Neutrophil extracellular traps: Is immunity the second function of chromatin? (original) (raw)
Abstract
Neutrophil extracellular traps (NETs) are made of processed chromatin bound to granular and selected cytoplasmic proteins. NETs are released by white blood cells called neutrophils, maybe as a last resort, to control microbial infections. This release of chromatin is the result of a unique form of cell death, dubbed “NETosis.” Here we review our understanding of how NETs are made, their function in infections and as danger signals, and their emerging importance in autoimmunity and coagulation.
Introduction
Neutrophils are terminally differentiated white blood cells that have a short life in circulation. If called into action, neutrophils leave the blood vessels and move toward the site of infection, following a chemotactic gradient produced by microbial or endogenous signals. At the inflammatory site, neutrophils are “activated” to perform several tasks, including cytokine secretion, degranulation, and phagocytosis. Elie Metchnikoff (Metchnikoff, 1893) and Paul Ehrlich (Ehrlich, 1880) were the first to show that phagocytes ingest and digest bacteria. This process is of paramount importance in immunology.
Neutrophils have two distinctive morphological characteristics: the shape of their nucleus and their granules (Fig. 1). The nucleus of neutrophils is split into three to five lobules, hence the alternative name of “polymorphonuclear” often given to these cells. The evolutionary advantages of having a lobulated nucleus are not clear. Granules are specialized vesicles that contain a specific load, including many toxic molecules. Depending on their contents, granules are canonically classified into four groups: primary or azurophilic, secondary or specific, and tertiary or gelatinase, as well as secretory vesicles. Eosinophils, basophils, and mast cells also have granules, and together with neutrophils they make up the “granulocyte” family.
Figure 1.
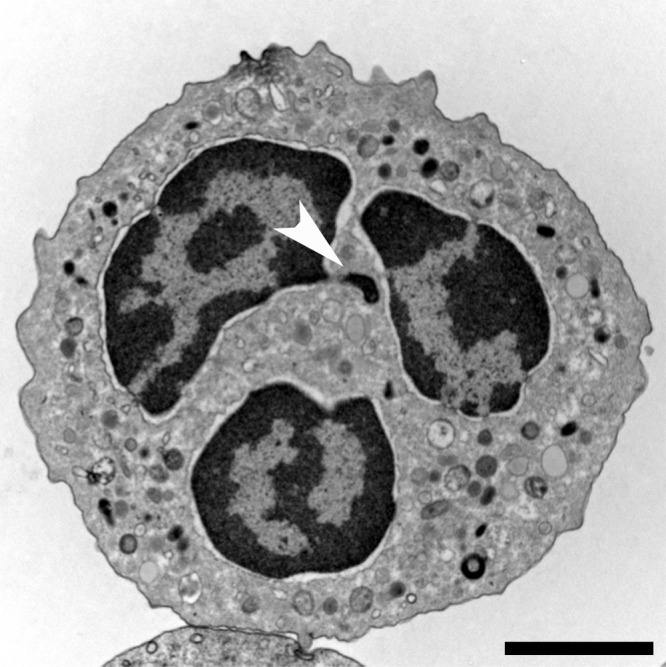
Neutrophil morphology. Transmission electron microscopy (TEM) of a naive human neutrophil. This cell contains various types of granules, clearly visible in the cytoplasm, as well as a lobulated nucleus. The highly condensed heterochromatin (dark) is neatly marginalized to the edge of the nucleus, only interrupted by euchromatic areas close to nuclear pores that mostly line the nuclear membrane. The brighter euchromatin is mostly in the center of the lobules. This neutrophil comes from a female donor and one inactivated x chromosome can be found as an extranuclear stretch of heterochromatin (arrowhead). These structures are termed Barr bodies, and in neutrophils “drum sticks.” Bar, 2 µm.
Neutrophils are efficient phagocytes and engulf microbes into phagosomes that rapidly fuse with the granules, creating an inhospitable environment. There, microbes are exposed to many enzymes, including lysozyme, which breaks the bacterial wall; proteases; and phospholipases. Also, very cationic peptides, like bactericidal permeability–increasing protein (BPI), defensins, and cathelicidins, are discharged into the phagolysosome. Simultaneously, reactive oxygen species (ROS), like superoxide and hydrogen peroxide, are generated by the NADPH oxidase complex at the phagosomal membrane and released into its lumen. The biological activity of many of these components under defined in vitro conditions has been demonstrated numerous times but the relative contribution of each of them to neutrophil function in vivo remains to be determined (Nathan, 2006; Borregaard, 2010; Amulic et al., 2012).
Neutrophils can also kill pathogens extracellularly by releasing neutrophil extracellular traps (NETs; Brinkmann et al., 2004). The impact of NETs derives from the combined antimicrobial activities of granular components, histones, and some cytoplasmic proteins. Eosinophils and mast cells, which are granulocytes closely related to neutrophils, granulocyte homologues in lower vertebrates, and even plants release extracellular traps. Hence, in addition to describing the function of NETs, we will also comment on the significance of extracellular traps in evolution.
NET morphology
The ultrastructure of NETs is unusual; NETs consist of smooth filaments with a diameter of ∼17 nm (Brinkmann et al., 2004), composed of stacked, and probably modified, nucleosomes (Urban et al., 2009). This backbone is studded with globular domains with a diameter of ∼50 nm made of granular proteins (Fig. 2; Brinkmann et al., 2004). This morphology in high-resolution scanning electron microscopy easily differentiates NETs from other fibrous structures such as fibrin. Interestingly, unfixed, fully hydrated NETs have a cloud-like appearance and occupy a space that is 10–15-fold bigger than the volume of the cells they originate from (Video 1), reflecting what they may look like in vivo when space is available, for example in the lung alveolus.
Figure 2.

Bacteria caught in NETs. Scanning electron microscopy of human neutrophils incubated with Salmonella, a bacterium that causes typhoid fever and gastroenteritis. The bacteria are trapped in NETs. Bar, 1 µm.
The mechanism of NET formation
NETs are the results of a unique form of cell death that morphologically is characterized by the loss of intracellular membranes before the integrity of the plasma membrane is compromised. Steinberg and Grinstein (2007) coined the term “NETosis” for neutrophil cell death that leads to the formation of NETs. To release NETs, activated neutrophils undergo dramatic morphological changes. Minutes after activation, they flatten and firmly attach to the substratum (Fig. 3 B). During the next hour, the nucleus loses its lobules, the chromatin decondenses, and the inner and outer nuclear membranes progressively detach from each other. Concomitantly, the granules disintegrate. After 1 h, the nuclear envelope disaggregates into vesicles and the nucleoplasm and cytoplasm form a homogenous mass (Fig. 3 C). Finally, the cells round up and seem to contract until the cell membrane ruptures and the interior of the cell is ejected into the extracellular space, forming NETs (Fig. 3 D and Video 2; Fuchs et al., 2007). Notably, despite the intermixing of cellular compartments, during the last phase of NETosis, <30 proteins are present in NETs. Most of them originate from granules, few are from the nucleus, and cytoplasmic NET components are rare (Urban et al., 2009). NETosis is morphologically quite different from apoptosis and other forms of cell death (Fuchs et al., 2007). Other investigators have proposed alternative processes to the one just described that will, because of space limitations, not be discussed further. For example, one interesting observation is that NETs can result from the release of nuclear fragments and then their chromatin without compromising the plasma membrane (Pilsczek et al., 2010). Also, Yousefi et al. (2008, 2009) have proposed that living eosinophils and neutrophils expel their mitochondria, which release their DNA into the extracellular space. Importantly, however, granulocytes are particularly poor in mitochondria, and mitochondrial DNA is 100,000 times less abundant in extracellular traps than nuclear DNA (Pilsczek et al., 2010); hence, the significance of this finding awaits further investigation.
Figure 3.

Schematic representation of the NETosis pathway. After stimulation of receptors (A), neutrophils adhere to the substrate (B) and mobilize granule components, namely NE and MPO (C). Granules are depicted as red circles. Histones in the nucleus get processed, and the intracellular membranes disintegrate. Finally, the cell membrane ruptures, and the mixture of cytoplasm and nucleoplasm gets expelled to form NETs (D).
Many physiological inducers (Fig. 3 A) of NETosis have been reported. Infections with bacteria, fungi, and HIV parasites (listed in Table 1) induce NETs. Other physiologically relevant stimuli are ROS like hydrogen peroxide (Fuchs et al., 2007). NET formation is also triggered, albeit inefficiently, by antibodies (Kessenbrock et al., 2009) and antibody–antigen complexes (Garcia-Romo et al., 2011; Lande et al., 2011), and by microbial components such as lipopolysaccharide (Neeli et al., 2009; Lim et al., 2011), M1 from Streptococcus pyogenes (Oehmcke et al., 2009), or lipophosphoglycans from Leishmania amazonensis (Guimarães-Costa et al., 2009). Rapid NET formation is also induced by platelets activated via Toll-like receptor 4 (TLR-4; Clark et al., 2007). NET formation appears to require attachment of neutrophils to a substrate that stimulates the MAC-1 integrin receptors (Neeli et al., 2008). In suspension, neutrophils make NETs poorly, probably preventing excessive formation of NETs in circulation and avoiding thrombus formation, which will be discussed later.
