Genes That Escape X-Inactivation in Humans Have High Intraspecific Variability in Expression, Are Associated with Mental Impairment but Are Not Slow Evolving (original) (raw)
Abstract
In female mammals most X-linked genes are subject to X-inactivation. However, in humans some X-linked genes escape silencing, these escapees being candidates for the phenotypic aberrations seen in polyX karyotypes. These escape genes have been reported to be under stronger purifying selection than other X-linked genes. Although it is known that escape from X-inactivation is much more common in humans than in mice, systematic assays of escape in humans have to date employed only interspecies somatic cell hybrids. Here we provide the first systematic next-generation sequencing analysis of escape in a human cell line. We analyzed RNA and genotype sequencing data obtained from B lymphocyte cell lines derived from Europeans (CEU) and Yorubans (YRI). By replicated detection of heterozygosis in the transcriptome, we identified 114 escaping genes, including 76 not previously known to be escapees. The newly described escape genes cluster on the X chromosome in the same chromosomal regions as the previously known escapees. There is an excess of escaping genes associated with mental retardation, consistent with this being a common phenotype of polyX phenotypes. We find both differences between populations and between individuals in the propensity to escape. Indeed, we provide the first evidence for there being both hyper- and hypo-escapee females in the human population, consistent with the highly variable phenotypic presentation of polyX karyotypes. Considering also prior data, we reclassify genes as being always, never, and sometimes escape genes. We fail to replicate the prior claim that genes that escape X-inactivation are under stronger purifying selection than others.
Keywords: X-inactivation, rate of evolution, expression evolution
Introduction
Mammals have evolved a mechanism to inactivate one of the female X chromosomes. Although in humans the majority of X-linked genes are subject to X-inactivation, at least 15% (Carrel and Willard 2005) are thought to escape X-inactivation being expressed from both the active X (Xa) and inactive X (Xi) chromosomes. Escape genes in human are distributed in clusters (Tsuchiya et al. 2004; Carrel and Willard 2005) and probably controlled at the chromatin domain level. The majority of escape genes have been shown to be located on the short arm of the X chromosome (Disteche 1999). This may reflect a mechanistic constraint, these genes being too distant from the X-inactivation center (Xic) in the long arm to be affected. They may also be protected from the spreading of XIST RNA, coded for by the XIST gene within the Xic, by centromeric heterochromatin.
Given the strong conservation of gene content on the mammalian X chromosome, it has been possible to ask whether the ability to escape X-inactivation might be an evolvable trait. Principally, this has been addressed by comparing mice and humans (Disteche et al. 2002; Carrel and Willard 2005; Yang et al. 2010). For example, Yang et al. (2010) used RNA sequencing technology, in combination with single nucleotide polymorphism (SNP) identification, to infer the escape profile in mice and compared this with human data. The profiles of escape in mice and humans show significant differences in the number of genes and overall status of inactivation with escape being more prevalent in humans for reasons unknown.
It is likely that this prevalence of escape from X-inactivation in humans is related to the relative severity of polyX karyotypes in humans (Yang et al. 2010). PolyX karyotypes are associated with numerous phenotypes, including mental retardation and growth effects (Rooman et al. 2002). Typically, when more than one X is present, all X chromosomes but one are inactivated (Lyon 1961; Belmont et al. 1986). Genes that escape X-inactivation are hence good candidates for dosage-mediated phenotypic disruptions associated with polyX karyotypes (Linden et al. 1995; Tartaglia et al. 2010; Berletch et al. 2011). Determining which genes escape X-inactivation is thus of potential clinical relevance.
Analysis of polyX karyotypes has also suggested that there is variability in phenotypic presentation between individuals with the same karyotype (Rooman et al. 2002; Otter et al. 2010; Tartaglia et al. 2010). Indeed although many XXX females go undiagnosed (Gustavson 1999; Tartaglia et al. 2010), many have immediately evident phenotypes (Otter et al. 2010). This may reflect differing degrees of mosaicism (Tartaglia et al. 2010). It might also, however, reflect variability between individuals as regards which genes escape X-inactivation. Consistent with this expectation, in humans genes that escape X-inactivation can have different expression levels in different individuals (Brown and Greally 2003; Carrel and Willard 2005), these variably expressed genes estimated to comprise 10% or more of X-linked genes.
In addition to clinical relevance, knowing which genes escape X-inactivation is important for molecular evolutionary inference, as genes escaping X-inactivation have different mean dominance to those not escaping and may be under different selective pressures (Park et al. 2010). Indeed, Park et al. (2010) report that genes that always escape X-inactivation have a lower _K_a/_K_s than those that sometimes do or never do. This, they suggest, may reflect differences in dominance. However, at first sight one might think that a dominance argument would make the opposite prediction: if most new mutations are recessive, as genes that never escape are haploid expressed, new mutations should be under stronger purifying selection than those diploid expressed (i.e., those that escape X-inactivation). Moreover, the class with the highest _K_a/_K_s are those that sometimes escape. A priori, all else being equal, one would expect this class to sit between the extremes of those that never and those that always escape. With these two caveats, it is worth asking whether the prior result is robust to reclassification of genes on addition of new data. In addition, it is necessary to address whether any result is robust to quantitative control for differences in absolute expression level (Pal et al. 2001; Drummond et al. 2006), the strongest predictor of rates of evolution.
The largest prior effort to determine the status of X-inactivation on human genes in a human cell line employed a quantitative assay based on fluorescent, single-nucleotide primer extension (Carrel and Willard 2005). This study examined a limited number (N = 94) of X-linked genes in fibroblasts, finding evidence for some form of escape for 35% of them, with 15% showing escape in all samples (Carrel and Willard 2005). Given the limited scale of this cell line-based assay, the same authors used a more systematic somatic cell hybrid system for more than 600 X-linked transcripts. This identified 94 transcripts that always escape inactivation and a further 61 that are heterogeneous.
Although the somatic cell hybrid data appear relatively consistent with the fibroblast data (Carrel and Willard 2005), it is worthwhile asking whether cell line-based data on a high-throughput scale can confirm or discover genes that escape X-inactivation. We address this issue by examining the RNA-Seq data of immortalized B-cells looking for evidence of heterozygosity within the transcriptome at X-linked loci. We identify a further 76 genes sometimes subject to some degree of escape from X-inactivation. With the same data we can also address the question of the level of heterogeneity. Are some individuals hyper-escapees, permitting significantly more genes to escape than others? Do populations differ in their profile of escape? To address these issues, we study the profile of escape between two populations, US residents with northern and western European ancestry (CEU) and Yoruban individuals of Nigeria (YRI). We find strong evidence for heterogeneity in escape, finding both between-population and between-individual differences. We find no evidence that genes that always escape X-inactivation have an unusually low rate of protein evolution, before or after control for expression level. These results potentially have ramifications for pharmacogenomics, for the etiology of X chromosome ploidy disruption phenotypes, and for molecular evolutionary inference.
Results
Identification of 76 New X-Inactivation Escapees
We located the biallelic sites in annotated genes, for which the transcript information was extracted from UCSC reference genes. Because the expression from the inactive X chromosome should be no higher than that of the active X chromosome, we considered the version of the gene with a smaller number of reads in heterozygosis to be the “silenced” allele from the inactive X chromosome and those with larger numbers as the active alleles. Assuming that incidences where fewer than 10% of alleles are from the silenced allele are not trustworthy to call heterozygosity (Carrel and Willard 2005), we obtained a total of 103 genes displaying evidence of escape from X-inactivation among 37 CEU individuals and 113 genes among 40 YRI individuals.
