Evolution and Functional Impact of Rare Coding Variation from Deep Sequencing of Human Exomes (original) (raw)
. Author manuscript; available in PMC: 2013 Jul 11.
Published in final edited form as: Science. 2012 May 17;337(6090):64–69. doi: 10.1126/science.1219240
Abstract
As a first step toward understanding how rare variants contribute to risk for complex diseases, we sequenced 15,585 human protein-coding genes to an average median depth of 111× in 2440 individuals of European (n = 1351) and African (n = 1088) ancestry. We identified over 500,000 single-nucleotide variants (SNVs), the majority of which were rare (86% with a minor allele frequency less than 0.5%), previously unknown (82%), and population-specific (82%). On average, 2.3% of the 13,595 SNVs each person carried were predicted to affect protein function of ∼313 genes per genome, and ∼95.7% of SNVs predicted to be functionally important were rare. This excess of rare functional variants is due to the combined effects of explosive, recent accelerated population growth and weak purifying selection. Furthermore, we show that large sample sizes will be required to associate rare variants with complex traits.
Understanding the spectrum of allelic variation in human genes and revealing the demographic and evolutionary forces that shape this variation within and among populations are major aims of human genetics research. Such information is critical for defining the architecture of common diseases, identifying functionally important variation, and ultimately facilitating the interpretation of personalized disease risk profiles (1–3). To date, surveys of human variation have been dominated by studies of single-nucleotide polymorphisms (SNPs) genotyped using high-density arrays composed of common variants (4–6). Although these projects have substantially improved our knowledge of common allelic variation and enabled genome-wide association studies (GWAS), they have been generally uninformative about the population genetics characteristics of rare variants, defined here as a minor allele frequency (MAF) of less than 0.5%.
Rare genetic variants are predicted to vastly outnumber common variants in the human genome (7, 8). By capturing and sequencing all protein-coding exons (i.e., the exome, which comprises ∼1 to 2% of the human genome), exome sequencing is a powerful approach for discovering rare variation and has facilitated the genetic dissection of unsolved Mendelian disorders and the study of human evolutionary history (9–14). Rare and low-frequency (MAF between 0.5 and 1%) variants have been hypothesized to explain a substantial fraction of the heritability of common, complex diseases (15). Because common variants explain only a modest fraction of the heritability of most traits (16, 17), the National Heart, Lung, and Blood Institute (NHLBI) recently sponsored the multicenter Exome Sequencing Project (ESP) to identify previously unknown genes and molecular mechanisms underlying complex heart, lung, and blood disorders by sequencing the exomes of a large number of individuals measured for phenotypic traits of substantial public health importance (e.g., early-onset myocardial infarction, stroke, and body mass index).
Data generation and variant discovery
A total of 63.4 terabases of DNA sequence was generated at two centers with three complementary definitions of the exome target and two different capture technologies (18). We sequenced samples from 15 different cohorts in the ESP to an average median depth of 111× (range of 23× to 474×). We found no evidence of cohort- and/or phenotype-specific effects, or other systematic biases, in the analysis of the filtered single-nucleotide variant (SNV) data (figs. S1 to S7). Exomes from related individuals were excluded from further analysis (fig. S8), resulting in a data set of 2440 exomes. We inferred genetic ancestry by using a clustering approach (18) and, unless otherwise noted, focused the remaining analyses on the inferred 1351 European-American (EA) and 1088 African-American (AA) individuals. We subjected the 563,698 variants in the intersection of all three capture targets to standard quality-control filters (18), resulting in a final data set of 503,481 SNVs identified in 15,585 genes and 22.38 Mb of targeted sequence per individual. We assessed data quality and error rates by several orthogonal methods (18). About 98% (941/961) of all variant sites that were experimentally tested were confirmed, including 98% (234/238) of singletons, 98% (678/693) of nonsingleton SNV sites with a MAF < 10%, and 97% (29/30) of SNV sites with a MAF ≥ 10%.