Table 1.
Pathogens that induce NETs
| Species | References |
|---|---|
| S. aureus | Fuchs et al., 2007; Pilsczek et al., 2010 |
| S. pyogenes | Buchanan et al., 2006 |
| Group A S. pyogenes | Lauth et al., 2009 |
| E. coli | Grinberg et al., 2008 |
| Shigella flexneri | Brinkmann et al., 2004 |
| Nontypeable H. influenzae | Hong et al., 2009 |
| Yersinia enterocolitica and Yersinia pseudotuberculosis | Casutt-Meyer et al., 2010 |
| Mannheimia haemolytica | Aulik et al., 2010 |
| Mycobacterium tuberculosis | Ramos-Kichik et al., 2009 |
| Candida albicans | Urban et al., 2006 |
| Aspergillus fumigatus | Bruns et al., 2010 |
| Aspergillus nidulans | Bianchi et al., 2011 |
| L. amazonensis | Guimarães-Costa et al., 2009 |
| Toxoplasma gondii | Abi Abdallah et al., 2012 |
| HIV-1 | Saitoh et al., 2012 |
Molecularly, the few events that have been shown to be required, sequentially, are the production of ROS, the migration of the protease neutrophil elastase (NE) and later myeloperoxidase (MPO) from granules to the nucleus, the processing of histones, and eventually the rupture of the cell. It is relevant to mention that the study of neutrophils is limited by the short life of these cells and the lack of established cell lines that faithfully reproduce granulocyte biology, which rules out many conventional molecular approaches. In this section, we review our current knowledge about the mechanism of NET formation.
NET formation requires the production of ROS. The NADPH oxidase enzyme complex (also called phagocytic oxidase; PHOX) assembles at the cell and phagosomal membrane and reduces molecular oxygen into superoxide anions by transferring electrons from NADPH. Superoxide dismutates into hydrogen peroxide, which in turn acts as substrate for one of the most abundant enzymes in the neutrophil’s granules: MPO. MPO reacts with hydrogen peroxide to generate hypohalous acids, such as hypochlorous acid (HOCl). ROS oxidize various types of molecules including nucleic acids, lipids, and proteins.
The requirement for ROS in NET formation was shown pharmacologically and, more relevantly, by testing the neutrophils of patients with immune deficiencies. Patients with mutations in any of the subunits of the PHOX complex cannot produce ROS or make NETs. These chronic granulomatous disease (CGD) patients suffer from life-threatening recurrent infections (Fuchs et al., 2007; Bianchi et al., 2009). Interestingly, when neutrophils of CGD patients are treated with H2O2, the cells produce NETs, showing that the pathway can be rescued downstream of PHOX (Fuchs et al., 2007).
The most potent inducer of PHOX activation is PMA, which directly stimulates PKC. Downstream of PKC but upstream of PHOX, the NET signaling cascade includes the Raf–MEK–ERK kinase pathway (Hakkim et al., 2011) and Rac2 (Ras-related C3 botulinum toxin substrate 2; Lim et al., 2011).
During NETosis, the segregation between eu- and heterochromatin is lost, and the nucleoplasm appears homogenous (Fuchs et al., 2007). This depends on the activity of NE and MPO, which are stored in azurophilic granules. NE is released, by an unknown mechanism, from granules and enters the nucleus, where it degrades the linker histone H1 and processes core histones (Papayannopoulos et al., 2010). NE activity is essential for NET formation because NE-deficient mice do not make NETs, which contributes to their immune deficiency (Papayannopoulos et al., 2010). MPO also migrates to the nucleus later than NE, where it enhances chromatin decondensation (Papayannopoulos et al., 2010). In agreement with this requirement, patients without MPO activity cannot produce NETs (Metzler et al., 2011), and hypochlorous acid, the product of MPO, is sufficient for NET release (Palmer et al., 2012). In addition to partial degradation by NE, histones undergo further modifications to decondense the chromatin structure. Upon neutrophil activation, the enzyme peptidylarginine deiminase 4 (PAD4) catalyzes the conversion of arginine residues to citrulline in three of the four core histones. In NETs and decondensed nuclei, but not in the nucleus of unstimulated neutrophils, histones are citrullinated (Neeli et al., 2008, 2009; Wang et al., 2009). The relevance of PAD4 was tested pharmacologically in cell lines, which make few NETs, if any, but not in neutrophils. In PAD4-null mice, hypercitrullination of H3 was not detectable, and the strain failed to produce NETs (Li et al., 2010; Hemmers et al., 2011). Interestingly, in a S. pyogenes infection model, PAD4-null mice developed larger lesions than their PAD4-expressing siblings (Li et al., 2010), but NET formation remains to be quantified in this model.
The autophagy pathway was recently proposed to be required for NETosis downstream of PHOX. When neutrophils are stimulated with PMA, they develop large vacuoles that are reminiscent of autophagosomes. Evidence for the involvement of this process in NETosis comes exclusively from pharmacological studies with wortmannin, which inhibits PI3Kinases and PI3K-like enzymes and has low specificity (Remijsen et al., 2011a). Experiments using genetic tools to implicate autophagy in NET formation have not been described.
Eventually, NETs are removed during the resolution of inflammation. NETs are susceptible to DNase1 (von Köckritz-Blickwede et al., 2009; Hakkim et al., 2010), an enzyme produced by the pancreas. It is not known what happens to the debris left by DNase1 but perhaps phagocytes, macrophages, and neutrophils newly recruited to the inflammatory site clean up the mess (Bratton and Henson, 2011).
Methods to quantify NETs
NETs are rather fragile structures, and some effort is required to unambiguously detect and quantify them. NET quantification should rely on their unique composition: chromatin tightly linked to neutrophil proteins such as NE, MPO, or calgranulin. This definition excludes chromatin released by other forms of cell death. Published methods of NET quantification include microscopy (Brinkmann et al., 2004; Hakkim et al., 2010; Papayannopoulos et al., 2010; Metzler et al., 2011; Remijsen et al., 2011b) and DNA detection either with membrane-impermeable DNA dyes (Brinkmann et al., 2004) or by staining the DNA in the supernatant after releasing the NETs with a mild nuclease treatment (Fuchs et al., 2007).
Immunostaining is an obvious way to detect NETs (Brinkmann et al., 2004, 2010) but is prone to biases introduced by the observer. Automatic microscopy (Hakkim et al., 2011) is an objective and quantitative method (Papayannopoulos et al., 2010; Metzler et al., 2011) to measure NET formation. Changes in nuclear morphology (loss of lobules and expansion of the nucleus) and composition (migration of NE and MPO to the nucleus) are specific and quantitative markers of the progress of NETosis. Anti-chromatin antibodies stain the compact nuclei of unstimulated neutrophils weakly, but the signal increases as the chromatin relaxes (Fig. 4; Ermert et al., 2009). In tissue sections (Brinkmann et al., 2004) and in secretions (Papayannopoulos et al., 2011; Manzenreiter et al., 2012), NETs have been identified using the same markers mentioned here. Computer-assisted analysis of the overlap between chromatin and neutrophil markers can quantify NETs in tissue sections. Although more technically challenging, NETs can also be identified in vitro and in vivo by measuring their size and detecting their antigens by scanning or transmission electron microscopy (Brinkmann et al., 2004; Krautgartner and Vitkov, 2008; Urban et al., 2009; Manzenreiter et al., 2012).
Figure 4.

Visualizing NETs using chromatin antibodies or DNA-intercalating dyes. Human neutrophils were activated in vitro and then processed for immunofluorescence. An antibody directed against the subnucleosomal complex of H2A, H2B, and DNA stains intact, compact chromatin only weakly, but reacts strongly with relaxed chromatin in the NETs (A, red in D). In contrast, DNA-intercalating dyes provide the brightest staining at sites of high DNA concentrations, as is the case in compact nuclei, whereas NETs are stained rather weakly (A, Hoechst 33342; blue in D). (C, green in D) The granular marker NE, which can be observed in granules in cells that are not yet activated, as well as in NETs. A projection of confocal z-stack is shown. Bar, 10 µm.
Methods that rely on non-cell-permeable DNA dyes like SYTOX green are simple and can be used for automatic screening of unfixed cells (Fuchs et al., 2007). In pure neutrophil cultures, quantification of extracellular DNA reflects the amount of NETs if the experimental setup excludes other forms of cell death. Special care must be taken when performing experiments that require prolonged incubation times (>4 h for human neutrophils), cocultivation with microbes, or direct application of toxins or drugs to the cells, as under these conditions, mechanisms of DNA release other than NETosis can contribute to the experimental readout.
Another method that demonstrates the binding between chromatin and neutrophil proteins, and can be diagnostic for NET, consists of resolving partially digested NETs electrophoretically. Very cationic proteins such as NE, MPO, and histones that, when pure, migrate to the anode, are dragged toward the cathode when complexed to DNA. These proteins, however, move toward the anode when the DNA in the NETs is degraded. Immune or enzymatic quantification of these proteins was used to demonstrate NETs in sputum from cystic fibrosis (CF) patients (Papayannopoulos et al., 2011).
