Interaction of human papillomavirus type 16 particles with heparan sulfate and syndecan-1 molecules in the keratinocyte extracellular matrix plays an active role in infection (original) (raw)
Abstract
Oncogenic human papillomaviruses (HPVs) attach predominantly to extracellular matrix (ECM) components during infection of cultured keratinocytes and in the rodent vaginal challenge model in vivo. However, the mechanism of virion transfer from the ECM to receptors that mediate entry into host cells has not been determined. In this work we strove to assess the role of heparan sulfate (HS) chains in HPV16 binding to the ECM and determine how HPV16 release from the ECM is regulated. We also assessed the extent to which capsids released from the ECM are infectious. We show that a large fraction of HPV16 particles binds to the ECM via HS chains, and that syndecan-1 (snd-1) molecules present in the ECM are involved in virus binding. Inhibiting the normal processing of snd-1 and HS molecules via matrix metalloproteinases and heparanase dramatically reduces virus release from the ECM, cellular uptake and infection. Conversely, exogenous heparinase activates each of these processes. We confirm that HPV16 released from the ECM is infectious in keratinocytes. Use of a specific inhibitor shows furin is not involved in HPV16 release from ECM attachment factors and corroborates other studies showing only the intracellular activity of furin is responsible for modulating HPV infectivity. These data suggest that our recently proposed model, describing the action of HS proteoglycan processing enzymes in releasing HPV16 from the cell surface in complex with the attachment factor snd-1, is also relevant to the release of HPV16 particles from the ECM to promote efficient infection of keratinocytes.
Introduction
Human papillomaviruses (HPVs) are small DNA-containing viruses that initiate benign and malignant epithelial tumours. HPV16 is the prototype of the oncogenic HPVs, causing genital and oral infections, and is a major cause of premalignant lesions and cancers in the same mucosal tissues (Chesson et al., 2012; Forman et al., 2012). The entry of HPV particles into human keratinocyte (HK) host cells is a multistep process initiated by binding to primary attachment factors, most commonly the heparan sulfate (HS) chains of proteoglycans (HSPGs) (Joyce et al., 1999; Combita et al., 2001; Giroglou et al., 2001). The interaction of HPV16 major capsid protein (L1) with the HS chain of proteoglycans is well characterized, but generally considered to play a passive role in infection (Knappe et al., 2007; Dasgupta et al., 2011).
Syndecan-1 (snd-1), the most abundant HSPG in keratinocytes, serves as an HPV attachment receptor (Selinka et al., 2002; Shafti-Keramat et al., 2003). Snd-1 is expressed primarily on epithelial cell basolateral surfaces (Rapraeger et al., 1986; Hayashi et al., 1987; Kim et al., 1994), and binds tightly to the extracellular matrix (ECM) components collagen (Koda et al., 1985), the fibronectins (Saunders & Bernfield, 1988) and laminin-332 (LN-332, formerly LN-5) (Okamoto et al., 2003; Rousselle & Beck, 2013). The HS chains of snd-1 proteoglycans accumulate various active ligands, including ECM proteins, proteinases, growth factors (GFs), cytokines, chemokines, and present these ligands to high-affinity cell-surface receptors (Yoneda & Couchman 2003; Choi et al., 2011). The ectodomains of all HS-chain-enriched snd-1 molecules are constitutively released (i.e. shed) from cell membranes as part of normal physiology (Bishop et al., 2007). Shed ectodomains bound to bioactive ligands (e.g. GFs) in soluble form also deliver ligands to their high-affinity, cognate receptors (Elenius & Jalkanen, 1994; Bernfield et al., 1999; Okamoto et al., 2003; Fears & Woods, 2006; Choi et al., 2010, 2011), or they can attach to and accumulate within the ECM (Bayer-Garner et al., 2001; Ding et al., 2005). Proteases, including matrix metalloproteinases (MMPs) and ADAM sheddases, mediate the ectodomain shedding of membrane-bound proteins (such as snd-1) (Flannery, 2006; Lambaerts et al., 2009; Choi et al., 2010; Pruessmeyer et al., 2010); HS moieties on snd-1 are processed by heparanase, which cleaves HS to release fragments that are 10–20 sugar residues long. Processed HS molecules are more bioactive than their native HS chains, and act as potent promoters of GF activity (Kato et al., 1998; Elkin et al., 2001).
Previously, we showed snd-1 shedding plays an active role in HPV16 infection in cultured HKs (Surviladze et al., 2012). Instead of dissociating from HS chains, HPV particles are released from the cell surface during normal HSPG processing. HPV particles remain complexed with HS and GFs, particularly ligands specific to epidermal GF receptor (EGFR). We reported that inhibiting MMP activity and subsequent HSPG shedding significantly decreases virus release and infection. The specificity of the GFs appears to bridge the virus to interact with cognate cellular receptors (i.e. a receptor tyrosine kinase/GFR), where subsequent signalling promotes infection. The importance of EGFR signalling in HPV infectious entry into HKs was subsequently confirmed (Schelhaas et al., 2012).
Considering HSPG biology, it is logical that HSPG-bound HPV particles would associate with the ECM. Membrane-bound and shed syndecan forms attach tightly to the ECM via their HS chains. Additionally, the ECM functions as a scaffold for secreted polysaccharides (Bishop et al., 2007). Indeed, many studies demonstrated that HPV particles accumulate on the ECM, and LN-332, an ECM-resident, is a proposed HPV attachment factor (Culp et al., 2006a, b; Smith et al., 2008). Recent work suggests direct protein–protein interaction between HPV16 and LN-332 (Cerqueira et al., 2013). HS was found to be important for HPV16 and HPV18 binding to the ECM (Selinka et al., 2007), but was unnecessary for HPV11 (Culp et al., 2006a). Although these studies used various cell lines and HPV types, ECM-attached virus was deemed infectious. Our previous work showed HPV virions bound to keratinocyte ECM are immobile, but that bound virus disappeared over time coincident with particle uptake by filopodia (Smith et al., 2008). Nevertheless, whether HS chains play a specific role in HPV binding to the ECM has been unclear, and the mechanism of HPV release from the ECM and transfer to the entry receptors on the keratinocyte surface has remained elusive.
Herein we investigated the mechanics of HPV16 binding to and release from the ECM to facilitate infection of HKs. We used non-carcinogenic HPV16 pseudovirions (PsVs), which are structurally indistinguishable, and deliver a reporter plasmid (pseudogenome) to the cell nucleus as a surrogate marker of infection. Our findings indicate that ECM-attached HPV16 particles are released via the activity of proteases and heparanase, in a manner similar to that which we described for cell-attached virus (Surviladze et al., 2012).
Results
Furin inhibitor does not alter HPV16 binding to or release from the ECM
The protein convertase furin is proposed to play a role in HPV release from primary attachment factors, especially on the ECM or basement membrane (Kines et al., 2009; Schiller et al., 2010). If furin is important for the transfer of ECM-bound HPV particles to HKs, then furin inhibition should decrease not only release of bound HPV particles from the ECM but also HPV internalization into cells and total infectivity. To investigate how furin activity influences HPV16-ECM interactions, we used furin inhibitor 1 (also called dec.-RVKR-CMK), initially testing the effect on HPV16 binding to cell-free ECM (isolated as described in Methods and Fig. S1, available in the online Supplementary Material). HPV16 PsVs were bound to cell-free ECM in the presence or absence of the furin inhibitor. After washing away unbound virus, analysis revealed that furin inhibitor 1 did not significantly alter HPV16 binding to the ECM (P = 0.989; Figs 1a and S1a); however, the furin inhibitor reduced furin activity in the cell media >60 % (data not shown). Next, we examined in two ways whether furin activity impacted HPV16 PsV release from the ECM. First, we utilized recombinant His-furin (Prepro Tech), which we verified was active and prone to deactivation by furin inhibitor 1 (Fig. S2). When His-furin was incubated with ECM-bound HPV16, there was no difference in the amount of HPV16 released from the ECM in two independent experiments (Figs 1b and S1b). Second, ECM-bound HPV16 was incubated with complete medium (CM) [Dulbecco's minimal essential medium (DMEM)+10 % FCS] in the presence or absence of the furin inhibitor for 4 h. HPV16 released into the conditioned medium was quantified revealing no significant difference in HPV liberation from the ECM (P = 0.7682; Figs 1c and S1c). These data demonstrate that furin activity has no effect on the release of HPV16 bound to ECM in vitro.