The vast majority of coding variation is rare and previously unknown
We observed a total of 503,481 SNVs and 117 fixed non-reference sites, of which 325,843 and 268,903 were found in AAs and EAs, respectively (fig. S9A). Excluding singletons, ∼58% of SNVs were population-specific (93,278 and 32,552 variants were uniquely observed in AAs and EAs, respectively), and the vast majority of these variants were rare (fig. S9B). Most SNVs (292,125 or 58%) were nonsynonymous, including 285,960 missense variants and 6165 nonsense variants (fig. S9C). Synonymous variants accounted for 38% (188,975) of all SNVs (fig. S9C), with the remaining 4% of SNVs (22,381) located in either splice sites or targeted noncoding regions. The majority of SNVs (411,084; 82%) were previously unknown, with more novel SNVs observed in AAs (240,341) than in EAs (204,415), although the proportion of SNVs that were novel was higher in EAs compared with AAs (76.0% versus 73.8%; χ2 = 398.3, df =1, P < 10−16). About 98% (402,813) of novel SNVs were rare, and 48.9% of all novel, rare SNVs were nonsynonymous.
The AA and EA sample sizes provided ∼90% power to detect variants with a MAF ≥ 0.1% and nearly 100% power to detect common variants (MAF ≥ 5%) (tables S1 and S2 and fig. S10). With our large sample size, the proportion of singletons identified rapidly decreased to a nearly constant rate of new singleton discovery such that each additional exome contributed ∼200 novel SNVs (fig. S11). The number of SNVs per gene rapidly stabilized for common variants in small sample sizes (∼100 individuals), whereas the number of rare variants continued to increase linearly (fig. S12).
Of the total SNVs, 57% (285,857) were singletons, and SNVs with three or fewer minor alleles accounted for 72% of all variants (fig. S9D). The proportion of singletons observed in AAs (49.3%, n = 140,818) was lower than that observed in EAs (50.7%, n = 144,821), but the overall site frequency spectra (SFS) and the SFS for both AAs and EAs are highly skewed, exhibiting a large excess of rare variants relative to the standard neutral model (19) (fig. S9D). The skew of the SFS was greater for EAs than AAs, and, at equal sample sizes, the odds that a SNV was a singleton were 1.7 times greater for EAs than AAs. Consistent with these observations, Tajima's D was highly negative for both EAs (−2.12) and AAs (−2.10) and dropped precipitously as sample size increased (fig. S9E), highlighting that sequencing a large number of individuals provides unique information about recent demographic history (13, 20, 21).
To identify putatively functional variation, we applied four popular methods applicable to non-synonymous variants (PolyPhen2, SIFT, a likelihood ratio test, and MutationTaster) and three conservation-based methods applicable to all types of variants [GERP, PhyloP, and a novel population genetics approach that combines conservation information with the SFS that we designate SFS-Del (18)]. About 47% of all SNVs (74% of nonsynonymous and 6% of synonymous variants) are predicted to be deleterious by one or more method (Fig. 1A); however, overlap among methods is modest. For example, only 1% of nonsynonymous variants are predicted to be functional by all seven methods, and variants predicted by any single approach are likely to have a high false-positive rate (Fig. 1A). Therefore, we used a conservative majority rule approach and deemed nonsynonymous variants predicted by at least four of the seven applicable methods and synonymous sites predicted by at least two of the three applicable methods (fig. S13) to be putatively functional. In total, 16.9% of SNVs (85,224) meet this criteria, of which 81,170 were nonsynonymous SNVs. About 95.7% (81,555) of all SNVs conservatively predicted to be functional were rare, and the odds ratio (OR) that rare variants are functional compared with variants with a MAF > 0.5% is 4.2 [95% confidence interval (CI) from 4.0 to 4.3; Fisher's exact test; P < 10−15), underscoring the potential impact of rare variants on health-related traits.
Fig. 1.
