using an N-terminal anti-ADAM10 antibody. RFUs were determined. Statistical significance was determined using Student’s t test; values are provided as highly significant (**p < 0.01). Abbreviation: relative fluorescence units (RFU). II: Representative overlay of the FACS analysis performed
TSPAN15 expression increases the ectodomain sheddase activity of ADAM10
The increased surface localization of the mature form of ADAM10 after TSPAN15 expression suggested that well-characterised ADAM10 shedding substrates such as N-cadherin [13] and the APP [18] are subjects for increased surface shedding. In support of this assumption the coexpression of ADAM10 and TSPAN15 in Cos7 cells led to a clearly increased generation of the carboxyterminal membrane stub of endogenously expressed N-cadherin (Fig. 4a-I). Additionally single transfections of TSPAN15 in Cos7 cells were performed, and no apparent additional increase in the level of endogenous and mature ADAM10 was obvious (Suppl. Fig. 3). However, TSPAN15 expression in these cells increased the CTF generation of N-cadherin compared to untransfected and EGFP-transfected cells, indicating that more ADAM10 was transported to the cell surface (Fig. 4a-II), where cleavage is supposed to occur. In murine neuronal cells the α-secretase cleavage of APP resulted in the generation of the C83 membrane stub. The expression of TSPAN15 in these cells significantly increased the activity of the α-secretase leading to 3.7-fold higher levels of C83-APP carboxyterminal fragment as compared to control cells (Fig. 4b). ELISA-based analysis of the endogenously produced soluble APP alpha (sAPPα) revealed a threefold increase after expression of TSPAN15 (Fig. 4c). Also in human cells (HEK293) the single transfection of human C-terminal EGFP-tagged TSPAN15 led to an accumulation of the C83-APP fragment (Fig. 4d) together with an increased level of mature ADAM10, providing evidence for a role of TSPAN15 in ADAM10 activity modulation also in the human system. Together these data strongly suggest that TSPAN15 not only affects the cellular localization of ADAM10, but also modulates its protease activity.
Fig. 4.
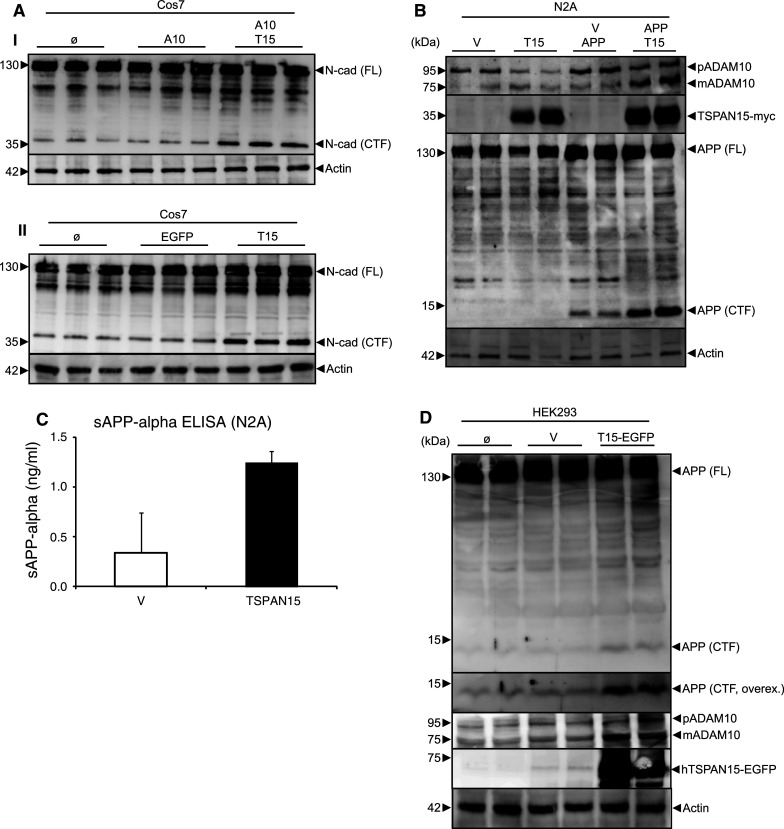
Activity analysis of ADAM10. a-I: murine (m)ADAM10 expressed or coexpressed with TSPAN15-myc in Cos7 cells. Activity of ADAM10 was studied through the analysis of ADAM10-specific shedding events using immunoblot detection of N-cadherin [full-length (Fl) N-cadherin (130 kDa), C-terminal fragment (CTF) N-cadherin (37 kDa)] applying a C-terminal-specific N-cadherin antibody. II: EGFP and TSPAN15-myc were transiently expressed in Cos7 cells, and the processing of N-cadherin was analyzed by Western blot. b N2A cells were transfected with pcDNA3.1 or TSPAN15-myc and APP/pcDNA3.1 or APP/TSPAN15-myc. Processing of APP was analyzed using a C-terminal-specific anti-APP antibody. Actin served as protein loading control. c Cell culture supernatants of N2A cells transfected with EGFP or TSPAN15-myc and sAPPalpha content were determined using sandwich ELISAs. d HEK293 cells were transfected with human TSPAN15-EGFP or vector control, and endogenous APP processing was analyzed. APP (FL) APP full-length protein; APP (CTF) APP C-terminal C83 fragment; overex overexposed image of the upper panel. ADAM10, TSPAN15 and actin expression analysis was included
TSPAN15 knockdown influences endogenous ADAM10 maturation and localization
