Central Nervous System Stromal Cells Control Local CD8+ T Cell Responses during Virus-Induced Neuroinflammation (original) (raw)
Abstract
Stromal cells generate a complex cellular scaffold that provides specialized microenvironments for lymphocyte activation in secondary lymphoid organs. Here, we assessed whether local activation of stromal cells in the central nervous system (CNS) is mandatory to transfer immune recognition from secondary lymphoid organs into the infected tissue. We report that neurotropic virus infection in mice triggered the establishment of such stromal cell niches in the CNS. CNS stromal cell activation was dominated by a rapid and vigorous production of CC-motif chemokine receptor (CCR) 7 ligands CCL19 and CCL21 by vascular endothelial cells and adjacent fibroblastic reticular cell (FRC)-like cells in the perivascular space. Moreover, CCR7 ligands produced by CNS stromal cells were crucial to support recruitment and local re-activation of antiviral CD8+ T cells and to protect the host from lethal neuroinflammatory disease, indicating that CNS stromal cells generate confined microenvironments that control protective T cell immunity.
Keywords: Inflammation, encephalitis, virus infection, stromal cells
Graphical Abstract
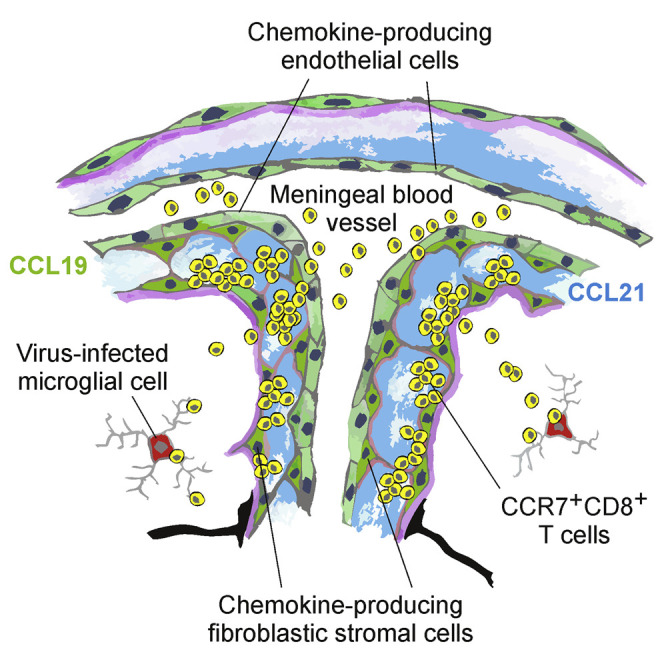
Highlights
- •
CNS stromal cells swiftly generate CCR7 ligands during neurotropic virus infection - •
CCR7-expressing antiviral CD8+ T cells prevent lethal CNS disease - •
Stromal cell-derived CCR7 ligands guide CD8+ T cells to infected target cells
Whether CNS stromal cells regulate protective T cell immunity is unclear. Ludewig and colleagues demonstrate that local chemokine production by activated endothelial and fibroblastic stromal cells is required to transfer protective CD8+ T cell immunity from the draining lymph node to virus-infected areas of the central nervous system.
Introduction
Successful adaptive immune responses against pathogens rely on the activation of lymphocytes in secondary lymphoid organs (SLOs) and the recruitment of effector cells to sites of pathogen replication (Junt et al., 2008). Within SLOs, appropriately stimulated fibroblastic reticular cells (FRCs) generate a cellular scaffold that maintains immune responsiveness (Chai et al., 2013), for example through fostering dendritic cell-T cell interaction (Bajénoff et al., 2006) or supporting B cell activation (Cremasco et al., 2014). Other SLO stromal cells such as blood endothelial cells (BECs) of high endothelial venules (HEVs) regulate SLO cellularity via stringent control of lymphocyte migration (Browning et al., 2005, Mionnet et al., 2011, Onder et al., 2013). The establishment of different cellular compartments within SLOs is mediated to a large extent through the generation of homeostatic chemokines such as the chemokine CC-motif receptor 7 (CCR7) ligands CCL19 and CCL21 (Randall et al., 2008, Förster et al., 2008). Podoplanin (PDPN)- and extracellular matrix protein ER-TR7-expressing FRCs represent a major lymph node (LN) stromal cell population involved in the production of CCR7 ligands (Link et al., 2007, Scandella et al., 2008). In addition, BECs in HEVs and lymphatic endothelial cells (LECs) contribute to production of CCL21 (Gunn et al., 1998). Moreover, CCR7 ligands can be generated in peripheral, non-lymphoid organs (Christopherson et al., 2003, Lo et al., 2003, Krumbholz et al., 2007, Columba-Cabezas et al., 2003, Kivisäkk et al., 2004) where their expression further increases during inflammatory reactions (Pashenkov et al., 2003, Krumbholz et al., 2007, Rangel-Moreno et al., 2007, Burke et al., 2010). These data suggest that CCR7 ligands expressed in non-lymphoid organs might serve as inducible chemoattractants to foster immune surveillance.
Tight control of lymphocyte entry and effector function is particularly important for organs such as the CNS because immunopathological damage of this organ during active immune responses has to be kept at a minimum (Ransohoff and Engelhardt, 2012, Moseman and McGavern, 2013). The finding that T cells migrating into the inflamed CNS express CCR7 (Alt et al., 2002) suggests that T cell entry to the CNS might be controlled via this molecule. Moreover, chronic inflammatory diseases of the CNS such as multiple sclerosis (MS) are associated with elevated CCL19 levels in the cerebrospinal fluid (Pashenkov et al., 2003, Krumbholz et al., 2007) and significantly higher expression in active and inactive lesions (Krumbholz et al., 2007). In an experimental model of MS, both CCL19 and CCL21 can be detected in ECs of inflamed venules and cells that accumulate in the perivascular space (Alt et al., 2002, Columba-Cabezas et al., 2003). However, it has remained elusive whether local activation of fibroblastic and/or endothelial stromal cells in the CNS is required to transfer immune recognition from SLOs into the infected tissue and to what extent these processes control local immune responsiveness during virus-induced neuroinflammatory CNS disease.
To address these questions, we utilized intranasal infection with a neurotropic coronavirus that leads to a transient encephalitis in immunocompetent animals but causes chronic demyelinating and lethal disease when the host is immunodeficient (Bergmann et al., 2006, Gil-Cruz et al., 2012). Here, we focused on the events that take place early during establishment of the infection when viral replication has caused a rapid and more than 1,000-fold upregulation of Ccl19 and Ccl21 gene expression in the olfactory bulb. We found that two distinct stromal cell populations, i.e., ECs of meningeal and sub-meningeal venules and PDPN+ER-TR7+ FRC-like cells in the perivascular space, served as major source for CCR7 ligands. CCR7 expression on antiviral T cells was mandatory to prevent lethal neuroinflammatory disease, while provision of extra-lymphatic CCL21 ensured sufficient CD8+ T cell recruitment to and re-activation in the CNS to protect the host from severe disease. We therefore conclude that activation of CNS stromal cells is critical for optimal control of neurotropic viral infection and that this function is executed—at least partially—through the swift provision of CCR7 ligands.