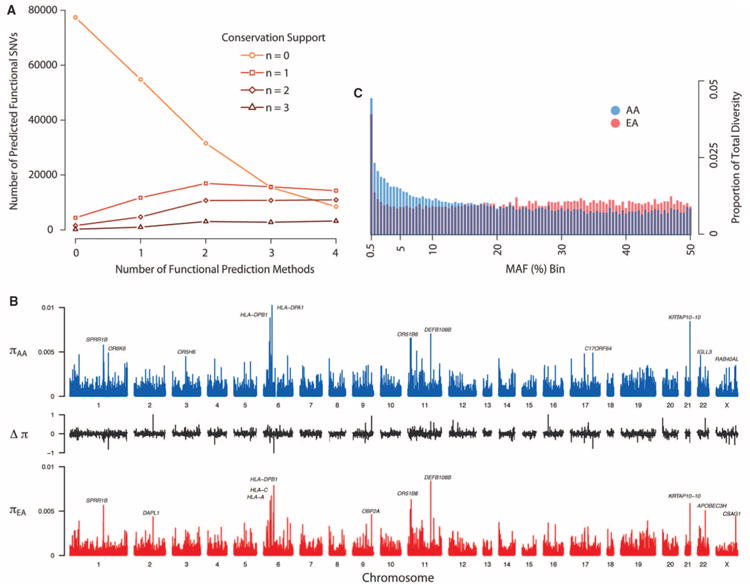
Characteristics of protein-coding variation in humans. (A) Number of nonsynonymous SNVs predicted to be functionally important as a function of seven different methods (18). (B) Distributions of π across the exome in AAs (blue) and EAs (red). The value of π for each gene is shown as a vertical line. The middle section shows the difference in diversity between EA and AA (Δπ = πEA − πAA), scaled between 0 and 1. (C) Distributions of the proportion of total diversity, π, attributable to SNVs with different MAFs in the EA and AA samples. The x axis is binned in increments of 0.5%.
Patterns of coding variation by gene and pathway
The median number of SNVs per gene was 24, ranged between 0 and 761, and was significantly different (Wilcoxon-rank sum test; P < 10−15) between AAs (median of 16, range from 0 to 566) and EAs (median of 13, range from 0 to 410). Mutational target size plays an important role in governing differences in polymorphism across loci, because gene length accounts for 76.6% of variation in the number of SNVs across genes (95% bootstrap CI = 73.9 to 79.1%; P < 10−15).
The proportion of rare variants per base pair (bp) in each gene was higher (mean = 2.015%; 95% range = 0.621 to 4.057%) than that of common variants (0.334%; 95% range = 0.000 to 1.143%), and the average ratio of rare to common alleles per bp was ∼6:1. We identified 110 genes that showed an unusually high proportion of rare variants, including six histone genes that are likely subject to greater selective constraint (table S3). The number of putatively functional variants also varied widely across genes (fig. S14B), ranging from 0 to >100, with a median of two in both EA and AA samples.
Nucleotide diversity (π) varied considerably among genes, ranging from ∼0 to 1.319% per bp (mean = 0.042%; Fig. 1B). Mean π in AAs (0.047%) was significantly higher (P < 10−15, paired t test) than π in EAs (0.035%), and π per gene was modestly correlated (_r_2 = 63%; P value < 10−15) between AAs and EAs (fig. S15). Rare variants account for 4% of total diversity, more than any other MAF bin (of width 0.5%) in both EAs and AAs (Fig. 1C). Rare and low-frequency SNVs comprise ∼13 and 20% of total diversity in the EA and AA samples, respectively (Fig. 1C). In both samples, estimates of π were highest for human lymphocyte antigen (HLA) loci and other genes related to immune function, such as DEFB108B, and olfactory receptors (Fig. 1B). When genes were grouped into functional categories by KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway, estimates of π were highest for pathways related to immune function and olfaction and lowest for pathways involved in basic cellular processes (fig. S16).
Abundance of rare variation explained by human demographic history
The excess of rare variation across the exome is consistent with explosive human population growth (22). To investigate this further, we used an out-of-Africa (OOA) demographic model (23) to predict the expected joint distribution of allele frequencies between EA and AA samples via a diffusion approximation (18). The OOA model, modified to account for admixture, captures prominent features of the joint frequency distribution. However, both populations contain more rare variants than predicted by this model (18) (Fig. 2), most likely because of rapid population growth in the past few thousand years that is undetectable with smaller sample sizes (fig. S9E). We revisited the demographic model from Gravel et al. (23), allowing for a reduced initial European expansion that is compensated for by accelerated growth starting after the split of European and Asian populations. Similarly, we introduced a phase of exponential growth in the African population starting at the same time. The resulting demographic model is an improved fit to the synonymous site-frequency spectrum (18) (Fig. 2B) and strongly supports a recent, dramatic acceleration of population growth. The maximum-likelihood time for accelerated growth was 5115 years ago (Fig. 2B).