We then addressed the question if reduced expression of TSPAN15 also affects the maturation state and surface levels of ADAM10. Due to the lack of a specific anti-murine TSPAN15 antibody, we evaluated the knockdown of endogenous TSPAN15 in N2A cells by qRT-PCR analysis. TSPAN15-specific siRNA led to a reduction of TSPAN15 mRNA expression to about 14 % remaining expression compared to expression levels after control siRNA treatment (Fig. 5a). Consequently, the reduction of TSPAN15 in N2A cells led to increased levels of the proform of ADAM10 and a decreased amount of the mature form of the protease (Fig. 5b), indicating less cell surface transport of endogenous ADAM10. This is also reflected by an approximate two-fold reduction of the endogenous surface expression of ADAM10 in comparison to the control siRNA-treated cells as visualized by flow cytometry (Fig. 5c-I, II). Interestingly, the knockdown of TSPAN15 did not significantly influence the level of sAPPalpha in the cell culture supernatants (Suppl. Fig. 4). To analyze if there is a possible compensatory activity of other proteases, which have previously been implicated in influencing sAPPalpha production, we performed qRT-PCR experiments. These experiments revealed that also ADAM9 is highly expressed in N2A cells, whereas ADAM17 expression levels were below our detection limit. In addition, TSPAN12, which was first described to influence ADAM10 activity and APP processing, is also highly expressed in these cells (Suppl. Fig. 5). In conclusion, both overexpression experiments as well as the knockdown of TSPAN15 confirmed the central role of TSPAN15 in the regulation of the subcellular localization of ADAM10.
Fig. 5.

Knockdown of TSPAN15. a Knockdown of TSPAN15 in N2A cells was verified through qRT-PCR analysis. b N2A cells were transfected with siRNA against TSPAN15 and control siRNA. After lysis and immunoblot analysis, ADAM10 was detected. Actin served as protein-loading control. c-I: Surface expression of ADAM10 was analyzed through FACS analysis using a N-terminal anti ADAM10 antibody. Remaining surface expression of ADAM10 was determined (%). II: Representative overlay of FACS analysis of ADAM10 surface protein after TSPAN15 knockdown
TSPAN15 binds to ADAM10 within the ER promoting the exit into the secretory route
To clarify the mechanism of TSPAN15/ADAM10 interaction on a functional level we decided to analyze the association of ADAM10 when TSPAN15 is retained in the ER through a C-terminal addition of an ER retention motif [36], thereby preventing the tetraspanin's exit from this compartment. After pulldown of TSPAN15 or the TSPAN15-ER-retention mutant, the proform of ADAM10 was predominantly coprecipitated, and almost no mature form of the protease was detectable (Fig. 6a). In addition, immunofluorescence analysis confirmed the ER localization of the mutant TSPAN15 and revealed that ADAM10 was also completely retained in the ER (Fig. 6b-I, II). These data suggest that the interaction between both proteins already occurs in the ER and is essential for proper maturation and activation of ADAM10. To more precisely understand the TSPAN15-dependent transport of ADAM10, we metabolically labeled N2A cells and followed the endogenous ADAM10 maturation and processing. Both events take place in a post-ER compartment [5]. In control vector-transfected cells and a 2-h chase period about, 32 % (46 % at 6 h) of ADAM10 has undergone furin-mediated prodomain cleavage, whereas 67 % (83 % at 6 h) of the pro-ADAM10 was processed when TSPAN15 was co-transfected (Fig. 6c). This experimental approach clearly reveals that expression of TSPAN15 accelerates the generation of the mature form of ADAM10. It also leads to a faster shedding of ADAM10 itself, reflected by the ADAM10-CTF accumulation in the 18-h chase sample, an event that is known to occur at the plasma membrane mediated by the cleavage activity of ADAM9 and ADAM15 [39], respectively. Analyzing long chase periods (18 and 30 h), it also appears that TSPAN15 stabilizes the mature form of ADAM10 in contrast to control vector-transfected cells where the labeled pool of mature ADAM10 is already decreasing. Taken together, TSPAN15 associates with ADAM10 early after synthesis, and the complex is able to leave the ER-triggering delivery of the protease to the plasma membrane where the tetraspanin also contributes to the stabilization and a longer halflife of the protease.