Antimicrobial activity
The main job of neutrophils is to eliminate microbes. It is probable that NETs evolved to rein in infections by, as their name indicates, “trapping,” preventing dissemination, inactivating virulence factors, and exterminating microbes. Trapping microbes prevents their dissemination from the initial infection site. Microbes most likely stick to NETs through charge interactions (Urban et al., 2009; Bartneck et al., 2010). Indeed, pathogens mask themselves with a capsule or by changing their surface charge, thus preventing binding to NETs (Wartha et al., 2007). Bacteria also attach nucleases to their surfaces to disengage themselves from NETs (Sumby et al., 2005). Group A S. pyogenes (Buchanan et al., 2006), pneumococcus, and Staphylococcus aureus (Berends et al., 2010) encode endonucleases that liberate them from NETs, permitting the invasion of deeper organs (Beiter et al., 2006). The antimicrobial activity of NETs depends on their structure, which provides a high local concentration of antimicrobials in direct proximity to trapped microorganisms and is lost after DNase digestion (Brinkmann et al., 2004). Accordingly, expression of these DNases is essential for these bacteria to be pathogenic (Buchanan et al., 2006).
NETs can inactivate microbial proteins, called “virulence factors,” that modify the function of host cells. NE on the NETs specifically cleaves virulence factors of Shigella flexneri, Salmonella typhimurium, and Yersinia enterocolitica (Weinrauch et al., 2002; Brinkmann et al., 2004). NETs also contain Cathepsin G and Proteinase 3, which are closely related to NE and might cleave virulence factors of a different class of pathogens (Averhoff et al., 2008). NETs contain several proteins that kill or inhibit microbes. These include enzymes (lysozyme, proteases), antimicrobial peptides (BPI, defensins), ion chelators (calgranulin), and, interestingly, histones. The antimicrobial activity of NETs is likely the result from the combination of these components, their effects enhanced by the high local concentrations achieved on the NETs. Also, some of the NET components work solo. Parker et al. (2012) showed that the activity of MPO on NETs is essential to kill S. aureus. The antifungal activity of NETs has been assigned to Calgranulin (Urban et al., 2009; Bianchi et al., 2011), which chelates Zinc, a cation required for fungal growth. Lastly, antibodies against histones prevent NET-mediated killing of various microorganisms (Brinkmann et al., 2004), underlining the finding that these abundant proteins kill microbes very efficiently, as discussed next.
The antimicrobial activity of NETs can be measured with different methods. The simplest one is to induce the formation of NETs, add microbes, and assess the number of surviving bacteria after an incubation period by plating. This experiment is controlled with cultures where the NETs are degraded by DNases before infection. Any lingering live neutrophil capable of phagocytosis can be inhibited with Cytochalasin D. This method might not distinguish between microbial killing and microbial “clumping”; i.e., several bacteria sticking together in a piece of NET could form a single colony. The issue of clumping is resolved by adding yet another step in the experiment by incubating the bacteria with DNases again just before plating to dissolve the clumps. Dissolving these “clumps” allows the adequate enumeration of bacteria (Urban et al., 2006, 2009; Wartha et al., 2007; Lauth et al., 2009; Bruns et al., 2010; Cogen et al., 2010; Parker et al., 2012); see also the editorial by Nauseef (2012). Clumping is a reflection of one of the other essential functions of NETs: trapping (Menegazzi et al., 2012).
A second method to measure microbial killing is to prevent killing by blocking NET components with antibodies (Brinkmann et al., 2004) or cation chelators, for example Zinc (Urban et al., 2009). Microbial killing can also be demonstrated using commercially available fluorescent dyes that report viability (Hong et al., 2009; Lauth et al., 2009) or indicators of metabolic activity. Also, recombinant microbes that express enzymes like luciferase (Gabriel et al., 2010) were reported.
Using these methods, investigators have shown that NETs kill Gram-positive and -negative bacteria, parasites, and fungi in vitro, but do NETs contribute to microbial killing in vivo? Interestingly, newborns are highly susceptible to infections, and their formation of NETs is delayed (Fadeel, 2009; Yost et al., 2009; Yost and Zimmerman, 2009). More relevantly, patients with inherited deficiencies in genes involved in NET formation, namely PHOX, MPO, and NE, suffer from repeated infections, as do mice in which these genes were ablated. These proteins are important for many neutrophil functions, though some of the symptoms could be caused by defects in NET formation. Thus, experiments will have to be designed to tease apart the different functions of neutrophils to measure these enzymes’ contributions to NET formation, phagocytosis, and degranulation.
The importance of NETs in microbial defense is underscored by their presence in pus. Superficial infections produce pus. For centuries, as Pisetsky (2011) pointed out in a recent review, pus with a high viscosity was regarded as “good” because it resolved the infection. Now we know that pus consists mostly of neutrophils surrounded by NETs (Fig. 5).
Figure 5.

NETs are abundant in Pus. Pus consists of numerous neutrophils in various stages of NETosis (arrowheads) surrounded by NETs. Semithin cryosection of pus from a Molluscum contagiosum lesion stained for NE (green) and chromatin (red). Bar, 20 µm.
The relevance of NETs was indirectly demonstrated in a CGD patient with severe Aspergillosis (Bianchi et al., 2009). CGD is an inherited disease caused by nonfunctional PHOX. This defect interferes with phagocytic killing and prevents the formation of NETs. As expected, neutrophils isolated from this patient did not kill hyphae of the Aspergillus strain isolated from his lungs because the neutrophils did not make NETs, and hyphae are too large to be phagocytosed. The life of the patient was in danger and, after other therapies failed, the patient was treated with gene therapy. This therapy partially restored the activity of PHOX, as well as the capacity to make NETs. Notably, after gene therapy, the patient’s neutrophils, like those of a healthy donor, killed Aspergillus poorly by phagocytosis but effectively through NETs. The infection resolved only a couple of weeks after treatment and the patient was cured. Because the infectious agent was not susceptible to phagocytosis, this recovery is likely caused by the regained capacity to make NETs.
Besides being active against bacteria, fungi, and parasites, NETs are also antiviral. Recently, Saitoh et al. (2012) found that NET formation can be triggered by human immunodeficiency virus 1 (HIV-1), probably through TLR-7 and TLR-8, endosomal receptors that sense viral RNA. Interestingly, HIV-1 virions bound to NETs and were inactivated, a process that was blocked if DNase was present and NETs were degraded (Saitoh et al., 2012).
Histones: organizing life, inducing death
Histones are indispensable for eukaryotic and archaeal life. Two of each of the core histones, H2A, H2B, H3, and H4, form the nucleosome core particle, a disc of ∼10 nm in diameter that is wrapped by 147 base pairs of DNA. Histone H1 links these core particles, forming a stack that coils around itself. This architecture allows chromatin to condense, for example during mitosis, and relax to allow transcription.
Around the middle of the previous century, investigators found that histones, unexpectedly, are potent antibiotics (Miller et al., 1942). They kill bacteria at nanomolar concentrations (Hirsch, 1958) far more effectively than most other antimicrobials. Much later, driven by the hypothesis that animals living in aquatic biotopes brimming with microorganisms need protection, histone fragments were isolated as antimicrobials from the stomachs of toads (Cho et al., 2009) and from other aquatic animals (Park et al., 1998; Birkemo et al., 2003, 2004; Kawasaki et al., 2003; Patat et al., 2004; Lüders et al., 2005; Dorrington et al., 2011; Noga et al., 2011). Histone fragments, as opposed to whole histones, kill microbes only in simple solutions, like saline; therefore, the role of these fragments in vivo should be investigated further. In mammals, extranuclear histones are found in the cytoplasm and on the surface of cells (for review see Parseghian and Luhrs, 2006) and are released abundantly in NETs (Urban et al., 2009).
Histones kill Gram-positive and -negative bacteria (Hirsch, 1958) and parasites (Wang et al., 2011). One mole of histones kills ∼100-fold more bacteria than other antimicrobials, such as defensins. The mechanism of histone toxicity is poorly understood, although, like many other antimicrobials, eukaryotic histones are cationic, probably allowing them to bind to microbial membranes, either destroying them or making them permeable enough for small factors, including histones themselves. Histone fragments might bind to prokaryotic DNA (Kawasaki et al., 2008) and interfere with gyrase activity (Lemaire et al., 2008). Notably, Charles Esmon’s group (Xu et al., 2009) and others (Gupta et al., 2010; Saffarzadeh et al., 2012) showed that histones also kill mammalian cells, and implicated this toxicity in the pathogenesis of sepsis (Xu et al., 2009). This could be another example of immune effectors inducing collateral damage. The mechanisms behind histone toxicity should be investigated further.