Fig. 2.
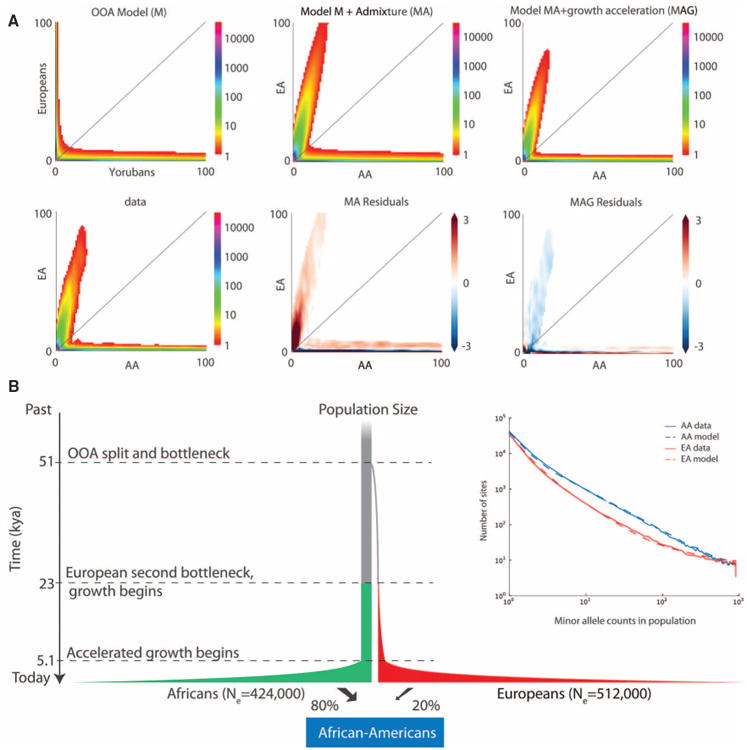
Deep sequencing reveals increases of recent population size. (A) Joint SFS predicted from different demographic models (top) compared with the observed data (bottom), displaying allele counts between 0 and 100 chromosomes. The three models are (left) an OOA model without admixture derived from the 1000 Genomes data, (middle) the same model with the AA panel modeled as an 80%:20% admixture between African and European lineages, and (right) the same model further modified to account for recent growth acceleration. Anscombe residuals are displayed, with regions showing more variants than predicted by the model in blue and less in red. Bins with expected counts <1 are displayed as white in all graphs. (B) Schematic representation (not to scale) of the inferred demographic model and parameters (18). kya, thousand years ago. (Inset) Comparison of the observed SFS to that predicted by the demographic model incorporating recent accelerated growth.
The EA population growth, previously estimated at 0.38% per generation, is now modeled at the first step as 0.307% (SD of ±0.003%), followed by explosive growth of 1.95% (SD ±0.03%) over the past 5115 years. The growth in the AA sample during this same period is estimated to be 1.66% (SD ±0.03%). The estimated standard deviations (18) are quite small, and, for data sets of this scale, it is likely that other sources of uncertainty (e.g., mutation rate or model specification) play a more important role than finite genome fluctuations. The final population sizes in this model are lower than current census sizes, and we speculate that larger sample sizes will be necessary to fully capture the signature recent growth-rate expansion imparted on patterns of DNA sequence variation.
Impact of natural selection on rare coding variation
To investigate the effect of purifying selection on nonsynonymous variants, we examined the relationship between MAFs of non-synonymous SNVs and functional prediction scores from SIFT, Polyphen2, a likelihood ratio test statistic, and MutationTaster (18). Each prediction score showed a significant (P < 10−16) negative correlation with MAF in the combined sample (Fig. 3A) as well as in each sample separately (18). Moreover, the proportion of predicted deleterious changes was negatively correlated with MAF (Fig. 3B). We next mapped 31,115 nonsynonymous SNVs to known protein structures and classified them into different structural categories (Fig. 3C) (18). Nonsynonymous SNVs in all categories, except sites that contact other protein chains, showed a significant excess of rare variants compared with synonymous sites (Fig. 3C), as expected if subjected to purifying selection. The relative effect sizes show that categories of variants with direct functional importance (e.g., active sites, enrichment of 2.8%; ligand-binding residues, enrichment of 1.7%) are under stronger constraint than categories important for structural stability. The exception is residues that form side-chain hydrogen bonds, which show a 2.8% enrichment of rare variants, suggesting that hydrogen bonds make a large contribution to protein folding and stability.
