Phosphatidylserine vesicles enable efficient en bloc transmission of multiple enteroviruses (original) (raw)
. Author manuscript; available in PMC: 2019 Aug 21.
Abstract
A central paradigm within virology is that each viral particle largely behaves as an independent infectious unit. Here we demonstrate that clusters of enteroviral particles are packaged within phosphatidylserine (PS) lipid-enriched vesicles that are non-lytically released from cells and provide greater infection efficiency than free single viral particles. We show that vesicular PS lipids are co-factors to the relevant enterovirus receptors in mediating subsequent infectivity and transmission, in particular to primary human macrophages. We demonstrate that clustered packaging of viral particles within vesicles enables multiple viral RNA genomes to be collectively transferred into single cells. This study reveals a novel mode of viral transmission, where enteroviral genomes are transmitted from cell-to-cell en bloc in membrane-bound PS vesicles instead of single independent genomes. This has implications for facilitating genetic cooperativity among viral quasispecies as well as enhancing viral replication.
Graphical abstract
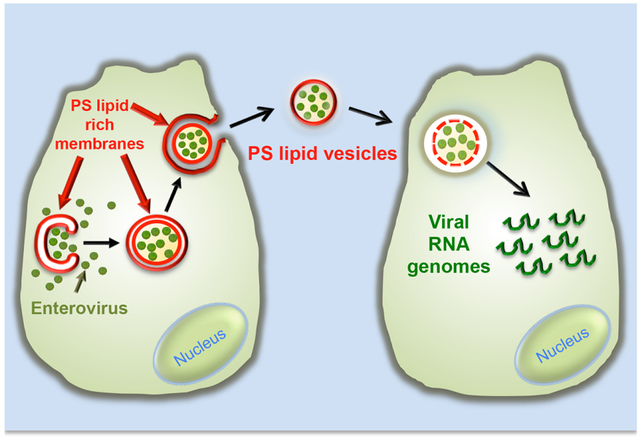
Introduction
Enteroviruses are a large genus of single positive strand RNA viruses whose members including Poliovirus (PV), Coxsackievirus, Rhinovirus, Enterovirus 68 are the causative agents of a number of important and widespread human diseases including poliomyelitis, myocarditis, hand foot and mouth disease, the common cold and more recently a severe respiratory disease with paralytic symptoms. In addition to greater than 70 enteroviral serotypes identified in humans, enteroviral quasispecies are common largely as a result of inherent error making and lack of proofreading mechanisms of viral RNA dependent RNA polymerases (RdRp).
Enteroviral RNA genomes serve as templates for both translation and replication and these processes take place on host intracellular membranes (de Boon and Ahlquist, 2010; Hsu et al., 2010). After enteroviruses have bound their specific host receptors either at the cell surface or within endocytic vesicles (Brandenburg et al., 2007), the capsid undergoes a conformational change that allows the viral RNA to be transferred across the endosomal membrane into the cytoplasm through a yet completely defined mechanism (Strauss et al., 2013). In the cytoplasm the enteroviral RNA is first translated into non-structural proteins and structural proteins, where the former makes up the RNA genome replication machinery and the latter the nucleocapsid. The viral RNA replication machinery are then assembled on the cytoplasmic membrane leaflet of ER derived membranes which are subsequently modified by viral and host proteins to have a specific lipid blueprint of enrichment in phosphatidylinositol-4-phosphate and cholesterol lipids. These lipids regulate the membrane association, assembly and activity of the viral replication protein complex, including the RdRp, and thus facilitate viral RNA synthesis (Hsu et al., 2010; Ilnytska et al., 2013; Nchoutmboube et al., 2013).
Once the enteroviral RNA is synthesized, little is known about where in the host cell it is packaged in capsids and how these capsids are released from cells. While enteroviruses have historically been considered non-enveloped (i.e. lacking a host-derived membrane bilayer around their capsids) and thus rely on cell lysis to exit, a recent report of extracellular Coxsackievirus B3 (CVB3) being present in vesicles (Robinson et al., 2014) and PV being able to spread non-lytically among host cells (Bird et al., 2014) have raised important questions regarding the extracellular nature of enteroviral particles and the significance of non-lytic exit in the viral lifecycle. Moreover Hepatitis A virus, another plus strand RNA virus long considered to be non-enveloped has been reported to be surrounded by a membrane (Feng et al., 2013).
A central paradigm in virology is that viruses behave as independent infectious units. While there are exceptions to this, such as Vaccinia virus particles preventing superinfection by inducing the host cell to repel other virions (Doceul et al., 2010), it is largely accepted that the fate of individual viral genomes are not dependent on one another during exit from one cell and entry into another (Brandenburg and Zhuang 2007). Here we investigate the assembly, exit and subsequent infection processes of enteroviral particles using a combination of imaging techniques including confocal microscopy, super-resolution light microscopy, correlative light electron microscopy along with single molecule RNA fluorescence in situ hybridization (FISH), proteomic and biochemical approaches. We show that infectious enteroviral particles are clustered within phosphatidylserine (PS) lipid enriched vesicles and non-lytically secreted out of cells. These viral particles in vesicles are more efficient in establishing infection than free viral particles. We demonstrate that vesicles encapsulate and traffic large numbers of mature infectious viral particles between cells and consequently enable the transfer of multiple viral RNA genomes collectively into new host cells by a mechanism that is dependent on both the virus specific receptor at the host cell as well as the vesicular PS lipids.
Results
Assembled Poliovirus capsids are localized to viral RNA replication organelles.
We first investigated the intracellular spatio-temporal dynamics of newly synthesized PV particles. The generation of PV particles, as well as enterovirus assembly in general, comprises a multistep process where capsid subunits (VP0, VP1, VP3) form pentamers, which polymerize into capsids (Liu et al., 2010). Once RNA is packaged, the VP0 subunits get cleaved into VP2 and VP4 to generate mature infectious virions (Liu et al., 2010). From screening a large number of neutralizing antibodies, we identified the A12 antibody, that binds deep inside the canyon bridging both rims of two adjacent pentamers and thus recognizing assembled nucleocapsids (Chen et al., 2011; Chen et al., 2013), to visualize PV capsids within infected cells. We fixed cells at various intervals after PV infection and co-immunolabeled with A12, anti-viral VP1 to detect individual VP1 subunits and anti-viral 3AB antibodies, the latter to detect viral 3AB to localize replication organelles where viral RNA is synthesized (Hsu et al., 2010). Newly assembled capsids were clearly detectable from 3-4 hr p.i. (post-infection) and onward and they were localized to the replication organelles (Figure 1A). Note that in contrast to A12 labeling, VP1 was localized to both the replication organelles and dispersed across the cytoplasm, consistent with its cytosolic nature. At 6-7hr p.i., capsids were dispersed from the replication sites to the cytoplasm and sequestered in puncta (Figure 1A, 6-7hr p.i. inset). At this time there is a known cessation in viral RNA synthesis (Ehrenfeld et al., 1970) and the timing of capsid release from replication organelles was coupled to viral RNA synthesis since prematurely inhibiting viral RNA synthesis with the inhibitor Guanidine HCl (Barton and Flanegan, 1977) triggered capsids to rapidly disperse into the cytoplasm (Figure S1A).