Fig. 6.

Early interaction of TSPAN15 with ADAM10. a Murine ADAM10 was either expressed with TSPAN15-myc or TSPAN15-ER-myc. Cells were lysed, and myc-tagged proteins were precipitated using an anti-myc antibody. After immunoblotting coprecipitated ADAM10 was detected using a C-terminal-specific antibody (B42.1). **b-**I, II: Cos7 cells were transfected with TSPAN15-ER-myc or TSPAN15-ER-myc/ADAM10. Confocal immunofluorescence pictures were taken using an anti-myc antibody, an anti-KDEL antibody and an N-terminal-specific ADAM10 antibody and adequate secondary antibody pairs. Scale bar 100 μm. c N2A cells were transfected with EGFP or TSPAN15-myc, pulsed for 1 h with 35S methionine/cysteine and chased for 0, 2, 6, 18 and 30 h. After cell lysis equal amounts of protein were used, and ADAM10 was precipitated using a C-terminal-specific anti-ADAM10 antibody, subjected to SDS-PAGE and analyzed by fluorographics. TSPAN15-myc expression was analyzed by Western blot, and actin served as protein-loading control for the lysates
Discussion
Recent studies analyzing the role of the metalloproteinase ADAM10 in mice suggested that it plays a major role in embryonic development, but also in the homeostasis of adult tissues such as the brain, skin, intestine and immune system [17, 18, 40, 41]. The severe phenotypical consequences due to a lack of ADAM10 can be mainly explained by impaired Notch-signaling. However, it was suggested that a transgenic overexpression of ADAM10 in postmitotic neurons of the CNS is well tolerated and may provide an efficient tool to prevent the excessive production of the neurotoxic amyloid beta peptide [14, 42].
Hence, a fine-tuned regulation of the activity of ADAM10 will be required to increase the potential therapeutic specificity and efficacy, and to reduce possible side effects due to unwanted biological effects of generally altered ectodomain shedding events. Apart from the direct modulation of ADAM10 expression levels through activation of its transcription (20–23), a possible manipulation of the intracellular trafficking of this protease may be another way to determine the strength of proteolytic activity and specificity towards selected substrates. In the present study we performed a split-ubiquitin-based yeast two-hybrid screen, which allowed us to identify membrane interactors of ADAM10. In this screen TSPAN15 was isolated several times, and due to the reported interaction of ADAM10 with other tetraspanins such as CD9, CD81, CD82 and TSPAN12 [23, 24], we decided to study this interaction more carefully. In our screen against mouse brain cDNAs we also identified TSPAN3 once (data not shown), further confirming the potential importance of tetraspanin interactions with ADAM proteases. Interestingly, the tetraspanins that have been shown to interact with ADAM10 recently were identified using different experimental approaches, e.g., after characterization of a CD81 coprecipitating antibody [24] or after mass-spectroscopy identification of a TSPAN12 interacting proteins [23]. In the latter study palmitoylation sites and the C-terminal region of TSPAN12 seemed to influence the interaction with ADAM10 [23]. In the future it will be interesting to study more thoroughly the role of the structural domains important for mediating interaction with ADAM10 and the regulation and mechanisms of protein-protein interaction between ADAM10 and tetraspanins.