Fig. 3.
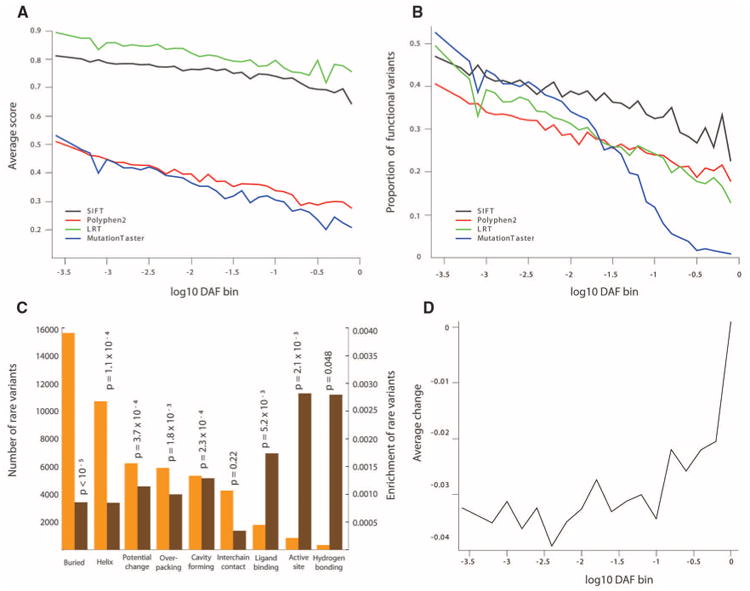
Signatures of purifying selection in protein-coding SNVs. (A) Relationship between the evidence that a variant is functionally important and MAF for four different methods. (B) Relationship between the proportion of putatively functional variants and MAF for the same predictions as in (A). (C) Comparison of the number of rare SNVs (orange) and enrichmentofrareornon-synonymous SNVs (brown) located in different protein structural categories [_P_ values were calculated by a permutation test (18)]. (D) Relationship between average change of w score of synonymous variants and DAF.
To investigate selective constraint acting on synonymous variants, we calculated the correlation between the derived allele frequency (DAF) of synonymous variants and their corresponding change in the relative adaptiveness value, or w score (24). The w score summarizes information about selective constraints on the efficiency of codon-anticodon coupling and the number of tRNA gene copies in the genome. Negative values indicate synonymous variants that may decrease translational efficiency or accuracy. We found a weak but significant positive correlation between DAF and change in w score (r = 0.03; P < 10−16), consistent with the action of purifying selection (Fig. 3D).
We examined selective sweeps by identifying genes with high ratios of divergence (human-specific lineage substitutions relative to chimp and macaque) compared with polymorphism within humans, which are predicted to increase between-species divergence and decrease within-population diversity. We identified genes in which the ratio of nonsynonymous to synonymous divergence was high relative to the ratio of non-synonymous to synonymous SNVs (25). We also identified genes with either a high or low ratio of π in AAs relative to π in EAs and genes with diversity estimates in the bottom 20th percentile in which at least one SNV had an _F_ST ≥ 0.3. In total, 114 genes met one or more of these criteria (table S4). About 25% of these genes have been implicated as targets of positive selection (26). The 114 candidate selection genes were significantly enriched (false discovery rate ≤ 5%) for five KEGG pathways, including olfactory transduction and metabolic pathways (table S5).