Furin activity is not required for HPV16 internalization into HKs
To examine furin's role during HPV internalization versus later steps of HK infection, HaCaT cells were seeded atop ECM-bound PsVs in the absence or presence of the furin inhibitor. After 4 h (37 °C), virus uptake by the cells was stopped by transfer to ice. Extracellular HPV16 was removed with a widely used trypsin/EDTA treatment (Pauza & Price, 1988; Campos et al., 2012; Burkard et al., 2014; Ren et al., 2014) combined with a mild acid and high ionic strength wash (Sorkin & Duex, 2010) (Fig. S3). We found the furin inhibitor had no influence on the levels of HPV16 internalized by HKs grown on ECM-bound PsVs (P = 0.954; Figs 1d and S1d), a result consistent with a report that furin inhibitor 1 did not prevent BPV1 or HPV16 PsV cellular entry (Richards et al., 2006).
Intracellular furin activity regulates HPV16 infection
Richards et al. (2006) previously showed furin inhibitor 1 blocks HPV16 pseudoinfection. To validate this finding in our hands, we pre-bound HPV16 PsVs to the ECM in the presence of furin inhibitor and then seeded HaCaT cells atop the ECM-bound PsVs as above. The furin inhibitor reduced infection by 99.4 % compared with infection without the inhibitor (Figs 1e and S1e). In this experiment as well as prior work (Richards et al., 2006), the cell permeability of furin inhibitor 1 makes it difficult to specifically identify the stage(s) of HPV infection at which furin is needed. To focus the inhibitor at infection steps post entry, HaCaT cells were pre-incubated with cell-permeable furin inhibitor 1 for 1 h. Separately, HPV16 PsVs were attached to ECM without furin inhibitor. After thoroughly washing away extracellular furin inhibitor, HaCaT cells were seeded atop the ECM-bound PsVs. Infection was still inhibited ∼70 % (Fig. 1e), with no statistical difference between the two furin-treatment conditions (P = 0.128), providing further evidence that furin's intracellular action is important for HPV16 infectivity of keratinocytes.
Confocal microscopy studies indicated that furin inhibitor 1 blocked HPV infection after capsids arrived in the endosomal compartment (Richards et al., 2006). We investigated this biochemically by isolating cell fractions enriched in endosomes/lysosomes (EL fraction) and the marker CD63 (Fig. 1f). PsV-exposed HaCaT cells grown in the presence of the furin inhibitor for 16 h revealed substantial increases in both L1 and L2 capsid proteins in the EL fraction compared with cells without inhibitor (Fig. 1f). We verified that the furin inhibitor reduced furin activity in the EL fractions after 3 h (Fig. 1g). The lower level of furin inhibition by this assay (30 %) compared with >95 % infection inhibition most likely reflects differences in incubation times, substrates and/or the presence of other proteases in the EL fractions. Although we find that furin activity does not regulate HPV16 release from the ECM or virus internalization, our data support the published microscopy data indicating that furin is needed for endosomal escape of viral components. We concur that furin's intracellular action is important for HPV16 infection of HKs.
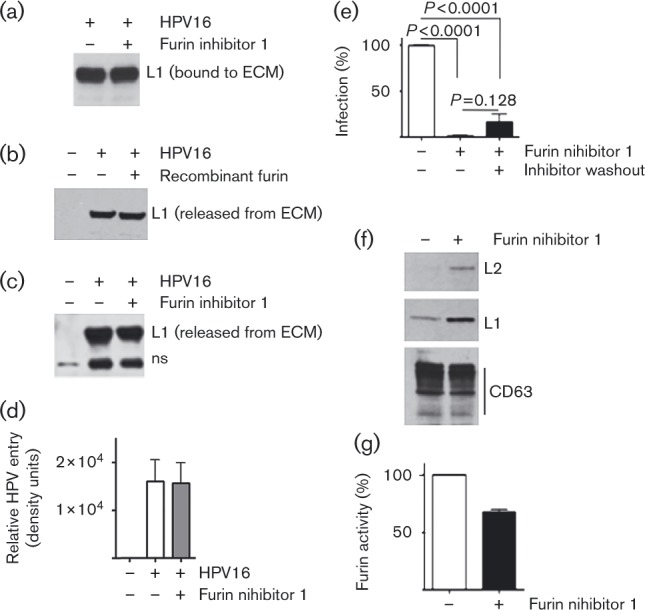
Fig. 1. Effect of furin inhibitor on HPV16 binding to and release from ECM, internalization by HKs and infection. (a, c–e) HPV16 PsVs were bound to cell-free ECM in the presence or absence of 1.3 μM furin inhibitor. Unbound virus particles were intensively washed away; treatments were as indicated. (a–d) The final products were extracted with RIPA buffer, fractionated by SDS-PAGE, and analysed by immunoblot for HPV16 L1 (ns is a non-specific cellular protein). Results in each panel are representative of two or three experiments; error bars represent the sem. (a) HPV16 PsVs bound to the ECM. (b) HPV16 release in the presence of furin. ECM-bound virus was incubated with 1.5 ml complete medium (CM), with or without 3 nM His-furin at 37 °C for 3 h. Virus released into the medium was recovered by pull-down then analysed as in (a). (c) HPV16 PsV release from ECM. ECM-bound virus was incubated with 1.5 ml CM at 37 °C for 4 h with and without furin inhibitor. Virus released into the medium was recovered and analysed as in (a, b). (d) HPV16 entry assay. HaCaT cells were seeded atop ECM-bound HPV16 and incubated for 4 h at 37 °C in the presence or absence of furin inhibitor. Extracellular virus was removed as detailed in Methods, and intracellular HPV levels analysed by immunoblot. Graphed is the mean of four independent experiments. (e) Infectious entry of HPV16. HPV16 PsVs were bound to ECM; infectious transfer of virus to HaCaT cells in the absence or presence of furin inhibitor was measured at 24 h post-infection. HaCaT cells were pre-incubated at 37 °C for 1 h with furin inhibitor. Extracellular furin inhibitor was removed by intensive washing and cells were added to the ECM-bound HPV16. Relative infectivity was scored 24 h post-infection. (f, g) Furin activity in EL fractions. HPV16-exposed HaCaT cells were incubated for 16 h with or without 3 μM furin inhibitor 1. An equal amount of purified EL fraction lysate was fractionated by SDS-PAGE and analysed for capsid proteins using rabbit polyclonal HPV16 antibody. CD63 (an EL fraction-specific protein detected with anti-CD63 mAb) was detected as a loading control (f). (g) Furin activity in EL fractions of HKs growing in the presence or absence of 3 μM furin inhibitor 1 for 3 h.
Heparanase and MMPs affect HPV16 infectious transfer from the ECM to keratinocytes
We previously demonstrated that HSPG processing enzymes, the MMPs, are actively involved in HPV16 release from primary attachment factors and HK infection (Surviladze et al., 2012). Heparanase, an endoglycosidase that cleaves HS side chains and liberates the HSPG-bound GFs, is also active in regulation of HSPG shedding (Purushothaman et al., 2008; Ramani et al., 2013). We hypothesized that heparanase is involved in HPV release from primary attachment factors and, thus, regulates HPV infectivity. We performed experiments similar to those detailed for the furin inhibitor (Fig. 1) using an MMP inhibitor (batimastat) and a heparanase inhibitor (OGT211; Tocris Bioscience). Whereas these agents had no measurable effect on HPV16 PsV binding to the ECM, they both reduced virus release from the ECM (Fig. 2a, b). We also found that MMPs and heparanases are important in HPV16 infectious transfer from the ECM (Fig. 2c). After binding PsVs to cell-free ECM, HaCaT cells were seeded atop the two. Infectivity assays showed both batimastat and OGT2115 significantly reduced the ability of ECM-bound HPV16 to infect cells.