Interestingly, the human gene locus on chromosome 10 for TSPAN15 (NET7) was also reported to be genetically linked with the late onset of AD [43], suggesting that the modulation of the gene activity of TSPAN15 may influence the onset of APP processing and AD pathology. Studies in human colon cancer cell lines revealed that both TSPAN15 and ADAM10 are part of the tetraspanin web in highly metastatic active cancer cells [44]. However, the functional relevance of this interaction remained obscure. Our biochemical and cell biological experiments suggest that TSPAN15 has a transport-promoting effect on ADAM10. Furthermore, overexpression of only ADAM10 has only minor effects towards reaching the goal of increased surface expression and activity. Importantly, co-transfection with TSPAN15 significantly increased the shedding activity of ADAM10. ADAM10 is synthesized as a zymogen in the ER, and at steady state it is mainly found within the Golgi compartment where the activation of the proteinase through furin/PC7-mediated proteolysis also takes place. It is thought that, after transport to the plasma membrane, shedding of the substrates occurs [45]. TSPAN15 is involved in this sequence of maturation events. Our data suggest that both ADAM10 and TSPAN15 physically interact early after synthesis, which is supported by the fact that TSPAN15 preferentially binds to the proform of ADAM10. This scenario seems similar to the situation described for synapse-associated protein 97 (SAP97) where early interactions between ADAM10 and SAP97 due to binding at a SH3 domain at the C-terminus of the protease were found to be able to increase ADAM10-mediated APP cleavage [46]. It is interesting to note that tetraspanin12 (TSPAN12) apparently also leads to an increased maturation of ADAM10 [23], suggesting that both tetraspanins equally contribute to the regulation of the cellular transport of the protease or maybe fulfill identical roles in different tissues.
How do these interactions affect the half-life of ADAM10 in the endoplasmic reticulum? An arginine-rich ER-retention motif at the C-terminus of ADAM10 was shown to inhibit transport to the plasma membrane [47]. It is conceivable and supported by our pulse-chase experiments that this motif is masked through the TSPAN15-ADAM10 interaction and therefore ER-exit of the complex is facilitated and ADAM10 is stabilized on the cell surface (Fig. 7). This assumption is further supported by the detected increase in immunoblot studies of the mature form of ADAM10 after TSPAN15-myc transfection when compared with control cells 48-h post transfection. It seems likely that after overexpression of both proteins the ER exit becomes saturated at a certain point leading to an accumulation of the proform of ADAM10. Limiting the amount of ADAM10, which reaches the secretory pathway, may provide a means of keeping the enzyme in an inactive state. The expression and distribution of different tetraspanins including TSPAN15 appear as major checkpoints for the regulated ER exit, enabling processing and activation and regulating the supply of active ADAM10 to the cell surface. Since increased ADAM10 expression is suggested to be effective to prevent the excessive production of the neurotoxic amyloid beta in AD [14], it may be interesting to study the transcriptional control of TSPAN15 and other ADAM10-promoting tetraspanins in more detail in order to find ways of specifically increasing their expression levels. This approach is especially of interest since the localization of the γ-secretase complex is also dependent on the expression of certain tetraspanins [32]. On the other hand, downregulation of ADAM10 may be advised in certain types of cancer [48, 49], and lowering the expression level of tetraspanins will most likely also decrease ADAM10 activity. However, many questions about possible side effects of such a tetraspanin modulation, about possible influences on other ADAM proteases, about the exact mode of interaction and regulation of these fascinating scaffold proteins still need to be answered.
Fig. 7.

Model of TSPAN15 function. (1) ADAM10 is synthesized as an inactive precursor (pro-ADAM10) in the ER, and TSPAN15 accelerates its ER exit. (2) After ER exit the immature ADAM10 is activated through removal of the inhibitory prodomain by furin or proprotein convertase (PC7), and is then transported to the plasma membrane together with TSPAN15. (3) At the plasma membrane TSPAN15 facilitates the integration of ADAM10 in the tetraspanin web. (4, 5) ADAM10 is stabilized, and the web composition enables ADAM10 to get access to its substrates and subsequent cleavage events in the juxtamembrane regions of the substrates take place. (6) Afterwards remaining membrane-bound fragments are cleaved through ripping proteases (γ-secretase complex/SPPLs) in the transmembrane regions, thereby liberating soluble intracellular domains (ICD), which might have signaling functions
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft Sonderforschungsbereich 877 A3 (P.S.) and B8 (M.S.) and GRK1459. We are also grateful to Wim Annaert for providing us with the B42.1/B63.3 anti-mouse ADAM10 or APP antibody, and Eric Rubinstein for the human TSPAN15 expression construct.