Chromatin sensing: NETs are a danger signal
The innate immune system evolved to detect and react to the disruption of homeostasis. This is sensed through receptors that detect microbe-specific molecules called “pathogen-associated molecular patterns” (PAMPS) and endogenous molecules that signal danger, “danger-associated molecular patterns” (DAMPs). PAMPs are common to many microbes, for example lipopolysaccharide, peptidoglycan, and flagellin, which are of bacterial origin, as well as RNA and DNA, which can be of viral, bacterial, or parasitic origin. Some examples of DAMPs are heat shock proteins and high-mobility group box 1 (HMGB1) protein, as well as RNA and DNA of host origin.
DNA can be sensed by extra- and intracellular receptors. Extracellular DNA activates TLR-9, which resides in the phagosomes of monocytes and dendritic cells (DCs). TLR-9 is preferentially activated by nonmethylated DNA rich in cytidine and guanosine, or CpG, which is more abundant in microbes than in eukaryotes. Lande et al. (2007) showed that DNA complexed with the antimicrobial LL37 (processed cathelicidin of 37 amino acids) or HMGB1 forms stable structures that activate DCs more potently than naked DNA. Importantly, DNA is rarely, if ever, naked; it complexes with “histone-like” proteins in bacteria and with histones in eukaryotes and archea. Hence, in vivo TLR-9 is likely exposed to DNA–protein complexes. The groups of both Michelle Gilliet and Virginia Pascual showed that NETs activate TLR-9 in DCs (Garcia-Romo et al., 2011; Lande et al., 2011). NETs can also prime T cells, although it is not known through which receptor (Tillack et al., 2012). DNA is not the only component of chromatin or of NETs that can activate the innate immune system. It was also reported that histones activate TLR-2 and TLR-4, which suggests that NETs serve as innate immune activators through different receptors (Semeraro et al., 2011).
DNase1, DNase1-like 2, and DNase1-like 3 are three similar secreted enzymes that cleave DNA and dispose of extracellular DNA, including NETs as mentioned in “Mehods to quantify NETs.” It is possible that these nucleases prevent TLR-9 and other receptor activation by NETs, an option that is substantiated by the role they play in autoimmunity.
In summary, during inflammation, NETs are likely to contribute to alerting the immune system of a danger by activating DNA receptors such as TLR-9. This activation might turn out to help in the recruitment of immune cells to mount an acquired immune response or to resolve the inflammation. The activation of DNA receptors by NETs can also have negative effects as reviewed in the “Autoimmunity” section.
The dark side of NETs
Paracelsus wrote, “All things are poison, and nothing is without poison; only the dose permits something not to be poisonous.” NETs can either fight disease or cause disease depending on the place, time, and dose. Making too many or not disposing of NETs at the right time and in the right place is pathogenic. Here we describe some pathologies where NETs play a role.
CF.
CF is the most common severe inherited disease among people of European origin. CF patients produce large quantities of a tenacious mucus that facilitates colonization of the lungs with bacteria like S. aureus, Haemophilus influenzae, and Pseudomonas aeruginosa. The CF patients suffer from a persistent neutrophil-rich inflammation that destroys their lungs. Recently, it was found that NETs are present in sputum from CF patients (Manzenreiter et al., 2012). The abundance of NETs, and specifically their chromatin backbone, contributes to the viscosity of CF sputum. Indeed, CF patients are palliatively treated with recombinant DNase to liquefy the sputum and facilitate mucociliar clearance. During this process, NE is released from NETs and is thought to induce tissue damage. NE might play a paradoxical role and also be beneficial for the patient because this enzyme processes core histones, relaxes chromatin, and fluidizes the sputum (Papayannopoulos et al., 2011), which may explain the failure of NE inhibitors in the clinics.
Preeclampsia.
Preeclampsia is a late pregnancy disorder affecting between 5 and 7% of pregnant women. It is characterized by hypertension and proteinuria. Often, acute kidney and liver failure as well as hemolysis are life threatening for the mother, whereas the fetus can suffer severe hypoxia. The outermost layer of the placenta, the trophoblast, is a multinucleated syncytium covered with numerous microvilli. It continuously sheds membranous particles of various sizes. In vitro, these particles induce NETs. For unknown reasons, in pre-eclamptic but not in normal placentas, NETs are in close contact with the syncytiotrophoblast. NETs could obstruct the intervillous space, reduce the blood flow, and lead to hypoxia in the fetus (Gupta et al., 2005). A second pathomechanism of the disease could be the induction of NETosis by activated endothelial cells. The resulting NETs could, in turn, damage the endothelium, establishing a vicious circle leading to more severe preeclampsia (Gupta et al., 2010).
Coagulation.
Coagulation is a way to reduce blood loss after injury, but it also represents a primitive innate immune response that limits microbial spreading (Esmon et al., 2011). Coagulation is an example of how the amount of NET formation can determine a “good” or “bad” outcome. NETs participate in timely clot formation, but if present in excess they induce massive coagulation that can stop the blood supply of organs, causing severe ischemia.
Arterial blood clots are often induced by damage to the endothelium. In contrast, venous thrombi mainly develop when the blood flow is reduced for several hours. In both situations, neutrophils accumulate and adhere tightly to the endothelium. There, neutrophils produce NETs that serve as a scaffold for the stimulation of thrombus formation (Fuchs et al., 2010). Both NE and cathepsin G, two serine proteases that are in the NETs, degrade inhibitors of coagulation. In mice deficient in both enzymes, during arterial thrombosis, fibrin deposition and clot formation are reduced, as is the case when the mice are treated with an anti-NET antibody (Massberg et al., 2010). Interestingly, in an Escherichia coli systemic infection, the proportion of bacteria sequestered in the microvasculature of the liver was higher in animals with functional NETs than in animals treated with an anti-chromatin antibody that blocks NET function (Massberg et al., 2010), underlining the fact that coagulation also reduces bacterial spread to other organs. Together, these data indicate that clotting is enhanced by NETs, promoting defense against pathogens.
Although the vessel is not damaged at the onset of venous thrombogenesis, numerous neutrophils and macrophages are recruited and play a major role during formation of the thrombus. There, activated platelets stimulate neutrophils to form NETs (Clark et al., 2007; Caudrillier et al., 2012), which serve as a prothrombotic scaffold and bind and activate FXII (von Brühl et al., 2012). Consequently, NETs can be detected in venous thrombi (Brill et al., 2012).
Periodontitis.
Periodontitis, an inflammatory disease of the tissue supporting the teeth, is caused by bacteria, such as Porphyromonas gingivalis (Farquharson et al., 2012), that recruit neutrophils into the gingival crevice, where they produce NETs (Vitkov et al., 2009). Chronic periodontitis and hypercoagulation are epidemiologically associated to atherothrombosis (Demmer and Desvarieux, 2006), which can cause abdominal aneurysms. In a rat model, repeated injections with Porphyromonas gingivalis led to abdominal aneurysms (Delbosc et al., 2011). The intraluminal thrombi observed in this model were rich in NETs, which may damage the endothelium underlying the thrombus. In addition to the proteolytic enzymes present in NETs, histones were shown to directly kill endothelial cells (Gupta et al., 2010; Saffarzadeh et al., 2012). Luminal NETs were also found at atherosclerotic sites in a mouse model and in human samples (Megens et al., 2012).
Autoimmunity.
Autoimmunity is an immune response against self, an aberration that causes debilitating diseases. Systemic lupus erythematosus (SLE) patients, mostly women, often make antibodies against DNA, histones, and neutrophil proteins; i.e., the components of the NETs. Although the etiology of SLE is not known, and it is likely that different diseases are subsumed under the same name, it is clear that this disease is exacerbated by inflammation. Neutrophils play a pivotal, but until recently undefined, role in SLE (Baechler et al., 2003; Bennett et al., 2003).
Neutrophils isolated from SLE patients are more prone to making NETs, particularly in response to antibody complexes; i.e. antibodies bound to their antigens (Craft, 2011; Garcia-Romo et al., 2011; Lande et al., 2011; Villanueva et al., 2011) These immune complexes isolated from other autoimmune diseases, such as small vessel vasculitis or Wegener’s disease, also induce NET formation, albeit with lower efficiency (Kessenbrock et al., 2009). Indeed, neutrophils isolated from healthy donors only respond to these immune complexes if primed in the dish. Interestingly, NETs activate TLR-9 in DCs, the translators of the innate to the acquired immune response, to make interferons (Garcia-Romo et al., 2011; Lande et al., 2011). This exacerbates the disease, suggesting that NETs might initiate autoimmune responses.
An inherited form of SLE was linked to a mutation in DNase1 (Yasutomo et al., 2001) or to DNase1-like 3 (Al-Mayouf et al., 2011), an enzyme that degrades NETs, as described earlier. This suggests that persistence of NETs is also linked to SLE. Furthermore, in a European cohort, lack of NET degradation in the sera of a subpopulation of SLE patients was due either to the presence of DNase1 inhibitors or a high titer of anti-NET antibodies (Hakkim et al., 2010). In either case, low NET degradation correlates with lupus nephritis, a severe consequence of the disease. The inability to degrade NETs could be caused by complement activation and increased deposition of the complement protein C1q, which inhibits DNase1 (Leffler et al., 2012). It appears that inappropriate production or prolonged exposure to NETs could circumvent tolerance and lead to the production of autoantibodies.