We consider only genes with replicate evidence as “validated” escapees. Replication means either two or more individuals or two or more SNPs within one individual, providing evidence of escape (table 1) (for the set of 33 genes with prima facie evidence of escape but without replication, see supplementary table S1, Supplementary Material online). Allowing for overlap between the methods for replications, we find that we can replicate 38 of the previously reported escape genes based on the rodent/human somatic cell hybrids assay and the primary human cell line assay (Carrel and Willard 2005). In addition, we observed a further 76 validated genes that escape inactivation in B lymphocyte cell lines from normal individuals (table 1), giving a total of 114 robustly described escape genes. Of the newly validated escape genes, 62 were reported not to be escapees in the prior analysis (rather than simply not studied). Of these, 19 were doubly replicated in our sample, both by escape being detected in multiple individuals and through multiple SNPs within one individual.
Table 1.
The 114 Escape Genes and the Nature of the Replication Evidence.
| Genes | SNPs | Persons | Reported | Genes | SNPs | Persons | Reported | Genes | SNPs | Persons | Reported |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ABCB7 | Yes | Yes | Heter | HAUS7 | No | Yes | SEPT6 | Yes | Yes | Inactive | |
| AIFM1 | No | Yes | Escape | HCFC1 | No | Yes | Heter | SH3BGRL | No | Yes | Escape |
| ALG13 | No | Yes | Heter | HDHD1 | Yes | Yes | Escape | SH3KBP1 | Yes | Yes | Heter |
| APEX2 | Yes | Yes | Inactive | HUWE1 | No | Yes | Inactive | SLC25A43 | Yes | Yes | |
| APOO | No | Yes | Inactive | IDS | Yes | Yes | Inactive | SLC25A5 | No | Yes | Inactive |
| ARHGAP4 | No | Yes | IGBP1 | No | Yes | Inactive | SLC38A5 | No | Yes | Inactive | |
| ARMCX3 | No | Yes | Inactive | IRAK1 | Yes | Yes | Inactive | SMC1A | No | Yes | Heter |
| ATP6AP1 | No | Yes | Inactive | LAMP2 | Yes | Yes | Inactive | SNX12 | No | Yes | Inactive |
| ATP6AP2 | Yes | Yes | Inactive | LOC550643 | No | Yes | STS | Yes | Yes | Escape | |
| ATP7A | No | Yes | Heter | MAGED1 | No | Yes | SUV39H1 | No | Yes | Inactive | |
| BCOR | No | Yes | Heter | MAGED2 | Yes | Yes | Inactive | SYN1 | No | Yes | Inactive |
| BTK | Yes | Yes | Heter | MAGEH1 | No | Yes | Inactive | TAZ | No | Yes | |
| CCDC22 | No | Yes | Inactive | MAP7D2 | No | Yes | Inactive | TBC1D25 | No | Yes | Inactive |
| CD99L2 | Yes | Yes | Inactive | MAP7D3 | Yes | Yes | Inactive | TBL1X | Yes | Yes | Heter |
| CDK16 | No | Yes | Escape | MBNL3 | No | Yes | Inactive | TCEAL4 | No | Yes | Heter |
| CTPS2 | No | Yes | Escape | MED12 | No | Yes | Inactive | TLR7 | No | Yes | |
| CXORF21 | Yes | Yes | MED14 | No | Yes | Escape | TMEM187 | Yes | Yes | Heter | |
| CXORF38 | Yes | Yes | Escape | MID1IP1 | No | Yes | Inactive | TRAPPC2 | No | Yes | Escape |
| CXORF40A | No | Yes | Inactive | MORF4L2 | Yes | Yes | Heter | TSIX | Yes | Yes | |
| CYBB | No | Yes | MPP1 | No | Yes | Inactive | TSR2 | No | Yes | Inactive | |
| DDX26B | No | Yes | Inactive | MSL3 | Yes | Yes | Heter | TXLNG | Yes | Yes | Escape |
| DDX3X | No | Yes | Escape | MTMR1 | No | Yes | Inactive | UBA1 | Yes | Yes | Escape |
| DKC1 | No | Yes | Inactive | NSDHL | No | Yes | Inactive | UBL4A | Yes | Yes | Inactive |
| DMD | Yes | Yes | Inactive | P2RY10 | No | Yes | USP9X | Yes | Yes | Escape | |
| DNASE1L1 | Yes | Yes | Inactive | PDHA1 | Yes | Yes | Inactive | UTP14A | No | Yes | Heter |
| DOCK11 | No | Yes | Heter | PDK3 | Yes | No | Inactive | VBP1 | Yes | Yes | Inactive |
| EBP | No | Yes | Inactive | PGK1 | No | Yes | Inactive | WWC3 | Yes | Yes | Inactive |
| EDA2R | No | Yes | Heter | PIM2 | No | Yes | Inactive | XIAP | No | Yes | Inactive |
| EIF1AX | No | Yes | Escape | PIN4 | No | Yes | Heter | XIST | No | Yes | Escape |
| EIF2S3 | Yes | Yes | Escape | PIR | Yes | Yes | Escape | ZC4H2 | No | Yes | Inactive |
| ELF4 | Yes | Yes | PJA1 | No | Yes | Inactive | ZFX | Yes | Yes | Escape | |
| ELK1 | No | Yes | Inactive | PLXNA3 | Yes | No | Inactive | ZMYM3 | No | Yes | Inactive |
| FAM3A | No | Yes | Inactive | PQBP1 | No | Yes | Inactive | ZNF275 | Yes | Yes | Inactive |
| FLNA | Yes | Yes | Inactive | PRKX | Yes | Yes | Heter | ZNF75D | Yes | No | Inactive |
| FTSJ1 | No | Yes | Inactive | RBM3 | Yes | Yes | Inactive | ||||
| G6PD | Yes | Yes | Inactive | RENBP | Yes | Yes | Heter | ||||
| GDI1 | No | Yes | RNF113A | No | Yes | Inactive | |||||
| GEMIN8 | No | Yes | Escape | RPL10 | Yes | No | Inactive | ||||
| GPR174 | No | Yes | SASH3 | No | Yes | Inactive | |||||
| GRIPAP1 | No | Yes | Inactive | SAT1 | No | Yes | Inactive |
Considering instances where we could in principle have provided additional support for escape (i.e., we have polymorphic markers passing transcriptome level quality control), there are 23 genes at a minimum 7× coverage for which prior evidence (Carrel and Willard 2005) suggested escape from inactivation to some degree that we could not confirm (here we include our 33 nonreplicated escapees as providing support). Even if we permit a minimum of 20× coverage to consider a gene, we still find ten that we fail to replicate. As coverage increases, so decreasing false-negative calls of haploid expression, there is an approximately constant ratio of the number of genes whose escape we can confirm to the number we cannot confirm (with 7× coverage, the ratio of the number of those we cannot replicate to the number we can replicate is 0.47, whereas at 50× it is 0.45).
Of our 114 escapees, there are 110 incidences where two or more individuals across the whole sample show evidence that a given gene escapes X-inactivation. There are 60 that escape in at least two different individuals within the CEU population and 80 genes that escape in at least two different individuals within the YRI population (fig. 1), a total of 103 different genes with within-population replication. There are in total 45 genes confirmed heterozygous with multiple SNPs (see table 1) of which 27 were previously not known to escape X-inactivation and 41 of which are confirmed by between-individual replication as well. A few (three in CEU and four in YRI) of the replicated 114 genes contain heterozygosity with most, but not all, of the sites being consistent across several individuals. We considered these as replicated as 1) most sites were consistent and 2) as noted above, a lack of evidence for consistency of heterozygosity within a gene is not unexpected as it could reflect either SNPs having different coverage (in some individuals some potentially heterozygous sites could then not be interrogated) or owing to some falling below the 10% threshold that we set, in which case they would still be called homozygous.
Fig. 1.

The location of escape genes in CEU and YRI cluster in similar chromosomal locations. The genes found in more than three individuals and in greater than 50% of the potentially informative samples are considered to be common escape genes (red) in each population, whereas the others are rare escape genes (blue) in the populations. The genes solely replicated via more than one SNPs per gene are not included. Their inclusion makes no difference to qualitative trends. The x axis refers to the count of individuals with evidence for escape in the corresponding genes. Note Xist is within the Xic domain.