Footnotes
J. Prox, M. Willenbrock, P. Saftig and M. Schwake contributed equally.
Contributor Information
Paul Saftig, Phone: +49-431-8802216, FAX: +49-431-8802238, Email: psaftig@biochem.uni-kiel.de.
Michael Schwake, Phone: +49-431-8802218, FAX: +49-431-8802238, Email: mschwake@biochem.uni-kiel.de.
References
- 1.Reiss K, Saftig P. The “a disintegrin and metalloprotease” (ADAM) family of sheddases: physiological and cellular functions. Semin Cell Dev Biol. 2009;20(2):126–137. doi: 10.1016/j.semcdb.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 2.Saftig P, Reiss K. The “A disintegrin and metalloproteases” ADAM10 and ADAM17: novel drug targets with therapeutic potential? Eur J Cell Biol. 2011;90(6–7):527–535. doi: 10.1016/j.ejcb.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 3.Lopez-Perez E, Zhang Y, Frank SJ, Creemers J, Seidah N, Checler F. Constitutive alpha-secretase cleavage of the beta-amyloid precursor protein in the furin-deficient LoVo cell line: involvement of the pro-hormone convertase 7 and the disintegrin metalloprotease ADAM10. J Neurochem. 2001;76(5):1532–1539. doi: 10.1046/j.1471-4159.2001.00180.x. [DOI] [PubMed] [Google Scholar]
- 4.Lopez-Perez E, Seidah NG, Checler F. Proprotein convertase activity contributes to the processing of the Alzheimer’s beta-amyloid precursor protein in human cells: evidence for a role of the prohormone convertase PC7 in the constitutive alpha-secretase pathway. J Neurochem. 1999;73(5):2056–2062. [PubMed] [Google Scholar]
- 5.Anders A, Gilbert S, Garten W, Postina R, Fahrenholz F. Regulation of the alpha-secretase ADAM10 by its prodomain and proprotein convertases. FASEB J. 2001;15(10):1837–1839. doi: 10.1096/fj.01-0007fje. [DOI] [PubMed] [Google Scholar]
- 6.Lin J, Luo J, Redies C. Differential expression of five members of the ADAM family in the developing chicken brain. Neuroscience. 2008;157(2):360–375. doi: 10.1016/j.neuroscience.2008.08.053. [DOI] [PubMed] [Google Scholar]
- 7.Hattori M, Osterfield M, Flanagan JG. Regulated cleavage of a contact-mediated axon repellent. Science. 2000;289(5483):1360–1365. doi: 10.1126/science.289.5483.1360. [DOI] [PubMed] [Google Scholar]
- 8.Janes PW, Saha N, Barton WA, Kolev MV, Wimmer-Kleikamp SH, Nievergall E, Blobel CP, Himanen JP, Lackmann M, Nikolov DB. Adam meets Eph: an ADAM substrate recognition module acts as a molecular switch for ephrin cleavage in trans. Cell. 2005;123(2):291–304. doi: 10.1016/j.cell.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 9.Vincent B. ADAM proteases: protective role in Alzheimer’s and prion diseases? Curr Alzheimer Res. 2004;1(3):165–174. doi: 10.2174/1567205043332072. [DOI] [PubMed] [Google Scholar]
- 10.Altmeppen HC, Prox J, Puig B, Kluth MA, Bernreuther C, Thurm D, Jorissen E, Petrowitz B, Bartsch U, De Strooper B, Saftig P, Glatzel M. Lack of a-disintegrin-and-metalloproteinase ADAM10 leads to intracellular accumulation and loss of shedding of the cellular prion protein in vivo. Mol Neurodegener. 2011;6:36. doi: 10.1186/1750-1326-6-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hinkle CL, Diestel S, Lieberman J, Maness PF. Metalloprotease-induced ectodomain shedding of neural cell adhesion molecule (NCAM) J Neurobiol. 2006;66(12):1378–1395. doi: 10.1002/neu.20257. [DOI] [PubMed] [Google Scholar]
- 12.Gutwein P, Mechtersheimer S, Riedle S, Stoeck A, Gast D, Joumaa S, Zentgraf H, Fogel M, Altevogt DP. ADAM10-mediated cleavage of L1 adhesion molecule at the cell surface and in released membrane vesicles. FASEB J. 2003;17(2):292–294. doi: 10.1096/fj.02-0430fje. [DOI] [PubMed] [Google Scholar]