Results
Rapid Production of CCR7 Ligands during Neurotropic Virus Infection
Viral infection of the CNS triggers activation of several inflammatory cascades including the generation of chemokines (Hosking and Lane, 2010). To assess whether infection of mice with neurotropic viruses triggers a distinct program of stromal cell activation including expression of chemokines, we infected mice with the mouse hepatitis virus (MHV) strain A59 via the intranasal route. As shown in Figure 1A, infectious viral particles were first detectable in olfactory bulbs and cervical LNs (cLNs) on day 2 post infection (p.i.). By day 4, viral titers had reached peak values in olfactory bulbs and the virus had spread to distal CNS areas, including the spinal cord. After 6 days, the virus had been eliminated from draining cLNs, while clearance from CNS tissues was not achieved until day 10 (Figure 1A). In order to visualize viral dissemination at the peak of the infection and to identify infected cells, we analyzed paramedian sagittal sections of infected brains (day 6 p.i.) by confocal laser scanning microscopy using antibodies against the MHV nucleoprotein (MHV-N), activated microglial cells (ionized calcium-binding adaptor molecule 1/Iba-1) and neurons (neuronal nuclei, NeuN) (Figure 1B, i). Strong viral antigen expression was detectable in rostral regions of the brain such as the olfactory bulb, anterior olfactory nucleus, and the piriform cortex (Figure 1B, i and Figure S1A). Infected microglial cells revealed by anti-Iba-1 and anti-MHV-N co-staining, were found, for example, in the olfactory bulb (Figure 1B, ii). Moreover, infected NeuN+ neurons were detected in the anterior olfactory nucleus (Figure 1B, iii) and aspartoacylase (ASPA)- (Figure 1B, iv) or Olig2-expressing (data not shown) oligodendrocytes staining positive for MHV-N were found in the locus coeruleus. The finding that both neurons and glial cells stained positive for MHV-N supports the notion that the virus utilizes axonal transport (Perlman et al., 1990) and cell-to-cell spread (Gallagher and Buchmeier, 2001) as means of dissemination. To assess whether the viral infection had triggered the generation of microenvironments that support lymphocyte activation, we performed quantitative RT-PCR analysis of olfactory bulb tissue at the peak of the infection. Interestingly, mRNA expression of inflammatory chemokines and several genes involved in lymphoid organogenesis such as Ltb, Ltbr, and Il7 were substantially upregulated in the infected olfactory bulb (Figure 1C). Likewise, expression of the homeostatic chemokines Ccl19, Ccl21, and Cxcl13 increased substantially in inflamed CNS tissue compared to naive controls (Figure 1C). Since the increase in relative expression of CCR7 ligands was most prominent in olfactory bulb and cLN, we reasoned that processes driving production of these chemokines in the cLN were mirrored during local inflammatory reactions in the CNS. As shown in Figures 1D and 1E, expression of both Ccl19 and Ccl21 mRNA increased roughly 1,000-fold at their peak on day 20 in the olfactory bulb and the pattern of chemokine activation in different CNS areas was accompanied by the infiltration of inflammatory cells (Figures S1B and S1C). Elevated chemokine expression was only transient and followed the resolution of the viral infection with substantially lower expression on day 20 post infection (Figures 1D and 1E). Thus, the presence of actively replicating virus within the CNS, particularly in the olfactory bulb as virus entry point, drives a rapid increase in transcriptional activity of Ccl19/Ccl21 genes, a process that is paralleled by infiltration of immune cells and upregulation of genes associated with stromal cell activation.
Figure 1.

Viral Replication in the CNS Drives Strong Induction of CCR7 Ligand Expression
(A) C57BL/6 (WT) mice were infected i.n. with 5 × 104 pfu MHV A59. Viral titers in cervical lymph nodes (cLN) and the indicated CNS regions were determined at different days post infection. Values indicate mean of log transformed values ± SEM from two independent experiments (n = 6 mice per time point).
(B) Distribution of MHV infected cells visualized by staining against MHV-N, Iba-1 and NeuN on paramedian sagittal brain section (day 6 p.i.) (i); high resolution analysis of infected Iba-1+ microglial cells in olfactory bulb (ii), NeuN+ neurons of the anterior olfactory nucleus (iii) and ASPA+ oligodendrocytes of the locus coeruleus (iv); scale bars equal 1 mm (i) and 10 μm (ii–iv); (representative images from two experiments with four mice).
(C) Quantitative RT-PCR analysis of genes encoding for inducible and homeostatic chemokines and genes associated with lymphoid organogenesis in cervical lymph nodes (cLN) and olfactory bulb of infected mice (day 6 p.i.) compared to naive controls. Spatial and temporal regulation of (D) Ccl19 and (E) Ccl21 mRNA was analyzed at the indicated time points by quantitative RT-PCR. Values indicate mean ± SEM from two independent experiments (n = 4 mice per time point). See also Figure S1.
Activated CNS Stromal Cells Produce CCL19 and CCL21
While CCL21 decorates extracellular matrix proteins, CCL19 is rapidly internalized and degraded following binding to CCR7 (Comerford et al., 2013). Hence, to localize CCL19-producing cells in situ, we took advantage of the recently described Ccl19-cre bacterial artificial chromosome transgenic mouse that facilitates expression of the Cre recombinase in FRCs of secondary lymphoid organs (Chai et al., 2013). We crossed Ccl19-cre mice with Rosa26-eyfp mice (referred to as Ccl19 eyfp) to label cells exhibiting Ccl19 promoter activity with the yellow fluorescent protein. Combination of this detection method with anti-CCL21 immunostaining in confocal microscopic analysis of olfactory bulb tissue of naive Ccl19 eyfp mice revealed only faint CCL21 staining and few cells that exhibited Ccl19 promoter activity (Figure S2A). In contrast, cells showing Ccl19-cre activity and anti-CCL21 staining were clearly detectable at day 6 post infection and were mainly confined to meningeal and sub-meningeal areas (Figures 2A and 2B, magnified in boxed areas). Nevertheless, PDPN expression, which serves as a marker for FRC activation (Chai et al., 2013), was strongly increased in regions of elevated CCR7 ligand production (Figure 2B). While mRNA expression analysis revealed rather strong induction of Cxcl13 transcript levels (Figure 1C), expression of CXCL13 protein was found only in very few cells (Figures S2B and S2C).
Figure 2.

Viral Infection Drives Local Activation of CNS Stromal Cells
Confocal microscopic analysis of Ccl19 gene expression, CCL21 production, and ICAM1 upregulation MHV-infected olfactory bulbs of _Ccl19_eyfp mice. (A) Olfactory bulbs of infected _Ccl19_eyfp mice stained with the indicated antibodies at day 6 p.i. Sagittal overview of the olfactory bulb. Scale bar represents 200 μm. Boxed areas show magnified meningeal and sub-meningeal regions, scale bar represents 20 μm.
(B) Ccl19 gene activity as revealed by EYFP expression on day 6 p.i. Olfactory bulb sections were stained with antibodies against PDPN and CCL21 and counterstained with DAPI. Merged channels are shown in the upper left panel; scale bar represents 200 μm. Boxed areas show magnified meningeal and sub-meningeal regions; scale bar represents 20 μm.
(C–F) High-resolution analysis of meningeal and submeningeal areas in virus-infected olfactory bulb tissue using antibodies against the indicated molecules and counterstaining with DAPI; arrows indicate BECs, arrowheads indicate FRC-like cells. (C) Meningeal blood vessel is shown; scale bar represents 30 μm. (D) Submeningeal postcapillary venule is shown; scale bar represents 10 μm. (E) ICAM1 upregulation in meningeal blood vessel from day 6 infected animal with inlet showing a comparable blood vessel from naive control; scale bars represent 30 μm. (F) ICAM1 expression of PDPN+EYFP+ FRC-like perivascular cells; scale bars represent 30 μm. Representative images from three independent experiments (n = 6 mice). See also Figure S2.