Implications for disease and personal genomics
We evaluated gene-specific power of rare variant association studies in the EA and AA samples. We used Fisher's exact test, a robust approach for aggregate testing of rare variation at a locus (27), to determine the power to detect an association for each gene harboring rare causal variants with ORs of 1.5 or 5 in 400 cases and 400 controls (18). In both the EA and AA samples, cases and controls were sampled from 1000 individuals selected to minimize any confounding effects of population stratification (fig. S17), with power calculations assuming a type I error rate of α = 0.001. In each sample, power varies widely across loci, and <5% of genes achieve 80% power even when relatively strong effects (OR = 5) are modeled (Fig. 4A); when causal variants are assumed to have an OR of 1.5, no genes achieve 80% power (fig. S18). Furthermore, although the AA sample has uniformly higher power per gene relative to the EA sample (Fig. 4A), caution is warranted because this is largely a function of our modeling assumptions (18).
Fig. 4.
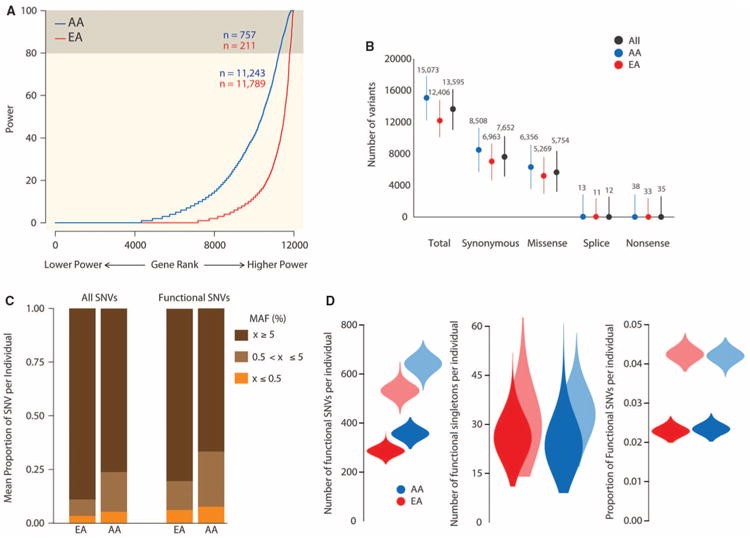
Power of rare variant association mapping and personal genomics characteristics of protein-coding SNVs. (A) Distribution of gene-specific estimates of power to map causal rare variants across 12,000 protein-coding genes with at least three SNVs in the EA (red) or AA (blue) samples. Power varied widely across loci, and <5% of genes (beige) achieve 80% power even when relatively strong effects (OR = 5) are modeled. (B) Average number (points) and range (vertical lines) of synonymous, missense, splice site, and nonsense SNVs. (C) Average proportion of SNVs per individual that are rare (MAF ≤ 0.5%), intermediate (0.5% < MAF < 5%), or common (MAF ≥ 5%) in the population from which they were sampled. The proportions of rare and intermediate frequency variants per individual are significantly higher (Wilcoxon-rank sum test; P < 10−15) for putatively functional SNVs. (D) Violin plots showing the distribution of number of functional SNVs, number of functional singletons, and proportion of functional SNVs per individual in the EA and AA samples. Darker and lighter shaded plots correspond to conservative and more liberal definitions of functional variation, respectively.
The mean number of SNVs per exome (homozygous nonreference and heterozygous genotypes) was 13,595, and ∼66% (8893) of these sites were heterozygous. As expected, AAs had significantly more SNVs per exome than EAs (15,073 versus 12,406, Mann-Whitney test, P < 10−16), which is true for all classes of sites (Fig. 4B). Moreover, on average, each individual possessed 35 nonsense variants and was homozygous for at least one nonreference nonsense variant; 318 individuals (181 AAs and 137 EAs) were compound heterozygotes for nonsense SNVs. The mean number of novel SNVs per individual was 549 overall, but AAs had more than twice the number of novel SNVs compared with EAs (762 versus 362, respectively; _P_= 1.9 × 10−7 correcting for differences in the mean number of SNVs between populations). The fraction of overall variation that was novel in AAs was higher than in EAs (5 and 3%, respectively; P < 10−16). Lastly, although most protein-coding variants were rare in the full AA and EA population samples, the majority of SNVs found in an average individual were common (Fig. 4C).