- 13.Reiss K, Maretzky T, Ludwig A, Tousseyn T, de Strooper B, Hartmann D, Saftig P. ADAM10 cleavage of N-cadherin and regulation of cell–cell adhesion and beta-catenin nuclear signalling. EMBO J. 2005;24(4):742–752. doi: 10.1038/sj.emboj.7600548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Postina R, Schroeder A, Dewachter I, Bohl J, Schmitt U, Kojro E, Prinzen C, Endres K, Hiemke C, Blessing M, Flamez P, Dequenne A, Godaux E, van Leuven F, Fahrenholz F. A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J Clin Invest. 2004;113(10):1456–1464. doi: 10.1172/JCI20864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hooper NM, Turner AJ. The search for alpha-secretase and its potential as a therapeutic approach to Alzheimer s disease. Curr Med Chem. 2002;9(11):1107–1119. doi: 10.2174/0929867023370121. [DOI] [PubMed] [Google Scholar]
- 16.Asai M, Hattori C, Szabo B, Sasagawa N, Maruyama K, Tanuma S, Ishiura S. Putative function of ADAM9, ADAM10, and ADAM17 as APP alpha-secretase. Biochem Biophys Res Commun. 2003;301(1):231–235. doi: 10.1016/S0006-291X(02)02999-6. [DOI] [PubMed] [Google Scholar]
- 17.Hartmann D, de Strooper B, Serneels L, Craessaerts K, Herreman A, Annaert W, Umans L, Lubke T. Lena Illert A, von Figura K, Saftig P. The disintegrin/metalloprotease ADAM 10 is essential for notch signalling but not for alpha-secretase activity in fibroblasts. Hum Mol Genet. 2002;11(21):2615–2624. doi: 10.1093/hmg/11.21.2615. [DOI] [PubMed] [Google Scholar]
- 18.Jorissen E, Prox J, Bernreuther C, Weber S, Schwanbeck R, Serneels L, Snellinx A, Craessaerts K, Thathiah A, Tesseur I, Bartsch U, Weskamp G, Blobel CP, Glatzel M, De Strooper B, Saftig P. The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J Neurosci. 2010;30(14):4833–4844. doi: 10.1523/JNEUROSCI.5221-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kopan R, Ilagan MX. The canonical notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137(2):216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holback S, Adlerz L, Iverfeldt K. Increased processing of APLP2 and APP with concomitant formation of APP intracellular domains in BDNF and retinoic acid-differentiated human neuroblastoma cells. J Neurochem. 2005;95(4):1059–1068. doi: 10.1111/j.1471-4159.2005.03440.x. [DOI] [PubMed] [Google Scholar]
- 21.Prinzen C, Muller U, Endres K, Fahrenholz F, Postina R. Genomic structure and functional characterization of the human ADAM10 promoter. FASEB J. 2005;19(11):1522–1524. doi: 10.1096/fj.04-3619fje. [DOI] [PubMed] [Google Scholar]
- 22.Tippmann F, Hundt J, Schneider A, Endres K, Fahrenholz F. Up-regulation of the alpha-secretase ADAM10 by retinoic acid receptors and acitretin. FASEB J. 2009;23(6):1643–1654. doi: 10.1096/fj.08-121392. [DOI] [PubMed] [Google Scholar]
- 23.Xu D, Sharma C, Hemler ME. Tetraspanin12 regulates ADAM10-dependent cleavage of amyloid precursor protein. FASEB J. 2009;23(11):3674–3681. doi: 10.1096/fj.09-133462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arduise C, Abache T, Li L, Billard M, Chabanon A, Ludwig A, Mauduit P, Boucheix C, Rubinstein E, Le Naour F. Tetraspanins regulate ADAM10-mediated cleavage of TNF-alpha and epidermal growth factor. J Immunol. 2008;181(10):7002–7013. doi: 10.4049/jimmunol.181.10.7002. [DOI] [PubMed] [Google Scholar]
- 25.Berditchevski F, Odintsova E. Tetraspanins as regulators of protein trafficking. Traffic. 2007;8(2):89–96. doi: 10.1111/j.1600-0854.2006.00515.x. [DOI] [PubMed] [Google Scholar]
- 26.Stipp CS, Kolesnikova TV, Hemler ME. Functional domains in tetraspanin proteins. Trends Biochem Sci. 2003;28(2):106–112. doi: 10.1016/S0968-0004(02)00014-2. [DOI] [PubMed] [Google Scholar]
- 27.Levy S, Shoham T. Protein-protein interactions in the tetraspanin web. Physiology (Bethesda) 2005;20:218–224. doi: 10.1152/physiol.00015.2005. [DOI] [PubMed] [Google Scholar]
- 28.Hemler ME. Specific tetraspanin functions. J Cell Biol. 2001;155(7):1103–1107. doi: 10.1083/jcb.200108061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maecker HT, Todd SC, Levy S. The tetraspanin superfamily: molecular facilitators. FASEB J. 1997;11(6):428–442. [PubMed] [Google Scholar]
- 30.Hemler ME. Tetraspanin functions and associated microdomains. Nat Rev Mol Cell Biol. 2005;6(10):801–811. doi: 10.1038/nrm1736. [DOI] [PubMed] [Google Scholar]
- 31.Yunta M, Lazo PA. Tetraspanin proteins as organisers of membrane microdomains and signalling complexes. Cell Signal. 2003;15(6):559–564. doi: 10.1016/S0898-6568(02)00147-X. [DOI] [PubMed] [Google Scholar]
- 32.Wakabayashi T, Craessaerts K, Bammens L, Bentahir M, Borgions F, Herdewijn P, Staes A, Timmerman E, Vandekerckhove J, Rubinstein E, Boucheix C, Gevaert K, De Strooper B. Analysis of the gamma-secretase interactome and validation of its association with tetraspanin-enriched microdomains. Nat Cell Biol. 2009;11(11):1340–1346. doi: 10.1038/ncb1978. [DOI] [PubMed] [Google Scholar]
- 33.Yanez-Mo M, Gutierrez-Lopez MD, Cabanas C. Functional interplay between tetraspanins and proteases. Cell Mol Life Sci. 2011;68(20):3323–3335. doi: 10.1007/s00018-011-0746-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gunther W, Luchow A, Cluzeaud F, Vandewalle A, Jentsch TJ. ClC-5, the chloride channel mutated in Dent’s disease, colocalizes with the proton pump in endocytotically active kidney cells. Proc Natl Acad Sci USA. 1998;95(14):8075–8080. doi: 10.1073/pnas.95.14.8075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blanz J, Groth J, Zachos C, Wehling C, Saftig P, Schwake M. Disease-causing mutations within the lysosomal integral membrane protein type 2 (LIMP-2) reveal the nature of binding to its ligand beta-glucocerebrosidase. Hum Mol Genet. 2010;19(4):563–572. doi: 10.1093/hmg/ddp523. [DOI] [PubMed] [Google Scholar]
- 36.Reczek D, Schwake M, Schroder J, Hughes H, Blanz J, Jin X, Brondyk W, Van Patten S, Edmunds T, Saftig P. LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase. Cell. 2007;131(4):770–783. doi: 10.1016/j.cell.2007.10.018. [DOI] [PubMed] [Google Scholar]
- 37.Behnke J, Eskelinen EL, Saftig P, Schroder B. Two dileucine motifs mediate late endosomal/lysosomal targeting of transmembrane protein 192 (TMEM192) and a C-terminal cysteine residue is responsible for disulfide bond formation in TMEM192 homodimers. Biochem J. 2011;434(2):219–231. doi: 10.1042/BJ20101396. [DOI] [PubMed] [Google Scholar]
- 38.Wang B, Pelletier J, Massaad MJ, Herscovics A, Shore GC. The yeast split-ubiquitin membrane protein two-hybrid screen identifies BAP31 as a regulator of the turnover of endoplasmic reticulum-associated protein tyrosine phosphatase-like B. Mol Cell Biol. 2004;24(7):2767–2778. doi: 10.1128/MCB.24.7.2767-2778.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tousseyn T, Thathiah A, Jorissen E, Raemaekers T, Konietzko U, Reiss K, Maes E, Snellinx A, Serneels L, Nyabi O, Annaert W, Saftig P, Hartmann D, De Strooper B. ADAM10, the rate-limiting protease of regulated intramembrane proteolysis of Notch and other proteins, is processed by ADAMS-9, ADAMS-15, and the gamma-secretase. J Biol Chem. 2009;284(17):11738–11747. doi: 10.1074/jbc.M805894200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weber S, Niessen MT, Prox J, Lullmann-Rauch R, Schmitz A, Schwanbeck R, Blobel CP, Jorissen E, de Strooper B, Niessen CM, Saftig P. The disintegrin/metalloproteinase Adam10 is essential for epidermal integrity and notch-mediated signaling. Development. 2011;138(3):495–505. doi: 10.1242/dev.055210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tian L, Wu X, Chi C, Han M, Xu T, Zhuang Y. ADAM10 is essential for proteolytic activation of notch during thymocyte development. Int Immunol. 2008;20(9):1181–1187. doi: 10.1093/intimm/dxn076. [DOI] [PubMed] [Google Scholar]
- 42.Endres K, Fahrenholz F. Upregulation of the alpha-secretase ADAM10–risk or reason for hope? FEBS J. 2010;277(7):1585–1596. doi: 10.1111/j.1742-4658.2010.07566.x. [DOI] [PubMed] [Google Scholar]
- 43.Grupe A, Li Y, Rowland C, Nowotny P, Hinrichs AL, Smemo S, Kauwe JS, Maxwell TJ, Cherny S, Doil L, Tacey K, van Luchene R, Myers A, Wavrant-De Vrieze F, Kaleem M, Hollingworth P, Jehu L, Foy C, Archer N, Hamilton G, Holmans P, Morris CM, Catanese J, Sninsky J, White TJ, Powell J, Hardy J, O’Donovan M, Lovestone S, Jones L, Morris JC, Thal L, Owen M, Williams J, Goate A. A scan of chromosome 10 identifies a novel locus showing strong association with late-onset Alzheimer disease. Am J Hum Genet. 2006;78(1):78–88. doi: 10.1086/498851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Le Naour F, Andre M, Greco C, Billard M, Sordat B, Emile JF, Lanza F, Boucheix C, Rubinstein E. Profiling of the tetraspanin web of human colon cancer cells. Mol Cell Proteomics. 2006;5(5):845–857. doi: 10.1074/mcp.M500330-MCP200. [DOI] [PubMed] [Google Scholar]
- 45.Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F. Constitutive and regulated alpha-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci USA. 1999;96(7):3922–3927. doi: 10.1073/pnas.96.7.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marcello E, Gardoni F, Mauceri D, Romorini S, Jeromin A, Epis R, Borroni B, Cattabeni F, Sala C, Padovani A, Di Luca M. Synapse-associated protein-97 mediates alpha-secretase ADAM10 trafficking and promotes its activity. J Neurosci. 2007;27(7):1682–1691. doi: 10.1523/JNEUROSCI.3439-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marcello E, Gardoni F, Di Luca M, Perez-Otano I. An arginine stretch limits ADAM10 exit from the endoplasmic reticulum. J Biol Chem. 2010;285(14):10376–10384. doi: 10.1074/jbc.M109.055947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Duffy MJ, McKiernan E, O’Donovan N, McGowan PM. The role of ADAMs in disease pathophysiology. Clin Chim Acta. 2009;403(1–2):31–36. doi: 10.1016/j.cca.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 49.Armanious H, Gelebart P, Anand M, Belch A, Lai R. Constitutive activation of metalloproteinase ADAM10 in mantle cell lymphoma promotes cell growth and activates the TNF{alpha}/NF{kappa}B pathway. Blood. 2011;117(23):6237–6246. doi: 10.1182/blood-2010-10-313940. [DOI] [PubMed] [Google Scholar]
- 50.Suter B, Auerbach D, Stagljar I. Yeast-based functional genomics and proteomics technologies: the first 15 years and beyond. Biotechniques. 2006;40(5):625–644. doi: 10.2144/000112151. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.