Fig. 2. Effect of MMP and heparanase inhibitors on ECM binding and release, and infectious transfer of HPV16 from the ECM to keratinocytes. (a) HaCaT cell ECM was incubated with HPV16 PsVs in the presence of 1 μM batimastat, 20 μM OGT2115, or with a mixture of both. After 1 h at room temperature, unbound PsVs were removed by intensive washing, and ECM-bound virus was solubilized and analysed as described in Fig. 1(a). (b) HPV-bound ECM was incubated at 37 °C for 3 h with complete medium (CM), or with CM supplemented with 1 μM batimastat or 20 μM OGT2115. Conditioned media were analysed for released HPV16 as described in Fig. 1(b). (c) HaCaT cells were seeded atop ECM-bound HPV16 PsVs and incubated at 37 °C in the presence of 1 μM batimastat or 20 μM OGT2115. Relative pseudoinfection was scored after 24 h. Error bars represent the sem of three independent experiments.
HPV16 particles attach to the ECM via HS chains
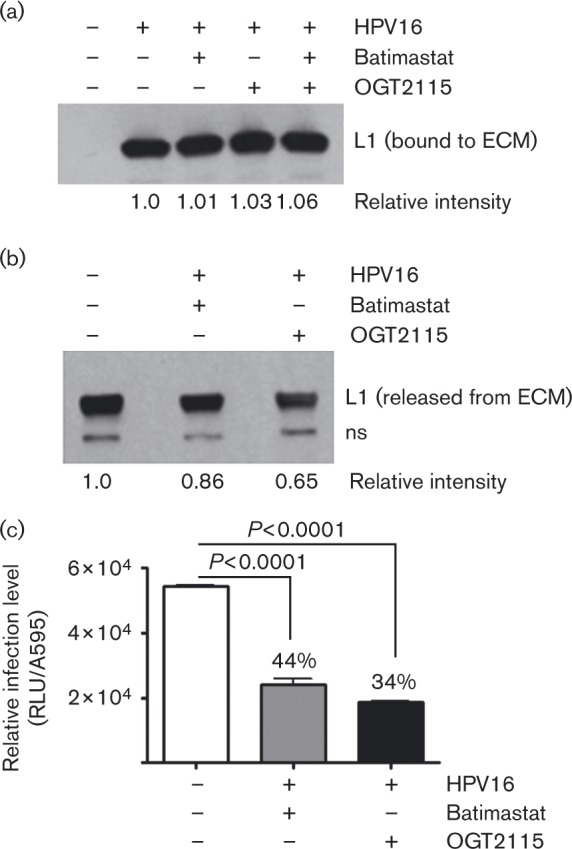
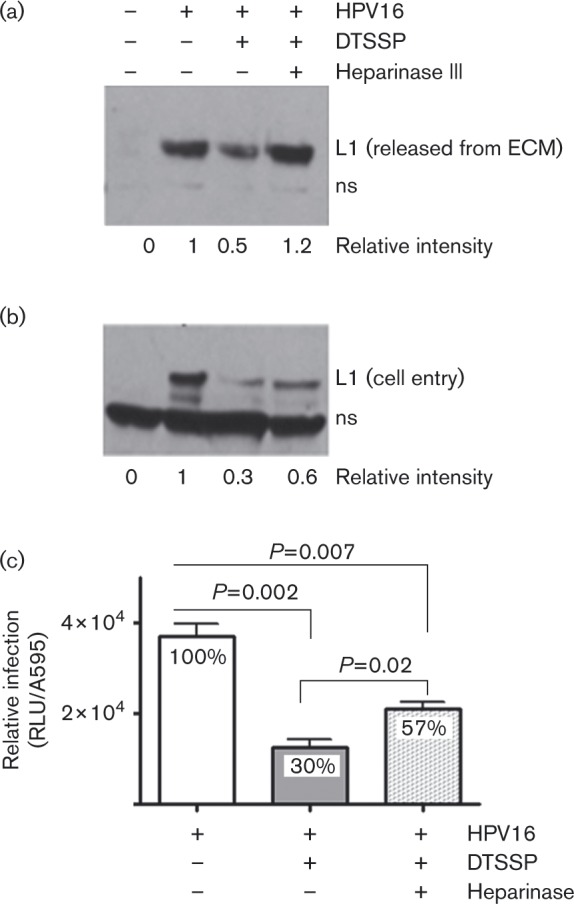
As heparanase degrades polymeric HS at the cell surface and within the ECM, reduced HPV infection via the heparanase inhibitor suggests HS processing is involved in infectious transfer of HPV16 from the ECM to cells. ECM is a depot for shed snd-1 ectodomains containing HS chains (Bayer-Garner et al., 2001). Thus, it is reasonable that HS chains could affix virus to the ECM. Thus, we treated ECM-bound HPV16 with heparinase III, an enzyme from Flavobacterium heparinum that specifically cleaves HS chains at pH 7, and quantified the virus released. To reduce the role of proteolysis in virus release, we used DTSSP [3,3′-dithiobis(sulfosuccinimidylpropionate)] to specifically cross-link proteins in the ECM prior to HPV16 binding. DTSSP forms stable amine bonds; thus, it should cross-link and prevent protein release, but have little effect on sugar chains like HS. We first verified that there was no difference between the ability of HPV16 to bind to native ECM versus DTSSP-cross-linked ECM (xECM; Fig. 3a). We confirmed that cross-linking protects ECM-resident proteins from proteolytic processing by incubating native and xECM with complete medium for 20 h. After washing to remove unbound proteins, we found DTSSP cross-linking inhibited LN-332 degradation by ∼2-fold (Fig. 3b). At the same time, HPV16 release from the xECM decreased ∼50 % compared with the amount of virus released from native ECM (Fig. 3c). This suggests that some PsVs bind to ECM-resident proteins, or that binding avidity increased in large cross-linked structures. Nevertheless, heparinase III treatment doubled the release of PsVs bound to xECM (Fig. 3d), indicating a sizeable proportion of HPV16 particles bind the ECM via HS chains.
Fig. 3. Effect of ECM-protein cross-linking on HPV16-ECM interactions. ECM proteins were cross-linked with DTSSP prior to HPV16 PsV exposure. Unbound particles were removed by intensive washing and ECM-bound virions were subjected to various treatments. The products were analysed by SDS-PAGE and immunoblot. Relative protein levels (shown below the blots) determined by densitometry. (a) HPV16 PsVs bound to native or DTSSP xECM for 1 h. (b) Native or xECM analysed for levels of intact LN-332. (c) Native or xECM pre-bound with HPV16 were incubated at 37 °C for 20 h. Conditioned medium was subjected to the soluble HPV16 pull-down assay. (d) HPV16 bound to native and xECM was incubated at 37 °C for 2 h in the presence or absence of 0.1 U heparinase III. Virus released into the conditioned medium was analysed for HPV16 L1 content as in (c).
ECM-resident snd-1 is involved in HPV16 binding to the ECM
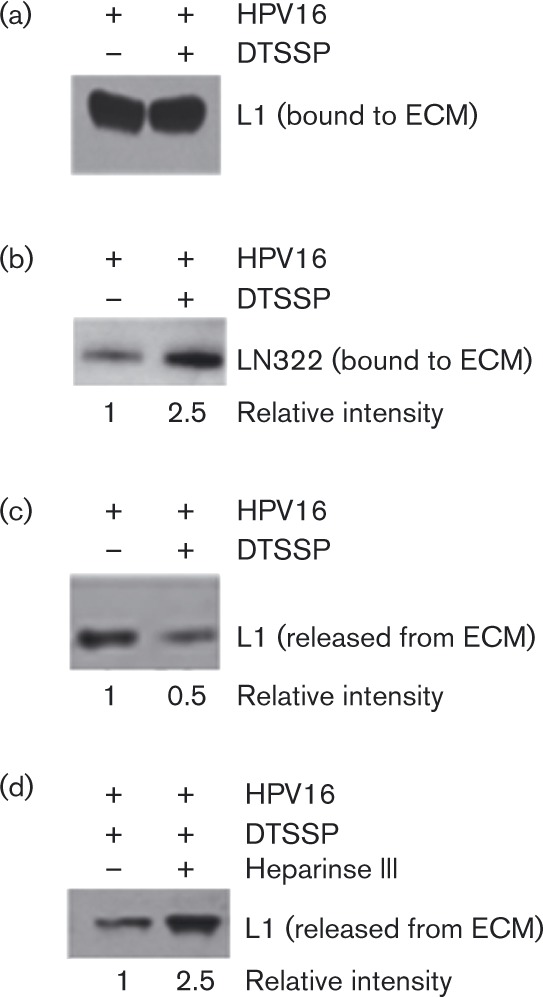
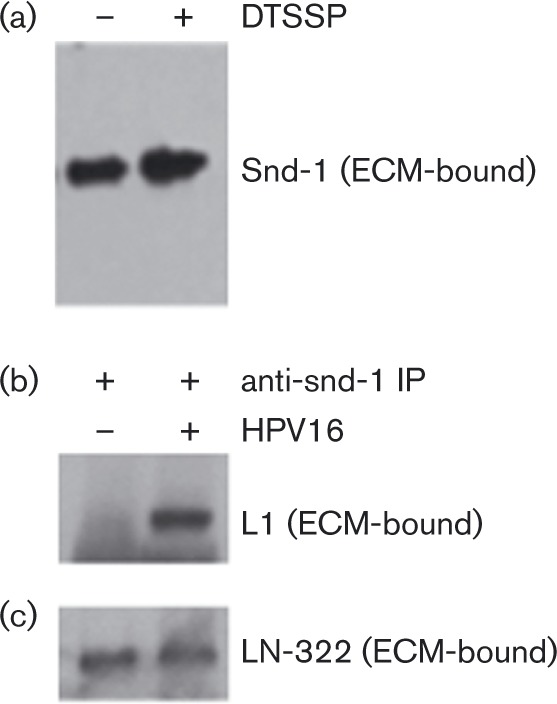
Shed snd-1 molecules accumulate in the ECM (Bayer-Garner et al., 2001; Ding et al., 2005) and are also primary cell-surface receptors for HPV16 (Selinka et al., 2002; Shafti-Keramat et al., 2003). Thus, we hypothesized that ECM-resident, shed snd-1 molecules (containing HS chains) are involved in ECM binding by HPV16. We first confirmed the presence of snd-1 proteoglycans in native and xECM (Fig. 4a). We then used immunoprecipitation (IP) for snd-1 (Fig. S4) to show that L1 interacts with ECM-associated snd-1 (Fig. 4b). The stringent IP conditions suggest high-affinity interactions similar to those between L1 and HSPG (Joyce et al., 1999). HPV16 attachment to ECM, however, does not affect snd-1 binding to LN-332 (Fig. 4c).
Fig. 4. Analysis of HK-secreted ECM for shed forms of snd-1.Native or xECM solubilized directly in Laemmli buffer, fractionated by SDS-PAGE and analysed by immunoblot. (a) Snd-1 content in ECM. (b, c) HPV16 PsVs were bound to native ECM for 1 h; unbound virus was removed by intensive washing and extracted with RIPA buffer. IP was performed using anti-snd-1 rabbit polyclonal antibody coupled to Protein A-magnetic beads. Immunoblot was performed for snd-1 (see Fig. S4), HPV16 L1 (b) and LN-332 (c). Three independent experiments verified that PsVs associate with snd-1 in the ECM.
Cleavage of HS chains increases HPV16 release from the ECM concomitant with increased HK entry and infection
A role for HS chain-processing enzymes in HPV infection has not been investigated. We predicted ECM-resident HS chains should be free from DTSSP protein cross-linking and remain susceptible to heparinase. We then determined that heparinase III increases HPV16 release from xECM to promote infection (Fig. 5). In a variation of experiments shown in Fig. 3(c, d), HKs were seeded atop native or xECM-bound virus. Consistent with results in Fig. 3(c), xECM reduced the release of bound HPV16 PsVs by ∼65 % compared with virus release from native ECM (Fig. 5a). However, heparinase III restored HPV16 release from xECM (Fig. 5a), similar to data in Fig. 3(d) without cells. This further confirms that HPV16 particles interact with HS moieties bound in the ECM. As anticipated, cross-linking led to proportionately (∼70 %) lower HPV internalization and infection (P = 0.002) in the cells seeded atop xECM containing bound HPV16 compared with cells on native ECM with bound HPV16 (Fig. 5b, c). Similar to increased HPV16 release from xECM in the presence of heparinase III, the presence of heparinase III doubled the amount of internalized virus as well as infection levels (P = 0.02) in cells compared with samples without heparinase (Fig. 5b, c). In summary, ECM-protein cross-linking did not affect HPV16 binding, but decreased HPV release from the ECM, cellular internalization and infection levels in HaCaT cells. Promoting HS chain processing in protein xECM proportionately restored HPV16 released from the ECM, HK entry and infection. These findings provide strong evidence that HPV particles interact with HS chains in the ECM, and that infection is strongly affected by HS chain processing in the ECM.
Fig. 5. Effect of ECM cross-linking on HPV16 release from the ECM, and internalization into and infection of keratinocytes. HaCaT cells were seeded over virus-bound native or xECM in the presence or absence of 0.1 U heparinase III, and incubated at 37 °C. (a) Conditioned media were collected after 20 h and analysed for the content of released virus. (b) HPV16 entry into HaCaT cells grown atop ECM-bound virus was measured after 3.5 h, as described in Figs. 1(d) and S2. The cellular non-specific band (ns) served as a loading control. L1 band intensities were quantified by densitometry (a, b). (c) HPV16 PsVs were bound to native and xECM. Unbound virus was removed and HaCaT cells were seeded atop HPV-ECM in the presence or absence of 0.1 U heparinase III. Infection was assayed 24 h later. Error bars indicate the sem from two independent experiments performed in triplicate.
HPV16 PsV released from ECM is infectious
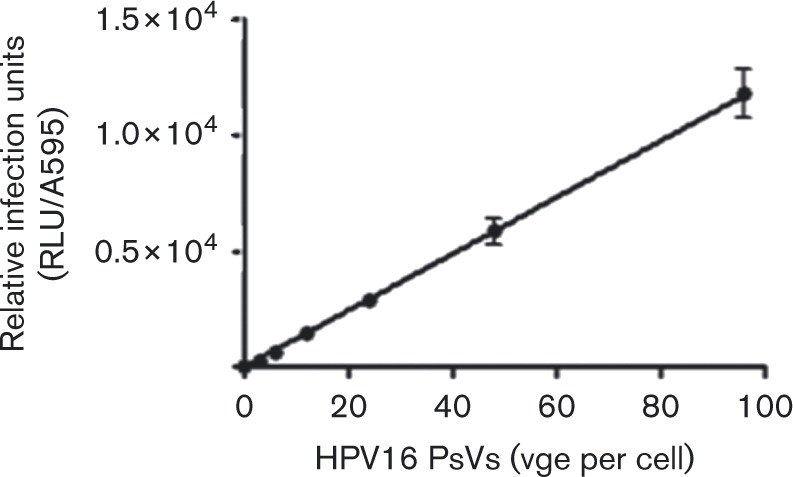
Our data show a strong positive correlation among HPV16 PsV release from the ECM, cellular internalization of virus particles, and infection when HaCaT cells were seeded atop ECM-containing virus. We verified infection is directly proportionate to the amount of HPV16 released from the ECM by collecting released virus and testing dose-dependent infection. Analysis revealed a linear dose–response relationship between the concentration of HPV16 PsV released from the ECM and HK infectivity (Fig. 6).