Genes Newly Identified to Escaping X-Inactivation Cluster in Known Domains of Escape
The previously described escapees tend to be distant from the X-inactivation centre. The same is seen with the new inventory of escapees, in which we also see that escapees defined within each population locate to the same regions of the X chromosome (fig. 1), with the majority of escape genes being located in the short arm and the distal portion of the long arm of the X chromosome (region of PAR2). This is consistent with the previously reported control of chromatin domains in human X-inactivation (Tsuchiya et al. 2004; Carrel and Willard 2005; Yang et al. 2010) and with the related claim (Lahn and Page 1999) that escape genes are more common in the relatively recently added strata of the X chromosome (strata 3–5) compared with the more ancient strata (S1 and S2) (χ2 test, P = 0.02; strata data from Kelkar et al. [2009]). S1, the most ancient stratum, dominates the long arm of the X and has the lowest proportion of genes escaping X-inactivation. For a frequency plot of escapees by strata, see supplementary figure S1, Supplementary Material online. The difference in escape regularity between the short and long arms of the X chromosome is not obviously explained as an artifact of expression level, read coverage, or density of heterozygous sites, as there is no significant difference between genes on two arms of the X, neither in expression level (Mann–Whitney U test, P = 0.3779), coverage at polymorphic sites (Mann–Whitney U test, P = 0.120), nor in density of heterozygous sites (229/391 in short arm vs. 363/619 in long arm).
There may also be a small cluster of genes escaping inactivation in the immediate vicinity of Xic (fig. 1). This cluster is visible only in the YRI population but this population has a greater extent of DNA heterozygosity, making it easier to identify escape genes. If this cluster is real, it is associated with few genes and, controlling for the degree of heterozygosis, there is no significant difference between the two populations. However, a similar cluster of escape was observed in prior analysis (Carrel and Willard 2005). The possibility that Xist may have weaker effects in the immediate vicinity of the X-inactivation center Xic (chrX: 65,000,000–80,000,000) (fig. 1) is, we suggest, worthy of deeper scrutiny. As regard the issues of sites in the vicinity of Xic, there is no significant expression difference between genes closer to Xic and those on the short arm (Mann–Whitney U test, P = 0.6558). Read coverage also shows no difference in this region (Mann–Whitney U test, P = 0.676).
Genes Escaping X-Inactivation Are Commonly Related to Mental Impairment
It is notable that many X chromosome ploidy alterations (including XXY and XXX, XXXX, XXXXX) are associated with learning impairments (Rooman et al. 2002). Indeed, this may be the only consistent feature of polyX karyotypes (Rooman et al. 2002). As typically all but one X is inactivated, the phenotype of X polysomies is often thought to reflect the action of genes that escape X-inactivation. Do we find any evidence that genes escaping inactivation are commonly associated with mental retardation?
We can define X-linked mental retardation (XLMR) or intellectual disability (ID) genes as those genes, mutation within which are associated with disturbance of normal intellectual functioning (Gecz et al. 2009; Stevenson and Schwartz 2009). A list of such genes is available from Greenwood Genetic Centre (Gecz et al. 2009). Among the 114 replicated escape genes, there are 22 genes (supplementary table S2, Supplementary Material online) involved in the diseases of XLMR or ID. There are 833 examinable genes covered with reads, including 91 XLMR/ID genes and 114 escapees. To determine whether 22 is significantly greater than expected, we randomly selected 114 of 833 and recorded how often the number of XLMR genes found is ≥22. The observed number is indeed more than expected by chance (P = 0.0025, from 10,000 simulants). These 22 genes would be good candidates for further analysis in this context, as impaired intellectual functioning may reflect higher dosage of these genes. There is considerable between-individual variation in the number of XLMR escape genes (supplementary fig. S2, Supplementary Material online). It would be instructive to know whether this variation correlates with any mental functioning parameters in XX females as well as polyX subjects.
A common, but not universal, phenotype of X polysomies is an effect on stature, typically manifested as rapid growth (Rooman et al. 2002). In no small part this is owing to overexpression of the pseudoautosomal gene SHOX (Rao et al. 1997). Linkage analysis has suggested, however, that Xq24 might also harbor such stature genes (Deng et al. 2002; Liu et al. 2004). This has been replicated in some (Liu et al. 2006) but not all (Visscher et al. 2007) studies. We find 6 of the 50 genes that reside within Xq24 escape X-inactivation (these being LAMP2, SLC25A5, DOCK11, RNF113A, SEPT6, and SLC25A43). This is no more than expected by chance (randomization test, as above, P > 0.05). Text mining for any association with growth phenotypes (via http://diseases.jensenlab.org/Search) suggests no evident connections.
The Profile of Escape Differs between CEU and YRI
In this study, we find a total of 66 genes that escape X-inactivation in both the CEU and YRI populations, including several well-known escape genes (HDHD1, STS, ZFX, EIF2S3, CXorf38, DDX3X). However, we are especially interested in the differences between populations rather than the common escape genes of the two populations. Table 2 presents all of the replicated escapee genes that are genetically polymorphic in both the populations and hence potentially identifiable as escapees, as well as the escape status of these genes.
Table 2.
Escape Genes and Their Proportion of Escape among Individuals in CEU and YRI.
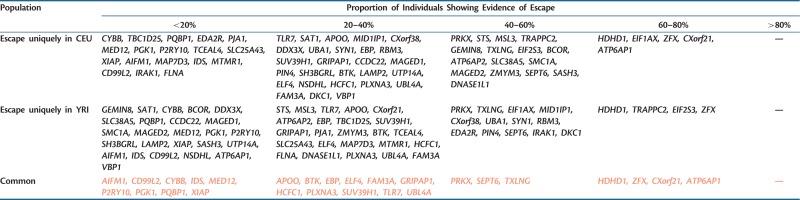
We address whether there are differences between the populations by a randomization test (see Materials and Methods). The answer to the question as to whether the difference in the profiles is due to chance is unambiguous: the two groups of populations are considerably different (observed χ2 = 196.56, expected = 119.94 ± 16.07 [SD]; from randomization P < 0.0001). This is unlikely to be an artifact of coverage differences between the two populations as the coverage is not significantly different between the two (Mann–Whitney U test, P = 0.37). Moreover, if we exclude from analysis any heterozygous sites with a less than 10× coverage, so giving more confidence in calling a lack of escape but also identifying fewer escaping genes, we still observe a significant difference between the populations (randomization test described in Materials and Methods, P = 0.016). P value is reduced not least because the sample size is reduced. At this cutoff, 82 genes in CEU and 90 genes in YRI are retained as escapees. Using only single-end data rather than single-end and paired-end data also makes no difference to the conclusion of between-population differences (randomization test described in Materials and Methods, P = 0.006). A difference in the profile of escape can be both because the genes escaping in the two populations are different and because the proportions of individuals showing escape for a given gene are different.
Analysis of Which Genes Are Variable in Their Propensity for Escape Is of Low Power
The analysis considering all genes en mass demonstrates striking variation between the populations. But can we identify which genes are different between the two populations? Regarding the individual genes, we identified a number of cases with significant differences between the two groups (table 3 and supplementary fig. S3, Supplementary Material online). Owing to the different numbers of informative individuals for each gene, the P values for each gene are not strictly comparable. Importantly, with low sample sizes (few heterozygous individuals), P can never be very low. As the sample size varies between genes, the usual consideration of how to estimate the true number of significant instances, by examination of the form of the distribution of 1 − P versus rank order (Lai 2007), is not valid. As such we consider those that are significant as candidates for genes showing differences between populations, but this would require experimental confirmation, not least because no incidence passes multi-test correction.
Table 3.