We next examined the distribution of functionally important variation, functionally important singletons, and the proportion of functionally important SNVs per individual (Fig. 4D) by using both conservative and more liberal criteria (18). On average, individuals possess between 318 and 580 predicted functional protein-coding SNVs depending on how functional variants are defined, with slightly more in AA than in EA individuals (Fig. 4D). The average number of predicted functional singletons per individual was more robust to the definition of functional variants, ranged from 25 to 31, and was slightly higher in AA compared to EA individuals (Fig. 4D). In both cases, however, there was more variation among individuals than between populations.
Lastly, the average proportion of predicted functional SNVs per individual varied between 2.3 and 4.2% (Fig. 4D). When the more liberal definition of functional SNVs was used, EA individuals have a significantly higher proportion of predicted functional SNVs compared with AA individuals (Fig. 4D; Wilcoxon-rank sum test; P < 10−15), consistent with empirical estimates and theoretical expectations because of the lower EA effective population size (28, 29). However, when the more conservative definition was used, this pattern was reversed, and AA individuals have a significantly higher proportion of predicted functional SNVs compared with EA individuals (Fig. 4D; Wilcoxon-rank sum test; P < 10−15). These results highlight how the definition of functional variants can influence inferences and underscore the importance of continued methodological development to robustly identify functionally important variation. Nonetheless, there was considerable rare genetic variation among individuals that is predicted to be functional, which could explain variability in disease risk and adverse drug response.
Conclusion
Our results have several important implications for human disease gene mapping and personal genomics. In particular, the vast majority of protein-coding variation is evolutionarily recent, rare, and enriched for deleterious alleles. Thus, rare variation likely makes an important contribution to human phenotypic variation and disease susceptibility. However, detecting the effects of rare variants requires very large sample sizes, because the power to detect an association is low for most human genes. Accounting for the SFS on a gene-by-gene basis should facilitate the development of more powerful association tests. Additionally, because most rare SNVs are population-specific, replication of disease associations across populations may be challenging. Lastly, as whole-genome sequencing at high coverage becomes increasingly feasible, statistical and experimental methods that accurately identify functionally important protein-coding and regulatory variation are needed to empower association studies, identify variants causally related to disease, and provide clinically actionable information.
Supplementary Material
Supplementary info
Acknowledgments
We acknowledge the support of the NHLBI and the contributions of the research institutions, study investigators, field staff, and study participants in creating this resource for biomedical research; and the Population Genetics Project Team. Funding for GO ESP was provided by NHLBI grants RC2 HL-103010 (HeartGO), RC2 HL-102923 (LungGO), and RC2 HL-102924 (Women's Health Initiative Exome Sequencing Project, WHISP). The exome sequencing was performed through NHLBI grants RC2 HL-102925 (BroadGO) and RC2 HL-102926 (SeattleGO). Filtered sets of annotated variants and their allele frequencies are available at http://evs.gs.washington.edu/EVS/ and have been deposited in dbSNP (www.ncbi.nlm.nih.gov/snp; local batch ID, ESP2500). Genotypes and phenotypes from a large subset of individuals are also available via dbGaP (www.ncbi.nlm.nih.gov/gap) using the following accession information: NHLBI GO-ESP: Women's Health Initiative Exome Sequencing Project (WHI) – WHISP, WHISP_Subject_Phenotypes, pht002246.v2. p2, phs000281.v2.p2; NHLBI GO-ESP: Heart Cohorts Exome Sequencing Project (JHS), ESP_HeartGO_JHS_LDLandEOMI_ Subject_Phenotypes, pht002539.v1.p1, phs000402.v1.p1; NHLBI GO-ESP: Heart Cohorts Exome Sequencing Project (FHS), HeartGO_FHS_LDLandEOMI_PhenotypeDataFile, pht002476.v1.p1, phs000401.v1.p1; NHLBI GO-ESP: Heart Cohorts Exome Sequencing Project (CHS), HeartGO_CHS_LDL_ PhenotypeDataFile, pht002536.v1.p1, phs000400.v1.p1; NHLBI GO-ESP: Heart Cohorts Exome Sequencing Project (ARIC), ESP_ARIC_LDLandEOMI_Sample, pht002466.v1.p1, phs000398. v1.p1; NHLBI GO-ESP: Lung Cohorts Exome Sequencing Project (Cystic Fibrosis), ESP_LungGO_CF_PA_Culture_Data, pht002227. v1.p1, phs000254.v1.p1; NHLBI GO-ESP: Early-Onset Myocardial Infarction (Broad EOMI), ESP_Broad_EOMI_Subject_Phenotypes, pht001437.v1.p1, phs000279.v1.p1; NHLBI GO-ESP: Lung Cohorts Exome Sequencing Project (Pulmonary Arterial Hypertension), PAH_Subject_Phenotypes_Baseline_Measures, pht002277.v1.p1, phs000290.v1.p1; NHLBI GO-ESP: Lung Cohorts Exome Sequencing Project (Lung Health Study of Chronic Obstructive Pulmonary Disease), LHS_COPD_Subject_ Phenotypes_Baseline_Measures, pht002272.v1.p1, phs000291. v1.p1. C.D.B. is on the scientific advisory board for Personalis, Incorporated; Mubadala Medical Holding Company; 23andme “Roots into the future” project; and Ancestry.com. M.J.R. owns stock in Illumina.