It remains to be determined whether NETs present antigens that are altered relative to their tolerized counterparts. One example is ulcerative colitis, a severe inflammation of the colon with formation of autoantibodies against the granular protein lactoferrin bound to DNA, a complex that is present in NETs (Teegen et al., 2009). Posttranslational histone modifications have been proposed to induce formation of autoantibodies (Neeli et al., 2008; Liu et al., 2012). Recently, it was shown that patients with Felty’s syndrome, a form of rheumatoid arthritis, produce autoantibodies against citrullinated histones (Dwivedi et al., 2012). It is possible that proteolytic and oxidative processing of proteins during NETosis generates neoantigens.
Evolution
Infection and immunity are two of the driving forces of evolution of both host and pathogen. NETs contribute to these processes. Pathogenic bacteria have evolved surface nucleases that detach them from the NETs to permit dissemination, as is the case with group A Streptococci (Buchanan et al., 2006) and pneumococci (Beiter et al., 2006). This was further, and elegantly, shown by the dependence on selection for a DNase for the global dissemination of a hypervirulent strain of invasive group A Streptococcus (Walker et al., 2007).
On the host side, the principal components of NETs—DNA and histones—are ancient. These two are shared between archea and eukaryotes. In humans, extracellular traps are not exclusive to neutrophils; mast cells (von Köckritz-Blickwede et al., 2008) also release their chromatin decorated with cytoplasmic proteins. Neutrophils and neutrophil-like cells of mammals, fish (Palić et al., 2007a,b), and birds (Chuammitri et al., 2009) make NETs through similar mechanisms. Even moths react to extracellular nucleic acids in a way reminiscent of NETs, but these structures have an RNA backbone (Altincicek et al., 2008). Surprisingly, upon infection, specialized cells in the surface of a plant’s root release their chromatin in a process that requires production of ROS (Hawes et al., 2011). These NET-like structures have a defense function, as degrading them with DNases makes the plant more susceptible to fungal infections.
These examples from the plant and animal kingdoms suggest that chromatin evolved with two functions: first to organize large pieces of DNA and second to be used as a weapon to defend the integrity of genomes. Chromatin is regarded as the safeguard and regulator of genetic information. NETs could be one of the configurations where chromatin is used in defense. The expulsion of chromatin as a weapon might well be an ancient tool conserved in evolution in the form of extracellular traps (ETs). Exploring how ETs are made and testing their relevance in sickness and in health could enhance our understanding of this novel aspect of immunity. ETs could, on the host side, help organisms survive in an environment where predation and parasitism by microbes are a threat. However, ETs drive the evolutionary selection of more pathogenic strains of microorganisms.
Online supplemental material
Two supplemental videos are available at http://www.jcb.org/cgi/content/full/jcb.201203170/DC1.
Supplementary Material
Supplemental Material
Acknowledgments
We thank Christan Goosmann for preparing the pus sample and Diane Schad for drawing Fig. 3. We are grateful to Kathleen Metzler, Borko Amulic, Lars Kuhn, Alf Herzig, and Constance Scharff for critically reading the manuscript.
Footnotes
Abbreviations used in this paper:
CF
cystic fibrosis
CGD
chronic granulomatous disease
DC
dendritic cell
ET
extracellular traps
HIV-1
human immunodeficiency virus 1
MPO
myeloperoxidase
NE
neutrophil elastase
NET
neutrophil extracellular trap
PAD4
peptidylarginine deiminase 4
PHOX
phagocytic oxidase
ROS
reactive oxygen species
SLE
Systemic lupus erythematosus
TLR
Toll-like receptor
References
- Abi Abdallah D.S., Lin C.Y., Ball C.J., King M.R., Duhamel G.E., Denkers E.Y. 2012. Toxoplasma gondii triggers release of human and mouse neutrophil extracellular traps. Infect. Immun. 80:768–777 10.1128/IAI.05730-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Mayouf S.M., Sunker A., Abdwani R., Abrawi S.A., Almurshedi F., Alhashmi N., Al Sonbul A., Sewairi W., Qari A., Abdallah E., et al. 2011. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat. Genet. 43:1186–1188 10.1038/ng.975 [DOI] [PubMed] [Google Scholar]
- Altincicek B., Stötzel S., Wygrecka M., Preissner K.T., Vilcinskas A. 2008. Host-derived extracellular nucleic acids enhance innate immune responses, induce coagulation, and prolong survival upon infection in insects. J. Immunol. 181:2705–2712 [DOI] [PubMed] [Google Scholar]
- Amulic B., Cazalet C., Hayes G.L., Metzler K.D., Zychlinsky A. 2012. Neutrophil function: from mechanisms to disease. Annu. Rev. Immunol. 30:459–489 10.1146/annurev-immunol-020711-074942 [DOI] [PubMed] [Google Scholar]
- Aulik N.A., Hellenbrand K.M., Klos H., Czuprynski C.J. 2010. Mannheimia haemolytica and its leukotoxin cause neutrophil extracellular trap formation by bovine neutrophils. Infect. Immun. 78:4454–4466 10.1128/IAI.00840-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Averhoff P., Kolbe M., Zychlinsky A., Weinrauch Y. 2008. Single residue determines the specificity of neutrophil elastase for Shigella virulence factors. J. Mol. Biol. 377:1053–1066 10.1016/j.jmb.2007.12.034 [DOI] [PubMed] [Google Scholar]
- Baechler E.C., Batliwalla F.M., Karypis G., Gaffney P.M., Ortmann W.A., Espe K.J., Shark K.B., Grande W.J., Hughes K.M., Kapur V., et al. 2003. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA. 100:2610–2615 10.1073/pnas.0337679100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartneck M., Keul H.A., Zwadlo-Klarwasser G., Groll J. 2010. Phagocytosis independent extracellular nanoparticle clearance by human immune cells. Nano Lett. 10:59–63 10.1021/nl902830x [DOI] [PubMed] [Google Scholar]
- Beiter K., Wartha F., Albiger B., Normark S., Zychlinsky A., Henriques-Normark B. 2006. An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr. Biol. 16:401–407 10.1016/j.cub.2006.01.056 [DOI] [PubMed] [Google Scholar]
- Bennett L., Palucka A.K., Arce E., Cantrell V., Borvak J., Banchereau J., Pascual V. 2003. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 197:711–723 10.1084/jem.20021553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berends E.T.M., Horswill A.R., Haste N.M., Monestier M., Nizet V., von Köckritz-Blickwede M. 2010. Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J. Innate Immun. 2:576–586 10.1159/000319909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi M., Hakkim A., Brinkmann V., Siler U., Seger R.A., Zychlinsky A., Reichenbach J. 2009. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood. 114:2619–2622 10.1182/blood-2009-05-221606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi M., Niemiec M.J., Siler U., Urban C.F., Reichenbach J. 2011. Restoration of anti-Aspergillus defense by neutrophil extracellular traps in human chronic granulomatous disease after gene therapy is calprotectin-dependent. J. Allergy Clin. Immunol. 127:1243–1252e7 10.1016/j.jaci.2011.01.021 [DOI] [PubMed] [Google Scholar]
- Birkemo G.A., Lüders T., Andersen O., Nes I.F., Nissen-Meyer J. 2003. Hipposin, a histone-derived antimicrobial peptide in Atlantic halibut (Hippoglossus hippoglossus L.). Biochim. Biophys. Acta. 1646:207–215 10.1016/S1570-9639(03)00018-9 [DOI] [PubMed] [Google Scholar]
- Birkemo G.A., Mantzilas D., Lüders T., Nes I.F., Nissen-Meyer J. 2004. Identification and structural analysis of the antimicrobial domain in hipposin, a 51-mer antimicrobial peptide isolated from Atlantic halibut. Biochim. Biophys. Acta. 1699:221–227 [DOI] [PubMed] [Google Scholar]
- Borregaard N. 2010. Neutrophils, from marrow to microbes. Immunity. 33:657–670 10.1016/j.immuni.2010.11.011 [DOI] [PubMed] [Google Scholar]
- Bratton D.L., Henson P.M. 2011. Neutrophil clearance: when the party is over, clean-up begins. Trends Immunol. 32:350–357 10.1016/j.it.2011.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brill A., Fuchs T.A., Savchenko A.S., Thomas G.M., Martinod K., De Meyer S.F., Bhandari A.A., Wagner D.D. 2012. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemost. 10:136–144 10.1111/j.1538-7836.2011.04544.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann V., Reichard U., Goosmann C., Fauler B., Uhlemann Y., Weiss D.S., Weinrauch Y., Zychlinsky A. 2004. Neutrophil extracellular traps kill bacteria. Science. 303:1532–1535 10.1126/science.1092385 [DOI] [PubMed] [Google Scholar]
- Brinkmann V., Laube B., Abu Abed U., Goosmann C., Zychlinsky A. 2010. Neutrophil extracellular traps: how to generate and visualize them. J. Vis. Exp. 36:1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns S., Kniemeyer O., Hasenberg M., Aimanianda V., Nietzsche S., Thywissen A., Jeron A., Latgé J.-P., Brakhage A.A., Gunzer M. 2010. Production of extracellular traps against Aspergillus fumigatus in vitro and in infected lung tissue is dependent on invading neutrophils and influenced by hydrophobin RodA. PLoS Pathog. 6:e1000873 10.1371/journal.ppat.1000873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan J.T., Simpson A.J., Aziz R.K., Liu G.Y., Kristian S.A., Kotb M., Feramisco J., Nizet V. 2006. DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr. Biol. 16:396–400 10.1016/j.cub.2005.12.039 [DOI] [PubMed] [Google Scholar]
- Casutt-Meyer S., Renzi F., Schmaler M., Jann N.J., Amstutz M., Cornelis G.R. 2010. Oligomeric coiled-coil adhesin YadA is a double-edged sword. PLoS ONE. 5:e15159 10.1371/journal.pone.0015159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caudrillier A., Kessenbrock K., Gilliss B.M., Nguyen J.X., Marques M.B., Monestier M., Toy P., Werb Z., Looney M.R. 2012. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J. Clin. Invest. 122:2661–2671 10.1172/JCI61303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho J.H., Sung B.H., Kim S.C. 2009. Buforins: histone H2A-derived antimicrobial peptides from toad stomach. Biochim. Biophys. Acta. 1788:1564–1569 10.1016/j.bbamem.2008.10.025 [DOI] [PubMed] [Google Scholar]
- Chuammitri P., Ostojić J., Andreasen C.B., Redmond S.B., Lamont S.J., Palić D. 2009. Chicken heterophil extracellular traps (HETs): novel defense mechanism of chicken heterophils. Vet. Immunol. Immunopathol. 129:126–131 10.1016/j.vetimm.2008.12.013 [DOI] [PubMed] [Google Scholar]
- Clark S.R., Ma A.C., Tavener S.A., McDonald B., Goodarzi Z., Kelly M.M., Patel K.D., Chakrabarti S., McAvoy E., Sinclair G.D., et al. 2007. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 13:463–469 10.1038/nm1565 [DOI] [PubMed] [Google Scholar]
- Cogen A.L., Yamasaki K., Muto J., Sanchez K.M., Crotty Alexander L., Tanios J., Lai Y., Kim J.E., Nizet V., Gallo R.L. 2010. Staphylococcus epidermidis antimicrobial delta-toxin (phenol-soluble modulin-gamma) cooperates with host antimicrobial peptides to kill group A Streptococcus. PLoS ONE. 5:e8557 10.1371/journal.pone.0008557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft J.E. 2011. Dissecting the immune cell mayhem that drives lupus pathogenesis. Sci. Transl. Med. 3:73ps9 10.1126/scitranslmed.3002138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delbosc S., Alsac J.M., Journe C., Louedec L., Castier Y., Bonnaure-Mallet M., Ruimy R., Rossignol P., Bouchard P., Michel J.B., Meilhac O. 2011. Porphyromonas gingivalis participates in pathogenesis of human abdominal aortic aneurysm by neutrophil activation. Proof of concept in rats. PLoS ONE. 6:e18679 10.1371/journal.pone.0018679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demmer R.T., Desvarieux M. 2006. Periodontal infections and cardiovascular disease: the heart of the matter. J. Am. Dent. Assoc. 137:14S–20S, quiz:38S [DOI] [PubMed] [Google Scholar]
- Dwivedi N., Upadhyay J., Neeli I., Khan S., Pattanaik D., Myers L., Kirou K.A., Hellmich B., Knuckley B., Thompson P.R., et al. 2012. Felty’s syndrome autoantibodies bind to deiminated histones and neutrophil extracellular chromatin traps. Arthritis Rheum. 64:982–992 10.1002/art.33432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrington T., Villamil L., Gómez-chiarri M. 2011. Upregulation in response to infection and antibacterial activity of oyster histone H4. Fish Shellfish Immunol. 30:94–101 10.1016/j.fsi.2010.09.006 [DOI] [PubMed] [Google Scholar]
- Ehrlich P. 1880. Methodologische Beiträge zur Physiologie und Pathologie der verschiedenen Formen der Leukocyten. Z. Klin. Med. 1:553–560 [Google Scholar]
- Ermert D., Urban C.F., Laube B., Goosmann C., Zychlinsky A., Brinkmann V. 2009. Mouse neutrophil extracellular traps in microbial infections. J. Innate Immun. 1:181–193 10.1159/000205281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esmon C.T., Xu J., Lupu F. 2011. Innate immunity and coagulation. J. Thromb. Haemost. 9:182–188 10.1111/j.1538-7836.2011.04323.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadeel B. 2009. Babies born without safety NET. Blood. 113:6270–6271 10.1182/blood-2009-03-210328 [DOI] [PubMed] [Google Scholar]
- Farquharson D., Butcher J.P., Culshaw S. 2012. Periodontitis, Porphyromonas, and the pathogenesis of rheumatoid arthritis. Mucosal Immunol. 5:112–120 10.1038/mi.2011.66 [DOI] [PubMed] [Google Scholar]
- Fuchs T.A., Abed U., Goosmann C., Hurwitz R., Schulze I., Wahn V., Weinrauch Y., Brinkmann V., Zychlinsky A. 2007. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 176:231–241 10.1083/jcb.200606027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs T.A., Brill A., Duerschmied D., Schatzberg D., Monestier M., Myers D.D., Jr, Wrobleski S.K., Wakefield T.W., Hartwig J.H., Wagner D.D. 2010. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA. 107:15880–15885 10.1073/pnas.1005743107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel C., McMaster W.R., Girard D., Descoteaux A. 2010. Leishmania donovani promastigotes evade the antimicrobial activity of neutrophil extracellular traps. J. Immunol. 185:4319–4327 10.4049/jimmunol.1000893 [DOI] [PubMed] [Google Scholar]
- Garcia-Romo G.S., Caielli S., Vega B., Connolly J., Allantaz F., Xu Z., Punaro M., Baisch J., Guiducci C., Coffman R.L., et al. 2011. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 3:73ra20 10.1126/scitranslmed.3001201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinberg N., Elazar S., Rosenshine I., Shpigel N.Y. 2008. Beta-hydroxybutyrate abrogates formation of bovine neutrophil extracellular traps and bactericidal activity against mammary pathogenic Escherichia coli. Infect. Immun. 76:2802–2807 10.1128/IAI.00051-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimarães-Costa A.B., Nascimento M.T.C., Froment G.S., Soares R.P.P., Morgado F.N., Conceição-Silva F., Saraiva E.M. 2009. Leishmania amazonensis promastigotes induce and are killed by neutrophil extracellular traps. Proc. Natl. Acad. Sci. USA. 106:6748–6753 10.1073/pnas.0900226106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A.K., Hasler P., Holzgreve W., Gebhardt S., Hahn S. 2005. Induction of neutrophil extracellular DNA lattices by placental microparticles and IL-8 and their presence in preeclampsia. Hum. Immunol. 66:1146–1154 10.1016/j.humimm.2005.11.003 [DOI] [PubMed] [Google Scholar]
- Gupta A.K., Joshi M.B., Philippova M., Erne P., Hasler P., Hahn S., Resink T.J. 2010. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 584:3193–3197 10.1016/j.febslet.2010.06.006 [DOI] [PubMed] [Google Scholar]
- Hakkim A., Fürnrohr B.G., Amann K., Laube B., Abed U.A., Brinkmann V., Herrmann M., Voll R.E., Zychlinsky A. 2010. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA. 107:9813–9818 10.1073/pnas.0909927107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakkim A., Fuchs T.A., Martinez N.E., Hess S., Prinz H., Zychlinsky A., Waldmann H. 2011. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat. Chem. Biol. 7:75–77 10.1038/nchembio.496 [DOI] [PubMed] [Google Scholar]
- Hawes M.C., Curlango-Rivera G., Wen F., White G.J., Vanetten H.D., Xiong Z. 2011. Extracellular DNA: the tip of root defenses? Plant Sci. 180:741–745 10.1016/j.plantsci.2011.02.007 [DOI] [PubMed] [Google Scholar]
- Hemmers S., Teijaro J.R., Arandjelovic S., Mowen K.A. 2011. PAD4-mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PLoS ONE. 6:e22043 10.1371/journal.pone.0022043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch J.G. 1958. Bactericidal action of histone. J. Exp. Med. 108:925–944 10.1084/jem.108.6.925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong W., Juneau R.A., Pang B., Swords W.E. 2009. Survival of bacterial biofilms within neutrophil extracellular traps promotes nontypeable Haemophilus influenzae persistence in the chinchilla model for otitis media. J. Innate Immun. 1:215–224 10.1159/000205937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki H., Isaacson T., Iwamuro S., Conlon J.M. 2003. A protein with antimicrobial activity in the skin of Schlegel’s green tree frog Rhacophorus schlegelii (Rhacophoridae) identified as histone H2B. Biochem. Biophys. Res. Commun. 312:1082–1086 10.1016/j.bbrc.2003.11.052 [DOI] [PubMed] [Google Scholar]
- Kawasaki H., Koyama T., Conlon J.M., Yamakura F., Iwamuro S. 2008. Antimicrobial action of histone H2B in Escherichia coli: evidence for membrane translocation and DNA-binding of a histone H2B fragment after proteolytic cleavage by outer membrane proteinase T. Biochimie. 90:1693–1702 10.1016/j.biochi.2008.07.003 [DOI] [PubMed] [Google Scholar]
- Kessenbrock K., Krumbholz M., Schönermarck U., Back W., Gross W.L., Werb Z., Gröne H.J., Brinkmann V., Jenne D.E. 2009. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 15:623–625 10.1038/nm.1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krautgartner W.D., Vitkov L. 2008. Visualization of neutrophil extracellular traps in TEM. Micron. 39:367–372 10.1016/j.micron.2007.03.007 [DOI] [PubMed] [Google Scholar]
- Lande R., Gregorio J., Facchinetti V., Chatterjee B., Wang Y.H., Homey B., Cao W., Wang Y.H., Su B., Nestle F.O., et al. 2007. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 449:564–569 10.1038/nature06116 [DOI] [PubMed] [Google Scholar]
- Lande R., Ganguly D., Facchinetti V., Frasca L., Conrad C., Gregorio J., Meller S., Chamilos G., Sebasigari R., Riccieri V., et al. 2011. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci. Transl. Med. 3:73ra19 10.1126/scitranslmed.3001180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauth X., von Köckritz-Blickwede M., McNamara C.W., Myskowski S., Zinkernagel A.S., Beall B., Ghosh P., Gallo R.L., Nizet V. 2009. M1 protein allows Group A streptococcal survival in phagocyte extracellular traps through cathelicidin inhibition. J. Innate Immun. 1:202–214 10.1159/000203645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leffler J., Martin M., Gullstrand B., Tydén H., Lood C., Truedsson L., Bengtsson A.A., Blom A.M. 2012. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J. Immunol. 188:3522–3531 10.4049/jimmunol.1102404 [DOI] [PubMed] [Google Scholar]
- Lemaire S., Trinh T.T., Le H.T., Tang S.C., Hincke M., Wellman-Labadie O., Ziai S. 2008. Antimicrobial effects of H4-(86-100), histogranin and related compounds—possible involvement of DNA gyrase. FEBS J. 275:5286–5297 10.1111/j.1742-4658.2008.06659.x [DOI] [PubMed] [Google Scholar]
- Li P., Li M., Lindberg M.R., Kennett M.J., Xiong N., Wang Y. 2010. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 207:1853–1862 10.1084/jem.20100239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim M.B.H., Kuiper J.W.P., Katchky A., Goldberg H., Glogauer M. 2011. Rac2 is required for the formation of neutrophil extracellular traps. J. Leukoc. Biol. 90:771–776 10.1189/jlb.1010549 [DOI] [PubMed] [Google Scholar]
- Liu C.L., Tangsombatvisit S., Rosenberg J.M., Mandelbaum G., Gillespie E.C., Gozani O.P., Alizadeh A.A., Utz P.J.U. 2012. Specific post-translational histone modifications of neutrophil extracellular traps as immunogens and potential targets of lupus autoantibodies. Arthritis Res. Ther. 14:R25 10.1186/ar3707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüders T., Birkemo G.A., Nissen-Meyer J., Andersen O., Nes I.F. 2005. Proline conformation-dependent antimicrobial activity of a proline-rich histone h1 N-terminal Peptide fragment isolated from the skin mucus of Atlantic salmon. Antimicrob. Agents Chemother. 49:2399–2406 10.1128/AAC.49.6.2399-2406.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzenreiter R., Kienberger F., Marcos V., Schilcher K., Krautgartner W.D., Obermayer A., Huml M., Stoiber W., Hector A., Griese M., et al. 2012. Ultrastructural characterization of cystic fibrosis sputum using atomic force and scanning electron microscopy. J. Cyst. Fibros. 11:84–92 10.1016/j.jcf.2011.09.008 [DOI] [PubMed] [Google Scholar]
- Massberg S., Grahl L., von Bruehl M.-L., Manukyan D., Pfeiler S., Goosmann C., Brinkmann V., Lorenz M., Bidzhekov K., Khandagale A.B., et al. 2010. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 16:887–896 10.1038/nm.2184 [DOI] [PubMed] [Google Scholar]
- Megens R.T.A., Vijayan S., Lievens D., Döring Y., van Zandvoort M.A.M.J., Grommes J., Weber C., Soehnlein O. 2012. Presence of luminal neutrophil extracellular traps in atherosclerosis. Thromb. Haemost. 107:597–598 10.1160/TH11-09-0650 [DOI] [PubMed] [Google Scholar]
- Menegazzi R., Decleva E., Dri P. 2012. Killing by neutrophil extracellular traps: fact or folklore? Blood. 119:1214–1216 10.1182/blood-2011-07-364604 [DOI] [PubMed] [Google Scholar]
- Metchnikoff E. 1893. Lecons sur la pathologie comparee de l’inflammation. Bibliothéque des annales de l’Institut Pasteur. 7:348–357 [Google Scholar]
- Metzler K.D., Fuchs T.A., Nauseef W.M., Reumaux D., Roesler J., Schulze I., Wahn V., Papayannopoulos V., Zychlinsky A. 2011. Myeloperoxidase is required for neutrophil extracellular trap formation: implications for innate immunity. Blood. 117:953–959 10.1182/blood-2010-06-290171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller B.F., Abrams R., Dorfman A., Klein M. 1942. Antibacterial properties of protamine histone. Science. 96:428–430 10.1126/science.96.2497.428 [DOI] [PubMed] [Google Scholar]
- Nathan C. 2006. Neutrophils and immunity: challenges and opportunities. Nat. Rev. Immunol. 6:173–182 10.1038/nri1785 [DOI] [PubMed] [Google Scholar]
- Nauseef W.M. 2012. Editorial: Nyet to NETs? A pause for healthy skepticism. J. Leukoc. Biol. 91:353–355 10.1189/jlb.1011495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neeli I., Khan S.N., Radic M. 2008. Histone deimination as a response to inflammatory stimuli in neutrophils. J. Immunol. 180:1895–1902 [DOI] [PubMed] [Google Scholar]
- Neeli I., Dwivedi N., Khan S., Radic M. 2009. Regulation of extracellular chromatin release from neutrophils. J. Innate Immun. 1:194–201 10.1159/000206974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noga E.J., Borron P.J., Hinshaw J., Gordon W.C., Gordon L.J., Seo J.-K. 2011. Identification of histones as endogenous antibiotics in fish and quantification in rainbow trout (Oncorhynchus mykiss) skin and gill. Fish Physiol. Biochem. 37:135–152 10.1007/s10695-010-9422-7 [DOI] [PubMed] [Google Scholar]
- Oehmcke S., Mörgelin M., Herwald H. 2009. Activation of the human contact system on neutrophil extracellular traps. J. Innate Immun. 1:225–230 10.1159/000203700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palić D., Andreasen C.B., Ostojić J., Tell R.M., Roth J.A. 2007a. Zebrafish (Danio rerio) whole kidney assays to measure neutrophil extracellular trap release and degranulation of primary granules. J. Immunol. Methods. 319:87–97 10.1016/j.jim.2006.11.003 [DOI] [PubMed] [Google Scholar]
- Palić D., Ostojić J., Andreasen C.B., Roth J.A. 2007b. Fish cast NETs: neutrophil extracellular traps are released from fish neutrophils. Dev. Comp. Immunol. 31:805–816 10.1016/j.dci.2006.11.010 [DOI] [PubMed] [Google Scholar]
- Palmer L.J., Cooper P.R., Ling M.R., Wright H.J., Huissoon A., Chapple I.L.C. 2012. Hypochlorous acid regulates neutrophil extracellular trap release in humans. Clin. Exp. Immunol. 167:261–268 10.1111/j.1365-2249.2011.04518.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papayannopoulos V., Metzler K.D., Hakkim A., Zychlinsky A. 2010. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 191:677–691 10.1083/jcb.201006052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papayannopoulos V., Staab D., Zychlinsky A. 2011. Neutrophil elastase enhances sputum solubilization in cystic fibrosis patients receiving DNase therapy. PLoS ONE. 6:e28526 10.1371/journal.pone.0028526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park I.Y., Park C.B., Kim M.S., Kim S.C. 1998. Parasin I, an antimicrobial peptide derived from histone H2A in the catfish, Parasilurus asotus. FEBS Lett. 437:258–262 10.1016/S0014-5793(98)01238-1 [DOI] [PubMed] [Google Scholar]
- Parker H., Albrett A.M., Kettle A.J., Winterbourn C.C. 2012. Myeloperoxidase associated with neutrophil extracellular traps is active and mediates bacterial killing in the presence of hydrogen peroxide. J. Leukoc. Biol. 91:369–376 10.1189/jlb.0711387 [DOI] [PubMed] [Google Scholar]
- Parseghian M.H., Luhrs K.A. 2006. Beyond the walls of the nucleus: the role of histones in cellular signaling and innate immunity. Biochem. Cell Biol. 84:589–604 10.1139/o06-082 [DOI] [PubMed] [Google Scholar]
- Patat S.A., Carnegie R.B., Kingsbury C., Gross P.S., Chapman R., Schey K.L. 2004. Antimicrobial activity of histones from hemocytes of the Pacific white shrimp. Eur. J. Biochem. 271:4825–4833 10.1111/j.1432-1033.2004.04448.x [DOI] [PubMed] [Google Scholar]
- Pilsczek F.H., Salina D., Poon K.K.H., Fahey C., Yipp B.G., Sibley C.D., Robbins S.M., Green F.H.Y., Surette M.G., Sugai M., et al. 2010. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J. Immunol. 185:7413–7425 10.4049/jimmunol.1000675 [DOI] [PubMed] [Google Scholar]
- Pisetsky D.S. 2011. Pus: the Rodney Dangerfield of immunology. Arthritis Res. Ther. 13:131 10.1186/ar3477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Kichik V., Mondragón-Flores R., Mondragón-Castelán M., Gonzalez-Pozos S., Muñiz-Hernandez S., Rojas-Espinosa O., Chacón-Salinas R., Estrada-Parra S., Estrada-García I. 2009. Neutrophil extracellular traps are induced by Mycobacterium tuberculosis. Tuberculosis (Edinb.). 89:29–37 10.1016/j.tube.2008.09.009 [DOI] [PubMed] [Google Scholar]
- Remijsen Q., Vanden Berghe T., Wirawan E., Asselbergh B., Parthoens E., De Rycke R., Noppen S., Delforge M., Willems J., Vandenabeele P. 2011a. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 21:290–304 10.1038/cr.2010.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remijsen Q., Kuijpers T.W., Wirawan E., Lippens S., Vandenabeele P., Vanden Berghe T. 2011b. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ. 18:581–588 10.1038/cdd.2011.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffarzadeh M., Juenemann C., Queisser M.A., Lochnit G., Barreto G., Galuska S.P., Lohmeyer J., Preissner K.T. 2012. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS ONE. 7:e32366 10.1371/journal.pone.0032366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T., Komano J., Saitoh Y., Misawa T., Takahama M., Kozaki T., Uehata T., Iwasaki H., Omori H., Yamaoka S., et al. 2012. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe. 12:109–116 10.1016/j.chom.2012.05.015 [DOI] [PubMed] [Google Scholar]
- Semeraro F., Ammollo C.T., Morrissey J.H., Dale G.L., Friese P., Esmon N.L., Esmon C.T. 2011. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood. 118:1952–1961 10.1182/blood-2011-03-343061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg B.E., Grinstein S. 2007. Unconventional roles of the NADPH oxidase: signaling, ion homeostasis, and cell death. Sci. STKE. 2007:pe11 10.1126/stke.3792007pe11 [DOI] [PubMed] [Google Scholar]
- Sumby P., Barbian K.D., Gardner D.J., Whitney A.R., Welty D.M., Long R.D., Bailey J.R., Parnell M.J., Hoe N.P., Adams G.G., et al. 2005. Extracellular deoxyribonuclease made by group A Streptococcus assists pathogenesis by enhancing evasion of the innate immune response. Proc. Natl. Acad. Sci. USA. 102:1679–1684 10.1073/pnas.0406641102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teegen B., Niemann S., Probst C., Schlumberger W., Stöcker W., Komorowski L. 2009. DNA-bound lactoferrin is the major target for antineutrophil perinuclear cytoplasmic antibodies in ulcerative colitis. Ann. N. Y. Acad. Sci. 1173:161–165 10.1111/j.1749-6632.2009.04752.x [DOI] [PubMed] [Google Scholar]
- Tillack K., Breiden P., Martin R., Sospedra M. 2012. T lymphocyte priming by neutrophil extracellular traps links innate and adaptive immune responses. J. Immunol. 188:3150–3159 10.4049/jimmunol.1103414 [DOI] [PubMed] [Google Scholar]
- Urban C.F., Reichard U., Brinkmann V., Zychlinsky A. 2006. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell. Microbiol. 8:668–676 10.1111/j.1462-5822.2005.00659.x [DOI] [PubMed] [Google Scholar]
- Urban C.F., Ermert D., Schmid M., Abu-Abed U., Goosmann C., Nacken W., Brinkmann V., Jungblut P.R., Zychlinsky A. 2009. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 5:e1000639 10.1371/journal.ppat.1000639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva E., Yalavarthi S., Berthier C.C., Hodgin J.B., Khandpur R., Lin A.M., Rubin C.J., Zhao W., Olsen S.H., Klinker M., et al. 2011. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J. Immunol. 187:538–552 10.4049/jimmunol.1100450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitkov L., Klappacher M., Hannig M., Krautgartner W.D. 2009. Extracellular neutrophil traps in periodontitis. J. Periodontal Res. 44:664–672 10.1111/j.1600-0765.2008.01175.x [DOI] [PubMed] [Google Scholar]
- von Brühl M.-L., Stark K., Steinhart A., Chandraratne S., Konrad I., Lorenz M., Khandoga A., Tirniceriu A., Coletti R., Köllnberger M., et al. 2012. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 209:819–835 10.1084/jem.20112322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Köckritz-Blickwede M., Goldmann O., Thulin P., Heinemann K., Norrby-Teglund A., Rohde M., Medina E. 2008. Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood. 111:3070–3080 10.1182/blood-2007-07-104018 [DOI] [PubMed] [Google Scholar]
- von Köckritz-Blickwede M., Chow O.A., Nizet V. 2009. Fetal calf serum contains heat-stable nucleases that degrade neutrophil extracellular traps. Blood. 114:5245–5246 10.1182/blood-2009-08-240713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker M.J., Hollands A., Sanderson-Smith M.L., Cole J.N., Kirk J.K., Henningham A., McArthur J.D., Dinkla K., Aziz R.K., Kansal R.G., et al. 2007. DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat. Med. 13:981–985 10.1038/nm1612 [DOI] [PubMed] [Google Scholar]
- Wang Y., Li M., Stadler S., Correll S., Li P., Wang D., Hayama R., Leonelli L., Han H., Grigoryev S.A., et al. 2009. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 184:205–213 10.1083/jcb.200806072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Chen Y., Xin L., Beverley S.M., Carlsen E.D., Popov V., Chang K.-P., Wang M., Soong L. 2011. Differential microbicidal effects of human histone proteins H2A and H2B on Leishmania promastigotes and amastigotes. Infect. Immun. 79:1124–1133 10.1128/IAI.00658-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wartha F., Beiter K., Albiger B., Fernebro J., Zychlinsky A., Normark S., Henriques-Normark B. 2007. Capsule and D-alanylated lipoteichoic acids protect Streptococcus pneumoniae against neutrophil extracellular traps. Cell. Microbiol. 9:1162–1171 10.1111/j.1462-5822.2006.00857.x [DOI] [PubMed] [Google Scholar]
- Weinrauch Y., Drujan D., Shapiro S.D., Weiss J., Zychlinsky A. 2002. Neutrophil elastase targets virulence factors of enterobacteria. Nature. 417:91–94 10.1038/417091a [DOI] [PubMed] [Google Scholar]
- Xu J., Zhang X.M., Pelayo R., Monestier M., Ammollo C.T., Semeraro F., Taylor F.B., Esmon N.L., Lupu F., Esmon C.T. 2009. Extracellular histones are major mediators of death in sepsis. Nat. Med. 15:1318–1321 10.1038/nm.2053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasutomo K., Horiuchi T., Kagami S., Tsukamoto H., Hashimura C., Urushihara M., Kuroda Y. 2001. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat. Genet. 28:313–314 10.1038/91070 [DOI] [PubMed] [Google Scholar]
- Yost C.C., Zimmerman G.A. 2009. Response: Gestational age as a factor in neutrophil extracellular trap formation. Blood. 114:4911–4912 10.1182/blood-2009-10-243048 [DOI] [Google Scholar]
- Yost C.C., Cody M.J., Harris E.S., Thornton N.L., McInturff A.M., Martinez M.L., Chandler N.B., Rodesch C.K., Albertine K.H., Petti C.A., et al. 2009. Impaired neutrophil extracellular trap (NET) formation: a novel innate immune deficiency of human neonates. Blood. 113:6419–6427 10.1182/blood-2008-07-171629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefi S., Gold J.A., Andina N., Lee J.J., Kelly A.M., Kozlowski E., Schmid I., Straumann A., Reichenbach J., Gleich G.J., Simon H.-U. 2008. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat. Med. 14:949–953 10.1038/nm.1855 [DOI] [PubMed] [Google Scholar]
- Yousefi S., Mihalache C., Kozlowski E., Schmid I., Simon H.U. 2009. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 16:1438–1444 10.1038/cdd.2009.96 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material