Genes That Potentially Show Differences between the Two Populations in Escape Profile.
| Gene | CEU | YRI | P Value |
|---|---|---|---|
| USP9X | 4/14 | 0/34 | 0.00729 |
| ATP6AP1 | 3/5 | 1/23 | 0.02046 |
| MPP1 | 3/8 | 0/20 | 0.02228 |
| SASH3 | 7/13 | 5/35 | 0.02229 |
| TBL1X | 4/17 | 0/21 | 0.03858 |
| HUWE1 | 3/16 | 0/29 | 0.04549 |
| MORF4L2 | 4/13 | 0/14 | 0.05364 |
| CXorf21 | 7/9 | 3/14 | 0.05575 |
| LOC550643 | 2/9 | 0/28 | 0.05874 |
| LAMP2 | 3/12 | 1/33 | 0.06052 |
| SMC1A | 6/13 | 3/24 | 0.07425 |
| VBP1 | 4/12 | 1/19 | 0.07787 |
| SLC38A5 | 2/4 | 2/26 | 0.08774 |
| BCOR | 3/6 | 1/13 | 0.09626 |
| SLC25A5 | 3/12 | 0/14 | 0.09837 |
| CA5BP1 | 1/2 | 0/18 | 0.09976 |
Despite the above caveats, there is one potentially notable observation. In all examples of a potential difference between the two populations (at P < 0.05), the prevalence of escape is higher in CEU than in YRI. This remains true if we consider also incidences where P lies between 0.05 and 0.1. This, however, is likely to be an artifact of higher diversity in YRI which leads to more potentially informative samples (heterozygous at the DNA level). In the samples where we detect a difference, the mean sample size in YRI is around 20 and around 10 in CEU. If we consider a case where 4/10 are escapees in CEU and 2/20 are escapees in YRI (around the average that we observe in the cases of significant difference) and compare this with the symmetrical case (1/10 in CEU and 8/20 in YRI), it is indeed the case that the χ2 values are higher for the former case (χ2 = 3.75) than in the symmetrical case (χ2 = 2.85). Thus, with the sorts of sample sizes and the sorts of ratios of escape to non-escape that we are looking at, we might expect to see more significant examples when the higher proportion of escapees is seen in the population with the lower number of informative examples.
Although we cannot be confident in having identified genes that show between-population within-species differences, it is worth asking whether there might be any commonality of those that are potentially different. On the X chromosome, the six genes that have significant escape variation (P < 0.05) are not clustered together (supplementary fig. S3, Supplementary Material online). Some of their neighboring genes with escape from X-inactivation do not have an escape profile showing significant differences between the two populations. This result might suggest that the between-population divergence, in regard to X-inactivation escape, is not owing to chromatin domain regulation. It could also mean, however, that owing to statistical limitations, we have incorrectly classified genes as to whether they differ in the escape propensity between different populations.
Evidence for Between-Individual Differences
The above data suggest that the two populations differ in their propensity to permit escape from X-inactivation. But might there also be females that are more or less prone to permitting escape? To address this, we can ask how often an individual has escape genes at potentially informative genes. To this end we calculated how many genes show escape and how many potential informative genes that could be heterozygous, but not show transcriptome level heterozygosity, and compared each individual with the total of others by the χ2-like test through simulation (see Materials and Methods). Of the 77 individuals, 5 in CEU and 8 in YRI show more escape than expected by chance, what we term hyper-escapee females (P < 0.05) (table 4). After Holm’s correction, four in CEU and one in YRI remain as hyper-escapees. This is consistent with the notion that even within populations individuals differ in their propensity to allow genes to escape inactivation (Carrel and Willard 2005). If we look only at these 13 individuals (significant before Holm’s correction), we still detect the significant differences between the two populations (from randomization, P = 0.028). As before, this can be both because the genes escaping in the two populations are different and because the proportions of individuals showing escape for a given gene are different. In addition, we find evidence for five and six hypo-escapee females, in CEU and YRI, respectively, but only one (in YRI) is significant after multi-test correction (Holm’s correction). Taken together, these results suggest that there are both between-individual and between-population differences in the propensity to escape.
Table 4.
Females Differ in Their Propensity to Allow Genes to Escape Inactivation.
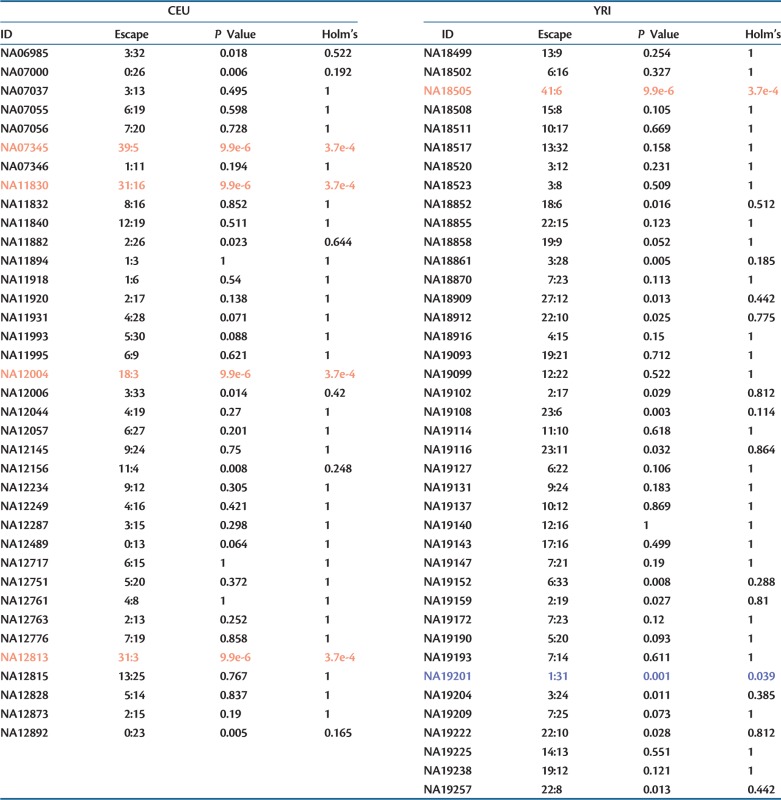
Although these results prima facie suggest that 6–17% of females are hyper-escapees and 1–14% are hypo-escapees, this analysis comes with a caveat. As the individuals differ as regards which genes are potentially informative (heterozygous at the DNA level) and genes differ as regards their propensity to escape inactivation, some of the between-individual heterogeneity may reflect differences in the set of informative genes rather than escape tendencies per se. However, if for each person we consider only those genes that are informative in other individuals, five and one incidences of hyper- and hypo-escape are still evident after Holm’s correction.
No Evidence That Permanently Escaping Genes Evolve Slowly
It has been reported that genes that always escape X-inactivation are under stronger purifying selection than either those that sometimes escape and those that never escape (Park et al. 2010), this being reflected in significantly lower _K_a/_K_s values. This was interpreted as possibly being due to differences in dominance. However, the group with the highest _K_a/_K_s were those that sometimes escape. A priori, all else being equal, from dominance arguments one would expect this class to sit between the extremes of those never and always escaping. Moreover, if most mutations are recessive, we might have expected that genes that never escape should be the ones under the stronger purifying selection as they are haploid expressed. With our new compendium of genes with replicated evidence for escape from X-inactivation, we can add to the prior data set to define new groupings of genes to examine the robustness of the prior claim.
The new merged data set comprises 446 genes (supplementary table S3, Supplementary Material online). We find evidence for heterogeneity between the three classes in _K_a/_K_s (Kruskall–Wallis test: P = 0.016). However, unlike what was previously described, when comparing between the different classes, the only robust result is that the heterogeneous group has a higher _K_a/_K_s than either those that always escape or those that never escape (fig. 2). Eliminating any genes for which _K_a/_K_s > 1 does not affect these conclusions and if anything makes the results more robust (Kruskal–Wallis test, P = 0.010; P for comparison of heterogeneous to inactive = 0.013, comparing heterogenous to escape = 0.019, and escape to inactive = 0.37). We thus cannot replicate the prior result that those genes that always escape have unusually low _K_a/_K_s. Genes that are heterogenous in expression appear to have higher _K_a/_K_s ratios.
Fig. 2.

_K_a/_K_s ratios of genes in the three X-inactivation classes in the merged data set. P values indicate significance on pairwise Mann–Whitney U tests. There are 35 that always escape, 206 always inactivated, and 205 heterogeneous (N = 446). Evolutionary rates are from the human–macaque orthologous genes with numbers taken from Ensembl or from Park et al. (2010). Outliners are not shown. Transverse lines indicate the median value.
Our data set requiring a minimum 7× coverage can legitimately report a new incidence of escape but may have a false-negative problem, i.e., genes that really do escape are categorized as not escaping just because coverage at the relevant heterozygous sites was not high enough to detect the rarely expressed allele. In this context, we would have forced some genes into the “sometimes escape” class when they should be in the “always escape” class. However, considering genes that ever escape X-inactivation as a single class (the union of sometimes and always, for which there should be no classification issue), there is no evidence that these evolve any slower than those that never escape (Mann–Whitney U test, P = 0.11) with those escaping having the higher median rate (_K_a/_K_s = 0.15 for genes that never escape and 0.22 for those that always or sometimes escape). Moreover, if the slow evolution of genes that always escape is real, then by miscalling some genes as being haploid expressed when before they were considered to be escapees, we would have moved slow evolving genes from the always escape class into the sometimes escape class. This bias would make it less likely that we would have obtained the result that the sometimes escape class are the fastest evolving. Were the sample of genes that were reclassified the faster evolving genes within the always escape class (possibly because they are low coverage hence lowly expressed and fast evolving), then this should have acted to exaggerate the slow evolution of the always escape class.
Our analysis and the prior one have a potential major artifact problem. While Park et al. (2010) compared genes that appear to show dosage compensation and those that do not, there was no quantitative control for differences in absolute expression level, the strongest predictor of rates of evolution (Pal et al. 2001; Drummond et al. 2006). If we allow for this covariate, can we recover any differences between the three classes? To address this, we reconsidered the merged data and obtained expression data from Su et al. (2004) where available (see Materials and Methods). This resulted in a data set of 262 genes (supplementary table S3, Supplementary Material online). For each gene with expression data, we calculated the mean expression level across tissues. As expected, median expression rate is a predictor of _K_a/_K_s (Spearman correlation, ρ = −0.15, P = 0.017). However, the three gene classes show no evidence of differing in their median expression level (Kruskall Wallis test, P = 0.57). In the smaller sample set for which expression data are available, the Kruskall–Wallis statistic, comparing _K_a/_K_s values between genes belonging to different X-inactivation status classes, remains significant (P = 0.03). However, after control for expression level (by considering the residuals from the loess regression of _K_a/_K_s against log[expression level]), the Kuskall–Wallis test is marginally weaker and nonsignificant (P = 0.071). This result, however, is sensitive to the exclusion of genes with _K_a/_K_s > 1 (P = 0.018).
Discussion
Our analysis increases by approximately 50% the number of genes showing evidence of escaping X-inactivation in humans. These escapees cluster with others in the domains thought to be relatively protected from the spreading of XIST. Consistent with a common finding of mental impairment in polyX individuals, there is an excess of genes associated with mental impairment among the escapees. We also found evidence for between-individual and between-population differences in the propensity to permit escape. This is consistent with the observation that polyX karyotype bearers are highly heterogeneous in presentation (Rooman et al. 2002).
The true extent of variation in escape from X-inactivation is likely to be greater than that witnessed here. For example, while we examined one high-resolution high-quality data set from one cell lineage, variation between tissues/cells within an individual (Lopes et al. 2010; Berletch et al. 2011) may also be relevant. Assuming the variation to be real, it is not unexpected that we both find new candidates and fail to replicate a few prior instances (even though we had informative samples). Indeed, it is striking that we report 62 new examples of escape, where the prior effort had information but found no evidence of escape, and only 23 examples where we could not replicate escape.
Given the ability of RNA-Seq to falsely report haploid expression (DeVeale et al. 2012), false-negative calls of haploid expression must be considered an alternative explanation for our inability to replicate some instances of escape. Similarly, as false inference of haploid expression is increasing unlikely as coverage/expression level goes up, so too we might expect that genes with haploid expression might be skewed toward the low coverage end. Indeed, the coverage of genes whose escape we can replicate (N = 38) is higher than that of genes whose escape we could not replicate (N = 23) (Mann–Whitney U test, P < 0.001). Although consistent with some of the failure to replicate being an artifact of low coverage, the same result is consistent with lower expression level owing to haploid expression. Arguing against the latter is the evidence that the genes that appear to be haploid expressed are, when analyzed across multiple tissues, no different in median expression level than those presenting evidence of escape. Some of the inability to replicate prior evidence for escape appears relatively solid as many genes appear to be haploid expressed even with >50× coverage.
While RNA-Seq artifacts (DeVeale et al. 2012) are less likely to lead to false positives, can we be confident that we have not overinterpreted the data? Our method to infer escape from X-inactivation via heterozygosity could be misleading or detecting something other than escape from X-inactivation. We showed (see Materials and Methods) that mapping errors appear not to be a serious issue with very few cases of X-linked “heterozygosity” seen in males and few instances of there being more than three alleles detected in any given female-derived cell line (and these potentially misleading SNPs being removed from analysis). However, as the analysis is done en mass (not at the single cell level), it might be that our inference of escape from X-inactivation is wrong.
A key possibility is that each cell in a given cell culture is not uniformly inactivating the same X chromosome (intra-cell lineage heterogeneity). While eliminating SNPs at lower than 10% frequency will eliminate any instances where there is rare cell lineage heterogeneity, could it be that some higher proportion of cells, at least in some samples, are inactivating the paternally derived X but the remaining cells are inactivating the maternal X? In principle, this could lead us to misclassify intra-lineage heterogeneity for escape from X-inactivation. This is a priori unlikely, not least because the silencing of X-linked genes is achieved during early embryogenesis (Brown et al. 1991; Heard and Disteche 2006), so in a given cell line we would expect only one X to be active. More importantly, the possibility of intra-cell lineage heterogeneity in the filtered data is strongly rejected on three counts. First, if a cell line is heterogeneous for which X chromosome is inactivated, we should expect that all, or nearly all, genomically heterozygous genes should show evidence of escape by our method. This we never observe (see fig. 3). Second, and related, were any cell lines heterogeneous, we would not expect to be unable to “replicate” all prior examples of escape. Third, were there heterogeneity for which X to inactivate in the cell population, we should detect escape genes all along the chromosome and not in proximity to known escapees. In contrast to this expectation, the great majority of our escapees map to the same genomic locations, ones known previously to harbor escapees and in evolutionarily modern strata, where escape is expected (Lahn and Page 1999). As we noted, several more cluster around Xic, a cluster hinted at before. For the above reasons, we can confidently reject the possibility of false attribution of escape owing to intra-cell lineage heterogeneity.
Fig. 3.

Location of escape genes and haploid expressed genes on the X chromosome of one individual of CEU (NA12004) and YRI (NA18511). Genes marked as a “potential site” are those where there is exonic heterozygozity at the DNA level and transcripts that pass the coverage threshold but that do not show evidence of escape (i.e., no evidence of biallelic expression). Those marked in blue/red show evidence of escape. The sum height of the colored bar indicates the net read depth summing over both alleles. The proportion of blue to red indicates the proportion of expression from the inactive X chromosome (blue) and the active X chromosome (we always presume the minority allele is from the inactive X chromosome). The data for the pattern of escape from the remaining individuals are shown in supplementary figure S5, Supplementary Material online.