High resolution microscopic analysis of day 6 olfactory bulb tissue revealed considerable EYFP expression in inflamed ECs of meningeal blood vessels (Figure 2C, arrows). Moreover, PDPN+ER-TR7+ fibroblastic cells lined the perivascular space around meningeal blood vessels and in the subarachnoid space that surrounds blood vessels protruding into the CNS parenchyma (also known as Robin-Virchow space; Ransohoff and Engelhardt, 2012) (Figure 2C, arrowheads). Likewise, CD31+ ECs of inflamed blood vessels in the olfactory bulb parenchyma (Figure 2D, arrows) and fibroblastic cells lining the perivascular space (Figure 2D, arrowheads) displayed Ccl19 promoter-driven EYFP activity and CCL21 expression. Stromal cell activation with expression of CCL21 was most pronounced in the vicinity of virus-infected cells (Figure S2D). In addition, ECs of meningeal blood vessels (Figure 2E, arrows) and perivascular EYFP+ fibroblastic cells (Figure 2F, arrowheads) had upregulated expression of the intercellular adhesion molecule 1 (ICAM1, CD54), whereas only BECs showed weak ICAM1 expression in the absence of CNS inflammation (Figure 2E, inlet). The numbers of EYFP-expressing cells in infected olfactory bulbs increased almost 10-fold within the first 4 days of the infection (Figure S2E) with a substantial expansion of both PDPN+ fibroblastic and CD31+ endothelial stromal cells (Figure S2F). Moreover, we found that only very few of the EYFP+ stromal cells expressed the proliferation marker Ki67 (data not shown), indicating that the increase in EYFP+ stromal cells was a result of their activation with enhanced expression of homeostatic chemokines.
Further flow cytometric analysis of CNS stromal cells (Figures 3A–3C) confirmed that both stromal cell populations responded to the infection with substantial upregulation of ICAM1, vascular cell adhesion molecule 1 (VCAM1, CD106) and MHC class I (Figures 3D and 3E). Moreover, fibroblastic stromal cells from infected CNS tissue expressed substantially less CD44 molecules, while expression of markers such as CD140a and CD140b was not affected (Figures 3D and 3E). CCL21 expression was detectable in both stromal cell populations during the infection (Figure 3F), while other CNS cells such as NeuN+ neurons, CD11b+ macrophages and/or microglia, ASPA+ oligodendrocytes, and GFAP+ astrocytes did not express CCL21 at day 6 post infection (Figure S3). These data indicate that two distinct stromal cell populations serve as the main source of CCR7 ligands at hotspots of neurotropic virus replication in the infected CNS.
Figure 3.

Virus Infection-Induced Activation and Chemokine Expression of CNS Stromal Cells
Flow cytometric analysis of myelin- and CD45-depleted cells from olfactory bulbs of C57BL/6 mice.
(A–C) Fibroblastic and endothelial stromal cells were distinguished using gating on CD45-negative (A) and glial fibrillary acidic protein (GFAP)-negative (B) cells and staining for CD31 and PDPN expression (C); representative dot plot analysis with quadstat values for CD31 and PDPN expression.
(D) Expression of fibroblastic stromal markers ICAM1, VCAM1, MHC I, CD140a, CD140b, and CD44 on PDPN+CD31– fibroblastic CNS stromal cells from naive (upper panels) and day 6 infected animals (lower panels).
(E) Expression of ICAM1, VCAM1, and MHC I on PDPN–CD31+ endothelial stromal populations from naive (upper panels) and infected animals (lower panels).
(F) Intracellular CCL21 production by PDPN+CD31– fibroblastic (left panel) and PDPN–CD31+ endothelial (right panel) stromal cells. Values indicate mean percentage of positive cells ± SEM compared to isotype control staining (gray shaded histograms); representative plots from two independent experiments (n = 4–6 mice per group). See also Figure S3.
Lethal Neuroinflammatory CNS Disease in the Absence of CCR7
The vigorous but locally restricted activation of stromal cells with dominant production of CCL19 and CCL21 during the response to neurotropic coronavirus infection suggested that these chemokines could contribute to the control of the virus. To test this hypothesis, we assessed whether mice lacking either CCR7 or CCR7 ligand expression in SLOs display differences in their defense against neurotropic virus infection. The latter mouse strain, known as plt/plt (paucity of lymph node T cells) (Gunn et al., 1999) shows expression of the CCL21b isoform in extra-lymphatic tissues (Nakano and Gunn, 2001). Notably, both mouse strains exhibit impaired T cell zone development in SLOs due to migration defects of DCs and lymphocytes (Gunn et al., 1999, Förster et al., 1999), hence showing a comparable loss of immunocompetence in response to systemic virus infection (Junt et al., 2002, Junt et al., 2004). While _Ccr7_-sufficient mice successfully controlled the infection and experienced transient weight loss during the first 2 weeks, _Ccr7_-deficient mice presented with rapid weight loss early after infection, succumbing to neuroinflammatory disease by day 10 (Figure 4A). In sharp contrast, all plt/plt mice survived the phase of transient weight loss (Figure 4A) and successfully controlled the infection as shown by significantly lower viral titers in brain tissues compared to Ccr7 −/− mice (Figure 4B). Impaired viral control of _Ccr7_-deficient and plt/plt mice was associated with a pronounced reduction in antiviral CD8+ T cell numbers (Figure 4C) and function (Figure 4D), as well as a reduction in antiviral CD4+ T cell function (Figure 4E) in the whole brain on day 10 p.i. Moreover, recruitment of CD8+ T cells into the olfactory bulb was affected already on day 4 by the _Ccr7_-deficiency (Figure S4A). The lack of CCR7 almost completed abolished the appearance of interferon (IFN)-γ-producing antiviral CD8+ T cells in all CNS regions (Figures S4B and S4C), while CD4+ T cell recruitment was affected to a lesser extent (Figures S4D and S4E). Importantly, _Ccr7_-deficient mice showed a stronger impairment of antiviral CD8+ T cell responses compared to plt/plt mice (Figures 4C and 4D). The lack of chemokine receptor CXCR3 that binds the inflammatory chemokines CXCL9 and CXCL10 and that has been implicated in the regulation of T cell migration in several models of neuro-inflammation (Bagai et al., 2005), did not affect survival following intranasal MHV A59 infection (Figure S4F). Likewise, the absence of CXCR5, i.e., the receptor for the B cell zone cytokine CXCL13, did not increase the susceptibility of the animals to MHV CNS infection (Figure S4G). Therefore, it appears that the ability of T cells to sense extra-lymphatic expression of the chemokines CCL19 and CCL21 through CCR7 plays a major role in the rapid control of neurotropic coronavirus infection.
Figure 4.

High Susceptibility to Neurotropic Viral Infection in _Ccr7_-Deficient Mice
(A) Weight loss of _Ccr7_-deficient, plt/plt, and WT mice was recorded during the indicated time period following infection with MHV A59. Values indicate mean percentage of the initial weight ± SEM from two independent experiments (n = 7–10 mice per group).
(B) Viral titers in CNS tissues as determined at day 10 after infection. Data indicate means of log transformed values ± SEM from two independent experiments (n = 8 mice per group).
(C) Tetramer-binding and IFN-γ production of (D) CD8+ and (E) CD4+ T cells from the whole brain was determined by flow cytometry on day 10 after infection. Representative dot plots analysis with percentage of virus-specific T cell population indicated. Graphs indicate mean percentage ± SEM of the respective virus-specific T cell populations from two independent experiments (n = 8 mice per group). Statistical analysis was performed using the Student’s t test (∗∗p < 0.01; ∗∗∗p < 0.001; n.s. not significant). See also Figure S4.
Rescue from Lethal Neuroinflammatory Disease by CCR7-Expressing CD8+ T Cells
Viral replication kinetics and the fast disease course suggested that appropriate activation of antiviral CD8+ T cells and elimination of the highly cytopathic pathogen from the CNS is of utmost importance to prevent lethal disease. To assess the activation and migration pattern of MHV-specific CD8+ T cells in vivo, we utilized a transgenic mouse expressing a H2-Kb-restricted TCR that recognizes the immunodominant epitope s598–605 of the MHV spike protein (TCR-S or Spiky). Adoptive transfer of congenic carboxy-fluorescein succimidyl ester (CFSE)-labeled Spiky cells into infected recipients revealed first activation in cLNs at 72 hr p.i. followed by strong proliferation (Figure 5A). Interestingly, accumulation of activated, CFSElow Spiky cells in the infected CNS could be detected on day 4 at the earliest (Figure 5B), indicating that the cells had to undergo at least 8 rounds of proliferation in the cLN before they gained the ability to migrate to the CNS. As expected, activation of Spiky cells in cLNs resulted in downregulation of CCR7 (Figure 5C). Activated Spiky cells circulating in blood and those that had infiltrated the CNS expressed CCR7 (Figure 5C) indicating their ability to sense CCR7 ligands produced by ECs and FRC-like cells in inflamed regions of the CNS. CCR7 expression detected on CNS-infiltrating Spiky cells was comparable to polyclonal CD8+ T cells that had infiltrated different regions of the CNS (Figure S5). Moreover, Spiky cells derived from cLNs, blood and the brain on days 4 and 8 post infection had downregulated CD62L (Figure 5D). Functional characterization of CNS-infiltrating Spiky cells revealed that these cells were fully activated effector T cells as shown by secretion of the cytokines IFN-γ and tumor necrosis factor alpha (TNF-α), and expression of the degranulation marker CD107a (lysosomal-associated membrane protein 1, LAMP1) (Figures 5E and 5F). These data support the notion that access of polyfunctional effector CD8+ T cells to the CNS is important for successful elimination of the virus from this highly vulnerable tissue.
Figure 5.

CCR7-Dependent Activation and Recruitment of Antiviral CD8+ T Cells Protects from Virus-Induced CNS Disease
Thy1.2+ WT mice received 106 TCR transgenic Thy 1.1+ Spiky cells labeled with the intracellular dye carboxy-fluorescein succimidyl ester (CFSE) and were subsequently infected i.n. with MHV. CFSE dilution of transferred Thy1.1+ CD8+ T cells from the cLNs (A) and brain tissue (B) was determined by flow cytometry at the indicated time points post infection. Values in histograms indicate percentage of proliferated cells (black line) compared to naive controls (gray histogram); representative data from two independent experiments (n = 5 mice).
(C) CCR7 and (D) CD62L expression on Thy1.1+CD8+ T cells from cLNs, blood and the brain was performed at the indicated time points post infection. Values in histograms indicate mean fluorescent intensity (MFI) values of the total Thy1.1+CD8+ T population (black lines) minus MFI of isotype controls (gray shaded lines) ± SEM. Data from two independent experiments (n = 5 mice). Expression of effector cytokines TNF-α and IFN-γ and the degranulation marker Lamp-1 by CNS infiltrating Thy1.1+CD8+ T cells (day 8 p.i.) following in vitro peptide restimulation is shown in representative dot plots (E) and summarized in a bar graph (F) with mean percentage ± SEM pooled from two independent experiments (n = 5 mice). _Ccr7_-deficient mice were adoptively transferred with a total of 107 of CD3+_Ccr7_-proficient, CD3+_Ccr7_-deficient, or 106 of transgenic Spiky cells and infected with MHV.
(G) Survival and (H) weight loss of the mice were recorded during the indicated period of time. Values in (H) indicate mean percentage of the initial weight ± SEM pooled from three independent experiments (n = 9 mice per group). See also Figure S5.
To investigate whether CCR7 expression on virus-specific CD8+ T cells is sufficient to secure control of neurotropic virus infection, we adoptively transferred _Ccr7_-proficient Spiky cells into _Ccr7_-deficient mice and infected the recipients with MHV. Controls included adoptive transfer of sorted polyclonal CD3+ cells from _Ccr7_-proficient or -deficient mice. As expected, _Ccr7_-deficient mice that had received _Ccr7_-deficient CD3+ cells were not protected from MHV CNS infection (Figures 5G and 5H, open red circles), while _Ccr7_-proficient CD3+ T cells provided protection (Figures 5G and 5H, gray circles). _Ccr7_-deficient mice receiving transfer of _Ccr7_-proficient Spiky CD8+ T cells showed a mild disease course and survived the infection (Figures 5G and 5H, white circles) indicating that protection against this neurotropic viral infection depends on the ability of antiviral CD8+ T cells to respond to CCR7 ligands.
Stromal Cell-Derived CCR7 Ligands Promote Recruitment of CD8+ T Cells to and Their Re-activation in the CNS
To determine whether the effects of CCR7 ligands on CD8+ T cell performance was due to the production of CCL19 and CCL21 in the draining cLN or a consequence of the substantial generation of these chemokines in the CNS, we used transfer of _Ccr7_-proficient and -deficient CD8+ Spiky cells into plt/plt mice. These mice are well-suited to address this question because they lack CCL19 and CCL21 expression in lymphoid organs (Gunn et al., 1999, Nakano and Gunn, 2001) but show production of CCL21 by stromal cells in MHV-infected olfactory bulbs (Figure 6A). To assess early CD8+ T cell activation, we transferred _Ccr7_-proficient or -deficient TCR Spiky cells into plt/plt mice and compared their activation status in cLNs and the brain 4 days after infection. We found that expansion and activation of antiviral CD8+ cells in cLNs was not dependent on CCR7 (Figure 6B). Importantly, accumulation of _Ccr7_-deficient TCR Spiky cells in the CNS was significantly reduced, while _Ccr7_-proficient or -deficient Spiky cells showed comparable activation marker profiles (Figure 6C). By day 6 p.i., the numbers of _Ccr7_-proficient Spiky CD8+ T cells infiltrating the CNS were still significantly higher compared to _Ccr7_-deficient cells (Figure 6D). Importantly, the presence or absence of CCR7 on antiviral CD8+ T cells did not affect their numbers in cLNs (Figure 6E) nor their proliferation (Figure 6F) or apoptosis (Figure 6G) in the CNS. To more closely mirror the kinetics of polyclonal CD8+ T cell activation, only very low numbers of Spiky cells (i.e., 104) that expressed CCR7 and the congenic marker Ly5.1 or were _Ccr7_-deficient and labeled with the Thy1.1 marker were co-transferred into plt/plt mice. During the peak of the CNS infection, i.e., at day 6, frequencies of both cell types were comparable in the draining LN (Figures 6H and 6I) and their activation was not affected by the _Ccr7_-deficiency (Figure 6J). Again, lack of CCR7 in Spiky cells had a pronounced effect on their accumulation in the virus-infected CNS resulting in a roughly 80% reduced frequencies compared to _Ccr7_-proficient cells (Figures 6H and 6I). Moreover, virus-specific, _Ccr7_-deficient CD8+ T cells that had reached the CNS exhibited impaired IFN-γ production (Figure 6J). These experiments show that local expression of CCR7 ligands not only supports the recruitment of virus-specific CD8+ T cells to the CNS, but is also required for the execution of their antiviral activity.
Figure 6.

Expression of CCR7 Ligands in the CNS Controls CD8+ T Cell Recruitment and Function
(A) Intracellular CCL21 production by PDPN+CD31− cells from cLNs and olfactory bulbs of WT (black lines) and plt/plt (red lines) mice either infected with MHV (solid lines) or left naive (dashed lines). Data are representative of two independent experiments (n = 4 mice per group).
(B and C) 105 TCR Spiky _Ccr7_-proficient or _Ccr7_-deficient CD8+ T cells were adoptively transferred into plt/plt mice 12 hr before infection. Absolute numbers of _Ccr7_-proficient and _Ccr7_-deficient Spiky transgenic cells in (B) cLNs and (C) brain were recorded on day 4 post infection. Representative contour plots of T cell activation markers CD44 and CD62L expressed by _Ccr7_-proficient (black) and _Ccr7_-deficient (red) Spiky cells compared to LN-derived naive control cells (gray contour). Values in the plots represent percentage of activated cells (i.e., CD44high or CD62Llow, respectively). At day 6 post infection, absolute numbers of _Ccr7_-proficient and _Ccr7_-deficient Spiky transgenic cells in (D) brain and (E) cLNs were determined. Frequencies of brain infiltrating Spiky cells expressing markers of (F) proliferation (Ki67) and (G) apoptosis (active caspase 3) were analyzed using flow cytometry day 6 after the infection. Data represent mean value ± SEM from two independent experiments (n = 5–6 mice per group).
(H–J) A mix of 104 of Ly5.1+_Ccr7_-proficient and 104 of Thy1.1+_Ccr7_-deficient Spiky TCR transgenic CD8+ T cells was adoptively transferred into plt/plt mice 12 hr prior to infection. (H) Representative dot plots showing the accumulation of _Ccr7_-proficient or _Ccr7_-deficient Spiky CD8+ T cells in cLNs (left panel) and brain tissue (right panel) of plt/plt mice on 6 day post infection. Values indicate the percentage of the respective CD8+ T cell population. (I) Frequencies of _Ccr7_-proficient and _Ccr7_-deficient TCR transgenic CD8+ T cells in cLNs and CNS of MHV infected plt/plt mice. (J) IFN-γ production by _Ccr7_-proficient and -deficient TCR transgenic CD8+T cells in cLNs and CNS. Data represent mean value ± SEM from two independent experiments (n = 5 mice). Statistical analysis was performed using the Student’s t test (∗∗p < 0.01;∗∗∗p < 0.001; n.s. not significant).
High resolution histological analysis revealed that activated CD8+ T cells were in intimate contact with endothelial cells of inflamed meningeal blood vessels that had upregulated Ccl19 expression (Figures 7A and 7B, arrowheads). Moreover, CD8+ T cells were found to interact with CCL19+PDPN+ perivascular fibroblastic stromal cells (Figures 7A and 7B, arrows). Next, we specifically ablated CCL19-expressing stromal cells using low doses of diphtheria toxin (DT) applied to Ccl19 eyfp mice that express the DT receptor in Cre recombinase-positive cells (Ccl19 eypf/idtr) (Buch et al., 2005, Cremasco et al., 2014). Low dose DT treatment led to a substantial loss of EYFP+ cells in the infected CNS (Figure 7C), but left the FRC network in the cLNs intact (Figure 7C, inlets). Importantly, CNS-restricted ablation of EYFP+ stromal cells resulted in reduced CD8+ T cell accumulation (Figure 7D), while CD4+ T cell recruitment was not altered (Figure 7E). As a consequence, CNS viral titers were significantly elevated in DT-treated Ccl19 eypf/idtr mice on day 8 p.i. (Figure 7F). CD8+ and CD4+ T cell compartments in cLNs were not changed by the DT treatment (Figures 7G and 7H). Moreover, we found that the local stimulation of antiviral CD8+ T cells via CCR7 not only improved the ability of CD8+ T cells to exit meningeal blood vessels, but substantially fostered their access to infected areas in the CNS parenchyma (Figures 7I and 7J). Taken together, these data show that activated CNS stromal cells are potent regulators of local antiviral CD8+ T cell activity and that this interaction depends to a large extent on the CCR7-CCR7 ligand axis.
Figure 7.

Activated CNS Stromal Cells Guide Antiviral CD8+ T Cells to the CNS Parenchyma
(A and B) C57BL/6 (A) and Ccl19 eyfp mice (B) were infected with MHV intranasally and brains were harvested for histological analysis on day 6 post infection. Meningeal and submeningeal areas were inspected for interactions of CD8+ T cells with endothelial cells (arrowheads) and perivascular fibroblasts (arrows). Scale bar represents 10 μm in (A) and 20 μm in (B).
(C) _Ccl19_eypf/idtr mice were infected with MHV intranasally, treated with PBS or diphtheria toxin (DT, 1 ng/g body weight) on days 1 and 3 post infection, and brains and cLNs (inlets) were examined by histological analysis on day 8 post infection using the indicated antibodies. Scale bar represents 30 μm.
(D and E) Absolute numbers of CD8+ (D) and CD4+ (E) T cells, and viral titers (F) in the brains of DT-treated mice.
(G and H) Absolute numbers of CD8+ (G) and CD4+ (H) T cells in cLNs of DT-treated mice. Data are shown as mean ± SEM (n = 4–6 mice per group, pooled from two independent experiments).
(I) Positioning of antiviral CD8+ T cells in olfactory bulb tissue in Ccr7 +/+ and Ccr7 −/− mice on day 6 post infection. Scale bar represents 10 μm.
(J) Quantification of CD8+ T cell density in meningeal/submeningeal (M/SM) and parenchymal (PAR) areas as determined by histological analysis in the olfactory bulbs of Ccr7 +/+ and Ccr7 −/− mice. Data are shown as mean ± SEM (n = 3–4 mice per group, two independent experiments); statistical analysis was performed by Student’s t test (∗p < 0.05; ∗∗p < 0.01).
Discussion
The CNS is considered an immunologically privileged site that becomes visible to immune effector cells under particular inflammatory conditions such as infection with neurotropic viruses (Galea et al., 2007, Moseman and McGavern, 2013). Because the CNS is vital and highly vulnerable, immune responses against neurotropic infectious agents need to be well-controlled, i.e., the efficacy of pathogen elimination has to be high, while immunopathological damage has to be kept minimal. The present study shows that activated antiviral CD8+ T cells utilize CCR7 to optimize migration into the inflamed CNS and to control their functional activity during neurotropic viral infection. Moreover, our findings indicate that CCR7 ligands produced by vascular and perivascular stromal cells in affected CNS regions function as part of a multi-layered control network that secures spatiotemporal focusing of the immune response to areas of viral replication.
Production of CCL19 and CCL21 by stromal cells in LNs and other SLOs is essential to recruit lymphocytes to these sites of immune activation and to induce adaptive immune responses (Förster et al., 2008). CNS infection with a neurotropic coronavirus, as shown here, triggered a moderate and transient increase in Ccl19 and Ccl21 gene expression in cLNs. It is possible that virus and viral products have been drained via lymphatic vessels that connect the dura to the cLNs (Aspelund et al., 2015). Within the CNS, both ECs and perivascular FRC-like cells showed swift and substantial production of CCR7 ligands. The expression of CCR7 ligands can be induced via the ligation of the LTβR in fibroblastic (Chai et al., 2013) and in endothelial stromal cells (Onder et al., 2013). Moreover, acute inflammatory mediators such as TNF can foster activation of the CCR7/CCL19 axis (Bosè et al., 2013). It is likely that the initial upregulation of Ccl19 and Ccl21 mRNA expression in MHV infected CNS is triggered via inflammatory cytokines such as TNF. Notably, microglial cells are a prominent source of TNF during viral encephalitis (Ghoshal et al., 2007). Thus, infection of microglia cells, a major target cell population of MHV in the CNS, could trigger secretion of TNF that leads to the initial low-level induction of CCL19/CCL21 expression in stromal cells. With the immigration of activated, lymphotoxin-expressing T cells, as demonstrated in our study, fibroblastic and endothelial stromal cells would receive additional activating signals to change into activation mode including massive production of CCR7 ligands.
In addition to their enhanced chemokine production, the phenotype of ECs and FRC-like cells in the affected regions adapted properties that are typical for SLO stromal cells with high level expression of adhesion molecules and upregulation of MHC class I (Malhotra et al., 2012). Because expression of selectins and integrins by inflamed brain vessels reduces the rolling velocity of lymphocytes and permits prolonged interactions with the endothelial wall (Engelhardt, 2006), focal activation of blood vessel ECs most likely fosters recruitment of T cells to sites of viral replication. FRC-like cells in the perivascular space in the vicinity of virus-infected areas exhibited not only elevated expression of ICAM1, and VCAM1, but also of other markers that are typical for LN FRCs including ER-TR7 and PDPN (Cremasco et al., 2014). Importantly, ER-TR7+ reticular networks have been observed during CNS infection with the lymphocytic choriomeningitis virus (LCMV) (Kim et al., 2009) or during chronic Toxoplasma gondii infection (Wilson et al., 2009). Thus, it is likely that brain vessel ECs and the surrounding FRC-like cells are programmed by the local inflammatory milieu to generate those structures that foster and maintain lymphocyte recruitment and activation.
The presence of CCR7 was mandatory to protect mice from severe neuroinflammatory disease as _Ccr7_-deficient animals succumbed to infection in only 10 days. Notably, presence of CCR7 on virus-specific CD8+ T cells was sufficient to rescue _Ccr7_-deficient animals from lethal disease clearly indicating that CCR7 signaling can be decisive for optimal recruitment of activated CD8+ T cells into the inflamed CNS. Adoptive transfer experiments using _Ccr7_-proficient and -deficient antiviral CD8+ T cells suggested that the lack of CCR7 signals in SLOs can be compensated by other stimuli, but that focal, high-level production of CCR7 ligands at sites of peripheral inflammation is critical for both recruitment and performance of antiviral T cells. The finding that _Ccr7_-deficient splenic CD8+ T cells did not show impaired IFN-γ production during systemic lymphocytic choriomeningitis virus infection (Junt et al., 2004) supports our interpretation that the SLO environment guarantees sufficient immune stimulation to generate protective antiviral immunity. Interestingly, systemic Toxoplasma gondii infection of _Ccr7_-deficient mice leads to rapid replication of tachyzoites and dissemination to several peripheral organs including the brain (Noor et al., 2010). It is striking that parasite infection of the CNS provokes generation of perivascular reticular fibers that are coated with CCL21 and that anti-parasitic CD8+ T cells migrate along these fibers (Wilson et al., 2009). Because viral CNS infection efficiently triggers activation of perivascular FRC-like cells, it is reasonable to assume that the perivascular infrastructure supporting antimicrobial immunity is built by FRC-like stromal cells. As predicted by Perry and colleagues (Galea et al., 2007) these processes therefore substantially diminish immune privilege of the CNS and permit efficient immune surveillance.
In conclusion, our study shows that the rapid generation of CCR7 ligands by stromal cells of the CNS is critical for the containment of neurotropic virus infection. The chemokines can be generated rapidly and at high levels by meningeal and sub-meningeal blood vessel ECs and perivascular FRC-like cells. Once appropriately activated, CNS stromal cells generate a confined micro-environment that permits optimal T cell recruitment and activation. It remains to be determined whether some areas in the CNS provide such T cell activation hubs in a constitutive fashion and thereby foster activation of self-reactive T cells by perivascular dendritic cells (Greter et al., 2005). Moreover, it will be important to assess whether and how immune cells that have entered the CNS via the stromal cell-determined entry points leave the organ after the pathogen has been cleared. It is likely that the recently discovered lymphatic connection to the cLNs (Aspelund et al., 2015, Louveau et al., 2015) serve as exit points for immune cells that had accumulated in the inflamed tissue. Accordingly, dysregulation of immune cell influx versus efflux might foster self-perpetuation of focal immune cell aggregations with development of ectopic lymphoid-like structures (Pitzalis et al., 2014). Thus, stromal cells function as critical gate-keepers for activated immune cells and neuroinflammation might be exacerbated if activation of stromal cells following resolution of CNS infection is not restrained. Further dissection of cellular and molecular mechanisms that control stromal cell activation in the CNS is warranted to delineate potential avenues for attenuation of inflammatory processes in this vulnerable organ.
Experimental Procedures
Mice
The TCR transgenic mouse strain C57BL/6N-Tg(Tcra,Tcrb)577Biat (Spiky) was generated as described previously (Nindl et al., 2012). Ccr7 −/−, Cxcr3 −/−, Cxcr5 −/−, iDTR and plt/plt mice were obtained from the Institute for Laboratory Animal Sciences at the University of Zürich. BAC-transgenic C57BL/6N-Tg(Ccl19-Cre)489Biat (Ccl19-Cre) mice have been previously described (Chai et al., 2013). C57BL/6N (B6) mice were purchased from Charles River Laboratories. All mice were maintained in individually ventilated cages and were used in the age of 6 to 9 weeks. Experiments were performed in accordance with federal and cantonal guidelines (Tierschutzgesetz) under the permission numbers SG09/87 and SG13/04 following review and approval by the Veterinary Office of the Canton of St. Gallen.
Virus Infection and Determination of Virus-Induced Pathology
Mice were infected intranasally using 5 × 104 pfu MHV A59. At indicated time points mice were sacrificed, organs were collected, weighed and stored at −80°C until further analysis. Virus titers were determined from homogenized organs using standard plaque assay on L929 cells.
Adoptive T Cell Transfer
Single-cell suspensions from spleens were prepared by mechanical disruption of the organ and subjected to hypotonic red blood cell lysis. For in vivo proliferation, splenocytes were labeled using CFSE (Molecular Probes) according to the manufacturer’s protocol, and 107 cells (corresponding to 1 × 106 CD8+ TCR transgenic T cells) were transferred intravenously (i.v.) into recipient mice. For in vivo protection and co-transfer study, CD8+ T cells or CD3+ cells from spleens were obtained using anti-CD8 or anti-CD3 MACS beads (Miltenyi Biotec). For assessment of in vivo protection, recipients received 106 cells, while 105 and 104 cells were transferred to plt/plt mice in separate or competitive transfer assays (respectively). At twelve hours post adoptive transfer, recipient mice were infected with MHV A59.
Cell Isolation from Brain Tissues
Mice were sacrificed at the indicated time points and immediately perfused with PBS. CNS-infiltrating lymphocytes were isolated using mechanical disruption of the organ followed by enrichment based on 70%–30% Percoll gradients (GE Healthcare) and centrifugation for 25 min at 800 × g. For isolation of stromal cells from olfactory bulbs, the tissue was cut into small pieces, transferred into a 24-well dish filled with RPMI 1640 medium containing 2% FCS, 20 mM HEPES (all from Lonza), 1 mg/ml Collagenase Type P (Sigma-Aldrich), and 25 μg/ml DNaseI (AppliChem), and incubated at 37°C for 30 min. After enzymatic digestion, cell suspensions were washed with PBS containing 0.5% FCS and 10 mM EDTA (MACS buffer). To enrich the stromal cell fraction, we removed myelin-producing cells using 30% Percoll and 15 min centrifugation at 700 × g followed by hematopoietic cell depletion using MACS anti-CD45 and anti-Ter119 Microbeads (Miltenyi).
Statistical Analyses
All statistical analyses were performed with Prism 4.0 (GraphPad). Data were analyzed with the unpaired Student’s t test. A p value of < 0.05 was considered significant.
Author Contributions
J.C. and L.O. designed and conducted the experiments and wrote the paper; C.G.-C., E.W., S.C.-F., and C.P.-S. conducted experiments and analyzed data; T.R. generated transgenic mice; I.B. analyzed and discussed data; B.L. designed experiments, supervised the study, and wrote the paper.
Acknowledgments
We would like to thank Rita De Giuli and Manuela Schegg for technical support. This study was funded by grants from the Swiss MS Society (to B.L.), the Helmut Horten Foundation (to B.L.) and the Swiss National Science Foundation (146133, to B.L.) and by the Austrian GEN-AU III project Austromouse (to T.R.).
Published: February 23, 2016
Footnotes
Supplemental Information
Document S1. Figures S1–S5 and Supplemental Experimental Procedures
Document S2. Article plus Supplemental Information
References
- Alt C., Laschinger M., Engelhardt B. Functional expression of the lymphoid chemokines CCL19 (ELC) and CCL 21 (SLC) at the blood-brain barrier suggests their involvement in G-protein-dependent lymphocyte recruitment into the central nervous system during experimental autoimmune encephalomyelitis. Eur. J. Immunol. 2002;32:2133–2144. doi: 10.1002/1521-4141(200208)32:8<2133::AID-IMMU2133>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Aspelund A., Antila S., Proulx S.T., Karlsen T.V., Karaman S., Detmar M., Wiig H., Alitalo K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 2015;212:991–999. doi: 10.1084/jem.20142290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagai R., Valujskikh A., Canaday D.H., Bailey E., Lalli P.N., Harding C.V., Heeger P.S. Mouse endothelial cells cross-present lymphocyte-derived antigen on class I MHC via a TAP1- and proteasome-dependent pathway. J. Immunol. 2005;174:7711–7715. doi: 10.4049/jimmunol.174.12.7711. [DOI] [PubMed] [Google Scholar]
- Bajénoff M., Egen J.G., Koo L.Y., Laugier J.P., Brau F., Glaichenhaus N., Germain R.N. Stromal cell networks regulate lymphocyte entry, migration, and territoriality in lymph nodes. Immunity. 2006;25:989–1001. doi: 10.1016/j.immuni.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann C.C., Lane T.E., Stohlman S.A. Coronavirus infection of the central nervous system: host-virus stand-off. Nat. Rev. Microbiol. 2006;4:121–132. doi: 10.1038/nrmicro1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosè F., Petti L., Diani M., Moscheni C., Molteni S., Altomare A., Rossi R.L., Talarico D., Fontana R., Russo V. Inhibition of CCR7/CCL19 axis in lesional skin is a critical event for clinical remission induced by TNF blockade in patients with psoriasis. Am. J. Pathol. 2013;183:413–421. doi: 10.1016/j.ajpath.2013.04.021. [DOI] [PubMed] [Google Scholar]
- Browning J.L., Allaire N., Ngam-Ek A., Notidis E., Hunt J., Perrin S., Fava R.A. Lymphotoxin-beta receptor signaling is required for the homeostatic control of HEV differentiation and function. Immunity. 2005;23:539–550. doi: 10.1016/j.immuni.2005.10.002. [DOI] [PubMed] [Google Scholar]
- Buch T., Heppner F.L., Tertilt C., Heinen T.J., Kremer M., Wunderlich F.T., Jung S., Waisman A. A Cre-inducible diphtheria toxin receptor mediates cell lineage ablation after toxin administration. Nat. Methods. 2005;2:419–426. doi: 10.1038/nmeth762. [DOI] [PubMed] [Google Scholar]
- Burke M.L., McManus D.P., Ramm G.A., Duke M., Li Y., Jones M.K., Gobert G.N. Co-ordinated gene expression in the liver and spleen during Schistosoma japonicum infection regulates cell migration. PLoS Negl. Trop. Dis. 2010;4:e686. doi: 10.1371/journal.pntd.0000686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Q., Onder L., Scandella E., Gil-Cruz C., Perez-Shibayama C., Cupovic J., Danuser R., Sparwasser T., Luther S.A., Thiel V. Maturation of lymph node fibroblastic reticular cells from myofibroblastic precursors is critical for antiviral immunity. Immunity. 2013;38:1013–1024. doi: 10.1016/j.immuni.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson K.W., 2nd, Hood A.F., Travers J.B., Ramsey H., Hromas R.A. Endothelial induction of the T-cell chemokine CCL21 in T-cell autoimmune diseases. Blood. 2003;101:801–806. doi: 10.1182/blood-2002-05-1586. [DOI] [PubMed] [Google Scholar]
- Columba-Cabezas S., Serafini B., Ambrosini E., Aloisi F. Lymphoid chemokines CCL19 and CCL21 are expressed in the central nervous system during experimental autoimmune encephalomyelitis: implications for the maintenance of chronic neuroinflammation. Brain Pathol. 2003;13:38–51. doi: 10.1111/j.1750-3639.2003.tb00005.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comerford I., Harata-Lee Y., Bunting M.D., Gregor C., Kara E.E., McColl S.R. A myriad of functions and complex regulation of the CCR7/CCL19/CCL21 chemokine axis in the adaptive immune system. Cytokine Growth Factor Rev. 2013;24:269–283. doi: 10.1016/j.cytogfr.2013.03.001. [DOI] [PubMed] [Google Scholar]
- Cremasco V., Woodruff M.C., Onder L., Cupovic J., Nieves-Bonilla J.M., Schildberg F.A., Chang J., Cremasco F., Harvey C.J., Wucherpfennig K. B cell homeostasis and follicle confines are governed by fibroblastic reticular cells. Nat. Immunol. 2014;15:973–981. doi: 10.1038/ni.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt B. Molecular mechanisms involved in T cell migration across the blood-brain barrier. J Neural Transm (Vienna) 2006;113:477–485. doi: 10.1007/s00702-005-0409-y. [DOI] [PubMed] [Google Scholar]
- Förster R., Schubel A., Breitfeld D., Kremmer E., Renner-Müller I., Wolf E., Lipp M. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99:23–33. doi: 10.1016/s0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- Förster R., Davalos-Misslitz A.C., Rot A. CCR7 and its ligands: balancing immunity and tolerance. Nat. Rev. Immunol. 2008;8:362–371. doi: 10.1038/nri2297. [DOI] [PubMed] [Google Scholar]
- Galea I., Bechmann I., Perry V.H. What is immune privilege (not)? Trends Immunol. 2007;28:12–18. doi: 10.1016/j.it.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Gallagher T.M., Buchmeier M.J. Coronavirus spike proteins in viral entry and pathogenesis. Virology. 2001;279:371–374. doi: 10.1006/viro.2000.0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoshal A., Das S., Ghosh S., Mishra M.K., Sharma V., Koli P., Sen E., Basu A. Proinflammatory mediators released by activated microglia induces neuronal death in Japanese encephalitis. Glia. 2007;55:483–496. doi: 10.1002/glia.20474. [DOI] [PubMed] [Google Scholar]
- Gil-Cruz C., Perez-Shibayama C., Firner S., Waisman A., Bechmann I., Thiel V., Cervantes-Barragan L., Ludewig B. T helper cell- and CD40-dependent germline IgM prevents chronic virus-induced demyelinating disease. Proc. Natl. Acad. Sci. USA. 2012;109:1233–1238. doi: 10.1073/pnas.1115154109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greter M., Heppner F.L., Lemos M.P., Odermatt B.M., Goebels N., Laufer T., Noelle R.J., Becher B. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat. Med. 2005;11:328–334. doi: 10.1038/nm1197. [DOI] [PubMed] [Google Scholar]
- Gunn M.D., Tangemann K., Tam C., Cyster J.G., Rosen S.D., Williams L.T. A chemokine expressed in lymphoid high endothelial venules promotes the adhesion and chemotaxis of naive T lymphocytes. Proc. Natl. Acad. Sci. USA. 1998;95:258–263. doi: 10.1073/pnas.95.1.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn M.D., Kyuwa S., Tam C., Kakiuchi T., Matsuzawa A., Williams L.T., Nakano H. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J. Exp. Med. 1999;189:451–460. doi: 10.1084/jem.189.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosking M.P., Lane T.E. The role of chemokines during viral infection of the CNS. PLoS Pathog. 2010;6:e1000937. doi: 10.1371/journal.ppat.1000937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junt T., Nakano H., Dumrese T., Kakiuchi T., Odermatt B., Zinkernagel R.M., Hengartner H., Ludewig B. Antiviral immune responses in the absence of organized lymphoid T cell zones in plt/plt mice. J. Immunol. 2002;168:6032–6040. doi: 10.4049/jimmunol.168.12.6032. [DOI] [PubMed] [Google Scholar]
- Junt T., Scandella E., Förster R., Krebs P., Krautwald S., Lipp M., Hengartner H., Ludewig B. Impact of CCR7 on priming and distribution of antiviral effector and memory CTL. J. Immunol. 2004;173:6684–6693. doi: 10.4049/jimmunol.173.11.6684. [DOI] [PubMed] [Google Scholar]
- Junt T., Scandella E., Ludewig B. Form follows function: lymphoid tissue microarchitecture in antimicrobial immune defence. Nat. Rev. Immunol. 2008;8:764–775. doi: 10.1038/nri2414. [DOI] [PubMed] [Google Scholar]
- Kim J.V., Kang S.S., Dustin M.L., McGavern D.B. Myelomonocytic cell recruitment causes fatal CNS vascular injury during acute viral meningitis. Nature. 2009;457:191–195. doi: 10.1038/nature07591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivisäkk P., Mahad D.J., Callahan M.K., Sikora K., Trebst C., Tucky B., Wujek J., Ravid R., Staugaitis S.M., Lassmann H., Ransohoff R.M. Expression of CCR7 in multiple sclerosis: implications for CNS immunity. Ann. Neurol. 2004;55:627–638. doi: 10.1002/ana.20049. [DOI] [PubMed] [Google Scholar]
- Krumbholz M., Theil D., Steinmeyer F., Cepok S., Hemmer B., Hofbauer M., Farina C., Derfuss T., Junker A., Arzberger T. CCL19 is constitutively expressed in the CNS, up-regulated in neuroinflammation, active and also inactive multiple sclerosis lesions. J. Neuroimmunol. 2007;190:72–79. doi: 10.1016/j.jneuroim.2007.07.024. [DOI] [PubMed] [Google Scholar]
- Link A., Vogt T.K., Favre S., Britschgi M.R., Acha-Orbea H., Hinz B., Cyster J.G., Luther S.A. Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nat. Immunol. 2007;8:1255–1265. doi: 10.1038/ni1513. [DOI] [PubMed] [Google Scholar]
- Lo J.C., Chin R.K., Lee Y., Kang H.S., Wang Y., Weinstock J.V., Banks T., Ware C.F., Franzoso G., Fu Y.X. Differential regulation of CCL21 in lymphoid/nonlymphoid tissues for effectively attracting T cells to peripheral tissues. J. Clin. Invest. 2003;112:1495–1505. doi: 10.1172/JCI19188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louveau A., Smirnov I., Keyes T.J., Eccles J.D., Rouhani S.J., Peske J.D., Derecki N.C., Castle D., Mandell J.W., Lee K.S. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523:337–341. doi: 10.1038/nature14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra D., Fletcher A.L., Astarita J., Lukacs-Kornek V., Tayalia P., Gonzalez S.F., Elpek K.G., Chang S.K., Knoblich K., Hemler M.E., Immunological Genome Project Consortium Transcriptional profiling of stroma from inflamed and resting lymph nodes defines immunological hallmarks. Nat. Immunol. 2012;13:499–510. doi: 10.1038/ni.2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mionnet C., Sanos S.L., Mondor I., Jorquera A., Laugier J.P., Germain R.N., Bajénoff M. High endothelial venules as traffic control points maintaining lymphocyte population homeostasis in lymph nodes. Blood. 2011;118:6115–6122. doi: 10.1182/blood-2011-07-367409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moseman E.A., McGavern D.B. The great balancing act: regulation and fate of antiviral T-cell interactions. Immunol. Rev. 2013;255:110–124. doi: 10.1111/imr.12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano H., Gunn M.D. Gene duplications at the chemokine locus on mouse chromosome 4: multiple strain-specific haplotypes and the deletion of secondary lymphoid-organ chemokine and EBI-1 ligand chemokine genes in the plt mutation. J. Immunol. 2001;166:361–369. doi: 10.4049/jimmunol.166.1.361. [DOI] [PubMed] [Google Scholar]
- Nindl V., Maier R., Ratering D., De Giuli R., Züst R., Thiel V., Scandella E., Di Padova F., Kopf M., Rudin M. Cooperation of Th1 and Th17 cells determines transition from autoimmune myocarditis to dilated cardiomyopathy. Eur. J. Immunol. 2012;42:2311–2321. doi: 10.1002/eji.201142209. [DOI] [PubMed] [Google Scholar]
- Noor S., Habashy A.S., Nance J.P., Clark R.T., Nemati K., Carson M.J., Wilson E.H. CCR7-dependent immunity during acute Toxoplasma gondii infection. Infect. Immun. 2010;78:2257–2263. doi: 10.1128/IAI.01314-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onder L., Danuser R., Scandella E., Firner S., Chai Q., Hehlgans T., Stein J.V., Ludewig B. Endothelial cell-specific lymphotoxin-β receptor signaling is critical for lymph node and high endothelial venule formation. J. Exp. Med. 2013;210:465–473. doi: 10.1084/jem.20121462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pashenkov M., Söderström M., Link H. Secondary lymphoid organ chemokines are elevated in the cerebrospinal fluid during central nervous system inflammation. J. Neuroimmunol. 2003;135:154–160. doi: 10.1016/s0165-5728(02)00441-1. [DOI] [PubMed] [Google Scholar]
- Perlman S., Evans G., Afifi A. Effect of olfactory bulb ablation on spread of a neurotropic coronavirus into the mouse brain. J. Exp. Med. 1990;172:1127–1132. doi: 10.1084/jem.172.4.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitzalis C., Jones G.W., Bombardieri M., Jones S.A. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat. Rev. Immunol. 2014;14:447–462. doi: 10.1038/nri3700. [DOI] [PubMed] [Google Scholar]
- Randall T.D., Carragher D.M., Rangel-Moreno J. Development of secondary lymphoid organs. Annu. Rev. Immunol. 2008;26:627–650. doi: 10.1146/annurev.immunol.26.021607.090257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangel-Moreno J., Moyron-Quiroz J.E., Hartson L., Kusser K., Randall T.D. Pulmonary expression of CXC chemokine ligand 13, CC chemokine ligand 19, and CC chemokine ligand 21 is essential for local immunity to influenza. Proc. Natl. Acad. Sci. USA. 2007;104:10577–10582. doi: 10.1073/pnas.0700591104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff R.M., Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol. 2012;12:623–635. doi: 10.1038/nri3265. [DOI] [PubMed] [Google Scholar]
- Scandella E., Bolinger B., Lattmann E., Miller S., Favre S., Littman D.R., Finke D., Luther S.A., Junt T., Ludewig B. Restoration of lymphoid organ integrity through the interaction of lymphoid tissue-inducer cells with stroma of the T cell zone. Nat. Immunol. 2008;9:667–675. doi: 10.1038/ni.1605. [DOI] [PubMed] [Google Scholar]
- Wilson E.H., Harris T.H., Mrass P., John B., Tait E.D., Wu G.F., Pepper M., Wherry E.J., Dzierzinski F., Roos D. Behavior of parasite-specific effector CD8+ T cells in the brain and visualization of a kinesis-associated system of reticular fibers. Immunity. 2009;30:300–311. doi: 10.1016/j.immuni.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. Figures S1–S5 and Supplemental Experimental Procedures
Document S2. Article plus Supplemental Information