Figure 1. Poliovirus capsids are captured by phosphatidylserine lipid enriched autophagosome-like organelles and released non-lytically from cells.

(A) Capsids undergo dynamic spatial transitions during infection. HeLa cells infected with PV and immunolabeled with A12, anti-VP1 and anti-3AB antibodies. Scale bars, 5μm.
(B) Capsids (A12) colocalized with autophagosome marker LC3-II. PV Infected HeLa cells were immunolabeled with A12 and anti-LC3-II antibodies. Scale bar 5μm.
(C) Electron micrographs of PV infected cells show PV capsids in double-membrane autophagosome-like organelles. Scale bars, 5μm and 200nm (Inset).
(D) PV infected cells at 4hr p.i. expressing GFP-LactC2 and FAPP1mRFP imaged by structured illumination microscopy. Region of interest is magnified in right panel. Scale bar 5μm.
(E) PV infected cells at 7hr p.i. expressing GFP-LactC2 were immunolabeled with anti-GFP and A12 antibodies. Region of interest is magnified in right panel. Arrows indicate A12 positive autophagosome-like organelles co labeled with GFP-LactC2. Scale bar 5μm.
(F) Cells co-expressing GFP-LactC2 and LC3-RFP were infected with PV and imaged by confocal microscopy at 7hr pi. Region of interest is magnified in right panel. Scale bar 5μm.
(G) Capsid distribution between 7 and 8hr p.i. PV infected cells were immunolabeled with A12, anti-VP1 and anti-3AB antibodies. Scale bar 5μm.
(H) Plasma membrane integrity remains intact when PV exits cells. Trypan Blue diffusion across the plasma membrane was measured concurrently with sampling of extracellular medium for PV particles.
Poliovirus capsids are captured by phosphatidylserine lipid enriched autophagosomelike organelles and released non-lytically from cells.
Between 6 and 7hr p.i., we found that >85% of capsids (n=10 cells) were on punctate cytoplasmic structures that colocalized with the autophagosomal membrane protein, LC3-II (Figure 1B). By transmission electron microscopy we observed numerous double-membraned autophagosome-like organelles containing capsids (Figure 1C). Previous reports (Taylor et al., 2009; Kirkegaard and Jackson, 2005; Jackson et al., 2005) had found that perturbation of the host autophagy pathway led to a decrease in PV release from infected cells. Consistent with that, either disrupting autophagy by siRNA depletion of autophagy machinery LC3 or beclin 1, or acutely stimulating autophagy by treating cells with tat-beclin 1 peptides, blocked or enhanced PV release by ~ 10 fold respectively (Figure S1B and S1C). However these capsid containing autophagosome-like organelles did not follow the conventional autophagy pathway and fuse their contents with lysosomes as inhibiting lysosomal enzymes did not further increase LC3-II levels beyond the 4-fold increase observed in PV infected cells (Supplementary Figure 1D and 1E) and none of the A12/LC3-II co-labeled structures contained lysosomal enzymes at any point during infection (Figure S1F and S1G). Notably the SNARE protein Syntaxin 17, normally localized to autophagosomes and required for fusion with lysosomes (Itakura et al., 2012), did not localize to these A12/LC3-II co-labeled structures (Figure S1H).
However we found that the membranes of both the replication organelles and the autophagosome-like organelles contained negatively charged phosphatidylserine (PS) lipids. PS lipids in uninfected cells are primarily located at the cytoplasmic leaflets of the plasma and endosomal membranes as well as at the lumenal leaflet of the endoplasmic reticulum membrane (Leventis and Grinstein, 2010; Kay et al., 2012). Cells were co-transfected with GFP-LactC2 and FAPP1mRFP, cytosolic live cell reporters for PS (Kay et al., 2012) and PI4P lipids respectively, the latter to report on the location of the ER-derived replication organelle membranes (Hsu et al., 2010). At 4hr p.i., cells were imaged live by structured illumination microscopy. Highly localized PS-rich membrane domains exposed to the cytoplasm were found distributed across the replication organelles (Figure 1D, arrows). Later between 6 and 7hr p.i., cells were fixed and immunolabeled with anti-GFP and A12 antibodies and imaged by confocal microscopy. Numerous A12 positive structures were found also co-labeled with GFP-LactC2 indicating the presence of PS lipids on their membrane leaflets exposed to the cytoplasm (Figure 1E, arrows). Consistent with this, in PV infected live cells co-expressing LC3-mRFP and GFP-LactC2, >90% (organelles measured across 10 cells) of the LC3-mRFP labeled autophagosome-like organelles were co-labeled with GFP-LactC2 (Figure 1F, arrows).
We then investigated the fate of these capsid-containing organelles during the rest of the infection time period. Between 7hr and 8hr p.i., there was a 70%±10% (n=15) decrease in the number of capsids within the cytoplasm (Figure 1G). Using the cell impermeable Trypan blue dye, we found that the PM remained intact during this time while there was an ~6-fold increase in extracellular viral titers (Figure 1H). This lack of plasma membrane permeability during PV infection had also been previously observed (Taylor et al., 2009; Bird et al., 2014). Although by 12hr p.i. cells eventually lysed (not shown), this data indicated that the majority of PV particles were released prior to cell lysis.
Extracellular PV particles are found in uniformly large vesicles.
Scanning electron microscopy (SEM) of 7hr p.i. cells revealed numerous vesicular structures of similar size docked at the extracellular side of the intact plasma membrane (Figure 2A and inset). While these SEM images may not reflect the true shape and size of these vesicles within live cells, they do point to a striking uniformity in size. Measurements of the cross-section diameter from 100 randomly selected extracellular vesicles from 4 different cells, showed that ~90% of the vesicles were between 250nm and 350nm in diameter (Figure 2B). Correlative fluorescence imaging in conjunction with SEM confirmed that these vesicles contained A12 labeled capsids (Figure 2C).
Figure 2. Extracellular PV particles are found in large uniform sized vesicles.