We note that our evidence for escape does not preclude the possibility that the genes are haploid expressed in any given cell. It is possible that our escape genes are subject to allelic exclusion, permitting haploid expression in any given cell, but with the two alleles being each expressed in different cells within the cell lineage: some of the time the paternally derived allele is expressed, sometimes the maternally derived one, but not necessarily both in any given cell, at any given time. In this instance, the genes escape X-inactivation, in the sense that in some cells the genes are not subject to the usual inactivation that affects the rest of the chromosome. As these genes, although haploid expressed, are not subject to X-inactivation, we consider them a bona fide possible instance of escape. We note, however, that the inference of escape (in this and prior en mass analyses) need not imply diploid expression in any given cell. We suggest that single cell transcriptomics would be a sensible follow-up analysis, both to confirm our findings and to resolve whether escapees are subject to mono- or bi-allelic expression within any given cell.
That the prior finding of strong purifying selection on genes that always escape X-inactivation (Park et al. 2010) is not robust to addition of one set of extra data (and that from potentially more “natural” cell lines rather than inter-specific hybrids) leads us to suggest that it is better to withhold firm statements about the mode of evolution of genes in the three classes until more cell types are sampled. We do not wish to conclude that genes in the heterogeneous class are under weaker purifying selection, just that with the limited data available this is currently the best tentative conclusion. That the between-class heterogeneity is possibly sensitive to control for gene expression level provides further reason to be cautious in interpretation. We do wish to suggest that the prior claims (Park et al. 2010) for especially strong purifying selection on genes that always escape X-inactivation, and the concomitant interpretation of this in terms of dominance, should not be considered as robust. Given too that the sometimes escape class are not intermediate in their evolutionary rate between the always and never class (in neither the original nor this subsequent analysis) suggests that a simple interpretation in terms of dominance is not immediately attractive. The difference between the rate of evolution of genes that sometimes escape and those that always escape is unlikely to be owing to masking by Y-linked homologs as for both cases the presence of a Y-linked homolog is equally unlikely (supplementary table S4, Supplementary Material online). Y linked homologs are considerably more common for genes that always escape X-inactivation (supplementary table S4, Supplementary Material online).
If the genes that sometimes escape are those fastest evolving why might this be? Here we can only conjecture. Given that escape genes are strong candidates for sex-biased genes (Ellegren and Parsch 2007) and given faster evolution of sex-biased genes, differential strengths of purifying selection or positive selection associated with differential involvement in sex-biased expression would be a possibility worthy of future scrutiny. A further quandary is why it is that the X-inactivation status can be variable within a species but classes of gene appear to have characteristic evolutionary rates between species. One possibility is that the classificatory status (always escape, never escape, and sometimes escape) is relatively well conserved. Park et al. (2010) assert from unpublished work that X-inactivation status is conserved across primates. With cross-species data on X-inactivation status, this suggestion can be scrutinized further.
Materials and Methods
Data Collection
We used data generated by RNA sequencing of immortalized B-cells obtained from CEU and YRI individuals (Cheung et al. 2010). The RNA sequencing data were downloaded from the NCBI GEO database (Barrett et al. 2009) (CEU: GSE16921 and GSE25030, YRI: GSE19480). We used all of female individuals in the CEU and YRI data sets and randomly chose males as controls. Single-end sequencing data of GSE16921 and paired-end sequencing data of GSE25030 were aligned to the genome, and mapped files were combined to identify genes that escape inactivation. Samples NA10847 and NA12414 in GSE25030 were removed because the genotypes of these individuals were not available in the published version of dbSNP provided by the HapMap project. Gene and exon annotation data were obtained from the UCSC annotation database (hg19, GRCh37).
Coverage Analysis
We used the program BEDtools (Quinlan and Hall 2010) to calculate the genome-wide alignment coverage.
Mapping of the Reads to the Reference Genomes
Reads were mapped to the reference chromosomes sequence (build hg19) using Tophat (Trapnell et al. 2009). The retrieved reads were split so that they could be mapped against a collection of splice junctions, by which the RNA sequencing data can effectively be managed. We used the default settings of Tophat to analyze reads produced by the Illumina Genome Analyzer. These settings allowed no more than two mismatches on the high-quality (left) end of the reads with a sum of the Phred quality values at all mismatch positions not exceeding 70.
Heterozygous Allele Calling and Identification
We used the program SAMTOOLS (Li et al. 2009), which uses Bayesian inference to detect SNP sites in one individual. All possible bialleles at variant sites according to the reference genome were collected, whereas heterozygous sites with a QUAL value of 20 or less (Phred quality of sequencing) and a mapped depth of 6 or less were excluded from consideration.
Regarding the nonuniformity of single-end reads with different biases on the 5′- and 3′-end of fragments, we considered the regions in which the reads mapped to both the forward and reverse strands to improve the accuracy of the fragment tail determined by sequencing. The called biallelic sites that appeared only at the tail of reads and with reads mapped against only a forward or reverse strand were removed because this variant site may have been produced due to sequencing error. To improve the confidence of the heterozygosis identification, genotype data published by the International HapMap Project were used as a reference.
Strategy and Quality Control
As X-inactivation occurs early in embryogenesis (Brown et al. 1991; Heard and Disteche 2006), all cells from a given cell line derived from a postpartum subject should express only one of two alleles. This should be true regardless of whether the cell line has one or multiple founding cells, so long as all founding cells belong to the same lineage and the time to common ancestry of cells within that lineage is post the time of X-inactivation determination. Heterozygosity of X-linked markers in the transcriptome of a cell line is thus a possible indication of escape from X-inactivation. To identify genes that express both the maternal and paternal X chromosomes, we used high-throughput RNA sequencing data from normal female individuals in the CEU and YRI groups (see Materials and Methods). RNA sequencing reads were mapped against human reference genomes. The mapped reads reflected the status of expression (Wang et al. 2009). Expression from both alleles at an X-linked locus was evidenced from validated SNP sites in the mapped reads. Homozygosity in the transcriptome of genes heterozygous at the DNA level we define as genes lacking evidence for escape from X-inactivation. However, these genes could also be imprinted or subject to allelic exclusion, these both being forms of haploid expression that need not be mechanistically coupled to X-inactivation.
Although the approach is in principle straightforward, the sequencing fold-coverage and breadth-of-coverage can, however, influence the reliability and apparent extent of biallelic expression in our data. To minimize noise, information from regions with an insufficient coverage of mapped reads should be omitted. To this end, we calculated the coverage of the mapped reads based on the exons of all genes on the X chromosome (supplementary fig. S4, Supplementary Material online). The coverage of the mapped reads in YRI was slightly less than that in CEU (but not significantly so), which could impede observation of the most informative sites in YRI. However, the normalized abundance of the X chromosome and autosomes did not show a significant bias. The prior NGS study in mice (Yang et al. 2010) considered 5× coverage an adequate minimum to call escape from X-inactivation. We prefer that 7× or greater depth of coverage is the minimum level sufficient to find transcript level heterozygosis in our study, as, if a biallelic site is expressing equally from both alleles, then 7× coverage is adequate to incorrectly infer a lack of biallelic expression less than 5% of the time. Regions with lower coverage were excluded.
To avoid the identification of false-positive heterozygosis with low numbers of silenced alleles (potentially owing to cell line heterogeneity with a rare cell lineage having the opposite X-inactivation profile or owing to sequencing artifact), we required at least a 10% ratio of rare transcript variant versus common transcript variant, this being a previously employed threshold used to identify escape genes in humans (Carrel and Willard 2005). Note that rare/common here refers to the frequency of the alleles in the transcriptome of an individual not within the population. Although by this definition we exclude leaky or artifactual signals of heterozygosity, we may in turn incorrectly increase the number non-escapees (false negatives).
The variant sites in CEU and YRI obtained from dbSNP134 published by the International HapMap Project (Altshuler et al. 2010) were used as the validated variant sites to identify our heterozygous sites detected in mapped reads based on sequencing. A total of 73,792 and 89,732 X-linked SNP sites were detected in the CEU and YRI, respectively. Of these 21,087 SNPs and 26,413 are SNPs inside genes in CEU and YRI, respectively (31.24 and 37.41 SNPs per gene). However, most of these are intronic and hence of no utility for detection of escape from X-inactivation. Of the 1,001 X-linked genes (which include 823 known human protein-coding genes and 178 non-protein-coding genes [Hsu et al. 2006]), 675 and 706 X-linked genes identified in CEU and YRI, respectively, were considered to be potentially informative containing at least one well resolved exonic SNP in our sample.
Quality Control of Data: Mapping Errors are Rare
Before considering the derivation of genes potentially subject to escape from X-inactivation, as evidenced by heterozygosity in RNA-Seq samples, we investigate the quality of the data. Even with the quality control that we impose mapping errors may yet be an issue. This could be acute in the case of missing duplicate genes. Imagine we focus on an X-linked gene. Imagine too that this X-linked gene has, at least in some individuals, a paralog elsewhere in the genome but that this paralog does not feature in the reference genome. Under this circumstance, we would be forced to map the transcript from the non-focal gene back to the focal gene. If the two duplicates are allelically different, then we might incorrectly infer escape from X-inactivation. Ensuring that we employ only well-described SNPs from HapMap for the focal genes should mitigate this problem to a large degree (any random mutation in the non-focal gene we would not consider as evidence for heterozygosity) but need not necessarily eliminate it entirely. This could be considered one specific manifestation of the more general problem of incorrect mapping of RNA-Seq reads to the genome.
We can examine this problem by employing expression in male-derived cell lines as a negative control. If incorrect mapping is the issue and both the focal X-linked gene and the non-focal gene are expressed in males, then males too should appear “heterozygous” on the X chromosome. We detect very few instances (three polymorphic sites in CEU and two in YRI) of heterozygosity for X-linked genes in males suggesting that our female sample is largely free of mapping error. These sites are found in genes STS, FTX, PLXNA3, CXorf4B, and MTMR1. STS PLXNA3 and MTMR1 appeared in both of CEU and YRI and CXorf40B appeared only in YRI. Only one site shows heterozygosis in each of five males. This is most likely to be a mapping error possibly resulting from reads being derived from the undescribed areas or CNVs.
Note too that the presence of these heterozygous X-linked genes in males need not imply a mapping issue. It could be the case that there is one X-linked gene that within the cell culture has mutated and is polymorphic for a previously identified SNP (although this is unlikely to explain repeated heterozygosity). As the RNA-Seq data are from cell cultures en mass (not at the single cell), we therefore expect some low residual rate of mutationally derived heterozygosity. We removed from further analysis the sites that are heterozygous in males and could have misled analysis in females.
The robust nature of the evidence is confirmed by a further negative control. If mapping is a real problem, we should also detect X-linked loci in females with three or more alleles. We detect only 26 sites in 285 genes from 37 CEU females and only 14 sites in 510 genes from 40 YRI females with more than two alleles in a given female per X-linked gene. These sites too were removed from further analysis.
In principle, analysis of pseudoautosomal genes could provide a positive control. Unfortunately, the reference SNPs in HapMap used as the validated sites were not represented by any of 19 pseudoautosomal genes (Helena Mangs and Morris 2007), with the exception of XG; however, there was insufficient read coverage support for XG. Prior analysis of the same RNA-Seq data set has demonstrated its ability to detect autosomal heterozygosity (Cheung et al. 2010).
With the above quality controls we would, in addition, expect that signals of heterozygosity or homozygosity should be consistent between SNPs from the same gene. In both populations, we have several examples (32 and 44 genes in CEU and YRI, respectively) of instances where an individual has more than one polymorphic site in each population. Within the genes containing multiple informative sites, the majority (90.3% in CEU and 90.9% in YRI) of the RNA-Seq reads are consistent, i.e., the RNA-Seq reads were either all heterozygous or all homozygous at all potentially heterozygous sites. Many of the exceptions were instances where one site is heterozygous but the other site is not called heterozygous as the read coverage was not high enough. Considering instances where there are multiple potentially informative sites (read coverage high enough), there are 1,643 cases (genes in individuals) which have multiple potential heterozygous sites as well as sufficient read coverage. Of them, there are only 75 cases (<5%) where at least one site is not consistent with others.
Randomization to Determine Significance of Between-Population Variation in X-Inactivation
To determine whether there is between-population variation in escape tendency, in the two populations we calculated, for each gene, how many individuals have escaped inactivation (not necessarily replicated) and how many individuals could have been informative because they are heterozygous at the DNA level. The data from individuals whose genes lack coverage of sufficiently supported reads were excluded. We performed a χ2-like test using P values derived from Monte Carlo simulations. The significance test was based on the null expectation that for any given gene the proportion of escapees is identical in CEU and YRI and dependent on the amassed proportion of escapees for that gene. To this end, we took the total observed number of escapees and randomly reallocated them to the two groups as a function of the relative number of potentially informative individuals within each group. For each gene we could then calculate a χ2 value, which could be compared against the distribution from the simulations. With low sample sizes in some instances, this Monte Carlo method is preferable to derivation of P from χ2 tables. For the overall difference between the two populations, we consider the sum χ2 over all genes.
Molecular Evolutionary Rate Consideration and Merging of Data Sets
We downloaded from Ensembl a list of human macaque X-linked orthologs and associated _K_a and _K_s values. DAVID (http://david.abcc.ncifcrf.gov/conversion.jsp, last accessed September 20, 2013) was employed to convert Ensembl IDs to Refgene names. We then considered the genes that were informative in our sample (had SNPs and sufficient read coverage) and asked for how many we had rate estimation. We identified 291 such genes.
To consider the relationship between escape status and rate of evolution, we merge our data with that from the prior analysis (data from supplementary table S7, Supplementary Material online, of Park et al. [2010]). We apply the rule that if a gene has information from only one of the two data sets, then that data are preserved. If both sets agree on the status (always escape, heterogeneous, never escape), then the status is preserved. If the data sets disagree, then the gene is regarded as being in the heterogeneous class (i.e., sometimes escaping). Thus, some of the genes previously considered to always escape X-inactivation can now be considered in the sometimes escape class and some previously in the “never escape” class can also be reclassified as sometimes escape.
Rate of Gene Expression
The mean expression of 11,449 genes in 28 human tissues was derived from BioGPS, this corresponding to the data from the Affimetrix array analyzed by Su et al. (2004). We summarized GCRMA normalized probe intensity levels to Ensembl IDs corresponding to protein coding genes. All probes matching to more than one Ensembl gene ID were removed. We applied a mask to all expression values lower than the average of the expression of the negative controls in each tissue, transforming them to 0. Any gene that had expression values lower than the average of the negative controls in every tissue was removed. Expression values were then normalized against the total signal level in each tissue. Only after all the filtering did we extract only those genes that are X-linked.
Supplementary Material
Supplementary figures S1–S5 and tables S1–S4 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Supplementary Data
Acknowledgments
L.D.H. is a Wolfson Royal Society research merit Award Holder. A.O.U. is a Royal Society Dorothy Hodgkin Research Fellow. This work is supported by the National Basic Research Program of China (No. 2011CBA00400), the National Natural Science Foundation of China (No. 81030015), and the E-Institutes of Shanghai Municipal Education Commission.
References
- Altshuler DM, Gibbs RA, Peltonen L, et al. (69 co-authors) Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett T, Troup DB, Wilhite SE, et al. (14 co-authors) NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res. 2009;37:D885–D890. doi: 10.1093/nar/gkn764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmont AS, Bignone F, Ts'o PO. The relative intranuclear positions of barr bodies in XXX non-transformed human fibroblasts. Exp Cell Res. 1986;165:165–179. doi: 10.1016/0014-4827(86)90541-0. [DOI] [PubMed] [Google Scholar]
- Berletch J, Yang F, Xu J, Carrel L, Disteche C. Genes that escape from X inactivation. Hum Genet. 2011;130:237–245. doi: 10.1007/s00439-011-1011-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CJ, Ballabio A, Rupert JL, Lafreniere RG, Grompe M, Tonlorenzi R, Willard HF. A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature. 1991;349:38–44. doi: 10.1038/349038a0. [DOI] [PubMed] [Google Scholar]
- Brown CJ, Greally JM. A stain upon the silence: genes escaping X inactivation. Trends Genet. 2003;19:432–438. doi: 10.1016/S0168-9525(03)00177-X. [DOI] [PubMed] [Google Scholar]
- Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434:400–404. doi: 10.1038/nature03479. [DOI] [PubMed] [Google Scholar]
- Cheung VG, Nayak RR, Wang IX, Elwyn S, Cousins SM, Morley M, Spielman RS. Polymorphic cis- and trans-regulation of human gene expression. PLoS Biol. 2010;8:e1000480. doi: 10.1371/journal.pbio.1000480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng HW, Xu FH, Liu YZ, et al. (11 co-athors) A whole-genome linkage scan suggests several genomic regions potentially containing QTLs underlying the variation of stature. Am J Med Genet. 2002;113:29–39. doi: 10.1002/ajmg.10742. [DOI] [PubMed] [Google Scholar]
- DeVeale B, van der Kooy D, Babak T. Critical evaluation of imprinted gene expression by RNA-Seq: a new perspective. PLoS Genet. 2012;8:e1002600. doi: 10.1371/journal.pgen.1002600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disteche CM. Escapees on the X chromosome. Proc Natl Acad Sci U S A. 1999;96:14180–14182. doi: 10.1073/pnas.96.25.14180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disteche CM, Filippova GN, Tsuchiya KD. Escape from X inactivation. Cytogenet. Genome Res. 2002;99:36–43. doi: 10.1159/000071572. [DOI] [PubMed] [Google Scholar]
- Drummond DA, Raval A, Wilke CO. A single determinant dominates the rate of yeast protein evolution. Mol Biol Evol. 2006;23:327–337. doi: 10.1093/molbev/msj038. [DOI] [PubMed] [Google Scholar]
- Ellegren H, Parsch J. The evolution of sex-biased genes and sex-biased gene expression. Nat Rev Genet. 2007;8:689–698. doi: 10.1038/nrg2167. [DOI] [PubMed] [Google Scholar]
- Gecz J, Shoubridge C, Corbett M. The genetic landscape of intellectual disability arising from chromosome X. Trends Genet. 2009;25:308–316. doi: 10.1016/j.tig.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Gustavson KH. Triple X syndrome deviation with mild symptoms. The majority goes undiagnosed. Lakartidningen. 1999;96:5646–5647. [PubMed] [Google Scholar]
- Heard E, Disteche CM. Dosage compensation in mammals: fine-tuning the expression of the X chromosome. Genes Dev. 2006;20:1848–1867. doi: 10.1101/gad.1422906. [DOI] [PubMed] [Google Scholar]
- Helena Mangs A, Morris BJ. The human pseudoautosomal region (PAR): origin, function and future. Curr Genomics. 2007;8:129–136. doi: 10.2174/138920207780368141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu F, Kent WJ, Clawson H, Kuhn RM, Diekhans M, Haussler D. The UCSC known genes. Bioinformatics. 2006;22:1036–1046. doi: 10.1093/bioinformatics/btl048. [DOI] [PubMed] [Google Scholar]
- Kelkar A, Thakur V, Ramaswamy R, Deobagkar D. Characterisation of inactivation domains and evolutionary strata in human X chromosome through Markov segmentation. PLoS One. 2009;4:e7885. doi: 10.1371/journal.pone.0007885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahn BT, Page DC. Four evolutionary strata on the human X chromosome. Science. 1999;286:964–967. doi: 10.1126/science.286.5441.964. [DOI] [PubMed] [Google Scholar]
- Lai Y. A moment-based method for estimating the proportion of true null hypotheses and its application to microarray gene expression data. Biostatistics. 2007;8:744–755. doi: 10.1093/biostatistics/kxm002. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden M, Bender B, Robinson A. Sex chromosome tetrasomy and pentasomy. Pediatrics. 1995;96:672–682. [PubMed] [Google Scholar]
- Liu Y-Z, Xiao P, Guo YF, et al. (13 co-athors) Genetic linkage of human height is confirmed to 9q22 and Xq24. Hum Genet. 2006;119:295–304. doi: 10.1007/s00439-006-0136-y. [DOI] [PubMed] [Google Scholar]
- Liu YZ, Xu FH, Shen H, et al. (15 co-authors) Genetic dissection of human stature in a large sample of multiplex pedigrees. Ann Hum Genet. 2004;68:472–488. doi: 10.1046/j.1529-8817.2003.00117.x. [DOI] [PubMed] [Google Scholar]
- Lopes A, Burgoyne P, Ojarikre A, Bauer J, Sargent C, Amorim A, Affara N. Transcriptional changes in response to X chromosome dosage in the mouse: implications for X inactivation and the molecular basis of Turner Syndrome. BMC Genomics. 2010;11:82. doi: 10.1186/1471-2164-11-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon MF. Gene action in the X-chromosome of the mouse (Mus musculus L.) Nature. 1961;190:372–373. doi: 10.1038/190372a0. [DOI] [PubMed] [Google Scholar]
- Otter M, Schrander-Stumpel CT, Curfs LM. Triple X syndrome: a review of the literature. Eur J Hum Genet. 2010;18:265–271. doi: 10.1038/ejhg.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal C, Papp B, Hurst LD. Highly expressed genes in yeast evolve slowly. Genetics. 2001;158:927–931. doi: 10.1093/genetics/158.2.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C, Carrel L, Makova KD. Strong purifying selection at genes escaping X chromosome inactivation. Mol Biol Evol. 2010;27:2446–2450. doi: 10.1093/molbev/msq143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao E, Weiss B, Fukami M, et al. (17 co-athors) Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat Genet. 1997;16:54–63. doi: 10.1038/ng0597-54. [DOI] [PubMed] [Google Scholar]
- Rooman RP, Van Driessche K, Du Caju MV. Growth and ovarian function in girls with 48,XXXX karyotype—patient report and review of the literature. J Pediatr Endocrinol Metab. 2002;15:1051–1055. doi: 10.1515/jpem.2002.15.7.1051. [DOI] [PubMed] [Google Scholar]
- Stevenson RE, Schwartz CE. X-linked intellectual disability: unique vulnerability of the male genome. Dev Disabil Res Rev. 2009;15:361–368. doi: 10.1002/ddrr.81. [DOI] [PubMed] [Google Scholar]
- Su AI, Wiltshire T, Batalov S, et al. (13 co-authors) A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci U S A. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia N, Howell S, Sutherland A, Wilson R, Wilson L. A review of trisomy X (47,XXX) Orphanet J Rare Dis. 2010;5:8. doi: 10.1186/1750-1172-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya KD, Greally JM, Yi Y, Noel KP, Truong JP, Disteche CM. Comparative sequence and X-inactivation analyses of a domain of escape in human xp11.2 and the conserved segment in mouse. Genome Res. 2004;14:1275–1284. doi: 10.1101/gr.2575904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visscher PM, Macgregor S, Benyamin B, et al. (14 co-authors) Genome partitioning of genetic variation for height from 11,214 sibling pairs. Am J Hum Genet. 2007;81:1104–1110. doi: 10.1086/522934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Babak T, Shendure J, Disteche CM. Global survey of escape from X inactivation by RNA-sequencing in mouse. Genome Res. 2010;20:614–622. doi: 10.1101/gr.103200.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data