Footnotes
References and Notes
- 1.Bamshad MJ, et al. Nat Rev Genet. 2011;12:745. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- 2.Ajay SS, Parker SC, Abaan HO, Fajardo KV, Margulies EH. Genome Res. 2011;21:1498. doi: 10.1101/gr.123638.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sobreira NL, et al. PLoS Genet. 2010;6:e1000991. doi: 10.1371/journal.pgen.1000991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.International HapMap Consortium. Nature. 2005;437:1299. [Google Scholar]
- 5.Frazer KA, et al. Nature. 2007;449:851. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li JZ, et al. Science. 2008;319:1100. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- 7.Fu YX. Theor Popul Biol. 1995;48:172. doi: 10.1006/tpbi.1995.1025. [DOI] [PubMed] [Google Scholar]
- 8.Marth GT, et al. Genome Biol. 2011;12:R84. doi: 10.1186/gb-2011-12-9-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ng SB, et al. Nature. 2009;461:272. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O'Roak BJ, et al. Nat Genet. 2011;43:585. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ng SB, et al. Nat Genet. 2010;42:790. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ng SB, et al. Nat Genet. 2010;42:30. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tennessen JA, Madeoy J, Akey JM. Genome Res. 2010;20:1327. doi: 10.1101/gr.106161.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yi X, et al. Science. 2010;329:75. [Google Scholar]
- 15.McClellan J, King MC. Cell. 2010;141:210. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- 16.Manolio TA, et al. Nature. 2009;461:747. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gibson G. Nat Rev Genet. 2011;13:135. doi: 10.1038/nrg3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Supplementary materials are available on Science Online.
- 19.Kimura M. Nature. 1968;217:624. doi: 10.1038/217624a0. [DOI] [PubMed] [Google Scholar]
- 20.Ramírez-Soriano A, Nielsen R. Genetics. 2009;181:701. doi: 10.1534/genetics.108.094060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akey JM, et al. PLoS Biol. 2004;2:e286. doi: 10.1371/journal.pbio.0020286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coventry A, et al. Nat Commun. 2010;1:131. doi: 10.1038/ncomms1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gravel S, et al. Proc Natl Acad Sci USA. 2011;108:11983. [Google Scholar]
- 24.dos Reis M, Savva R, Wernisch L. Nucleic Acids Res. 2004;32:5036. doi: 10.1093/nar/gkh834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McDonald JH, Kreitman M. Nature. 1991;351:652. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- 26.Akey JM. Genome Res. 2009;19:711. doi: 10.1101/gr.086652.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Asimit J, Zeggini E. Annu Rev Genet. 2010;44:293. doi: 10.1146/annurev-genet-102209-163421. [DOI] [PubMed] [Google Scholar]
- 28.Kryukov GV, Shpunt A, Stamatoyannopoulos JA, Sunyaev SR. Proc Natl Acad Sci USA. 2009;106:3871. doi: 10.1073/pnas.0812824106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lohmueller KE, et al. Nature. 2008;451:994. doi: 10.1038/nature06611. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary info