Recent advances in understanding viral evasion of type I interferon (original) (raw)
Summary
The type I interferon (IFN) system mediates a wide variety of antiviral effects and represents an important first barrier to virus infection. Consequently, viruses have developed an impressive diversity of tactics to circumvent IFN responses. Evasion strategies can involve preventing initial virus detection, via the disruption of the Toll‐like receptors or the retinoic acid inducible gene I (RIG‐I) ‐like receptors, or by avoiding the initial production of the ligands recognized by these receptors. An alternative approach is to preclude IFN production by disarming or degrading the transcription factors involved in the expression of IFN, such as interferon regulatory factor 3 (IRF3)/IRF7, nuclear factor‐κB (NF‐κB), or ATF‐2/c‐jun, or by inducing a general block on host cell transcription. Viruses also oppose IFN signalling, both by disturbing the type I IFN receptor and by impeding JAK/STAT signal transduction upon IFN receptor engagement. In addition, the global expression of IFN‐stimulated genes (ISGs) can be obstructed via interference with epigenetic signalling, and specific ISGs can also be selectively targeted for inhibition. Finally, some viruses disrupt IFN responses by co‐opting negative regulatory systems, whereas others use antiviral mechanisms to their own advantage. Here, we review recent developments in this field.
Keywords: antiviral, evasion, inhibition, interferon, virus
Introduction
Despite almost constant exposure to pathogens, mammals are only rarely infected to the point where disease becomes evident. The first line of defence consists of the interferon (IFN) family of soluble cytokines. The IFNs have anti‐cancer, anti‐proliferative, anti‐viral and immunomodulatory functions1 through the expression of more than 300 IFN‐stimulated genes (ISGs).2 There are three classes of IFNs which are produced by different cell types, bind unique receptors and have distinctive biological actions.3 Here, we focus on the type I IFNs, which are produced by most cell types and have potent, inherent antiviral activity.4 The type I IFN response is bimodal: first, detection of an invading virus leads to IFN production and secretion and second, IFN acts in an autocrine and paracrine manner to induce ISGs, the products of which work collectively to disrupt viral replication and spread.
To generate a productive infection, viruses must overcome antiviral responses, and accordingly, every aspect of these defences is targeted for inhibition. Here, we describe the IFN response and viral immune evasion strategies. As this topic has been extensively reviewed previously, we will focus on the most recent advances.
Virus recognition
In the first step of the biphasic type I IFN response, virus is detected through the recognition of pathogen‐associated molecular patterns (PAMPs), highly conserved structural features found in broad classes of pathogens. PAMPs are sensed by pattern recognition receptors (PRRs), including the toll‐like receptors (TLRs).5 The TLRs recognize viral components including glycoproteins and nucleic acids such as dsRNA or CpG DNA. Via their cytoplasmic Toll/interleukin‐1 receptor (TIR) domains, TLRs recruit TIR‐containing adaptors such as MyD88, TIR‐domain‐containing adapter‐inducing IFN‐β (TRIF), Mal and TRIF‐related adaptor molecule (TRAM), leading to the activation of nuclear factor‐κB (NF‐κB) and interferon regulatory factor 3 (IRF3) (Fig. 1).
Figure 1.
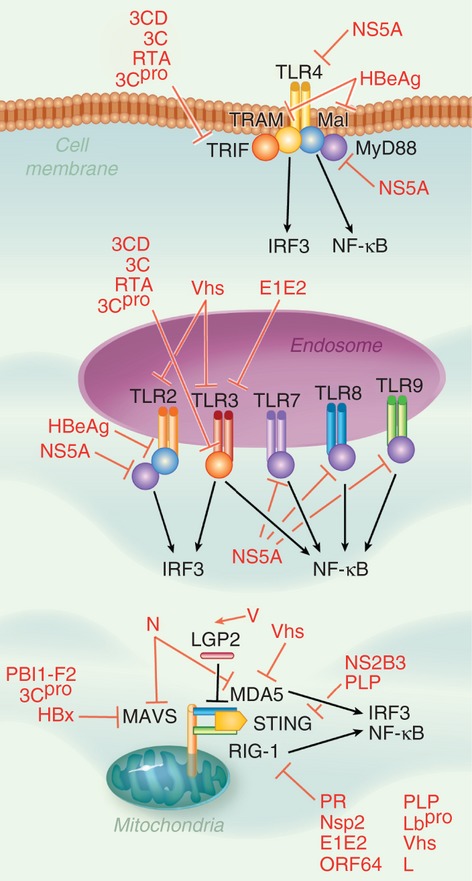
Pathways to virus detection. Viral pathogen‐associated molecular patterns (PAMPs) are identified by various pattern recognition receptors (PRRs), such as the Toll‐like receptors (TLRs) and retinoic acid inducible gene I (RIG‐I)‐like receptors (RLRs). TLRs are found both at the plasma membrane and in endosomes, detect a variety of virus‐associated ligands, and signal through adaptors proteins Toll/interleukin‐1 receptor‐domain‐containing adapter‐inducing IFN‐β (TRIF), TRIF‐related adaptor molecule (TRAM), Mal and MyD88 to lead to the activation of transcription factors interferon regulatory factor 3 (IRF3) and nuclear factor‐κB (NF‐κB). RLRs retinoic acid inducible gene I (RIG‐I) and melanoma differentiation‐associated gene 5 (MDA5), which are negatively regulated by LGP2, detect viral dsRNA, and signal through the adaptors mitochondrial antiviral signalling protein (MAVS) and STING to cause IRF3 and NF‐κB activation. Virtually every step in this process can be impeded by viral proteins.
Recently, several viruses have been found to disrupt TLR signalling by interfering with the adaptor molecule TRIF. For example, 3CD protease‐polymerase, an intermediate in the polyprotein processing cascade of hepatitis A virus,6 and 3C protein of enterovirus 71,7 use protease activity to cleave TRIF. Replication and transcription activator (RTA) from Kaposi's sarcoma‐associated herpesvirus (KSHV) also reduces TRIF levels, likely through a proteasome‐mediated pathway.8 Other TLR adaptor proteins are also affected – the hepatitis B virus HBeAg protein uses its precore specific sequence, which shows homology to the TIR motif, to compete with TIR‐containing proteins Mal and TRAM to impede their interactions with downstream signalling molecules.9
A second class of PRRs is the retinoic acid inducible gene I (RIG‐I)‐like receptor (RLR) family, including RIG‐I and melanoma differentiation‐associated gene 5 (MDA5).10 The RLRs detect cytoplasmic dsRNA, interact with the adaptor mitochondrial antiviral signalling protein (MAVS) and activate NF‐κB and IRF3. Like TLRs, RLRs are hindered by viruses. For instance, the N protein from human respiratory syncytial virus (RSV) inhibits MDA5 and MAVS,11 whereas the HIV protease decreases cytoplasmic RIG‐I levels by targeting the sensor to the lysosome.12 In contrast, the V proteins of several paramyxoviruses promote an interaction between RIG‐I and LGP2,13 an RLR that lacks signalling capacity.14 Several viruses target RIG‐I via viral de‐ubiquitinating enzymes (DUBs), such as Arterivirus non‐structural protein 2, Nairovirus L protein,15 KSHV ORF64,16 severe acute respiratory syndrome coronavirus (SARS‐CoV) papain‐like proteases,17 and foot‐and‐mouth disease virus (FMDV) Lbpro.18 These DUBs remove K63‐linked ubiquitin on RIG‐I, preventing its interaction with MAVS.19
MAVS is also a popular focus of viral antagonists. The influenza A protein PB1‐F2 binds the transmembrane domain of MAVS, causing a drop in the mitochondrial membrane potential,20 which is required for MAVS function.21 Coxsackievirus B3 encodes the cysteine protease 3Cpro, which directly cleaves both TRIF and MAVS, impeding both the TLR3 and RLR pathways, respectively.22 Finally, the hepatitis B virus protein HBx associates with and blocks the action of MAVS.23
The adaptor protein STING, which interacts with RIG‐I and MAVS and is involved in the detection of cytosolic DNA,24 is also affected by viral proteins, such as the protease complex NS2B3 of Dengue virus, which cleaves STING into inactive fragments.25 Interestingly, the papain‐like proteases from human coronavirus NL63 and SARS‐CoV, which possess protease and DUB enzyme activities, disrupt the dimerization of STING by decreasing its level of ubiquitination.17
Several viral proteins target both TLRs and RLRs at the expression level. The virion host shutoff (vhs) endoribonuclease from herpes simplex virus type 2 (HSV‐2) specifically reduces TLR2, TLR3, RIG‐I and MDA5 mRNA,26 whereas hepatitis C virus (HCV) uses E1E2 to down‐regulate the mRNA levels of TLR3 and RIG‐I via E1E2,27 and NS5A to decrease TLR4, MyD88, IRF3 and NF‐κB2.28
The most straightforward mechanism of viral evasion of the IFN response is to avoid detection in the first place. Several viruses conceal or degrade dsRNA, a by‐product of viral replication. For example, tick‐borne encephalitis virus delays antiviral signalling by sequestering RNA molecules into cytoplasmic membrane‐defined compartments, where they are inaccessible to PRR recognition.29 Similarly, Japanese encephalitis virus (JEV) conceals its dsRNA among intracellular membranes.30 Amazingly, species‐specific differences in the timing of the release of viral dsRNA into the cytosol account for the drastically different pathogenesis of JEV in humans compared with pigs.30 Rather than hide it, Lassa fever virus uses the 3′–5′ exonuclease activity of its NP protein to degrade its dsRNA,31 whereas the C protein from human parainfluenza virus type 1 is thought to regulate viral RNA production in such a way as to prevent dsRNA from accumulating at all.32
Transcription factor activation to IFN expression
Viral sensing by PRRs activates three main transcription factor complexes involved in IFN‐β production: NF‐κB, IRF3/IRF7 and ATF2/c‐jun (Fig. 2).33 In resting cells, NF‐κB is held as an inactive complex in the cytoplasm by its inhibitor, IκBα.34 PRR activation stimulates IκBα phosphorylation and degradation, releasing NF‐κB to translocate to the nucleus and induce target genes. A recent example of viral disruption of NF‐κB activation involves the V protein from measles virus, which binds to the nuclear location signal of the NF‐κB subunit p65, impairing its nuclear translocation.35 The NF‐κB essential modulator (NEMO), a regulatory component involved in the phosphorylation of IκBα,36 is also targeted, as it is cleaved into inactive fragments by the FMDV protease 3Cpro.37
Figure 2.
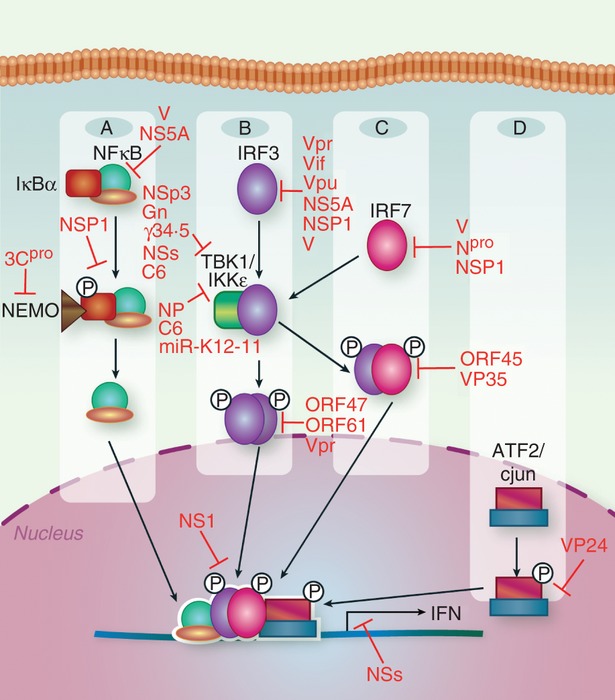
Transcription factor activation to interferon (IFN) production. Detection of viral components by pattern recognition receptors (PRRs) leads to the activation of a variety of transcription factors. (a) Nuclear factor‐κB (NF‐κB) is held in an inactive cytoplasmic complex via interaction with its inhibitor, IκBα. Upon virus recognition, IκBα is phosphorylated in a process involving the regulatory component NEMO, leading to degradation of the inhibitor, freeing NF‐κB to translocate to the nucleus and bind the IFN promoter. (b) Upon virus detection, constitutively expressed interferon regulatory factor 3 (IRF3) is phosphorylated by the kinases TBK1 and IKKε, leading to its dimerization and nuclear translocation. (c) Although IRF7 is minimally expressed in most resting cell types, low level IFN production induces IRF7 expression, leading to its phosphorylation by TBK1/IKKε, heterodimerization with IRF3, nuclear translocation, and increased IFN expression. (d) The constitutively nuclear ATF2/cjun is phosphorylated upon virus detection by stress‐activated members of the mitogen‐activated protein (MAP) kinase superfamily, leading to the activation of the complex. Cooperative binding of NF‐κB, IRF3/IRF7 and ATF2/cjun to the IFN promoter leads to full expression of type I IFN genes. Each of these pathways is subject to inhibition by viruses.
Less is understood about ATF2/c‐Jun. This complex is constitutively nuclear, even in its inactive form, and is stimulated by phosphorylation of its activation domains.38 Virus infection triggers the stress‐activated members of the mitogen‐activated protein (MAP) kinase superfamily, which phosphorylate and activate ATF2/cJun. For the first time, a viral protein blocking this complex has been described; the Zaire ebola virus protein VP24 prevents the phosphorylation of p38 MAP kinase and the downstream activation of ATF2.39
Critical factors involved in IFN expression include IRF3 and IRF7.40 IRF3, which is constitutively expressed in resting cells, is phosphorylated upon PRR signalling by the IκB kinase (IKK)‐related kinases IKKε and TBK‐1, causing IRF3 to homodimerize and translocate to the nucleus. There, IRF3 interacts with the histone acetyl transferases CBP and p300, and associates with the IFN‐β promoter. IRF3 can also directly activate a subset of ISGs in the absence of IFN.41,42 Accordingly, IRF3 is a popular target for viral inhibition. The V protein of Sendai virus directly binds IRF3, impairing its function.43 Varicella zoster virus induces atypical, TBK1‐independent phosphorylation of IRF3 that blocks downstream dimerization and activity, via the viral serine‐threonine protein kinase ORF47.44 Additionally, varicella zoster virus ORF61 interacts specifically with activated, phosphorylated IRF3, and uses its RING finger E3 ubiquitin ligase domain to ubiquitinate and degrade IRF3 via the proteasome pathway.45 HIV immune evasion is complex and cell‐type dependent; in T cells, it has previously been shown that viral proteins Vpr and Vif disrupt the IFN response via the degradation of IRF3,46,47 whereas in dendritic cells (DCs), IRF3 has recently been found to remain intact, but its activation and nuclear translocation are impeded by Vpr.48 The HIV protein Vpu also degrades IRF3, by binding and directing it to the lysosome.49 Instead of interfering with IRF3 activation, NS1 from RSV associates with both IRF3 and its co‐activator CBP, impeding their interaction and impairing promoter binding.50
Several viral proteins indirectly disrupt IRF3 activation by interfering with the kinases TBK1 or IKKε. The papain‐like protease domain 2 of NSp3 from mouse hepatitis virus (MHV) A59 has been found to de‐ubiquitinate TBK1, decreasing its kinase activity and stabilizing it in an inactive conformation.51 Although the mechanisms are currently unclear, the severe fever with thrombocytopenia syndrome virus NSs protein52 and the HSV‐1 γ34.5 protein associate with and inhibit TBK1,53 while the Tula virus glycoprotein Gn disrupts IFN production at the level of the TBK1 complex.54 Although they do not impede TBK1, the the NP proteins of several arenaviruses associate with the kinase domain of IKKε, impairing its binding to MAVS and preventing it from phosphorylating IRF3.55 KSHV also inhibits IKKε signalling by encoding an miRNA known as miR‐K12‐11, which down‐regulates IKKε mRNA translation.56 Lastly, the C6 protein from vaccinia virus interferes with the activation of IRF3 and IRF7 at the level of TBK1/IKKε, via interaction with the kinase scaffold proteins TANK, NAP1 or SINTBAD.57 As the exact contribution of these scaffold proteins to antiviral signalling is unclear, elucidation of C6 activity could provide valuable insight into IFN production.
Unlike IRF3, IRF7 is basally expressed at very low to undetectable levels in most cells. IFN‐β production by IRF3, NF‐κB and ATF2/c‐jun induces the expression of IRF7. Like IRF3, IRF7 is phosphorylated by TBK1 and IKKε, causing it to heterodimerize with IRF3 and stimulate full type I IFN expression.58 KSHV ORF45 impedes the phosphorylation and activation of IRF7 (but not IRF3) by competitive inhibition, as it is phosphorylated by IKKε and TBK1 more efficiently than IRF7.59 ORF45 may also block IRF7 by associating with its inhibitory domain, stabilizing autoinhibitory intramolecular interactions to keep the protein in a closed, inactive conformation.60 The V proteins of several paramyxoviruses associate with and disrupt IRF7,61 as does Npro from classical swine fever virus.62 Some strains of rotavirus use their NSP1 protein to cause IRF7 degradation via the proteasome, whereas other strains target IRF3, IRF5 or β‐transducin repeat‐containing protein (β‐TrCP), a component of the E3 ubiquitin ligase complex that activates NF‐κB.63 Finally, the ebolavirus VP35 protein represents an interesting example of IRF7 inhibition: in macrophages and conventional DCs, VP35 interferes with IRF7 activation via the RLR pathway, whereas in plasmacytoid DCs, VP35 does not block IFN production, because this cell type activates IRF7 through the TLR pathway.64 Hence, non‐redundant IFN induction pathways can help an organism to counteract specific virus evasion mechanisms.
Viruses can also impair IFN gene expression by inducing a general disruption of host cell transcription. The NSs protein from La Crosse encephalitis virus does just this, exploiting specific components of the DNA‐damage response to cause the proteasomal degradation of the hyperphosphorylated form of RPB1, a component of cellular RNA polymerase II (RNAP II), allowing it to selectively silence elongating RNAP II complexes. This does not impede the virus itself, as RNAP II is not required for the transcription or replication of the La Crosse encephalitis virus genome.65
IFN signalling
The second step of the biphasic IFN response, where secreted IFN binds its receptor (IFNAR) and activates ISG induction, is also actively disrupted by viruses. Although the exact mechanism is unknown, ORF54, a functional dUTPase from murine γ‐herpesvirus‐68, causes the degradation of the IFNAR1 protein, even in the absence of dUTPase enzymatic activity.66 Several other viruses indirectly target IFNAR, by activating alternative signalling. For instance, HCV induces the Ras/Raf/MEK pathway, which increases the phosphorylation of a destruction motif in the cytoplasmic tail of IFNAR1, leading to its ubiquitin‐dependent endocytosis.67 The Kunjin strain of West Nile virus may employ a similar strategy, as the viral proteins NS4A and NS4B block IFN signalling by stimulating the unfolded protein response,68 possibly via IFNAR degradation.69
Interferon binding to IFNAR activates the Janus family protein kinases (JAKs) Tyk2 and Jak1, inducing site‐specific phosphorylation of tyrosine residues in signal transducers and activation of transcription 1 (STAT1) and STAT2, leading to their activation and formation of a heterotrimeric complex containing IRF9, known as IFN‐stimulated gene factor‐3 (ISGF3) (Fig. 3).70 Each stage of the JAK/STAT signalling pathway is disrupted by viral proteins. Human metapneumovirus reduces Jak1 and Tyk2 mRNAs and proteins,71 leading to decreased IFNAR cell surface expression by way of increased internalization but not degradation, possibly through the loss of Tyk2.72 The E6 and E7 proteins from human papillomaviruses specifically interfere with the STAT1 promoter to block its transcription, in a mechanism that may involve histone deacetylation and DNA methylation.73 The C protein of human parainfluenza virus type 1 impedes the nuclear translocation of STAT1 by physically retaining it in the cytoplasm in perinuclear aggregates associated with late endosomal markers.74 RSV NS‐1 and NS‐2 prevent the phosphorylation and nuclear translocation of STAT1 and STAT2 after IFN‐β treatment of bone‐marrow‐derived DCs,75 whereas in the respiratory epithelium, NS2 causes the degradation of STAT2.76,77 Viral interferon regulatory factor 2 (vIRF2) from KSHV decreases STAT1 and IRF9 levels to impair ISGF3 function.78 HSV‐2 causes the selective loss of STAT2 transcripts and proteins in some cell types, whereas in others, STAT2 levels remain constant but its phosphorylation and nuclear translocation are inhibited.79 The papain‐like protease from SARS‐CoV has a complex mechanism of interference: it is a de‐ubiquitinating enzyme that up‐regulates the expression of ubiquitin‐conjugating enzyme E2‐25k, leading to the ubiquitin‐dependent proteasomal degradation of extracellular signal‐regulated kinase (ERK) 1, which interferes with ERK1‐mediated STAT1 phosphorylation.80 Interestingly, adenovirus stabilizes tyrosine‐phosphorylated, activated STAT1, sequestering it at viral replication centres, potentially through binding with viral DNA.81 Adenovirus also impairs the dephosphorylation of STAT1 by obstructing its interaction with the protein tyrosine phosphatase TC45.81
Figure 3.
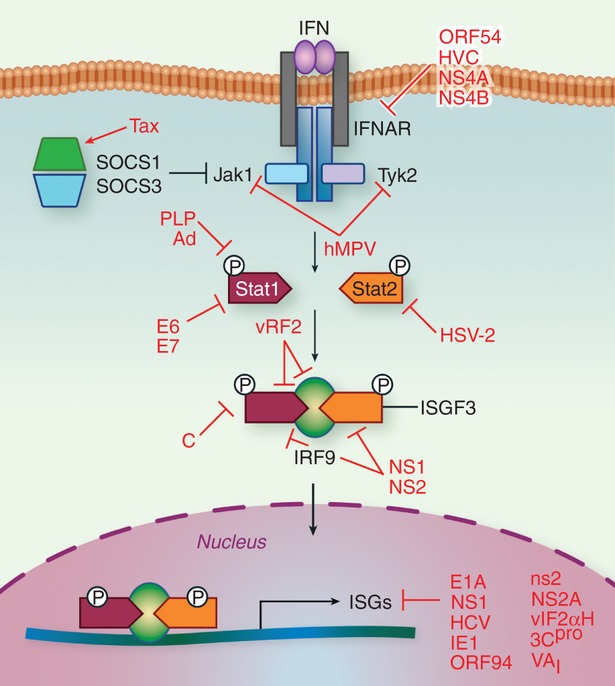
Interferon (IFN) signalling to ISG expression. Secreted IFN binds to the cell‐surface receptor IFNAR, leading to the activation of kinases Tyk2 and Jak1, which phosphorylate and activate proteins signal transducer and activator of transcription 1 (STAT1) and STAT2. This results in the formation of a heterotrimeric complex containing interferon regulatory factor 9 (IRF9), known as IFN‐stimulated gene factor‐3 (ISGF3). Jak protein activation is negatively regulated by the IFN‐inducible proteins SOCS1 and SOCS3. Binding of ISGF3 to the promoters of ISGs leads to their transcriptional activation, and the collective actions of the hundreds of ISGs induced by IFN inhibit both virus replication and spread. Each aspect of this process can be disrupted by viral processes.
ISG expression and function
Once activated, ISGF3 binds the promoters of ISGs, leading to their transcriptional activation.70 While investigating how the human adenovirus protein E1A evades the type I IFN response, Fonseca et al.82 furthered our understanding of this process, demonstrating how studying the virus leads to a better understanding of the host. They found that IFN‐mediated antiviral activity requires the mono‐ubiquitination of histone 2B (H2B) at lysine 120, a post‐translational modification associated with transcriptionally active chromatin, in both the transcribed regions and the promoters of ISGs. This finding is a novel and unexpected aspect of antiviral signalling. Additionally, they found that E1A disrupts the hBre1 complex responsible for H2B mono‐ubiquitination, preventing the expression of ISGs, and allowing viral escape of antiviral signalling.82 In another elegant study, Marazzi et al.83 demonstrated how viruses exploit epigenetic signalling to regulate antiviral gene expression. They found that the NS1 protein of influenza A strain H3N2 contains a short sequence that mimics the histone H3 tail. This permits histone‐modifying enzymes to act on NS1; accordingly, NS1 is both acetylated and methylated in infected cells.83 Modified NS1 associates with the human PAF1 transcription elongation complex, allowing the virus to hijack the host transcriptional elongation machinery. NS1 also disrupts transcriptional elongation at sites of active antiviral gene transcription, selectively impairing the expression of ISGs).83
Instead of globally obstructing ISG expression, some viruses target particular ISGs. For example, HCV infection up‐regulates a microRNA that specifically decreases the expression of the ISG IFITM1.84 The immediate‐early 1 (IE1) protein of human cytomegalovirus (HCMV) down‐regulates IFN‐inducible Sp100 protein levels. While IE1 interacts with and causes proteasome‐mediated degradation of Sp100A, it is unclear how IE1 affects additional Sp100 isoforms.85
Although the antiviral functions of many ISGs are not clearly understood,86 those of 2’‐5’‐oligoadenylate synthetase (OAS) and protein kinase R (PKR) are well elucidated.4 In response to dsRNA, OAS produces 2’‐5’‐linked oligoadenylates (2‐5A) from ATP, which activate latent RNase L, leading to degradation of host and viral mRNAs, while PKR phosphorylates the eukaryotic protein synthesis initiation factor‐2α subunit (eIF‐2α), disrupting protein synthesis. HCMV ORF94 blocks the expression and therefore the activity of OAS.87 Adenoviruses have an unusual mechanism for impeding OAS; they generate large amounts of virus‐associated RNA (VAI), which is processed by the host cell enzyme Dicer, producing small interfering RNAs.88 VAI molecules act as pseudo‐inhibitors, because they strongly bind, but poorly induce, OAS1.89 Instead of interfering with OAS directly, MHV uses its ns2 protein, a phosphodiesterase, to cleave 2‐5A molecules, preventing RNase L activation.90 JEV NS2A physically interacts with PKR to impede its activation in response to various stimuli.91 Poliovirus overcomes the PKR‐mediated translational inhibition by cleaving an additional eukaryotic initiation factor, eIF5B, via the viral proteinase 3Cpro, creating a cleavage fragment that is able to rescue viral translation under conditions of eIF2α phosphorylation.92 Interestingly, the Ambystoma tigrinum virus, which infects ectotherms such as amphibians, reptiles and fish, was found to encode a protein homologous to eIF2α, called vIF2αH, which impairs eIF2α phosphorylation through the degradation of fish PKZ, a homologue of PKR. Although the exact mechanism for this process is not known, it is intriguing that the activity of PKZ was found to be required for vIF2αH to cause its degradation.93
In some cases, viruses turn the tables completely, using particular ISGs to their own advantage. For instance, MxA is a 76 000 molecular weight ISG, which interferes with the replication of HSV‐1. Remarkably, HSV‐1 stimulates the expression of a 56 000 molecular weight MxA isoform via alternative splicing, in the absence of type I IFN. This novel isoform of MxA, which associates with virion components and nuclear viral replication compartments, increases virus replication.94 HCMV has long been known to directly induce the expression of the ISG viperin in the absence of IFN production.95 Recently, it has been shown that via interaction with the viral protein vMIA, viperin is re‐localized to the mitochondria, where it disrupts the actin cytoskeleton and enhances viral infection.96 In fact, HCMV replication is decreased in cells lacking viperin. Rotavirus infection of intestinal epithelial cells leads to a strong induction of the type I IFN response, but instead of limiting virus growth, IFN signalling promotes rotavirus replication, particularly at the early stages.97 The proposed mechanism is that type I IFN increases PKR levels, which the virus somehow exploits for its own replication.97
If a virus fails to completely block IFN production, a final subversion strategy is to modulate the negative regulation of the IFN response, which normally functions to turn off antiviral signalling upon viral clearance. The suppressor of cytokine signalling proteins SOCS1 and SOCS3 are induced by IFN, and directly interact with and inhibit JAK function in a negative feedback loop.98 The human T‐cell leukaemia virus type 1 takes advantage of this, using its Tax protein to both up‐regulate SOCS1 expression through NF‐κB activation and to stabilize the SOCS1 protein.99 Surprisingly, SOCS was found to be required for Tax to impair IFN production, but was dispensable for Tax to block IFN signalling. Interleukin‐6 up‐regulates SOCS3; intriguingly, amino acid substitutions in the core region of HCV both produce interleukin‐6 via activation of the unfolded protein response and render HCV more resistant to type I IFN.100
Conclusion
The number and diversity of viral targets for the disruption of the type I IFN response is staggering, as every step in this process can be inhibited in some way by viral proteins. Although developments in this field are rapidly accumulating, there is much still to learn. Each step taken to characterize how viruses manipulate these pathways helps to further our understanding of antiviral signalling, truly exemplifying the saying: know thy enemy, know thyself.
References
- 1.Der SD, Zhou A, Williams BR, Silverman RH. Identification of genes differentially regulated by interferon α, β, or γ using oligonucleotide arrays. Proc Natl Acad Sci U S A. 1998;95:15623–8. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Veer MJ, Holko M, Frevel M, Walker E, Der S, Paranjape JM, Silverman RH, Williams BR. Functional classification of interferon‐stimulated genes identified using microarrays. J Leukoc Biol. 2001;69:912–20. [PubMed] [Google Scholar]
- 3.Donnelly RP, Kotenko SV. Interferon‐λ: a new addition to an old family. J Interferon Cytokine Res. 2010;30:555–64. doi: 10.1089/jir.2010.0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev. 2001;14:778–809. doi: 10.1128/CMR.14.4.778-809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawai T, Akira S. Toll‐like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–50. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 6.Qu L, Feng Z, Yamane D, Liang Y, Lanford RE, Li K, Lemon SM. Disruption of TLR3 signaling due to cleavage of TRIF by the hepatitis A virus protease‐polymerase processing intermediate, 3CD. PLoS Pathog. 2011;7:e1002169. doi: 10.1371/journal.ppat.1002169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lei X, Sun Z, Liu X, Jin Q, He B, Wang J. Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by Toll‐like receptor 3. J Virol. 2011;85:8811–8. doi: 10.1128/JVI.00447-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahmad H, Gubbels R, Ehlers E, Meyer F, Waterbury T, Lin R, Zhang L. Kaposi sarcoma‐associated herpesvirus degrades cellular Toll‐interleukin‐1 receptor domain‐containing adaptor‐inducing β‐interferon (TRIF) J Biol Chem. 2011;286:7865–72. doi: 10.1074/jbc.M110.191452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lang T, Lo C, Skinner N, Locarnini S, Visvanathan K, Mansell A. The hepatitis B e antigen (HBeAg) targets and suppresses activation of the toll‐like receptor signaling pathway. J Hepatol. 2011;55:762–9. doi: 10.1016/j.jhep.2010.12.042. [DOI] [PubMed] [Google Scholar]
- 10.Maelfait J, Beyaert R. Emerging role of ubiquitination in antiviral RIG‐I signaling. Microbiol Mol Biol Rev. 2012;76:33–45. doi: 10.1128/MMBR.05012-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lifland AW, Jung J, Alonas E, Zurla C, Crowe JE, Jr, Santangelo PJ. Human respiratory syncytial virus nucleoprotein and inclusion bodies antagonize the innate immune response mediated by MDA5 and MAVS. J Virol. 2012;86:8245–58. doi: 10.1128/JVI.00215-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Solis M, Nakhaei P, Jalalirad M. RIG‐I‐mediated antiviral signaling is inhibited in HIV‐1 infection by a protease‐mediated sequestration of RIG‐I. J Virol. 2011;85:1224–36. doi: 10.1128/JVI.01635-10. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Childs K, Randall R, Goodbourn S. Paramyxovirus V proteins interact with the RNA helicase LGP2 to inhibit RIG‐I‐dependent interferon induction. J Virol. 2012;86:3411–21. doi: 10.1128/JVI.06405-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoneyama M, Kikuchi M, Natsukawa T. The RNA helicase RIG‐I has an essential function in double‐stranded RNA‐induced innate antiviral responses. Nat Immunol. 2004;5:730–7. doi: 10.1038/ni1087. et al. [DOI] [PubMed] [Google Scholar]
- 15.van Kasteren PB, Beugeling C, Ninaber DK, Frias‐Staheli N, van Boheemen S, Garcia‐Sastre A, Snijder EJ, Kikkert M. Arterivirus and nairovirus ovarian tumor domain‐containing deubiquitinases target activated RIG‐I to control innate immune signaling. J Virol. 2012;86:773–85. doi: 10.1128/JVI.06277-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inn KS, Lee SH, Rathbun JY, Wong LY, Toth Z, Machida K, Ou JH, Jung JU. Inhibition of RIG‐I‐mediated signaling by Kaposi's sarcoma‐associated herpesvirus‐encoded deubiquitinase ORF64. J Virol. 2011;85:10899–904. doi: 10.1128/JVI.00690-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun L, Xing Y, Chen X. Coronavirus papain‐like proteases negatively regulate antiviral innate immune response through disruption of STING‐mediated signaling. PLoS ONE. 2012;7:e30802. doi: 10.1371/journal.pone.0030802. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang D, Fang L, Li P. The leader proteinase of foot‐and‐mouth disease virus negatively regulates the type I interferon pathway by acting as a viral deubiquitinase. J Virol. 2011;85:3758–66. doi: 10.1128/JVI.02589-10. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gack MU, Shin YC, Joo CH. TRIM25 RING‐finger E3 ubiquitin ligase is essential for RIG‐I‐mediated antiviral activity. Nature. 2007;446:916–20. doi: 10.1038/nature05732. et al. [DOI] [PubMed] [Google Scholar]
- 20.Varga ZT, Grant A, Manicassamy B, Palese P. The influenza virus protein PB1‐F2 inhibits the induction of type I interferon by binding to MAVS and decreasing the mitochondrial membrane potential. J Virol. 2012;86:8359–66. doi: 10.1128/JVI.01122-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koshiba T, Yasukawa K, Yanagi Y, Kawabata S. Mitochondrial membrane potential is required for MAVS‐mediated antiviral signaling. Sci Signal. 2011;4:ra7. doi: 10.1126/scisignal.2001147. [DOI] [PubMed] [Google Scholar]
- 22.Mukherjee A, Morosky SA, Delorme‐Axford E, Dybdahl‐Sissoko N, Oberste MS, Wang T, Coyne CB. The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog. 2011;7:e1001311. doi: 10.1371/journal.ppat.1001311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar M, Jung SY, Hodgson AJ, Madden CR, Qin J, Slagle BL. Hepatitis B virus regulatory HBx protein binds to adaptor protein IPS‐1 and inhibits the activation of β interferon. J Virol. 2011;85:987–95. doi: 10.1128/JVI.01825-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakhaei P, Hiscott J, Lin R. STING‐ing the antiviral pathway. J Mol Cell Biol. 2010;2:110–2. doi: 10.1093/jmcb/mjp048. [DOI] [PubMed] [Google Scholar]
- 25.Yu CY, Chang TH, Liang JJ, Chiang RL, Lee YL, Liao CL, Lin YL. Dengue virus targets the adaptor protein MITA to subvert host innate immunity. PLoS Pathog. 2012;8:e1002780. doi: 10.1371/journal.ppat.1002780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yao XD, Rosenthal KL. Herpes simplex virus type 2 virion host shutoff protein suppresses innate dsRNA antiviral pathways in human vaginal epithelial cells. J Gen Virol. 2011;92(Pt 9):1981–93. doi: 10.1099/vir.0.030296-0. [DOI] [PubMed] [Google Scholar]
- 27.Eksioglu EA, Zhu H, Bayouth L, Bess J, Liu HY, Nelson DR, Liu C. Characterization of HCV interactions with Toll‐like receptors and RIG‐I in liver cells. PLoS ONE. 2011;6:e21186. doi: 10.1371/journal.pone.0021186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tamura R, Kanda T, Imazeki F. Hepatitis C virus nonstructural 5A protein inhibits lipopolysaccharide‐mediated apoptosis of hepatocytes by decreasing expression of Toll‐like receptor 4. J Infect Dis. 2011;204:793–801. doi: 10.1093/infdis/jir381. et al. [DOI] [PubMed] [Google Scholar]
- 29.Miorin L, Albornoz A, Baba MM, D'Agaro P, Marcello A. Formation of membrane‐defined compartments by tick‐borne encephalitis virus contributes to the early delay in interferon signaling. Virus Res. 2012;163:660–6. doi: 10.1016/j.virusres.2011.11.020. [DOI] [PubMed] [Google Scholar]
- 30.Espada‐Murao LA, Morita K. Delayed cytosolic exposure of Japanese encephalitis virus double‐stranded RNA impedes interferon activation and enhances viral dissemination in porcine cells. J Virol. 2011;85:6736–49. doi: 10.1128/JVI.00233-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hastie KM, Kimberlin CR, Zandonatti MA, MacRae IJ, Saphire EO. Structure of the Lassa virus nucleoprotein reveals a dsRNA‐specific 3’–5’ exonuclease activity essential for immune suppression. Proc Natl Acad Sci U S A. 2011;108:2396–401. doi: 10.1073/pnas.1016404108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boonyaratanakornkit J, Bartlett E, Schomacker H. The C proteins of human parainfluenza virus type 1 limit double‐stranded RNA accumulation that would otherwise trigger activation of MDA5 and protein kinase R. J Virol. 2011;85:1495–506. doi: 10.1128/JVI.01297-10. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wathelet MG, Lin CH, Parekh BS, Ronco LV, Howley PM, Maniatis T. Virus infection induces the assembly of coordinately activated transcription factors on the IFN‐β enhancer in vivo. Mol Cell. 1998;1:507–18. doi: 10.1016/s1097-2765(00)80051-9. [DOI] [PubMed] [Google Scholar]
- 34.Pfeffer LM. The role of nuclear factor κB in the interferon response. J Interferon Cytokine Res. 2011;31:553–9. doi: 10.1089/jir.2011.0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schuhmann KM, Pfaller CK, Conzelmann KK. The measles virus V protein binds to p65 (RelA) to suppress NF‐κB activity. J Virol. 2011;85:3162–71. doi: 10.1128/JVI.02342-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gautheron J, Courtois G. “Without Ub I am nothing”: NEMO as a multifunctional player in ubiquitin‐mediated control of NF‐κB activation. Cell Mol Life Sci. 2010;67:3101–13. doi: 10.1007/s00018-010-0404-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang D, Fang L, Li K. Foot‐and‐mouth disease virus 3C protease cleaves NEMO to impair innate immune signaling. J Virol. 2012;86:9311–22. doi: 10.1128/JVI.00722-12. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ford E, Thanos D. The transcriptional code of human IFN‐β gene expression. Biochim Biophys Acta. 2010;1799:328–36. doi: 10.1016/j.bbagrm.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 39.Halfmann P, Neumann G, Kawaoka Y. The Ebolavirus VP24 protein blocks phosphorylation of p38 mitogen‐activated protein kinase. J Infect Dis. 2011;204(Suppl. 3):S953–6. doi: 10.1093/infdis/jir325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol. 2008;26:535–84. doi: 10.1146/annurev.immunol.26.021607.090400. [DOI] [PubMed] [Google Scholar]
- 41.Mossman KL, Macgregor PF, Rozmus JJ, Goryachev AB, Edwards AM, Smiley JR. Herpes simplex virus triggers and then disarms a host antiviral response. J Virol. 2001;75:750–8. doi: 10.1128/JVI.75.2.750-758.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo J, Peters KL, Sen GC. Induction of the human protein P56 by interferon, double‐stranded RNA, or virus infection. Virology. 2000;267:209–19. doi: 10.1006/viro.1999.0135. [DOI] [PubMed] [Google Scholar]
- 43.Ye J, Maniatis T. Negative regulation of interferon‐β gene expression during acute and persistent virus infections. PLoS ONE. 2011;6:e20681. doi: 10.1371/journal.pone.0020681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vandevenne P, Lebrun M, El Mjiyad N. The varicella‐zoster virus ORF47 kinase interferes with host innate immune response by inhibiting the activation of IRF3. PLoS ONE. 2011;6:e16870. doi: 10.1371/journal.pone.0016870. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu H, Zheng C, Xing J, Wang S, Li S, Lin R, Mossman KL. Varicella‐zoster virus immediate‐early protein ORF61 abrogates the IRF3‐mediated innate immune response through degradation of activated IRF3. J Virol. 2011;85:11079–89. doi: 10.1128/JVI.05098-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Doehle BP, Hladik F, McNevin JP, McElrath MJ, Gale M., Jr Human immunodeficiency virus type 1 mediates global disruption of innate antiviral signaling and immune defenses within infected cells. J Virol. 2009;83:10395–405. doi: 10.1128/JVI.00849-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Okumura A, Alce T, Lubyova B, Ezelle H, Strebel K, Pitha PM. HIV‐1 accessory proteins VPR and Vif modulate antiviral response by targeting IRF‐3 for degradation. Virology. 2008;373:85–97. doi: 10.1016/j.virol.2007.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harman AN, Lai J, Turville S. HIV infection of dendritic cells subverts the IFN induction pathway via IRF‐1 and inhibits type 1 IFN production. Blood. 2011;118:298–308. doi: 10.1182/blood-2010-07-297721. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Doehle BP, Chang K, Rustagi A, McNevin J, McElrath MJ, Gale M., Jr Vpu mediates IRF3 depletion during HIV infection by a lysosomal‐dependent mechanism. J Virol. 2012;86:8367–74. doi: 10.1128/JVI.00423-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ren J, Liu T, Pang L, Li K, Garofalo RP, Casola A, Bao X. A novel mechanism for the inhibition of interferon regulatory factor‐3‐dependent gene expression by human respiratory syncytial virus NS1 protein. J Gen Virol. 2011;92(Pt 9):2153–9. doi: 10.1099/vir.0.032987-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang G, Chen G, Zheng D, Cheng G, Tang H. PLP2 of mouse hepatitis virus A59 (MHV‐A59) targets TBK1 to negatively regulate cellular type I interferon signaling pathway. PLoS ONE. 2011;6:e17192. doi: 10.1371/journal.pone.0017192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qu B, Qi X, Wu X. Suppression of the interferon and NF‐κB responses by severe fever with thrombocytopenia syndrome virus. J Virol. 2012;86:8388–401. doi: 10.1128/JVI.00612-12. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ma Y, Jin H, Valyi‐Nagy T, Cao Y, Yan Z, He B. Inhibition of TANK binding kinase 1 by herpes simplex virus 1 facilitates productive infection. J Virol. 2012;86:2188–96. doi: 10.1128/JVI.05376-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matthys V, Gorbunova EE, Gavrilovskaya IN, Pepini T, Mackow ER. The C‐terminal 42 residues of the Tula virus Gn protein regulate interferon induction. J Virol. 2011;85:4752–60. doi: 10.1128/JVI.01945-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pythoud C, Rodrigo WW, Pasqual G, Rothenberger S, Martinez‐Sobrido L, de la Torre JC, Kunz S. Arenavirus nucleoprotein targets interferon regulatory factor‐activating kinase IKKε. J Virol. 2012;86:7728–38. doi: 10.1128/JVI.00187-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liang D, Gao Y, Lin X, He Z, Zhao Q, Deng Q, Lan K. A human herpesvirus miRNA attenuates interferon signaling and contributes to maintenance of viral latency by targeting IKKε. Cell Res. 2011;21:793–806. doi: 10.1038/cr.2011.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Unterholzner L, Sumner RP, Baran M. Vaccinia virus protein C6 is a virulence factor that binds TBK‐1 adaptor proteins and inhibits activation of IRF3 and IRF7. PLoS Pathog. 2011;7:e1002247. doi: 10.1371/journal.ppat.1002247. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ning S, Pagano JS, Barber GN. IRF7: activation, regulation, modification and function. Genes Immun. 2011;12:399–414. doi: 10.1038/gene.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liang Q, Fu B, Wu F, Li X, Yuan Y, Zhu F. ORF45 of Kaposi's sarcoma‐associated herpesvirus inhibits phosphorylation of IRF7 by IKKε and TBK1 as an alternative substrate. J Virol. 2012;86:10162–72. doi: 10.1128/JVI.05224-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sathish N, Zhu FX, Golub EE, Liang Q, Yuan Y. Mechanisms of autoinhibition of IRF‐7 and a probable model for inactivation of IRF‐7 by Kaposi's sarcoma‐associated herpesvirus protein ORF45. J Biol Chem. 2011;286:746–56. doi: 10.1074/jbc.M110.150920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kitagawa Y, Yamaguchi M, Zhou M. A tryptophan‐rich motif in the human parainfluenza virus type 2 V protein is critical for the blockade of toll‐like receptor 7 (TLR7)‐ and TLR9‐dependent signaling. J Virol. 2011;85:4606–11. doi: 10.1128/JVI.02012-10. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fiebach AR, Guzylack‐Piriou L, Python S, Summerfield A, Ruggli N. Classical swine fever virus N(pro) limits type I interferon induction in plasmacytoid dendritic cells by interacting with interferon regulatory factor 7. J Virol. 2011;85:8002–11. doi: 10.1128/JVI.00330-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Arnold MM, Patton JT. Diversity of interferon antagonist activities mediated by NSP1 proteins of different rotavirus strains. J Virol. 2011;85:1970–9. doi: 10.1128/JVI.01801-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Leung LW, Park MS, Martinez O, Valmas C, Lopez CB, Basler CF. Ebolavirus VP35 suppresses IFN production from conventional but not plasmacytoid dendritic cells. Immunol Cell Biol. 2011;89:792–802. doi: 10.1038/icb.2010.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Verbruggen P, Ruf M, Blakqori G, Overby AK, Heidemann M, Eick D, Weber F. Interferon antagonist NSs of La Crosse virus triggers a DNA damage response‐like degradation of transcribing RNA polymerase II. J Biol Chem. 2011;286:3681–92. doi: 10.1074/jbc.M110.154799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Leang RS, Wu TT, Hwang S, Liang LT, Tong L, Truong JT, Sun R. The anti‐interferon activity of conserved viral dUTPase ORF54 is essential for an effective MHV‐68 infection. PLoS Pathog. 2011;7:e1002292. doi: 10.1371/journal.ppat.1002292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang Q, Gong R, Qu J. Activation of the Ras/Raf/MEK pathway facilitates hepatitis C virus replication via attenuation of the interferon‐JAK‐STAT pathway. J Virol. 2012;86:1544–54. doi: 10.1128/JVI.00688-11. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ambrose RL, Mackenzie JM. West Nile virus differentially modulates the unfolded protein response to facilitate replication and immune evasion. J Virol. 2011;85:2723–32. doi: 10.1128/JVI.02050-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu J, HuangFu WC, Kumar KG. Virus‐induced unfolded protein response attenuates antiviral defenses via phosphorylation‐dependent degradation of the type I interferon receptor. Cell Host Microbe. 2009;5:72–83. doi: 10.1016/j.chom.2008.11.008. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pitha PM, Kunzi MS. Type I interferon: the ever unfolding story. Curr Top Microbiol Immunol. 2007;316:41–70. doi: 10.1007/978-3-540-71329-6_4. [DOI] [PubMed] [Google Scholar]
- 71.Ren J, Kolli D, Liu T, Xu R, Garofalo RP, Casola A, Bao X. Human metapneumovirus inhibits IFN‐β signaling by downregulating Jak1 and Tyk2 cellular levels. PLoS ONE. 2011;6:e24496. doi: 10.1371/journal.pone.0024496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gauzzi MC, Barbieri G, Richter MF, Uze G, Ling L, Fellous M, Pellegrini S. The amino‐terminal region of Tyk2 sustains the level of interferon α receptor 1, a component of the interferon α/β receptor. Proc Natl Acad Sci U S A. 1997;94:11839–44. doi: 10.1073/pnas.94.22.11839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hong S, Mehta KP, Laimins LA. Suppression of STAT‐1 expression by human papillomaviruses is necessary for differentiation‐dependent genome amplification and plasmid maintenance. J Virol. 2011;85:9486–94. doi: 10.1128/JVI.05007-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schomacker H, Hebner RM, Boonyaratanakornkit J, Surman S, Amaro‐Carambot E, Collins PL, Schmidt AC. The C proteins of human parainfluenza virus type 1 block IFN signaling by binding and retaining Stat1 in perinuclear aggregates at the late endosome. PLoS ONE. 2012;7:e28382. doi: 10.1371/journal.pone.0028382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jie Z, Dinwiddie DL, Senft AP, Harrod KS. Regulation of STAT signaling in mouse bone marrow derived dendritic cells by respiratory syncytial virus. Virus Res. 2011;156:127–33. doi: 10.1016/j.virusres.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ramaswamy M, Shi L, Monick MM, Hunninghake GW, Look DC. Specific inhibition of type I interferon signal transduction by respiratory syncytial virus. Am J Respir Cell Mol Biol. 2004;30:893–900. doi: 10.1165/rcmb.2003-0410OC. [DOI] [PubMed] [Google Scholar]
- 77.Ramaswamy M, Shi L, Varga SM, Barik S, Behlke MA, Look DC. Respiratory syncytial virus nonstructural protein 2 specifically inhibits type I interferon signal transduction. Virology. 2006;344:328–39. doi: 10.1016/j.virol.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 78.Mutocheluh M, Hindle L, Areste C. Kaposi's sarcoma‐associated herpesvirus viral interferon regulatory factor‐2 inhibits type 1 interferon signalling by targeting interferon‐stimulated gene factor‐3. J Gen Virol. 2011;92(Pt 10):2394–8. doi: 10.1099/vir.0.034322-0. et al. [DOI] [PubMed] [Google Scholar]
- 79.Kadeppagari RK, Sanchez RL, Foster TP. HSV‐2 inhibits type‐I interferon signaling via multiple complementary and compensatory STAT2‐associated mechanisms. Virus Res. 2012;167:273–84. doi: 10.1016/j.virusres.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li SW, Lai CC, Ping JF, Tsai FJ, Wan L, Lin YJ, Kung SH, Lin CW. Severe acute respiratory syndrome coronavirus papain‐like protease suppressed α interferon‐induced responses through downregulation of extracellular signal‐regulated kinase 1‐mediated signalling pathways. J Gen Virol. 2011;92(Pt 5):1127–40. doi: 10.1099/vir.0.028936-0. [DOI] [PubMed] [Google Scholar]
- 81.Sohn SY, Hearing P. Adenovirus sequesters phosphorylated STAT1 at viral replication centers and inhibits STAT dephosphorylation. J Virol. 2011;85:7555–62. doi: 10.1128/JVI.00513-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fonseca GJ, Thillainadesan G, Yousef AF, Ablack JN, Mossman KL, Torchia J, Mymryk JS. Adenovirus evasion of interferon‐mediated innate immunity by direct antagonism of a cellular histone posttranslational modification. Cell Host Microbe. 2012;11:597–606. doi: 10.1016/j.chom.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 83.Marazzi I, Ho JS, Kim J. Suppression of the antiviral response by an influenza histone mimic. Nature. 2012;483:428–33. doi: 10.1038/nature10892. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bhanja Chowdhury J, Shrivastava S, Steele R, Di Bisceglie AM, Ray R, Ray RB. Hepatitis C virus infection modulates interferon stimulatory gene IFITM1 by up‐regulating miR‐130a. J Virol. 2012;86:10221–5. doi: 10.1128/JVI.00882-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kim YE, Lee JH, Kim ET. Human cytomegalovirus infection causes degradation of Sp100 proteins that suppress viral gene expression. J Virol. 2011;85:11928–37. doi: 10.1128/JVI.00758-11. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liu SY, Sanchez DJ, Cheng G. New developments in the induction and antiviral effectors of type I interferon. Curr Opin Immunol. 2011;23:57–64. doi: 10.1016/j.coi.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tan JC, Avdic S, Cao JZ, Mocarski ES, White KL, Abendroth A, Slobedman B. Inhibition of 2’,5’‐oligoadenylate synthetase expression and function by the human cytomegalovirus ORF94 gene product. J Virol. 2011;85:5696–700. doi: 10.1128/JVI.02463-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Andersson MG, Haasnoot PC, Xu N, Berenjian S, Berkhout B, Akusjarvi G. Suppression of RNA interference by adenovirus virus‐associated RNA. J Virol. 2005;79:9556–65. doi: 10.1128/JVI.79.15.9556-9565.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Meng H, Deo S, Xiong S, Dzananovic E, Donald LJ, van Dijk CW, McKenna SA. Regulation of the interferon‐inducible 2’‐5’‐oligoadenylate synthetases by adenovirus VA(I) RNA. J Mol Biol. 2012;422:635–49. doi: 10.1016/j.jmb.2012.06.017. [DOI] [PubMed] [Google Scholar]
- 90.Zhao L, Jha BK, Wu A, Elliott R, Ziebuhr J, Gorbalenya AE, Silverman RH, Weiss SR. Antagonism of the interferon‐induced OAS‐RNase L pathway by murine coronavirus ns2 protein is required for virus replication and liver pathology. Cell Host Microbe. 2012;11:607–16. doi: 10.1016/j.chom.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tu YC, Yu CY, Liang JJ, Lin E, Liao CL, Lin YL. Blocking dsRNA‐activated protein kinase PKR by Japanese Encephalitis Virus nonstructural protein 2A. J Virol. 2012;86:10347–58. doi: 10.1128/JVI.00525-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.White JP, Reineke LC, Lloyd RE. Poliovirus switches to an eIF2‐independent mode of translation during infection. J Virol. 2011;85:8884–93. doi: 10.1128/JVI.00792-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jancovich JK, Jacobs BL. Innate immune evasion mediated by the Ambystoma tigrinum virus eukaryotic translation initiation factor 2α homologue. J Virol. 2011;85:5061–9. doi: 10.1128/JVI.01488-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ku CC, Che XB, Reichelt M. Herpes simplex virus‐1 induces expression of a novel MxA isoform that enhances viral replication. Immunol Cell Biol. 2011;89:173–82. doi: 10.1038/icb.2010.83. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhu H, Cong JP, Shenk T. Use of differential display analysis to assess the effect of human cytomegalovirus infection on the accumulation of cellular RNAs: induction of interferon‐responsive RNAs. Proc Natl Acad Sci U S A. 1997;94:13985–90. doi: 10.1073/pnas.94.25.13985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Seo JY, Yaneva R, Hinson ER, Cresswell P. Human cytomegalovirus directly induces the antiviral protein viperin to enhance infectivity. Science. 2011;332:1093–7. doi: 10.1126/science.1202007. [DOI] [PubMed] [Google Scholar]
- 97.Frias AH, Jones RM, Fifadara NH, Vijay‐Kumar M, Gewirtz AT. Rotavirus‐induced IFN‐β promotes anti‐viral signaling and apoptosis that modulate viral replication in intestinal epithelial cells. Innate Immun. 2012;18:294–306. doi: 10.1177/1753425911401930. [DOI] [PubMed] [Google Scholar]
- 98.Strebovsky J, Walker P, Dalpke AH. Suppressor of cytokine signaling proteins as regulators of innate immune signaling. Front Biosci. 2012;17:1627–39. doi: 10.2741/4008. [DOI] [PubMed] [Google Scholar]
- 99.Charoenthongtrakul S, Zhou Q, Shembade N, Harhaj NS, Harhaj EW. Human T cell leukemia virus type 1 Tax inhibits innate antiviral signaling via NF‐κB‐dependent induction of SOCS1. J Virol. 2011;85:6955–62. doi: 10.1128/JVI.00007-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Funaoka Y, Sakamoto N, Suda G. Analysis of interferon signaling by infectious hepatitis C virus clones with substitutions of core amino acids 70 and 91. J Virol. 2011;85:5986–94. doi: 10.1128/JVI.02583-10. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]