(A) Scanning electron microscopy of a PV infected cell at 7hr p.i.. Scale bar 3um. Inset shows higher magnification of uniform size vesicles docked on the extracellular side of the plasma membrane. Scale bar 1um.
(B) Extracellular vesicle size distribution in PV infected cells. Cross section diameter of a 100 randomly selected extracellular vesicles from 4 different cells, were measured from scanning electron micrographs and plotted. Data represented as mean ± SEM.
(C) Correlative fluorescence and Scanning Electron Microscopy (SEM). PV infected cell was immunolabeled with A12 at 7hr pi, epifluorescence image was obtained (right) and then sample was processed for SEM (left). Arrows point to A12 labeled extracellular vesicles. Scale bar 1μm.
Mature enteroviral particles are released in PS enriched vesicles.
We next investigated whether the extracellular vesicles containing PV retained the PS lipids that had been components of the autophagosome-like organelles (Figures 1D-1F). Incubation of PV infected cells with Alexa 568 coupled Annexin V, a non-cell permeable fluorescent reporter protein for PS lipids (Koopman et al., 1994) revealed numerous fluorescent puncta dotting the surface of the cell at 7hr p.i. (Figure 3A). Similar results were also observed with CVB3 (at 7hr p.i.) and human Rhinovirus infections (at 12hr p.i.) (Figure 3A). Note that this pattern of Annexin V labeling of enterovirus-infected cells was different from both apoptotic and mock infected cells: In the former, the entire plasma membrane was labeled with Annexin V as a result of PS lipids being on the extracellular membrane leaflet of the cell, a hallmark of apoptosis (Figure 3B, apoptosis) while in the latter there was no labeling since PS lipids were on the cytosolic leaflet of the plasma membrane (Figure 3B, mock).
Figure 3. Mature PV, CVB3 and Rhinovirus particles are released in phosphatidylserine lipid vesicles.

(A) Cells infected with PV, CVB3 or Rhinovirus were incubated with Alexa568-annexin V and imaged by confocal/DIC at 7hrs pi. Scale bar 5μm for PV and Rhinovirus, 10μm for CVB3.
(B) Mock and Apoptotic HeLa cells labeled with Alexa568-Annexin V. Scale bar 10μm.
(C) Dynamics of Alexa568-annexin V labeled PS vesicle release from PM projections of PV infected cells at 7hr p.i.
(D) Quantification of PS vesicles released from enterovirus infected cells.
(E) VP2/VP0 ratio of whole cell lysate and PS vesicles in PV infected cells. Isolated PS vesicles from PV infected cells at 7hr p.i. were analyzed by SDS-PAGE/Western with anti-PV VP2 antibody.
(F) Isolated PS vesicles from human Rhinovirus infected cells contain mature Rhinoviral particles. Isolated PS vesicles were processed for SDS-PAGE/Western analysis with anti-HRV2/VP2 (neutralizing) antibodies.
We performed time-lapse confocal/DIC imaging on PV infected cells in the presence of Alexa568-Annexin V to determine if these vesicles were being released. We observed Annexin V labeled vesicles emerging at the cell surface at discrete domains and being rapidly released into the extracellular medium (Figure 3C; arrow; Movie 1). We then quantified the amount of PS vesicles released during PV, CVB3 or Rhinovirus infection relative to mock-infected cells (for each respective virus). We collected the extracellular medium, removed large cell debris and enriched for vesicles of size range 100nm-500nm using differential centrifugation. This size range was chosen based on our previous light and EM data (Figure 2). The enrichment for vesicles of this size range was confirmed by transmission electron microscopy (Figure S2A). The vesicles were incubated with Alexa 565-Annexin V and following the wash to remove any unbound Annexin V were placed in a spectrofluorometer. Fluorescence measurements revealed a net ~9-fold, ~3-fold and ~3-fold increase in amounts of PS vesicles collected from the supernatants of PV, CVB3 and Rhinovirus infected cells respectively compared to mock-infected cells (Figure 3D).
We subsequently enriched for PS vesicles from the collected vesicle fraction using magnetic separation with Annexin V conjugated magnetic microbeads. In parallel, vesicles were also incubated with magnetic microbeads lacking Annexin V to control for nonspecific binding. Post magnetic separation, the samples were processed for SDS-PAGE/Western analysis. We found that the extracellular vesicles pre and post Annexin V isolation, had a VP2/VP0 ratio ~ 2-fold greater than the whole cell lysate (Figure 3E). This indicated that these extracellular released PS vesicles were not non-specific shedding of host membrane but rather conduits for the selective release of mature PV particles. Similar results were also obtained from human Rhinovirus 2 infected cells where PS vesicles were found to be containing mature viral particles (Figure 3F).
Infection by PV particles in vesicles is dependent on both the virus specific receptor and the PS lipids.
We next compared the infection capability of PV particles within vesicles released from cells compared to free viral particles. Free viral particles were obtained by 3-cycles of quick freeze-thaw of the vesicle fraction. Freeze-thaw does not significantly impact PV infectivity (Strazynski et al., 2002), and no difference in infectivity was observed by plaque assay between equivalent numbers of PV particles isolated by freeze thaw or collection from the supernatant of post vesicle enrichment fractions (data not shown). The collected extracellular vesicles from PV infected cells or free PV particles were then incubated with a confluent layer of HeLa cells (Figure 4A). After 4 hours of infection with either vesicles or free viral particles, numerous PV replicating (based on immunofluorescence labeling of VP1) infected cells were found in both cell populations (Figure 4A).
Figure 4. Infection by PV particles in vesicles is more efficient than free viral particles and is dependent on both the Poliovirus receptor and PS lipids.

(A) Free or vesicle associated PV particles were incubated with new recipient cells and replication was detected at 4hr p.i., by immunolabeling with anti-VP1 antibodies. Scale bar 500μm.
(B) CD155/PVR neutralizing antibodies block infection PV particles in vesicles.
(C) Blocking PS lipids on vesicles containing PV particles block infection. Vesicles collected by differential centrifugation were incubated with different amounts of Annexin V protein prior to incubation with recipient HeLa cells. Replication was measured at 4hr p.i., by SDS-PAGE/Western analysis with anti-3AB antibody.
(D) Quantification of Western results in (C).
(E) HeLa cells were incubated with equal titers of free and vesicle associated PV particles. Infection efficiency was determined by quantifying viral 3AB protein levels at peak replication time (4hr p.i.). Data represented as mean ± SD.
(F) Primary human macrophages were incubated with equal titers of free and vesicle associated PV particles. Infection efficiency was determined by quantifying viral 3AB protein levels at peak replication time (8hr p.i.). Data represented as mean ± SD.
To determine if the PV particles within vesicles still required the PV receptor CD155 on the host cell for infection, cells were incubated with CD155 neutralizing antibodies prior to exposure to vesicles and viral 3AB replication protein levels were measured after 4 hours of infection. In the presence of neutralizing antibodies, vesicle infectivity was inhibited by >95% indicating that PV vesicles were not just simply “fusing” with cells but that the viral particles within the vesicles still required binding to PV receptor for transfer of viral RNA into the host cell cytoplasm (Figure 4B).
We next determined if the infection was dependent on PS lipids of the vesicles. Vesicles isolated by differential centrifugation were incubated with different amounts of Annexin V protein, which binds and masks the PS head-groups on the lipids (Swairjo et al., 1995). After removing any unbound Annexin V, the vesicles were added to HeLa cells and replication measured after 4 hours. Strikingly, masking of the vesicle-associated PS lipids by Annexin V, inhibited PV infection of the host cells, in a dose-dependent manner (Figures 4C and 4D). These data indicated that PS lipids are cofactors for PV infection and that mature infectious PV particles are predominantly in the PS vesicle fraction of vesicles collected by differential centrifugation. These findings also suggest a possible mechanism of infection where PS lipids enable binding and endoyctic uptake of the vesicle followed by lysis/permeabilization of the vesicular membrane to allow release and binding of PV particles to the PV receptor.
Infection by PV particles in vesicles is more efficient than free PV particles.
We next measured and compared the level of infection of host cells when infected with equivalent numbers of PV particles either in vesicles or as free. As a measure of infection efficiency we quantified and plotted the levels of viral 3AB replication proteins at peak replication times (Figure 4E). Due to viral RNA synthesis feeding back on viral RNA translation (and vice versa), 3AB levels reflect viral RNA levels (Hsu et al., 2010). We found that at 4hr p.i. viral 3AB levels were ~40% greater in cells incubated with PV particles in vesicles than free PV particles (Figure 4E). This difference was even more striking when primary human macrophages, cells that are specialized to recognize and take up PS lipid containing cells and vesicles (Fadok et al., 1992), were used as the recipient host. Here vesicle-enclosed PV particles were almost 2-fold greater in infection efficiency compared to free PV particles (Figure 4F).
Unilamellar PS vesicles released from PV infected cells contain multiple viral particles.
To quantify the clustering of viral particles within vesicles, we collected free and vesicle associated PV particles and immunolabeled them with A12 antibodies. We then imaged the coverglass immobilized PV particles by total-internal reflection fluorescence (TIRF) and by Differential intensity Contrast (DIC) microscopy (Figure 5A). Viruses were deposited on coverslip in two formats: vesicle-free fraction (“Free”), or vesicle-embedded fraction (“Vesicular”). Imaging revealed cluster of viruses within vesicles, while free viruses yielded a more diffuse distribution (Figure 5A). We quantified the difference in distribution of measured fluorescence by computing the radial autocorrelation functions g(r) for the intensity map I(u→):g(r)=〈∮‖v→‖=rI(u→)I(u→+v→)du→〉u→.
Figure 5. PS vesicles contain clustered PV particles, which enable multiple viral RNA genomes to be collectively transferred into a new host cell.

(A) TIRF and DIC images of free and vesicle associated PV particles labeled with Atto488 labeled A12 antibody.
(B) Difference in distribution of fluorescence in (A) by computation of the radial autocorrelation function g(r).
(C) dSTORM imaging free and vesicle associated viral particles labeled with Atto 488-A12 antibody.
(D) Calculation of Ripleys K function to assess the degree of clustering of vesicle associated PV particles relative to free particles. Data represented as mean ± SEM.
(E) PS vesicles isolated from PV infected cells using Annexin V microbeads were imaged by TEM. Note the numerous electron dense viral particles in each vesicle and the unilamellar surrounding membrane. Asterisk shows annexin V microbead attached on the exterior of one vesicle. Scale Bars 100 nm.
(F) Collected intact vesicles or free viral particles from PV infected cells were incubated with a confluent layer of new host cells at different viral titers. Viral RNAs were monitored by single molecule RNA FISH and imaged by dual confocal/DIC microscopy at 1.5hr p.i. Shown are images of single HeLa cells infected with either free viral particles or vesicle associated viral particles. Images presented were acquired with the same microscopy settings. Scale bar 2μm.
(G) Collected vesicles were incubated with cells with/without GuHCL for 1.5hr and viral RNA molecules were monitored by single molecule RNA FISH (see also Figure S3 for quantification). Scale bar 2μm.
(H) Quantification of the number of viral RNA molecules per cell in cells infected with either free (n=55 cells) or vesicle associated PV particles (n=55 cells). (***: p<2.10−3; ****: p<10−4)
(I) Percent of cells with 15 or greater PV RNA puncta was quantified for cells infected with either free or vesicle-associated PV particles for each viral titer shown. Data represented as mean ± SEM.
The observed exponential decay of these autocorrelation functions enabled us to quantify the length scales that characterize the imaged aggregates (Figure 5B). We found that vesicle-embedded viruses yielded a characteristic clustering scale of 2.0±0.1μm (n=4 images), while free virus yielded a five-time smaller characteristic scale of 0.4±0.1 μm (n=2 images). However, these estimates are too close to the spatial resolution of TIRF imaging and thus only qualitative. To circumvent this diffraction limit, we then applied the direct stochastic optical reconstruction (dSTORM) methodology to achieve super-resolution of these viral particles (Figure 5C). dSTORM relies on sequential imaging and fitting to achieve a spatial resolution of individual dyes within 30nm (Bates et al., 2007; Baddeley et al., 2009).
In order to assess the degree of clustering within these viral spreads we used the Ripley’s K statistical test. By definition,
K(r)=An2∑i≠jδ(∕rdij),withδ(x)={1forx≤10forx>0}
with dij being the distance between the _i_th and _j_th points, A is the image area. By definition, ∕πr2K(r)is around 1 for a homogenous distribution of points, and larger than 1 for clustered spatial distribution (Veatch et al., 2012; Termini et al., 2014). Based on the electron microscopy images (Figures 2A and 2B; Figure S2A), we anticipated clusters of viruses within vesicles of 200nm-400nm diameter. Hence, we calculated K(r=200nm) from the dSTORM data to test whether vesicles contained clusters of viruses: we found that K(r=200nm)=2.4±0.9 for free viruses and K(r=200nm)=13.7±6.7 for viruses within vesicles (Figure 5D). Hence, super-resolution microscopy does confirm that these viral vesicles do pack large numbers of viruses.