Fig. 6. Dose-responsive infection of HKs by HPV16 PsVs released from ECM. HaCaT cell-derived ECM was incubated with ∼3 × 108 viral genome equivalents (vge) per well HPV16 PsVs at 37 °C for 2.5 h. Conditioned medium was collected and quantified for HPV16 PsVs. HaCaT cells were incubated with serial dilutions of the ECM-released HPV16 and infection was scored after 24 h. Raw data were normalized to protein content and infectivity plotted as a function of exposure dose. Error bars represent the sem of three replicates.
Discussion
HPVs have long been known to interact with cell-surface HSPGs (Joyce et al., 1999; Combita et al., 2001; Giroglou et al., 2001), and to bind strongly to ECM secreted by keratinocytes and other epithelial cells (Culp et al., 2006a, b; Day et al., 2007). Yet, ECM moieties responsible for HPV interaction and the mode of virus transfer from these molecules to plasma membrane receptors responsible for virus entry into host keratinocytes have not been clearly defined. Herein we demonstrate that HS chain-processing enzymes augment the release and infectivity of ECM-bound HPV PsVs, indicating that considerable numbers of virus particles attach to the ECM via these HS chains. We found shed, ECM-resident snd-1 interacts with HPV16 and with LN-332, demonstrating snd-1 is an ECM attachment factor for HPV16, in addition to its role binding HPVs on the plasma membrane (Shafti-Keramat et al., 2003; Surviladze et al., 2012). The interaction of snd-1 with LN-332 is expected on the basis of reports that shed snd-1 molecules with their native HS chains are concentrated in the ECM (Bayer-Garner et al., 2001; Ding et al., 2005), and that LN-332 binds plasma membrane resident snd-1 with high affinity and specificity via HS chains (Rapraeger et al., 1986; Bernfield et al., 1999; Okamoto et al., 2003). LN-332 has been identified as an ECM-resident HPV attachment factor (Culp et al., 2006a,b; Selinka et al., 2007; Cerqueira et al., 2013). As LN-332 intrinsically lacks HS chains, but contains HS-binding domains (Sung et al., 1997; Carulli et al., 2012), it seems more likely that HS chains bridge the HPV-LN-332 interaction, and this could account for ECM co-localization between HPV and LN-332 (Culp et al., 2006; Cerqueira et al., 2013). Indeed, our findings indicate that the normally processed HS chains of snd-1, and perhaps other HSPGs, comprise ECM-resident HPV receptors. However, we cannot rule out the existence of other non-HSPG receptors in the ECM, including LN-332. Highly charged HPV particles may associate with many other molecules.
We found no evidence that furin plays a role in extracellular HPV–keratinocyte interactions or transfer of ECM-attached virions to receptors responsible for virus entry. Our results are consistent with previous reports, showing furin's activity is dispensable for HPV endocytic entry, and that furin inhibitor 1 caused HPV accumulation in late endosomal compartments (Richards et al., 2006; Day & Schelhaas, 2014). Extracellular furin is suggested to catalyse HPV capsid conformational changes by cleaving L2, which then promotes dissociation from HSPGs and interaction with a non-HSPG cell-surface receptor that promotes entry (Day et al., 2008). However, our data showing equivalent entry in the presence or absence of furin inhibitor fail to support this idea.
This work reveals two aspects about ECM binding by HPV. First, ECM-protein cross-linking had no measurable outcome on HPV16 binding. This means that either HPV attachment to ECM proteins fails to require conformational changes or that HPVs largely bind non-proteinaceous molecules (HS chains of ECM-integrated proteins), which were not DTSSP cross-linked. Second, it seems unlikely that HPV capsid conformational changes are required for release from ECM-factors and subsequent infectious host cell entry. This is based on finding similar binding efficiencies of HPV16 to native and xECM, strongly indicating that the same attachment molecules are engaged by virus on the two matrices. Similar virus–ECM binding should result in equivalent capsid conformational changes, which, if essential for virus release and infectivity, should cause equal HPV release from primary attachment factors, and similar infectivity of the cells grown over native or xECM. Yet, comparing activities following binding to native or xECM, virus release from xECM, HK entry and cell infectivity was reduced by ∼50–70 %. We cannot exclude the possibility that conformational changes in HPV L1 and L2 proteins occur during interaction with plasma membrane components. However, to our knowledge there is no direct evidence that capsid conformational changes are necessary for the release of HPV16 from an attachment factor, particularly HSPGs.
Models have been proposed to explain the mechanism of HPV release from HSPGs and particle movement to receptor(s) responsible for virus entry. One suggests that conformational changes in the virus capsid structure are brought about by HSPG binding, which then allows furin cleavage of HPV L2 protein, and subsequently triggers HPV release from attachment factors that passively accumulate virus particles on their HS chains (Schiller et al., 2010). We proposed a model based upon the physiological processing of HSPG molecules, wherein HPV particles are released from the cell surface still in complex with HS and GFs, and signal via GFRs to promote infection (Surviladze et al., 2012). Neither of these models addressed the release and infectivity of ECM-bound virus, but our findings herein suggest the applicability of our model where HPVs highjack normal HSPG processing to gain infectious entry into keratinocytes (Fig. 7). The inhibited viral release from the ECM, cellular entry and infectivity from xECM can be readily explained by our model where proteases and heparanase play an important role in HPV release from primary receptors.
Fig. 7. Schematic presentation of ECM-bound HPV16 release and infectious entry in HKs. (a) Highly charged HPV molecules may bind many ECM constituents (e.g. snd-1 or LN-332) by direct protein–protein interaction; however, a considerable amount of virus binds via the proteoglycan HS chains. Heparanase (i) or proteases (ii) process the ECM, resulting in release of virus attached to fragments of ECM-resident HS or proteins, respectively. Soluble virus can attach to HKs via the specificity of ligands in complex with HPV16 binding to cognate receptors (iii), or less specifically (iv). (b) Cross-linking of ECM-resident proteins with DTSSP (X) does not affect HPV16 binding, but inhibits virus release by preventing protease digestion of the ECM. HPV binds to the HS in the ECM or to GF-decorated HS chain of shed snd-1. Heparinase III catalyses virus particle released still in complex with some ligands (e.g. GFs) attached to HS chains (v) or with fragments of HS (vi). Liberated HPV could attach to HKs by direct protein–protein interaction of charged capsids with non-specific receptors (vii). Virus released by HS processing could bind to HS-binding HK receptors (viii) or cognate receptors of HS-interacting ligands/GFs complexed with HPV16 (ix).

Our model wherein HPVs usurp HSPG processing and GFR/RTK signalling to promote infection resonates with a role for epithelial wounding in mediating papillomavirus infections in vivo (Shope & Hurst, 1933). Epithelial breaks result in an influx of GFs and cytokines involved in regulating syndecan shedding (Choi et al., 2011). Snd-1 expression is strongly upregulated in migrating and proliferating keratinocytes, and shed syndecans present in wound fluids regulate GFs (Kato et al., 1998) and MMP activities (Tokumaru et al., 2000). Thus, HPVs appear to have evolved to commandeer the epithelial wound, not only to gain access to mitotically active basal cells, but also to usurp wound-responsive factors and architecture that promote infection. Many intracellular pathogens of the female genital tract (HIV, herpesviruses, Chlamydia, Neisseria), interact with cellular HSPGs (Liu & Thorp, 2002). It is thus tempting to speculate these pathogens also appropriate HSPG biology during infection. In summary, our work provides new insights into the transmission of oncogenic HPVs and underscores that pathogens highjack normal cell functions during infection of their hosts. These findings may point to additional targets for preventing HPV infections, and potentially those of similarly acting pathogens.
Methods
Cell culture, pseudovirion production and infections
HaCaT cells were grown as reported previously (Surviladze et al., 2012). All 37 °C incubations were in 5 % CO2. Luciferase-expressing HPV16 PsVs were produced, isolated and quantified as described previously (Campos & Ozbun, 2009; Surviladze et al., 2012). Infections were initiated using PsVs at 100–200 viral genome equivalents per cell and quantified 24 h post exposure as reported previously (Surviladze et al., 2012). Raw data were normalized by protein content.
HPV particle internalization assay
Extracellular virus was removed by treatment with 0.25 % trypsin/5 mM EDTA for 15 min, 37 °C. Pelleted cells were washed with ice-cold PBS and treated with ice-cold 0.1 M sodium acetate buffer (pH 4.5) containing 0.5 M NaCl for 2 min, 4 °C. Cells were washed three times with PBS and lysed with RIPA buffer.
Immunoprecipitation (IP), SDS-PAGE and immunoblotting
ECM lysates were prepared in RIPA buffer [50 mM Tris (pH 7.5), 0.1 % SDS, 1 % sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 % Triton X-100, 1 mM sodium vanadate, 1 mM PMSF, 10 ng leupeptin ml− 1, 10 ng aprotinin ml− 1]. Supernatants (post 10 min at 17 000 g, 4 °C) were subjected to IP or dissolved in Laemmli sample buffer containing 2 % SDS and 5 % β-mercaptoethanol. Affinity purified polyclonal anti-snd-1 antibody coupled to protein A magnetic beads was used for snd-1 IP. After 1 h of incubation at 4 °C, beads were washed with RIPA buffer and PBS and resuspended in Laemmli sample buffer. Samples were boiled and fractionated by SDS-PAGE, transferred onto Immobilon-P membranes (Millipore), and analysed with various antibodies: anti-HPV16 L1 mouse mAb (CAMVIR-1); anti-LN-332 alpha chain mouse mAb (clone P3E4); anti-actin goat polyclonal antibody; anti-CD63 mouse mAb, anti-syndecan-1 mouse mAb and goat polyclonal Ab (all from Santa Cruz Biotech); affinity purified anti-syndecan-1 rabbit polyclonal antibody (Millipore). Antigen–antibody interactions and visualizations were as reported previously (Surviladze et al., 2012). Stripped blots were reprobed with actin antibody; after scanning, the intensity of individual bands was quantified using AlphaEaseFC (Alpha Innotech).
Preparation of HK ECM
HaCaT cell-derived ECM was prepared as described by Kariya et al. (2012). Briefly, subconfluent cells were removed from plates by incubation with 10 mM EDTA and washing three times with PBS. Cells remaining on the plates were completely removed with 20 mM NH4OH for 5 min, room temperature (RT) and additional PBS washing. ECM preparations were verified microscopically to be cell-free. ECM was incubated with DMEM/FCS or BSA for 1 h, RT to reduce non-specific binding.
Keratinocyte-to-ECM add-back experiments
ECM was prepared as described above. PsVs were added and incubated for 1 h, RT. Unbound PsVs were removed by washing three times with PBS and twice with complete medium (CM). HaCaT cells were seeded to be 60 % confluent atop ECM-bound PsVs.
Soluble HPV16 pull-down assay
ECM-bound HPV16 was washed intensively and incubated at 37 °C in CM. Media were collected after various time points, centrifuged to remove cell debris, and released PsVs were captured with heparin-agarose beads (Sigma-Aldrich) for 1 h, 4 °C (Joyce et al., 1999; Kim et al., 2009; Faust et al., 2010). Beads were washed three times with PBS, resuspended with SDS sample buffer, and analysed by SDS-PAGE and immunoblot for HPV16 L1.
ECM cross-linking
ECM components were cross-linked with 1 mM DTSSP (Thermo Scientific) following the manufacturer's instructions; reactions were quenched with 50 mM Tris pH 7.5 for 15 min, RT. After three PBS washes, 1 % BSA was added for 1 h, RT to reduce non-specific interactions on the plate.
Isolation of EL fraction
HPV16-bound HaCaT cells were cultivated for 24 h in the presence of 3 μM furin inhibitor 1 (dec.-RVKR-CMK; Millipore), then washed with PBS. Fractionation buffer (10 mM Tris, pH 7.0, 250 mM sucrose) was added, cells were dislodged manually, then passed ten times through a 27-gauge needle. The EL fraction was obtained as described by Schröter et al. (1999), resuspended in furin assay buffer (100 mM HEPES, pH 7.4, 0.5 % Triton X-100, 1 mM CaCl2 and 1 mM β-mercaptoethanol).
Furin activity
Fluorometric assays were performed as reported by Molloy et al. (1992). Briefly, the reaction contained 75 μM Boc-RVRR-AMC furin substrate (Enzo Biochem) in assay buffer and 3 nM recombinant furin, conditioned medium or EL lysate for 3 h, 30 °C. In some cases 3 μM furin inhibitor 1 was present. The AMC cleavage peptide was quantified by fluorometry (380 nm excitation, 460 nm emission).
Acknowledgements
This work was supported by a RAC grant (Z. S.) from the University of New Mexico and a US National Institutes of Health grant (CA132136 to M. A. O.). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. We thank N. Fusenig for HaCaT cells, M. Müller for the HPV16-L1/L2 plasmid and C. Buck for 293TT cells. We are grateful to N. Patterson for excellent technical assistance. We offer special appreciation to T. Roitbak for insightful discussions and reagent sharing.
Supplementary Data
Supplementary Data
References
- Bayer-Garner I.B., Sanderson R.D., Dhodapkar M.V., Owens R.B., Wilson C.S. (2001). Syndecan-1 (CD138) immunoreactivity in bone marrow biopsies of multiple myeloma: shed syndecan-1 accumulates in fibrotic regions Mod Pathol 14 1052–1058 10.1038/modpathol.3880435 . [DOI] [PubMed] [Google Scholar]
- Bernfield M., Götte M., Park P.W., Reizes O., Fitzgerald M.L., Lincecum J., Zako M. (1999). Functions of cell surface heparan sulfate proteoglycans Annu Rev Biochem 68 729–777 10.1146/annurev.biochem.68.1.729 . [DOI] [PubMed] [Google Scholar]
- Bishop J.R., Schuksz M., Esko J.D. (2007). Heparan sulphate proteoglycans fine-tune mammalian physiology Nature 446 1030–1037 10.1038/nature05817 . [DOI] [PubMed] [Google Scholar]
- Burkard C., Bloyet L.-M., Wicht O., van Kuppeveld F.J., Rottier P.J.M., de Haan C.A.M., Bosch B.J. (2014). Dissecting virus entry: replication-independent analysis of virus binding, internalization, and penetration using minimal complementation of β-galactosidase PLoS One 9 e101762 10.1371/journal.pone.0101762 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos S.K., Ozbun M.A. (2009). Two highly conserved cysteine residues in HPV16 L2 form an intramolecular disulfide bond and are critical for infectivity in human keratinocytes PLoS One 4 e4463 10.1371/journal.pone.0004463 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos S.K., Chapman J.A., Deymier M.J., Bronnimann M.P., Ozbun M.A. (2012). Opposing effects of bacitracin on human papillomavirus type 16 infection: enhancement of binding and entry and inhibition of endosomal penetration J Virol 86 4169–4181 10.1128/JVI.05493-11 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carulli S., Beck K., Dayan G., Boulesteix S., Lortat-Jacob H., Rousselle P. (2012). Cell surface proteoglycans syndecan-1 and -4 bind overlapping but distinct sites in laminin α3 LG45 protein domain J Biol Chem 287 12204–12216 10.1074/jbc.M111.300061 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerqueira C., Liu Y., Kühling L., Chai W., Hafezi W., van Kuppevelt T.H., Kühn J.E., Feizi T., Schelhaas M. (2013). Heparin increases the infectivity of Human Papillomavirus type 16 independent of cell surface proteoglycans and induces L1 epitope exposure Cell Microbiol 15 1818–1836 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesson H.W., Ekwueme D.U., Saraiya M., Watson M., Lowy D.R., Markowitz L.E. (2012). Estimates of the annual direct medical costs of the prevention and treatment of disease associated with human papillomavirus in the United States Vaccine 30 6016–6019 10.1016/j.vaccine.2012.07.056 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S., Lee H., Choi J.R., Oh E.S. (2010). Shedding; towards a new paradigm of syndecan function in cancer BMB Rep 43 305–310 10.5483/BMBRep.2010.43.5.305 . [DOI] [PubMed] [Google Scholar]
- Choi Y., Chung H., Jung H., Couchman J.R., Oh E.-S. (2011). Syndecans as cell surface receptors: unique structure equates with functional diversity Matrix Biol 30 93–99 10.1016/j.matbio.2010.10.006 . [DOI] [PubMed] [Google Scholar]
- Combita A.L., Touzé A., Bousarghin L., Sizaret P.-Y., Muñoz N., Coursaget P. (2001). Gene transfer using human papillomavirus pseudovirions varies according to virus genotype and requires cell surface heparan sulfate FEMS Microbiol Lett 204 183–188 10.1111/j.1574-6968.2001.tb10883.x . [DOI] [PubMed] [Google Scholar]
- Culp T.D., Budgeon L.R., Christensen N.D. (2006a). Human papillomaviruses bind a basal extracellular matrix component secreted by keratinocytes which is distinct from a membrane-associated receptor Virology 347 147–159 10.1016/j.virol.2005.11.025 . [DOI] [PubMed] [Google Scholar]
- Culp T.D., Budgeon L.R., Marinkovich M.P., Meneguzzi G., Christensen N.D. (2006b). Keratinocyte-secreted laminin 5 can function as a transient receptor for human papillomaviruses by binding virions and transferring them to adjacent cells J Virol 80 8940–8950 10.1128/JVI.00724-06 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta J., Bienkowska-Haba M., Ortega M.E., Patel H.D., Bodevin S., Spillmann D., Bishop B., Sapp M., Chen X.S. (2011). Structural basis of oligosaccharide receptor recognition by human papillomavirus J Biol Chem 286 2617–2624 10.1074/jbc.M110.160184 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day P.M., Schelhaas M. (2014). Concepts of papillomavirus entry into host cells Curr Opin Virol 4 24–31 10.1016/j.coviro.2013.11.002 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day P.M., Thompson C.D., Buck C.B., Pang Y.Y., Lowy D.R., Schiller J.T. (2007). Neutralization of human papillomavirus with monoclonal antibodies reveals different mechanisms of inhibition J Virol 81 8784–8792 10.1128/JVI.00552-07 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day P.M., Gambhira R., Roden R.B., Lowy D.R., Schiller J.T. (2008). Mechanisms of human papillomavirus type 16 neutralization by l2 cross-neutralizing and l1 type-specific antibodies J Virol 82 4638–4646 10.1128/JVI.00143-08 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding K., Lopez-Burks M., Sánchez-Duran J.A., Korc M., Lander A.D. (2005). Growth factor-induced shedding of syndecan-1 confers glypican-1 dependence on mitogenic responses of cancer cells J Cell Biol 171 729–738 10.1083/jcb.200508010 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elenius K., Jalkanen M. (1994). Function of the syndecans - a family of cell surface proteoglycans J Cell Sci 107 2975–2982 . [DOI] [PubMed] [Google Scholar]
- Elkin M., Ilan N., Ishai-Michaeli R., Friedmann Y., Papo O., Pecker I., Vlodavsky I. (2001). Heparanase as mediator of angiogenesis: mode of action FASEB J 15 1661–1663 . [DOI] [PubMed] [Google Scholar]
- Faust H., Knekt P., Forslund O., Dillner J. (2010). Validation of multiplexed human papillomavirus serology using pseudovirions bound to heparin-coated beads J Gen Virol 91 1840–1848 10.1099/vir.0.019349-0 . [DOI] [PubMed] [Google Scholar]
- Fears C.Y., Woods A. (2006). The role of syndecans in disease and wound healing Matrix Biol 25 443–456 10.1016/j.matbio.2006.07.003 . [DOI] [PubMed] [Google Scholar]
- Flannery C.R. (2006). MMPs and ADAMTSs: functional studies Front Biosci 11 544–569 10.2741/1818 . [DOI] [PubMed] [Google Scholar]
- Forman D., de Martel C., Lacey C.J., Soerjomataram I., Lortet-Tieulent J., Bruni L., Vignat J., Ferlay J., Bray F., other authors (2012). Global burden of human papillomavirus and related diseases Vaccine 30 (Suppl 5), F12–F23 10.1016/j.vaccine.2012.07.055 . [DOI] [PubMed] [Google Scholar]
- Giroglou T., Florin L., Schäfer F., Streeck R.E., Sapp M. (2001). Human papillomavirus infection requires cell surface heparan sulfate J Virol 75 1565–1570 10.1128/JVI.75.3.1565-1570.2001 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K., Hayashi M., Jalkanen M., Firestone J.H., Trelstad R.L., Bernfield M. (1987). Immunocytochemistry of cell surface heparan sulfate proteoglycan in mouse tissues. A light and electron microscopic study J Histochem Cytochem 35 1079–1088 10.1177/35.10.2957423 . [DOI] [PubMed] [Google Scholar]
- Joyce J.G., Tung J.-S., Przysiecki C.T., Cook J.C., Lehman E.D., Sands J.A., Jansen K.U., Keller P.M. (1999). The L1 major capsid protein of human papillomavirus type 11 recombinant virus-like particles interacts with heparin and cell-surface glycosaminoglycans on human keratinocytes J Biol Chem 274 5810–5822 10.1074/jbc.274.9.5810 . [DOI] [PubMed] [Google Scholar]
- Kariya Y., Sato H., Katou N., Kariya Y., Miyazaki K. (2012). Polymerized laminin-332 matrix supports rapid and tight adhesion of keratinocytes, suppressing cell migration PLoS One 7 e35546 10.1371/journal.pone.0035546 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M., Wang H., Kainulainen V., Fitzgerald M.L., Ledbetter S., Ornitz D.M., Bernfield M. (1998). Physiological degradation converts the soluble syndecan-1 ectodomain from an inhibitor to a potent activator of FGF-2 Nat Med 4 691–697 10.1038/nm0698-691 . [DOI] [PubMed] [Google Scholar]
- Kim C.W., Goldberger O.A., Gallo R.L., Bernfield M. (1994). Members of the syndecan family of heparan sulfate proteoglycans are expressed in distinct cell-, tissue-, and development-specific patterns Mol Biol Cell 5 797–805 10.1091/mbc.5.7.797 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.J., Lim S.J., Kim J.Y., Kim S.Y., Kim H.J. (2009). A method for removing contaminating protein during purification of human papillomavirus type 18 L1 protein from _Saccharomyces cerevisiae_Arch Pharm Res 32 1759–1766 10.1007/s12272-009-2214-x . [DOI] [PubMed] [Google Scholar]
- Kines R.C., Thompson C.D., Lowy D.R., Schiller J.T., Day P.M. (2009). The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding Proc Natl Acad Sci U S A 106 20458–20463 10.1073/pnas.0908502106 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knappe M., Bodevin S., Selinka H.-C., Spillmann D., Streeck R.E., Chen X.S., Lindahl U., Sapp M. (2007). Surface-exposed amino acid residues of HPV16 L1 protein mediating interaction with cell surface heparan sulfate J Biol Chem 282 27913–27922 10.1074/jbc.M705127200 . [DOI] [PubMed] [Google Scholar]
- Koda J.E., Rapraeger A., Bernfield M. (1985). Heparan sulfate proteoglycans from mouse mammary epithelial cells. Cell surface proteoglycan as a receptor for interstitial collagens J Biol Chem 260 8157–8162 . [PubMed] [Google Scholar]
- Lambaerts K., Wilcox-Adelman S.A., Zimmermann P. (2009). The signaling mechanisms of syndecan heparan sulfate proteoglycans Curr Opin Cell Biol 21 662–669 10.1016/j.ceb.2009.05.002 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Thorp S.C. (2002). Cell surface heparan sulfate and its roles in assisting viral infections Med Res Rev 22 1–25 10.1002/med.1026 . [DOI] [PubMed] [Google Scholar]
- Molloy S.S., Bresnahan P.A., Leppla S.H., Klimpel K.R., Thomas G. (1992). Human furin is a calcium-dependent serine endoprotease that recognizes the sequence Arg-X-X-Arg and efficiently cleaves anthrax toxin protective antigen J Biol Chem 267 16396–16402 . [PubMed] [Google Scholar]
- Okamoto O., Bachy S., Odenthal U., Bernaud J., Rigal D., Lortat-Jacob H., Smyth N., Rousselle P. (2003). Normal human keratinocytes bind to the alpha3LG4/5 domain of unprocessed laminin-5 through the receptor syndecan-1 J Biol Chem 278 44168–44177 10.1074/jbc.M300726200 . [DOI] [PubMed] [Google Scholar]
- Pauza C.D., Price T.M. (1988). Human immunodeficiency virus infection of T cells and monocytes proceeds via receptor-mediated endocytosis J Cell Biol 107 959–968 10.1083/jcb.107.3.959 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruessmeyer J., Martin C., Hess F.M., Schwarz N., Schmidt S., Kogel T., Hoettecke N., Schmidt B., Sechi A., other authors (2010). A disintegrin and metalloproteinase 17 (ADAM17) mediates inflammation-induced shedding of syndecan-1 and -4 by lung epithelial cells J Biol Chem 285 555–564 10.1074/jbc.M109.059394 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purushothaman A., Chen L., Yang Y., Sanderson R.D. (2008). Heparanase stimulation of protease expression implicates it as a master regulator of the aggressive tumor phenotype in myeloma J Biol Chem 283 32628–32636 10.1074/jbc.M806266200 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani V.C., Purushothaman A., Stewart M.D., Thompson C.A., Vlodavsky I., Au J.L., Sanderson R.D. (2013). The heparanase/syndecan-1 axis in cancer: mechanisms and therapies FEBS J 280 2294–2306 10.1111/febs.12168 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapraeger A., Jalkanen M., Bernfield M. (1986). Cell surface proteoglycan associates with the cytoskeleton at the basolateral cell surface of mouse mammary epithelial cells J Cell Biol 103 2683–2696 10.1083/jcb.103.6.2683 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X.-X., Ma L., Liu Q.-W., Li C., Huang Z., Wu L., Xiong S.-D., Wang J.-H., Wang H.-B. (2014). The molecule of DC-SIGN captures enterovirus 71 and confers dendritic cell-mediated viral trans-infection Virol J 11 47 10.1186/1743-422X-11-47 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards R.M., Lowy D.R., Schiller J.T., Day P.M. (2006). Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection Proc Natl Acad Sci U S A 103 1522–1527 10.1073/pnas.0508815103 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousselle P., Beck K. (2013). Laminin 332 processing impacts cellular behavior Cell Adhes Migr 7 122–134 10.4161/cam.23132 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders S., Bernfield M. (1988). Cell surface proteoglycan binds mouse mammary epithelial cells to fibronectin and behaves as a receptor for interstitial matrix J Cell Biol 106 423–430 10.1083/jcb.106.2.423 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schelhaas M., Shah B., Holzer M., Blattmann P., Kühling L., Day P.M., Schiller J.T., Helenius A. (2012). Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis PLoS Pathog 8 e1002657 10.1371/journal.ppat.1002657 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller J.T., Day P.M., Kines R.C. (2010). Current understanding of the mechanism of HPV infection Gynecol Oncol 118 S12–S17 10.1016/j.ygyno.2010.04.004 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröter C.J., Braun M., Englert J., Beck H., Schmid H., Kalbacher H. (1999). A rapid method to separate endosomes from lysosomal contents using differential centrifugation and hypotonic lysis of lysosomes J Immunol Methods 227 161–168 10.1016/S0022-1759(99)00079-4 . [DOI] [PubMed] [Google Scholar]
- Selinka H.-C., Giroglou T., Sapp M. (2002). Analysis of the infectious entry pathway of human papillomavirus type 33 pseudovirions Virology 299 279–287 10.1006/viro.2001.1493 . [DOI] [PubMed] [Google Scholar]
- Selinka H.-C., Florin L., Patel H.D., Freitag K., Schmidtke M., Makarov V.A., Sapp M. (2007). Inhibition of transfer to secondary receptors by heparan sulfate-binding drug or antibody induces noninfectious uptake of human papillomavirus J Virol 81 10970–10980 10.1128/JVI.00998-07 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafti-Keramat S., Handisurya A., Kriehuber E., Meneguzzi G., Slupetzky K., Kirnbauer R. (2003). Different heparan sulfate proteoglycans serve as cellular receptors for human papillomaviruses J Virol 77 13125–13135 10.1128/JVI.77.24.13125-13135.2003 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shope R.E., Hurst E.W. (1933). Infectious papillomatosis of rabbits: with a note on the histopathology J Exp Med 58 607–624 10.1084/jem.58.5.607 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J.L., Lidke D.S., Ozbun M.A. (2008). Virus activated filopodia promote human papillomavirus type 31 uptake from the extracellular matrix Virology 381 16–21 10.1016/j.virol.2008.08.040 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorkin A., Duex J.E. (2010). Quantitative analysis of endocytosis and turnover of epidermal growth factor (EGF) and EGF receptor Chapter 15, Unit 15.14 In Current Protocols in Cell Biology. Edited by Bonifacino J. S., Dasso M., Harford J. B., Lippincott-Schwartz J., Yamada K. M. New York: Wiley. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung U., O'Rear J.J., Yurchenco P.D. (1997). Localization of heparin binding activity in recombinant laminin G domain Eur J Biochem 250 138–143 10.1111/j.1432-1033.1997.00138.x . [DOI] [PubMed] [Google Scholar]
- Surviladze Z., Dziduszko A., Ozbun M.A. (2012). Essential roles for soluble virion-associated heparan sulfonated proteoglycans and growth factors in human papillomavirus infections PLoS Pathog 8 e1002519 10.1371/journal.ppat.1002519 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokumaru S., Higashiyama S., Endo T., Nakagawa T., Miyagawa J.I., Yamamori K., Hanakawa Y., Ohmoto H., Yoshino K., other authors (2000). Ectodomain shedding of epidermal growth factor receptor ligands is required for keratinocyte migration in cutaneous wound healing J Cell Biol 151 209–220 10.1083/jcb.151.2.209 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneda A., Couchman J.R. (2003). Regulation of cytoskeletal organization by syndecan transmembrane proteoglycans Matrix Biol 22 25–33 10.1016/S0945-053X(03)00010-6 . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data