Consistent with these findings, electron micrographs of PS vesicles (after isolation by Annexin V coupled microbeads) revealed multiple clustered viral particles surrounded by a single membrane bilayer (Figure 5E). Per 200nm cross-section, a single vesicle contained on average 19 ± 3 PV particles (n=6 vesicles).
The presence of a single bilayer, as opposed to multiple bilayers also indicated that the double-membraned autophagosome-like organelles had fused with the plasma membrane, rather than budded, in order to release the PS vesicles. Thus, the PS lipids on the extracellular membrane leaflet of the vesicle are in a compartment that is topologically equivalent to the lumenal membrane leaflet of the double-membraned autophagosome-like organelle. The ER is a major membrane source for autophagosomes (Hanasaki et al., 2013), and has luminal membrane leaflets enriched in PS lipids (Kay et al., 2010). Given that the isolated PS vesicles also contain ER resident proteins (Figure S2B), has luminal membrane leaflets enriched in PS lipids (Kay et al., 2010) it is highly likely that the autophagosome-like organelles and thereby the released PS vesicles originated from ER and/or ER derived replication organelle membranes.
Vesicles allow multiple viral RNA molecules to be collectively transferred into cells.
Given our dSTORM and TEM findings we conjectured that multiple PV particles within a single vesicle might allow multiple viral genomes to be simultaneously transferred into a single cell. To test this hypothesis we performed single molecule RNA fluorescence in situ hybridization (single molecule RNA FISH) (Raj et al., 2008). Multiple oligonucleotide fluorescent probes hybridizing to an RNA molecule allow sufficient sensitivity to detect single RNA molecules within cells (Shaffer et al., 2013; Lubeck and Cai 2012).
Forty-eight fluorescently labeled nucleic acid probes, each 22 nucleotides in length, complementary to the PV genome, were synthesized. Fixed numbers of HeLa cells were incubated with different titers of either free PV particles or vesicle associated PV particles for 1.5hrs and processed for FISH labeling as described previously (Shaffer et al., 2013) (Figure 5F). Note that we could exclude the possibility of any viral RNA synthesis occurring during this incubation time because cells with and without Guanidine HCL, an inhibitor of viral RNA synthesis, showed similar amounts of viral RNA molecules per cell (Figure 5G and Figure S3).
For cells incubated with free PV particles, discrete fluorescent puncta, spatially segregated from one another in the cytoplasm were detected in individual cells (Figure 5F, free virus). In contrast for cells incubated with equivalent titers of PV particles in vesicles, there were many more fluorescent puncta, spatially juxtaposed (Figure 5F, virus in vesicle). The lowest multiplicity of infection with free viral particles was observed at 1.5 ×107 pfu/ml where there was on average only 1 fluorescent puncta per cell (Figure 5F and 5H, free virus). Hence the size of these puncta was approximated as a single viral RNA molecule and used in subsequent quantification and analysis of FISH data. At each titer, for either free virus or vesicle-associated virus, 55 cells were randomly chosen and viral RNA molecules counted in each cell. The number of viral RNA molecules per cell were subsequently plotted in Figure 5H.
From our quantification we found that at 12 × 107 pfu/ml and 6 × 107 pfu/ml, there were ~ 40% and ~75% respectively more viral RNA molecules per cell when PV particles were presented in vesicles than as free virus (Figure 5H). Indeed for cells incubated with free PV particles, we found that as the viral titer decreased 4-fold (from 12 × 107 pfu/ml to 3 × 107 pfu/ml), there was a ~ 90% decrease in the number of PV RNA molecules per cell whereas for cells incubated with PV particles in vesicles, this decrease was significantly less, only ~40% (Figure 5H). Furthermore at any given titer there were significantly more cells with >15 PV RNA molecules within them when they had been infected with PV in vesicles as opposed to free PV (Figure 5I). These data are consistent with our dSTORM findings (Figures 5A-5D) and transmission electron micrographs of isolated PS vesicles (Figure 5E) and indicate that vesicles contain multiple PV particles, which enable multiple viral RNA genomes to be transferred en bloc into a cell. Note that there was a large variation in the number of viral RNA genomes per cell when cells were infected with PV particles in vesicles as opposed to free PV particles (Figures 5H and 5I). Since de novo viral RNA synthesis can be ruled out, this variation is likely due to differences in the number of viral particles packaged per vesicle as well as contamination from free particles due to possible vesicle lysis.
Discussion
Here we have shown that multiple mature infectious enteroviral particles are released in single unilamellar PS lipid vesicles, which in turn enables multiple viral genomes to be collectively transferred to an individual cell in a new round of infection. These PS vesicles containing multiple viral genomes appear to be significantly more efficient in infection and likely to facilitate genetic exchange among viral genomes.
We first detected assembled PV capsids at replication organelles where viral RNA was synthesized (Figure 1A, Figure 6). This close juxtaposition of capsids with viral RNA synthesis sites would serve to immediately encapsidate the viral RNA and thereby facilitate efficient genome packaging as well as protection of viral genomes from host defenses. These viral particles then translocated into the cytoplasm, which was not only temporally coincident with a cessation of viral RNA synthesis but also could be prematurely triggered by inhibiting RNA synthesis with GuHCL (Figure S1A). Enteroviral 2C proteins may modulate the close coupling between RNA synthesis kinetics and capsid release from replication organelles as 2C proteins are not only localized to the replication organelles and required for viral RNA synthesis but also physically interact with capsids (Liu et al., 2010).
Figure 6. Model.

Assembled mature enteroviruses are released from the replication organelles into the cytoplasm. Clusters of multiple viral particles are selectively captured by double-membraned organelles that originate from the ER and ER derived replication organelles. These double-membraned organelles, which resemble autophagosomes, contain PS lipids on both the lumenal and cytoplasmic leaflets of their membranes. They fuse with the plasma membrane and release a unilamellar PS-lipid enriched vesicle, containing multiple viral particles, into the extracellular medium. This vesicle then facilitates infection in a PS-lipid and viral receptor dependent mechanism resulting in the collective transfer to a new recipient host cell of multiple viral RNA genomes. This mode of viral transmission enhances infection efficiency and potentially allows for genetic complementation among quasispecies.
Once the capsids dissociated from the replication organelles they were sequestered within double-membraned LC3-II positive autophagosome-like organelles. How the viral cargo is recognized and captured within these organelles, the capsid associated determinants and whether LC3-II proteins play a role in these processes as they do in canonical autophagy pathways (Rogov et al., 2014) is currently unknown. Interestingly unlike canonical autophagosomes which fuse with lysosomes and degrade their cytoplasmic cargo, these capsid-containing organelles fused with the plasma membrane to non-lytically release >85% of the PV particles into the extracellular environment within unilamellar vesicles of size range 200-400 nm. Notably, the SNARE protein syntaxin 17 was not localized to the autophagosome-like organelles to regulate their fusion with lysosomes, but instead was found sequestered away at the replication organelles (Figure S1H). Whether specific enteroviral proteins actively modulate its subcellular localization or it is an indirect consequence of the affinity of the syntaxin 17 hydrophobic hairpin tail for the PI4P/cholesterol rich replication organelle membranes remains to be investigated. This type of non-conventional secretion of autophagosomal membranes from the cell has never been reported and identifying the machinery regulating this process, including determining which cytoskeletal components and SNARE proteins are utilized for movement out to the periphery and fusion with the plasma membrane, may provide novel therapeutic targets to block enterovirus release from cells.
PS vesicles non-lytically released from cells were selectively enriched in mature enteroviral particles (Figure 3). While we cannot exclude the possibility that non-PS vesicles may also contribute to the non-lytic release of enteroviral particles, the observed significant inhibition of subsequent infection when PS is blocked (Figures 4C and 4D) suggest that PS vesicles constitute a large fraction of the non-lytic conduit for enteroviral release from cells. Live-cell time-lapse imaging in the presence of fluorescently labeled Annexin V protein revealed that the outer membrane leaflet of the released vesicle and hence topologically equivalent to the lumenal membrane leaflet of the double membraned organelle pre-fusion, contained PS lipids (Figures 3A-3D; Movie S1). The ER and/or the ER derived replication organelles both have PS lipids on their lumenal and cytoplasmic leaflets (Leventis and Grinstein, 2010; Lev, 2012) (Figure 1D) and moreover the PS can flip between these two leaflets (Clark, 2011). Thus it is likely that the host source for the autophagosome-like organelles is the ER and/or ER derived replication organelles. Supporting this conclusion the released PS vesicles indeed contained ER markers including the integral ER membrane protein calnexin (Figure S2B).
Super-resolution imaging and transmission EM revealed extracellular PS vesicles to be containing multiple viral particles, at least 20 per 200 nm cross-section (Figures 5A-5E). Supporting these data, in single molecule RNA FISH experiments, we found that infection by PV particles in vesicles allowed the collective transfer of multiple viral genomes into a single host cell (Figures 5F-5I). One important implication of these findings is that it ties the fate of individual viral genomes from previous rounds of replication to each other and thereby may provide selective advantages, in terms of replication kinetics and genetic diversity, to individual genomes compared to the case for which each genome infected cells separately from the others. Indeed infection efficiency was significantly higher when cells were infected with PV particles in vesicles as opposed to an equivalent number of free virus particles (Figures 4E and 4F). Enteroviruses as well as all other positive strand RNA viruses, are enormously diverse in genomic variety due to the inherent error rates and lack of proof reading in their RNA polymerases which can generate large numbers of viral quasispecies after even a single round of infection (Borderia et al., 2011). Vesicular transfer of multiple particles among cells would increase the chances of genetic complementation among viral quasispecies, potentially benefiting the replication efficiency of otherwise attenuated or weaker/unsuitable genomes and enabling them to maintain a presence in the genetic pool. Indeed our findings may provide a mechanism explaining Andino and colleagues’ results of cooperative interactions between neurotropic and non-neurotropic PV quasispecies (Vignuzzi et al., 2006). Secondly, when multiple viral genomes are transferred to a single cell, the likelihood of one or more genomes surviving the hostile host environment to override host defenses and replicate may be higher. Thirdly, for positive strand RNA viruses, rather than a single genome having to switch between RNA translation and RNA synthesis (until sufficient levels of viral RNA have been synthesized to take over some of those functions), multiple genomes could right from the start of infection partition RNA translation and RNA synthesis functions among themselves and enhance overall replication kinetics and viral protein levels. Finally the PS lipids on the vesicles themselves could enhance infection efficiency by attracting PS scavenging cells, such as macrophages and dendritic cells to take up the viral genomes and provide a host environment for replication. In particular we found that PV particles within vesicles could replicate significantly more efficiently within primary macrophages than free PV particles (Figure 4F).
What is the mechanism whereby viral particles within vesicles infect host cells? We found infection to be dependent not only on the virus specific receptor expressed by the host but also on the PS lipids associated with the vesicle having access to the host cell (Figures 4C and 4D). One potential mechanism to explain these findings is that PS lipids on vesicles engage PS receptors on the recipient host cell prior to the viral particles engaging their own receptors. The binding to PS receptors can trigger phagocytic uptake of vesicles (Hoffmann et al., 2001) followed by lysis or permeabilization of the vesicle within the endosomal compartment to then enable viral particles to engage their specific receptors. Equally possible, binding to PS receptors may lyse the vesicle on the cell surface, resulting in the release of a concentrated bolus of viral particles in the immediate vicinity of the cell. Reliance on PS lipids and PS receptors for infection has been documented for a number of other viruses including Vaccinia, Dengue, Ebola, Hepatitis A, and HIV (Sui et al., 2006; Mercer and Helenius, 2009; Feng et al., 2014; Morizano and Chen, 2014). The specific use of PS lipids by enteroviruses as well as other viruses to traffic between cells may have significant in vivo implications for viral pathogenesis and tissue tropism. In particular, the infection of primary macrophages, the major PS sensing cells in the body, may provide enteroviruses with the ability to target a key cell subset of the immune system while suppressing its inflammatory responses, as PS lipids have been well documented to inhibit inflammatory cytokine production by macrophages (Hochreiter-Hufford and Ravichandran, 2013). PS-lipid vesicles may also help enteroviruses exploit the natural motility of macrophages and help spread them distant sites including perhaps the central nervous system (Ousman and Kubes, 2012).
In summary we report here a novel mode of viral transmission among cells whereby viral particles are collectively released within PS-lipid enriched vesicles, which then provide both greater infection efficiency and potentially an opportunity for cooperation and complementation among viral quasispecies. This mode of transmission links the fate of multiple viral particles to one another and may have implications for maintaining viral genetic diversity within viral evolution.
Experimental Procedures (See Supplementary Online information for detailed methods)
All detailed protocols and information regarding plasmids, antibodies, cell culture, virus infection and propagation are provided in the Supplemental Online Materials.
Immunofluorescence
Cells were plated on glass coverslips and fixed with 4% PFA for 15 minutes at RT. Cells were permeabilized with either 0.2% Saponin or 0.1% Triton X-100 and sequentially incubated with primary and fluorophore-tagged secondary antibodies. Coverslips were mounted in Fluoromount-G (Southern Biotech) and imaged.
Confocal Microscopy
All confocal images were obtained with an LSM780 laser scanning confocal microscope system (Carl Zeiss USA) and images were analyzed with either Zen (Carl Zeiss) or Image J (NIH) software.
Direct Stochastic Optical Reconstruction Microscopy (dSTORM)
Free PV particles and PV particles in vesicles were plated on gridded glass bottom dishes and fixed with 4% PFA for 15 minutes at room temperature. Subsequently they were permeabilized with 0.2% saponin and incubated with Atto488 conjugated A12 antibodies. An oxygen-scavenging PBS solution (10mM NaCl, 0.5mg/ml glucose oxidase, 40g/ml catalase, 2% glucose and 10mM MEA) was used for imaging. dSTORM images were obtained on a Zeiss ELYRA PS.1 system (Carl Zeiss, USA). Images were acquired with a Plan-Apochromat 100x/1.46 oil immersion objective and an Andor iXon 885 EMCCD camera. 20,000 images were acquired per sample with an exposure time of 33 ms. Raw images were reconstructed and analyzed with ZEN software (Carl Zeiss, USA) and MatLab (MathWorks Inc. USA) using methodology from Veatch et al., 2012 and Termini et al., 2014.
Structured Illumination Microscopy (SIM)
Super-resolution 3D-SIM imaging was performed on a Zeiss ELYRA PS.1 system (Carl Zeiss, USA). Images were acquired with a Plan-Apochromat 63x/1.40 oil immersion objective and an Andor iXon 885 EMCCD camera. Fifteen images per plane (five phases, three rotations) and 0.125 mm z section of 3 mm height were required for generating super resolution images. Raw images were reconstructed and processed to demonstrate structure with greater resolution by the ZEN 2011 software (Carl Zeiss, USA).
Single molecule RNA FISH
We performed single molecule RNA FISH according to Shaffer et al. 2013. Cells were incubated with either free or vesicle associated PV particles for 1.5hrs. Cells were subsequently fixed in pre-chilled methanol (−20°C) for 10 minutes. The methanol was removed and the cells were hybridized with 10μL of hybridization buffer containing 4μM PV probe, 10% formamide, 2X SSC, and 10% dextran sulfate for 10 minutes at 37°C. Next, the samples were washed three times with pre-warmed wash buffer (10% formamide and 2X SSC) at 37°C for one minute and imaged with Zeiss LSM780 confocal microscope.
Annexin V labeling
Cells for live imaging were grown on coverslip-bottomed Lab-Tek chambers (Thermo Fisher, NY) and infected with PV for 7 hours. Cells were then replaced in imaging media (DMEM Phenol Red free supplemented with 10% FBS and 50mM HEPES pH 7.3). Alexa 568-Annexin V was added on the cells and imaging was performed on a Zeiss LSM780 Confocal Laser Scanning microscope (Carl Zeiss, USA) equipped with 458nm, 488nm, 514nm, 565nm and 633nm laser lines and detecting system for fluorescence and DIC imaging. The microscope was additionally equipped with a heating stage and incubator with temperature, humidity, and CO2 control for live-cell imaging.
Supplementary Material
1
2
3
4
5
Acknowledgements
The authors would like to thank Yasmine Belkaid, Kostya Chumakov, Ana Maria Cuervo, Jim Hogle, Gerald Feldman, Jennifer Jones, Jennifer Lippincott-Schwartz, Sanford Simon, Radek Dobrowolski, Wilma Friedman, Ellie Ehrenfeld and members of the Altan-Bonnet lab for fruitful discussions. The authors would like to especially thank Wen-Chin Tseng for help in constructing the graphical cartoons. The authors also thank Frank Macaluso and Geoffrey Perumal of the Albert Einstein College of Medicine Analytical Imaging facility for technical support. Extramural NIH RO1AI091985-01A1 (to AC) and intramural NIH funds supported this work.
References
- Baddeley D, Jayasinghe ID, Cremer C, Cannell MB, and Soeller C (2009). Light-induced dark states of organic fluochromes enable 30 nm resolution imaging in standard media. Biophys J 96, L22–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates M, Huang B, Dempsey GT, and Zhuang X. (2007) Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science. 317, 1749–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton DJ, and Flanegan JB (1997). Synchronous replication of poliovirus RNA: initiation of negative-strand RNA synthesis requires the guanidine-inhibited activity of protein 2C. J Virol 71, 8482–8489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird SW, Maynard ND, Covert MW, and Kirkegaard K (2014). Nonlytic viral spread enhanced by autophagy components. Proc Natl Acad Sci U S A 111, 13081–13086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borderia AV, Stapleford KA, and Vignuzzi M. (2011) RNA virus population diversityu:implications for interspecies transmission. Curr Opin Virol 1, 643–648 [DOI] [PubMed] [Google Scholar]
- Brandenburg B and Zhuang X (2007). Virus trafficking-learning from single virus trafficking. Nat Rev Microbiol 5, 197–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandenburg B, Lee LY, Lakadamyali M, Rust MJ, Zhuang X, and Hogle JM (2007). Imaging poliovirus entry in live cells. PLoS Biol 5, e183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Chumakov K, Dragunsky E, Kouiavskaia D, Makiya M, Neverov A, Rezapkin G, Sebrell A, and Purcell R (2011). Chimpanzee-human monoclonal antibodies for treatment of chronic poliovirus excretors and emergency postexposure prophylaxis. J Virol 85, 4354–4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Fischer ER, Kouiavskaia D, Hansen BT, Ludtke SJ, Bidzhieva B, Makiya M, Agulto L, Purcell RH, and Chumakov K (2013). Cross-neutralizing human anti-poliovirus antibodies bind the recognition site for cellular receptor. Proc Natl Acad Sci U S A 110, 20242–20247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MR. (2011) Flippin’ Lipids. Nature Immunol 12, 373–375 [DOI] [PubMed] [Google Scholar]
- Dempsey GT, Vaughan JC, Chen KH, Bates M, and Zhuang X (2011). Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat Methods 8, 1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Boon JA, and Ahlquist P (2010). Organelle-like membrane compartmentalization of positive-strand RNA virus replication factories. Annu Rev Microbiol 64, 241–256. [DOI] [PubMed] [Google Scholar]
- Doceul V, Hollinshead M, van der Linden L, Smith GL. (2010) Repulsion of superinfecting virions: a mechanism for rapid virus spread. Science 327, 873–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenfeld E, Maizel JV, and Summers DF (1970). Soluble RNA polymerase complex from poliovirus-infected HeLa cells. Virology 40, 840–846. [DOI] [PubMed] [Google Scholar]
- Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, and Henson PM (1992). Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol 148, 2207–2216. [PubMed] [Google Scholar]
- Feng Z, Hensley L, McKnight KL, Hu F, Madden V, Ping L, Jeong SH, Walker C, Lanford RE, and Lemon SM (2013). A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 496, 367–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Li Y, McKnight KL, Hensley L, Lanford RE, Walker CM, and Lemon SM (2014). Human pDCs preferentially sense enveloped hepatitis A virions. J Clin Invest. pii: 77527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, et al. (2013). Autophagosomes form at ER-mitochondria contact sites. Nature 495, 389–393. [DOI] [PubMed] [Google Scholar]
- Hochreiter-Hufford A, and Ravichandran KS (2013). Clearing the dead: apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harb Perspect Biol 5, a008748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann PR, deCathelineau AM, Ogden CA, Leverrier Y, Bratton DL, Daleke DL, Ridley AJ, Fadok VA, and Henson PM. (2001) Phosphatidylserine (PS) induces PS receptor-mediated macropinocytosis and promotes clearance of apoptotic cells. J Cell Biol. 155, 649–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu NY, Ilnytska O, Belov G, Santiana M, Chen YH, Takvorian PM, Pau C, van der Schaar H, Kaushik-Basu N, Balla T, et al. (2010). Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 141, 799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilnytska O, Santiana M, Hsu NY, Du WL, Chen YH, Viktorova EG, Belov G, Brinker A, Storch J, Moore C, et al. (2013). Enteroviruses harness the cellular endocytic machinery to remodel the host cell cholesterol landscape for effective viral replication. Cell Host Microbe 14, 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura E, Kishi-Itakura C, Mizushima N (2012) The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151,1256–69. [DOI] [PubMed] [Google Scholar]
- Jackson WT, Giddings TH Jr., Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, and Kirkegaard K (2005). Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol 3, e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay JG, Koivusalo M, Ma X, Wohland T, and Grinstein S (2012). Phosphatidylserine dynamics in cellular membranes. Mol Biol Cell 23, 2198–2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkegaard K, and Jackson WT (2005). Topology of double-membraned vesicles and the opportunity for non-lytic release of cytoplasm. Autophagy 1, 182–184. [DOI] [PubMed] [Google Scholar]
- Koopman G, Reutelingsperger CP, Kuijten GA, Keehnen RM, Pals ST, and van Oers MH (1994). Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood 84, 1415–1420. [PubMed] [Google Scholar]
- Lev S (2012) Non-vesicular lipid transfer from the ER. Cold Spring Harbor Perspect Biol 4, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leventis PA, and Grinstein S (2010). The distribution and function of phosphatidylserine in cellular membranes. Annu Rev Biophys 39, 407–427. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wang C, Mueller S, Paul AV, Wimmer E, and Jiang P (2010). Direct interaction between two viral proteins, the nonstructural protein 2C and the capsid protein VP3, is required for enterovirus morphogenesis. PLoS Pathog 6, e1001066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubeck E, and Cai L (2012). Single-cell systems biology by super-resolution imaging and combinatorial labeling. Nat Methods 9, 743–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer J and Helenius A. (2009) Virus entry by macropinocytosis. Nat Rev Cell Biol 11, 510–520 [DOI] [PubMed] [Google Scholar]
- Morizano K and Chen IS (2014). Role of phosphatidlyserine receptors in enveloped virus infection. J Virol 88, 4275–4290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nchoutmboube JA, Viktorova EG, Scott AJ, Ford LA, Pei Z, Watkins PA, Ernst RK, and Belov GA (2013). Increased long chain acyl-Coa synthetase activity and fatty acid import is linked to membrane synthesis for development of picornavirus replication organelles. PLoS Pathog 9, e1003401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ousman SS, and Kubes P (2012). Immune surveillance in the central nervous system. Nat Neurosci 15, 1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, and Tyagi S (2008). Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 5, 877–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson SM, Tsueng G, Sin J, Mangale V, Rahawi S, McIntyre LL, Williams W, Kha N, Cruz C, Hancock BM, et al. (2014). Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS Pathog 10, e1004045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogov V, Dotsch V, Johansen T, and Kirkin V (2014) Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell. 53, 167–78 [DOI] [PubMed] [Google Scholar]
- Shaffer SM, Wu MT, Levesque MJ, and Raj A (2013). Turbo FISH: a method for rapid single molecule RNA FISH. PLoS One 8, e75120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss M, Levy HC, Bostina M, Filman DJ, and Hogle JM (2013). RNA transfer from poliovirus 135S particles across membranes is mediated by long umbilical connectors. J Virol 87, 3903–3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strazynski M, Kramer J, and Becker B (2002). Thermal inactivation of poliovirus type 1 in water, milk and yoghurt. Int J Food Microbiol 74, 73–78. [DOI] [PubMed] [Google Scholar]
- Sui L, Zhang W, Chen Y, Zheng Y, Wan T, Yang Y, Fang G, Mao J, and Cao X (2006). Human membrane protein Tim-3 facilitates hepatitis A virus entry into target cells. Int J Mol Med 17, 1093–1099. [PubMed] [Google Scholar]
- Swairjo MA, Concha NO, Kaetzel MA, Dedman JR, and Seaton BA (1995). Ca(2+)-bridging mechanism and phospholipid head group recognition in the membrane-binding protein annexin V. Nat Struct Biol 2, 968–974. [DOI] [PubMed] [Google Scholar]
- Taylor MP., Burgon TB, Kirkegaard K, and Jackson WT (2009) Role of microtubules in extracellular release of Poliovirus. J Virol. 83, 6599–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Termini CM, Cotter ML, Marjon KD, Buranda T, Lidke KA, and Gillette JM. (2014)The membrane scaffold CD82 regulates cell adhesion by altering α4 integrin stability and molecular density. Mol Biol Cell. 25, 1560–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch SL, Machta BB, Shelby SA, Chiang EN, Holowka DA, and Baird BA.(2012) Correlation functions quantify super-resolution images and estimate apparent clustering due to over-counting. PLoS One. 7, e31457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignuzzi M, Stone JK, Arnold JJ, Cameron CE, and Andino R (2006). Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 439, 344–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1
2
3
4
5